

Abstract
The only enzyme of the citric acid cycle for which no open reading frame (ORF) was found in the Helicobacter pylori genome is the NAD-dependent malate dehydrogenase. Here, it is shown that in this organism the oxidation of malate to oxaloacetate is catalyzed by a malate:quinone oxidoreductase (MQO). This flavin adenine dinucleotide-dependent membrane-associated enzyme donates electrons to quinones of the electron transfer chain. Similar to succinate dehydrogenase, it is part of both the electron transfer chain and the citric acid cycle. MQO activity was demonstrated in isolated membranes of H. pylori. The enzyme is encoded by the ORF HP0086, which is shown by the fact that expression of the HP0086 sequence from a plasmid induces high MQO activity in mqo deletion mutants of Escherichia coli or Corynebacterium glutamicum. Furthermore, this plasmid was able to complement the phenotype of the C. glutamicum mqo deletion mutant. Interestingly, the protein predicted to be encoded by this ORF is only distantly related to known or postulated MQO sequences from other bacteria. The presence of an MQO shown here and the previously demonstrated presence of a 2-ketoglutarate:ferredoxin oxidoreductase and a succinyl-coenzyme A (CoA):acetoacetyl-CoA transferase indicate that H. pylori possesses a complete citric acid cycle, but one which deviates from the standard textbook example in three steps.
A controversy with regard to the presence of malate dehydrogenase (MDH) in Helicobacter pylori became apparent when the genomic sequences of two strains of this organism were published (2, 29). Whereas biochemical measurements indicated that MDH activity (EC 1.1.1.37) was present in this organism, no open reading frame (ORF) for a possible MDH could be found in the genomic sequences (14, 17, 19, 24). In some organisms genes encoding MDH are more similar to genes for l-lactate dehydrogenases (6). However, such ORFs were also lacking in H. pylori, excluding the possibility that an mdh gene had been erroneously annotated as an ldh gene. H. pylori does have an ORF (dld) for a lactate dehydrogenase, but this is a membrane-bound d-lactate dehydrogenase.
The presence or absence of an MDH in H. pylori has implications for its central metabolism. Considerable confusion exists as to whether H. pylori possesses a complete citric acid cycle and whether this cycle can operate oxidatively or functions only in a branched mode. Physiological studies of lactate and pyruvate oxidation by well-aerated cells indicated that some oxidative citric acid cycle activity might be present (5). However, the original annotation of the genome indicated three omissions in the list of ORFs comprising a typical citric acid cycle (29). The omissions were α-ketoglutarate dehydrogenase, succinyl-coenzyme A (CoA) ligase, and MDH. Earlier biochemical studies showed that instead of α-ketoglutarate dehydrogenase H. pylori possesses α-ketoglutarate:ferredoxin oxidoreductase (EC 1.2.7.3) (18). Furthermore, a succinyl-CoA:acetoacetyl-CoA transferase could convert succinyl-CoA to succinate (11, 18). Some authors doubt whether the fumarate reductase of H. pylori can operate as a succinate dehydrogenase (SDH) (24). Biochemical assays, however, indicate that a relatively high level of SDH activity does exist (5, 7; see below). Thus, an ORF encoding an MDH is, in principle, the only omission in the list for a complete citric acid cycle (19).
We observed that the H. pylori genome contains an ORF encoding a protein with distant similarity to malate:quinone oxidoreductase (MQO), or the malate dehydrogenase (acceptor), EC 1.1.99.16, from Corynebacterium glutamicum (22). MQO is a citric acid cycle enzyme that, like MDH, converts malate to oxaloacetate but is a membrane-associated enzyme (or peripheral membrane enzyme) containing tightly bound flavin adenine dinucleotide (FAD) as a cofactor. In contrast to MDH, it donates the electrons from malate oxidation to quinones. The quinones are subsequently oxidized by the electron transfer chain. Cohn proved the existence of MQO in the 1950s in Micrococcus lysodeikticus (“Micrococcus luteus”), and the enzyme has since then been found in several gram-positive and gram-negative bacteria (8; see also references cited in reference 22). However, in the past decades MQO seems to have escaped the attention of most microbiologists. Recently, we were able to clone a gene from C. glutamicum encoding an MQO (22). Several homologues, with previously unknown functions, were observed in other bacteria. The protein hypothetically encoded by HP0086 from strain 26695 and its homologue in strain J99 are distantly related to these MQO sequences (Fig. 1). We wondered whether HP0086 might encode an MQO and thus complete the list of genes encoding citric acid cycle enzymes in H. pylori. Convincing experimental evidence for this hypothesis is presented here.
