

Abstract
A 72-year-old woman presented with gradually-worsening myalgia and muscle weakness of the proximal lower limbs as well as elevated serum creatine kinase level. Based on a clinicoseropathological examination including a muscle biopsy, she was diagnosed with anti-signal recognition particle (SRP) myopathy. Although the myopathy relapsed two times in two years under oral prednisolone and intravenous immunoglobulin therapy, the myopathy remained in remission for more than three years after resection of gastric cancer. Although the anti-SRP myopathy is not considered to be cancer-associated in general, we should note that some cases of anti-SRP myopathy may be ameliorated with appropriate cancer treatment.
Keywords: anti-signal recognition particle myopathy; gastric cancer; anti-glutamic acid decarboxylase antibody; intravenous immunoglobulin, muscle biopsy; MRI
Introduction
Idiopathic inflammatory myopathies (IIMs), also known as autoimmune myositis, are a heterogeneous group of potentially treatable myopathies. Initially, only dermatomyositis and polymyositis were classified as inflammatory myopathies, with a myositis with a characteristic skin rash considered to be dermatomyositis (1). At present, the accumulation of clinicopathological evidence in addition to the discovery of muscle-specific antibodies suggestively classifies IIMs into four major subgroups: dermatomyositis, inclusion-body myositis, immune-mediated necrotizing myopathy (IMNM), and antisynthetase syndrome (2). Under the latest criteria, most of the previously diagnosed polymyositis cases have been revised to IMNM and antisynthetase syndrome, and the existence of polymyositis has been deemed questionable (3).
Identification and characterization of anti-signal recognition particle (SRP) and anti-3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) antibodies allowed further classification of IMNM: anti-SRP myopathy, anti-HMGCR myopathy, and seronegative IMNM (4). IMNM is clinically characterized by elevated serum creatine kinase (CK) levels and proximal symmetrical muscle weakness and pathologically characterized by a large number of necrotic fibers with minimal inflammatory infiltrates (5).
Recently, the differences in clinical features and backgrounds between anti-SRP and anti-HMGCR myopathies have been intensely investigated. We previously reported that anti-SRP myopathy was associated with more severe muscle weakness and a poorer response to immunotherapy than anti-HMGCR myopathy (6). Furthermore, it is generally considered that anti-HMGCR myopathy and seronegative IMNM show a higher incidence of cancer than in the general population. However, because anti-SRP myopathy is not associated with an increased risk for cancer, cancer screening is unnecessary in patients with anti-SRP myopathy (4,7,8).
We herein report a 72-year-old woman with anti-SRP myopathy who remained in remission for more than 3 years after resection of gastric cancer with successful tapering of prednisolone (PSL). We believe that this case suggests that anti-SRP myopathy can be ameliorated with appropriate treatment of synchronous cancer.
Case Report
A 72-year-old Japanese woman with a history of hypertension and hyperlipidemia who had been taking medication, including statins, noted gradually worsening myalgia and muscle weakness of the bilateral proximal lower limbs that had started from 69 years old. She was referred to our hospital in October 2015. She did not suffer from dyspnea or dysphagia. At her first visit, a red skin rash resembling V neck sign was observed on the anterior chest. Her respiratory sounds were normal. She did not show edema in her face or extremities. Her Medical Research Council (MRC) grade was 4/5 power in the neck flexor and symmetrical upper and lower proximal limbs. Blood tests revealed elevated CK levels at 1,314 U/L. Anti-SRP antibody was positive, and anti-nuclear, HMGCR, mitochondrial M2, aminoacyl tRNA synthetase, SS-A, SS-B, Scl-70, Jo-1, cyclic citrullinated peptide, and double-stranded DNA antibodies were all negative. Her respiratory function was normal.
An electrocardiogram revealed premature ventricular contraction. Plain computed tomography (CT) of the chest and abdomen showed normal findings. Magnetic resonance imaging (MRI) showed a high intensity of the bilateral quadriceps femoris muscles on diffusion-weighted and short tau inversion recovery imaging and enhancement of the same muscle on T1-weighted imaging after gadolinium administration (Fig. 1). Electromyogram in the right biceps brachii and vastus lateralis muscles showed many fibrillation potentials and positive sharp waves at rest as well as low amplitude motor unit potentials with early recruitment at the maximum contraction. A muscle biopsy from the right rectus femoris muscle showed several necrotic and many regenerating fibers without lymphocyte infiltration or perifascicular atrophy.
