

Abstract
Accurate ring strain energies (RSEs) for three-membered symmetric inorganic rings El3 and organic dihetero-monocycles El2C and their silicon El2Si and germanium El2Ge analogues have been computed for group 14–16 “El” heteroatoms using appropriate homodesmotic reactions and calculated at the DLPNO-CCSD-(T)/def2-TZVPP//B3LYP-D4/def2-TZVP(ecp) level. Rings containing triels and Sn/Pb heteroatoms are studied as exceptions to the RSE calculation as they either do not constitute genuine rings or cannot use the general homodesmotic reaction scheme due to uncompensated interactions. Some remarkable concepts already related to the RSE such as aromaticity or strain relaxation by increasing the s-character in the lone pair (LP) of the group 15–16 elements are analyzed extensively. An appealing alternative procedure for the rapid estimation of RSEs using additive rules, based on contributions of ring atoms or endocyclic bonds, is disclosed.
Short abstract
Accurate ring strain energies (RSEs) are calculated for saturated three-membered homoatomic inorganic rings El3 and diheterotetreliranes El2Tt (Tt = C, Si, Ge). Aromaticity, lone-pair-induced strain relaxation for the group 15−16 elements, and bond stiffness are extensively discussed as main factors affecting the RSE. An attractive alternative procedure for fast estimation of the RSE using additive rules is proposed.
Introduction
Ring strain energy (RSE) constitutes a remarkable parameter assessing the instability of small rings whose atoms acquire unfavorable high-energy bond angles and/or bond distances. This parameter provides information about the energy necessary for ring-enlargement or ring-opening transformations and even affecting other properties such as the stereochemical stability of σ3λ3-pnictogen ring atoms.1 It is also a useful parameter to explain electronic properties and reactivity in organic and inorganic ring systems, such as combustion processes of cyclopropane and cyclobutane,2 ring-opening metathesis polymerization (ROMP) processes,3 key reactions such as the ring-opening polymerizations (ROP) undergone by aziridines4 and other strained small rings (e.g., phosphiranes),5 NMR chemical changes in norbornanes,6 or changes in the hapticity of cyclopentadienyl rings in metallocenophanes.7 Not surprisingly, many computational studies have focused on how the RSE varies with the count of the ring members, the type of atoms forming the ring, and the nature of the peripheral substituents.8
The attractiveness of three-membered rings (3MRs) has prompted extensive studies on their synthesis and characterization for both organoheterocycles9 and purely inorganic rings. Among inorganic rings, kinetically stabilized tritetreliranes (R2El)3 (El = Si,10 Ge,11 and Sn12) have been reported, including X-ray structures in some cases. Also, tripnictogeniranes (REl)3 such as the lighter triaziridines10j,13 (El = N) and their heavier congeners triphosphiranes10j,14 (El = P) and triarsiranes15 (El = As) were described. It is worth mentioning that the important open-chain molecule ozone O3 has a less stable (ΔΔE = 30.2 kcal/mol)16 cyclic allotrope (c-O3)10j although heavier trichalcogeniranes are, to the best of our knowledge, so far experimentally unreported. Three-membered heterocycles possessing two identical heteroatoms “El” in addition to one carbon atom, referred to as CEl2 for short, have prominent examples in dioxiranes17 and the related oxaziridines18 that can be considered as epoxy derivatives of carbonyl compounds and imines, respectively, and are widely used as oxidizing reagents. As for the heavier heteroatomic tritetreliranes Tt2Tt′ (Tt, Tt′ = Si, Ge, Sn), the thermolysis of siladigermirane (SiGe2),19 as well as the isolation of stable adducts derived from the disilagermirane (Si2Ge)20 and disilastannirane (Si2Sn) rings, was reported.21 Moreover, [1+2] cycloaddition of the heavier carbene analogues, silylene (R2Si:) and germylene (R2Ge:), to a phosphaalkene leading to phosphasilirane and phosphagermirane, respectively,22 as well as the synthesis and properties of diphosphasiliranes23 and phosphadisiliranes, was also described.24 Three-membered rings containing silicon introduce interesting perspectives in the development of new silicon-containing polymeric materials that could exhibit interesting properties, in line with the unique characteristics of the well-known silicones, with many different uses such as in oils, greases, rubber-like materials,25 electric insulators,26 hydraulic fluids, and membranes27 among others. Rivard et al.28 recently reviewed the small (three- and four-membered) inorganic rings (i.e., containing no C as ring atom) described to date, which open up a range of interesting possibilities with potential yet to be discovered. Noteworthy is that only 30 types of inorganic rings and 40 organic rings (containing at least one C atom) have been reported so far. Considering that there are only 20 different atoms among the main group elements (groups 13–16) from second to sixth rows, it is possible to obtain 1540 combinations with repetition of the different elements in three-membered rings. In other words, only around 4.5% of the rings have been reported so far, and not all possible applications have been exhausted for them. This is therefore an exciting field still to be explored. Moreover, RSE estimates were reported for only a very limited number of organoheterocycles. Recently, RSE data were published for the parent rings (CH2)2X, where X are group 13–16 elements (El) in their lowest oxidation state.29 The RSE for parent heterocycles C2O, C2S, C2N, C2P and disilaanalogues Si2El (El = C, N, O, Si, P, S),30 CPN,31 CPO,32 and CPS33 (using only ring atoms to refer to each ring type) and of inorganic ditetreliranes Si2El (El = Al, Si, P)34 and Ge2El (El = Ga, Ge, As)35 as well were reported. There is therefore an important gap with respect to the systematic evaluation of RSE for a wide range of small rings.
For this purpose, the RSE of a set of 54 3MRs are herein reported for symmetric (homocyclic) inorganic rings El3 (1), organoheterocycles with two identical heteroatoms CEl2 (2), and the heavier analogues SiEl2 (3) and GeEl2 (4), where El is an element belonging to groups 13–16 with its typical covalency (3, 4, 3, and 2 for groups 13, 14, 15, and 16, respectively) completed with bonds to hydrogen (i.e., parent rings) (Figure 1). Data corresponding to the already recently reported C2El rings29 (5) were also obtained to analyze possible trends in the factors affecting the ring strain.
Figure 1.

Three-membered heterocycles herein (1–4) and previously (5) studied. X is a group 13–16 element (El) with its typical covalency completed with bonds to H.
The interesting question of whether it is possible to determine how much strain each ring bond or atom contributes to the total RSE and if this can be calculated additively, is also addressed.
Results and Discussion
Accurate RSEs—Scope and Limitations
RSEs of all 3MRs herein investigated were calculated using suitable homodesmotic reactions (reaction class 4, or “RC4”). The latter are the second-best type in a hierarchy of increasingly accurate processes, due to the conservation of larger fragments, according to a recent classification and redefinition of the reaction types used in thermochemistry.36 The homodesmotic reactions used (Scheme 1) to evaluate RSEs were shown29 to be sufficiently accurate compared to the formally highest ranked, hyper-homodesmotic reactions, that have the disadvantage of being prone to unwanted interactions present in longer chains. The RSE was obtained as the average of the opposite of energetics (including zero-point energy correction) for the three endocyclic bond-cleavage reactions in each case, at the DLPNO-CCSD(T)/def2-TZVPP(ecp)//B3LYP-D4/def2-TZVP(ecp) level (see computational details) (Table 1).
Scheme 1. One of the Three Possible Homodesmotic (RC4) Ring-Opening Reactions Used for the Estimation of RSEs for 1–4El.

Table 1. Calculated (DLPNO-CCSD(T)/def2-TZVPP(ecp)) RSE Values (kcal/mol) for Compounds 1El, 2El, 3El, and 4El.
| 1Tl | 1.85a | 2Tl | 3Tl | 4Tl | |||
| 1C(10j,29,37) | 27.27 | 2C | 27.27 | 3C(24) | 36.32 | 4C(24) | 36.97 |
| 1Si(10j,37b,38) | 36.09 | 2Si(37b,38) | 39.32 | 3Si | 36.09 | 4Si | 37.09 |
| 1Ge(35) | 38.70 | 2Ge | 40.67 | 3Ge | 37.95 | 4Ge | 38.70 |
| 1Sn | 36.29a | 2Sn | 39.12 | 3Sn | 35.89 | 4Sn | 36.67a |
| 1Pb | 15.22b | 2Pb | 3Pb | 4Pb | |||
| 1N(10j) | 13.96 | 2N | 21.63 | 3N | 41.59 | 4N | 37.76 |
| 1P(10j) | 6.06 | 2P | 12.24 | 3P | 17.02 | 4P | 18.35 |
| 1As | 5.74 | 2As | 12.04 | 3As | 15.83 | 4As | 17.03 |
| 1Sb | 5.77 | 2Sb | 12.10 | 3Sb | 14.21 | 4Sb | 15.36 |
| 1Bi | 6.34 | 2Bi | 10.01 | 3Bi | 12.41 | 4Bi | 13.49 |
| 1O(10j) | 28.42 | 2O | 21.40 | 3O | 33.78 | 4O | 35.52 |
| 1S(10j) | 28.77 | 2S | 19.36 | 3S | 22.44 | 4S | 24.67 |
| 1Se | 25.71 | 2Se | 18.87 | 3Se | 20.33 | 4Se | 22.20 |
| 1Te | 26.52 | 2Te | 20.49 | 3Te | 19.86 | 4Te | 21.71 |
| 1Po | 24.98 | 2Po | 19.62 | 3Po | 18.55 | 4Po | 20.14 |
In case of the homocyclic inorganic rings, 1El, no energy minimum was found for group 13 elements. Instead, a variety of previously reported El3H3 hydride isomers were obtained (El = B,39 Al, Ga39,40), whose interest stems from their potential use as reversible hydrogen carrier devices at low and medium temperatures41 and formation of clusters.42 El3H3 hydrides for the heaviest triels In and Tl have not been previously reported and their calculated structures I–VIII are collected together with the corresponding relative energies (Figure S1). As they do not contain three identical ElH units (structure IVTl being an exception, vide infra), no appropriate homodesmotic reactions were found that could allow the estimation of their RSE (Scheme 1).
