

Introduction
Observations that specific brain regions are important in specific behavioral patterns have been known at least since the remarkable case of Phineas Gage,1 and the ways that use of alcohol and other substances can negatively impact behavior has been noted for centuries (or even millennia). What was missing until recent decades has been a comprehensive understanding of the neurobiological underpinnings of addiction; yet, this knowledge is essential for clinicians and patients.
Advances in neuroscience research at the anatomical, electrophysiological, and neuroimaging levels have all contributed to establishing central nervous system models that help to explain the signs, symptoms, and course of addictive disorders in the context of relevant environmental contributing factors. Both obvious and counter-intuitive aspects of addiction can now be understood through research into the neurobiology of decision-making, choice, habit formation, and brain development. This evolving and growing body of work can be intimidating for clinicians and patients, but exploration of the basic concepts can help to reduce the mystery of addictive behaviors and provide frameworks for prevention and treatment of substance use disorders (SUDs).
Although there are multiple frameworks through which to view and consider addiction (a term corresponding to the definition of moderate to severe SUD in DSM 5),2 including philosophical and psychosocial frameworks, most researchers in addiction medicine consider addiction to be a chronic, relapsing, but treatable biopsychosocial disorder of the brain. What this means is that the clinical components of addictive disorders are reflected in changes to specific brain neurotransmitter systems and functional networks, knowledge of which can help clinicians ground their practice in neuroscience. Shared effects of various substances are apparent for major neurobiological models of addiction, such as those delineated by Koob and Volkow,3 but it is also important to understand unique neurobiological effects of different classes of drugs.
In this review, we focus on the key elements of addiction and describe neurobiological underpinnings of these features. A full review of neurobiology is beyond the scope of this overview. Instead, we focus on the major maturational and neuroplastic processes that underlie the onset and natural history of SUD both in general populations, where SUDs may improve without access to formal treatment, and in treatment-seeking cases, which tend to have a chronic, relapsing course. We then describe the alterations in brain circuitry that help explain the loss of control over drug use in those who have developed an addiction as they relate to the three stages of the addiction cycle: binge/intoxication, withdrawal/negative affect, and preoccupation/anticipation (or craving).4 We lastly describe areas where future research is needed to fill in gaps in knowledge.
Onset and Natural History of Substance Use Disorders
The science of human development underpins our understanding of the onset of SUD.5 Environmental factors that influence an individual’s risk for substance use and developing an SUD include not only substance exposures themselves but also characteristics of the family, neighborhood, school, and cultural context in which the individual grows up and lives. These environmental influences interact in complex ways with genes as well as brain maturation.6 Propensity to use substances may arise prior to the first use of alcohol, nicotine, or other substances. Early-life environments that are stressful have been demonstrated to increase subsequent likelihood of use of substances and SUD, in both preclinical (animal) and clinical research.7 For example, the Adverse Childhood Experiences (ACES) retrospective cohort study of primary care patients (N=~8600) demonstrated that patients who reported more early childhood abuse and neglect experiences were more likely to report using substances during teen or adult years, to have an SUD, and to have initiated drug use at an early age.8