Figure 1.
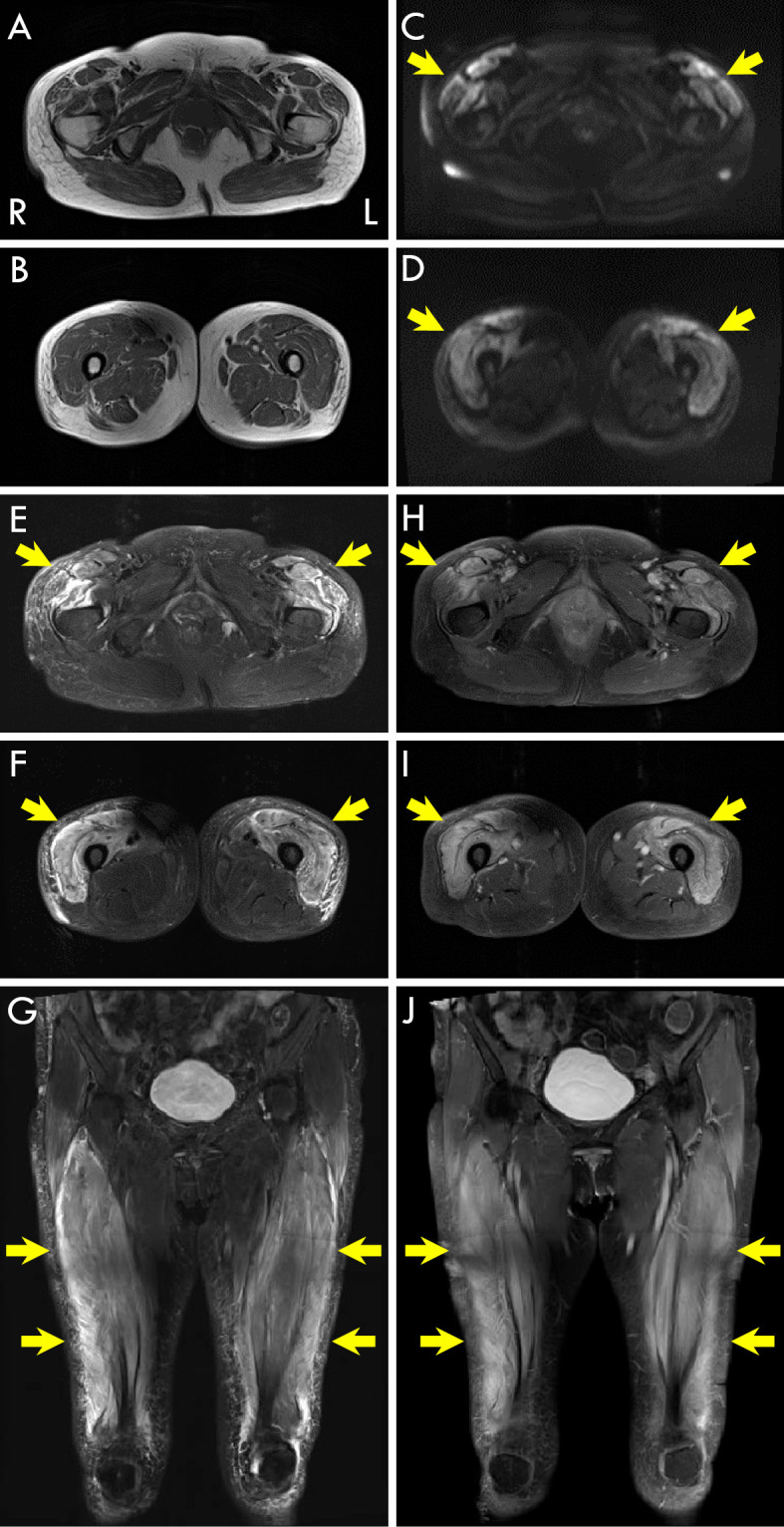
3T magnetic resonance imaging (MRI) of the pelvic girdle and the distal thigh level. Axial T1-weighted MRI of the pelvic girdle (A) and the distal thigh level (B). The bilateral quadriceps femoris muscles show hyperintensity (arrows) on diffusion-weighted imaging (C and D: DWI) and short tau inversion recovery imaging (E-G: STIR) of the pelvic girdle (C and E) and the distal thigh level (D and F). (G) Coronal STIR also showed the hyperintensity of the bilateral quadriceps femoris muscles (arrows). Axial T1-weighted MRI with gadolinium contrast (Gd) showed enhancement (arrows) in the bilateral quadriceps femoris muscles of the pelvic girdle (H) and the distal thigh level (I). (J) Coronal T1-weighted MRI with gadolinium contrast also showed the enhancement of the bilateral quadriceps femoris muscles (arrows).
