

Abstract
Huntington’s disease (HD) is caused by an expanded CAG repeat in huntingtin (HTT). Since HD is dominant and loss of HTT leads to neurological abnormalities, safe therapeutic strategies require selective inactivation of mutant HTT. Previously, we proposed a concept of CRISPR-Cas9 using mutant-specific PAM sites generated by SNPs to selectively inactivate mutant HTT. Aiming at revealing suitable targets for clinical development, we analyzed the largest HD genotype dataset to identify target PAM-altering SNPs (PAS) and subsequently evaluated their allele specificities. The gRNAs based on the PAM sites generated by rs2857935, rs16843804, and rs16843836 showed high levels of allele specificity in patient-derived cells. Simultaneous use of two gRNAs based on rs2857935-rs16843804 or rs2857935-rs16843836 produced selective genomic deletions in mutant HTT and prevented the transcription of mutant HTT mRNA without impacting the expression of normal counterpart or re-integration of the excised fragment elsewhere in the genome. RNA-seq and off-target analysis confirmed high levels of allele specificity and the lack of recurrent off-targeting. Approximately 60% of HD subjects are eligible for mutant-specific CRISPR-Cas9 strategies of targeting one of these three PAS in conjunction with one non-allele-specific site, supporting high applicability of PAS-based allele-specific CRISPR approaches in the HD patient population.
Keywords: Huntington’s disease, allele-specific CRISPR-Cas9, haplotype-targeting, PAM-altering SNP, genomic deletion, TP-CRISPR
Graphical abstract
CRISPR-Cas9 to selectively inactivate mutant huntingtin may produce robust therapeutic benefits in Huntington’s disease (HD). We revealed PAM-altering SNPs that permit allele-specific CRISPR-Cas9 in HD (rs2857935, rs16843804, and rs16843836). Targeting those sites in combinations using dual gRNAs prevented the transcription of mutant HTT mRNA without impacting the normal counterpart.
Introduction
Huntington’s disease (HD) (MIM 143100) is a dominantly inherited neurodegenerative disease.1, 2, 3 An expansion of a glutamine-encoding CAG trinucleotide repeat (>35) in the huntingtin gene (HTT) leads to neurodegeneration and premature death.2,4 Inheritance of one allele with an expanded CAG repeat is sufficient to produce changes in neurological domains such as involuntary movement, cognitive decline, and/or psychiatric changes.1,3,5, 6, 7, 8, 9, 10 In contrast, loss of one copy of HTT does not cause HD,11 implying that HD is due to dominant actions of mutant HTT rather than haploinsufficiency. Indeed, inheritance of two loss-of-function HTT alleles is associated with profound neurodevelopmental abnormalities,12, 13, 14 not HD. The disease-causing genetic mutation in HD was discovered more than 25 years ago,2 and subsequent studies using model systems provided insights into numerous underlying disease mechanisms.3,15,16 Despite extensive investigations, effective treatments have not yet been developed, warranting alternative approaches for drug development in HD.17,18
Given the known root cause of the disease,2 therapeutics that directly target HTT expression have been emerging as promising intervention strategies over the years.19, 20, 21, 22, 23, 24, 25, 26, 27 In support, numerous pre-clinical studies have showed that HTT-lowering approaches were feasible and could ameliorate phenotypes in HD animal models.17,19,22,28,29 The safety and target engagement of antisense oligonucleotide (ASO) designed to lower both mutant and normal huntingtin protein expression levels (https://clinicaltrials.gov/ct2/show/NCT02519036) also has been demonstrated in early manifest HD subjects.30 By contrast, a large phase 3 clinical trial testing the same ASO was halted recently due to unfavorable risk-benefit profile (https://www.roche.com/media/releases/med-cor-2021-03-22b.htm).31,32 Although the reasons for this negative outcome are not completely clear, it is conceivable that non-selective reduction of huntingtin might have been a contributing factor. Embryonic lethality in Htt knockout mice,33, 34, 35 neurodevelopmental problems due to hypomorphic HTT alleles in humans,12, 13, 14 and abnormalities caused by Htt knockout in adult mice36,37 all argue that HTT-targeting therapeutics that are allele specific may be safer and more effective. These findings and the lack of HD in a phenotypically normal human carrying only one copy of HTT11 imply that inactivating the disease producing CAG-expanded HTT allele in HD heterozygotes is likely to produce significant therapeutic benefits without producing huntingtin deficiency-associated problems.
Among various HTT-targeting approaches,18,28,38 we aim to develop allele-specific CRISPR-Cas9 strategies because of the robustness and flexibility of DNA-targeting strategies.39,40 We reasoned that developing allele-specific CRISPR-Cas9 strategies that directly target the CAG repeat would be technically challenging because of (1) the lack of a PAM (protospacer adjacent motif) site in the vicinity, (2) the presence of many CAG repeat-containing genes in the genome,6,41,42 and (3) the non-expanded CAG repeat in normal HTT allele. Therefore, we set out to develop allele-specific strategies of targeting the haplotype carrying the disease-causing mutation using DNA sequence variations that are different between mutant and normal HTT. Importantly, a single mismatch in the PAM site dramatically reduces the CRISPR-Cas9 editing efficiency,26,43,44 offering opportunities for allelic discrimination. Capitalizing on single-nucleotide polymorphisms (SNPs) that generate or eliminate PAM sites (namely, PAM-altering SNP [PAS]) on HTT, we and others proposed the concept of an allele-specific CRISPR-Cas9 strategy to inactivate the mutant HTT by excising a region that is important for gene expression.25,26 Still, the target PAS that can be applicable to the largest HD population without on-target/off-target toxicity are largely unknown. Given the potential of haplotype-targeting PAS-based allele-specific transcription prevention-CRISPR (TP-CRISPR) strategies in HD,25,26 identification of clinically relevant PAS with significant applicability in the patient population and comprehensive evaluation of their strengths and limitations has become very important to advance these promising intervention strategies through drug development pipelines. Therefore, we took advantage of the power of the largest HD genotype dataset ever assembled to date45,46 to identify target PAS that are relevant at the population level and performed comprehensive molecular characterization to demonstrate the safety of candidate allele-specific TP-CRISPR strategies that merit clinical development.
Results
Mutant specificities of PAS in HD subjects
A CRISPR-Casp9 strategy to generate small indels in an exon of the target gene has been widely used in the genome editing field. In many cases, such indels produced by non-homologous DNA end joining repair shift the frames of exons, resulting in nonsense-mediated decay (NMD) of the mRNA of the target gene. If applied to HD in an allele-specific manner, this approach may produce robust knockout effects. Thus, it will be important to identify exonic variations that permit mutant-specific CRISPR-Cas9 that aims at inducing NMD of the mutant HTT mRNA. However, if exon 1 HTT protein plays an important role in HD,47, 48, 49, 50 single gRNA CRISPR-Cas9 approaches for NMD may not address such putative toxic species because of the lack of haplotype-specific variations in the HTT exon 1.51,52 Alternatively, CRISPR-Cas9-mediated genomic deletion may address HD regardless of the identity of the toxic species because it can prevent the production of the mutant HTT mRNA by eliminating an important region for the gene transcription and the expanded repeat from the mutant locus. Therefore, we focused on identifying relevant genetic variations that permit allele-specific TP-CRISPR and subsequently validating them using patient-derived cells.
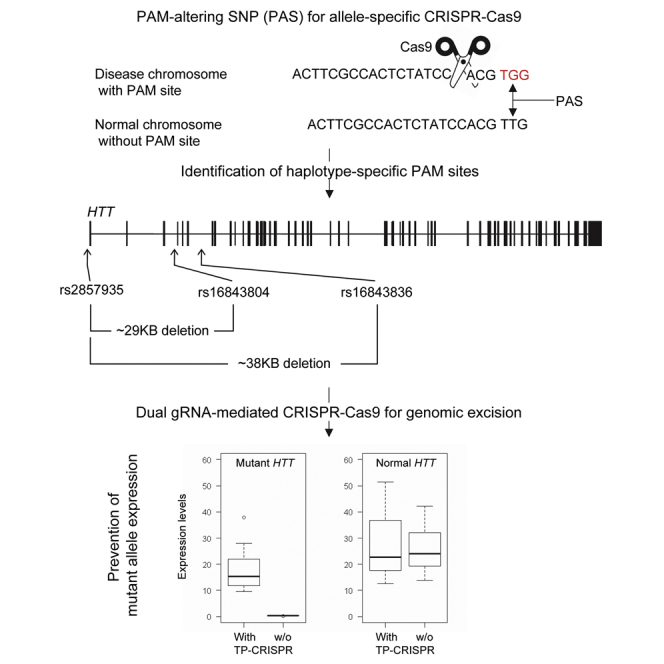
To identify mutant HTT-specific CRISPR-Cas9 PAM sites that can be used for the biggest proportion of HD subjects and to judge their levels of mutant specificity (i.e., the percentage of HD subjects who carry the PAS-generated PAM site only on the mutant HTT), we analyzed phased genotypes of 8,543 HD subjects with European ancestry who carry one mutant HTT with 40–55 CAG repeats.46 Considering the regulatory elements of neighboring genes and the size of a feasible genomic deletion, we analyzed 20 and 40 kb flanking regions upstream and downstream of the transcription start site (TSS), respectively. Previously, we provided a comprehensive list of SNP variations that alter 19 PAM sequences for various Cas9 orthologs.26 Given significantly decreased tolerance of the PAM site containing a single mismatch44 and data showing high levels of allele specificity of CRISPR-Cas9 strategies using PAS,25,26 genetic variations that alter PAM sites for other Cas9 orthologs offer wide opportunities for allele-specific genome engineering. In this study, we focused on an NGG PAM sequence that supports the most widely used Cas9. Among 1,045 SNPs in the region, alleles of 418 SNPs generate or eliminate the NGG PAM sequence (i.e., PAS). Frequency analysis identified 224 polymorphic PAS that can be used for allele-specific CRISPR-Cas9 targeting. Since (1) both reference and alternative alleles of 5 PAS, and (2) two alternative alleles of 1 PAS generate NGG PAM sites, 230 PAM-generating alleles (PGAs) are present in the region (Table S1). For each of 230 PGAs that were also polymorphic in the 8,543 HD subjects (frequency, neither 0% nor 100%), we counted HD subjects who carry the PAM site on the (1) mutant HTT, (2) normal HTT, (3) both, or (4) neither to calculate the percentage of HD subjects potentially eligible for allele-specific CRISPR-Cas9 treatment at that site (Table S2). The highest mutant specificity was observed at rs1313774 (38.5%) (Table S2). However, overall individual mutant specificities of the 230 PGAs were relatively low (Figure S1A). Therefore, for subsequent analyses, we applied a 10% mutant specificity threshold, revealing 10 PAS that are relevant at the population level; 4 and 6 PAS are located upstream and downstream of the TSS, respectively (Figure S1B). HD individuals who carry the PAM site on both mutant and normal chromosomes account for the largest proportions for the nine PAS sites (rs1313769, rs1313774, rs12506200, rs2857935, rs7659144, rs7688390, rs16843804, rs6828615, and rs16843836) (Figure S1B, yellow). In addition, rs28820097 showed the largest percentage for HD individuals who carry the PAM site only on the normal HTT (Figure S1B, green). These data indicate that different PAS must be targeted depending on the alleles present on the mutant and normal HTT haplotypes in a given HD subject. Of note, there remained HD individuals who were not eligible for an allele-specific targeting strategy based on any of the 10 PAS (32.4%).