While a complete, mechanistic explanation is not fully developed, it is apparent that genetic factors underlie significant risk for substance use disorders,9 and that interactions of biology and the environment play a key role in these trajectories from early childhood toward SUD in the teens and in adult life. Epigenetic modifications of gene expression have been shown to link early developmental adversity to sensitivity to stress and associated behavioral disorders.10 Importantly, altered brain development associated with adverse environments can sometimes be ameliorated.11 A study in rural Georgia of participants in a prevention trial found a strong correlation between number of teen years in poverty and diminished volume of brain structures that contribute to academic functioning, social development, learning, memory, mood, and stress reactivity (left dentate gyrus, CA3 subfield of the hippocampus, and left amygdala) by the early 20s among those who were in the study control group.12 In contrast, those volumetric declines were not seen in those in the group whose families had received an experimental family-focused intervention when they were in middle school (i.e. early adolescence).12 These results support the premise that family-based interventions may mitigate the developmental impacts of what otherwise would be a toxic social environment (poverty), even without changing other aspects of that environment.
Interventions as early as the prenatal and infancy periods have also been demonstrated to improve later behavioral, cognitive, and health outcomes. For instance, large randomized studies show that providing guidance to low-income, first-time mothers during pregnancy and in the first two years of a child’s life through home visitation by nurses can have a range of lasting positive impacts on the child—not only reduced abuse and neglect but also reduced substance use onset and improved cognitive and behavioral outcomes into the teen years.13 These and other longitudinal, prevention intervention studies document the importance of risk factors for the onset of substance use and the potential benefits of effective interventions that successfully address such risk factors.14
A key question is why substance use so typically starts during adolescence, a period of risk for many behavioral concerns.5,6 Developmental neuroscience has documented that brain development is uneven, with limbic structures involved in emotional responsivity and reward maturing earlier than prefrontal cortical areas involved in decision making, impulse control, and judgment (see Figure 1).15 Circuitry of the prefrontal cortex does not completely reach maturity until the mid-20s.16 One result of this developmental mismatch is an increased propensity for risk-taking among youth.17 While this can be seen as developmentally appropriate, since this time of life is when an individual is challenged to become independent from parents and fashion their self-identity, the propensity for novel experiences and risk-taking can have very negative outcomes. The uneven maturation of the adolescent brain also increases susceptibility to environmental influences dominant during the teen years, such as peer influence.18 The hazards are compounded by the fact that, since it is not fully mature, the adolescent brain is more vulnerable to lasting effects of substance use, including increased risk of addiction.19 The risk for developing a SUD is highest for those who initiate substances in the early teens, and addiction is most likely to begin in the late teen years.20
Figure 1:
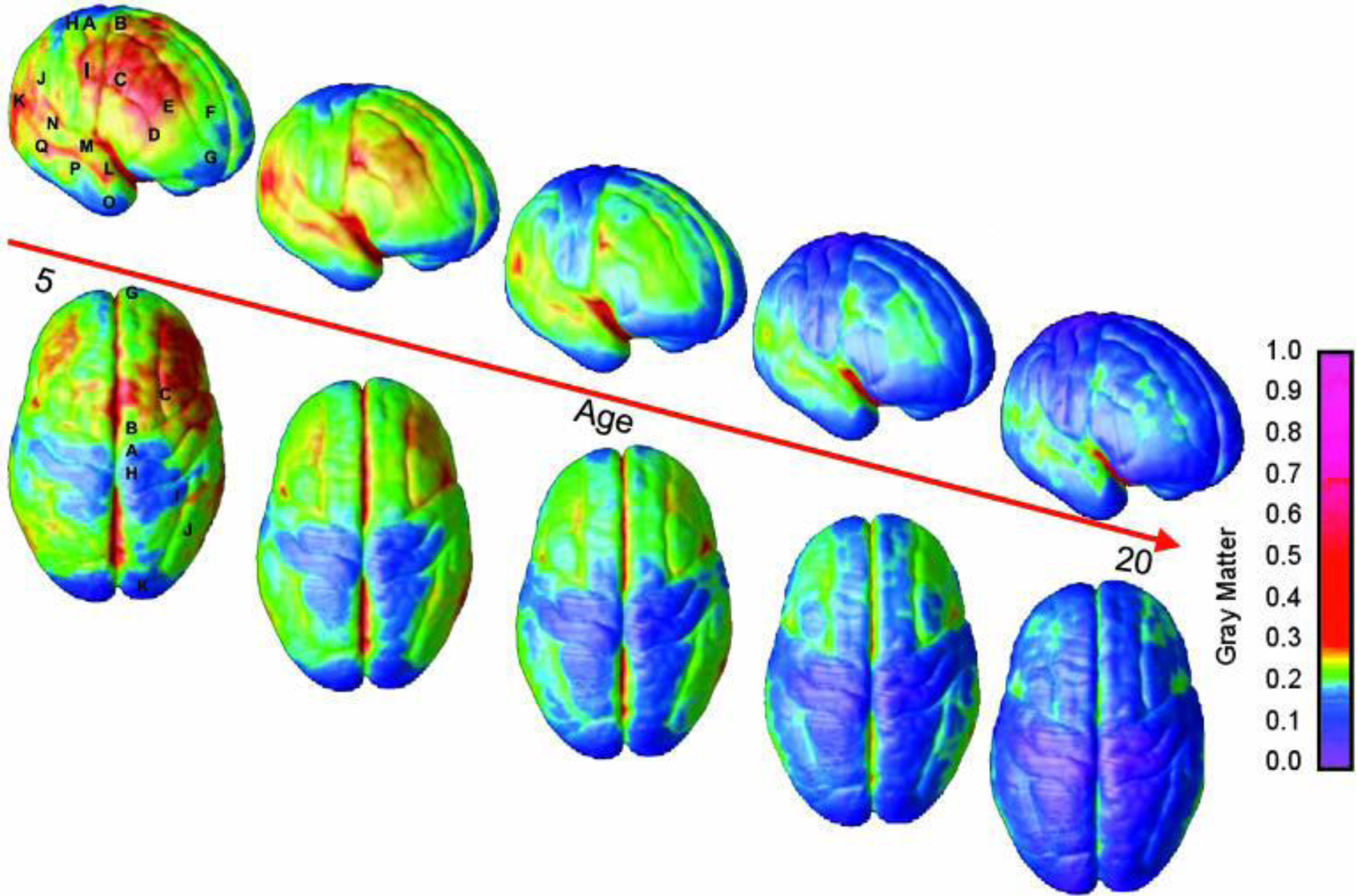
Right lateral and top views of the dynamic sequence of gray matter maturation over the cortical surface. The side bar shows a color representation in units of gray matter volume. The following regions were selected for analyses in each hemisphere: A, precentral gyrus and primary motor cortex; B, superior frontal gyrus, posterior end near central sulcus; C, inferior frontal gyrus, posterior end; D, inferior frontal sulcus, anterior end in the ventrolateral prefrontal cortex; E, inferior frontal sulcus in the dorsolateral prefrontal cortex; F, anterior limit of superior frontal sulcus; G, frontal pole; H, primary sensory cortex in postcentral gyrus; I, supramarginal gyrus (area 40); J, angular gyrus (area 39); K, occipital pole; L–N, anterior, middle, and posterior portions of STG; O–Q, anterior, middle, and posterior points along the inferior temporal gyrus anterior end.
(Reproduced from Gogtay N, Giedd JN, Lusk L, et al. Dynamic mapping of human cortical development during childhood through early adulthood. PNAS. 2004;101(21):8174–8179.)
