

Summary
Histone deacetylase (HDAC) proteins are epigenetic regulators that govern a wide variety of cellular events. With a role in cancer formation, HDAC inhibitors have emerged as anti-cancer therapeutics. Among the eleven metal-dependent class I, II and IV HDAC proteins targeted by inhibitor drugs, class IIa HDAC4, 5, 7, and 9 harbor low deacetylase activity and are hypothesized to be “reader” proteins, which bind to post-translationally acetylated lysine. However, evidence linking acetyllysine binding to a downstream functional event is lacking. Here, we report for the first time that HDAC4, 5 and 7 dissociated from corepressor NCoR in the presence of an acetyllysine-containing peptide, consistent with reader function. Documenting the biological consequences of this possible reader function, mutation of a critical acetylation site regulated Androgen Receptor (AR) transcriptional activation function through HDAC7-NCoR-HDAC3 dissociation. The data document the first evidence consistent with epigenetic reader functions of class IIa HDAC proteins.
Keywords: histone deacetylase, HDAC7, NCoR, androgen receptor, epigenetic reader
eTOC blurb
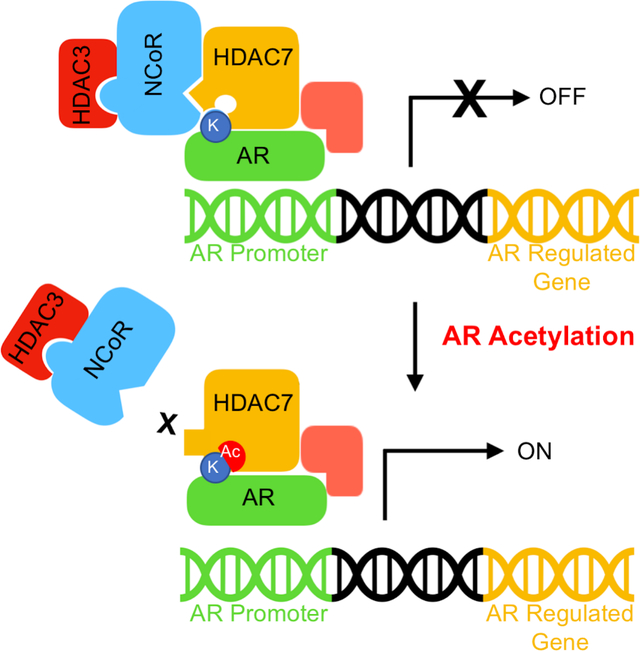
Zhang et al. report evidence for the epigenetic reader function of Histone Deacetylase 7 (HDAC7). A model is proposed where transcription by Androgen Receptor (AR) is activated by binding of the AR K630 acetylation site to HDAC7 and dissociation of the repressive NCoR-HDAC3 complex
Graphical Abstract
Introduction
Lysine acetylation has emerged as a prominent posttranslational modification with impact on numerous cellular functions (Choudhary et al., 2014). As a well-studied example, acetylation of nucleosomal histone proteins influences gene expression through chromatin remodeling (Hebbes et al., 1988), which is a key mechanism in epigenetic regulation. The removal of the acetyl group from acetyllysine is catalyzed by histone deacetylase (HDAC) proteins. Numerous studies showed a correlation between overexpression of HDAC proteins and human tumor formation (West and Johnstone, 2014). Elevated HDAC activities resulted in reduced global acetylation of histone H4 and altered gene expression, which is a hallmark of cancer onset (Fraga et al., 2005). Due to the role in cancer progression, four HDAC inhibitors are approved as cancer therapeutics, including SAHA (Vorinostat or suberoyl anilide hydroxamic acid) (Hesham et al., 2018).
The 18 human HDAC proteins comprise four classes (de Ruijter et al., 2003). Class I (HDAC1, 2, 3, and 8), II (HDAC4, 5, 6, 7, 9, and 10) and IV (HDAC11) bear a catalytic metal ion, whereas class III (Sirtuins 1–7) utilize NAD+ as the cosubstrate (Houtkooper et al., 2012). Class II is further subdivided into class IIa (HDAC4, 5, 7, and 9) and IIb (HDAC6 and 10) based on size and catalytic activity. Class IIa HDAC proteins bear a unique structural zinc binding site that is located adjacent to the active site (Bottomley et al., 2008b; Schuetz et al., 2008). Importantly, class IIa proteins harbor almost no deacetylase activity due to mutation of a catalytic Tyr to His (Clocchiatti et al., 2011; Jones et al., 2008; Lahm et al., 2007). We note that the mutation does not rule out the possibility that class IIa HDAC proteins can serve as a deacylase towards substrates yet to be identified. However, the current evidence suggests that class IIa proteins regulate histone deacetylation and gene expression by interacting with other proteins, including the multi-protein complex containing the nuclear receptor corepressor NCoR/SMRT and HDAC3 (Fischle et al., 2002b; Guenther et al., 2001; Li et al., 2000). Through NCoR/SMRT binding, class IIa HDAC proteins act as scaffolds to recruit active HDAC3 and regulate histone-mediated gene expression.
Whereas the scaffolding function of Class IIa HDAC proteins is well established, the purpose of the inactive catalytic domain in that scaffolding activity remains unclear. One hypothesis suggests that HDAC4, 5, 7, and 9 act as “reader” proteins, similar to bromodomain-containing proteins (Arrowsmith et al., 2012; Bradner et al., 2010; Lobera et al., 2013). With bromodomains, acetyllysine binding results in a downstream effect. For example, the bromodomain of acetyltransferase p300/CBP aids in acetyllysine-dependent histone substrate recruitment (Manning et al., 2001), which leads to elevate histone acetylation and transcriptional activation (Ebrahimi et al., 2019). As this example illustrates, the characterization of bromodomains as “readers” of acetylation has provided critical mechanistic insights into epigenetic regulation of gene expression. Importantly, bromodomains have emerged as targets for drug development (Smith and Zhou, 2016).
Despite speculation, little evidence supports the hypothesis that class IIa HDAC proteins maintain reader function. An acetyllysine-containing peptide inhibited class IIa deacylation of non-natural trifluoroacetyllysine substrates (Bradner et al., 2010), which suggests that acetyllysine binds to the active site of class IIa HDAC proteins. In addition, the presence of HDAC inhibitors or mutation of amino acids in the inactive catalytic domain disrupted the HDAC4-NCoR/SMRT complex (Bottomley et al., 2008b; Gaur et al., 2016; Kim et al., 2015), which suggests that active site binding influences the scaffolding activity of class IIa HDAC proteins. These combined results implicate a model where class IIa HDAC proteins act as readers through active site binding of acetyllysine-containing proteins to promote NCoR/SMRT dissociation. However, no experimental evidence yet documents acetyl-lysine dependent disruption of HDAC-NCoR/SMRT binding. More importantly, no acetyllysine-containing protein is known to bind to the active site of a class IIa HDAC in a “reader” manner. Therefore, no cellular mechanism links acetyllysine-containing protein binding to HDAC7-NCoR association and a downstream biological consequence. To date, any possible reader function of class IIa HDAC proteins remains unsubstantiated.
Androgen receptor (AR) is a transcriptional activator regulated by binding to a hormone ligand, such as dihydrotestosterone (DHT) (Gao et al., 2005). The transcriptional activity of AR is influenced by HDAC4 and HDAC7 (Karvonen et al., 2006; Yang et al., 2011). With HDAC4, the E3 ubiquitin ligase activity of HDAC4 facilitates sumoylation to inhibit AR activities (Yang et al., 2011). In contrast, HDAC7 regulates AR activity through co-localization with the NCoR-HDAC3 complex (Karvonen et al., 2006). Related, AR-mediated transcription hinges on the acetylation of K630 or K632/633, with loss of transactivation upon mutation of K630 or K632/633 (Fu et al., 2000; Fu et al., 2002). Moreover, wild type AR showed 10-fold reduced binding to NCoR than the K630A mutant (Fu et al., 2002). Taken together, prior data suggests that acetylation-dependent AR transcriptional activity involves the HDAC7-NCoR-HDAC3 complex, although a molecular mechanism linking AR function to HDAC7 and NCoR is unknown.
Based on the prior literature, we hypothesized that class IIa HDAC proteins act as reader proteins to influence the transcriptional activity of AR. Consistent with the reader hypothesis, HDAC4, 5, and 7, but not HDAC9, bound NCoR in an acetyl-lysine-dependent manner. An assessment of binding affinity confirmed the stable binding of an acetyllysine-containing peptide with HDAC7. To provide a biological context for the class IIa HDAC-NCoR reader function, dissociation of the HDAC7-NCoR complex was dependent on the K630 acetylation site of AR, and the transcriptional activity of AR required both an intact HDAC7 active site and K630 acetylation site. In total, the work here reports the first evidence for the epigenetic reader function of HDAC7, where AR transcriptional activity is regulated through acetyllysine-dependent HDAC7-NCoR complex dissociation.
Results
Inhibitor-dependent class IIa HDAC-NCoR binding
A key feature of epigenetic reader proteins, such as bromodomains, is that a binding event leads to a downstream function. Related to class IIa HDAC proteins, prior work documented that HDAC inhibitors disrupted the HDAC4-NCoR/SMRT-HDAC3 complex (Bottomley et al., 2008b; Gaur et al., 2016). In addition, gain-of-function (GOF) mutant HDAC4 bound more tightly to HDAC inhibitors and demonstrated more dramatic loss of NCoR binding than wild type (Gaur et al., 2016; Hudson et al., 2015). This prior data suggests a reader function for HDAC4, where active site binding results in dissociation of NCoR/SMRT. However, the other three class IIa HDAC isoforms, HDAC5, HDAC7, and HDAC9, had not yet been tested. To initially confirm and extend the prior data, SAHA inhibitor-dependent binding of both wild type and GOF mutants of all four class IIa HDAC isoforms to NCoR was assessed using a gel analysis.
First, GOF mutants of HDAC4, 5, 7, and 9 were generated using Quickchange mutagenesis (Figure S1A–B)(Lahm et al., 2007; Ling et al., 2012; Schuetz et al., 2008), and the deacetylase activity of GOF mutants of HDAC4 and HDAC7 was confirmed (Figure S1C, lanes 4 and 6). The GOF mutant is an ideal negative control for reader function because the active mutant is still capable of binding to a substrate, but only transiently, assuring that mutation maintains protein structure and function. Next, Flag-tagged WT or GOF mutant HDAC4, 5, 7 and 9 were overexpressed in HEK293 cells and then immunoprecipitated to assess NCoR binding. Consistent with prior data (Bottomley et al., 2008b; Gaur et al., 2016; Ling et al., 2012), both wild type and GOF mutant HDAC4 bound NCoR (Figure 1A, lanes 2 and 4), which was disrupted by SAHA (Figure 1A, lane 3 and 5). Similar to HDAC4, HDAC5 and 7 showed similar strong NCoR binding (Figure 1B and 1C, lanes 2 and 4), with reduction in the presence of SAHA (Figure 1B and 1C, lanes 3 and 5). GOF mutants of HDAC4, 5 and 7 showed more dramatic SAHA-dependent loss of HDAC-NCoR interaction (Figure 1, lanes 2 versus 3) compared with wild type (Figure 1, lanes 4 versus 5), consistent with prior work showing that SAHA is 300-fold more potent towards the HDAC7 GOF mutant than the wild type (Schuetz et al., 2008). In contrast to the other class IIa HDAC isoforms, neither the wild type nor GOF mutant of HDAC9 bound NCoR (Figure 1D, lanes 2–5). The inability for HDAC9 to bind NCoR is in agreement with a previous study (Joshi et al., 2013).
Figure 1. HDAC inhibitor-dependent HDAC-NCoR binding.
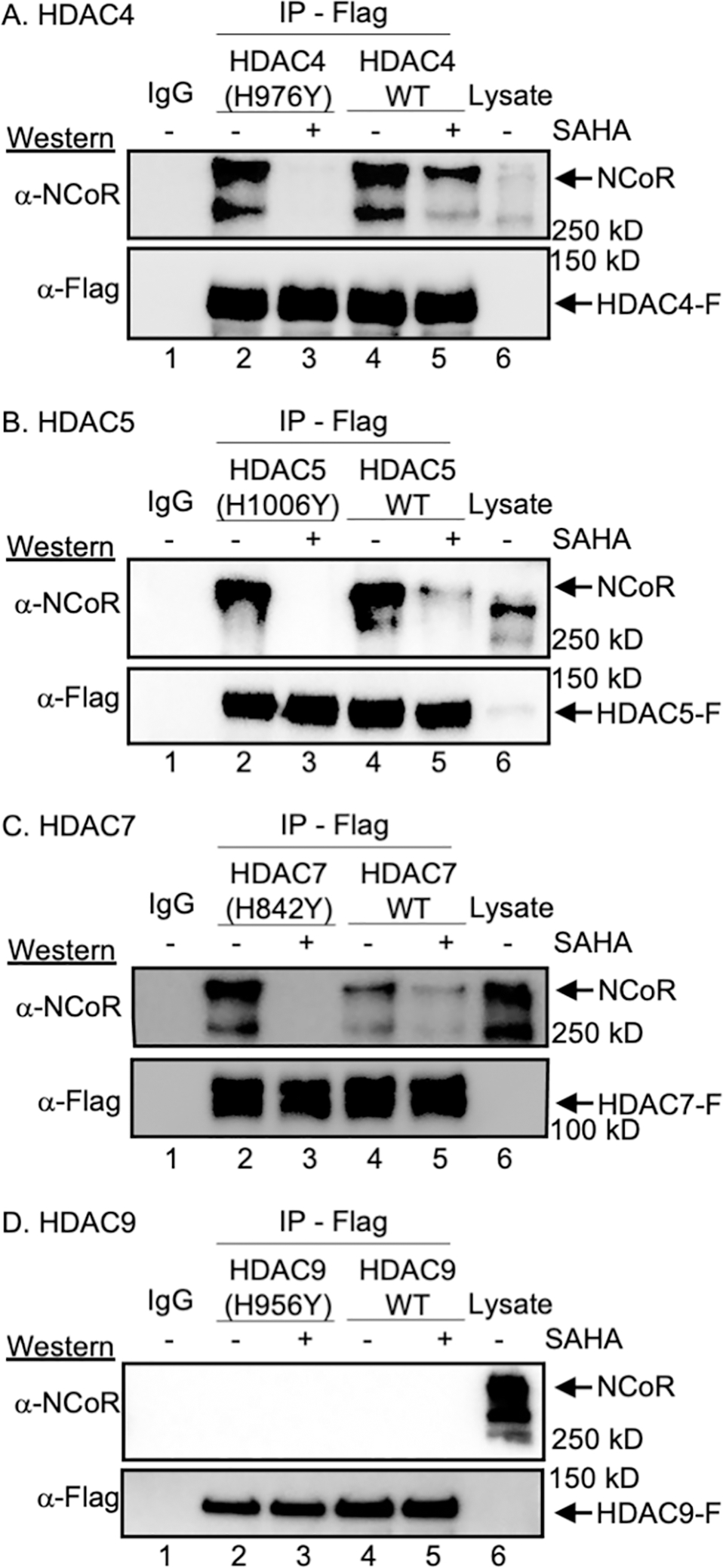
(A) Flag-tagged wild type (WT) or GOF HDAC4 (A), HDAC5 (B), HDAC7 (C), or HDAC9 (D) were overexpressed in HEK293 cells, which were then treated with SAHA (10 μM) to induce acetylation. After lysis, Flag-tagged HDAC proteins were immunoprecipitated (IP) in the presence or absence of SAHA. Bound proteins were resolved by SDS-PAGE, followed by western blot analysis with NCoR and Flag antibodies. As a gel migration control, lysate (Lys) from transfected cells were included. All trials include a bead binding control using lysates without expression of HDAC-Flag (IgG). Activity assays associated with the wild type and mutant HDAC proteins are shown in Figure S1. Additional independent trials and SAHA dose dependence are shown in Figure S2.