With regard to triel-containing dihetero-monocyclic species 2El, it is known that the incorporation of boron in small rings stabilizes the planar tetracoordination at carbon by π delocalization, in addition to less important σ donor effects.43 The diboracyclopropane (diborirane) 2BC2v conformer with tetrahedral C atom and its planar Cs conformer were reported to be second- and first-order saddle points, respectively, at the B3LYP/6-311+G* level. The energy minimum resulted to be a fully planar cyclic structure with a two-electron three-center B–H–B bond. However, among cyclic structures with the required connectivity (containing a CH2 group), the most stable one contains an asymmetric planar tetracoordinated carbon atom featuring different endocyclic C–B bond distances.44 The corresponding unreported heavier cyclic analogues with the appropriate cyclic −TrH–TrH–CH2– connectivity, for Tr = Al (2Al) and Ga (2Ga), show a distorted tetrahedral (i.e., nonplanar) geometry at C for the energy minima (I), and two other second-order saddle point cyclic structures with increasing energy for tetrahedral C2v (II) and planar Cs environments at C (III), respectively (Figure 2). It seems clear that, unlike B, the heavier triels do not favor planar tetrahedral geometries at the adjacent C atom. Not only in the abovementioned triel-containing 2El but also in 3El and 4El rings, the lack of undistorted cyclic minima precluded the design of suitable homodesmotic reactions considering these effects. Accordingly, these rings were excluded from the RSE calculations.
Figure 2.

Optimized structures for the distorted tetrahedral (I), tetrahedral C2v (II), and planar Cs (III) 2Al isomers, number of imaginary frequencies (NIMAG), and their respective relative ZPE-corrected energies (kcal/mol) for 2El (El: Al, Ga) isomers (values for E = Ga in parenthesis).
On the other hand, a triplumbirane, c-(PbR2)3 (R = 2,4,6-triethylphenyl), was isolated in 2003.45 The parent 1Pb species was shown to display a cyclic character, featuring elongated bond distances of ∼3.231 Å together with substituent twisting of ca. 50° outside ideal conditions (H–Pb–H plane bisecting the endocyclic bond angle) calculated at the HF/DZ(d) level. This reveals that the Pb–Pb bonding is characterized by donor–acceptor interactions and not arising from overlapping of identical atomic orbitals (AOs). Thus, the Pb–Pb bonds formally result from the interaction of plumbylene (R2Pb:) lone pairs with the empty p AO at P of one of the neighboring plumbylene units, thus forcing the plumbylene substituents to twist from their ideal (untwisted) position to maximize overlap (Figure 2b).46 At the working level of theory (B3LYP/def2-TZVPP(ecp)//B3LYP-D4/def2-TZVP(ecp)), the parent compound 1Pb presents a ring critical point (RCP) (Figure 3a) and the natural bond orbital47 (NBO) analysis reveals that each Pb–Pb bond is rather elongated and weak (dPb–Pb = 3.137 Å; WBIPb–Pb = 0.547) compared to related acyclic species (dPb–Pb = 2.895 Å; WBIPb–Pb = 0.854 for model H3Pb–PbH3). The endocyclic Pb–Pb linkage in 1Pb seems to be mostly formed as a donor–acceptor bond from a filled roughly sp2 AO at one Pb atom (Pb1) to an empty almost pure p orbital at the second Pb atom (Pb2), with remarkable SOPT (second-order perturbation theory) electron donation to the σ*(Pb1–Pb3) (ESOPT = 24.2 kcal/mol) and the two σ*(Pb1–H) molecular orbitals (MOs) (ESOPT = 29.4 kcal/mol each). The unsymmetrically located bond critical point (BCP), closer to the acceptor Pb2 atom, also supports this view (dPb1-BCP = 1.634 Å; dPb2-BCP = 1.503 Å), displaying an almost vanishing value for the Laplacian of the electron density at the BCP (∇2ρ = 0.0389 au) as one of the characteristic features of dative bonding.48 However, it was not possible to differentiate the two expected valence–shell concentration bands (VSCC) corresponding to the donor and acceptor atoms at the central part of the ∇2ρ function along the Pb–Pb path. Instead, only one broad band (superposition of two individual and similar VSCCPb bands), mostly located within the basin of the donor atom, as additional signature of dative bonding,48 could be observed (Figure S2). The same type of nonsymmetric donor–acceptor tetrel–tetrel bonds with twisted substituents were found in 1Sn.
Figure 3.
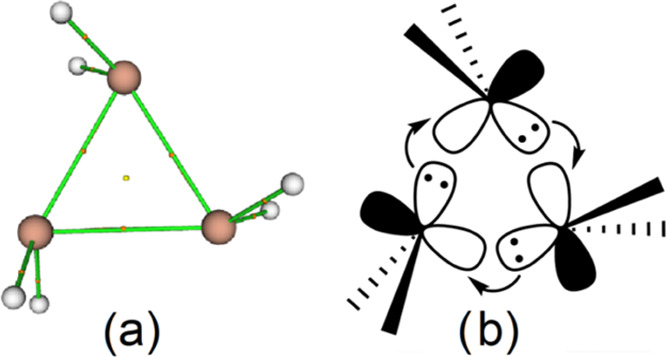
Computed (B3LYP/def2-TZVPP) BCP (small orange spheres), RCP (small yellow sphere), and bond paths for 1Pb; sketched representation of El–El bonds for 1Sn and 1Pb.
For these two rings 1Sn and 1Pb, the general homodesmotic reactions used for all other rings (Scheme 1) should not be used as it is not possible to compensate for this type of interactions in the open-chain ring-cleavage products, where donor–acceptor El→El bonds are not present. Therefore, different hyper-homodesmotic reactions were used (Scheme 2) taking advantage of the presence of donor–acceptor Pb→Pb bonds with twisted substituents in five- and six-membered rings (PbH2)n (n = 5, 6) and assuming negligible ring strain in these moderate-sized homocyclic rings. The RSE for 1Pb was obtained as the average of both RC5 reactions (per 1Pb unit). However, the H substituents are not twisted in the Sn-containing five- and six-membered homocyclic rings, which let the effect of the donor–acceptor bonds in 1Sn uncompensated. Similarly, 1Tl (structure IVTl, Figure S1) displays a twisted geometry corresponding to three donor–acceptor endocyclic Tl→Tl bonds, but larger sized (TlH)n rings (n = 5, 6) are not stable. Therefore, the corresponding obtained RSE values for both 1Sn and 1Tl (Table 1) were calculated according to Scheme 1 and should be taken with caution.
Scheme 2. Hyper-homodesmotic (RC5) Reactions Used for the Estimation of RSE for 1Pb.

A less pronounced substituent twist can be observed in 4Sn (Figure S3). In this case, since the Sn–Sn bond is not symmetrical, neither are the two Ge–Sn bonds which have slightly different bond distances (dGe–Sn1 = 2.655 Å, WBIGe–Sn1 = 0.942; dGe–Sn2 = 2.662 Å, WBIGe–Sn2 = 0.922). This results from a different p-character at Ge and Sn being 85.2 and 72.8%, respectively, in the first case, and reversed to 74.7 and 84.3% in the second one. Hence, due to uncompensated interactions in the homodesmotic cleavage reactions, the computed RSE for 4Sn should also be taken with caution.
The other lead-containing rings 2Pb, 3Pb, and 4Pb do not exist as minima and 5Pb was already reported29 as a pseudocyclic Dewar–Chatt–Duncanson-type structure.49
It is known that Se3 and Te3 show a slight preference for the cyclic D3h over the acyclic C2v (bent) geometry, which is the most stable isomer for the lighter chalcogens O and S.50 At the working level of theory, the same cyclic preference is observed for the heaviest Po3 and new linear species D∞h were found as most unstable isomers for O3 and Te3, whereas D∞h-Po3 is a transition state between two degenerated C2v structures (see Table S3 for relative energies of trichalcogen isomers).
Factors Affecting RSE
As observed for monoheteroatomic saturated29 and unsaturated51 3MRs, one of the main mechanisms of strain relaxation is the effect of lone electron pair (LP) hybridization. According to this effect, an increase in the s-character of the LP-containing AO enhances the p-character of the AOs used by El in the endocyclic bonds, giving rise to a spn-type hybridization with n > 3 (“high” p-character, beyond 75%) which fits better to small endocyclic bond angles (Figure 4).
Figure 4.

Plots of RSE vs p-character of the AO used by the heteroatom El for its endocyclic El–Y bonds in El2Y rings: (a) 1El (Y: El, black), 2El (Y: C, red), (b) 3El (Y: Si, black), and 4El (Y: Ge, red).