Alleles of candidate PAS on the common HTT haplotypes
The disease-causing CAG expansions are found on many diverse HTT haplotypes,51, 52, 53, 54, 55, 56, 57 posing a significant challenge in developing allele-specific haplotype-targeting strategies. We previously defined common HTT haplotypes to help with patient stratification in allele-specific gene targeting.51 In brief, the 16 common HTT haplotypes comprise the 10 most frequent disease haplotypes and the 10 most frequent normal haplotypes. The most common mutant (hap.01) and normal haplotype (hap.08) account for 38.5% and 25.5% of mutant and normal chromosomes, respectively.51 HD subjects carrying common HTT haplotypes showed relatively similar age at onset.56 Still, haplotypes can serve as a foundation for genomic medicine, facilitating the discovery of allele-specific targets for clinical trial recruitment. For example, once alleles of target SNP sites are assigned to HTT haplotypes, simply determining the mutant and normal HTT haplotypes in a given HD individual will immediately point to all other allele-specific targets in that individual. Therefore, we determined the alleles of the 10 candidate PAS present on each of 16 common HTT haplotypes. The most frequent HTT haplotype on the disease chromosomes (i.e., hap.01) carries PGAs at nine PAS sites (Figure 1, alleles in bold). In contrast, the most common HTT haplotype in the normal chromosomes carries PGAs at four PAS sites. Therefore, HD individuals with the most frequent diplotype (i.e., expanded CAG on hap.01 and non-expanded CAG on hap.08) carry mutant HTT-specific PAM sites at six candidate PAS sites (Figure 1; Table 1). Many HD individuals with different combinations of common haplotypes carry mutant HTT-specific PGAs at various locations, supporting the broad applicability of mutant HTT-specific CRISPR targeting strategies using PAS.
Figure 1.

Alleles of 10 candidate PAS on the 16 common HTT haplotypes
A total of 418 NGG PAS were identified from the analysis of the 60 kb region of chr4 in our HD modifier GWAS data. Subsequently, the levels of mutant specificity were calculated for each PAS, revealing 10 PAS that were polymorphic in 8,543 HD subjects and also showed mutant specificity values higher than 10%. Four and six PAS are located upstream and downstream of the transcription start site of HTT, respectively. Locations of those PAM sites are relative to the HTT RefSeq, NM_002111 (vertical bars represent exons). A mutant specificity value for a PAS represents the proportion (%) of HD subjects who carry the PAM site only on the mutant HTT. We also determined the alleles of 10 PAS on the most common HTT haplotypes based on phased genotype data of HD subjects. Alleles in bold represent NGG PAM-generating alleles. Labels of gRNA represent names of gRNA that we designed based on the PAM site generated by the PAS.
Table 1.
Editing efficiencies, and allele specificities of gRNAs based on candidate PAS
| PAS | gRNA label |
BP (GRCh37) | Distance to TSS | Ref | Alt | HD subjects with PAM site on (%) |
Editing efficiency (%) |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutant | Normal | Both | Neither | Mutant | Normal | ||||||
| rs1313769 | L1 | 3056975 | −19433 | T | G | 38.4 | 8.1 | 44.6 | 8.9 | 0 | 0 |
| rs1313774 | L2 | 3059649 | −16759 | T | C | 38.5 | 8.1 | 44.5 | 8.9 | 0.7 | 0.1 |
| rs12506200 | L3 | 3064053 | −12355 | G | A | 15.9 | 1.4 | 82.4 | 0.3 | 0 | 0 |
| rs2857935 | L4 | 3075691 | −717 | G | C | 27.6 | 2.7 | 68.2 | 1.5 | 6.6 | 0.2 |
| rs28820097 | R1 | 3097495 | 21087 | G | A | 14.8 | 35.5 | 27.4 | 22.2 | 0 | 10.5 |
| rs7659144 | R2 | 3098321 | 21913 | C | G | 32.5 | 3 | 62.4 | 2.2 | 0 | 0 |
| rs7688390 | R3 | 3098889 | 22481 | G | A | 10.9 | 4 | 84.4 | 0.6 | 8.4 | 8.4 |
| rs16843804 | R4 | 3104390 | 27982 | C | T | 28.7 | 2.8 | 66.7 | 1.8 | 14.1 | 0.8 |
| rs6828615 | R5 | 3113188 | 36780 | C | T | 10.9 | 4 | 84.5 | 0.6 | 0 | 0 |
| rs16843836 | R6 | 3113337 | 36929 | G | A | 28.7 | 2.9 | 66.7 | 1.8 | 11.8 | 0.1 |
We designed a gRNA for each of 10 candidate PAS, and tested these in 2 HD patient-derived iPSC lines carrying the most frequent diplotype (i.e., expanded CAG repeat on hap.01 and normal CAG repeat on hap.08). Both lines carry adult-onset CAG repeats (46 CAG in iPSC-A; 42 CAG in iPSC-B). The levels of editing efficiency and allele specificity were determined by transfection and subsequent MiSeq analysis. Labels of gRNAs represent the names of test gRNAs used in the study. L and R represent the left and right side of the TSS. Reference and alternative alleles are shown; PAM-generating allele (PGA) are highlighted by bold font. The frequency of PGA for a given SNP and the levels of allele specificity were based on the phased allele analysis of 8,543 HD subjects. PAM on the mutant, normal, and both represent the percentages of HD subjects who carry the PAM site on the mutant, normal, and both chromosomes, respectively. Table cells with italic numbers represent PAM sites in the HD subjects with the most common diplotype. For example, our test HD patient cells carry mutant-specific PAM sites at rs1313769, rs1313774, rs2857935, rs7659144, rs16843804, and rs16843836. In contrast, rs28820097 generates the PAM site only on the normal HTT in the HD subjects with the hap.01/hap.08 diplotype. Editing efficiency values represent means of at least four independent transfection experiments (without selection) and MiSeq analysis. BP, genomic coordinate in base pair (GRCh37/hg19).
CRISPR-Cas9 editing efficiency and allele specificity in patient-derived cells
Next, we determined editing efficiencies and allele specificities of gRNAs that were designed based on the 10 candidate PAS. We tested individual gRNA in two independent patient-derived iPSC lines, both carrying the most frequent diplotype (hap.01/hap.08). We transfected patient-derived lines with plasmids for Cas9 and an individual test gRNA without selection and subsequently performed MiSeq analysis to determine editing efficiency and allele specificity. Based on PGAs on the common HTT haplotypes, we predicted that gRNAs L1 (designed based on the PAM site generated by rs1313769), L2 (rs1313774), L4 (rs2857935), R2 (rs7659144), R4 (rs16843804), and R6 (rs16843836) (Table 1) would lead to preferential editing of the disease chromosome because the corresponding PAS generate PAM sites only on the mutant HTT in HD individuals who carry the most frequent diplotype. We also predicted that the gRNAs L3 (rs12506200), R3 (rs7688390), and R5 (rs6828615) would edit both the mutant and normal HTT; gRNA R1 (rs28820097) would selectively edit the non-expanded chromosome. MiSeq analysis showed that four gRNAs (L1, L3, R2, and R5) did not generate any detectable editing, while the remaining six yielded editing results. For example, gRNAs L4, R4, and R6 were highly mutant specific as these produced 33, 17.5, and 118 times more indels on the CAG-expanded chromosomes compared with its normal counterpart, respectively (Table 1). The gRNA R1 produced indels only on the normal HTT, and the gRNA R3 generated indels on both mutant and normal HTT as expected (Table 1).
Off-target effects
CRISPR-Cas9 editing efficiencies in iPSCs were modest in general (Table 1), which is consistent with previous observations.58, 59, 60 Although L1, L3, R2, and R5 did not produce any detectable editing in our iPSC lines, those gRNAs produced low but detectable editing in HEK293 cells, which show high transfection efficiencies (data not shown). The lack of editing at L1, L3, R2, and R5 target sites in HD iPSCs might be due to low transfection efficiencies, genomic structure of the target region, or off-targeting. Thus, we evaluated the likelihood of off-targeting effects for each of our 10 candidate gRNAs. The gRNAs that showed relatively good editing efficiencies in HD iPSC lines showed low levels of predicted off-targeting. For example, gRNAs L4, R4, and R6 did not have any predicted off-targets with up to two mismatches (Table S3A). However, some gRNAs that showed zero editing in HD iPSCs (e.g., L1, L3, R5) revealed large numbers of predicted off-targets. The gRNA R2 showed a relatively small number of off-targets but on-target editing efficiency was low; additional analysis is required to determine whether local genomic structure at rs7659144 inhibited efficient CRISPR-Cas9 gene editing. Subsequently, focusing on candidate sites L4, R4, and R6, we experimentally validated predicted off-targets that are located on the exons of protein-coding genes because the impact of small indels in intergenic regions or introns was expected to be minimal. When allowing 3 mismatches, 13, 6, and 5 off-targets were predicted for L4, R4, and R6, respectively (Table S3A). Among them, we performed MiSeq analysis for 5, 1, and 1 predicted off-targets that are located in coding exons. We did not detect any DNA modification at the predicted exonic off-target sites for R4 and R6 (Table S3B). However, MiSeq analysis of patient-derived iPSCs revealed small numbers of edited sequence reads in cells treated with L4 (Table S3B). Predicted off-target sites for L4 contain repetitive sequences and therefore, the small numbers of edited alleles at those off-target sites might be due to sequencing errors.