To thoroughly characterize the inhibitory effect of SAHA on NCoR binding, dose-dependent SAHA binding inhibition was assessed with HDAC4 and 7. HDAC4 and 7 were selected as representative class IIa HDAC proteins here, given their role in AR activity, as discussed later. Dose dependent reduction in NCoR binding was observed with both WT and GOF mutants of HDAC4 and 7 (Figure S2E and S2F), with 100 μM necessary to disrupt binding. These results suggest that the binding modes of NCoR to HDAC4 and 7 are similar, with dose dependent HDAC inhibitor-mediated disruption of NCoR binding.
Acetyllysine-dependent class IIa HDAC-NCoR binding
SAHA and acetyllysine bind to the active site of HDAC proteins in a similar manner, with coordination to the catalytic metal (Darkin-Rattray et al., 1996; Finnin et al., 1999; Vannini et al., 2007). Given their similar active site binding, acetyllysine-containing peptides might disrupt NCoR binding to the class IIa HDAC proteins similar to SAHA, which would be consistent with possible reader function. To explore the possibility that acetyllysine containing peptides cause HDAC-NCoR dissociation, similar to the SAHA inhibitor, peptides derived from histone 4 lysine 12 (H4K12) were synthesized, including acetylated Ac-Leu-Gly-Lys(Ac)-NH2 (LGKAc) and nonacetylated Ac-Leu-Gly-Lys-NH2 (LGK). These short histone-derived peptides are commonly used in commercial HDAC activity assays, making them an ideal general acetyllysine-containing substrate. NCoR binding experiments were then performed with HDAC4, 5 and 7 in the presences of the peptides. The LGKAc peptide caused a dramatic disruption of HDAC-NCoR complex (Figure 2A–C, lane 4) compared to untreated control samples (Figure 2A–C, lane 2), similar to SAHA (Figure 2A–C, lane 5). Importantly, unacetylated LGK failed to disrupt the HDAC-NCoR complex (Figure 2A–C, lane 3). Quantification of three independent trials showed significant reduction in NCoR binding to all three HDAC isoforms in the presence of the LGKAc peptide, but not the unacetylated LGK peptide (Figure 2E), showing the reproducibility of the experiment. Interestingly, the first version of the peptide that was tested, Ac-Ala-Lys(Ac)-Leu-OH (AKAcL-OH), which contains a free carboxylate terminus, did not disrupt the HDAC7-NCoR complex (Figure S3E). We speculate that electrostatic repulsion between Asp residues at the HDAC7 active site entrance (D759 and D626) (Fischle et al., 2002b; Guenther et al., 2001) and the C-terminal carboxylic acid prevented peptide binding, suggesting that class IIa HDAC proteins prefer binding to internal acetyllysine residues. These results demonstrate that HDAC4, 5 and 7 bind to acetyllysine to dissociate NCoR.
Figure 2. NCoR binds to HDAC4, 5, and 7 in an acetyllysine-dependent manner.
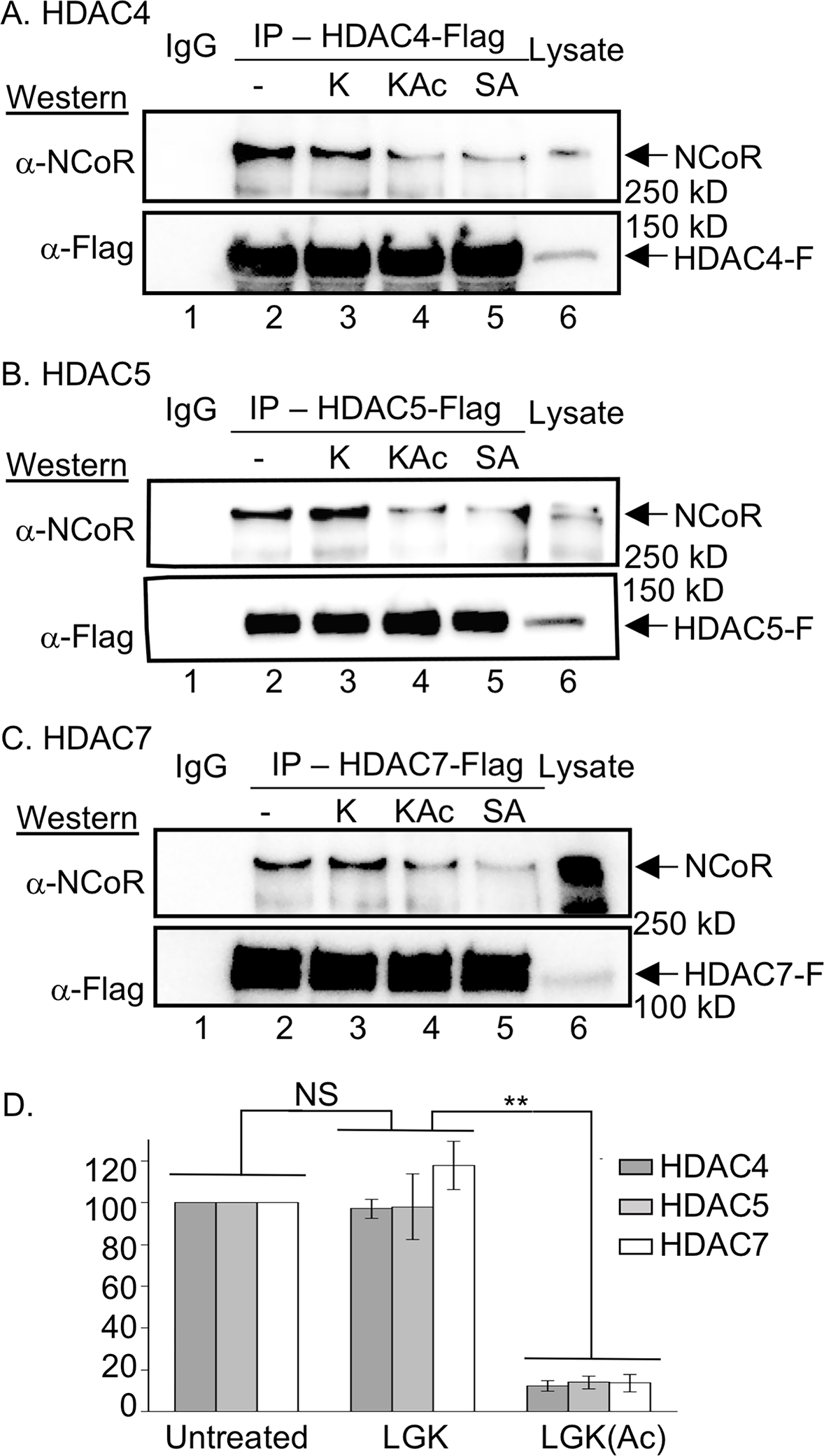
Flag-tagged HDAC4 (A), HDAC5 (B), and HDAC7 (C) were overexpressed in HEK293 cells, which were then treated with SAHA (10 μM) to induce acetylation. After lysis, anti-Flag beads were used to immunoprecipitate (IP) the HDAC proteins either in the absence or presence of 1 mM LGK (K), LGKAc (KAc), or SAHA (SA). Bound proteins from the IP were resolved by SDS-PAGE, followed by Western blot analysis with NCoR and Flag antibodies. Lysate (Lys) from transfected cells were included as a gel migration control. All trials include a bead binding control using lysates without expression of HDAC-Flag (IgG). Additional independent trials and dose dependence are shown in Figure S3A–C. Binding affinity studies are shown in Figure S4. (D) NCoR band intensities from three independent trials with HDAC4, 5 and 7 (parts A-C and S73A-C) were quantified and normalized to untreated (set to 100%), with the student t test applied to show significance. N.S. = not significant (p > 0.05), and ** = p < 0.01. Raw data are shown in Figure S3D.
To further characterize acetyllysine-mediated NCoR-HDAC disruption, dose-dependent binding experiments were performed, similar to SAHA. NCoR-HDAC dissociation was observed with 1 mM LGKAc peptide (Figure S3F–G). The reduced dissociation potency of LGKAc compared to SAHA (1 mM with LGKAc versus 0.1 mM with SAHA) is consistent with the low micromolar IC50 values reported for acetyllysine peptides as inhibitors (Bradner et al., 2010), but the mid-nanomolar KD and IC50 values reported for SAHA inhibition (Lauffer et al., 2013; Negmeldin et al., 2018; Negmeldin and Pflum, 2017).
Binding assessment of the HDAC7-acetyllysine peptide complex
A critical prerequisite of reader function is that acetyllysine-containing peptides or proteins bind stably to class IIa HDAC proteins, as was previously observed with bromodomain proteins (Jacobson et al., 2000). Acetyllysine-dependent disruption of the class IIa HDAC-NCoR interaction (Figure 2) suggests stable binding between class IIa HDAC proteins and acetyllysine-containing peptides. Likewise, prior work documenting that acetyllysine-containing peptides acted as inhibitors of class IIa deacetylation using an unnatural trifluoroacetyllysine substrate (Bradner et al., 2010) is also consistent with class IIa HDAC-acetyllysine interactions. However, no report to date characterizes the binding affinity of an acetyl-lysine peptide to a class IIa catalytic domain.
To provide evidence for stable interaction between class IIa HDAC proteins and acetyllysine-containing peptides, the binding affinity of the LGKAc peptide to HDAC7 was assessed. We selected HDAC7 for these biophysical studies given the focus of this project on the unique relationship between HDAC7 and AR, as discussed below. The catalytic domain of HDAC7 (cdHDAC7) was expressed in bacteria and purified (Figure S4A–B). Purified cdHDAC7, along with acetylated LGKAc or unacetylated LGK peptides, were subjected to analysis by Bio-Layer Interferometry (BLI). While the LGK peptide did not incur observable binding, even at 5 μM concentration (Figure S4C–E), the LGKAc peptide showed dose dependent association and dissociation with cdHDAC7. The dissociation constant (KD) from three independent trials was in the range from 0.72 ± 0.1 μM to 1.0 ± 0.1 μM (Figure S4C–E). The observed sub-micromolar binding affinity is considerably lower than the 1 mM concentration of LGKAc peptide necessary to dissociate NCoR from class IIa HDAC proteins in lysates (Figure S3F–G). We speculate that the LGKAc peptide might be susceptible to both degradation by proteases and deacetylation by active HDAC proteins in the cell lysate, resulting in the requirement for elevated concentrations.
AR K630 is Critical for HDAC7-NCoR Dissociation
In addition to stable acetyllysine binding, a critical aspect of reader proteins is that binding by an acetyllysine-containing protein leads to a downstream biological event. HDAC7 regulates AR activity through NCoR-HDAC3 binding (Karvonen et al., 2006), and the transcriptional activity of AR is dependent on acetylation at a critical K630 residue (Fu et al., 2000; Fu et al., 2002). Despite the connections between AR activity, NCoR binding, and K630 acetylation, a molecular mechanism accounting for these activities has not yet been tested. We hypothesize that acetyl-K630 of AR binds directly to the HDAC7 active site to dissociate the NCoR-HDAC3 complex and activate transcription, consistent with a reader function for HDAC7.
As a first test of this hypothesis, we assessed whether an acetyllysine-containing peptide derived from AR would disrupt the NCoR-HDAC7 complex, similar to the LGKAc peptide. Two peptides including residues 625 to 637 of the AR sequence were synthesized: acetylated TLGARKAcLKKLGNL (AR K630Ac) and unacetylated TLGARKLKKLGN (AR K630, where K630 is underlined in both sequences). AR K630Ac caused dramatic NCoR dissociation from HDAC7 (Figure 3A, lane 6), whereas AR K630 did not (Figure 3A, lane 2), consistent with a role for AR acetylation in HDAC7-NCoR complex disruption.
Figure 3. AR influenced HDAC7-NCoR association.