Most substance use begins with tobacco, alcohol, and then cannabis—before other substances like cocaine or opioids are used. Most who misuse opioids have already used these so-called “gateway substances” during adolescence.21,22 While multiple studies attest to overlapping onset of substance use,23,24 neurobiological gateway mechanisms for nicotine have the most evidence. By increasing midbrain dopamine neurons’ firing rate, nicotine can amplify the reinforcing effects of other substances and non-drug rewards (see below).22,25
Because of the complex interacting biological, psychosocial, and developmental risk factors and various protective factors that may mitigate them (such as attentive parenting and strong social supports), only a subset of people who use drugs—even in their teen years—go on to develop an SUD.26,27 For instance, using DSM-III-R or DSM-IV definitions, retrospective U.S. national studies have estimated that about 9% of cannabis users will ever develop cannabis dependence, with higher rates for those with early onset, frequent use, or use of more potent forms of cannabis. Similarly, 15–23% of alcohol users develop alcohol dependence, 17–21% of cocaine users develop cocaine dependence and 32–67% of tobacco users develop nicotine dependence.26,27
Of those who develop an SUD, many report current remission of their symptoms. Over 20 million persons in the USA report being in recovery from problematic substance use; most of these persons either did not avail themselves of formal treatment or it was inaccessible to them, yet they may have years or decades without symptoms of SUD.28–30 In some cases, SUDs resolve on their own as a result of maturation or because of changing life circumstances or environments—the famous desistence of opioid use among Vietnam war soldiers returning to the U.S. is one example.31 However, a recent longitudinal study following 11 early cohorts of the annual Monitoring the Future study found that the majority of high school seniors who had reported SUD symptoms when surveyed in the late 1970s or early 1980s still reported SUD symptoms more than three decades later, at age 50—indicating a long-term course for many SUDs that emerge early in life (i.e. in the teen years).32
Among persons admitted to treatment for SUD (especially severe SUD), a chronic, relapsing course is well documented.2,33,34 Use of substances in the year following treatment interventions varies but has been estimated to be 40–60%,35 with longer studies suggesting multiple cycles of heavy use, periodic abstinence, interspersed with incarceration for many.33,34,36,37 While treatment is a broad predictor of remission,28,35,36 the underlying chronic, relapsing pattern remains and may be understood based on the neurobiology of the addiction cycle (Figure 2).38,39
Figure 2:
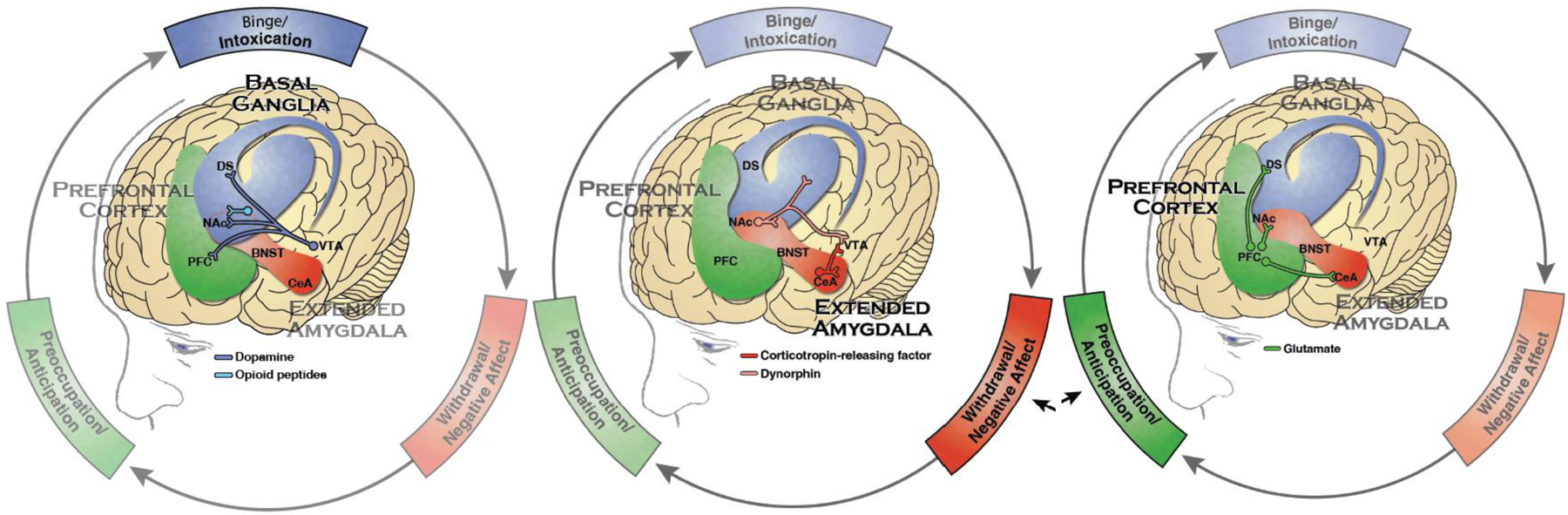
The three stages of addiction: Binge/Intoxication, the stage at which an individual consumes an intoxicating substance and experiences its rewarding or pleasurable effects, primarily involves basal ganglia structures; Withdrawal/Negative Affect, the stage at which an individual experiences a negative emotional state in the absence of the substance, involves stress hormone responses and the extended amygdala; and Preoccupation/Anticipation, the stage at which one seeks substances again after a period of abstinence, involving interactions of the prefrontal cortex, the extended amygdala, and the basal ganglia.
(Reproduced from U.S. Department of Health and Human Services (HHS), Office of the Surgeon General, Facing Addiction in America: The Surgeon General’s Report on Alcohol, Drugs, and Health. Washington, DC: HHS, 2016.)
Notes: Blue represents the basal ganglia involved in the Binge/Intoxication stage. Red represents the extended amygdala involved in the Negative Affect/Withdrawal stage. Green represents the prefrontal cortex involved in the Preoccupation/Anticipation stage. Not shown is the neurotransmitter norepinephrine which is also activated in the extended amygdala during withdrawal. PFC - prefrontal cortex, DS - dorsal striatum, NAc - nucleus accumbens, BNST - bed nucleus of the stria terminalis, CeA - central nucleus of the amygdala, VTA - ventral tegmental area.
Neuroplastic Changes in Addiction and the Addiction Cycle
Among the changes apparent with chronic drug intake is the formation of tolerance and physical dependence. While not synonymous with addiction, tolerance and dependence develop when the body adapts to the frequent presence of an exogenous substance by counteracting its intracellular signaling responses, such as the levels of adenyl cyclase, and by dialing down its own related endogenous signaling system—endorphins and dynorphins in the case of opioid drugs, or downregulating cannabinoid CB1 receptors in the case of cannabis, and so on. The resulting tolerance is the need for increasing doses of the drug to produce the desired effect; and the resulting physical dependence leads to the emergence of withdrawal symptoms when the drug is interrupted. While tolerance and withdrawal (i.e. physical dependence) are typically part of addiction, many substances, including multiple prescription medications, may produce dependence and tolerance without producing addiction. Furthermore, the severity and duration of withdrawal symptoms depend on the specific drug consumed.