On immunohistochemistry, major histocompatibility complex (MHC)-I was mildly expressed on the sarcolemma, and membrane attack complex (MAC) was deposited on the sarcolemma of the scattered fibers, findings consistent with IMNM (Fig. 2). The muscle specimen did not show perifascicular atrophy or sarcoplasmic immunoreactivity for myxovirus resistance protein A (MxA), both typical findings for dermatomyositis. Just before starting oral PSL at 65 mg [approximately 1 mg/kg body weight (BW)] daily in November 2015, the MRC grade in the lower proximal limbs had decreased to 2/5, and the serum CK level was the highest throughout the whole clinical course (4,046 U/L).
Figure 2.

Histopathology of the biopsied right rectus femoris muscle. Fiber sizes ranged from 10 to 70 μm, and several necrotic and many regenerating fibers were present (arrows). Lymphocyte infiltration or perifascicular atrophy was not seen. Fibers with internal nuclei (arrowheads) were identified at a rate of 6% (A: Hematoxylin and Eosin staining). No ragged-red fibers, rimmed vacuoles, or cytoplasmic bodies were present [B: modified Gomori trichrome (mGT) stain]. Intermyofibrillar networks were well-organized, except for necrotic and regenerating fibers [C: nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR) stain]. Type 1, 2A, 2B, and 2C fibers (arrow) accounted for 25%, 35%, 37%, and 3%, respectively (D: ATPase stain). No glycogen storage was found [E: periodic acid-Schiff (PAS) stain]. Enzymatic activity was observed in a few regenerating fibers (arrows) and perimysium [F: alkaline phosphatase (ALP) stain]. Diffuse major histocompatibility complex (MHC)-I expression on the sarcolemma was observed (G). Membrane attack complex (MAC) deposition on the sarcolemma was observed (H). Scale bar=100 μm in A-F and 50 μm in G and H.
Myalgia soon disappeared and muscle weakness in the lower proximal limbs gradually recovered to 4/5 power in September 2016 when the tapered PSL was at 4 mg. In October 2016, myalgia and muscle weakness in the lower proximal limbs recurred (MRC grade: 2/5; CK 3,517 U/L). Oral PSL was increased to 20 mg (approximately 0.3 mg/kg BW), and intravenous immunoglobulin (IVIg, 400 mg/kg BW, 5 days per month) was combined because the PSL had caused psilosis and BW gain. Myalgia and muscle weakness did not completely remit this time (MRC grade: 2-3/5; CK 206-539 U/L). In May 2017, oral tacrolimus was added but then stopped three months later due to renal injury. In November 2017, myalgia and muscle weakness again worsened (MRC grade: 2/5; CK 1,754 U/L) when the tapered PSL was at 4 mg, so the PSL was increased again to 20 mg.
A few days later, nausea and epigastralgia appeared after meal and continued for several days. Upper gastrointestinal fiberscopy revealed gastric cancer at the greater curvature in the antral zone. In January 2018, laparoscopic partial gastrectomy was performed, which revealed metastasis of the dissected lymph node. The pathology of the gastric specimen showed poorly differentiated adenocarcinoma of the antral zone without invasion into the muscularis propria vertically or the resected edges laterally (Fig. 3A, B). Because the anti-glutamate decarboxylase 65 (GAD65) antibody had turned positive eight months before the gastric cancer was found in November 2017, we further performed immunostaining for GAD65 of the resected stomach, which revealed the expression of GAD65 in both the cancer and normal region, such as the cancer mucosa, cancer submucosa, muscularis propria, and adjacent normal mucosa (Fig. 3C-G). Because the myopathy relapsed under IVIg therapy, IVIg therapy was not added after gastrectomy.
Figure 3.