FIG. 1.

Tree reflecting the similarity between MQOs from different organisms. Bar, expected change of 0.1 per amino acid. For details of the analysis, see Materials and Methods.
MATERIALS AND METHODS
Bacterial strains, plasmids, growth, and medium compositions.
The strains and plasmids used in this work are listed in Table 1. For isolation of membranes, Escherichia coli was routinely grown overnight on Luria-Bertani medium (25) at 37°C. When strains carried the kanamycin resistance marker, 50 μg of kanamycin ml−1 was added to the medium. C. glutamicum was grown overnight on 2× tryptone-yeast extract medium (25) at 30°C with, in the case of the strains carrying the marker, 25 μg of kanamycin ml−1.
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Characteristics | Source or reference(s) |
|---|---|---|
| Strains | ||
| C. glutamicum | ||
| ATCC 13032 | Wild type | 1 |
| Δmqo | mqo deletion derivative of ATCC 13032 | M. E. van der Rest, unpublished data |
| H. pylori ATCC 49503 | Wild type | 12 |
| E. coli | ||
| DH5α | supE44 ΔlacU169 (φ80dlacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | 25 |
| Δmqo | mqo (yojH) deletion derivative of MC4100 | 4, M. E. van der Rest, unpublished data |
| Plasmids | ||
| pBluescript II SK | AmprlacZ | 26 |
| pEKEx1 | Kanrlaclqtac | 15 |
| pHp-mqo | pEKEx1 with H. pylori mqo gene (ORF HP0086) | This paper |
The minimal medium agar used in this study for growth of C. glutamicum will be described elsewhere (D. Molenaar, unpublished data).
H. pylori ATCC 49503 was grown in Brucella broth supplemented with 5% fetal calf serum at 37°C in 100 ml of medium in cell-culture bottles shaken at 140 rpm. These bottles were kept in a 2.5-liter anaerobic jar in which microaerobic conditions were achieved using Anaerocult C (Merck). Cells grown for 24 to 48 h were subcultured at least three times in fresh, prewarmed medium.
Isolation of membrane fragments.
Membrane fragments of C. glutamicum or E. coli were isolated from overnight cultures (22). An overnight culture was centrifuged and washed twice with ice-cold buffer, consisting of 50 mM HEPES, 10 mM potassium-acetate, 10 mM CaCl2, and 5 mM MgCl2 titrated with NaOH to pH 7.5 (buffer A). The pellet was resuspended in approximately 10 ml of buffer A for every 100 ml of the original culture, and the suspension was passed through a French pressure cell three times at 30,000 lb/in2 (207 MPa) in the case of C. glutamicum cells or twice at 10,000 lb/in2 (69 MPa) in the case of E. coli cells. Cell debris was removed by centrifuging for 10 min at 10,000 × g and 4°C. The supernatant was centrifuged for 30 min at 75,000 × g and 4°C. The membrane pellet was resuspended in the same amount of buffer A and centrifuged again. The pellet was then resuspended in a small volume of buffer A, 100 to 200 μl for every 10 ml of the original extract. The final protein concentration was usually between 4 and 12 mg ml−1.
For the preparation of H. pylori membranes, cells grown for 48 h were harvested by centrifugation at 3,400 × g for 15 min. After being washed with buffer A, cells were resuspended in buffer A to an optical density at 578 nm of approximately 20 and sonicated four times for 1 min on ice (Branson cell disrupter B 15; output control, 5; 50% pulsed) under an N2 atmosphere, with the sonications separated by 1-min intervals of cooling. Debris was removed by centrifugation at 3,400 × g for 15 min. The membrane fraction was collected at 200,000 × g for 45 min and washed once with buffer A.
DNA manipulations and cloning of the gene encoding the MQO from H. pylori.