Rings containing a tetrel atom as heteroelement El lack LPs, thus preventing the corresponding strain relaxation mechanism, which makes them generally the most strained rings for all four ring types 1–4El. In the case of pnictogen- and chalcogen-containing rings, there is a clear ring strain relaxation from the second to the third row in the 1El and 2El rings (excepting 1O), although it remains roughly constant for the heaviest elements (Figure 4a). In turn, for the 3El and 4El rings (Figure 4b), the linear correlation between the increase of the p-character in El for the Si/Ge–El bond and the RSE relaxation is quite remarkable, as already reported for the case of the monohetero-monocyclic rings 5El (which also included group 13 heteroelements). It is important to note that 3N and 3O present RSE well beyond the heaviest congeners and in the range of group 14 rings. On the other hand, among the four types of rings studied, the least strained are homoatomic 1El rings, while the ring strain slightly increases on changing one El atom to C (2El), Si (3El), and Ge (4El).
The RSE also shows some correlation with the s-character of the LP-containing AO at the El heteroatom for groups 15 and 16 (Figure S4 and Table S1), although it does not vary significantly for the heaviest group 15 1El-type rings.
The unusual stability of cyclopropane 1C, with a RSE (27.5 kcal/mol) very close to that of cyclobutane (26.5 kcal/mol), contrasts with its silicon counterpart trisilacyclopropane (trisilirane, 1Si) whose RSE is beyond twice that of tetrasilacyclobutane.52 This, along with the exhibition of olefin-like properties, such as the formation of metal complexes or undergoing catalytic hydrogenation,53 has led to numerous studies attributing σ-aromaticity as the cause of this stabilization.54 However, based on “extracyclic resonance energy” (ECRE) calculations, the σ-aromaticity stabilization for 1C has been quantified in only 3.5 kcal/mol.55 Nucleus-independent chemical shifts (NICS)56 were reported to quantify aromaticity, so that the more negative the NICS value, the higher the aromatic character. The analysis of NICS values for a complete set of three- to six-membered inorganic rings derived from the main group elements unveils that the ring σ-(anti)aromaticity is mostly due to interactions among electrons making up the endocyclic El–El bonds rather than the El–H bonds or LPs.57 This study also points out the fact that the simple counting of electrons in endocyclic bonds follows Hückel’s rule so that the saturated three- and five-membered rings (4n+2 electrons) are aromatic whereas four- and six-membered rings (4n electrons) are antiaromatic. NICS analysis using localized molecular orbitals suggested that the σ-ring electrons of trichalcogeniranes 1El (El = O, S, Se, Te, Po) are chiefly responsible for their aromaticity.57 Moreover, a NICS variation study along the z-axis perpendicular to the ring plane revealed double (σ + π) aromaticity in heavier trichalcogeniranes, the induced diatropic ring current arising from Tx,y-allowed transitions involving both σ- (a1′ → e′) and π-type (a2″ → e″) molecular orbitals (MOs).58 The overall excitation energies were shown to decrease (aromaticity increase) on descending the group, with the π-aromaticity prevailing over the σ-component only in the case of S and Se. NICS(1) values were calculated for all herein studied 1El rings at the working level of theory, being in line with those already reported (root-mean-square error RMSE = 0.657 ppm). For group 14 1El rings, the increase (less negative) in NICS(1) values on descending the group entails a destabilization of the rings (higher RSE) (Figure 5). The decrease in aromaticity might be related to poorer spatial overlap among σ(El–H) MOs at both sides of the rings due to the increasing ring size (Figure S5). However, for group 15–16 elements, a remarkable increasing aromaticity (more negative NICS(1) values) on descending the groups does not significantly affect the RSE (except for N). This could be the result of the counteracting additional effect of a simultaneous increase in the flexibility of endocyclic bond angles (vide infra).
Figure 5.

Plot of RSE vs NICS(1) for 1El.
To try to explain this effect, the highest occupied molecular orbital–least unoccupied molecular orbital (HOMO–LUMO) gap, ΔεH–L, was studied. It has been used as an indicator of the kinetic stability of a compound so that a low-lying HOMO would indicate a difficult electron extraction and a high LUMO suggesting the unfavorable addition of electrons.59 This ΔεH–L represents the chemical hardness (η) of a molecule according to Pearson,60 large band gaps corresponding to stable structures, which is the normal situation for classical aromatics.
However, Fowler pointed out that the (large) HOMO–LUMO separation cannot be seen as an absolute criterion for the aromaticity or kinetic stability in the case of polycyclic aromatic hydrocarbons, as lower gaps facilitate electronic transitions related to delocalization.61 In fact, there exists a clear linear correlation between more negative NICS(1) values (aromaticity) with lower H–L gap values in 1El rings for groups 15 and 16 (Figure 6). The latter is most likely just reflecting a decrease in AO energy differences as the principal quantum number increases when moving down the group. On the other hand, the lack of LPs in group 14 1El rings entails higher gap between σ(El–El) and σ*(El–El), which is rather large for the scarcely σ-aromatic cyclopropane (vide supra) and decreases for heavier congeners, paralleling a decrease in σ-aromaticity (Figure 6).
Figure 6.

Plot of η vs NICS(1) for 1El. Oxygen is excluded from the linear correlation of group 16.
The increasing p-character in endocyclic bonds on descending the groups is expected to cause a decrease in bond stiffness. To evaluate this effect, relaxed force constants k0 were calculated for every endocyclic bond. The Hessian matrix in nonredundant internal coordinates is transformed into its inverse (or Moore–Penrose pseudo-inverse) providing the constant compliance matrix Cij from which the relaxed force constants are obtained as reciprocals of the diagonal elements, k0ii = 1/Cii. These are generally used as numerically stable and fully transferable parameters.62 The four ring types 1–4 show a remarkable decrease in the El–El bond strength, k0El–El, for El heteroatoms from the third row onward, in line with an increase in the p-character, especially for pnictogen and chalcogen El elements (Figure 7) where LPs strain relaxation29,51 is possible. As already observed for the p-character, for 1El and 2El rings, the expected decrease in k0El–El on moving down the group does not lead to a significant relaxation of the ring strain, except for 2Pn to some extent (Pn = pnictogen atoms). By contrast, in 3El and 4El rings, the RSE decreases significantly with the increase of bond flexibility with significant linear correlation (R2 = 0.476 and 0.529 for 3El and 4El, respectively).
Figure 7.

Plots of RSE against k0El–El in El2Y rings: (a) 1El (Y: El, black), 2El (Y: C, red), (b) 3El (Y: Si, black), and 4El (Y: Ge, red). Correlation lines for the different ring systems and groups (Tt: tetrels; Pn: pnictogens; Ch: chalcogens) excluding O and N.
The force constant for the El–El bond, k0El–El, is expected to be coupled to some extent to that for the El–Y–El (Y: El, C, Si or Ge) bond angle, k0El–Y–El, as indeed observed by the systematic linear increase in k0El–El when increasing k0El–Y–El for all tetrel-containing rings 2El, 3El, and 4El (Figure 8). However, in homoatomic 1El rings, only some correlation is observed by groups, after excluding the lightest elements.
Figure 8.

Plots of k0El–El against k0El–Y–El in El2Y rings: (a) 1El (Y: El, black), 2El (Y: C, red), (b) 3El (Y: Si, black), and 4El (Y: Ge, red). Correlation lines for the different ring systems and groups (Tt: tetrels; Pn: pnictogens; Ch: chalcogens) excluding O and N.
A decrease in k0El–El–El (increase of bond angle elastic behavior) on descending the groups leads to a strain relaxation in all four types of rings, excluding the lightest (second row) and group 14 elements (Figure S6). In the case of homoatomic 1El rings, the bond angle flexibility and aromatic stabilization are counteracting effects that cause the RSE to remain largely unchanged when moving down the groups 15 and 16, except for the lightest 1N and 1O rings, the latter displaying low aromaticity and (comparatively) high bond angle flexibility. Simple acyclic model molecules HEl–El–ElH lacking competing aromaticity effects clearly show higher bond flexibility than the corresponding cyclic analogues El3, as expected, as well as (nonlinear) increasing angle flexibility on descending the groups 14–16 (Figure S7).
Additive Estimation of RSEs
Although high-accuracy RSEs have been reported in the preceding section for a wide set of 3MRs, it would be desirable to have a quick method at hand for obtaining a rough estimation of RSEs for other unreported ring systems. Such methodology should consist of a summatory of contributions arising from the different constituent elements being either ring atoms and/or bonds. Therefore, as first approach, the RSE could be additively estimated (RSEad) using only atomic addends Ai extended to the three constituent atoms i (eq 1) at any given ring.
| 1 |
Out of the 58 RSE values herein calculated for rings 1–4 (Table 1), only 49 were used, excluding those with Sn heteroatoms (1–4Sn) because they introduce uncompensated effects (vide supra) in the RSE calculation, as well as the only herein described Pb- and Tl-containing ring (1Pb and 1Tl), which are not suitable for later stages. In addition, 12 previously reported RSE values29 were included at (approximately) the same level63 for monohetero-monocycles 5El. To calculate the contributions to the ring strain of the triels group, 5Al and 5Ga were also included, in addition to six new molecules Tt2Tr and CSiTr (where Tt: Si, Ge and Tr: Al, Ga)64 (vide infra). In this way, an oversized system of 67 equations (eq 1) with 15 A1El unknowns can be written.