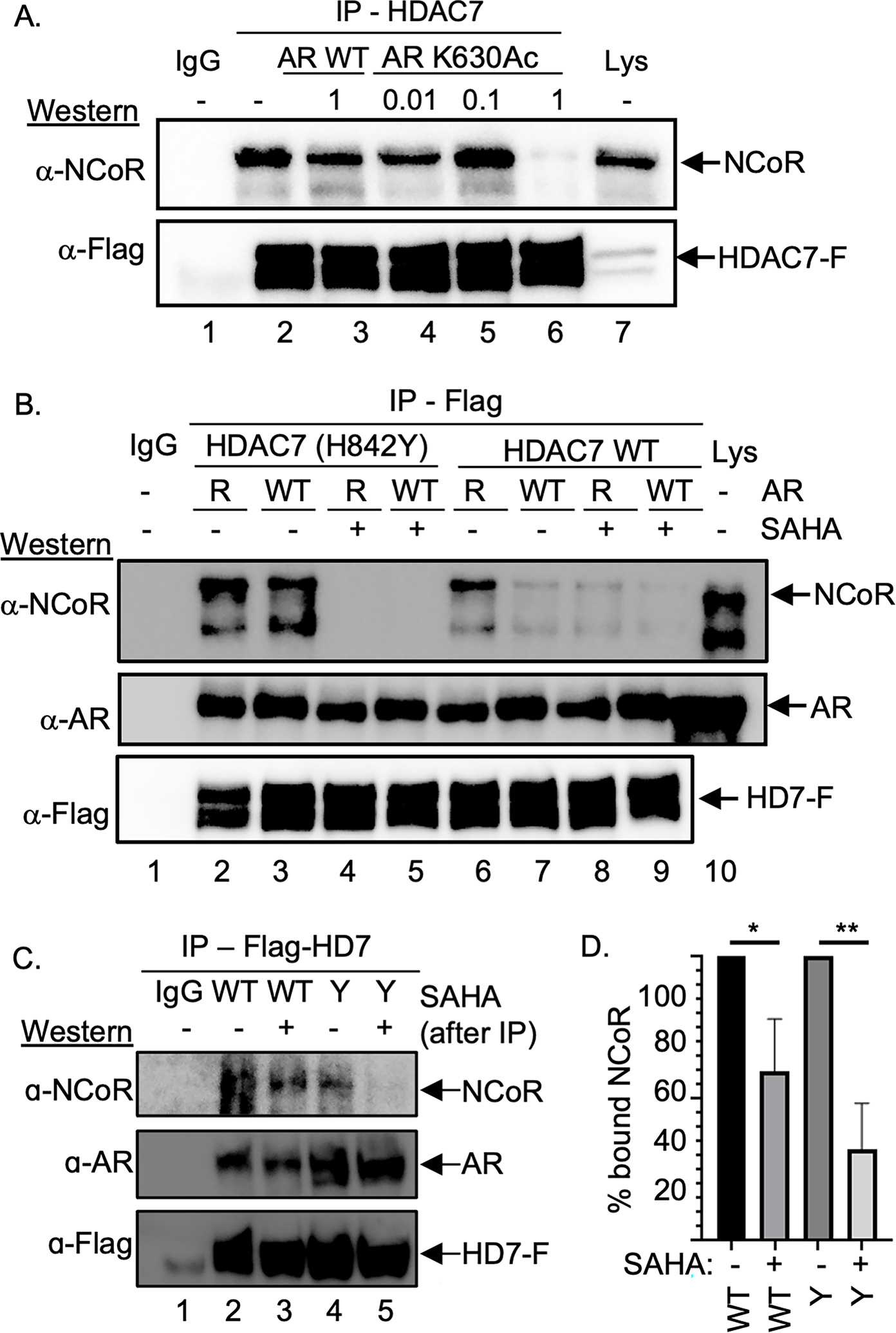
(A) Flag-tagged HDAC7 was expressed in HEK293 cells, which were then treated with SAHA (10 μM) to induce acetylation, followed by lysis, immunoprecipitation (IP) in the presence of different mM concentrations of AR K630Ac or AR WT peptide, SDS-PAGE separation, and western blot analysis with NCoR and Flag (HD7-F) antibodies. Repetitive independent trials are shown in Figure S5A. (B) HDAC7 wild type (WT) or GOF mutant were co-expressed with AR WT or K630R mutant in HEK293 cells, followed by lysis, immunoprecipitation (IP) of HDAC-Flag from the lysates, SDS-PAGE separation, and western blot analysis with NCoR, AR, or Flag (HD7-F) antibodies. As a gel migration control, lysate (Lys) from transfected cells were included. Repetitive independent trials are shown in Figure S5B. (C) HDAC7 wild type (WT) or GOF mutant (Y) were co-expressed with AR WT in HEK293 cells, which were then treated with SAHA (10 μM) to induce acetylation. After lysis and immunoprecipitation (IP), bound proteins were washed with a high salt (500 mM) buffer, before SAHA (100 μM) or DMSO vehicle was added and further washed. Bound proteins were separated by SDS-PAGE and visualized with NCoR, AR, or Flag (HD7-F) antibodies. Repetitive independent trials are shown in Figures S5C. (D) NCoR proteins levels from three independent trials from part C and Figures S5C were quantified, normalized to samples without SAHA (set to 100%), and plotted with mean and standard error shown (Figure S5D). * = p<0.05, ** = p < 0.01. All trials include a bead binding control using lysates without expression of HDAC7-Flag (IgG).
To further test the hypothesis that AR acetylation affects the HDAC7-NCoR complex, NCoR binding assays were performed in the presence of overexpressed full-length AR wild type or unacetylated K630R mutant. The presence of co-expressed wild type AR resulted in the loss of NCoR in the HDAC7-Flag immunoprecipitation (Figure 3B, lane 7). In contrast, co-expression of unacetylated AR K630R mutant did not disrupt the HDAC7-NCoR complex (Figure 3B, lane 6), consistent with the model where K630Ac binds the HDAC7 active site to dissociate NCoR. Likewise, NCoR remained bound to the GOF HDAC7 mutant in the presence of either wild type or K630R AR (Figure 3B, lanes 2 and 3) due to active deacetylation and loss of “reader” function. Interestingly, AR co-immunoprecipitated with HDAC7 in all samples (Figure 3B, lanes 2–9), which suggests that AR interacts with HDAC7 independently of acetylation-induced active site binding. These finding provide the first evidence that HDAC7 acts as a reader of AR acetylation to affect NCoR-HDAC3 association.
An alternative mechanistic hypothesis explaining the AR K630-dependent NCoR-HDAC7 dissociation is that cellular acetyllysine-binding factors, such as transcriptional coactivators, interact with acetylated-K630 AR to physically disrupt NCoR binding. In this case, SAHA or acetyllysine-containing peptides could alter AR acetylation through inhibition of cellular HDAC proteins to influence transcriptional coactivator interaction, instead of direct HDAC7 active site binding, to dissociate NCoR. To experimentally test this alternative hypothesis, overexpressed wild type or GOF mutant HDAC7 were immunoprecipitated in the presence of overexpressed AR and then washed with a high salt (500 mM) buffer to remove unbound cellular proteins, including coactivators and deacetylases. As expected, NCoR remained bound to the samples in this initial immunoprecipitation (Figure S5C), documenting the absence of NCoR-disrupting factors. Next, SAHA was added to the purified AR-HDAC7-NCoR complex to assess if NCoR dissociates through direct HDAC7 binding in the absence of cellular proteins. SAHA incubation resulted in the NCoR dissociation with both wild type (Figure 3C, compare lanes 2 and 3) and GOF mutant HDAC7 (Figure 3C, compare lanes 4 and 5), similar to earlier NCoR binding studies (Figure 1C and 3B). Quantification of NCoR protein levels from three independent trials documented reproducible SAHA-dependent NCoR loss (Figure 3D). Without the influence of cellular coactivators or deacetylases, the data are consistent with direct binding of SAHA to the HDAC7 active site, which further confirms the reader function of HDAC7.
Finally, to confirm the interaction between HDAC7 and AR in cells, their cellular localization was studied using immunohistochemistry and confocal microscopy. As expected, HDAC7 showed strong colocalization with AR in the absence and presence of dihydrotestosterone (DHT) ligand (Figure S6), which is consistent with earlier coimmunoprecipitation data (Figure 3B). Interestingly, AR and HDAC7 resided in both the nucleus and cytoplasm without DHT treatment, whereas they are found primarily in the nucleus with DHT treatment (Figure S6), similar to a previous report (Karvonen et al., 2006). The colocalization data is consistent with acetylation-independent interaction of HDAC7 and AR.
AR transcriptional activity is dependent on HDAC7
With AR acetylation-dependent disruption of the HDAC7-NCoR complex established, the next step to test the reader function of HDAC7 was to assess HDAC7-dependent AR transcriptional activation function. AR-mediated transcription was monitored in the presence of HDAC7 using a gene reporter system. HEK293 cells were selected due to the absence of endogenous AR expression (Alimirah et al., 2006), which allows expression of wild type and mutant AR. As expected, AR-dependent transcription increased roughly 10-fold in the presence of DHT ligand (100 ± 10 %, Figure 4A, lane 2), compared to the absence (10 ± 1%, Figure 4A, lane 1). Whereas co-expression of wild type HDAC7 did not influence DHT-induced transcription (98 ± 1%, Figure 4A, lanes 2 and 3), co-expression of GOF mutant HDAC7 significantly decreased transcription (56 ± 1%, Figure 4A, lane 4). These observations are consistent with earlier co-immunoprecipitation data where GOF mutant HDAC7 was unable to disrupt NCoR binding (Figure 3B), resulting in continued repression of transcription through HDAC3 recruitment. To confirm the importance of AR K630 acetylation in transcriptional activation, as observed previously (Fu et al., 2002), AR-mediated transactivation activity was reduced to basal levels with co-expression of AR K630R (Figure 4A, lanes 7–12), showing acetylation-dependent AR transcription. As a positive control, co-expression of HDAC4 inhibited AR activity (43 ± 1 %, Figure 4A, lane 6), likely due to AR sumoylation (Yang et al., 2011). As a negative control, the presence of HDAC9 GOF mutant did not affect AR transcriptional activity (93 ± 3 %, Figure 4A, lane 5). This transcriptional data document the dependence of AR gene expression activity on both HDAC7 and K630, consistent with a reader function for HDAC7.
Figure 4. GOF mutant HDAC7 decreased AR-mediated transcription.
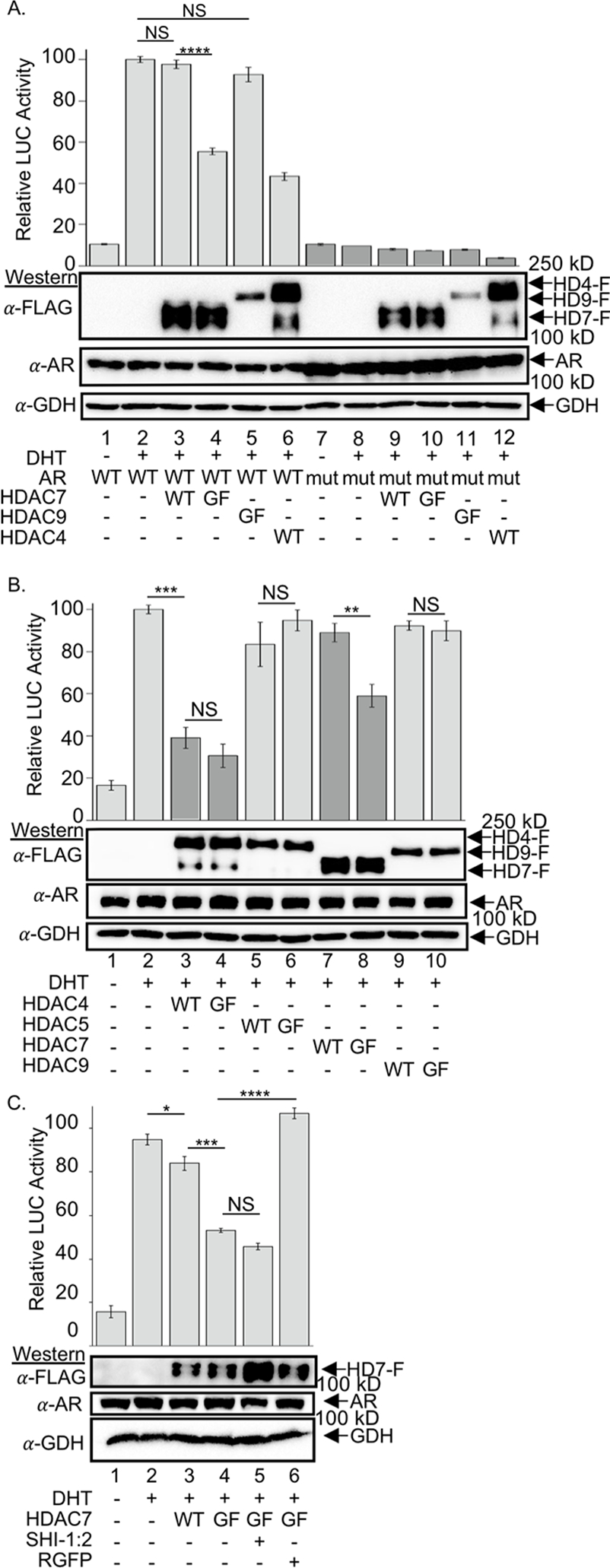
Wild type and GOF mutant HDAC4, HDAC5, HDAC7, or HDAC9 were co-expressed with wild type AR (A, B, and C) or K630R mutant AR (A only), as indicated, in the presence of the AR-dependent reporter gene constructs ARE-LUC and global expression control construct Ren-LUC (all samples). Cells were then treated without or with DHT (1 nM). HDAC1/2-selective inhibitor SHI-1:2 (10 μM) or HDAC3-selective inhibitor (RGFP996, 10 μM) were also included in part C. Light signal due to luciferase expression was measure 36 h after DHT treatment and normalized to Ren luciferase levels. Mean light intensity data and standard error normalized as a percentage of the DHT sample (lane 2, set to 100%) from at least three independent trials are shown, with the data provided in Figure S7. Student t-test analysis was applied, where NS = not significant (p > 0.05), * = p < 0.05, ** = p < 0.01, ***= p < 0.001, and ***= p < 0.0001. Representative gel images show protein expression levels in the samples, where GAPDH (GDH) was included as a load control.
To confirm the unique role of HDAC7 in AR-mediated transcription, all class IIa HDAC proteins were tested in the reporter assay. Whereas HDAC7 showed transcriptional signal dependent on deacetylase activity (Figure 4B, lanes 7 and 8), the other class IIa HDAC proteins did not (Figure 4B, lanes 3–6, 9–10). The reporter assay data provide evidence that HDAC7 uniquely acts as a reader of AR acetylation.