Addiction, per se, arises when the individual loses control over use of the substance (i.e. spending a great deal of time using a substance, using larger amounts than intended, etc.). It is driven by the formation of associations between the drug and environmental as well as internal cues that trigger craving, or incentive salience; neurobiological changes that lead to intensified dysphoria during withdrawal, as well as increased preoccupation with the drug; and reduction of self-inhibitory control over drug taking despite, in many cases, an awareness that the drug has assumed too much importance and is harming health and other aspects of the individual’s life like relationships, school, or work.2,38–41
As displayed in Figure 2, three areas of the brain are critical for these processes: the basal ganglia, the extended amygdala, and the prefrontal cortex.38 Changes to circuits in these brain areas play key roles in three repeating stages of the addiction cycle, respectively: binge/intoxication, the acute rewarding and reinforcing effects upon taking the drug; withdrawal/negative affect, the unpleasant physical and emotional symptoms associated with a period of abstinence; and preoccupation/anticipation, craving the drug in response to internal and external cues. Dynamic relationships across these three stages help explain patterns of heavy consumption, followed by abstinence (can be brief or extended), and then recurrent use to alleviate negative internal states or in response to anticipatory motivation. Classes of substances differ in how they produce their effects, especially in the binge/intoxication phase, as well as in how and to what extent they disrupt the relevant circuits, but the overall patterns are consistent across substances.
Binge/intoxication.
Either immediately or soon after taking a drug (rapidity depending on the substance’s specific properties and mode of administration), the individual experiences rewarding euphoria (i.e. positive reinforcement) and, in the case of a person with dependence or addiction, relief from withdrawal symptoms (i.e. negative reinforcement). The exact neurobiological correlates of experienced pleasure or the drug “high” are debated and may differ for different substances (see Future Directions, below), but well understood is the role of dopamine signaling in neurons projecting from the ventral tegmental area (VTA) to the nucleus accumbens (NAC) in building associations between drug-taking and reward, reinforcing those associations each time drug-taking is repeated, and reinforcing associations between the drug and internal and external cues.42 Multiple preclinical studies in rodents have identified the neuronal and neurochemical actions of drugs on this dopaminergic VTA-NAC circuit (see Koob and Volkow4 for a full description of these pathways). While the mechanisms of action vary by drug, enhancing dopamine activity in the basal ganglia is responsible for the reinforcing properties of all substances whose misuse can result in addiction.
When an association between dopamine and positive reinforcement was first discovered in rodent studies in the 1970s,43,44 it was believed that dopamine was the neurotransmitter responsible for rewarding feelings that drive drug seeking. Over subsequent decades, dopamine came to be thought of as the “pleasure chemical,” and this idea was expressed widely in the media and popular science explanations of drug addiction. In the past two decades, the science of dopamine has departed from this popular conception with the discovery that dopamine has more to do with reinforcement learning and motivated goal-seeking, and probably not drugs’ hedonic effects, which engage other neurotransmitter systems including endogenous opioids and cannabinoids.45 Dopamine signaling in the basal ganglia controls the salience of cues (external or internal) that engender action.46 It is released in the NAC both in anticipation of rewards and when rewards exceed expectation, and it is diminished when rewards do not materialize or are less than expected.
The development of incentive salience is a crucial neuroplastic response to natural rewards such as food and one that is hijacked by drugs. Through conditioning and expectation of reward, a memory is formed that will incentivize behaviors to consume the reward (e.g. food or drugs). The conditioned memories that drive incentive salience towards conditioned stimuli are fundamental to survival but also facilitate the development of an addiction. When drugs are repeatedly administered, dopamine comes to be released when the individual encounters or experiences the cues associated with the drug prior to its consumption, energizing the behaviors to obtain it.47 Incentive salience has the motivational effect of driving drug-seeking during the anticipation/preoccupation phase (described below).
Depending on the class of drug, dopaminergic signaling in the basal ganglia may be enhanced through several different mechanisms including: direct or indirect increases in activity of VTA-dopamine neurons, increasing terminal dopamine release, blocking dopamine reuptake, or enhancing the post-synaptic actions of dopamine on NAC neurons. An additional neuroplastic mechanism shared among most drugs with their repeated administration is the increase of the ratio of the glutamate receptor, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, to that of the N-methyl-D-aspartate (NMDA) receptor, which results in enhanced excitatory transmission onto VTA-dopamine neurons.48,49 See Box 1 for summaries of the specific mechanisms of action of major drug classes.
Box 1: Reinforcement (Dopamine Signaling) Mechanisms for Major Drug Classes.
OPIOIDS
Opioids, including morphine heroin, fentanyl, and prescription analgesics like oxycodone, increase dopamine signaling in the basal ganglia indirectly through their actions at specific opioid receptors, especially the mu opioid receptor. Preclinical research shows that activation of mu opioid receptors on gamma aminobutyric acid (GABA) cells in the VTA disinhibits dopamine neurons increasing their activity and enhancing dopamine release in the NAC.50
ALCOHOL
Alcohol’s reinforcing effects have been associated with processes involving multiple molecular targets, including enhancement of opioid signaling through mu opioid receptors. Alcohol also enhances GABAergic neurotransmission via its direct effects in GABA-A receptors which are believed to contribute to reward and to its anxiolytic effects.4
STIMULANTS (COCAINE AND AMPHETAMINE-LIKE SUBSTANCES)
The reinforcing effects of stimulants are mediated by their direct effects on dopamine neurons. Cocaine enhances dopamine levels primarily by inhibiting the dopamine transporter, thus reducing reuptake of dopamine from the synapse. Amphetamine-like substances, however, both inhibit the transporter and directly increase vesicular dopamine release.48 In either case, the net effect is to increase dopamine in the NAC. The effects of this class of drugs are also mediated by increases in the activation of the other monoamine systems, serotonin and norepinephrine.48 The bias towards a particular monoamine system depends on the specific stimulant. For example, cathinones (bath salts) have a greater effect on serotonin than does amphetamine, which is biased towards dopamine and norepinephrine systems.51
BENZODIAZEPINES
Benzodiazepines are allosteric modulators of GABA-A receptors, meaning that their binding shifts the way the receptor responds to its standard ligands. Although both GABA and dopamine neurons in the VTA express these receptors, benzodiazepines bind to those containing the alpha-1 subunit, which are only found on GABA neurons in the VTA and are lacking in VTA-dopamine neurons.52–54 The resulting inhibition of VTA-GABA neurons enhances dopamine release. In human studies, benzodiazepines enhance the subjective effects of opioids, including “high” and “liking,” indicating that these drugs’ rewarding properties may be synergistic, accounting for their common co-use.55 Combining the two classes of drugs has also been implicated in increasing the risk for overdose, due to their shared effect of inhibiting respiration.55–57
NICOTINE
The reinforcing effects of nicotine are mediated by multiple receptors in the VTA and NAC,4 and its actions at nicotinic acetyl choline receptors (nAChR) alpha-4 beta-2 subtype appear to be integral to these effects.58 In addition to increasing dopamine discharge rate, nicotine changes the pattern of discharge to favor a phasic or bursting mode.59 Dopamine bursting promotes the formation of associations between stimuli and rewards, and this may be the basis for reinforcement-enhancing effects of nicotine in combination with other substances.60,61 Analyzing data from two cohort studies, Kandel and Kandel found that cocaine dependence was highest in users who had first smoked cigarettes and that concurrent smoking around the time of cocaine initiation was associated with more persistent cocaine use and addiction—consistent with the priming effect they found in an animal model. Conversely, cocaine does not appear to prime the nicotine response.22 Nicotine’s linkages to later addiction to other classes of drugs like opioids are not fully established.