(A) Partially resected stomach of this case. Adenocarcinoma (size: 41×27 mm) was identified in the greater curvature of the antral zone (Or: oral side of the resected edge, An: anal side of the resected edge). (B-F) Representative histopathological images of the section that was made on the red line in the panel A. (B) Cancer tissue was identified within the two-way arrow, and the lesion did not invade the muscularis propria (Hematoxylin and Eosin staining). (C-F) Immunohistochemistry for glutamate decarboxylase 65 (GAD65) showed reactivity in cancer mucosa (D), cancer submucosa (E), and muscularis propria (F). (G) Adjacent normal mucosa also showed immunoreactivity for GAD65.
Oral PSL and azathioprine combination were administered after gastrectomy, and myopathy remained in remission for 3 years after the PSL was tapered to 1 mg every other day, with sequelae of MRC grade 4/5 in the lower proximal limbs without myalgia (Fig. 4). The red skin rash resembling V neck sign on her chest changed into pigmentation over the course of treatment. No evidence of relapse of the gastric cancer was found during the same follow-up period.
Figure 4.

Clinical course after visiting our hospital. CEA: carcinoembryonic antigen, CK: creatine kinase, Cre: creatinine, GAD: glutamate decarboxylase, PSL: prednisolone
Discussion
In this case, although the first relapse of anti-SRP myopathy was treated with 20 mg of oral PSL plus IVIg, which did not lead to complete remission, the second relapse remained in remission for more than three years after resection of gastric cancer with successful PSL tapering to 1 mg every other day. Gastric cancer was found due to the appearance of epigastralgia after the re-increase of oral PSL at the second relapse.
Because our case showed a V-neck-sign-like rash and long-term remission of myopathy with little immunotherapy after cancer resection, many may feel that this case should be classified under the historical criteria of dermatomyositis. However, the important point is that the present pathological criteria for dermatomyositis include perifascicular atrophy and sarcoplasmic immunoreactivity for MxA, a surrogate marker for type 1 interferon pathway activation (3,9), both of which were negative in this patient. Sarcoplasmic expression of MxA has been shown to be a highly sensitive and specific marker for dermatomyositis (10,11). In contrast, IMNM is not associated with the type 1 or type 2 interferon signature. Pathologically, active myofiber necrotic and regenerating processes with minimal inflammatory infiltrates, key findings of IMNM, were seen in this case (12). In addition, the myofiber expression of MHC-I and sarcolemmal deposition of MAC, which are believed to be pathognomonic of IMNM (13), were also observed in this case (Fig. 2G, H). Recently, purified IgG from the plasma of anti-SRP or anti-HMGCR autoantibody-positive IMNM patients was reported to reproduce the muscle weakness and myofiber necrosis and regeneration in mice, which suggested that humoral mechanisms could be key players in pathophysiology of IMNM (14). Because the muscle pathology gives us insights into the pathophysiology and clues about the development of novel therapies for IIMs, we emphasize the importance of making an accurate diagnosis based on a muscle biopsy.
Although the histopathological features of anti-SRP myopathy are similar to those of anti-HMGCR myopathy, the backgrounds, risk factors, and clinical features are different. We previously reported that human leukocyte antigen (HLA)-DRB1*08:03 was associated with anti-SRP myopathy and HLA-DRB1*11:01 with anti-HMGCR myopathy in Japanese populations (15). In Caucasians, although a specific HLA haplotype was not shown to be associated with anti-SRP myopathy, HLA-DRB1*11 was associated with anti-HMGCR myopathy (16). Although the significance has not been fully elucidated, statin exposure is often seen in anti-HMGCR myopathy, at rates ranging from 15% to 65% (17,18). At present, statin exposure is considered less relevant to anti-SRP myopathy than to anti-HMGCR myopathy. Although both seropositive IMNMs show severe muscle weakness and poor recovery, we previously reported that anti-SRP myopathy showed more prominent weakness than anti-HMGCR myopathy (6).
Malignancy is an important comorbidity associated with a poor life prognosis with IIM. Concerning IMNM, seronegative and anti-HMGCR-positive patients have been shown to have a higher incidence of cancers than the general population (seronegative: 29%; anti-HMGCR 17%) (7,19). However, anti-SRP myopathy patients have not been shown to have an increased risk of synchronous cancer (8.1%) (5,7). In those studies, malignancy that occurred within three years, either before or after, of the diagnosis of myopathy was counted when calculating the standardized incidence ratio, based on the commonly used criteria of cancer-associated myositis. No specific type of cancer has been shown to be relevant to IMNMs. Yamaguchi et al. reported a case of anti-HMGCR myopathy wherein remission was not induced by resection of gastric cancer, even after three weeks, but was later induced by combination therapy of PSL and IVIg for more than three and half years (20). This case of anti-HMGCR myopathy also suggests the possibility that some cases of IMNM may have a causative association between their pathophysiology and malignancy.