NCoR represses transcription primarily by recruiting HDAC3 to genomic DNA and deacetylating nucleosomal histones (Fischle et al., 2002b). To test the HDAC3 dependence of AR transcriptional activity, the same AR-dependent gene reporter assay was performed in the presence of HDAC3-selective inhibitor, RGFP966. Whereas GOF mutant HDAC7 reduced DHT-induced AR transcription (Figure 4C, lane 4) compared to wild type (Figure 4C, lane 3) or untreated (Figure 4C, lane 2) samples, RGFP966 restored AR-mediated transcription in the presence of GOF mutant HDAC7 to levels similar as untreated samples (Figure 4C, lane 6). As a control, the HDAC1/2 dual selective inhibitor SHI:12 did not influence GOF mutant repressed AR-mediated transcription (Figure 4C, lane 5), despite prior work showing that AR associates with HDAC1 or HDAC2 to repress transcription (Chng et al., 2012). The HDAC inhibitor study confirmed that AR-mediated gene repression by HDAC7 is dependent on HDAC3, consistent with recruitment of the NCoR-HDAC3 complex.
To extend the study of HDAC7-dependent AR transcriptional activity to a biologically-relevant context, mRNA levels of two AR-regulated genes, SPRF5 and Wnt16 (Tanner et al., 2011), were assessed in the presence of expressed AR and HDAC7 using reverse transcription/polymerase chain reaction (RT-PCR) analysis. Upon DHT treatment, both genes showed robust upregulation (Figure 5A, lanes 4–6) compared to the untreated control (Figure 5A, lanes 1–3). While the presence of wild type HDAC7 only modestly reduced mRNA levels of both genes (Figure 5A, lanes 7–9), the presence of HDAC7 GOF mutant significant reduced mRNA levels of both genes (Figure 5A, lanes 10–12). Quantification of triplicates from three independent trials showed that GOF mutant HDAC7 reduced SPRF5 expression to 44 ± 1 % (Figure 5B, column 4) and Wnt16 expression to 40 ± 1 % (Figure 5C, column 4) compared to untransfected cells (Figures 5B and 5C, column 2). The RT-PCR results confirmed that HDAC7 reader function is required for transcription of AR-dependent cellular genes.
Figure 5. HDAC7-dependent gene expression by AR using RT-PCR.
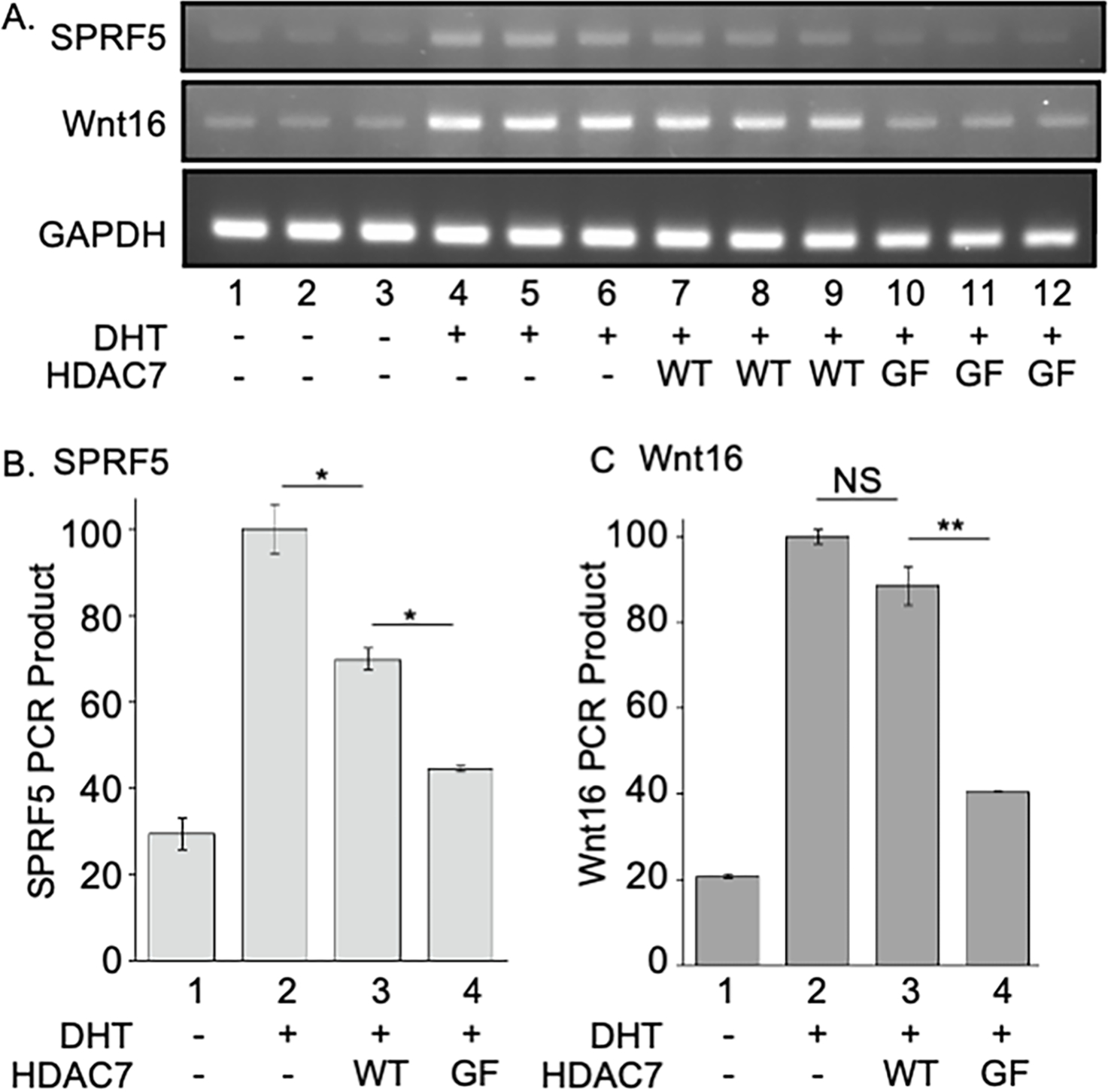
(A) Wild type AR was co-expressed with wild type (WT) or GOF mutant (GF) HDAC7. Cells were then untreated (0.01% DMSO only) or treated with DHT (1 nM in 0.01% DMSO) for 36 hours. After lysis, total cDNA was synthesized from mRNA with reverse transcriptase and mRNA levels of SPRF5 and Wnt16 were measure by RT-PCR reaction from the total cDNA. Representative gel images show triplicate reactions from three independent trials. (B and C) Signals were quantified from part A using ImageJ 1.47v and normalized to the DHT alone sample (lane 2, set to 100%) to calculate mean percentage and standard error. Mean light intensity data and standard error from at least three independent trials are shown, with the data provided in Figure S8. Student t-test analysis was applied, where NS = not significant (p > 0.05), * = p < 0.05, ** = p < 0.01.
ER and AR Similarly Interact with HDAC7 and NCoR
Finally, we extended these studies to a second nuclear receptors to test if HDAC7 reader function affects other nuclear receptor family members. Estrogen receptor α (ER) is a transcriptional activator regulated by binding to ligands, such as estradiol (E2) (Heldring et al., 2007). Similar to AR, ER transcriptional activity is influenced by HDAC4 and HDAC7 through direct interaction (Leong et al., 2005; Malik et al., 2010). Additionally, ER recruits NCoR/SMRT corepressors to regulate transcriptional function (Fu et al., 2003; Varlakhanova et al., 2010). Despite the connections between ER, class IIa HDAC proteins, and NCoR/SMRT corepressors, similar to AR, no study has yet directly probed their possible acetylation-dependent interactions.
Based on the AR data, we hypothesized that ER acetylation affects the HDAC7-NCoR complex. To probe this hypothesis, NCoR binding assays were performed in the presence of overexpressed full-length ER wild type and either HDAC7 WT or gain-of-function (GOF) mutant. NCoR bound HDAC7 GOF mutant to a greater extent than HDAC7 WT (Figure 6, lanes 2 vs 4), which was disrupted in the presence of SAHA (Figure 6, lanes 3 and 5). Similar to AR, ER robustly bound to HDAC7 WT or GOF in the absence or presence of SAHA treatment (Figure 6, lanes 2–5), suggesting that ER also interacts with HDAC7 independently of acetylation-induced active site binding. In fact, immunohistochemistry documented that HDAC7 colocalized with ER in the absence and presence of ligand (Figure S9B). In this case, ER predominantly resided in the nucleus with HDAC7 both in the presence and absence of E2 ligand, which is consistent with previous work (Monje et al., 2001). Based on the similar data, we speculated that both AR and ER interact with HDAC7 to regulate acetylation-dependent NCoR binding.
Figure 6. HDAC7-NCoR dissociation in the presence of ER.
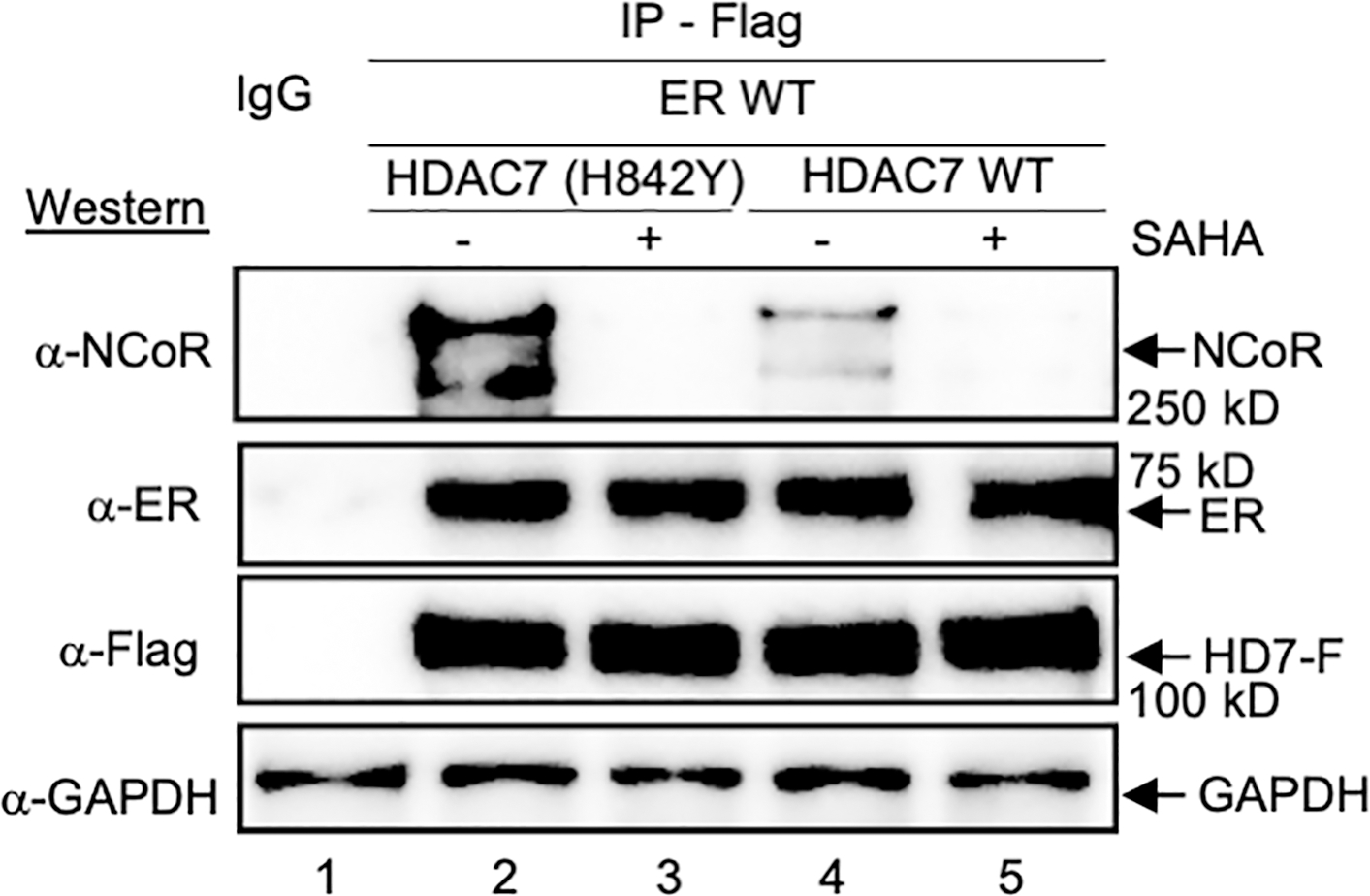
HDAC7 wild type (WT) or GOF mutant were co-expressed with wild type ER in HEK293 cells, followed by lysis, immunoprecipitation (IP) of HDAC-Flag from the lysates, and western blot analysis with NCoR, Flag (HD7-F), ER antibodies. Repetitive independent trials are shown in Figure S9. All trials include a bead binding control using lysates without expression of HDAC-Flag (IgG).
Discussion
HDAC proteins are epigenetic repressors due to deacetylation of nucleosomal histone substrates (Krämer et al., 2001). However, class IIa HDAC proteins cannot function directly as acetylation-dependent epigenetic enzymes since they are catalytically inactive (Bradner et al., 2010; Lahm et al., 2007; Schuetz et al., 2008). Prior work documented that class IIa HDAC4, 5 and 7 interact with the repressive NCoR/SMRT-HDAC3 complex to regulate transcription (Fischle et al., 2001; Mottis et al., 2013; Wong et al., 2014). Here we demonstrate for the first time that binding of HDAC4, 5 and 7 to NCoR is acetyllysine-dependent, which is consistent with possible “reader” function. The “reader” model proposes that binding of acetyllysine into the active sites of HDAC4, 5 and 7 releases the NCoR/SMRT-HDAC3 complex (Figure 7A). By controlling HDAC3-mediated deacetylation, HDAC4, 5 and 7 indirectly control gene expression.
Figure 7. Proposed model of class IIa HDAC “reader” function.