CANNABINOIDS
Cannabinoids activate type 1 cannabinoid receptors (CB1) in the VTA, but the mechanism by which this activation facilitates dopamine release are not well understood. Cannabis receptor pharmacology research is providing clues. For example, in light of the sometimes contradictory reinforcing and aversive effects of cannabis, Spiller and colleagues document the importance of balance of both CB1 and CB2 receptor activation in cannabis effects.62 Their work suggests that reinforcing and aversive effects may be mediated by differential CB1 and CB2 receptor expression.
Another important component of the binge/intoxication stage is release of both dopamine and glutamate (an excitatory neurotransmitter) in the dorsal striatum, which is implicated in behavioral reinforcement.63,64 This is another mechanism through which learning processes contribute to the development of compulsive drug use. Indeed, addiction is sometimes compared to a well-learned skill, one that is difficult to unlearn (see Future Directions below).
Withdrawal/Negative Affect.
Koob and Volkow highlight the importance of negative emotional states and memories in the addiction cycle, based on both the expressed responses of human subjects and from fMRI studies correlated with amygdala and hippocampus activation during craving episodes.4
As the drug leaves the system and its acute effects wane, an individual who has developed an addiction may experience not only the physical symptoms of withdrawal, which are usually relatively short term, but also protracted negative emotional withdrawal symptoms that are part of the long-lasting adaptations in addiction. The negative emotional state during the withdrawal stage of the addiction cycle is caused by increased sensitivity of the extended amygdala to stress, mediated in part through enhanced corticotropin-releasing factor (CRF), norepinephrine, and dynorphin signaling. The feelings of stress caused by this activity in the extended amygdala, compounded in some cases by intense physical symptoms of drug withdrawal, are one reason why it is incorrect to think of people with addiction as simply hedonically driven, motivated by the euphoria or high caused by drug intoxication. To a significant degree, people with addiction are motivated to experience relief, to escape the stress and suffering of withdrawal, which in many cases can be intense or even unbearable.
Preoccupation/Anticipation
A core feature of SUD, especially the more severe forms (i.e. addiction), is craving, sometimes described as an overwhelming need and urgent compulsion to use a substance after a period of abstinence (short or long).2,40,41 This preoccupation/anticipation stage in the addiction cycle involves reciprocal interaction of the dopaminergic pathways of the prefrontal cortex, the brain area most associated with goal-directed behaviors, self-inhibition, and judgment, with both the extended amygdala and the basal ganglia.
Signaling in dopaminergic and glutamate connections between the prefrontal cortex and basal ganglia motivate drug seeking in response to cues. Inhibitory connections between the prefrontal cortex and the dorsal striatum that have been weakened by chronic substance use let the habitual response—taking the drug—win out. These weakened inhibitory signals are especially powerless in the face of the dysphoric feelings generated by the stress neurotransmitters in the extended amygdala. This shift in the balance of what are colloquially called the “Go” and “Stop” systems coordinated by the prefrontal cortex helps explain addiction’s chronic, relapsing course.65
Future Directions
Much is known about the neurobiology of addiction, at least in broad terms—including the development of incentive salience and compromised self-inhibition that underpins compulsive drug use. But there are areas where our knowledge remains limited. One, oddly enough, is the neurobiology of pleasure itself. Greater understanding that dopamine has to do mainly with reinforcement but not with hedonic perception has left a gap in our understanding of just what is pleasurable about rewards. Some research points to so-called “hedonic hotspots” in the orbitofrontal cortex, insula, NAC, ventral pallidum, and pontine parabrachial nucleus that have been identified in conjunction with “liking” in animal models.66 Liking here is distinguished from the anticipatory (and dopamine-related) signal of “wanting.” Activity in hedonic hotspots during pleasurable experience involves endogenous opioid, endocannabinoid, and orexin (but not dopamine) signaling,45 and how different drugs interact with these hotspots remains to be seen. But it is almost certainly the case that the neurobiology of pleasure, including the drug high, will not be a simple story of one neurotransmitter flooding the brain with good feelings.
Another gap in our understanding is the neurobiology of recovery from addiction. We understand how the brain changes when an SUD develops, but we still know very little about how the brain restores balance among the systems disrupted during active drug use. Longitudinal clinical research suggests that stable recovery is more likely with lengthy periods of supported abstinence.34,67 Yet, susceptibility to relapse even after years of abstinence is not rare, strongly indicating that some neuroplastic changes, for instance learned associations between drugs and reward that were formed during the development of the disorder, weaken only very gradually. Addiction is often likened to riding a bicycle: You may not have ridden one in years; yet, if you get on one, you’ll be able to ride it quite well. Among the many avenues of addiction treatment research being pursued, one is to find ways of identifying and weakening those learned associations between a drug and its craving-inducing cues.68 It may also be the case that different drug use disorders may have different long-term impacts on those who have recovered from them, and it is likely the case that individual differences will play a role. There could be many neurobiological trajectories of recovery.