All common molecular biological techniques used have been described previously (25). Preparation of electrocompetent cells and electroporation of C. glutamicum were performed as described previously (30). PCR was performed using chromosomal DNA from H. pylori strain ATCC 49503 as the template, which was prepared by boiling cells for 5 min and removing debris by centrifugation. The oligonucleotides HpF1 (GATAGGGTGCTTGGAATG) and HpR1 (GCATGTAAAGGTTTATCA) were used to amplify a 1.6-kbp fragment containing the entire HP0086 ORF, starting from 125 bases upstream of the ORF to 113 bases downstream of it. The amplification was performed using Pfu polymerase (Promega, Madison, Wis.) and with the following thermocycler parameters: 1 min at 94°C (denaturation), 1 min at 51°C (primer annealing), 2 min at 72°C (extension), 30 cycles. The fragment was ligated into a pBluescript II SK vector opened with EcoRV and transformed in E. coli DH5α. The fragment containing the HP0086 ORF was isolated from this plasmid using the restriction enzymes PstI and SalI. The PstI-SalI fragment was cloned into the E. coli or C. glutamicum shuttle vector pEKEx1. This plasmid was called pHp-mqo. PCR and plasmid isolation were performed to confirm the presence of pHp-mqo in the transformants. Although in pHP-mqo ORF HP0086 is under the control of a lac promoter originating from pEKEx1, this promoter is leaky. The level of MQO activity was very high even in the absence of inducer, both in E. coli and C. glutamicum. Addition of 1 mM IPTG was, for both organisms, deleterious for growth and, in the case of C. glutamicum, for specific activity of MQO. Therefore, all experiments were performed with cells grown in the absence of inducer.
Measurement of MQO, SDH, and NADPH dehydrogenase activities.
MQO, SDH, and NADPH dehydrogenase activities in membrane fragments were measured by absorbance changes of the electron acceptor 2,6-dichlorophenolindophenol (DCPIP) in the presence of a 1 mM concentration of either malate, succinate, or NADPH (22). The NADPH dehydrogenase measurements included correction for the high rate of chemical reduction of DCPIP by NADPH. The existence of a coupled reaction of oxaloacetate reduction with NADH oxidation in isolated membranes of C. glutamicum/pHP-mqo was tested by measuring NADH oxidation at 340 nm in the presence of 10 μM stigmatellin and 1 mM oxaloacetate. These measurements were carried out at room temperature. Oxygen consumption activities in membranes of E. coli and H. pylori were determined in a biological oxygen demand cell equipped with a Clark-type electrode. The temperature of the cell was kept at 30°C in the case of E. coli membranes or at 37°C in the case of H. pylori membranes.
Measurement of malate formation by membranes.
A number of experiments were carried out to determine whether MQO from H. pylori when expressed in C. glutamicum Δmqo also catalyzes oxaloacetate reduction. Membranes were isolated from C. glutamicum Δmqo/pHp-mqo and resuspended at 4 to 4.5 mg of protein ml−1. Of this suspension 200 μl was added to 10 ml of buffer in a 100-ml conical flask and was stirred at 25°C. At time zero, 1 mM oxaloacetate, 10 μM stigmatellin, and 0.2 to 1 mM NADH were added, and 0.5-ml samples were drawn at several time intervals. Each sample was mixed immediately with 20 μl of perchloric acid solution (10 ml of 70% [wt/vol] perchloric acid mixed with 70 ml of water) and left on ice for 1 min. The pH was then neutralized with 2 M KOH. The sample was left on ice for 1 min and then centrifuged for 2 min in an Eppendorf centrifuge at full speed. The malate content of the supernatant was immediately measured by an MDH coupled assay (23).
Protein determination.
Protein was determined with bicinchoninic acid in the presence of 0.5% (mass/vol) sodium dodecyl sulfate, according to a protocol adapted from one provided elsewhere (27).
Sequence data sources and analysis.
Database searches using DNA and protein sequences were performed with the advanced BLAST service at the European Molecular Biology Laboratory searching the GenBank, EMBL, and Swissprot databases or with the BLAST service at the National Center for Biotechnology Information. Protein sequence alignment and clustering analysis were performed at the Multalin server of the Institut National de la Recherche Agronomique (Toulouse, France), using the blosum62 comparison matrix with the penalties for gap opening and gap extension set to 10 and 0, respectively (10). The sources for the MQO sequences were the Genbank, EMBL, and Swissprot databases (sequence accession numbers are as follows: C. glutamicum, O69282; Pseudomonas fluorescens, AF176206; H. pylori, AE000530; Bacillus halodurans, AB013369; E. coli, P33940; Mycobacterium tuberculosis, O05807; Campylobacter jejuni, AL111168), the Neisseria meningitidis Sequencing Group at the Sanger Centre (N. meningitidis, ORF NM0333, preliminary sequence data at the group's website [ftp://ftp.sanger.ac.uk/pub/pathogens/nm]), the Institute for Genomic Research (Staphylococcus aureus, preliminary sequence data at the institute's website [http://www.tigr.org]), and the Pseudomonas Genome Project (Pseudomonas aeruginosa, preliminary sequence data at the project website [http://www.pseudomonas.com]).