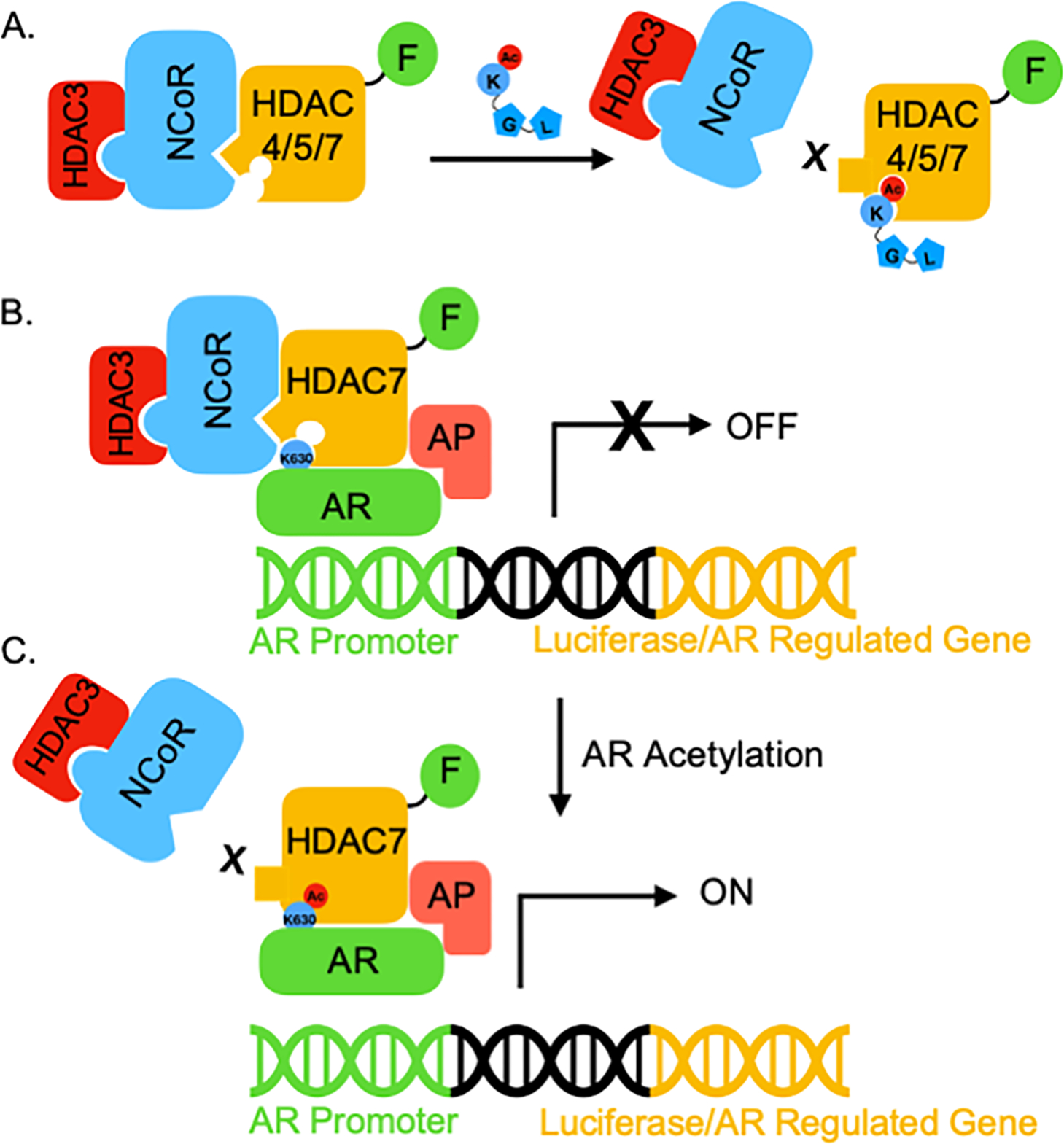
(A) HDAC4, 5, and 7 can act as epigenetic ‘readers’ that recruit the NCoR-HDAC3 complex, which is disrupted when acetyllysine binds to the inactive active site. (B) HDAC7 bridges unacetylated AR and NCoR to repress the activity of AR through HDAC3 recruitment. This acetylation-independent binding of AR to the HDAC7-NCoR-HDAC3 complex might be direct or indirect through associated proteins (AP), and is likely mediated outside of the HDAC7 active site. (C) Once AR is acetylated on its critical lysine K630, acetyl-K630 binds the HDAC7 active site to dissociate the NCoR-HDAC3 complex. Without epigenetic repression by HDAC3, AR-mediated transcription is active.
Several prior studies are consistent with the proposed model of HDAC7 reader function. An HDAC inhibitor disrupted class IIa HDAC binding to an NCoR/SMRT-derived peptide (Hudson et al., 2015), which suggests that active site binding molecules (either an inhibitor or acetyllysine) affect HDAC-NCoR association. Similarly, HDAC3 dissociated from HDAC4 after incubation with an HDAC inhibitor, which could be due to the loss of NCoR association after active site binding by the inhibitor (Bottomley et al., 2008b). Deacylation by class IIa HDAC proteins was inhibited by an acetyllysine-containing substrate (Bradner et al., 2010), which is consistent with HDAC active site binding by acetyllysine. Finally, chemoproteomics studies reported that class IIa HDAC proteins (HDAC4, 5, and 7), but not NCoR or HDAC3, were enriched by inhibitor-bound beads (Bantscheff et al., 2011; Lobera et al., 2013). In total, prior reports are consistent with the proposed model where HDAC7 acts as a reader of AR acetylation to modulate gene expression.
To thoroughly characterize the reader function of HDAC7, the dissociation constant of an acetyllysine peptide bound to HDAC7 was determined to be 0.7–1.0 μM using BLI analysis. Although no prior literature reports the binding affinity of a class IIa HDAC to an acetyllysine-containing peptide, the inhibitory potency (IC50) of acetyllysine-containing peptides in deacetylase assays with class IIa HDAC proteins is 0.64 and 2.3 μM (Bradner et al., 2010), similar to the binding data. To compare the affinities observed here with HDAC7 to bromodomain-containing reader proteins, dissociation constants of 1.4 – 39 μM were determined using isothermal calorimetry for acetyllysine-containing peptides bound to bromodomain-containing TAFII250, which showed a 1:1 stoichiometry (Jacobson et al., 2000). NMR titration was also used to assess the binding affinity of acetyllysine-containing peptides to bromodomain-containing P/CAF (350 μM), GCN5 (900 μM), and CBP (50 μM) proteins (Dhalluin et al., 1999; Hudson et al., 2000; Mujtaba et al., 2004). In total, the affinity of HDAC7 for an acetyllysine-containing peptide observed here is similar or better than those reported for bromodomain-containing reader proteins, further corroborating possible reader function.
Prior HDAC7 structural studies proposed that acetyllysine-containing substrates interact with both the active site and the nearby structural zinc metal region that binds SMRT/NCoR (Desravines et al., 2017; Schuetz et al., 2008). Similarly with HDAC4, a SMRT-derived peptide bound a cleft between the structural zinc domain and the active site (Kim et al., 2015; Park et al., 2018). The proximity of the structural zinc region to the active site suggests that SMRT/NCoR-class Ila HDAC protein complexes are sensitive to acetyllysine binding. In addition, inhibitor binding to the HDAC4 active site resulted in conformational changes in the structural zinc region, which could explain the inhibitor-dependent HDAC4-NCoR/SMRT disruption (Bottomley et al., 2008b). Based on these prior structural studies, a conformational change in the NCoR/SMRT interaction domain after active site binding, or a blocking of the active site cleft, or both, might be responsible for release of NCoR/SMRT. Further structural studies are needed to test this hypothesis.
Whereas HDAC4, 5, and 7 showed inhibitor and acetyllysine sensitive NCoR association (Mottis et al., 2013; Wong et al., 2014), HDAC9 did not. In fact, there is debate in the literature regarding the HDAC9-NCoR/SMRT interaction. Ectopically expressed HDAC9 bound NCoR by co-immunoprecipitation (Petrie et al., 2003), and HDAC9 bound an NCoR-derived peptide, similar to HDAC4, 5, and 7 (Hudson et al., 2015). However, proteomics analysis after immunoprecipitation documented that HDAC4, 5 and 7 exist in a cluster of proteins containing NCoR, whereas HDAC9 was in a separate cluster absent of NCoR (Joshi et al., 2013). Additional studies with HDAC9 are needed to identify reader activity, which might be distinct from HDAC4, 5, and 7
To demonstrate the biological relevance of HDAC7 reader function, we provide evidence that HDAC7 acts as a reader of AR acetylation to activate AR-mediated transcription. According to the proposed model, unacetylated AR binds to the HDAC7-NCoR-HDAC3 complex (Figure 7B). In this transcriptionally inhibited state, HDAC7 acts as a regulatory scaffold to recruit the repressive NCoR-HDAC3 complex to AR-bound genomic DNA. Upon acetylation of AR at K630, the newly formed acetyllysine residue binds into the active site of HDAC7 to dissociate the NCoR-HDAC3 complex from AR (Figure 7C). Without histone deacetylation by HDAC3, the presence of AR results in activation of transcription (Figure 7C), likely through binding to coactivator proteins, such as acetyltransferases. With this model, AR acts both as a DNA-binding transcriptional activator and as a recruiter of repressive HDAC7-NCoR-HDAC3 to influence transcription via two complementary mechanisms. In support of this model is the fact that activation of AR by DHT treatment induced nuclear localization of both AR and HDAC7 (Figure S6).
An interesting observation in these studies is that AR binds to HDAC7 independently of acetylation (Figure 3B). Similarly, a prior co-localization study demonstrating that AR and HDAC7 interact independently of NCoR (Karvonen et al., 2006). Taken together, HDAC7 reader function might be facilitated by “pre-binding” of substrates, which ensures that acetyllysine is in proximity to the active site for efficient binding. In fact, previous mass spectrometry data indicated that most acetylation occurs with a low median stoichiometric ratio of 0.02% (Hansen et al., 2019). Similarly, the acetyllysine levels of AR after immunoprecipitation were low or unobservable (Figure S11), consistent with a low stoichiometry. Given the low AR acetylation levels, yet prominent effects by K630 on transcription, we hypothesize that pre-binding of AR to HDAC7 promotes a high local concentration of acetylated K630 on AR near the HDAC7 active site to facilitate binding and NCoR release. In support of this hypothesis, prior work with bromodomains and HDAC8 documented the role of interactions outside of the acetyllysine binding domains in reader function (Castaneda et al., 2017; Mujtaba et al., 2002; Owen et al., 2000). Additional reader substrates of class IIa HDAC proteins will need to be identified to explore whether acetylation-independent pre-binding is a common feature of their acetyllysine binding function.
Acetylation is critical for AR-mediated transcription since K630R or K632/633R mutants of AR were weak transcriptional activators (Fu et al., 2003; Fu et al., 2000; Fu et al., 2002). In addition, wild type AR displayed 10 times worse NCoR binding than the K630A AR mutant (Fu et al., 2002), which is consistent with acetylation-dependent NCoR-AR interaction and the proposed model (Figure 7B). Related to the HDAC7-AR interaction, fluorescence staining showed that HDAC7 and AR colocalized upon treatment of testosterone, although without NCoR (Karvonen et al., 2006). Hollenberg et al. suggested a direct interaction between AR and NCoR (Cheng et al., 2002; Hodgson et al., 2005), although their study did not preclude the possibility that AR exerts its interaction with NCoR through scaffolding HDAC7. In total, prior data on AR-mediated transcriptional regulation is consistent with the proposed model involving HDAC7 reader activity.
Many nuclear receptors, including estrogen receptor (ER) and thyroid receptor (TR), share a common acetylation motif with AR (Wang et al., 2011). Acetylation is also critical to regulate transcription by ER and TR (Sanchez-Pacheco et al., 2009; Wang et al., 2001). We speculate that additional receptors interact with class IIa HDAC proteins to mediate acetylation-dependent transcriptional activation. In fact, here we provide evidence that HDAC7-NCoR disruption by SAHA occurs in the presence of ER (Figure 6), similar to AR. Like HDAC7, HDAC5 and HDAC9 directly interacted with ER to inhibit its transcriptional activity by recruiting NCoR/SMRT (van Rooij et al., 2010). Also similar to AR, ER requires K266 and K268 for full transcriptional activity (Wang et al., 2001), suggesting that one or both of these sites are involved in class IIa reader binding. TR also interacts with SMRT dependent on the presence of three lysine residues (Sanchez-Pacheco et al., 2009). Future studies are needed to fully understand the relationship between class IIa HDAC proteins and the transcriptional activity of the hormone receptor family.
HDAC4, 5, and 7 dissociated from NCoR dependent on both inhibitor and acetyllysine binding (Figures 1 and 2), consistent with prior work (Mottis et al., 2013; Wong et al., 2014), suggesting that all three isoforms have reader functions. However, only HDAC7 affected the transcriptional activity of AR as a function of reader activity (Figure 4B). The combined data suggest that HDAC7 uniquely collaborates with AR to regulate transcriptional activity. We hypothesize that HDAC4, 5, and 7 cooperate with different transcription factors to modulate epigenetic regulatory mechanisms. Additional studies are needed to identify the unique transcription factors and/or enhancer complexes coordinating with HDAC4, 5, and 7 for reader-mediated epigenetic regulation.
Significance
The first evidence of the epigenetic “reader” function of HDAC7 is reported. While prior work proposed an acetyllysine-dependent reader function for class IIa HDAC proteins (Arrowsmith et al., 2012; Bradner et al., 2010; Lobera et al., 2013), here experimental data documents that HDAC7 influences NCoR-HDAC3- and K630-dependent AR transcriptional activity, consistent with the proposed model (Figure 7). To further test this model of reader function, future work will focus on identifying additional acetyllysine-binding substrates of class IIa HDAC proteins. Given the biological importance of reader proteins, such as bromodomains (Smith and Zhou, 2016), and the use of HDAC inhibitors in the clinic (Eckschlager et al., 2017), a full understanding of the epigenetic reader functions of class IIa HDAC proteins will be critical to fully characterize the roles of HDAC proteins in cell biology, in addition to assisting HDAC-targeted drug development efforts.