Conclusions
The medical framing of addiction as a chronic, relapsing brain disorder has been slow to win adherents outside of medicine, and studies show that even clinicians cling to stigmatizing views that see people with addiction as bad or weak.69 The medical framing has been crucial to research explicating addiction neurobiology and has been essential to developing effective treatments, such as those that currently exist for opioid use disorder. However, since learning mechanisms are so central to the processes that make drug use compulsive in people with addiction, some critics argue that the medical framing is overly reductive and pessimistic, and even disempowering to persons who may hear “brain disorder” or “brain disease” and assume that it somehow means non-treatable or incurable, which is not the case.70, 71 These terminological (and in some cases, philosophical) questions will continue to be fruitful areas of debate and inquiry. If anything, framing addiction as a learned behavior (as well as a disease) helps make the case for greater societal and healthcare investment in prevention: It may be the case that the surest way to keep people from riding bikes is to keep them from learning to ride them in the first place.
Addiction occurs in a minority of people who use drugs repeatedly, a result of a combinations of biological and psychosocial risk factors. The pathways to addiction involve changes to neurocircuits in limbic and prefrontal regions, and some of these neuroplastic changes may be slow to resolve or “change back” even with months or years of abstinence, warranting calling addiction a chronic brain disorder with high risk of relapse. This understanding of addiction has been crucial to marshalling biomedical research to find and develop treatments. Educating the public, policymakers, and clinicians about the efficacy of these treatments is a crucial part of eroding the stigma that has prevented their wider adoption.
Key Points:
Developmental neuroscience helps to explain risk for onset of substance use and substance use disorders.
Severe substance use disorders involve changes to limbic and prefrontal brain areas after chronic drug exposure.
The addiction cycles involves reinforced, learned associations between drugs and cues that trigger anticipation of that reward (known as incentive salience), as well as heightened dysphoria during withdrawal, and weakened prefrontal cortical circuits needed for inhibiting habitual responses.
Synopsis:
While substance experimentation typically begins in adolescence, substance use disorders usually develop in late teens or early adulthood, often in individuals who are vulnerable because of biological and socioeconomic risk factors. Severe substance use disorders—synonymous with addiction—involve changes to limbic and prefrontal brain areas after chronic drug exposure. These changes involve learned associations between drug reward and cues that trigger anticipation of that reward (known as incentive salience), as well as heightened dysphoria during withdrawal and weakened prefrontal circuits needed for inhibiting habitual responses.
Clinical Care Points:
Developmental neuroscience helps to demonstrate both the negative power of risk environments and the positive power of prevention interventions.
Treatments that reduce the rewarding and reinforcing properties of substances address a key component of the neurobiological binge/intoxication phase of the addiction cycle.
Both acute withdrawal syndromes and longer-term withdrawal-related negative affect are key risk factors for relapse.
Providing support to patients in recovery in times of stress addresses a key risk factor for relapse, as elucidated in multiple neuroscience studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Publisher's Disclaimer: Disclaimer: The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Institute on Drug Abuse, the National Institutes of Health, or the U.S. Department of Health and Human Services.
Disclosures: Compton reports long-term stock holdings in General Electric Company, 3M Companies and Pfizer, Inc. unrelated to the manuscript. Other authors have no disclosures to report.
References:
- 1.O’Driscoll K, Leach JP. “No longer Gage”: an iron bar through the head. Early observations of personality change after injury to the prefrontal cortex. BMJ. 1998;317(7174):1673–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, Text Revision (DSM-5-TR). Washington, D.C.: American Psychiatric Press. 2022. [Google Scholar]
- 3.Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry. 2016;3(8):760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koob GF, Volkow ND. Neurocircuitry of Addiction. Neuropsychopharmacology. 2010;35:217–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.National Research Council and Institute of Medicine. From Neurons to Neighborhoods: The Science of Early Childhood Development. Committee on Integrating the Science of Early Childhood Development. Washington, D.C.: National Academy Press. 2000 [PubMed] [Google Scholar]
- 6.Volkow ND, Boyle M. Neuroscience of addiction: relevance to prevention and treatment. Am J Psychiat. 2018;175:729–740. [DOI] [PubMed] [Google Scholar]
- 7.Hughes K, Bellis MA, Hardcastle KA, et al. The effect of multiple adverse childhood experiences on health: a systematic review and meta-analysis. Lancet Public Health. 2017;2(8):e356–e366. [DOI] [PubMed] [Google Scholar]
- 8.Dube SR, Felitti VJ, Dong M, Chapman DP, Giles WH, Anda RF. Childhood abuse, neglect, and household dysfunction and the risk of illicit drug use: the Adverse Childhood Experiences study. Pediatrics. 2003;111(3):564–572. [DOI] [PubMed] [Google Scholar]
- 9.Walker DM, Nestler EJ. Neuroepigenetics and addiction. Handbook of clinical neurology. 2018;148:747–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Enoch M-A. The role of early life stress as a predictor for alcohol and drug dependence. Psychopharmacology. 2011;214(1):17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson CA, Zeanah CH, Fox NA, Marshall PJ, Smyke AT, Guthrie D. Cognitive recovery in socially deprived young children: the Bucharest Early Intervention Project. Science. 2007;318:1937–1940. [DOI] [PubMed] [Google Scholar]