RESULTS
Comparison of putative and experimentally established MQO sequences.
Two experimentally established MQO sequences from C. glutamicum and E. coli are known (22; M. E. van der Rest, C. Lange, and D. Molenaar, unpublished data). Furthermore, a number of putative MQO sequences, all of eubacterial origin, can be found in databases. The derived protein sequences, although originating from both gram-positive and gram-negative organisms, form a coherent cluster in an alignment analysis, with 42% or higher identity and 61% or higher similarity (Fig. 1). A distinct feature in these sequences is a putative nucleotide-binding fold close to the N terminus, where FAD is thought to bind (22). The hypothetical protein HP0086 from H. pylori strain 26695 and its homologue in strain J99 are only distantly related to this cluster of MQO sequences, with identity ranging from 15 to 18% and similarity from 31 to 34%. However, this similarity is concentrated in a number of regions distributed over the whole protein. In searches with the position-specific iterated BLAST algorithm (3) the HP0086 sequence of H. pylori was also found to be similar to glycerol-3-phosphate dehydrogenase (GPD) flavoprotein subunits of bacterial origin; for example, it was found to be similar to the GPD subunit B of Haemophilus influenzae (Swissprot accession no. P43800). Close examination of the alignment of HP0086 with these GPD's revealed, however, that the similarity is mainly concentrated in the N-terminal region, which is the putative cofactor-binding region. A gene (Cj0393c) in the genome of Campylobacter jejuni encoding a hypothetical protein with 48% identity to HP0086 is also distantly related to the cluster of MQO sequences.
Cloning, expression, and complementation studies of HP0086.
To demonstrate that the hypothetical protein encoded by ORF HP0086 from H. pylori is an MQO, the ORF was expressed from a plasmid in E. coli Δmqo and C. glutamicum Δmqo strains, which lack MQO activity. This plasmid, called pHP-mqo, was constructed by amplifying ORF HP0086 from H. pylori DNA by PCR and inserting the product in the E. coli or C. glutamicum shuttle vector pEKEx1. In this plasmid the ORF is under the control of a lac promoter. However, because of the leakiness of this promoter, no inducer had to be added to obtain very high expression (see Materials and Methods).
Malate oxidation by MQO in isolated membranes can be measured either by monitoring oxygen consumption or by observing reduction of the artificial acceptor dye DCPIP upon addition of malate. The DCPIP-reducing activity of the MQO from H. pylori in the hosts C. glutamicum and E. coli (Table 2) was very high compared to rates of the endogenous MQO's in E. coli or C. glutamicum wild types (0 to 50 or 100 to 400 nmol min−1 mg of protein−1, respectively [D. Molenaar et al., unpublished data; van der Rest et al., unpublished data]). Also, the oxygen consumption rate measured in membranes of E. coli was very high (Table 2). From the perspective of electron transfer the oxygen reduction rate would be equivalent to a DCPIP reduction rate of 1,820 (2 × 910) nmol min−1 mg of protein−1, which is similar to the measured DCPIP reduction rate. It shows, furthermore, that the MQO from H. pylori is fully coupled to the electron transfer chain of E. coli. d-Malate oxidation (8% of l-malate oxidation) was also observed in membranes from E. coli Δmqo/pHp-mqo but was not measurable in membranes from C. glutamicum Δmqo/pHp-mqo (data not shown).
TABLE 2.
MQO activities measured by DCPIP reduction or oxygen consumption in E. coli and C. glutamicum Δmqo strains expressing ORF HP0086 from a plasmid
| Strain | Activity (nmol min−1 mg of protein−1)
|
|||
|---|---|---|---|---|
| DCPIP reduction
|
O2 consumption
|
|||
| No plasmid | pHp-mqo | No plasmid | pHp-mqo | |
| E. coli Δmqo | <5 | 2,510 | <2 | 910 |
| C. glutamicum Δmqo | <5 | 927 | NDa | ND |
ND, not determined.