Limitations of the study
As the initial study reporting a reader function of a Class IIa HDAC protein, additional studies are needed to further test the proposed reader mechanism. In fact, a limitation of this study was the need to use overexpressed wild type and mutant HDAC and AR proteins. Overexpressed proteins are commonly used to overcome low abundance or allow use of mutants (Bottomley et al., 2008a; Fischle et al., 1999; Guenther et al., 2001; Karvonen et al., 2006; Kim et al., 2015; Yang et al., 2011), and results with endogenous or stably expressed proteins have reproduced those with overexpressed proteins (Fischle et al., 2002a; Karvonen et al., 2006; Yang et al., 2011). Future work exploring the reader functions of Class IIa HDAC proteins using endogenous proteins in additional biological contexts will provide necessary confirmation.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mary Kay H. Pflum (Pflum@wayne.edu)
Materials availability
Plasmids generated in this study are available upon request.
Data and Code availability
All repetitive trials and raw data are reported in the supplementary information file. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell line
HEK293 cells (human embryonic kidney cells, ATCC; authenticated annually using short tandem repeat profiling) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (Life technologies) and 1% antibiotic/antimycotic (Hyclone) at 37°C in a 5% CO2 incubator. Jetprime reagent (VWR) was used for transfection of plasmid DNA (2 μg) into HEK293 cells (2 × 106 cells) at 70% confluency. When HDAC7 and AR expression constructs were co-transfected, both plasmids (2 μg of each) were used with the same conditions. After a 24 h growth period, cells were treated with SAHA (10 μM) for 24 h to induce robust acetylation, and then harvested and washed once with DPBS (Dulbecco’s Phosphate Buffered Saline, Hyclone, 10 mM Na2HPO4, 1.8 mM KH2PO4 pH 7.4, 137 mM NaCl, 2.7 mM KCl). Cells were either used immediately or stored at −80 °C as a cell pellet.
METHOD DETAILS
Plasmids
pcDNA3.1-Flag HDAC4, 5, 7 and 9 were described earlier (Fischle et al., 1999; Yang et al., 2011; Yuan et al., 2010; Zhang et al., 2004). GOF mutants of HDAC4, 5, 7, and 9 were created using QuickChange site-directed mutagenesis (Agilent, 200521) with primers in Figure S1. Sequences were confirmed by DNA sequencing. pcDNA3-AR and pcDNA3-AR K630R were described earlier (Fu et al., 2002). pLR-ARE-LUC and pLR-Renilla-LUC plasmids were described earlier (Zhang et al., 2016). pcDNA-HA-ER WT was a gift from Sarat Chandarlapaty (Addgene plasmid number 49498).
HDAC Deacetylase Activity Assay
Transfection of plasmid DNA (pBJ5-Flag wild type HDAC1 or HDAC1 C151A (2 μg))(Wambua et al., 2014), pcDNA3.1-Flag wild type HDAC4 or HDAC4 H976Y (2 μg)), pcDNA3.1-Flag wild type HDAC7 or HDAC7 H842Y (2 μg)) into HEK293 cells (2×106) was carried out as described. Following transfection, cells were lysed using lysis buffer (500 μL; 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10% glycerol, and 0.5% Triton X-100) containing 1× protease inhibitor (GenDepot) for 30 min at 4 °C with rocking. Cell debris was removed by centrifugation (13.2 × 103 rpm) for 10 min at 4 °C and total protein concentrations from the soluble fraction was determine using Bradford reagent (Bio-Rad, cat# 5000205). Lysates (1 mg total protein) containing Flag-tagged HDAC proteins were immunoprecipitated using prewashed Anti-Flag M2 beads (25 μL) as described. Proteins after immunoprecipitation (20% of total) were assayed using HDAC-Glo™ kit (G648B-C, Promega) following the manufacturer’s instruction. Briefly, bound beads were washed with lysis buffer twice. The HDAC-Glo™ substrate (1 mL) and developer (1 μL) were first premixed to form the HDAC-Glo™ reagents. Then, to monitor deacetylase activity, the HDAC-Glo™ reagent (2 μL for HDAC1 and 1μL for HDAC4 and HDAC7) and HDAC-Glo™ buffer (48 μL) were added to the prewashed bound beads and incubated for 25 min at room temperature with rocking. Deacetylase activity was measured as luminescent signal using a GeniosPlus Fluorimeter (Tecan) at optimal gain. The remaining of the precipitated HDAC proteins (80% of total) was washed three times with lysis buffer, followed by 10% SDS-PAGE separation of proteins, transfer to a PVDF membrane, and visualization with Flag (F3165, Sigma) or an NCoR (5948S, Cell Signaling) antibodies using FluorChemQ gel imager (ProteinSimple).
Immunoprecipitation
Immunoprecipitation was performed as previously described with minor changes (36, 37). Following transfection as described above, cells were lysed using lysis buffer (500 μL; 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10% glycerol, and 0.5% Triton X-100) containing 1× protease inhibitor (GenDepot) for 30 min at 4 °C with rocking. Cell debris was removed by centrifugation (13.2 × 103 rpm) for 10 min at 4 °C and total protein concentrations from the soluble fraction was determine using Bradford reagent (Bio-Rad, cat#5000205). For the immunoprecipitation, lysates containing wild type or mutant Flag-tagged HDAC4, 5, 7 or 9 (2 mg of total protein, 1 mL of total volume) were mixed with pre-washed Flag M2 beads (25 μL bead slurry, Sigma) and rocked for 3 h at 4°C). For SAHA or acetyllysine competition during immunoprecipitation (Figures 1–3), an equal amount of the same lysate sample was immunoprecipitated as described in the presence of active site inhibitor SAHA (100 μM or concentration indicated in Figure S2 in <2% DMSO), Ac-LGKAc-NH2 peptide (1 mM or concentration indicated in Figure S3 in ultra-pure water; 95% purity; MALDI-TOF mass spectrometry - expected [M+H]+ = 400.45; observed [M+H]+ = 400.30), or Ac-LGK-NH2 peptide (1 mM or concentration indicated in Figure S3 in ultra-pure water; 95% purity; MALDI-TOF mass spectrometry - expected [M+H]+ = 358.45; observed [M+H]+ = 358.25), AR K630Ac peptide (1 mM or concentration indicated in Figure S5 in ultra-pure water; 95% purity; MALDI-TOF mass spectrometry - expected [M+H]+ = 1495.92; observed [M+H]+ = 1496.07), AR K630 peptide (1 mM or concentration indicated in Figure S5 in ultra-pure water; 96% purity; MALDI-TOF mass spectrometry - expected [M+H]+ = 1453.91; observed [M+H]+ = 1454.02). To assess AR acetylation levels (Figure S11), lysates (1 mg of total protein, 500 μL of total volume) were mixed with pre-washed Protein A/G beads (50 μL bead slurry, Santa Cruz Biotechnology) and primary AR antibody (sc-7305 Santa Cruz Biotechnology) diluted according to the manufacturer protocol. After immunoprecipitation, beads were washed three times with wash buffer (1 mL; 50 mM Tris-Cl pH 8.0, 500 mM NaCl, 10% glycerol, 0.5% triton-X-100) with centrifugation (5,000 rcf for 1 min) to collect the beads after each wash. For SAHA competition after immunoprecipitation (Figure 3C–D), beads after the initial washes were further washed with wash buffer (1 mL) supplemented with SAHA (100 μM in >0.002% DMSO) or DMSO carrier (>0.002%), with centrifugation (5,000 rcf for 1 min) to collect the beads after each of three washes. Following washing, bound proteins were eluted and denatured by boiling for 8 min at 95°C in SDS loading dye (Bio-Rad, Laemmli Sample Buffer, 65.8 mM Tris-HCl, pH 6.8, 26.3% (w/v) glycerol, 2.1% SDS and 0.01% bromophenol blue) supplemented with β-mercaptoethanol (10% v/v). Proteins were separated by 10% SDS-PAGE, followed by transfer to a PVDF membrane (Immobilon P, Millipore) and visualization with Flag (F3165, Sigma), NCoR (5948S, Cell Signaling), AR (5153S, Cell Signaling), ER (8644S, Cell Signaling), or acetyllysine (9441S, Cell Signaling) antibodies using the FluorChemQ gel imager (ProteinSimple).
Expression and Purification of the Catalytic Domain of Human Histone Deacetylase 7
The expression and purification of the Catalytic Domain of Human HDAC7 (cdHDAC7) procedure was adapted from a previously published protocol (Schuetz et al., 2008). The bacterial expression construct encoding His-tagged cdHDAC7 DNA in the pET28a-LIC backbone was obtained from Addgene (plasmid #51340) and was transformed into Rosetta2(DE3) competent cells using the heat shock method. Terrific broth (1 L, 1.2% Tryptone, 2.4% Yeast Extract and 0.5% Glycerol) supplemented with HEPES (100 mM, pH 7.5), kanamycin antibiotic (30 g/mL), and Zn(OAc)2 (100 μM) was inoculated with the transformed cells and grown at 37°C with shaking (250 rpm). When the optical density (OD) of the cell culture reached roughly 1.0, the temperature was lowered to 20°C and protein expression was induced by addition of isopropyl β-d-1-thiogalactopyranoside (IPTG, 0.4 mM). After 16 hr of protein expression at 37°C with shaking (250 rpm), the cell pellet was collected by centrifugation (5,000 rpm) at 4°C for 20 min and then suspended in lysis buffer (50 mL; 25 mM Tris pH 8, 500 mM NaCl, 5% Glycerol, 0.1% CHAPS, 1 mM DTT and 0.1 mM PMSF). Cells were lysed by sonication (6 × 30 s pulses with 30 s pauses in between), and then the cell debris after lysis was removed by centrifugation (40,000 rpm) for 30 min. For protein purification, Ni-NTA beads (4 mL, Qiagen, cat #30450) were first washed with washing buffer (40 mL; 25 mM Tris pH 8, 150 mM NaCl, 5 % Glycerol and 20 mM Imidazole). The soluble fraction of the lysate was added to the washed Ni-NTA beads and rocked for one hour at 4°C to allow protein binding. After binding, the unbound proteins from the lysate were allowed to flow through the column, followed by washing of the beads five times with washing buffer (10 mL) at room temperature. Bound proteins were eluted from the beads with elution buffer (4 mL; 25 mM Tris pH 8, 150 mM NaCl, 5% Glycerol and 300 mM Imidazole) five times at room temperature. The combined elution solution was loaded onto a HiTrap Q HP anion exchange column (GE Healthcare, cat #17-1154-01) using ÄKTA P-920 purification system. The flow rate was set to 1 mL/min and a gradient from 50 mM to 400 mM NaCl in HEPES was used to elute bound proteins. Protein purification was assessed using 10 % SDS-PAGE separation and visualization of proteins by Coomassie blue staining (Bio-Rad, cat #1610436). Fractions containing pure cdHDAC7 were combined and dialyzed (14 kDa cut off, Sigma Aldrich, cat #D9777) against HEPES buffer (25 mM HEPES pH 8, 200 mM NaCl) containing 10 % glycerol in preparation for biotinylation and bio-layer interferometry instrument (BLI) analysis.
Biotinylation of purified cdHDAC7
Purified cdHDAC7 was biotinylated to facilitate binding to the super streptavidin coated sensor (SSC sensor) for BLI analysis. The purified cdHDAC7 protein (0.5 mL of a 60 μM solution) was incubated with biotin-NHS-PEG4 (1.5 μL of a 20 mM solution, Thermo Fisher, cat #A39259) with a 1:1 protein-to-biotin ratio (0.3 μM of each) in a total of 0.5 mL to prevent over-biotinylation. The reaction was incubated on ice for 2 h before dialysis against HEPES buffer containing 10% glycerol using a slid-A-lyzer (10 kDa cut off, Thermo Fisher, cat #66380) to remove the biotin-NHS-PEG4 reagent. Dialyzed samples were fast frozen with liquid nitrogen in aliquots and stored at −80°C before use.
Binding Assessment of cdHDAC7-LGK(Ac) using bio-layer interferometry (BLI)
In preparation for BLI analysis, the biotinylated cdHDAC7 protein was thawed on ice and subjected to dialysis against HEPES buffer using a slid-A-lyzer (10 kDa cut off, Thermo Fisher, cat #66380) to remove glycerol. BLI analysis was carried out using an Octet Red 96 instrument (FortéBio). All kinetics experiments were performed at 25°C while stirred at 1000 rpm to create homogeneous mixtures. Biotinylated cdHDAC7 (0.1 mg/mL) was loaded onto the SSC sensor (FortéBio, cat #18–5057) until the binding signal reached 7 nm. Acetylated LGK(Ac) peptide was used at 20 nM, 1 μM, and 5 μM concentrations, whereas the unacetylated control peptide LGK was used only at a 5 μM concentration. The binding experiments were performed as follows: initial equilibration after loading was set to 1200 s and the baseline was then stabilized for 60 s. Peptide was then allowed to associate for 1200 s, followed by dissociation for 1200 s. FortéBio Octet Data Acquisition and Analysis Software version 7.0 was utilized to perform data acquisition and processing. Global fitting applied to acetylated LGK(Ac) peptide binding curves.