- 12.Brody GH, Gray J, Yu T, et al. Family-centered prevention ameliorates the longitudinal association of poverty with hippocampal and amygdalar volumes in adulthood. JAMA Pediatrics. 2017;171(1):46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olds DL, Kitzman HJ, Cole RE, et al. Enduring effects of prenatal and infancy home visiting by nurses on maternal life course and government spending: follow-up of a randomized trial among children at age 12 years. Arch Pediat Adol Med. 2010;164(5):419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catalano RF, Fagan AA, Gavin LE, et al. Worldwide application of prevention science in adolescent health. Lancet. 2012;379(9826):1653–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spear LP, The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav R. 2000;24:417–463. [DOI] [PubMed] [Google Scholar]
- 16.Gogtay N, Giedd JN, Lusk L, et al. Dynamic mapping of human cortical development during childhood through early adulthood. PNAS. 2004;101(21):8174–8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinberg L Risk taking in adolescence: new perspectives from brain and behavioral science. Curr Dir Psychol Sci. 2007;16(2):55–59. [Google Scholar]
- 18.Albert D, Chein J, Steinberg L. Peer influences on adolescent decision making. Curr Dir Psychol Sci. 2013;22(2):114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Squeglia LM, Jacobus J, Tapert SF. The influence of substance use on adolescent brain development. Clin EEG Neurosci. 2009;40(1):31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Compton WM, Thomas YF, Stinson FS, Grant BF. Prevalence, correlates, disability, and comorbidity of DSM-IV drug abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen PsychiatRY. 2007;64:566–576. [DOI] [PubMed] [Google Scholar]
- 21.Robins L, Compton W, Horton J. Is heroin the worst drug? Implications for drug policy. Addiction Research Theory. 2000;8(6):527–547. [Google Scholar]
- 22.Kandel DB, Kandel ER. The Gateway Hypothesis of substance abuse: developmental, biological and societal perspectives. Acta Paediatrica. 2015;104:130–137. [DOI] [PubMed] [Google Scholar]
- 23.Han B Compton WM, Blanco C, DuPont RL National Trends in Substance Use and Use Disorders Among Youth, J Am Acad Child Adol Psychiatry. 2017;56(9):747–754. [DOI] [PubMed] [Google Scholar]
- 24.DuPont RL, Han B, Shea CL, Madras BK. Drug use among youth: national survey data support a common liability of all drug use. Preventive Medicine. 2018;113:68–73. [DOI] [PubMed] [Google Scholar]
- 25.Perkins KA, Karelitz JL, Boldry MC. Nicotine acutely enhances reinforcement from non-drug rewards in humans. Front Psychiatry. 2017;8:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lopez-Quintero C, Hasin DS, de Los Cobos JP, Pines A, Wang S, Grant BF, Blanco C. Probability and predictors of remission from life-time nicotine, alcohol, cannabis or cocaine dependence: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Addiction. 2011;106(3):657–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anthony J, Warner L, Kessler R. Comparative epidemiology of dependence on tobacco, alcohol, controlled substances, and inhalants: Basic findings from the National Comorbidity Survey. Experimental and Clinical Psychopharmacology. 1994;2:244–268. [Google Scholar]
- 28.Jones CM, Noonan RK, Compton WM. Prevalence and correlates of ever having a substance use problem and substance use recovery status among adults in the United States, 2018. Drug Alcohol Depend. 2020;214:108169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Compton WM, Thomas YF, Stinson FS, Grant BF. Prevalence, correlates, disability, and comorbidity of DSM-IV drug abuse and dependence in the United States: results from the national epidemiologic survey on alcohol and related conditions. Arch Gen Psychiatry. 2007;64(5):566–76. [DOI] [PubMed] [Google Scholar]
- 30.Hasin DS, Stinson FS, Ogburn E, Grant BF. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2007;64(7):830–42. [DOI] [PubMed] [Google Scholar]
- 31.Robins LN, Helzer JE, Hesselbrock M, Wish E. Vietnam veterans three years after Vietnam: how our study changed our view of heroin. Am J Addict. 2010. May-Jun;19(3):203–11. [DOI] [PubMed] [Google Scholar]
- 32.McCabe SE, Schulenberg JE, Schepis TS, McCabe VV, Veliz PT. Longitudinal analysis of substance use disorder symptom severity at age 18 and substance use disorder in adulthood. JAMA Network Open, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dennis ML, Foss MA, Scott CK. An eight-year perspective on the relationship between the duration of abstinence and other aspects of recovery. Evaluation Review. 2007;31(6):585–612. [DOI] [PubMed] [Google Scholar]
- 34.Dennis ML, Scott CK, Funk R, Foss MA. The duration and correlates of addiction and treatment careers. J Sub Abuse Treat. 2005;28(2):S51–62. [DOI] [PubMed] [Google Scholar]
- 35.McLellan AT, Lewis DC, O’Brien CP, Kleber HD. Drug dependence, a chronic medical illness: implications for treatment, insurance, and outcomes evaluation. JAMA. 2000. Oct 4;284(13):1689–95 [DOI] [PubMed] [Google Scholar]
- 36.Scott CK, Foss MA, Dennis ML. Pathways in the relapse—treatment—recovery cycle over 3 years. Journal of Substance Abuse Treatment. 2005;28(2):S63–72. [DOI] [PubMed] [Google Scholar]
- 37.Hser YI, Longshore D, Anglin MD. The life course perspective on drug use: A conceptual framework for understanding drug use trajectories. Evaluation Review. 2007;31(6):515–47. [DOI] [PubMed] [Google Scholar]
- 38.U.S. Department of Health and Human Services (HHS), Office of the Surgeon General, Facing Addiction in America: The Surgeon General’s Report on Alcohol, Drugs, and Health. Washington, DC: HHS, 2016. [PubMed] [Google Scholar]
- 39.Feltenstein MW, See RE, Fuchs RA. Neural Substrates and Circuits of Drug Addiction. Cold Spring Harb Perspect Med. 2021;11(4):a039628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Q, Wang Y, Zhang Y, Li W, Yang W, Zhu J, et al. Craving correlates with mesolimbic responses to heroin-related cues in short-term abstinence from heroin: an event-related fMRI study. Brain Res. 2012;1469:63–72. [DOI] [PubMed] [Google Scholar]
- 41.Kakko J, Alho H, Baldacchino A, Molina R, Nava FA, Shaya G. Craving in Opioid Use Disorder: From Neurobiology to Clinical Practice. Front Psychiatry. 2019;10:592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koob GF. Hedonic valence, dopamine and motivation. Mol Psychiatry. 1996;1:186–189. [PubMed] [Google Scholar]