Molecular consequences of mutant HTT-specific TP-CRISPR
Based upon their applicability in the HD population, we narrowed the candidates to three PAS (L4, R4, and R6), which showed relatively good editing efficiencies compared with others, high levels of mutant specificity, and low likelihood of off-targeting. All candidate SNPs are annotated in the SNP databases. Also, the possibilities of allele-specific CRISPR-Cas9 using candidate PAS rs2857935, rs16843804, and rs16843836 have been reported previously.26 Furthermore, the feasibility of allele-specific CRISPR-Cas9 using rs2857935 and rs16843804 has been demonstrated in HD patient-derived fibroblast.25,26 The SNP rs16843836 has not been tested for allele-specific CRISPR-Cas9 in HD before. Our data in this study also showed the percentage of HD subjects who are eligible for a particular allele-specific CRISPR-Cas9 strategy (Table 1), providing their clinical relevancies at the population level. Next, we determined the molecular outcomes of mutant-specific TP-CRISPR using two allele-specific gRNAs simultaneously (e.g., L4-R4 and L4-R6) (Figure S2A). Since editing efficiency in iPSC lines is particularly low,59,60 molecular characterization of a bulk population with a small proportion of edited cells would lead to noisy data. Consequently, we characterized molecular outcomes of dual gRNA allele-specific TP-CRISPR in targeted clonal lines. We transfected two HD patient-derived iPSC lines with plasmids for Cas9 and two gRNAs (e.g., L4-R4 and L4-R6). Subsequently, transfected cells were subjected to dilution cloning to establish targeted clonal lines. Eventually, we analyzed 5 clonal lines from iPSC-A for L4-R4 and L4-R6 gRNA combinations, respectively (Figure S2B). Similarly, 10 targeted clonal lines were analyzed for iPSC-B. As expected, all targeted clonal lines showed (1) genomic deletion caused by simultaneous editing by L4-R4 or L4-R6 (Figure 2A) and (2) the lack of expanded CAG in DNA (Figure 2B). Sanger sequencing of DNA from the targeted clonal lines also confirmed the large genomic deletions (29 kb by L4-R4; 38 kb by L4-R6) on the mutant HTT chromosome (Figure 2C). As a result, targeted clonal lines did not produce mutant HTT mRNA containing the expanded CAG repeat (Figure 2D) or full-length mutant huntingtin protein (Figure 2E). Together, these data indicate that simultaneous use of the two mutant-specific gRNAs (L4-R4 or L4-R6) selectively induce genomic deletion of mutant HTT, preventing the production of mutant HTT mRNA and protein from the disease-causing gene.
Figure 2.

Molecular consequences of allele-specific TP-CRISPR
To unequivocally determine the molecular consequences of allele-specific TP-CRISPR, we treated two independent HD iPSC lines (iPSC-A and iPSC-B) with either empty vector (EV) or a gRNA combination (L4-R4 or L4-R6) and established targeted clonal lines for subsequent analyses. (A) All EV-treated or targeted clonal lines were checked by PCR assays that were designed to detect a large genomic deletion using primers (colored arrows) that are described in the Materials and methods to confirm the presence of a genomic deletion in the targeted clonal lines. A schematic diagram on the top panel summarizes the locations for allele-specific target sites (L4, R4, and R6) and primers (colored arrows) with the sizes of predicted genomic deletion. The bottom panel shows representative data, displaying PCR products of 653 and 645 BP, which indicate genomic deletion by L4-R4 and L4-R6, respectively. EV, empty vector; L4-R4, TP-CRISPR using a gRNA combination L4 and R4; L4-R6, TP-CRISPR using a gRNA combination L4 and R6. (B) The presence and absence of expanded CAG repeat in the genomic DNA were determined by the PCR assays. Top (red arrow) and bottom bands (blue arrow) represent mutant and normal HTT, respectively. (C) To determine the sequence of targeted clonal lines, we performed Sanger sequencing. Since genomic deletion by L4-R4 (designed to excise ∼29 kb) involved both rs2857935 (L4) and rs16843804 (R4), assigning the deletion alleles to the mutant HTT was based on a downstream SNP rs2024115, whose “A” is on the hap.01 mutant haplotype. For L4-R6 combination, which was expected to excise ∼38 kb from the mutant HTT, we confirmed the genomic deletion on the mutant HTT based on the “G” allele at rs16843836. Contiguous and dotted lines represent unmodified DNA sequence and deletion, respectively. (D) We also performed RT-PCR assays to detect expanded and normal CAG repeats in the RNA samples. Top (red arrow) and bottom bands (blue arrow) represent mutant and normal HTT RNA, respectively. (E) Whole-cell lysate was resolved by SDS-PAGE and probed by MAB2166 for immunoblot analysis of HTT protein. Top (red arrow) and bottom (blue arrow) bands represent mutant and normal huntingtin protein, respectively. Note, since adult-onset CAG repeats do not increase the size of huntingtin protein substantially, separation of full-length mutant and normal HTT protein on the gel is usually incomplete.
Lack of fragment re-integration
Inactivation of a gene of interest using two gRNAs has been gaining popularity in the CRISPR gene editing field.61 However, integration of the excised DNA elsewhere in the genome (namely, re-integration) has been a concern.62,63 This is particularly important for therapeutic CRISPR-Cas9 approaches. We thus determined whether genomic DNA of targeted clonal lines still contained the excised DNA. We determined the genotype of rs2285086, which is part of the genomic deletion and heterozygous in HD subjects with the most frequent diplotype. Due to the technical challenges and a limited sensitivity in methods using a mixed population of cells involving unedited cells, we alternatively analyzed multiple targeted clonal lines. Excision and re-integration in the targeted clones would maintain heterozygosity at rs2285086. As shown in Figure S3, empty vector (EV)-treated clonal lines showed heterozygosity at rs2285086 as expected. However, all 20 independent targeted clonal lines were hemizygous, indicating the lack of re-integration of the excised region (Figure S3). Since the excised DNA involves the TSS and entire HTT exon 1 with an expanded CAG repeat, the excised DNA could conceivably produce exon 1 huntingtin protein (without genomic re-integration), which has been suggested as an HD toxic species.48,49,64,65 Thus, we examined this question as an additional check on the absence of the excised fragment from the targeted cells. Using an N-terminal huntingtin antibody (N17 antibody)66 to detect huntingtin protein-containing exon 1 in immunoblot analysis, we observed reduced full-length huntingtin protein (mutant and normal huntingtin are not resolved in this gel system) in the targeted clonal lines compared with EV-treated clones without a smaller exon 1 encoded product (∼12 kDa; Figure S4). Since our original patient-derived iPSC lines did not produce exon 1 HTT protein (Figure S4, EV), it remains to be determined whether genomic deletion modifies the levels of exon 1 HTT protein in HD cells that normally produce the exon 1 HTT protein.
On-target gene specificity and effects on global transcriptome
For all 20 targeted clonal iPSC lines, we performed MiSeq analysis of cDNA, verifying that all targeted clonal lines expressed only normal HTT mRNA (Table S4). Subsequently, we performed RNA-seq analysis to determine the molecular consequences of allele-specific TP-CRISPR more comprehensively. To further confirm the allele specificity in RNA-seq data, we compared alleles of sequencing reads at 10 exonic SNPs that are heterozygous in HD subjects with the most frequent diplotype. As shown in Figures S5 and 3A, TP-CRISPR-treated clonal lines produced virtually zero mutant HTT-associated alleles at 10 heterozygous sites. In contrast, allele counts on the normal HTT were not different between TP-CRISPR-treated and EV-treated clones (Figure 3B). Finally, we carried out differential gene expression (DGE) analysis to identify genes significantly altered by TP-CRISPR using L4-R4 or L-R6 gRNA combinations. As shown in Figures S6A and S6B, HTT was the only significantly altered gene due to either L4-R4 or L4-R6 (black arrows). When we compared 10 clonal lines targeted by L4-R4 with 10 clonal lines targeted by L4-R6, none of the genes were significantly altered (Figure S6C), suggesting that strategies using L4-R4 and L4-R6 resulted in similar molecular outcomes. We then combined all 20 targeted clonal lines and compared them with 12 EV-treated clones to increase power and sensitivity. Still, HTT was the only gene that was statistically significant, supporting the high levels of on-target gene specificity (Figure 4). To check the sensitivity of our approach, we also compared 16 clonal lines from iPSC-A (EV and TP-CRISPR clonal lines) with 16 clonal lines from iPSC-B (EV and TP-CRISPR clonal lines), predicting that the patient-specific comparison would show numerous statistical significances. As expected, many genes were significantly different between the two lines by Bonferroni corrected p values (Figure S7). Together, our RNA-seq study had the power and sensitivity to detect significant differences, and therefore the lack of significantly altered genes except mutant HTT both reinforced the high levels of allele specificity and on-target gene specificity.
Figure 3.

High levels of mutant HTT specificity supported by RNA-seq analysis
Allele-specific expression (ASE) analysis was performed to evaluate the levels of allele specificity of our TP-CRISPR strategies. HD subjects with the most frequent diplotype (i.e., hap.01 and hap.08) are heterozygous at 10 exonic SNPs. Thus, we performed ASE using those 10 exonic SNP sites. Alleles of those 10 exonic SNPs on the mutant and normal HTT were based on our haplotype definitions and previous sequencing analysis. We counted the alleles on the mutant and normal HTT for a given SNP site, and then calculated average values. (A) Mean allele counts of 10 heterozygous exonic SNPs on the mutant HTT are summarized. Boxes on the left and right represent the distribution of alleles on the mutant HTT in EV-treated and targeted clonal lines, respectively. Student’s t test was performed (nominal p value, 3.53e−6). (B) The same analysis approach was applied to alleles of 10 heterozygous exonic SNPs that are on the normal HTT. Boxes on the left and right represent the distribution of alleles in EV-treated and targeted clonal lines, respectively. Student’s t test was performed (nominal p value, 0.67). Each box shows maximum, 75%, 50% (median), 75% quartile, and minimum.
Figure 4.

On-target gene specificity supported by RNA-seq analysis
To increase the sensitivity and the power to detect any small but significantly altered genes, we combined 10 targeted clonal lines targeted by L4-R4 and 10 clonal lines targeted by L4-R6 gRNA combinations to be compared with 12 EV-treated controls. (A) Significance values (uncorrected p values on the y axis) were compared with log2 (fold-change) (x axis) to highlight significantly altered genes. A horizontal and a vertical line represent Bonferroni-corrected significance and zero fold-change, respectively. (B) Expression levels of total HTT (mutant plus normal) based on the DGE analysis are summarized in a boxplot. Total HTT levels were decreased by 38% in TP-CRISPR targeted clonal lines.
Applicability of PAS-based allele-specific strategies in HD subjects
Confirming high levels of allele specificity and on-target gene specificity, we evaluated the cumulative mutant specificity of a set of PAS pairs. Based on the phased genotypes of 8,542 HD subjects, we first calculated the percentage of HD subjects carrying mutant-specific PAM sites both upstream and downstream of the TSS. The highest mutant specificities were observed for the L4-R4 and L4-R6 combinations because of strong linkage disequilibrium between rs16843804 (R4) and rs16843836 (R6); 27% of HD subjects carry mutant-specific PAM sites for rs2857935-rs16843804 and rs16843804-rs16843836 (Table S5). In addition, seven other pairs (such as L1-R2, L1-R4, L1-R6, L2-R2, L2-R4, L2-R6, L4-R2) also showed the mutant specificity greater than 20% (Table S5). Still, more than half of the HD population are not eligible for our candidate allele-specific CRISPR-Cas9 strategies targeting L4-R4 and L4-R6. Therefore, we were interested in finding out alternative allele-specific target sites for HD subjects who do not carry PAM sites at rs2857935-rs16843804 or rs2857935-rs16843836. Previously, Montsey et al. demonstrated allele-specific TP-CRISPR using one allele-specific and one non-allele-specific gRNA.25 Therefore, we also calculated mutant specificity of a set of PAS for an allele-specific TP-CRISPR strategy using one allele-specific gRNA. The proportion of HD subjects who are eligible for mutant-specific TP-CRISPR using rs1313774 and one non-allele-specific target site was 38.4%, representing the highest mutant specificity. Allele-specific TP-CRISPR-Cas9 strategies using one of the three candidate PAS in combination with one non-allele-specific target could be applied to approximately 60% of HD individuals. Overall, inclusion of additional PAS gradually increased the cumulative percentage of eligible HD individuals; 72.5% of HD subjects with European ancestry are eligible for mutant-specific TP-CRIPSR strategies using one of 10 PAS in combination with one non-allele-specific target site (Table S6; Figure 5).