Upon numerical resolution of this system, a set of parameters A1El (Table 2) with a high RMSE of 4.813 kcal/mol was obtained. This is in line with the highly dispersed plot of estimated RSEAad against the accurately (RC4-based) computed RSE (Table 1), with moderate R2 = 0.8175 (Figure S11a). A testbed of 13 new saturated 3MRs (Si2P, Si2S, Ge2N, CSiN, CSiP, CSiO, CSiS, CGeO, CGeAl, CGeGa, SiGeAl, SiGeGa, and SiGeO), not included in the set of rings belonging to the 1–5 categories (Figure 1), was used only for checking the performance of the additive method, by comparing the accurate RSE values resulting from the evaluation of RC4-type homodesmotic reactions (Scheme 1) with the RSEAad estimation (Table 3). The estimates arising from this approximation are not very close to the reference values but, despite being a crude method, it can give an idea of the RSE with a maximum unsigned absolute error (unsigned difference) of 10.63 kcal/mol. For instance, for the additive estimation of the RSE for alumasilagermirane (AlSiGe), the atomic-strain contributions A1 (in kcal/mol) for Al (19.87), Si (12.21), and Ge (12.85) are summed up, leading to RSEAad = 44.93 kcal/mol, which overestimates in 5.22 kcal/mol the reference most accurately computed RSE value of 39.71 kcal/mol obtained by evaluation of homodesmotic reactions.
Table 2. Calculated Atoms A1El and Bond-Strain Contributions B2El–El, B2C–El, B2Si–El, and B2Ge–El (kcal/mol) to RSEad.
| El | A1El | B2El–El | B2C–El | B2Si–El | B2Ge–El |
|---|---|---|---|---|---|
| Al | 19.87 | 17.76 | 13.07 | 13.57 | |
| Ga | 21.37 | 17.61 | 14.34 | 14.87 | |
| C | 8.31 | 8.48 | 8.48 | 13.71 | 13.93 |
| Si | 12.21 | 12.07 | 13.71 | 12.07 | 12.52 |
| Ge | 12.85 | 12.89 | 13.93 | 12.52 | 12.89 |
| N | 8.54 | 4.55 | 8.99 | 18.52 | 16.60 |
| P | 2.29 | 1.93 | 5.55 | 7.54 | 8.21 |
| As | 1.90 | 1.93 | 4.97 | 6.95 | 7.55 |
| Sb | 1.52 | 2.05 | 4.45 | 6.08 | 6.66 |
| Bi | 0.91 | 2.32 | 2.94 | 5.05 | 5.59 |
| O | 9.54 | 9.15 | 7.58 | 12.32 | 13.19 |
| S | 6.99 | 9.61 | 4.79 | 6.42 | 7.53 |
| Se | 6.03 | 8.71 | 4.45 | 5.81 | 6.75 |
| Te | 6.09 | 9.18 | 4.15 | 5.34 | 6.27 |
| Po | 5.44 | 8.77 | 3.46 | 4.89 | 5.69 |
Table 3. Calculated (DLPNO-CCSDT/def2TZVPPecp) Accurate (RC4) and Additively Estimated (ad) RSEs (kcal/mol) for a Testbed of 3MRsa.
| RSERC4 | RSEAad | RSEBad | |
|---|---|---|---|
| Si2P | 27.70 | 26.72 (−0.98) | 27.16 (−0.54) |
| Si2S | 24.68 | 31.42 (6.74) | 24.90 (0.22) |
| Ge2N | 41.76 | 34.23 (−7.53) | 46.10 (4.34) |
| CSiN | 39.69 | 29.07 (−10.63) | 41.22 (1.53) |
| CSiP | 26.79 | 22.82 (−3.97) | 26.80 (0.01) |
| CSiO | 38.70 | 30.07 (−8.63) | 33.60 (−5.10) |
| CSiS | 24.40 | 27.52 (3.11) | 24.91 (0.51) |
| CGeO | 37.81 | 30.70 (−7.11) | 34.69 (−3.12) |
| AlCGe | 43.45b | 41.03 (−2.42) | 45.25 (1.80) |
| GaCGe | 45.90 | 42.53 (−3.38) | 46.41 (0.50) |
| AlSiGe | 39.71 | 44.93 (5.22) | 39.16 (−0.56) |
| GaSiGe | 42.17 | 46.43 (4.26) | 41.74 (−0.43) |
| SiGeO | 42.58 | 34.60 (−7.98) | 38.02 (−4.56) |
Signed absolute errors are in parenthesis.
Calculated only with the contribution of the C–Al and C–Ge bond-cleavage homodesmotic reactions due to convergence problems in the third ring-opening reaction.
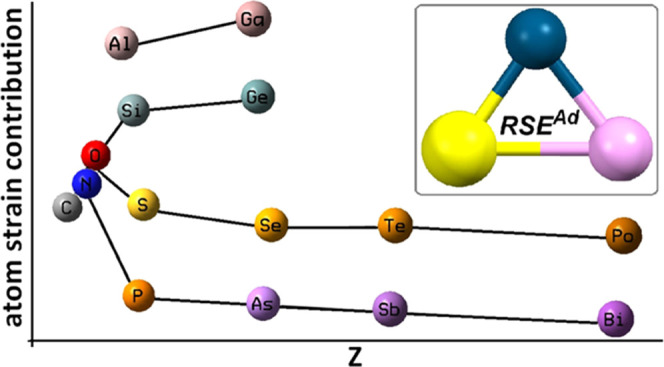
The variation of the obtained atom-strain contributions A1El (Figure 9) is in agreement with the general tendencies reported29 for heterocycles containing one heteroatom, 5El. Thus, second-row elements (C, N, and O) have similar values. On descending the groups, the atom-strain contributions decrease for elements having LPs (groups 15 and 16), especially for pnictogens, whereas they increase for those lacking LPs (groups 13 and 14), the highest values being observed for triels that bear an empty p orbital (Figure 9). This effect is most likely related to the already reported LP strain releasing effect,29,51 in turn affecting the p-character of the endocyclic bonds (see Figure 4).
Figure 9.

Variation of the calculated atom-strain contributions A1El (kcal/mol) to RSE1ad with the atomic number.
A refinement of the methodology consists of using bond-related addends B2i in the additive estimation RSEBad (eq 2), instead of atom-based parameters. Resolving the corresponding overdimensioned 67 equation system with 52 B2El–El′ variables (Table 2), the remarkably lower RMSE of 1.168 kcal/mol reflects the better estimation resulting from this approximation (Table 3). For the abovementioned example of the AlSiGe ring, the bond-based additive estimation would result from the summation of the strain contributions B2 (in kcal/mol) for Si–Al (13.07), Si–Ge (12.52), and Ge–Al (13.57), resulting in RSEBad = 39.16 kcal/mol, which only underestimates in 0.56 kcal/mol the reference value.
| 2 |
Analysis of the B2El–El′ bond-strain parameters reveals some interesting features (Figure 10), mostly in line with the abovementioned atom-strain contributions. As expected, bonds involving group 13–14 elements contribute the highest bond energy strain for all four types of rings 1–4El. Strain contributions are lower for bonds involving group 16 than group 15 elements, except for homoatomic El–El bonds (Figure 10). Remarkably high strain contributions are obtained for bonds of N with the heaviest tetrels (Si and Ge).
Figure 10.

Variation of the calculated bond-strain contributions B2 (kcal/mol) to RSE2ad with the atomic number.
Worth noting is that on plotting RSEBad versus RSE (Figure S11b), the scarcely dispersed linear correlation (R2 = 0.9893) becomes obvious, with three 5El rings (El = Po, Te, Bi) showing the highest positive deviations (Δ = RSEBad – RSE of 3.94, 3.02, and 1.81 kcal/mol, respectively), while the corresponding 2El rings feature the largest negative deviations (−3.94, −3.02, and −1.81 kcal/mol, respectively). This fact strongly suggests an overestimation of the three B2C–El and underestimation of the three B2El–El parameters for the three abovementioned El elements, assuming that B2C–C is properly estimated (as indeed evidenced by the correct additive estimation of all other 5El rings). The opposite effect is observed for 5O (Δ = −2.91 kcal/mol) and 2O (Δ = 2.91 kcal/mol), hinting at an underestimated B2C–O and overestimated B2O–O strain parameters (Figure 11).
Figure 11.

Plots of deviations Δ of additively estimated RSE based on (a) atom-strain and (b) bond-strain contributions vs the reference RSERC4.
One possibility for further refinement of the additive methodology would consist of using both atom- and bond-based addends in the third level of ring strain estimation, RSEA&Bad (eq 3).
| 3 |
With the 49 equations resulting from molecules 1–4 together with the 12 additional equations of type 5, an underdimensioned system of 67 (atom- and bond-based) unknowns with only 61 equations is obtained. Therefore, to balance the system, the abovementioned six rings (equations) (Tt2Tr and CSiTr, where Tt: Si, Ge and Tr: Al, Ga) had to be included, provided that they do not introduce new unknowns. In case of boron, no cyclic minima were found for Tt2B, whereas for Tt2In and Tt2Tl, all cyclic minima displayed unconventional geometries (vide supra) not allowing accurate calculation of RSE. Worth noting is that in the case of Si2Tr (Tr: Al, Ga), there exists a conformational preference (without significant variation in hybridization) for the H2Si–SiH2 moiety featuring two axial H atoms in relative antiperiplanar orientation and two equatorial H in a synclinal conformation.65
The resulting equidimensional system of 67 equations with 67 unknowns has not a single but an infinite number of mathematical solutions with the same RMSE (1.168 kcal/mol) which, in turn, does not essentially outperform that obtained with the only-bonds method (no variation up to the 13th decimal figure).