To verify that the MQO from H. pylori is also active in vivo, its ability to complement the phenotype of an mqo deletion strain of C. glutamicum was tested. C. glutamicum Δmqo in general grows slowly on several carbon substrates, but most distinctly it is unable to grow on a minimal medium designed for optimal growth of the wild type (D. Molenaar et al., unpublished data). Figure 2 shows this phenotype of C. glutamicum Δmqo and the fact that it can be complemented by expression of ORF HP0086 from plasmid pHp-mqo.
FIG. 2.
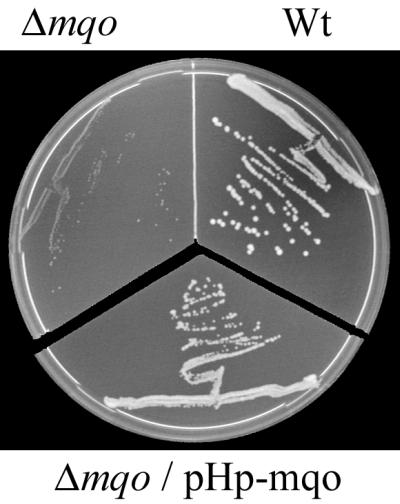
Complementation of the phenotype of an mqo deletion mutant of C. glutamicum by ORF HP0086 expressed from a plasmid. The C. glutamicum wild-type (Wt), Δmqo, and Δmqo/pHp-mqo strains were plated on minimal medium agar containing 1% (wt/vol) glucose. In the case of strain Δmqo/pHp-mqo the plate also contained 25 μg of kanamycin ml−1. The plates were incubated for 60 h at 30°C.
Evidence that MQO from H. pylori does not catalyze oxaloacetate reduction.
Two considerations motivated us to study whether the MQO of H. pylori, in addition to participating in malate oxidation, also catalyzes oxaloacetate reduction and would consequently interconvert malate and oxaloacetate reversibly. First, it might explain the observation of apparent MDH activity in assays in which oxaloacetate reduction was observed (17, 24). Second, it would indicate whether MQO is involved only in an oxidative citric acid cycle or might also be involved in the reductive branch of a branched citric acid cycle. An important factor in reversibility would be the nature of the quinone acceptor. The standard redox potential of ubiquinone redox couples would in general be too high (E0′ = +113 mV) (28) to reduce oxaloacetate to malate (E0′ = −172 mV). On the other hand, menaquinones, having a much lower redox potential (E0′ = −74 mV), may be able to reduce oxaloacetate. As H. pylori contains only menaquinones (20), reversibility of the MQO reaction could not be excluded beforehand. The MQO activities of membranes from H. pylori were expected to be too low to be able to detect malate formation. Therefore, it was decided to study this reaction with membranes from C. glutamicum Δmqo in which the MQO of H. pylori was expressed. C. glutamicum seems to be the ideal host, since it also possesses only menaquinones (9). The experiments were designed to induce the net reduction of oxaloacetate by NADH through the subsequent action of NADH:quinone oxidoreductase and MQO. The competing oxidation of NADH by oxygen was prevented by inhibiting the electron transfer chain with stigmatellin downstream of the dehydrogenases at the level of the cytochrome bc1 complex (22). In one type of experiment the NADH oxidation was observed by changes of absorbance at 340 nm. Membranes prepared from C. glutamicum/pHp-mqo oxidized NADH at a rate of 615 nmol min−1 mg of protein−1. This rate was inhibited by 68% by addition of 10 μM stigmatellin. Upon addition of oxaloacetate no stimulation of the inhibited NADH oxidation was observed, as might have been expected should electron transfer from NADH to oxaloacetate have taken place.
In a second experiment with the same membranes the formation of malate was tested at different time points after addition of oxaloacetate. No malate formation could be observed in the samples up to 2 h after the start of the experiment. On the other hand 50 μM malate was readily measured in control samples to which malate had been added before the reaction was stopped with perchloric acid. From these experiments it has to be concluded that MQO of H. pylori does not catalyze the reduction of oxaloacetate.
Dehydrogenase activities in membranes isolated from H. pylori.