Figure 5.

Cumulative mutant specificity of TP-CRISPR strategies using PAS
To calculate the proportion of HD subjects who are eligible for mutant HTT-specific TP-CRISPR strategies using one allele-specific gRNA and one non-allele-specific gRNA, we analyzed phased allele data for 224 PAS. For the initial coverage analysis, we identified HD subjects who carry the PAM site only on the mutant HTT and calculated the proportion of those HD subjects. Then, we re-calculated the mutant specificity in the remaining HD subjects to identify the PAS with the highest mutant specificity. We repeated these procedures 10 times to estimate the cumulative mutant specificity of the allele-specific TP-CRISPR strategies.
Discussion
Recently, a phase 3 clinical trial to determine the efficacy of a non-allele-specific ASO for HD was terminated prematurely based on the risk-benefit analysis of the interim data.31 Considering numerous preclinical studies supporting the therapeutic benefits of non-allele-specific HTT-lowering strategies in model systems,22 the lack of clinical benefits in the tominersen trial is quite striking. Currently, it is not known why an ASO that could significantly reduce mutant (and normal) huntingtin protein in humans without significant side effects30 did not perform well compared with the placebo. The “unfavorable efficacy trend” in the tominersen trial31 might be due to (1) treatment initiated too late in pathogenesis, (2) lack of allele specificity leading to insufficient total huntingtin,15 (3) inability to lower alternative toxic species, such as RAN translation or exon 1A huntingtin fragment,48,49,64,65 (4) insufficient delivery to striatum, (5) no changes in mutant/normal ratio,67 and/or (6) off-target toxicity. Among these, the timing of the treatment might have significantly contributed to the lack of clinical efficacy. The tominersen trial tested clinically manifest HD subjects defined by DCL4 (diagnostic confidence level 4) (https://clinicaltrials.gov/ct2/show/NCT03761849). Considering that changes are detectable prior to disease manifest,68 trial participants might have significant levels of neurodegeneration already. Since HTT-lowering drugs are not expected to stimulate neurogenesis, the best outcome of the tominersen trial might be maintaining the integrity of the surviving cells. If cells were significantly compromised already, lowering HTT might not lead to any functional improvements. In contrast, if applied early (at least prior to significant neurodegeneration), HTT-lowering drugs may be able to block HD pathogenesis in a sufficient number of neurons, leading to delay of clinical manifestation caused by the loss of neurons over certain levels.
Despite its benefits, early treatment (e.g., pre-symptomatic treatment) is challenging and must be applied carefully. For example, (1) complete Htt knockout causes embryo lethality and other deficits in mice,33, 34, 35, 36, 37,69 and (2) HTT deficiency is associated with developmental problems in humans,13,14 arguing against early applications of HTT-lowering drugs. However, one functional copy of HTT is sufficient for the survival of cells33,35 without causing HD or developmental problems,11,13,14 supporting mutant-specific targeting strategies as a means of early treatments in HD. One may support non-allele-specific lowering strategies to reduce the total HTT expression levels by half to generate therapeutic benefits without producing adverse effects due to deficiency. However, achieving specific levels of the target is technically challenging for lowering drugs due to the difficulty of achieving efficient delivery to target organs other than the liver.70 In contrast, allele-specific DNA-targeting strategies may overcome some of the limitations of lowering approaches, such as the requirement of repeated treatments,71,72 and off-target effects.73, 74, 75, 76, 77, 78
Still, barriers exist for DNA-targeting strategies for HD. For example, DNA-targeting strategies for HD must be highly allele specific because they produce permanent changes. Therefore, to develop therapeutic strategies that can be applied early without generating deficiency-associated problems, we focused on developing highly allele-specific CRISPR-Cas9 strategies capitalizing on PAS. Previously, we identified PAS by analyzing the 1000 Genomes Project data and tested a pair of PAS in patient-derived cells to selectively inactivate the mutant HTT. Similar approaches (i.e., genomic excision to inactivate mutant HTT) have been tested in patient-derived fibroblasts and a mouse model of HD.25,27 Consistent with previous findings,26 CRISPR-Cas9 approaches based on the candidate PAS were highly allele specific. Our data also showed that allele-specific genomic deletions did not produce chromosomal re-integration, which is a concern in therapeutic genome editing.63 In addition, the exon 1 huntingtin protein has been hypothesized as the pathogenic entity.47, 48, 49, 50 Regardless, if excised DNA is subject to degradation, our allele-specific genomic deletion strategies may represent the only means to block HD pathogenesis regardless of the identity of the toxic species (even DNA itself drives the HD pathogenesis). Our allele-specific TP-CRISPR-Cas9 strategies are expected to produce new DNA sequences at the mutant HTT locus. Our strategies targeting L4-R4 and L4-R6 sites respectively eliminate the first three and six exons, resulting in in-frame deletion. However, such a new DNA sequence is expected to generate knockout effects without producing truncated HTT protein because the edited mutant allele is not capable of generating mRNA due to the lack of promoter region and TSS. Together, the advantages of DNA-targeting strategies, high levels of allele specificity of PAS-based methods, and the robustness of genomic deletion approaches make our strategies suitable for early treatments in HD. Unlike late-onset neurodegenerative disorders, such as Alzheimer disease and Parkinson disease, HD is uniformly caused by a genetic change at the same site in the same gene in all affected individuals, making this disease potentially amenable to a therapeutic approach based directly on its root cause rather than on the subsequent deleterious consequences of the genetic defect. If the treatment permanently eliminated the disease-producing expanded CAG sequence, it could theoretically be applicable at any time in life, from the preimplantation embryo through to the adult. However, the period where such a treatment strategy is perhaps most urgently needed is that prior to extensive neurodegeneration, when the HD individual retains a large neuronal population that can still be saved. Recently, it has been demonstrated that CRISPR-Cas9-mediated HTT silencing delayed the onset of striatal atrophy and slowed the progression of the motor phenotype in zQ175 mice, supporting the feasibility and efficiency of pre-manifest CRISPR therapeutics.79
In this study, we focused on identifying relevant target sites for the patient population and evaluating their allele specificities subsequently. Non-HD cell lines that are commonly used in the CRISPR-Cas9 genome editing field, such as HEK293, are not optimal for evaluating of allele specificity due to the lack of the diplotype that is most frequent in HD. Therefore, we analyzed HD patient-derived iPSCs carrying the representative diplotype, revealing modest CRISPR-Cas9 editing efficiencies, which are in line with challenges in genome editing in iPSCs.58, 59, 60 Since modest editing efficiencies might be due to the cell type, not the target sites, our allele-specific CRISPR-Cas9 strategies may produce significant therapeutic benefits when using efficient delivery methods, which is supported by significant functional improvements in the mouse models of HD.27,79 We previously showed the proof-of-principle of allele-specific TP-CRISPR, using genetic data from the 1000 Genomes Project to select two PAS with flanking SNPs to permit unequivocal identification of targeted alleles. However, our previous approach did not permit assessment of relative editing efficiency or applicability in the HD population. In this study, we focused on the PAS that are frequent in the largest collection of nearly 10,000 genotyped HD individuals of European ancestry. Of the gRNAs targeting candidate PAS that were tested, five (three mutant HTT specific; one normal HTT specific; one non-allele specific in HD subjects with the most frequent diplotype) showed relatively higher editing efficiencies than the others. This lack of significant off-target effects was further supported by the absence of any gene expression alterations other than HTT in our 20 independent targeted clonal lines. Thus, the three mutant allele-specific PAS gRNAs provide strong candidates for the development of allele-specific HD therapeutics that would be applicable to approximately 30% of the European HD population.
Unfortunately, the feasibility of functional evaluation of our candidate strategies in animal models is very low. Since HD patient-derived iPSCs and neurons lack robust and consistent phenotypes,80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90 functional assessments of mutant-specific TP-CRISPR strategies in this cell type were also impractical. For example, neuronal induction/differentiation,83,84,90 the levels of nestin after reprogramming,85,87 characteristics of action potential,80,82,90 HTT protein aggregates,82,86,90 and CAG repeat instability82,88, 89, 90 were inconsistent between studies. In addition, most studies analyzed a small number of patient-derived cells carrying long CAG repeats and, therefore, it was uncertain whether differences between normal and HD neurons were due to the expanded CAG or other unknown factors. Advancing these candidates for testing of the allele-specific strategy in an accurate mouse system is not immediately possible since an appropriate mouse model does not exist. The Hu97/18 mouse model,91 which contains both CAG-expanded and non-expanded human HTT genes and no mouse Htt, is not suitable because the CAG-expanded transgene comprises approximately five copy-equivalents of mutant HTT (https://www.jax.org/strain/008197), and therefore, the application of a dual gRNA-mediated CRISPR-Cas9 strategy is expected to induce unpredictable larger deletions at multiple locations.92,93 Consequently, in vivo preclinical testing of the allele-specific strategy will require the development of HD mouse models with single copies of the CAG-expanded and non-expanded HTT genes, conferring heterozygosity for those PAS that are most frequently heterozygous in the HD population.
The generation of such genetically accurate, humanized models would also help to support the development and testing of efficient delivery systems to introduce CRISPR-Cas9 components to the brain and to achieve editing in target cells. While obtaining a broad distribution of targeting reagents is a challenge that faces both ASO and CRISPR approaches for neurological genetic disorders, the potential of CRISPR strategies to make permanent alterations that preclude the need for life-long episodic treatments is an attractive one if delivery, efficiency, and safety issues can be adequately addressed. Furthermore, a recent small-scale in vivo CRISPR-Cas9 gene editing for transthyretin amyloidosis showed robust target engagement with mild adverse effects in humans,94 supporting the feasibility of CRISPR therapeutics in HD. Our delineation and testing of allele-specific targets based on the actual HD population offers the promise that permanent therapeutic effects can be achieved without the potential side effects that could result from interfering with normal huntingtin expression. It is a critical step toward advancing this allele-specific strategy to benefit those carrying an expanded HTT CAG repeat and has implications for applying similar allele-specific strategies in other dominant human disorders.