It is possible to make a directed search to ascertain what is the most meaningful solution through a stepwise procedure. This requires a thermochemical evaluation of appropriate reactions for the estimation of electronic strain energy contribution of every X–Y endocyclic bond and using them as boundary conditions (see the SI). However, it resulted in not worth the great effort required for this much more elaborated methodology, as the ensuing set of A4El and bond B4El strain contributions (Table S5) do not represent any improvement regarding mathematical accuracy compared to the rather simple only-bonds additive estimation method for RSEs. Furthermore, it seems to overestimate the bond-strain contributions B4El (Figure S8) but at the price of compensating with increasingly negative atom-strain contributions A4El (Figure S9) for atoms typically involved in more strained rings.
Experimental Section
Density functional theory (DFT) calculations were performed with the ORCA program.66 All geometry optimizations were run in redundant internal coordinates in the gas phase, with tight convergence criteria, and using the B3LYP67 functional together with Ahlrichs segmented def2-TZVP basis set68 and the latest Grimme’s semiempirical atom-pairwise London dispersion correction (DFT-D4).69 From these geometries, all electronic data were obtained through single-point calculations (SP) using the same quality basis set but including additional polarization, def2-TZVPP.70 Energy values were corrected for the zero-point vibrational term at the optimization level and obtained by the newly developed DLPNO method71 for the “coupled-cluster” level with single, double, and triple perturbatively introduced excitations (CCSD(T)).72 Analysis of the hybridization in the AO used for the endocyclic bonds was performed with the NBO method.73 Properties derived from the topological analysis of the electronic density were obtained with the Multiwfn program,74 and MO was drawn with Visual Molecular Dynamics (VMD).75
Conclusions
Accurate high-level (DLPNO-CCSD(T)/def2TZVPP//B3LYP-D4/def2TZVP) values were provided for the ring strain energy (RSE) in three-membered symmetric inorganic rings El3 and organic dihetero-monocycles El2C and their silicon El2Si and germanium El2Ge analogues for group 14–16 heteroatoms El. The absence of undistorted cyclic minima prevented the design of suitable homodesmotic reactions for triel-containing 1–4El rings. For the 1Tl, 1Sn, and 1Pb rings containing endocyclic El–El donor–acceptor bonds, the general homodesmotic reactions used for other rings should be taken with caution due to uncompensated effects. Only in case of 1Pb, suitable alternative homodesmotic reactions with compensation of Pb–Pb donor–acceptor bonds with twisted substituents provided a good estimation of its RSE. With some exceptions in the 1–4El rings, pnictogen and chalcogen-containing derivatives exhibit a clear relaxation of the ring strain that increases on descending the groups, due to the increase of the s-character of the LP-containing orbital which promotes an increase of p-character of the AO used by El for the endocyclic bonds, in turn decreasing the bond stiffness. The simultaneous counteracting increase in aromaticity for 1El rings on moving down in groups 15–16 let the RSE almost unaffected. The latter has been analyzed based on the counter-intuitive correlation between increasing aromaticity and decreasing H–L gap, most likely arising from a decrease in AO energy differences as the principal quantum number increases down the group.
Finally, a new fast method of estimating the ring strain based on the additivity of atom- and/or endocyclic bond-strain contributions is proposed, thus providing an attractive and efficient alternative approximation to the RSE. The method of choice is based on a summatory of only-bonds addends and can be currently applied to any three-membered saturated ring containing only El–El and El–Tt (Tt = C, Si, Ge) bonds.
Acknowledgments
The authors thank the computation center at Servicio de Cálculo Científico (SCC—University of Murcia) for technical support and for the computational resources used.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.2c01777.
Plots of the relaxed force constants and RSE versus other parameters, additional tables with electronic, structural, and bond strength parameters, and Cartesian coordinates and energies for all calculated species (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Espinosa Ferao A.; García Alcaraz A. Benchmarking the inversion barriers in σ3λ3-phosphorus compounds: a computational study. New J. Chem. 2020, 44, 8763–8770. 10.1039/D0NJ01237H. [DOI] [Google Scholar]
- Lowry T. H.; Richardson K. S.. Mechanism and Theory in Organic Chemistry, 3rd ed.; Harper & Row: New York, 1987; Chapter 2.3, pp 294–298. [Google Scholar]
- Zuercher W. J.; Hashimoto M.; Grubbs R. H. Tandem Ring Opening-Ring Closing Metathesis of Cyclic Olefins. J. Am. Chem. Soc. 1996, 118, 6634–6640. 10.1021/ja9606743. [DOI] [Google Scholar]
- a Aziridines and Epoxides in Organic Synthesis, Yudin A. K., Ed.; Wiley-VCH: Weinheim, 2006. [Google Scholar]; b Padwa A.; Murphree S. S. Epoxides and aziridines—a mini review. Arkivoc 2006, 3, 6–33. [Google Scholar]
- Kobayashi S.; Kadokawa J.-I. Ring-opening polymerization of 1-(2,4,6-tri-tert-butylphenyl)-phosphirane: direct synthesis of a polyphosphine derivative. Macromol. Rapid Commun. 1994, 15, 567–571. 10.1002/marc.1994.030150618. [DOI] [Google Scholar]
- Marchand A. P.; Marchand N. W. NMR studies of rigid bicyclic systems. III. Assignment of chemical shift values in the NMR spectrum of norbornane. Tetrahedron Lett. 1971, 12, 1365–1368. 10.1016/S0040-4039(01)96711-5. [DOI] [Google Scholar]
- Tanabe M.; Bourke S. C.; Herbert D. E.; Lough A. J.; Manners I. Reversible, Strain-Controlled Haptotropic Shifts of Cyclopentadienyl Ligands in [1]- and [2]Metallocenophanes. Angew. Chem., Int. Ed. 2005, 44, 5886–5890. 10.1002/anie.200501155. [DOI] [PubMed] [Google Scholar]
- Höltzl T.; Nguyen M. T.; Veszpremi T. Mercury dications: linear form is more stable than aromatic ring. Phys. Chem. Chem. Phys. 2010, 12, 556–558. 10.1039/B916847H. [DOI] [PubMed] [Google Scholar]
- Bartok M.; Lang K. L.; Boyd D. R.; Jerina D. M.; Haddadin M. J.; Freeman J. P.; Adam W.; Yany F.; Dittmer D. C.; Sedergran T. C.. The Chemistry of Heterocyclic Compounds; Wiley: New York, 1985; Vol. 42. [Google Scholar]