To detect MQO activity by the dye reduction assay it is essential to isolate and wash the membranes in order to exclude contamination of the membrane preparation with cytoplasmic MDH and NAD or NADP. In principle, MDH, its cofactor NAD, and a membrane-associated NADH dehydrogenase might together also catalyze malate-dependent dye reduction. Considering the possibility that H. pylori also possesses an MDH, washed membranes of this organism were used for detection of enzyme activities. In such membranes MQO activity is readily detected by using DCPIP as an electron acceptor (Table 3). The route of electrons in this assay is unclear, but it probably leads from the enzyme either directly or via quinones to DCPIP. The malate-dependent DCPIP reduction rate catalyzed by H. pylori membranes could be stimulated by 30 to 50% by the addition of 60 μM ubiquinone-1. This suggests that quinones play, at least in part, an intermediary role in the reduction of the dye. The MQO activity in H. pylori membranes is in general low compared to, e.g., rates measured in membranes from wild-type C. glutamicum, which are on the order of 100 to 400 nmol min−1 mg of protein−1 (22). Relatively low dehydrogenase activities, however, seem to be generally the case for membranes of H. pylori (see also references 5 and 7) and may be connected with its slow growth or with partial inactivation of the electron transfer chain during membrane isolation.
TABLE 3.
Dehydrogenase activities in membrane fragments isolated from different batches of H. pylori cellsa
| Batch | OD at harvest | DCPIP-reducing activity (nmol min−1 mg protein−1)
|
||
|---|---|---|---|---|
| MQO | NADPH dehydrogenase | SDH | ||
| 1 | 0.33 | 33.7 | 14.0 (42) | 8.4 (25) |
| 2 | 0.33 | 45.1 | 46.0 (102) | 8.9 (20) |
| 3 | 1.1 | 55.8 | 20.1 (36) | 13.0 (23) |
| 4 | 0.65 | 67.0 | 6.1 (9) | 11.5 (17) |
| 5 | 0.54 | 69.6 | 0.0 (0) | 19.2 (28) |
| 6 | 1.0 | 106.6 | 84.6 (79) | 15.9 (15) |
| 7 | 1.0 | 136.9 | 43.6 (32) | 25.9 (19) |
Rows are arranged in order of ascending MQO activity. The numbers in parentheses are percentages showing the level of activity relative to that of MQO. OD, optical density at 578 nm.
Analyzing several batches for membrane-bound dehydrogenase activities by the DCPIP assay indicated that MQO and SDH activities are positively correlated. With Spearman's rank correlation test the absence of correlation was rejected when P was <0.01. The SDH activity equals 21% ± 4.5% (mean ± standard deviation of the sample) of the MQO activity. This correlation might be expected if both enzymes operate in the citric acid cycle. The NADPH dehydrogenase activity is not correlated with MQO or SDH activities. NADH dehydrogenase activity was not measurable with DCPIP as the acceptor. This is in accordance with the low NADH dehydrogenase activities determined by oxygen consumption rates (5).
In sharp contrast to the results obtained in experiments with E. coli membranes containing H. pylori MQO (Table 2), we could detect only a very low, but significant, malate-dependent O2 consumption rate of 7.5 nmol min−1 mg of protein−1 by membranes of H. pylori. Furthermore, this activity was detected only in the presence of a 60 μM concentration of the redox mediator ubiquinone-1. In the same preparation the rate of DCPIP reduction by MQO was 136.9 nmol min−1 mg of protein−1 (batch 6 in Table 3). In an experiment with batch 1 an oxygen consumption rate of 4.3 nmol min−1 mg of protein−1 was determined. If, as in the E. coli membranes, the rates of transfer of electrons from MQO into the electron transfer chain had been comparable to DCPIP reduction rates, an oxygen consumption rate of approximately 50 to 70 or 10 to 15 nmol min−1 mg of protein−1 might have been expected with batch 6 or 1, respectively. Since respiration is dependent on the integrity not only of the MQO but also of the subsequent redox enzymes, a possible explanation for the low rate of oxygen consumption may be that other components of the electron transfer chain are inactivated during membrane isolation.