Materials and methods
Analysis of HD GWAS data to identify candidate PAS
To produce a genomic deletion with the aim of preventing transcription of HTT using CRISPR-Cas9, two gRNAs must be used simultaneously: one targeting upstream and the other targeting downstream of the TSS. To identify mutant HTT-specific target sites upstream of the TSS, we analyzed up to 20 kb of the upstream region to avoid potential gene editing of a region that harbors transcription regulatory elements of a neighboring gene (i.e., GRK4). For downstream target sites, we analyzed up to 40 kb of the region downstream of the TSS because we previously demonstrated the feasibility of a genomic deletion of approximately 40 kb.26 Thus, our genetic analysis focused on a 60 kb genomic region around the TSS of HTT (chr4:3056408-3116408; GRCh37/hg19). To identify mutant HTT-specific CRISPR PAM sites for SpCas9 (i.e., NGG) and to directly calculate the proportion of HD subjects who carry a mutant HTT-specific PAM site for a given PAS, we analyzed HD GWAS data that were used to identify genetic modifiers of HD.46 Among 9,058 HD subjects (CAG 40-55) with European ancestry analyzed in our modifier GWAS, we analyzed the genotypes of 8,543 participants who are heterozygous HD subjects (i.e., carrying one CAG-expanded HTT) and showed unambiguous HTT haplotypes. Among 1,045 quality control-passed SNPs in the region,46 alleles of 418 SNPs generate or eliminate NGG PAM sites (i.e., PAS). We then calculated allele frequencies of those 418 PAS to exclude variations that are monomorphic in HD subjects, revealing 224 PAS that are polymorphic in our 8,543 GWAS participants. We identified a total of 230 PGAs from 224 PAS since both reference and alternative alleles of 5 PAS and 2 different alternative alleles of 1 PAS also generate NGG PAM sequence.
Calculating the levels of applicability of PAS in mutant HTT-specific targeting
We performed genotype phasing for HTT51,52,56 to determine (1) the HTT haplotypes and (2) alleles of 224 PAS on the mutant and normal HTT for a given HD subject. Subsequently, HTT haplotype data were used to determine the consensus alleles of candidate PAS on common HTT haplotypes.26 To evaluate the level of applicability of a PAS in mutant HTT-specific targeting, we calculated the percentage of HD subjects who carry the NGG PAM sequence on (1) the CAG-expanded HTT, (2) the non-expanded HTT, (3) both, or (4) neither for each of 224 PAS. The proportion of HD subjects who carry the PAM sequence only on the CAG-expanded HTT represents the levels of applicability of a PAS for allele-specific CRISPR therapeutics targeting (i.e., mutant specificity). We further used the mutant specificity data to narrow down to candidate PAS that can be utilized in a significant proportion of HD subjects. We applied a mutant specificity value 10%, yielding four and six PAS located upstream and downstream of the TSS of HTT, respectively.
Mapping alleles of 10 candidate PAS on the common HTT haplotypes
Definitions of HTT haplotypes and their frequencies on disease and normal chromosomes are described elsewhere.51,52,56 HTT haplotypes are defined by 21 SNPs, and the 16 common haplotypes account for more than 90% of CAG-expanded chromosomes in HD subjects with European ancestry.51 To determine the alleles of a given PAS on the HTT haplotypes, we grouped 17,086 chromosomes from 8,543 GWAS participants based on their HTT haplotypes. For a group of chromosomes of a given haplotype, we identified the consensus (i.e., most frequent) allele for a given PAS, and we repeated this procedure for each of the 10 candidate PAS and 16 common HTT haplotypes. Overall, alleles of PAS on the 16 common HTT haplotypes were determined at a 99.3% consensus rate (e.g., 90% consensus rate means 90% and 10% of the chromosomes, respectively, carry the most frequent and the other allele at a given site).
Cell culture, plasmids, and transfection
Two independent iPSC lines (iPSC-A and iPSC-B) carrying adult-onset CAG repeats (46/18 and 42/19 CAGs, respectively) were derived from our internal collection of lymphoblastoid cell lines by the Harvard Stem Cell Institute iPS Core Facility (http://ipscore.hsci.harvard.edu/). Both iPSC-A (female) and iPSC-B (male) carry expanded and normal CAG repeats on hap.01 and hap.08 haplotypes, respectively. The iPSC lines were cultured on Matrigel-coated plates (Corning) with mTeSR1 (STEMCELL Technologies) with 5% CO2 at 37°C. Plasmids for Cas9 (PX551; http://n2t.net/addgene:60957) and gRNA (PX552; http://n2t.net/addgene:60958) were obtained from Addgene. To express SpCas9 in iPSC lines, the pMecp2 promoter in the original PX551 plasmid was replaced by an EF-1α core promoter using a HindIII/AgeI fragment, generating the PX551 EFS plasmid. The hSyn promoter for EGFP marker was also replaced by an EF-1α core promoter using an ApaI/KpnI fragment (PX552 EFS plasmid). Cloning of the test gRNAs into the PX552 EFS plasmid was performed according to a recommended protocol (https://media.addgene.org/data/plasmids/60/60958/60958-attachment_wWVpb-8u9Mzp.pdf). Sequences of oligos to generate plasmids for test gRNAs are:
rs1313769, ACCGGCAGAGCTTGCAGTGAGCT and AACGGCATGCTGGCTCATGCCTG;
rs1313774, ACCGCATATAATCAAGAAATAAT and AACATTATTTCTTGATTATATGC;
rs2506200, ACCCAGGCATGAGCCAGCATGCC and AACGGCATGCTGGCTCATGCCTG;
rs2857935, ACCCCCGCTCCAGGCGTCGGCGG and AACCCGCCGACGCCTGGAGCGGG;
rs7688390, ACCAGAATGGACATCATAAAGAT and AACATCTTTATGATGTCCATTCT;
rs7659144, ACCCCCATGGGCCATGTGGAAAT and AACATTTCCACATGGCCCATGGG;
rs28820097, ACCCAACAACTAAAAGCACAACA and AACTGTTGTGCTTTTAGTTGTTG;
rs16843804, ACCGTCGATGATCTCTTTAACCG and AACCGGTTAAAGAGATCATCGAC;
rs6828615, ACCTGGGCTCACGCCTGTAATCC and AACGGATTACAGGCGTGAGCCCA;
rs16843836, ACCGCTATGTTTATCCTGCAACC and AACGGTTGCAGGATAAACATAGC.
For all candidate PAS, we used 20 nucleotide long gRNAs. Cells were transfected (72 h) with either (1) SpCas9 plasmid and EV for gRNA (EV control) or (2) SpCas9 plasmid and gRNA plasmid (treatment group) by Lipofectamine Stem Transfection Reagent (Invitrogen) according to the manufacturer’s instructions.
PCR amplification and Sanger sequencing
Genomic DNA was isolated from cells using a DNeasy Blood & Tissue Kit (QIAGEN). Total RNA was extracted using RNeasy Plus Mini Kits (QIAGEN). The quality and quantity of RNA were determined using a NanoDrop spectrophotometer (Thermo Scientific). cDNA was synthesized from 50 ng of total RNA using SuperScript IV Reverse Transcriptase (Invitrogen). PCR reactions were performed using Q5 High-Fidelity DNA Polymerase (NEB) or AccuPrime GC-Rich DNA Polymerase (Invitrogen) for high GC-rich template PCR. The PCR amplification using Q5 High-Fidelity DNA Polymerase comprised initial denaturation (3 min at 98°C), 35 cycles of denaturation (30 s at 98°C), annealing (30 s at 64°C), and extension (30 s at 72°C), and final extension for 2 min at 72°C followed by cooling down to 4°C. The reaction for AccuPrime GC-Rich DNA Polymerase consisted of an initial denaturation (3 min at 95°C), then 30 cycles of denaturation (30 s at 95°C), annealing (30 s at 59°C), and extension (30 s at 72°C), and then final extension for 10 min at 72°C, followed by cooling down to 4°C. PCR products were purified using a QIAquick PCR Purification Kit (QIAGEN) for subsequent analysis. For PCR assays to detect genomic deletion by L4-R4 combination, we used primers CTTCTCGCTGCACTAATCAC (a red arrow in Figure 2A) and ATTCCTCACAGCACATCTCT (a blue arrow in Figure 2A). For genomic deletion by L4-R6 combination, we used primers CTTCTCGCTGCACTAATCAC (a red arrow in Figure 2A) and AGAGGGTGAAATAGTGGCT (a cyan arrow in Figure 2A). Primers ATGAAGGCCTTCGAGTCCC and GGCTGAGGAAGCTGAGGA were used to amplify CAG repeat region in DNA and cDNA.
Determination of allele specificities and editing efficiencies by next generation sequencing
Upon ligation of Illumina adaptors and a unique identifier to the amplicon, paired-end sequencing (2 × 150 bp) was performed using the Illumina MiSeq platform. Deep-sequencing of PCR amplicons was performed by the DNA Core Facility at Massachusetts General Hospital (https://dnacore.mgh.harvard.edu/new-cgi-bin/site/pages/index.jsp). Sequence reads that could not be mapped were removed as part of quality control. Primers for PCR amplification for MiSeq analysis are:
rs1313769, CTGTAGTCCCAGCTACTCAGG and AACCCAGGGCTAGTTTGAGA;
rs1313774, CACCATGTTAGCCAGGATGG and CCTGGCCTAGAAGTTACCCT;
rs12506200, TGTAGGAATAGGGTCTCACTATGTGT and GGTGAGTACTCCAGGGGAA;
rs2857935, TGAGTATGGCTCTGGCCA and AGGAAGGTGAGAGGTGGG;
rs28820097, GCTGCATGTGAAATGGTGTAATAAGG and CCTCTTCCCCTATTTCTGGCTT;
rs7659144, CAGAGTATGTTTTCTGACCTCAGTATCATTAAG and CCATTTACTATGCTGTAACAGCACC;
rs7688390, CACTGTGTTCCATCCTGGG and GATGTTCTTGATGAACCAGCTTTTGG;
rs16843804, CAAGCTTGCTGACCCAATAGG and GAAGGGCTTCTCAAGCTAGG;
rs6828615, GGATTACAGGTGTGCACCAC and CCAGGGATACTAGCTGACTCAAG;
rs16843836, TGAGTCAGCTAGTATCCCTGGA and GCTGAGGACAGACGTGAGAT;
rs363099, GCCAGTAACCGTGTGTTCTC and CCTAGATGAACTCAGCCCAG.