In our case, the patient was treated with single therapy of high-dose PSL (1 mg/kg BW) at first but with low-dose PSL (0.3 mg/kg BW) plus IVIg therapy after the first relapse due to the psilosis and gain of BW. Although low-dose PSL plus IVIg did not lead to complete remission after the first relapse, resection of the gastric cancer after the second relapse led to immediate and complete remission under low-dose PSL single therapy for more than three years (Fig. 4). We added azathioprine to safely taper PSL four months after gastrectomy. Although this is a single case, the clinical course suggests that appropriate cancer therapy can ameliorate the condition of anti-SRP myopathy, which is associated with severe muscle weakness and a poor response to immunotherapy, and further suggests that we should perform a malignancy survey on patients with anti-SRP myopathy as well as anti-HMGCR myopathy and seronegative IMNM.
In the field of neurology, anti-GAD65 antibody is an established biomarker for stiff-person spectrum disorders, epilepsy, cerebellar ataxia, and limbic encephalitis (21). Anti-GAD65 antibody in this case turned positive eight months before the detection of gastric cancer and then again turned negative after gastrectomy. However, from another perspective, the period while the patient was anti-GAD65 antibody-positive overlapped with the period of IVIg treatment. Recently, 16 commercially available samples of IVIg preparations were tested, and 15 of them were positive for anti-GAD antibody in an enzyme-linked immunosorbent assay (ELISA) while none were positive for anti-GAD in cell-based assays (22). Because our patient was positive for anti-GAD antibody on an ELISA, care should be taken to avoid misunderstanding the pathophysiology under IVIg therapy. Although we detected immunoreactivity for GAD65 in both the cancer lesion and normal gastric tissues (Fig. 3), this is less likely to be relevant to the pathophysiology of the myopathy, as the anti-GAD65 antibody in this case turned positive several years after the onset of anti-SRP myopathy and showed a low titer even at its peak. We should note that anti-GAD antibody can turn positive under the use of IVIg preparations in patients.
In conclusion, the classification of IIMs has drastically changed with the accumulation of clinicoseropathological evidence. The accurate classification by a muscle biopsy remains important for the prediction of the prognosis and choice of treatment in such cases. Although it has been established that anti-SRP myopathy does not increase the risk of synchronous cancer, our case suggests that appropriate cancer treatment can ameliorate the condition of myopathy.
The authors state that they have no Conflict of Interest (COI).
Financial Support
The histopathological analysis of the muscle was supported by Intramural Research Grant (2-5, 29-4) for Neurological and Psychiatric Disorders of NCNP (IN).
Acknowledgement
The authors are grateful for the care provided by all those involved in the patient. The authors are also grateful for the pathological diagnosis provided by Taichiro Yoshimoto and Mio Sakaguchi and the technical assistance provided by Maho Fujii, at the Department of Pathology, Haga Red Cross Hospital.