- 43.Olds J, Milner P. Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol. 1954;47:419–27. [DOI] [PubMed] [Google Scholar]
- 44.Corbett D, Wise RA. Intracranial self-stimulation in relation to the ascending dopaminergic systems of the midbrain: a moveable electrode mapping study. Brain Res. 1980;185(1):1–15. [DOI] [PubMed] [Google Scholar]
- 45.Olney JJ, Warlow SM, Naffziger EE, Berridge KC. Current perspectives on incentive salience and applications to clinical disorders. Curr Opin Behav Sci. 2018;22:59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Viviani R, Dommes L, Bosch J et al. Signals of anticipation of reward and of mean reward rates in the human brain. Sci Rep 10, 4287 (2020). 10.1038/s41598-020-61257-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schultz W Multiple reward signals in the brain. Nat Rev Neurosci. 2000;1:199–207. [DOI] [PubMed] [Google Scholar]
- 48.Korpi ER, den Hollander B, Farooq U, Vashchinkina E, Rajkumar R, Nutt DJ, et al. Mechanisms of action and persistent neuroplasticity by drugs of abuse. Pharmacol Rev. 2015;67:872–1004. [DOI] [PubMed] [Google Scholar]
- 49.Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. [DOI] [PubMed] [Google Scholar]
- 50.Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992;12:483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verrico CD, Miller GM, Madras BK. MDMA (ecstasy) and human dopamine, norepinephrine, and serotonin transporters: implications for MDMA-induced neurotoxicity and treatment. Psychopharmacology (Berl). 2007;189:489–503. [DOI] [PubMed] [Google Scholar]
- 52.Kalivas PW, Duffy P, Eberhardt H. Modulation of A10 dopamine neurons by gamma-aminobutyric acid agonists. J Pharmacol Exp Ther. 1990;253:858–866. [PubMed] [Google Scholar]
- 53.Okada H, Matsushita N, Kobayashi K, Kobayashi K. Identification of GABAA receptor subunit variants in midbrain dopaminergic neurons. J Neurochem. 2004;89:7–14. [DOI] [PubMed] [Google Scholar]
- 54.Tan KR, Brown M, Labouebe G, Yvon C, Creton C, Fritschy JM, et al. Neural bases for addictive properties of benzodiazepines. Nature. 2010;463:769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones JD, Mogali S, Comer SD. Polydrug abuse: A review of opioid and benzodiazepine combination use. Drug Alcohol Depend. 2012;125(1–2):8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jones CM, McAninch JK. Emergency department visits and overdose deaths from combined use of opioids and benzodiazepines. Am J Prev Med. 2015;49(4):493–501. [DOI] [PubMed] [Google Scholar]
- 57.Afzal A, Kiyatkin EA. Interactions of benzodiazepines with heroin: respiratory depression, temperature effects, and behavior. Neuropharmacology. 2019;158:107677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, et al. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–177. [DOI] [PubMed] [Google Scholar]
- 59.Grenhoff J, Aston-Jones G, Svensson TH. Nicotinic effects on the firing pattern of midbrain dopamine neurons. Acta Physiol Scand. 1986;128:351–358. [DOI] [PubMed] [Google Scholar]
- 60.Palmatier MI, Evans-Martin FF, Hoffman A, Caggiula AR, Chaudhri N, Donny EC, et al. Dissociating the primary reinforcing and reinforcement-enhancing effects of nicotine using a rat self-administration paradigm with concurrently available drug and environmental reinforcers. Psychopharmacology (Berl). 2006;184:391–400. [DOI] [PubMed] [Google Scholar]
- 61.Perkins KA, Karelitz JL, Boldry MC. Nicotine acutely enhances reinforcement from non-drug rewards in humans. Front Psychiatry. 2017;8:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spiller KJ, Bi GH, He Y, Galaj E, Gardner EL, Xi ZX. Cannabinoid CB1 and CB2 receptor mechanisms underlie cannabis reward and aversion in rats. British journal of pharmacology. 2019;176(9):1268–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nature Reviews Neuroscience. 2009;10(8):561–572. [DOI] [PubMed] [Google Scholar]
- 64.Belin D, Jonkman S, Dickinson A, Robbins TW, Everitt BJ. Parallel and interactive learning processes within the basal ganglia: Relevance for the understanding of addiction. Behavioural Brain Research/ 2009;199(1):89–102. [DOI] [PubMed] [Google Scholar]
- 65.Volkow ND, Baler RD. NOW vs LATER brain circuits: implications for obesity and addiction. Trends Neurosci. 2015;38(6):345–52. [DOI] [PubMed] [Google Scholar]
- 66.Nguyen D, Naffziger EE, Berridge KC. Positive Affect: Nature and brain bases of liking and wanting. Curr Opin Behav Sci. 2021;39:72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DuPont RL, Compton WM, McLellan AT. Five-year recovery: A new standard for assessing effectiveness of substance use disorder treatment. J Subst Abuse Treat. 2015;58:1–5. [DOI] [PubMed] [Google Scholar]
- 68.Young EJ, Briggs SB, Miller CA. The Actin Cytoskeleton as a Therapeutic Target for the Prevention of Relapse to Methamphetamine Use. CNS Neurol Disord Drug Targets. 2015;14(6):731–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stone EM, Kennedy-Hendricks A, Barry CL, Bachhuber MA, McGinty EE. The role of stigma in U.S. primary care physicians’ treatment of opioid use disorder. Drug Alcohol Depend. 2021;221:108627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lewis M Brain change in addiction as learning, not disease. N Engl J Med. 2018;379(16):1551–1560. [DOI] [PubMed] [Google Scholar]
- 71.Satel S, Lilienfeld SO. Addiction and the brain-disease fallacy. Front Psychiatry 2013;4:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Key Additional Readings:
- Volkow ND, Koob GF, McLellan AT. Neurobiologic Advances from the Brain Disease Model of Addiction. New England Journal of Medicine. 2016;374(4):363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton WM, Valentino RJ, DuPont RL. Polysubstance use in the U.S. opioid crisis. Molecular Psychiatry. 2021;26(1):41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Boyle M. Neuroscience of addiction: relevance to prevention and treatment. American Journal of Psychiatry. 2018;175:729–740. [DOI] [PubMed] [Google Scholar]
- Feltenstein MW, See RE, Fuchs RA. Neural Substrates and Circuits of Drug Addiction. Cold Spring Harbor Perspectives in Medicine. 2021;11(4):a039628. [DOI] [PMC free article] [PubMed] [Google Scholar]