Off-target effects
CRISPR-Cas9 off-target prediction was based on Cas-OFFinder.95
Development of targeted single-cell clonal lines
To generate single-cell clones from iPSCs with adult-onset CAG repeats (iPSC-A and iPSC-B), cells were transfected with PX551 EFS and PX552 EFS vectors (containing either EV or the test gRNA) using the Lipofectamine Stem Transfection Reagent. Seventy-two hours after transfection without selection, 1.5 × 105 cells were seeded in a 60 mm dish with CloneR supplement (STEMCELL Technologies) and then incubated for an additional 2 weeks for dilution cloning. Visible colonies were picked, and individually maintained in 96-well plates for clonal expansion. Once cells reached approximately 80% confluence, they were sub-cultured in two 96-well plates, one for maintenance and the other for validation of on-target modification by Sanger sequencing analysis. Validated clonal lines were then expanded for molecular characterization and RNA-seq analysis. In summary, for iPSC-A, we picked 132 and 228 single cells after treatment for L4-R4 and L4-R6 strategies, respectively. Among them, 45 and 47 showed genomic deletion in PCR assays. Subsets of those candidate single cells were further subject to Sanger sequencing and MiSeq analysis to select the final 10 independent targeted clonal lines for RNA-seq analysis. Similarly, for iPSC-B, we picked 216 and 132 single cells after treatment for L4-R4 and L4-R6 strategies, respectively. Among them, 93 and 35 showed genomic deletion in PCR assays. Subsets of those candidate single cells were further evaluated to select 10 independent targeted clonal lines for RNA-seq analysis.
Detection of excision-reintegration
Genomic DNA of targeted clonal lines was subject to PCR using a primer set (GTTTAGTTAACACCCTTAGCAAC and CTAGCTTCTACCAGGAGAATAACA) to amplify a region encompassing rs2285086, which is located inside the genomic deletion and heterozygous in HD subjects with the most frequent diplotype. Consistent with HD subjects with the most frequent diplotype, our iPSC lines carry “A” and “G” alleles on the mutant and normal HTT, respectively. Seemingly homozygous genotype in the targeted clonal lines actually means hemizygous.
Immunoblot analysis of huntingtin
Cells were washed twice with cold PBS and lysed with RIPA buffer (Invitrogen) containing protease inhibitor (Roche). Cell debris was removed by centrifugation, and the supernatant was collected. Protein concentration was determined by BCA assays (Thermo Scientific), and samples were denatured by 2× SDS buffer (Invitrogen) with a reducing agent (Invitrogen) for 2 min at 80°C. Fifteen micrograms of whole cell lysate was resolved on a 6% Tris-glycine gel (Invitrogen) or 4%–20% gel (Invtrogen). Transferred membranes were probed by pan huntingtin antibody MAB2166 (EMD Millipore; amino acids 181–810) and N-terminal-specific antibody N17 antibody.66
RNA-seq analysis
To fully characterize the molecular consequences of our mutant-specific TP-CRISPR strategies, we performed RNA-seq analysis. Twelve EV-treated and 20 targeted clonal lines were further validated by Sanger sequencing and MiSeq analysis of genomic cDNA. Then, genome-wide RNA-seq analysis was performed by the Broad Institute. Sequence data were processed by STAR aligner96 as part of the Broad Institute’s standard RNA-seq analysis pipeline. For allele-specific expression analysis (ASE) of RNA-seq data focusing on HTT, we counted alleles harboring heterozygous exonic SNPs in targeted and control clonal lines. Our clonal lines derived from two HD iPSCs with hap.01/hap.08 diplotype carry 10 heterozygous exonic SNPs. Alleles of 10 heterozygous exonic SNPs on the mutant and normal HTT in hap.01/hap/08 diplotype were determined based on haplotype sequence data.52 Statistical significance in ASE of HTT was judged based on the Student’s t test, comparing counts of alleles on normal HTT in targeted clonal lines (n = 20) with those in controls (n = 12) for a given SNP site, followed by the Bonferroni multiple test correction. The same approach was applied to the number of alleles on the mutant HTT to compare targeted and control clones.
For DGE analysis, we used transcripts per million (TPM) data computed by the TPMCalculator (https://github.com/ncbi/TPMCalculator).97 Expression levels in 20,260 protein-coding genes based on Ensembl (ftp://ftp.ensembl.org/pub/release-75/gtf/homo_sapiens/) were normalized; 3,420 genes were excluded due to zero TPM values in at least one sample. Subsequently, we analyzed 16,840 genes expressed in all 32 samples. The DGE analysis was performed by the generalized linear model using a library of “glm” in R package v.3.3.1 (https://www.r-project.org/) after adjustment for four covariates including sex, batch, and two principal components based on RNA-seq data, followed by multiple test correction using the Bonferroni multiple test correction method. A multiple test corrected p value less than 0.05 was considered statistically significant in our DGE analysis.
Cumulative mutant specificity of a set of PAS
To identify a small set of PAS that can be applied to the maximum number of HD subjects, we calculated the mutant specificity of individual PAS in multiple iterations as similarly done previously.52 In brief, starting with all samples and PAS in the region, we calculated the mutant specificity to identify the SNP that showed the highest mutant specificity. We then excluded HD subjects who carry mutant-specific PAM at that PAS site, and re-calculated mutant specificity in the remaining HD subjects. This procedure was repeated 10 times. Alternative targets for each PAS were based on linkage equilibrium in HD GWAS data calculated by the PLINK program.98
Genomic coordinate
Genomic coordinate is based on GRCh37/hg19 unless otherwise specified.
Statistics and programs
Statistical significance was determined by Student’s t tests and linear regression analyses. Resulting nominal p values were corrected for multiple tests by the Bonferroni method for DGE and ASE analysis, respectively. Corrected p values < 0.05 were considered as statistically significant. R (v.3.3.1 and v.3.3.3) was used for statistical analyses and plotting.
Data availability
RNA-seq data have been deposited in Dryad (accession number, https://doi.org/10.5061/dryad.1ns1rn8vv).
Acknowledgments
J.-M.L. was supported by grants from Harvard NeuroDiscovery Center, NIH (NS105709; NS119471), and the CHDI Foundation.
Author contributions
J.W.S. and J.-M.L. conceptualized the study. J.W.S., D.E.C., S.Z., and S.S.P. performed the experiments. J.W.S., E.P.H., and J.-M.L. analyzed the data. J.W.S., R.Z.C., and J.-M.L. wrote the manuscript.
Declaration of interests
J.-M.L. serves in the advisory board of GenEdit Inc.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.08.005.
Supplemental information
References
- 1.Huntington G. On chorea. Med. Surg. Rep. 1872;26:320–321. [Google Scholar]
- 2.The Huntington's Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 3.Bates G.P., Dorsey R., Gusella J.F., Hayden M.R., Kay C., Leavitt B.R., Nance M., Ross C.A., Scahill R.I., Wetzel R., et al. Huntington disease. Nat. Rev. Dis. Primers. 2015:15005. doi: 10.1038/nrdp.2015.5. [DOI] [PubMed] [Google Scholar]
- 4.Keum J.W., Shin A., Gillis T., Mysore J.S., Abu Elneel K., Lucente D., Hadzi T., Holmans P., Jones L., Orth M., et al. The HTT CAG-expansion mutation determines age at death but not disease duration in Huntington disease. Am. J. Hum. Genet. 2016;98:287–298. doi: 10.1016/j.ajhg.2015.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bates G.P. History of genetic disease: the molecular genetics of Huntington disease - a history. Nat. Rev. Genet. 2005;6:766–773. doi: 10.1038/nrg1686. [DOI] [PubMed] [Google Scholar]
- 6.Gusella J.F., MacDonald M.E. Molecular genetics: unmasking polyglutamine triggers in neurodegenerative disease. Nat. Rev. Neurosci. 2000;1:109–115. doi: 10.1038/35039051. [DOI] [PubMed] [Google Scholar]
- 7.Killoran A., Biglan K.M. Current therapeutic options for Huntington's disease: good clinical practice versus evidence-based approaches? Movement disorders. Mov. Disord. 2014;29:1404–1413. doi: 10.1002/mds.26014. [DOI] [PubMed] [Google Scholar]
- 8.Peavy G.M., Jacobson M.W., Goldstein J.L., Hamilton J.M., Kane A., Gamst A.C., Lessig S.L., Lee J.C., Corey-Bloom J. Cognitive and functional decline in Huntington's disease: dementia criteria revisited. Mov. Disord. 2010;25:1163–1169. doi: 10.1002/mds.22953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stout J.C., Paulsen J.S., Queller S., Solomon A.C., Whitlock K.B., Campbell J.C., Carlozzi N., Duff K., Beglinger L.J., Langbehn D.R., et al. Neurocognitive signs in prodromal Huntington disease. Neuropsychology. 2011;25:1–14. doi: 10.1037/a0020937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thompson J.C., Harris J., Sollom A.C., Stopford C.L., Howard E., Snowden J.S., Craufurd D. Longitudinal evaluation of neuropsychiatric symptoms in Huntington's disease. J. Neuropsychiatry Clin. Neurosci. 2012;24:53–60. doi: 10.1176/appi.neuropsych.11030057. [DOI] [PubMed] [Google Scholar]
- 11.Ambrose C.M., Duyao M.P., Barnes G., Bates G.P., Lin C.S., Srinidhi J., Baxendale S., Hummerich H., Lehrach H., Altherr M., et al. Structure and expression of the Huntington's disease gene: evidence against simple inactivation due to an expanded CAG repeat. Somat. Cell Mol. Genet. 1994;20:27–38. doi: 10.1007/BF02257483. [DOI] [PubMed] [Google Scholar]
- 12.Jung R., Lee Y., Barker D., Correia K., Shin B., Loupe J., Collins R.L., Lucente D., Ruliera J., Gillis T., et al. Mutations causing Lopes-Maciel-Rodan syndrome are huntingtin hypomorphs. Hum. Mol. Genet. 2021;30:135–148. doi: 10.1093/hmg/ddaa283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopes F., Barbosa M., Ameur A., Soares G., de Sá J., Dias A.I., Oliveira G., Cabral P., Temudo T., Calado E., et al. Identification of novel genetic causes of Rett syndrome-like phenotypes. J. Med. Genet. 2016;53:190–199. doi: 10.1136/jmedgenet-2015-103568. [DOI] [PubMed] [Google Scholar]
- 14.Rodan L.H., Cohen J., Fatemi A., Gillis T., Lucente D., Gusella J., Picker J.D. A novel neurodevelopmental disorder associated with compound heterozygous variants in the huntingtin gene. Eur. J. Hum. Genet. 2016;24:1826–1827. doi: 10.1038/ejhg.2016.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cattaneo E., Zuccato C., Tartari M. Normal huntingtin function: an alternative approach to Huntington's disease. Nat. Rev. Neurosci. 2005;6:919–930. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- 16.Crook Z.R., Housman D.E. Surveying the landscape of Huntington's disease mechanisms, measurements, and medicines. J. Huntingtons Dis. 2013;2:405–436. doi: 10.3233/JHD-130072. [DOI] [PubMed] [Google Scholar]