References
- 1.Dalakas MC. Inflammatory muscle diseases. N Engl J Med 372: 1734-1747, 2015. [DOI] [PubMed] [Google Scholar]
- 2.Mariampillai K, Granger B, Amelin D, et al. Development of a new classification system for idiopathic inflammatory myopathies based on clinical manifestations and myositis-specific autoantibodies. JAMA Neurol 75: 1528-1537, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanboon J, Nishino I. Classification of idiopathic inflammatory myopathies: pathology perspectives. Curr Opin Neurol 32: 704-714, 2019. [DOI] [PubMed] [Google Scholar]
- 4.Tanboon J, Uruha A, Stenzel W, Nishino I. Where are we moving in the classification of idiopathic inflammatory myopathies? Curr Opin Neurol 33: 590-603, 2020. [DOI] [PubMed] [Google Scholar]
- 5.Allenbach Y, Benveniste O, Stenzel W, Boyer O. Immune-mediated necrotizing myopathy: clinical features and pathogenesis. Nat Rev Rheumatol 16: 689-701, 2020. [DOI] [PubMed] [Google Scholar]
- 6.Watanabe Y, Uruha A, Suzuki S, et al. Clinical features and prognosis in anti-SRP and anti-HMGCR necrotising myopathy. J Neurol Neurosurg Psychiatry 87: 1038-1044, 2016. [DOI] [PubMed] [Google Scholar]
- 7.Allenbach Y, Keraen J, Bouvier AM, et al. High risk of cancer in autoimmune necrotizing myopathies: usefulness of myositis specific antibody. Brain 139: 2131-2135, 2016. [DOI] [PubMed] [Google Scholar]
- 8.Anquetil C, Boyer O, Wesner N, Benveniste O, Allenbach Y. Myositis-specific autoantibodies, a cornerstone in immune-mediated necrotizing myopathy. Autoimmun Rev 18: 223-230, 2019. [DOI] [PubMed] [Google Scholar]
- 9.Allenbach Y, Mammen AL, Benveniste O, Stenzel W. 224th ENMC International Workshop: clinico-sero-pathological classification of immune-mediated necrotizing myopathies Zandvoort, The Netherlands, 14-16 October 2016. Neuromuscul Disord 28: 87-99, 2018. [DOI] [PubMed] [Google Scholar]
- 10.Uruha A, Nishikawa A, Tsuburaya RS, et al. Sarcoplasmic MxA expression: a valuable marker of dermatomyositis. Neurology 88: 493-500, 2017. [DOI] [PubMed] [Google Scholar]
- 11.Uruha A, Allenbach Y, Charuel JL, et al. Diagnostic potential of sarcoplasmic myxovirus resistance protein A expression in subsets of dermatomyositis. Neuropathol Appl Neurobiol 45: 513-522, 2019. [DOI] [PubMed] [Google Scholar]
- 12.Hoogendijk JE, Amato AA, Lecky BR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10-12 October 2003, Naarden, The Netherlands. Neuromuscul Disord 14: 337-345, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Allenbach Y, Arouche-Delaperche L, Preusse C, et al. Necrosis in anti-SRP+ and anti-HMGCR+ myopathies: role of autoantibodies and complement. Neurology 90: e507-e517, 2018. [DOI] [PubMed] [Google Scholar]
- 14.Bergua C, Chiavelli H, Allenbach Y, et al. In vivo pathogenicity of IgG from patients with anti-SRP or anti-HMGCR autoantibodies in immune-mediated necrotising myopathy. Ann Rheum Dis 78: 131-139, 2019. [DOI] [PubMed] [Google Scholar]
- 15.Ohnuki Y, Suzuki S, Shiina T, et al. HLA-DRB1 alleles in immune-mediated necrotizing myopathy. Neurology 87: 1954-1955, 2016. [DOI] [PubMed] [Google Scholar]
- 16. Rothwell S, Chinoy H, Lamb JA, et al. ; Myositis Genetics Consortium (MYOGEN). Focused HLA analysis in Caucasians with myositis identifies significant associations with autoantibody subgroups. Ann Rheum Dis 78: 996-1002, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 63: 713-721, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ge Y, Lu X, Peng Q, Shu X, Wang G. Clinical characteristics of anti-3-hydroxy-3-methylglutaryl coenzyme A reductase antibodies in Chinese patients with idiopathic inflammatory myopathies. PLoS One 10: e0141616, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kadoya M, Hida A, Hashimoto Maeda M, et al. Cancer association as a risk factor for anti-HMGCR antibody-positive myopathy. Neurol Neuroimmunol Neuroinflamm 3: e290, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamaguchi T, Matsunaga A, Ikawa M, Shirafuji N, Nishino I, Hamano T. [A case of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibody-positive paraneoplastic necrotizing myopathy associated with advanced gastric cancer that responded to intravenous immunoglobulin therapy]. Rinsho Shinkeigaku (Clin Neurol) 57: 118-123, 2017. (in Japanese). [DOI] [PubMed] [Google Scholar]
- 21.Budhram A, Sechi E, Flanagan EP, et al. Clinical spectrum of high-titre GAD65 antibodies. J Neurol Neurosurg Psychiatry 92: 645-654, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dimitriadou MM, Alexopoulos H, Akrivou S, Gola E, Dalakas MC. Anti-neuronal antibodies within the IVIg preparations: importance in clinical practice. Neurotherapeutics 17: 235-242, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]