- a Masamune S.; Hanzawa Y.; Murakami S.; Bally T.; Blount J. F. Cyclotrisilane (R2Si)3 and disilene (R2Si:SiR2) system: synthesis and characterization. J. Am. Chem. Soc. 1982, 104, 1150–1153. 10.1021/ja00368a065. [DOI] [Google Scholar]; b Masamune S.; Murakami S.; Tobita H.; Williams D. J. 1,1,2,2-Tetrakis(2,6-dimethylphenyl)-1,2-disilacyclopropane. J. Am. Chem. Soc. 1983, 105, 7776–7778. 10.1021/ja00364a069. [DOI] [Google Scholar]; c Schäfer A.; Weidenbruch M.; Peters K.; Schnering H. G. Hexa-tert-butylcyclotrisilane, a Strained Molecule with Unusually Long Si-Si and Si-C Bonds. Angew. Chem., Int. Ed. 1984, 23, 302–303. 10.1002/anie.198403021. [DOI] [Google Scholar]; d Ando W.; Hamada Y.; Sekiguchi A.; Ueno K. Oxasilacyclopropane. Isolation and characterization. Tetrahedron Lett. 1982, 23, 5323–5326. 10.1016/S0040-4039(00)85829-3. [DOI] [Google Scholar]; e Ando W.; Hamada Y.; Sekiguchi A.; Ueno K. Silathiacyclopropane. Isolation and characterization. Tetrahedron Lett. 1983, 24, 4033–4036. 10.1016/S0040-4039(00)88255-6. [DOI] [Google Scholar]; f West R.; De Young D. J.; Hailer K. J. Disilathiiranes: synthesis and crystal structure. J. Am. Chem. Soc. 1985, 107, 4942–4946. 10.1021/ja00303a020. [DOI] [Google Scholar]; g Yokelson H. B.; Millevolte A. J.; Gillette G. R.; West R. Disilaoxiranes: synthesis and crystal structure. J. Am. Chem. Soc. 1987, 109, 6865–6866. 10.1021/ja00256a058. [DOI] [Google Scholar]; h Grev R. S.; Schaefer H. F. III Cyclopolysilanes: Structure, Strain, and the Form of the Singly Occupied Molecular Orbital in Their Radical Anions. J. Am. Chem. Soc. 1987, 109, 6569–6577. 10.1021/ja00256a001. [DOI] [Google Scholar]; i Kitchen D. B.; Jackson J. E.; Allen L. C. Organosilicon Rings: Structures and Strain Energies. J. Am. Chem. Soc. 1990, 112, 3408–3414. 10.1021/ja00165a025. [DOI] [Google Scholar]; j Gimarc B. M.; Zhao M. Strain and resonance energies in main-group homoatomic rings and clusters. Coord. Chem. Rev. 1997, 158, 385–412. 10.1016/S0010-8545(97)90067-9. [DOI] [Google Scholar]
- a Masamune S.; Hanzawa Y.; Williams D. J. Synthesis of a cyclotrigermane and its conversion to a digermene. J. Am. Chem. Soc. 1982, 104, 6136–6137. 10.1021/ja00386a059. [DOI] [Google Scholar]; b Ando W.; Tsumuraya T. Synthesis of germathiiranes. Tetrahedron Lett. 1986, 27, 3251–3254. 10.1016/S0040-4039(00)84766-8. [DOI] [Google Scholar]; c Ando W.; Tsumuraya T. Digermirane and azadigermiridine. Synthesis and reactions. Organometallics 1988, 7, 1882–1883. 10.1021/om00098a036. [DOI] [Google Scholar]; d Tsumuraya T.; Sato S.; Ando W. Photolysis of cyclotrigermane. Synthesis and chemistry of digermiranes and digermetanes containing sulfur and selenium. Organometallics 1988, 7, 2015–2019. 10.1021/om00099a018. [DOI] [Google Scholar]; e Tsumuraya T.; Sato S.; Ando W. Synthesis and chemistry of thiagermiranes. Organometallics 1989, 8, 161–167. 10.1021/om00103a020. [DOI] [Google Scholar]
- Masamune S.; Sita L. R.; Williams D. J. Cyclotristannoxane (R2SnO)3 and cyclotristannane (R2Sn)3 systems. Synthesis and crystal structures. J. Am. Chem. Soc. 1983, 105, 630–631. 10.1021/ja00341a058. [DOI] [Google Scholar]
- a Leuenberger C.; Hoesch L.; Dreiding A. S. A Triaziridine. J. Chem. Soc., Chem. Commun. 1980, 1197–1198. 10.1039/c39800001197. [DOI] [Google Scholar]; b Hoesch L.; Leuenberger C.; Hilpert H.; Dreiding A. S. Triaziridine. II. Erste Beispiele: 2, 3-Dialkyl-triaziridin-1-carbonsäurealkylester. Helv. Chim. Acta 1982, 65, 2682–2696. 10.1002/hlca.19820650837. [DOI] [Google Scholar]; c Karahodza A.; Knaus K. J.; Ball D. W. Cyclic triamines as potential high energy materials. Thermochemical properties of triaziridine and triazirine. J. Mol. Struct.: THEOCHEM 2005, 732, 47–53. 10.1016/j.theochem.2005.07.004. [DOI] [Google Scholar]
- a Baudler M.; Carlsohn B.; Böhm W.; Reuschenbach G. Beiträge zur Chemie des Phosphors, 621 Zur Frage des 31P-kernresonanzspektroskopischen Nachweises von monomerem und dimerem Phenylphosphor und der Ringgröße von Organyl-cyclophosphanen. Z. Naturforsch., B: Anorg. Chem., Org. Chem. 1976, 31, 558–564. 10.1515/znb-1976-0506. [DOI] [Google Scholar]; b Baudler M.; Carlsohn B.; Kloth B.; Koch D. Beiträge zur Chemie des Phosphors. 67. Zur Kenntnis der Cyclotriphosphane (PC6H5)3, (PC6H5)2(PC2H5) und (PC6H5)2(PCH3). Z. Anorg. Allg. Chem. 1977, 432, 67–78. 10.1002/zaac.19774320108. [DOI] [Google Scholar]; c Chitnis S. S.; Musgrave R. A.; Sparkes H. A.; Pridmore N. E.; Annibale V. T.; Manners I. Influence of Ring Strain and Bond Polarization on the Ring Expansion of Phosphorus Homocycles. Inorg. Chem. 2017, 56, 4521–4537. 10.1021/acs.inorgchem.7b00086. [DOI] [PubMed] [Google Scholar]; d Schumann A.; Reiss F.; Jiao H.; Rabeah J.; Siewert J.-E.; Krummenacher I.; Braunschweig H.; Hering-Junghans C. A selective route to aryl-triphosphiranes and their titanocene-induced fragmentation. Chem. Sci. 2019, 10, 7859–7867. 10.1039/C9SC02322D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Thiele G.; Zoubek G.; Lindner H. A.; Ellermann J. Molecular and Crystal Structure of an Organocyclotriarsane. Angew. Chem., Int. Ed. 1978, 17, 135–136. 10.1002/anie.197801351. [DOI] [Google Scholar]; b Baudler M.; Bachmann P. Tri-tert-butylcyclotriarsane. Angew. Chem., Int. Ed. 1981, 20, 123–124. 10.1002/anie.198101231. [DOI] [Google Scholar]; c Spang C.; Edelmann F. T.; Noltemeyer M.; Roesky H. W. Anorganische Ringsysteme mit Ferrocenyl-Substituenten. Chem. Ber. 1989, 122, 1247–1254. 10.1002/cber.19891220706. [DOI] [Google Scholar]; d Schumann A.; Bresien J.; Fischer M.; Hering-Junghans C. Aryl-substituted triarsiranes: synthesis and reactivity. Chem. Commun. 2021, 57, 1014–1017. 10.1039/D0CC07533G. [DOI] [PubMed] [Google Scholar]
- Xantheas S. S.; Atchity G. J.; Elbert S. T.; Ruedenberg K. Potential energy surfaces of ozone. I. J. Chem. Phys. 1991, 94, 8054–8069. 10.1063/1.460140. [DOI] [Google Scholar]
- a Murray R. W.; Jeyaraman R. Dioxiranes: Synthesis and Reactions of Methyldioxiranes. J. Org. Chem. 1985, 50, 2847–2853. 10.1021/jo00216a007. [DOI] [Google Scholar]; b Mello R.; Fiorentino M.; Sciacovelli O.; Curci R. On the Isolation and Characterization of Methyl(trifluoromethyl)dioxirane. J. Org. Chem. 1988, 53, 3890–3891. 10.1021/jo00251a053. [DOI] [Google Scholar]; c Adam W.; Zhao C.-G.; Saha-Möller C. R.; Jakka K.. Oxidation of Organic Compounds by Dioxiranes; John Wiley & Sons Inc., 1990. [Google Scholar]; d Zou L.; Paton R. S.; Eschenmoser A.; NewHouse T. R.; Baran P. S.; Houk K. N. Enhanced Reactivity in Dioxirane C–H Oxidations via Strain Release: A Computational and Experimental Study. J. Org. Chem. 2013, 78, 4037–4048. 10.1021/jo400350v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson K. S.; Michaelis D. J.; Yoon T. P. Advances in the Chemistry of Oxaziridines. Chem. Rev. 2014, 114, 8016–8036. 10.1021/cr400611n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines K. M.; Cooke J. A. Thermolysis of a Siladigermirane: Evidence for the Formation of a Germasilene. Organometallics 1991, 10, 3421–3423. 10.1021/om00056a004. [DOI] [Google Scholar]
- Walewska M.; Baumgartner J.; Marschner C.; Albers L.; Müller T. Alkyne Addition and Insertion Reactions of [(Me3Si)3Si]2Ge·PMe3. Chem. - Eur. J. 2016, 22, 1–11. 10.1002/chem.201603317. [DOI] [PubMed] [Google Scholar]
- Abersfelder K.; Nguyen Tl.; Scheschkewitz D. Stannylsubstituted Disilenes and a Disilastannirane. Z. Anorg. Allg. Chem. 2009, 635, 2093–2098. 10.1002/zaac.200900234. [DOI] [Google Scholar]
- a Du Mont W.-W.; Seppälä E.; Gust T.; Mahnke J.; Müller L. Some new nucleophile-induced reactions involving GeCl2, SiCl2 AND GeMe2 transfer. Main Group Met. Chem. 2001, 24, 609–612. 10.1515/MGMC.2001.24.9.609. [DOI] [Google Scholar]; b Kimel B. K.; Tumanov V. V.; Egorov M. P.; Nefedov O. M. The first phosphagermacyclopropane prepared via cycloaddition of dimethylgermylene to the C=P double bond of phosphaalkene. Mendeleev Commun. 2001, 11, 85–86. 10.1070/MC2001v011n03ABEH001445. [DOI] [Google Scholar]
- Baudler V. M.; Jongebloed H. Synthese und Eigenschaften der Diphosphasilirane (t- BuP)2SiMe2 und (t-BuP)2SiPh2. Z. Anorg. Allg. Chem. 1979, 458, 9–21. 10.1002/zaac.19794580102. [DOI] [Google Scholar]
- Driess M.; Rell S.; Pritzkow H.; Janoschek R.. R2Si=P-SiR2F: 1,3-Sigrnatropic Migration of Fluorine in a 2-Phospha-l,3-disilaallyl Derivative Capable of Conjugation and Its Conversion to Phosphadisilacyclopropanes Angew. Chem., Int. Ed. 1997, 36 (12), .