- 17.Tabrizi S.J., Flower M.D., Ross C.A., Wild E.J. Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020;16:529–546. doi: 10.1038/s41582-020-0389-4. [DOI] [PubMed] [Google Scholar]
- 18.Tabrizi S.J., Ghosh R., Leavitt B.R. Huntingtin lowering strategies for disease modification in Huntington's disease. Neuron. 2019;101:801–819. doi: 10.1016/j.neuron.2019.01.039. [DOI] [PubMed] [Google Scholar]
- 19.Aronin N., DiFiglia M. Huntingtin-lowering strategies in Huntington's disease: antisense oligonucleotides, small RNAs, and gene editing. Mov. Disord. 2014;29:1455–1461. doi: 10.1002/mds.26020. [DOI] [PubMed] [Google Scholar]
- 20.Bonini N.M., La Spada A.R. Silencing polyglutamine degeneration with RNAi. Neuron. 2005;48:715–718. doi: 10.1016/j.neuron.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 21.Davidson B.L., Boudreau R.L. RNA interference: a tool for querying nervous system function and an emerging therapy. Neuron. 2007;53:781–788. doi: 10.1016/j.neuron.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 22.Keiser M.S., Kordasiewicz H.B., McBride J.L. Gene suppression strategies for dominantly inherited neurodegenerative diseases: lessons from Huntington's disease and spinocerebellar ataxia. Hum. Mol. Genet. 2016;25:R53–R64. doi: 10.1093/hmg/ddv442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.La Spada A.R. Getting a handle on Huntington's disease: silencing neurodegeneration. Nat. Med. 2009;15:252–253. doi: 10.1038/nm0309-252. [DOI] [PubMed] [Google Scholar]
- 24.Lu X.H., Yang X.W. Huntingtin holiday": progress toward an antisense therapy for Huntington's disease. Neuron. 2012;74:964–966. doi: 10.1016/j.neuron.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Monteys A.M., Ebanks S.A., Keiser M.S., Davidson B.L. CRISPR/Cas9 editing of the mutant huntingtin allele in vitro and in vivo. Mol. Ther. 2017;25:12–23. doi: 10.1016/j.ymthe.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin J.W., Kim K.H., Chao M.J., Atwal R.S., Gillis T., MacDonald M.E., Gusella J.F., Lee J.M. Permanent inactivation of Huntington's disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet. 2016 doi: 10.1093/hmg/ddw286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang S., Chang R., Yang H., Zhao T., Hong Y., Kong H.E., Sun X., Qin Z., Jin P., Li S., Li X.J. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington's disease. J. Clin. Invest. 2017;127:2719–2724. doi: 10.1172/JCI92087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wild E.J., Tabrizi S.J. Therapies targeting DNA and RNA in Huntington's disease. Lancet Neurol. 2017;16:837–847. doi: 10.1016/S1474-4422(17)30280-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto A., Lucas J.J., Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 30.Tabrizi S.J., Leavitt B.R., Landwehrmeyer G.B., Wild E.J., Saft C., Barker R.A., Blair N.F., Craufurd D., Priller J., Rickards H., et al. Phase 1–2a IONIS-HTTRx Study Site Teams Targeting huntingtin expression in patients with Huntington's disease. N. Engl. J. Med. 2019;380:2307–2316. doi: 10.1056/NEJMoa1900907. [DOI] [PubMed] [Google Scholar]
- 31.Sheridan C. Questions swirl around failures of disease-modifying Huntington's drugs. Nat. Biotechnol. 2021;39:650–652. doi: 10.1038/s41587-021-00955-y. [DOI] [PubMed] [Google Scholar]
- 32.Saft C. Huntington's disease: disappointments and new beginnings. Lancet Neurol. 2022;21:582–584. doi: 10.1016/S1474-4422(22)00189-2. [DOI] [PubMed] [Google Scholar]
- 33.Nasir J., Floresco S.B., O'Kusky J.R., Diewert V.M., Richman J.M., Zeisler J., Borowski A., Marth J.D., Phillips A.G., Hayden M.R. Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- 34.White J.K., Auerbach W., Duyao M.P., Vonsattel J.P., Gusella J.F., Joyner A.L., MacDonald M.E. Huntingtin is required for neurogenesis and is not impaired by the Huntington's disease CAG expansion. Nat. Genet. 1997;17:404–410. doi: 10.1038/ng1297-404. [DOI] [PubMed] [Google Scholar]
- 35.Zeitlin S., Liu J.P., Chapman D.L., Papaioannou V.E., Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat. Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- 36.Dietrich P., Johnson I.M., Alli S., Dragatsis I. Elimination of huntingtin in the adult mouse leads to progressive behavioral deficits, bilateral thalamic calcification, and altered brain iron homeostasis. PLoS Genet. 2017;13:e1006846. doi: 10.1371/journal.pgen.1006846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang G., Liu X., Gaertig M.A., Li S., Li X.J. Ablation of huntingtin in adult neurons is nondeleterious but its depletion in young mice causes acute pancreatitis. Proc. Natl. Acad. Sci. USA. 2016;113:3359–3364. doi: 10.1073/pnas.1524575113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson C.D., Davidson B.L. Huntington's disease: progress toward effective disease-modifying treatments and a cure. Hum. Mol. Genet. 2010;19:R98–R102. doi: 10.1093/hmg/ddq148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heidenreich M., Zhang F. Applications of CRISPR-Cas systems in neuroscience. Nat. Rev. Neurosci. 2016;17:36–44. doi: 10.1038/nrn.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doudna J.A., Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 41.Depienne C., Mandel J.L. 30 years of repeat expansion disorders: what have we learned and what are the remaining challenges? Am. J. Hum. Genet. 2021;108:764–785. doi: 10.1016/j.ajhg.2021.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Orr H.T., Zoghbi H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 43.Scott D.A., Zhang F. Implications of human genetic variation in CRISPR-based therapeutic genome editing. Nat. Med. 2017;23:1095–1101. doi: 10.1038/nm.4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Genetic Modifiers of Huntington’s Disease GeM-HD Consortium Identification of genetic factors that modify clinical onset of Huntington's disease. Cell. 2015;162:516–526. doi: 10.1016/j.cell.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Genetic Modifiers of Huntington’s Disease GeM-HD Consortium CAG repeat not polyglutamine length determines timing of Huntington's disease onset. Cell. 2019;178:887–900.e14. doi: 10.1016/j.cell.2019.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barbaro B.A., Lukacsovich T., Agrawal N., Burke J., Bornemann D.J., Purcell J.M., Worthge S.A., Caricasole A., Weiss A., Song W., et al. Comparative study of naturally occurring huntingtin fragments in Drosophila points to exon 1 as the most pathogenic species in Huntington's disease. Hum. Mol. Genet. 2015;24:913–925. doi: 10.1093/hmg/ddu504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neueder A., Landles C., Ghosh R., Howland D., Myers R.H., Faull R.L.M., Tabrizi S.J., Bates G.P. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington's disease patients. Sci. Rep. 2017;7:1307. doi: 10.1038/s41598-017-01510-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sathasivam K., Neueder A., Gipson T.A., Landles C., Benjamin A.C., Bondulich M.K., Smith D.L., Faull R.L.M., Roos R.A.C., Howland D., et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. USA. 2013;110:2366–2370. doi: 10.1073/pnas.1221891110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang H., Yang S., Jing L., Huang L., Chen L., Zhao X., Yang W., Pan Y., Yin P., Qin Z.S., et al. Truncation of mutant huntingtin in knock-in mice demonstrates exon1 huntingtin is a key pathogenic form. Nat. Commun. 2020;11:2582. doi: 10.1038/s41467-020-16318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chao M.J., Gillis T., Atwal R.S., Mysore J.S., Arjomand J., Harold D., Holmans P., Jones L., Orth M., Myers R.H., et al. Haplotype-based stratification of Huntington's disease. Eur. J. Hum. Genet. 2017;25:1202–1209. doi: 10.1038/ejhg.2017.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee J.M., Kim K.H., Shin A., Chao M.J., Abu Elneel K., Gillis T., Mysore J.S., Kaye J.A., Zahed H., Kratter I.H., et al. Sequence-level analysis of the major European Huntington disease haplotype. Am. J. Hum. Genet. 2015;97:435–444. doi: 10.1016/j.ajhg.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baine F.K., Kay C., Ketelaar M.E., Collins J.A., Semaka A., Doty C.N., Krause A., Greenberg L.J., Hayden M.R. Huntington disease in the South African population occurs on diverse and ethnically distinct genetic haplotypes. Eur. J. Hum. Genet. 2013;21:1120–1127. doi: 10.1038/ejhg.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kay C., Collins J.A., Skotte N.H., Southwell A.L., Warby S.C., Caron N.S., Doty C.N., Nguyen B., Griguoli A., Ross C.J., et al. Huntingtin haplotypes provide prioritized target panels for allele-specific silencing in Huntington disease patients of European ancestry. Mol. Ther. 2015;23:1759–1771. doi: 10.1038/mt.2015.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kay C., Tirado-Hurtado I., Cornejo-Olivas M., Collins J.A., Wright G., Inca-Martinez M., Veliz-Otani D., Ketelaar M.E., Slama R.A., Ross C.J., et al. The targetable A1 Huntington disease haplotype has distinct Amerindian and European origins in Latin America. Eur. J. Hum. Genet. 2017;25:332–340. doi: 10.1038/ejhg.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee J.M., Gillis T., Mysore J.S., Ramos E.M., Myers R.H., Hayden M.R., Morrison P.J., Nance M., Ross C.A., Margolis R.L., et al. Common SNP-based haplotype analysis of the 4p16.3 Huntington disease gene region. Am. J. Hum. Genet. 2012;90:434–444. doi: 10.1016/j.ajhg.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Warby S.C., Montpetit A., Hayden A.R., Carroll J.B., Butland S.L., Visscher H., Collins J.A., Semaka A., Hudson T.J., Hayden M.R. CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup. Am. J. Hum. Genet. 2009;84:351–366. doi: 10.1016/j.ajhg.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li X.L., Li G.H., Fu J., Fu Y.W., Zhang L., Chen W., Arakaki C., Zhang J.P., Wen W., Zhao M., et al. Highly efficient genome editing via CRISPR-Cas9 in human pluripotent stem cells is achieved by transient BCL-XL overexpression. Nucleic Acids Res. 2018;46:10195–10215. doi: 10.1093/nar/gky804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang L., Guell M., Byrne S., Yang J.L., De Los Angeles A., Mali P., Aach J., Kim-Kiselak C., Briggs A.W., Rios X., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013;41:9049–9061. doi: 10.1093/nar/gkt555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mali P., Aach J., Stranges P.B., Esvelt K.M., Moosburner M., Kosuri S., Yang L., Church G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vidigal J.A., Ventura A. Rapid and efficient one-step generation of paired gRNA CRISPR-Cas9 libraries. Nat. Commun. 2015;6:8083. doi: 10.1038/ncomms9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bothmer A., Gareau K.W., Abdulkerim H.S., Buquicchio F., Cohen L., Viswanathan R., Zuris J.A., Marco E., Fernandez C.A., Myer V.E., Cotta-Ramusino C. Detection and modulation of DNA translocations during multi-gene genome editing in T cells. The CRISPR journal. 2020;3:177–187. doi: 10.1089/crispr.2019.0074. [DOI] [PubMed] [Google Scholar]