Immunohistochemistry Staining
HEK293 cells (3 × 106) were seeded onto coverslips in a 6-well plate and grown in DMEM (2 mL) supplemented with 10% fetal bovine serum (Life technologies) and 1% antibiotic/antimycotic (Hyclone) at 37°C in a 5% CO2 incubator. After 24 h, cells were co-transfected with expression plasmids encoding WT HDAC7-Flag (0.64 μg) and either pcDNA3-AR or pcDNA-HA-ER plasmids (0.64 μg) using Jetprime reagent (VWR) according to the manufacturer protocol. After another 24 h, cells transfected with AR were incubated with DHT ligand (1 nM in 0.1% DMSO) or with the DMSO vehicle (0.1% v/v). Similarly, cells transfected with ER were incubated with E2 ligand (10 pM in 0.1% ethanol) or with the ethanol vehicle (0.1% v/v). Following 24 h incubation with ligands or vehicle, cells were washed with DPBS (2 mL; Thermo Fisher, SH30028FS) once, fixed with 4% paraformaldehyde (2 mL; Santa Cruz, sc-281692) for 10 minutes, washed three times with DPBS (2 mL), and fixed with ice cold methanol (2 mL) for 3 minutes. Fixed cells were washed three times with DPBS (2 mL) and permeabilized with permeabilization buffer (2 mL; 0.3% Triton X-100 in PBS, DPBS) for 10 minutes at room temperature. Following permeabilization, cells were washed three times with DPBS (2 mL) and blocked with SuperBlock™ Buffer (2 mL; Thermo Fisher, cat # 37515) for one hour at room temperature. After the blocking buffer was removed, cells were incubated with mouse anti-Flag (Sigma Aldrich, cat #F3156) and either rabbit anti-AR (Cell Signaling, cat #5153) or rabbit anti-ER antibody (Cell Signaling, cat #8644) overnight in DPBST (2 mL; 0.1% Triton X-100 in DPBS). After incubation with the primary antibodies, cells were washed three times with DPBS and incubated with anti-mouse Alexa Fluor 488 (Thermo Fisher, cat #A-11029) and anti-rabbit Alexa Fluor 594 secondary antibody (Thermo Fisher, cat #A-11037) mix in PBST (2 mL) for one hour at room temperature. Antibody-stained cells were washed with DPBS (2 mL) three times and counterstained with DAPI (2 mL; 300 nM in PBS; Thermo Fisher, cat #D21490) for five minutes. Cells were then washed three times with DPBS (2 mL), mounted on a glass slide with a drop of ProLong® Gold Antifade Reagent (Thermo Fisher, cat #P10144) and visualized using a Leica TCS SP8 inverted confocal microscope at a resolution of 2048 × 2048 and a line averaging of 32. Images were obtained using an Acousto Optical Beam Splitter (AOBS) and an Acousto Optical Tunable Filter (AOTF) with excitation lasers of 405, 488, 552 and 632 nm. Pictures were acquired using the LAS_X acquisition software with detection ranges of 415 to 485 nm for DAPI, 555 to 625 nm for Alexa Fluor 594, and 695–765 nm for Alexa Fluor 488. Pictures in the manuscript and supplemental information are reported exactly as they were obtained.
Reporter Gene Assay
HEK293 cells (200,000) were seeded in 6-well cell culture plates and grown in phenol red-free DMEM supplemented with 10% charcoal-stripped fetal bovine serum (A3382101, Fisher) at 37 °C in a 5% CO2 incubator. Transfections were performed using JetPrime reagent according to manufacturer instructions using pcDNA3-AR (0.32 μg), pLR-ARE-LUC (0.64 μg), pLR-Ren-LUC (0.32 μg) and HDAC-Flag expression construct (0.64 μg). Five hours after transfection, cells were washed once with DPBS (1 mL). Then, media (3 mL) in the absence (0.1% DMSO) or presence of DHT (1 nM on 0.1% DMSO) was added. Some reactions also included small molecule inhibitor (10 μM of SAHA, SHI:1–2, or RGFP966). Cells were incubated for 36 h before harvesting using passive lysis buffer (500 μL, E1960, Promega) by rocking at room temperature for 15 min. Lysates were generated based on manufacture instruction and immediately assayed using the Dual Luciferase Assay (E1960, Promega) using a Geniosplus Fluorimeter (Tecan). Activity is reported in relative light unit (RLU), which were determined as the ratio of inducible firefly luciferase luminescence from pLR-ARE-LUS divided by the luminescence of the renilla luciferase control from pLR-Ren-LUC. Mean and standard error were calculated from three independent trials.
RT-PCR of AR-Regulated Gene Expression
HEK293 cells were seeded at a density of 200,000 cells per well in 6-well cell culture plates and grown in phenol red-free DMEM supplemented with 10% charcoal-stripped fetal bovine serum (Fisher, cat #A3382101) at 37 °C in a 5% CO2 incubator. After growth of 24 h, cells were transfected with pcDNA3-AR (0.32 μg) and either pcDNA3.1-Flag HDAC7 or HDAC7 H842Y (0.64 μg), as described. After 5 h of growth, the media was removed and replaced with media in the absence (0.1% DMSO) or presence of DHT (1 nM on 0.1% DMSO). Cells were grown for another 36 h before media was removed and TRIzol™ reagent (800 μL, Thermo Fisher, cat #15596026) was added to the cell directly to denature the protein. The resulting mixture was diluted with chloroform (200 μL), followed by gentle shaking and incubation for 5 min. Cell debris was precipitated by centrifuging at 12,000 rpm for 10 min at 4°C. The supernatant after centrifugation (300 μL) was diluted with isopropanol (300 μL) and cooled at −20°C for 1 hr, followed by centrifugation at 4°C at 12,000 rpm for 10 min. The supernatant was removed carefully followed by washing of the RNA pellet with 70% ethanol (700 μL). The precipitated RNA was dried at RT for 5 min, followed by reconstitution with dd water (150 μL). RNA quantity was determined at 260 nm using a NanoDrop™ Spectrophotometer. RNA (700 ng) was mixed with random primer (1 μL, New England Biolabs, cat #S1330S), dNTP (1 μL, 10 mM), and ddH2O in a total volume of 10 μL. The RNA/Primer mixture was denatured for 5 min at 65°C, followed by addition of 10X amplification buffer (2 μL, New England Biolabs, cat #B0537S), RNase inhibitor (0.5 μL, New England Biolabs, cat #M0314S), and reverse transcriptase (0.25 μL, New England Biolabs, cat #M0380S) in a total volume of 20μL. Reverse transcription occurred by incubating the mixture for 5 min at 25°C for annealing and 30 min at 55°C for synthesis. The resulting cDNA (1 μL) was mixed with forward and reverse primers (1.5 μL of each, 10 μM for each primer) for the indicated genes (Table S1) and Taq 2X Master Mix (7.5 μL, New England Biolabs, cat #M0270L) in a total volume of 15 μL. The PCR protocol entailed initial denaturation for 5 min at 95°C, followed by cycles of 1 min at 55°C for annealing, 1 min at 68°C for extension, and 1 min at 95°C for denaturation. After 35 cycles for SFRP5 and Wnt16, or 25 cycles for GAPDH, a 7 min final extension at 68°C was performed. The PCR product was resolved on a 2% agarose gel supplemented with ethidium bromide (Sigma Aldrich, E1510) and visualized with a ChemiDoc gel imager (Bio-Rad). Band intensities were analyzed using ImageJ (Schneider et al., 2012). Intensities of each band was normalized against the GAPDH band by dividing the target gene RT-PCR band intensity by the corresponding GAPDH band intensity. DHT induction sample bands were averaged and set to 100% for comparison. All the other samples were normalized based on the DHT induction samples to give the relative intensity.
QUANTIFICATION AND STATISTICAL ANALYSIS
To quantify protein levels, bands in the gel images were quantified using ImageJ (Schneider et al., 2012); the whole lane was selected and plotted to show the intensity of the protein bands in that lane, which was then quantified by measuring the area under the curve above the background. The band intensities were normalized to one of the samples to generate normalized percentage values, with the raw data shown in the supplementary information. For the gene expression assays, mean light intensity data were normalized as a percentage of the DHT sample, with the raw data shown in the supplementary information. As indicated in the figure legends, the mean and standard error of at least 3 independent trials (all trials are provided as supplementary information) are shown in all figures. Student t-test analysis was applied to the normalized percentage values using Prism software (GraphPad Software, Inc.). One-tailed (Figure 3D) or two-tailed (all other figures) p values were determined from at least 3 three independent experiments using a 95% confidence interval with N.S. = not significant (p > 0.05), * = p < 0.05, ** = p < 0.01, *** = p < 0.001, and *** = p < 0.0001.
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Flag | Sigma | F3165 |
| anti-NCoR | Cell Signaling | 5948 |
| anti-AR | Cell Signaling | 5153 |
| anti-ER | Cell Signaling | 8644 |
| anti-acetyllysine | Cell Signaling | 9441S |
| anti-mouse Alexa Fluor 488 | Thermo Fisher | A-11029 |
| anti-rabbit Alexa Fluor 594 | Thermo Fisher | A-11037 |
| Anti-Flag M2 beads | Sigma | A2220 |
| Bacterial and virus strains | ||
| Rosetta2(DE3) | Sigma | 70954 |
| Chemicals, peptides, and recombinant proteins | ||
| LGKAc peptide | GenScript USA, Inc. | Custom synthesis |
| LGK peptide | GenScript USA, Inc. | Custom synthesis |
| DAPI | Thermo Fisher | D21490 |
| ProLong® Gold Antifade Reagent | Thermo Fisher | P10144 |
| Jetprime transfection reagent | VWR | 89129-924 |
| Critical commercial assays | ||
| QuickChange site-directed mutagenesis | Agilent | 200521 |
| HDAC-Glo™ assay | Promega | G648B-C |
| Bradford reagent | Bio-Rad | 5000205 |
| Dual Luciferase Assay | Promega | E1960 |
| Experimental models: Cell lines | ||
| HEK293 | ATCC | CRL-1573 |
| Oligonucleotides | ||
| See Figure S1A and S8A for oligonucleotide sequences | This paper | N/A |
| Recombinant DNA | ||
| pcDNA3.1-HDAC4-Flag | Addgene | Addgene #13821 |
| pcDNA3.1-Flag HDAC5 | Addgene | Addgene #13822 |
| pcDNA3.1-Flag HDAC7 | Addgene | Addgene #13824 |
| 2NVR (pET28a-LIC-HDAC7 catalytic domain) | Addgene | Addgene #51340 |
| pcDNA3.1-Flag HDAC9 | Zhang et al., 2004 | N/A |
| pcDNA3.1-Flag HDAC1 C151A | Wambua et al. 2014 | N/A |
| pcDNA3.1-Flag HDAC4 H976Y | This paper | N/A |
| pcDNA3.1-Flag HDAC5 H1006Y | This paper | N/A |
| pcDNA3.1-Flag HDAC7 H842Y | This paper | N/A |
| pcDNA3.1-Flag HDAC9 H956Y | This paper | N/A |
| pcDNA3-AR | Fu et al., 2022 | N/A |
| pcDNA3-AR(K630A) | Fu et al., 2022 | N/A |
| pLR-ARE-LUC | Zhang et al., 2016 | N/A |
| pLR-Renilla-LUC | Zhang et al., 2016 | N/A |
| pcDNA-HA-ER WT | Addgene | Addgene # 49498 |
| Other | ||
| FluorChemQ gel imager | ProteinSimple | |
| Octet Red 96 BLI instrument | FortéBio | |
| SSC sensor for BLI instrument | FortéBio | 18-5057 |
| TCS SP8 inverted confocal microscope | Leica | |
| Geniosplus Fluorimeter | Tecan | |
| ChemiDoc gel imager | Bio-Rad | |
Highlights.
HDAC4, 5 and 7 dissociated from corepressor NCoR dependent on acetyllysine peptides
Dissociation of the HDAC7-NCoR complex depended on the K630 acetylation site of AR
AR transcriptional activity required an intact HDAC7 active site and AR K630
The evidence is consistent with Class IIa HDAC7 epigenetic reader function
Acknowledgement:
We thank the National Institutes of Health (GM121061 and GM131821) and Wayne State University for funding, X. Zhang (Kamanos Cancer Institute, Wayne State University) for HDAC4, 5, 7, and 9 expression plasmids and technical support, R. G. Pestell (Pennsylvania Cancer and Regenerative Medicine Center) for the AR and AR K630R expression plasmids, J. T. Koh (University of Delaware) for the ARE-LUC and Ren-LUC gene reporter plasmids, and I. Gomes, K. Herath, and J. Knoff for comments on the manuscript.
Footnotes
Declaration of interest: Authors declare no competing interests.