- Szanka A.; Szanka I.; Kennedy J. P. Breathable rubbery skin protectors: Design, synthesis, characterization, and properties of cyanoacrylated silicone rubber networks. J. Polym. Sci. 2016, 54, 1367–1372. 10.1002/pola.27984. [DOI] [Google Scholar]
- Bele A.; Stiubianu G.; Vlad S.; Tugui C.; Varganici C. D.; Matricala L.; Ionita D.; Timpu D.; Cazacu M. Aging behavior of the silicone dielectric elastomers in a simulated marine environment. RSC Adv. 2016, 6, 8941–8955. 10.1039/C5RA22780A. [DOI] [Google Scholar]
- Postel S.; Schneider C.; Wessling M. Solvent dependent solute solubility governs retention in silicone based organic solvent nanofiltration. J. Membr. Sci. 2016, 497, 47–54. 10.1016/j.memsci.2015.09.014. [DOI] [Google Scholar]
- He G.; Shynkaruk O.; Lui M. W.; Rivard E. Small Inorganic Rings in the 21st Century: From Fleeting Intermediates to Novel Isolable Entities. Chem. Rev. 2014, 114, 7815–7880. 10.1021/cr400547x. [DOI] [PubMed] [Google Scholar]
- Planells A. R.; Espinosa Ferao A. Accurate Ring Strain Energy Calculations on Saturated Three-Membered Heterocycles with One Group 13–16 Element. Inorg. Chem. 2020, 59, 11503–11513. 10.1021/acs.inorgchem.0c01316. [DOI] [PubMed] [Google Scholar]
- a Boatz J. A.; Gordon M. S. Theoretical Studies of Three-Membered Ring Compounds Y2H4X (Y = C, Si; X = CH2, NH, O, SiH2, PH, S). J. Phys. Chem. A 1989, 93, 3025–3029. 10.1021/j100345a031. [DOI] [Google Scholar]; b Bach R. D.; Dmitrenko O. The Effect of Carbonyl Substitution on the Strain Energy of Small Ring Compounds and Their Six-Member Rings Reference Compounds. J. Am. Chem. Soc. 2006, 128, 4598–4611. 10.1021/ja055086g. [DOI] [PubMed] [Google Scholar]; c Morgan K. M.; Ellis K. A.; Lee J.; Fulton A.; Wilson S. L.; Dupart P. S.; Dastoori R. Thermochemical Studies of Epoxides and Related Compounds. J. Org. Chem. 2013, 78, 4303–4311. 10.1021/jo4002867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Espinosa A.; Gómez C.; Streubel R. Single Electron Transfer-Mediated Selective endo- and exocyclic Bond Cleavage Processes in Azaphosphiridine Chromium(0) Complexes: A Computational Study. Inorg. Chem. 2012, 51, 7250–7256. 10.1021/ic300522g. [DOI] [PubMed] [Google Scholar]; b Espinosa A.; Streubel R. Computational Studies on Azaphosphiridines, or How to Effect Ring-Opening Processes through Selective Bond Activation. Chem. - Eur. J. 2011, 17, 3166–3178. 10.1002/chem.201002565. [DOI] [PubMed] [Google Scholar]; c Villalba Franco J. M.; Schnakenburg G.; Sasamori T.; Espinosa Ferao A.; Streubel R. Stimuli-Responsive Frustrated Lewis-Pair-Type Reactivity of a Tungsten Iminoazaphosphiridine Complex. Chem. - Eur. J. 2015, 21, 9650–9655. 10.1002/chem.201501628. [DOI] [PubMed] [Google Scholar]
- a Krahe O.; Neese F.; Streubel R. The Quest for Ring Opening of Oxaphosphirane Complexes: A Coupled-Cluster and Density Functional Study of CH3PO Isomers and Their Cr(CO)5 Complexes. Chem. - Eur. J. 2009, 15, 2594–2601. 10.1002/chem.200801494. [DOI] [PubMed] [Google Scholar]; b Albrecht C.; Schneider E.; Engeser M.; Schnakenburg G.; Espinosa A.; Streubel R. Synthesis and DFT calculations of spirooxaphosphirane complexes. Dalton Trans. 2013, 42, 8897–8906. 10.1039/c3dt50556a. [DOI] [PubMed] [Google Scholar]
- Streubel R.; FaBbender J.; Schnakenburg G.; Espinosa Ferao A. Formation of Transient and Stable 1,3-Dipole Complexes with P,S,C and S,P,C Ligand Skeletons. Organometallics 2015, 34, 3103–3106. 10.1021/acs.organomet.5b00386. [DOI] [Google Scholar]
- Allard C. L.; Gautier P.; Gille A. L.; Thomas G. E.; Gilbert T. M. Optimized Structures and Ring Strain Energies of Isoelectronic Homo- and Heterosiliranes c-AX2SiR2SiR2q (A/q = Al/1–, Si/0, P/1+): Unexpected Effects of Charge and Size. Organometallics 2013, 32, 2558–2566. 10.1021/om400038z. [DOI] [Google Scholar]
- Allard C. L.; Gill S. T.; Gilbert T. M. Optimized structures and ring strain energies of isoelectronic homo- and heterogermiranes c-AX2GeH2GeH2q (A/q = Ga/–1, Ge/0, As/+1): Variable trends across triads. J. Organomet. Chem. 2013, 740, 110–115. 10.1016/j.jorganchem.2013.04.057. [DOI] [Google Scholar]
- Wheeler S. E.; Houk K. N.; Schleyer P. R.; Allen W. D. A Hierarchy of Homodesmotic Reactions for Thermochemistry. J. Am. Chem. Soc. 2009, 131, 2547–2560. 10.1021/ja805843n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lowry T. H.; Richardson K. S.. Mechanism and Theory in Organic Chemistry, 3rd ed.; Harper & Row: New York, 1987; Chapter 2.3. [Google Scholar]; b Skancke A.; Van Vechten D.; Liebman J. F.; Skancke P. N. Strain energy of three-membered rings: a new ultradiagonal definition as applied to silicon- and carbon-containing species. J. Mol. Struct. 1996, 376, 461–468. 10.1016/0022-2860(95)09062-2. [DOI] [Google Scholar]
- a Cremer D.; Gauss J.; Cremer E. Strain in three-membered rings containing silicon: The inability of silicon to form flexible hybrid orbitals. J. Mol. Struct.: THEOCHEM 1988, 169, 531–561. 10.1016/0166-1280(88)80281-1. [DOI] [Google Scholar]; b Liebman J. F.; Skancke P. N. Evaluation of strain in heterosiliranes: Systematics, surprises, and problems. Int. J. Quantum Chem. 1996, 58, 707–715. . [DOI] [Google Scholar]
- Srinivas G. N.; Anoop A.; Jemmis E. D.; Hamilton T. P.; Lammertsma K.; Leszczynski J.; Schaefer H. F. III Nonplanarity at Tri-coordinated Aluminum and Gallium: Cyclic Structures for X3Hnm (X = B, Al, Ga). J. Am. Chem. Soc. 2003, 125, 16397–16407. 10.1021/ja036868h. [DOI] [PubMed] [Google Scholar]
- Moc J.; Bober K.; Mierzwicki K. Trimers and tetramers of MH and MH3 (M = Al, Ga): Theoretical study. Chem. Phys. 2006, 327, 247–260. 10.1016/j.chemphys.2006.04.015. [DOI] [Google Scholar]
- Schüth F.; Bogdanović B.; Felderhoff M. Light metal hydrides and complex hydrides for hydrogen storage. Chem. Commun. 2004, 2249–2258. 10.1039/B406522K. [DOI] [PubMed] [Google Scholar]
- a Tan L.-P.; Die D.; Zheng B.-X. Growth mechanism, electronic properties and spectra of aluminum clusters. Spectrochim. Acta, Part A 2022, 267, 120545–120554. 10.1016/j.saa.2021.120545. [DOI] [PubMed] [Google Scholar]; b Mai D. T. T.; Tan Pham H.; Minh Tam N.; Nguyen M. T. Geometry and bonding of small binary boron-aluminum clusters BnAln (n = 1–7): Electron donation and interlocking aromaticity. Chem. Phys. Lett. 2019, 714, 87–93. 10.1016/j.cplett.2018.10.065. [DOI] [Google Scholar]
- Krogh-Jespersen K.; Cremer D.; Poppinger D.; Pople J. A.; Schleyer P. R.; Chandrasekhar J. Molecular Orbital Theory of the Electronic Structure of Molecules. 39. Highly Unusual Structures of Electron-Deficient Carbon Compounds. Reversal of van’t Hoff Stereochemistry in BBC Ring Systems1. J. Am. Chem. Soc. 1979, 101, 4843–4851. 10.1021/ja00511a011. [DOI] [Google Scholar]
- Sorger K.; Schleyer P. R. Planar and inherently non-tetrahedral tetracoordinate carbon: a status report. J. Mol. Struct.: THEOCHEM 1995, 338, 317–346. 10.1016/0166-1280(95)04233-V. [DOI] [Google Scholar]
- Stabenow F.; Saak W.; Marsmann H.; Weidenbruch M. Hexaarylcyclotriplumbane: A Molecule with a Homonuclear Ring System of Lead. J. Am. Chem. Soc. 2003, 125, 10172–10173. 10.1021/ja036299d. [DOI] [PubMed] [Google Scholar]
- a Nagase S. Interesting properties of the heavier group 14 analogues of aromatic and polycyclic carbon compounds. A theoretical study. Polyhedron 1991, 10, 1299–1309. 10.1016/S0277-5387(00)86105-7. [DOI] [Google Scholar]; b Driess M.; Grützmacher H. Main Group Element Analogues of Carbenes, Olefins, and Small Rings. Angew. Chem., Int. Ed. 1996, 35, 828–856. 10.1002/anie.199608281. [DOI] [Google Scholar]; c Weidenbruch M. From a Cyclotrisilane to a Cyclotriplumbane: Low Coordination and Multiple Bonding in Group 14 Chemistry. Organometallics 2003, 22, 4348–4360. 10.1021/om034085z. [DOI] [Google Scholar]; d Koch R.; Bruhn T.; Weidenbruch M. Theoretical Group 14 Chemistry. 4. Cyclotriplumbanes: Relativistic and Substituents Effects. J. Chem. Theory Comput. 2005, 1, 1298–1303. 10.1021/ct050144+. [DOI] [PubMed] [Google Scholar]