- 63.Brunet E., Jasin M. Induction of chromosomal translocations with CRISPR-Cas9 and other nucleases: understanding the repair mechanisms that give rise to translocations. Adv. Exp. Med. Biol. 2018;1044:15–25. doi: 10.1007/978-981-13-0593-1_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang S., Yang H., Huang L., Chen L., Qin Z., Li S., Li X.J. Lack of RAN-mediated toxicity in Huntington's disease knock-in mice. Proc. Natl. Acad. Sci. USA. 2020;117:4411–4417. doi: 10.1073/pnas.1919197117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bañez-Coronel M., Ayhan F., Tarabochia A.D., Zu T., Perez B.A., Tusi S.K., Pletnikova O., Borchelt D.R., Ross C.A., Margolis R.L., et al. RAN translation in Huntington disease. Neuron. 2015;88:667–677. doi: 10.1016/j.neuron.2015.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sapp E., Valencia A., Li X., Aronin N., Kegel K.B., Vonsattel J.P., Young A.B., Wexler N., DiFiglia M. Native mutant huntingtin in human brain: evidence for prevalence of full-length monomer. J. Biol. Chem. 2012;287:13487–13499. doi: 10.1074/jbc.M111.286609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bečanović K., Nørremølle A., Neal S.J., Kay C., Collins J.A., Arenillas D., Lilja T., Gaudenzi G., Manoharan S., Doty C.N., Beck J., et al. A SNP in the HTT promoter alters NF-kappaB binding and is a bidirectional genetic modifier of Huntington disease. Nat. Neurosci. 2015;18:807–816. doi: 10.1038/nn.4014. [DOI] [PubMed] [Google Scholar]
- 68.Paulsen J.S., Langbehn D.R., Stout J.C., Aylward E., Ross C.A., Nance M., Guttman M., Johnson S., MacDonald M., Beglinger L.J., et al. Predict-HD Investigators and Coordinators of the Huntington Study Group Detection of Huntington's disease decades before diagnosis: the Predict-HD study. J. Neurol. Neurosurg. Psychiatry. 2008;79:874–880. doi: 10.1136/jnnp.2007.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dragatsis I., Levine M.S., Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat. Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- 70.Roberts T.C., Langer R., Wood M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020;19:673–694. doi: 10.1038/s41573-020-0075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mercuri E., Darras B.T., Chiriboga C.A., Day J.W., Campbell C., Connolly A.M., Iannaccone S.T., Kirschner J., Kuntz N.L., Saito K., et al. CHERISH Study Group Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 2018;378:625–635. doi: 10.1056/NEJMoa1710504. [DOI] [PubMed] [Google Scholar]
- 72.Finkel R.S., Mercuri E., Darras B.T., Connolly A.M., Kuntz N.L., Kirschner J., Chiriboga C.A., Saito K., Servais L., Tizzano E., et al. ENDEAR Study Group Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017;377:1723–1732. doi: 10.1056/NEJMoa1702752. [DOI] [PubMed] [Google Scholar]
- 73.Michel S., Schirduan K., Shen Y., Klar R., Tost J., Jaschinski F. Using RNA-seq to assess off-target effects of antisense oligonucleotides in human cell lines. Mol. Diagn. Ther. 2021;25:77–85. doi: 10.1007/s40291-020-00504-4. [DOI] [PubMed] [Google Scholar]
- 74.Yoshida T., Naito Y., Yasuhara H., Sasaki K., Kawaji H., Kawai J., Naito M., Okuda H., Obika S., Inoue T. Evaluation of off-target effects of gapmer antisense oligonucleotides using human cells. Gene Cell. 2019;24:827–835. doi: 10.1111/gtc.12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jackson A.L., Burchard J., Schelter J., Chau B.N., Cleary M., Lim L., Linsley P.S. Widespread siRNA "off-target" transcript silencing mediated by seed region sequence complementarity. RNA. 2006;12:1179–1187. doi: 10.1261/rna.25706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Scacheri P.C., Rozenblatt-Rosen O., Caplen N.J., Wolfsberg T.G., Umayam L., Lee J.C., Hughes C.M., Shanmugam K.S., Bhattacharjee A., Meyerson M., Collins F.S. Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc. Natl. Acad. Sci. USA. 2004;101:1892–1897. doi: 10.1073/pnas.0308698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Persengiev S.P., Zhu X., Green M.R. Nonspecific, concentration-dependent stimulation and repression of mammalian gene expression by small interfering RNAs (siRNAs) RNA. 2004;10:12–18. doi: 10.1261/rna5160904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jackson A.L., Bartz S.R., Schelter J., Kobayashi S.V., Burchard J., Mao M., Li B., Cavet G., Linsley P.S. Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 2003;21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- 79.Liu H., Zhang C., Xu J., Jin J., Cheng L., Miao X., Wu Q., Wei Z., Liu P., Lu H., et al. Huntingtin silencing delays onset and slows progression of Huntington's disease: a biomarker study. Brain. 2021;144:3101–3113. doi: 10.1093/brain/awab190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Le Cann K., Foerster A., Rösseler C., Erickson A., Hautvast P., Giesselmann S., Pensold D., Kurth I., Rothermel M., Mattis V.B., et al. The difficulty to model Huntington's disease in vitro using striatal medium spiny neurons differentiated from human induced pluripotent stem cells. Sci. Rep. 2021;11:6934. doi: 10.1038/s41598-021-85656-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koyuncu S., Saez I., Lee H.J., Gutierrez-Garcia R., Pokrzywa W., Fatima A., Hoppe T., Vilchez D. Author Correction: the ubiquitin ligase UBR5 suppresses proteostasis collapse in pluripotent stem cells from Huntington's disease patients. Nat. Commun. 2020;11:985. doi: 10.1038/s41467-020-14726-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.HD iPSC Consortium Induced pluripotent stem cells from patients with Huntington's disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell. 2012;11:264–278. doi: 10.1016/j.stem.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chae J.I., Kim D.W., Lee N., Jeon Y.J., Jeon I., Kwon J., Kim J., Soh Y., Lee D.S., Seo K.S., et al. Quantitative proteomic analysis of induced pluripotent stem cells derived from a human Huntington's disease patient. Biochem. J. 2012;446:359–371. doi: 10.1042/BJ20111495. [DOI] [PubMed] [Google Scholar]
- 84.Conforti P., Besusso D., Bocchi V.D., Faedo A., Cesana E., Rossetti G., Ranzani V., Svendsen C.N., Thompson L.M., Toselli M., et al. Faulty neuronal determination and cell polarization are reverted by modulating HD early phenotypes. Proc. Natl. Acad. Sci. USA. 2018;115:E762–E771. doi: 10.1073/pnas.1715865115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.HD iPSC Consortium Developmental alterations in Huntington's disease neural cells and pharmacological rescue in cells and mice. Nat. Neurosci. 2017;20:648–660. doi: 10.1038/nn.4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jeon I., Lee N., Li J.Y., Park I.H., Park K.S., Moon J., Shim S.H., Choi C., Chang D.J., Kwon J., et al. Neuronal properties, in vivo effects, and pathology of a Huntington's disease patient-derived induced pluripotent stem cells. Stem Cell. 2012;30:2054–2062. doi: 10.1002/stem.1135. [DOI] [PubMed] [Google Scholar]
- 87.Mattis V.B., Tom C., Akimov S., Saeedian J., Østergaard M.E., Southwell A.L., Doty C.N., Ornelas L., Sahabian A., Lenaeus L., et al. HD iPSC-derived neural progenitors accumulate in culture and are susceptible to BDNF withdrawal due to glutamate toxicity. Hum. Mol. Genet. 2015;24:3257–3271. doi: 10.1093/hmg/ddv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Niclis J.C., Trounson A.O., Dottori M., Ellisdon A.M., Bottomley S.P., Verlinsky Y., Cram D.S. Human embryonic stem cell models of Huntington disease. Reprod. Biomed. Online. 2009;19:106–113. doi: 10.1016/s1472-6483(10)60053-3. [DOI] [PubMed] [Google Scholar]
- 89.Seriola A., Spits C., Simard J.P., Hilven P., Haentjens P., Pearson C.E., Sermon K. Huntington's and myotonic dystrophy hESCs: down-regulated trinucleotide repeat instability and mismatch repair machinery expression upon differentiation. Hum. Mol. Genet. 2011;20:176–185. doi: 10.1093/hmg/ddq456. [DOI] [PubMed] [Google Scholar]
- 90.Victor M.B., Richner M., Olsen H.E., Lee S.W., Monteys A.M., Ma C., Huh C.J., Zhang B., Davidson B.L., Yang X.W., Yoo A.S. Striatal neurons directly converted from Huntington's disease patient fibroblasts recapitulate age-associated disease phenotypes. Nat. Neurosci. 2018;21:341–352. doi: 10.1038/s41593-018-0075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Southwell A.L., Warby S.C., Carroll J.B., Doty C.N., Skotte N.H., Zhang W., Villanueva E.B., Kovalik V., Xie Y., Pouladi M.A., et al. A fully humanized transgenic mouse model of Huntington disease. Hum. Mol. Genet. 2013;22:18–34. doi: 10.1093/hmg/dds397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang L., Jia R., Palange N.J., Satheka A.C., Togo J., An Y., Humphrey M., Ban L., Ji Y., Jin H., et al. Large genomic fragment deletions and insertions in mouse using CRISPR/Cas9. PLoS One. 2015;10:e0120396. doi: 10.1371/journal.pone.0120396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cullot G., Boutin J., Toutain J., Prat F., Pennamen P., Rooryck C., Teichmann M., Rousseau E., Lamrissi-Garcia I., Guyonnet-Duperat V., et al. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun. 2019;10:1136. doi: 10.1038/s41467-019-09006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gillmore J.D., Gane E., Taubel J., Kao J., Fontana M., Maitland M.L., Seitzer J., O'Connell D., Walsh K.R., Wood K., et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 2021;385:493–502. doi: 10.1056/NEJMoa2107454. [DOI] [PubMed] [Google Scholar]
- 95.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vera Alvarez R., Pongor L.S., Mariño-Ramírez L., Landsman D. TPMCalculator: one-step software to quantify mRNA abundance of genomic features. Bioinformatics. 2019;35:1960–1962. doi: 10.1093/bioinformatics/bty896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data have been deposited in Dryad (accession number, https://doi.org/10.5061/dryad.1ns1rn8vv).