Inclusion and Diversity: One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper self-identifies as living with a disability.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alimirah F, Chen J, Basrawala Z, Xin H, and Choubey D (2006). DU-145 and PC-3 human prostate cancer cell lines express androgen receptor: implications for the androgen receptor functions and regulation. FEBS Lett 580, 2294–2300. [DOI] [PubMed] [Google Scholar]
- Arrowsmith CH, Bountra C, Fish PV, Lee K, and Schapira M (2012). Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov 11, 384–400. [DOI] [PubMed] [Google Scholar]
- Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y, Becher I, Bergamini G, et al. (2011). Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nature biotechnology 29, 255–265. [DOI] [PubMed] [Google Scholar]
- Bottomley MJ, Lo Surdo P, Di Giovine P, Cirillo A, Scarpelli R, Ferrigno F, Jones P, Neddermann P, De Francesco R, Steinkuhler C, et al. (2008a). Structural and Functional Analysis of the Human HDAC4 Catalytic Domain Reveals a Regulatory Structural Zinc-binding Domain. The Journal of biological chemistry 283, 26694–26704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottomley MJ, Lo Surdo P, Di Giovine P, Cirillo A, Scarpelli R, Ferrigno F, Jones P, Neddermann P, De Francesco R, Steinkuhler C, et al. (2008b). Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J Biol Chem 283, 26694–26704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, and Mazitschek R (2010). Chemical phylogenetics of histone deacetylases. Nature chemical biology 6, 238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaneda CA, Wolfson NA, Leng KR, Kuo YM, Andrews AJ, and Fierke CA (2017). HDAC8 substrate selectivity is determined by long- and short-range interactions leading to enhanced reactivity for full-length histone substrates compared with peptides. The Journal of biological chemistry 292, 21568–21577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Brzostek S, Lee SR, Hollenberg AN, and Balk SP (2002). Inhibition of the dihydrotestosterone-activated androgen receptor by nuclear receptor corepressor. Mol Endocrinol 16, 1492–1501. [DOI] [PubMed] [Google Scholar]
- Chng KR, Chang CW, Tan SK, Yang C, Hong SZ, Sng NY, and Cheung E (2012). A transcriptional repressor co-regulatory network governing androgen response in prostate cancers. The EMBO journal 31, 2810–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Weinert BT, Nishida Y, Verdin E, and Mann M (2014). The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol 15, 536–550. [DOI] [PubMed] [Google Scholar]
- Clocchiatti A, Florean C, and Brancolini C (2011). Class IIa HDACs: from important roles in differentiation to possible implications in tumourigenesis. J Cell Mol Med 15, 1833–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darkin-Rattray SJ, Gurnett AM, Myers RW, Dulski PM, Crumley TM, Allocco JJ, Cannova C, Meinke PT, Colletti SL, Bednarek MA, et al. (1996). Apicidin: a novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc Natl Acad Sci U S A 93, 13143–13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, and van Kuilenburg AB (2003). Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 370, 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desravines DC, Serna Martin I, Schneider R, Mas PJ, Aleksandrova N, Jensen MR, Blackledge M, and Hart DJ (2017). Structural Characterization of the SMRT Corepressor Interacting with Histone Deacetylase 7. Sci Rep 7, 3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, and Zhou MM (1999). Structure and ligand of a histone acetyltransferase bromodomain. Nature 399, 491–496. [DOI] [PubMed] [Google Scholar]
- Ebrahimi A, Sevinc K, Gurhan Sevinc G, Cribbs AP, Philpott M, Uyulur F, Morova T, Dunford JE, Goklemez S, Ari S, et al. (2019). Bromodomain inhibition of the coactivators CBP/EP300 facilitate cellular reprogramming. Nature chemical biology 15, 519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckschlager T, Plch J, Stiborova M, and Hrabeta J (2017). Histone Deacetylase Inhibitors as Anticancer Drugs. Int J Mol Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, and Pavletich NP (1999). Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401, 188–193. [DOI] [PubMed] [Google Scholar]
- Fischle W, Dequiedt F, Fillion M, Hendzel MJ, Voelter W, and Verdin E (2001). Human HDAC7 histone deacetylase activity is associated with HDAC3 in vivo. J Biol Chem 276, 35826–35835. [DOI] [PubMed] [Google Scholar]
- Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, and Verdin E (2002a). Enzymatic Activity Associated with Class II HDACs Is Dependent on a Multiprotein Complex Containing HDAC3 and SMRT/N-CoR. Molecular cell 9, 45–57. [DOI] [PubMed] [Google Scholar]
- Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, and Verdin E (2002b). Enzymatic activity associated with class IIHDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Molecular Cell 9, 45–57. [DOI] [PubMed] [Google Scholar]
- Fischle W, Emiliani S, Hendzel MJ, Nagase T, Nomura N, Voelter W, and Verdin E (1999). A new family of human histone deacetylases related to Saccharomyces cerevisiae HDA1p. The Journal of biological chemistry 274, 11713–11720. [DOI] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, et al. (2005). Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37, 391–400. [DOI] [PubMed] [Google Scholar]
- Fu M, Rao M, Wang C, Sakamaki T, Wang J, Di Vizio D, Zhang X, Albanese C, Balk S, Chang C, et al. (2003). Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Mol Cell Biol 23, 8563–8575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M, Wang C, Reutens AT, Wang J, Angeletti RH, Siconolfi-Baez L, Ogryzko V, Avantaggiati ML, and Pestell RG (2000). p300 and p300/cAMP-response element-binding protein-associated factor acetylate the androgen receptor at sites governing hormone-dependent transactivation. J Biol Chem 275, 20853–20860. [DOI] [PubMed] [Google Scholar]
- Fu M, Wang C, Wang J, Zhang X, Sakamaki T, Yeung YG, Chang C, Hopp T, Fuqua SA, Jaffray E, et al. (2002). Androgen receptor acetylation governs trans activation and MEKK1-induced apoptosis without affecting in vitro sumoylation and trans-repression function. Mol Cell Biol 22, 3373–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao WQ, Bohl CE, and Dalton JT (2005). Chemistry and structural biology of androgen receptor. Chemical Reviews 105, 3352–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaur V, Connor T, Sanigorski A, Martin SD, Bruce CR, Henstridge DC, Bond ST, McEwen KA, Kerr-Bayles L, Ashton TD, et al. (2016). Disruption of the Class IIa HDAC Corepressor Complex Increases Energy Expenditure and Lipid Oxidation. Cell reports 16, 2802–2810. [DOI] [PubMed] [Google Scholar]
- Guenther MG, Barak O, and Lazar MA (2001). The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol 21, 6091–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen BK, Gupta R, Baldus L, Lyon D, Narita T, Lammers M, Choudhary C, and Weinert BT (2019). Analysis of human acetylation stoichiometry defines mechanistic constraints on protein regulation. Nature Communications 10, 1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebbes TR, Thorne AW, and Crane-Robinson C (1988). A direct link between core histone acetylation and transcriptionally active chromatin. The EMBO journal 7, 1395–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Ström A, Treuter E, Warner M, et al. (2007). Estrogen Receptors: How Do They Signal and What Are Their Targets. Physiological Reviews 87, 905–931. [DOI] [PubMed] [Google Scholar]
- Hesham HM, Lasheen DS, and Abouzid KAM (2018). Chimeric HDAC inhibitors: Comprehensive review on the HDAC-based strategies developed to combat cancer. Med Res Rev 38, 2058–2109. [DOI] [PubMed] [Google Scholar]
- Hodgson MC, Astapova I, Cheng S, Lee LJ, Verhoeven MC, Choi E, Balk SP, and Hollenberg AN (2005). The androgen receptor recruits nuclear receptor CoRepressor (N-CoR) in the presence of mifepristone via its N and C termini revealing a novel molecular mechanism for androgen receptor antagonists. J Biol Chem 280, 6511–6519. [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, and Auwerx J (2012). Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson BP, Martinez-Yamout MA, Dyson HJ, and Wright PE (2000). Solution structure and acetyl-lysine binding activity of the GCN5 bromodomain. Journal of molecular biology 304, 355–370. [DOI] [PubMed] [Google Scholar]
- Hudson GM, Watson PJ, Fairall L, Jamieson AG, and Schwabe JW (2015). Insights into the Recruitment of Class IIa Histone Deacetylases (HDACs) to the SMRT/NCoR Transcriptional Repression Complex. J Biol Chem 290, 18237–18244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson RH, Ladurner AG, King DS, and Tjian R (2000). Structure and function of a human TAF(II)250 double bromodomain module. Science 288, 1422–1425. [DOI] [PubMed] [Google Scholar]
- Jones P, Altamura S, De Francesco R, Gallinari P, Lahm A, Neddermann P, Rowley M, Serafini S, and Steinkuhler C (2008). Probing the elusive catalytic activity of vertebrate class IIa histone deacetylases. Bioorg Med Chem Lett 18, 1814–1819. [DOI] [PubMed] [Google Scholar]
- Joshi P, Greco TM, Guise AJ, Luo Y, Yu F, Nesvizhskii AI, and Cristea IM (2013). The functional interactome landscape of the human histone deacetylase family. Mol Syst Biol 9, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karvonen U, Janne OA, and Palvimo JJ (2006). Androgen receptor regulates nuclear trafficking and nuclear domain residency of corepressor HDAC7 in a ligand-dependent fashion. Exp Cell Res 312, 3165–3183. [DOI] [PubMed] [Google Scholar]
- Kim GS, Jung HE, Kim JS, and Lee YC (2015). Mutagenesis Study Reveals the Rim of Catalytic Entry Site of HDAC4 and −5 as the Major Binding Surface of SMRT Corepressor. PLoS One 10, e0132680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer OH, Göttlicher M, and Heinzel T (2001). Histone deacetylase as a therapeutic target. Trends in Endocrinology & Metabolism 12, 294–300. [DOI] [PubMed] [Google Scholar]
- Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, Sambucini S, Bottomley MJ, Lo Surdo P, Carfi A, et al. (2007). Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci U S A 104, 17335–17340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffer BE, Mintzer R, Fong R, Mukund S, Tam C, Zilberleyb I, Flicke B, Ritscher A, Fedorowicz G, Vallero R, et al. (2013). Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J Biol Chem 288, 26926–26943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong H, Sloan JR, Nash PD, and Greene GL (2005). Recruitment of histone deacetylase 4 to the N-terminal region of estrogen receptor alpha. Mol Endocrinol 19, 2930–2942. [DOI] [PubMed] [Google Scholar]
- Li JW, Wang J, Wang JX, Nawaz Z, Liu JM, Qin J, and Wong JM (2000). Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. Embo Journal 19, 4342–4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling H, Peng L, Seto E, and Fukasawa K (2012). Suppression of centrosome duplication and amplification by deacetylases. Cell Cycle 11, 3779–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobera M, Madauss KP, Pohlhaus DT, Wright QG, Trocha M, Schmidt DR, Baloglu E, Trump RP, Head MS, Hofmann GA, et al. (2013). Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nature chemical biology 9, 319–325. [DOI] [PubMed] [Google Scholar]
- Malik S, Jiang S, Garee JP, Verdin E, Lee AV, O’Malley BW, Zhang M, Belaguli NS, and Oesterreich S (2010). Histone deacetylase 7 and FoxA1 in estrogen-mediated repression of RPRM. Mol Cell Biol 30, 399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning ET, Ikehara T, Ito T, Kadonaga JT, and Kraus WL (2001). p300 forms a stable, template-committed complex with chromatin: role for the bromodomain. Mol Cell Biol 21, 3876–3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje P, Zanello S, Holick M, and Boland R (2001). Differential cellular localization of estrogen receptor alpha in uterine and mammary cells. Mol Cell Endocrinol 181, 117–129. [DOI] [PubMed] [Google Scholar]
- Mottis A, Mouchiroud L, and Auwerx J (2013). Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev 27, 819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujtaba S, He Y, Zeng L, Farooq A, Carlson JE, Ott M, Verdin E, and Zhou M-M (2002). Structural Basis of Lysine-Acetylated HIV-1 Tat Recognition by PCAF Bromodomain. Molecular Cell 9, 575–586. [DOI] [PubMed] [Google Scholar]
- Mujtaba S, He Y, Zeng L, Yan S, Plotnikova O, Sachchidanand, Sanchez R, Zeleznik-Le NJ, Ronai Z, and Zhou MM (2004). Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Molecular cell 13, 251–263. [DOI] [PubMed] [Google Scholar]
- Negmeldin AT, Knoff JR, and Pflum MKH (2018). The structural requirements of histone deacetylase inhibitors: C4-modified SAHA analogs display dual HDAC6/HDAC8 selectivity. Eur J Med Chem 143, 1790–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negmeldin AT, and Pflum MKH (2017). The structural requirements of histone deacetylase inhibitors: SAHA analogs modified at the C5 position display dual HDAC6/8 selectivity. Bioorg Med Chem Lett 27, 3254–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DJ, Ornaghi P, Yang JC, Lowe N, Evans PR, Ballario P, Neuhaus D, Filetici P, and Travers AA (2000). The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. The EMBO journal 19, 6141–6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Kim GS, Hwang HJ, Nam TH, Park HS, Song J, Jang TH, Lee YC, and Kim JS (2018). Structural basis of the specific interaction of SMRT corepressor with histone deacetylase 4. Nucleic Acids Res 46, 11776–11788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie K, Guidez F, Howell L, Healy L, Waxman S, Greaves M, and Zelent A (2003). The histone deacetylase 9 gene encodes multiple protein isoforms. J Biol Chem 278, 16059–16072. [DOI] [PubMed] [Google Scholar]
- Sanchez-Pacheco A, Martinez-Iglesias O, Mendez-Pertuz M, and Aranda A (2009). Residues K128, 132, and 134 in the thyroid hormone receptor-alpha are essential for receptor acetylation and activity. Endocrinology 150, 5143–5152. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz A, Min J, Allali-Hassani A, Schapira M, Shuen M, Loppnau P, Mazitschek R, Kwiatkowski NP, Lewis TA, Maglathin RL, et al. (2008). Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J Biol Chem 283, 11355–11363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SG, and Zhou MM (2016). The Bromodomain: A New Target in Emerging Epigenetic Medicine. ACS Chem Biol 11, 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner MJ, Welliver RC Jr., Chen M, Shtutman M, Godoy A, Smith G, Mian BM, and Buttyan R (2011). Effects of androgen receptor and androgen on gene expression in prostate stromal fibroblasts and paracrine signaling to prostate cancer cells. PLoS One 6, e16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E, Fielitz J, Sutherland LB, Thijssen VL, Crijns HJ, Dimaio MJ, Shelton J, De Windt LJ, Hill JA, and Olson EN (2010). Myocyte enhancer factor 2 and class II histone deacetylases control a gender-specific pathway of cardioprotection mediated by the estrogen receptor. Circ Res 106, 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfi A, De Francesco R, Steinkuhler C, and Di Marco S (2007). Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep 8, 879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varlakhanova N, Snyder C, Jose S, Hahm JB, and Privalsky ML (2010). Estrogen receptors recruit SMRT and N-CoR corepressors through newly recognized contacts between the corepressor N terminus and the receptor DNA binding domain. Mol Cell Biol 30, 1434–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wambua MK, Nalawansha DA, Negmeldin AT, and Pflum MK (2014). Mutagenesis Studies of the 14 Å Internal Cavity of Histone Deacetylase 1: Insights towards the Acetate Escape Hypothesis and Selective Inhibitor Design. Journal of medicinal chemistry 57, 642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Fu M, Angeletti RH, Siconolfi-Baez L, Reutens AT, Albanese C, Lisanti MP, Katzenellenbogen BS, Kato S, Hopp T, et al. (2001). Direct acetylation of the estrogen receptor alpha hinge region by p300 regulates transactivation and hormone sensitivity. J Biol Chem 276, 18375–18383. [DOI] [PubMed] [Google Scholar]
- Wang C, Tian L, Popov VM, and Pestell RG (2011). Acetylation and nuclear receptor action. J Steroid Biochem Mol Biol 123, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AC, and Johnstone RW (2014). New and emerging HDAC inhibitors for cancer treatment. J Clin Invest 124, 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MM, Guo C, and Zhang J (2014). Nuclear receptor corepressor complexes in cancer: mechanism, function and regulation. Am J Clin Exp Urol 2, 169–187. [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Tse AK, Li P, Ma Q, Xiang S, Nicosia SV, Seto E, Zhang X, and Bai W (2011). Inhibition of androgen receptor activity by histone deacetylase 4 through receptor SUMOylation. Oncogene 30, 2207–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, Peng L, Radhakrishnan R, and Seto E (2010). Histone deacetylase 9 (HDAC9) regulates the functions of the ATDC (TRIM29) protein. J Biol Chem 285, 39329–39338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wharton W, Yuan Z, Tsai SC, Olashaw N, and Seto E (2004). Activation of the growth-differentiation factor 11 gene by the histone deacetylase (HDAC) inhibitor trichostatin A and repression by HDAC3. Mol Cell Biol 24, 5106–5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Mantravadi PK, Jobbagy S, Bao W, and Koh JT (2016). Antagonizing the Androgen Receptor with a Biomimetic Acyltransferase. ACS Chem Biol 11, 2797–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All repetitive trials and raw data are reported in the supplementary information file. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.