- a Reed A. E.; Weinhold F. Natural bond orbital analysis of near-Hartree–Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. 10.1063/1.445134. [DOI] [Google Scholar]; b Reed A. E.; Weinstock R. B.; Weinhold F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. 10.1063/1.449486. [DOI] [Google Scholar]
- a Stanford M. W.; Schweizer J. I.; Menche M.; Nichol G. S.; Holthausen M. C.; Cowley M. J. Intercepting the Disilene-Silylsilylene Equilibrium. Angew. Chem., Int. Ed. 2019, 58, 1329–1333. 10.1002/anie.201810056. [DOI] [PubMed] [Google Scholar]; b Nesterov V.; Baierl R.; Hanusch F.; Espinosa Ferao A.; Inoue S. N-Heterocyclic Carbene-Stabilized Germanium and Tin Analogues of Heavier Nitriles: Synthesis, Reactivity, and Catalytic Application. J. Am. Chem. Soc. 2019, 141, 14576–14580. 10.1021/jacs.9b08741. [DOI] [PubMed] [Google Scholar]; c Espinosa Ferao A.; García Alcaraz A.; Zaragoza Noguera S.; Streubel R. Terminal Phosphinidene Complex Adducts with Neutral and Anionic O-Donors and Halides and the Search for a Differentiating Bonding Descriptor. Inorg. Chem. 2020, 59, 12829–12841. 10.1021/acs.inorgchem.0c01874. [DOI] [PubMed] [Google Scholar]
- Chatt J.; Duncanson L. A. Olefin Co-ordination Compounds. J. Chem. Soc. 1953, 2939–2947. 10.1039/jr9530002939. [DOI] [Google Scholar]
- Toscano M.; Russo N.; Rubio J. Calculation of proton affinities and absolute gas basicities of X3 group VI triatomics: a density functional study. J. Chem. Soc., Faraday Trans. 1996, 92, 2681–2684. 10.1039/ft9969202681. [DOI] [Google Scholar]
- Rey Planells A.; Espinosa Ferao A. Accurate Ring Strain Energies of Unsaturated Three-Membered Heterocycles with One Group 13–16 Element. Inorg. Chem. 2022, 61, 6459–6468. 10.1021/acs.inorgchem.2c00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleyer P. R.Substituent Effects in Radical Chemistry, Viehe H. G.; Janousek Z.; Merenyi R., Eds.; Reidel: Dordrecht, 1986; pp 69–81. [Google Scholar]
- a de Meijere A. Bonding Properties of Cyclopropane and Their Chemical Consequences. Angew. Chem. 1979, 91, 867–884. 10.1002/ange.19790911104. [DOI] [Google Scholar]; b Bindungseigenschaften des Cyclopropans und chemische Konsequenzen. Angew. Chem., Int. Ed. 1979, 18, 809 - 826.10.1002/anie.197908093
- Fowler P. W.; Baker J.; Lillington M. The ring current in cyclopropane. Theor. Chem. Acc. 2007, 118, 123–127. 10.1007/s00214-007-0253-2. [DOI] [Google Scholar]
- Wu W.; Ma B.; Wu J. I.-C.; Schleyer P. R.; Mo Y. Is Cyclopropane Really the σ-Aromatic Paradigm?. Chem. - Eur. J. 2009, 15, 9730–9736. 10.1002/chem.200900586. [DOI] [PubMed] [Google Scholar]
- Schleyer P. R.; Maerker C.; Dransfeld A.; Jiao H. J.; van Eikema Hommes N. J. R. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. 10.1021/ja960582d. [DOI] [PubMed] [Google Scholar]
- Li Z.-H.; Moran D.; Fan K.-N.; Schleyer P. R. σ-Aromaticity and σ-Antiaromaticity in Saturated Inorganic Rings. J. Phys. Chem. A 2005, 109, 3711–3716. 10.1021/jp048541o. [DOI] [PubMed] [Google Scholar]
- Tsipis A. C.; Depastas I. G.; Tsipis C. A. Diagnosis of the σ-, π- and (σ+π)-Aromaticity by the Shape of the NICSzz-Scan Curves and Symmetry-Based Selection Rules. Symmetry 2010, 2, 284–319. 10.3390/sym2010284. [DOI] [Google Scholar]
- Manolopoulos D. E.; May J. C.; Down S. E. Theoretical studies of the fullerenes: C34 to C70. Chem. Phys. Lett. 1991, 181, 105–111. 10.1016/0009-2614(91)90340-F. [DOI] [Google Scholar]
- a Pearson R. G. Hard and soft acids and bases-the evolution of a chemical concept. Coord. Chem. Rev. 1990, 100, 403–425. 10.1016/0010-8545(90)85016-L. [DOI] [Google Scholar]; b Zhou Z.; Parr R. G.; Garst J. F. Absolute hardness as a measure of aromaticity. Tetrahedron Lett. 1988, 29, 4843–4846. 10.1016/S0040-4039(00)80623-1. [DOI] [Google Scholar]; c Zhou Z.; Parr R. G. New measures of aromaticity: absolute hardness and relative hardness. J. Am. Chem. Soc. 1989, 111, 7371–7379. 10.1021/ja00201a014. [DOI] [Google Scholar]; d Parr R. G.; Zhou Z. Absolute hardness: unifying concept for identifying shells and subshells in nuclei, atoms, molecules, and metallic clusters. Acc. Chem. Res. 1993, 26, 256–258. 10.1021/ar00029a005. [DOI] [Google Scholar]; e Makov G. Chemical Hardness in Density Functional Theory. J. Phys. Chem. A 1995, 99, 9337–9339. 10.1021/j100023a006. [DOI] [Google Scholar]; f Huang Y.-Z.; Yang S.-Y.; Li X.-Y. An investigation of the aromaticity of transition metal heterocyclic complexes by conventional criteria and indices of aromaticity. J. Organomet. Chem. 2004, 689, 1050–1056. 10.1016/j.jorganchem.2004.01.002. [DOI] [Google Scholar]
- Fowler P. Aromaticity revisited. Nature 1991, 350, 20–21. 10.1038/350020a0. [DOI] [Google Scholar]
- a Brandhorst K.; Grunenberg J. How strong is it? The interpretation of force and compliance constants as bond strength descriptors. Chem. Soc. Rev. 2008, 37, 1558–1567. 10.1039/b717781j. [DOI] [PubMed] [Google Scholar]; b Büschel S.; Jungton A.-K.; Bannenberg T.; Randoll S.; Hrib C. G.; Jones P. G.; Tamm M. Secondary Interactions in Phosphane-Functionalized Group 4 Cycloheptatrienyl–Cyclopentadienyl Sandwich Complexes. Chem. - Eur. J. 2009, 15, 2176–2184. 10.1002/chem.200802518. [DOI] [PubMed] [Google Scholar]; c Espinosa A.; de las Heras É.; Streubel R. Oxaphosphirane-Borane Complexes: Ring Strain and Migratory Insertion/Ring-Opening Reactions. Inorg. Chem. 2014, 53, 6132–6140. [DOI] [PubMed] [Google Scholar]
- DLPNO-CCSD(T)/def2-TZVPP(ecp)//B3LYP-D3/def2-TZVP(ecp) level.
- RSERC4 (kcal/mol): C2Al 44.42; C2Ga 43.84; Si2Al 38.64; Si2Ga 40.91; Ge2Al 40.03; Ge2Ga 42.64; CSiAl 43.68; CSiGa 45.36.
- Conformational Si-Si bond rotation barrier ΔErel = 0.40 and 0.64 kcal/mol for Si2Al and Si2Ga, respectively.
- Neese F.ORCA—An Ab initio, DFT and Semiempirical SCF-MO Package, version 4.2.1; Max Planck Institute for Bioinorganic Chemistry, 2019. [Google Scholar]
- a Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]; b Becke A. D. Density functional thermochemistry. III. The role of exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Caldeweyher E.; Mewes J.-M.; Ehlert S.; Grimme S. Extension and evaluation of the D4 London-dispersion model for periodic systems. Phys. Chem. Chem. Phys. 2020, 22, 8499–8512. 10.1039/D0CP00502A. [DOI] [PubMed] [Google Scholar]
- a Schafer A.; Huber C.; Ahlrichs R. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar]; basis sets may be obtained from the Basis Set Exchange (BSE) software and the EMSL Basis Set Library: https://bse.pnl.gov/bse/portal.; b Feller D. The Role of Databases in Support of Computational Chemistry Calculations. J. Comput. Chem. 1996, 17, 1571–1586. . [DOI] [Google Scholar]
- Riplinger C.; Sandhoefer B.; Hansen A.; Neese F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101 10.1063/1.4821834. [DOI] [PubMed] [Google Scholar]
- Pople J. A.; Head-Gordon M.; Raghavachari K. Quadratic configuration interaction. A general technique for determining electron correlation energies. J. Chem. Phys. 1987, 87, 5968–5975. 10.1063/1.453520. [DOI] [Google Scholar]
- a Reed A. E.; Weinhold F. Natural bond orbital analysis of near-Hartree–Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. 10.1063/1.445134. [DOI] [Google Scholar]; b Reed A. E.; Weinstock R. B.; Weinhold F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. 10.1063/1.449486. [DOI] [Google Scholar]
- Lu T.; Chen F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.