DISCUSSION
It has been suggested before by others that H. pylori might possess MQO activity (13, 20) (MQO was referred to as dye-linked malate dehydrogenase). However, the experimental details underlying this assertion were not published. The results presented in this paper clearly show that H. pylori does possess MQO and that it is encoded by the HP0086 ORF. This also implies that an MQO previously detected in C. jejuni is probably encoded by the gene Cj0393c (16). It is apparent from Fig. 1 and the alignment analysis that the H. pylori and C. jejuni MQOs form a separate group of MQOs. Like all other MQO sequences, H. pylori and C. jejuni MQOs contain a conserved hydrophobic sequence at the N terminus in the proximity of the αβα motif of a putative Rossmann fold involved in binding the ADP moiety of the FAD cofactor (22, 31). Most MQOs can easily be dissociated from the membrane by washing with chelators or low concentrations of detergent (21). The enzyme seems to be loosely associated to the membrane by hydrophobic patches or ionic interactions. In accordance with this, the amino acid sequence contains no hydrophobic stretches which could form a transmembrane helical anchor. Preliminary data obtained with H. pylori MQO solubilized with detergent show that the enzyme is activated by adding FAD but not flavin mononucleotide or NAD or NADP (B. Kather, unpublished results). The native membrane-associated enzyme shows no such dependency, probably because FAD is tightly bound in this conformation.
The presence of an MQO in H. pylori cannot explain the results obtained by others indicating that this organism possesses an MDH (17, 24). The common MDH assay, also used by these authors, is based on the measurement of NAD reduction or NADH oxidation in the presence of malate or oxaloacetate. MQO, however, does not donate electrons to or accept them from NAD or NADH, neither directly nor, as was shown above, by mediation of the electron transfer chain. Thus, the question remains what the meaning of these observations is. In one case the observed MDH activity was very low compared to the activities of the other citric acid cycle enzymes (17). In the other case higher activities were detected (24). Additionally, the conversion of malate to oxaloacetate was observed by proton nuclear magnetic resonance. This latter observation would, however, also be compatible with MQO activity. In view of the fact that no mdh gene can be found in the genome of H. pylori, the question of whether H. pylori possesses, in addition to an MQO, a possibly new type of NAD-dependent MDH can only be answered clearly by purification of such an enzyme. Raw cell extracts contain metabolites and also contain many enzymes that use NAD as a cofactor. Thus, one runs the risk of observing artifacts instead of the supposed enzyme activities.
One of the reasons for organisms to use an MQO for malate oxidation might be that the oxidation of malate by an NAD-dependent MDH has a very unfavorable standard free energy difference (ΔG°′ = +28.5 kJ mol−1). In contrast, the oxidation of malate by MQO has a very favorable standard free energy difference (ΔG°′ = −18.5 kJ mol−1 with menaquinone as the electron acceptor) (22). This difference should allow MQO to oxidize malate under circumstances where an MDH may not be able to do so, for example, when cytoplasmic [oxaloacetate]/[malate] or [NADH]/[NAD] ratios are high. Most MDHs are, when assayed under the right circumstances, capable of both malate oxidation and oxaloacetate reduction. However, as was shown above, MQO from H. pylori does not catalyze the reduction of oxaloacetate. This implies that MQO can only operate in the oxidative direction, which consequently strongly suggests that the citric acid cycle of H. pylori should, at least under some circumstances, operate oxidatively. It is an argument against a purely reductive function of the left branch (MDH or MQO, fumarase, and fumarate reductase or succinate dehydrogenase) (Fig. 3) of the citric acid cycle, as was asserted by others (24). If MQO were the only malate dehydrogenase in H. pylori, which at present seems uncertain, it would even be incompatible with such a function of the left branch.
FIG. 3.

Tentative scheme for the citric acid cycle of H. pylori based on genome sequence data and biochemical data. Unusual enzymes are labeled with an asterisk. Pyr, pyruvate; AcCoA, acetyl-CoA; Cit, citrate; Icit, isocitrate; Kg, 2-ketoglutarate; Suc-CoA, succinyl-CoA; Suc, succinate; Fum, fumarate; Mal, malate; Oaa, oxaloacetate; Fdox and Fdred: oxidized and reduced ferredoxin, respectively; MQ and MQH2, oxidized and reduced menaquinone, respectively. Enzymes: 1, pyruvate:ferredoxin oxidoreductase; 2, citrate synthase; 3, aconitase; 4, isocitrate dehydrogenase; 5, α-ketoglutarate:ferredoxin oxidoreductase; 6, succinyl-CoA acetoacetyl-CoA transferase; 7, fumarate reductase (SDH); 8, fumarase; 9, malate:quinone oxidoreductase. EC numbers and corresponding H. pylori genes of all enzymes except the MQO can be found elsewhere (19).
Figure 3 shows a scheme of the proposed citric acid cycle of H. pylori. For all enzymes displayed, corresponding genes are found in the chromosome of H. pylori. In this scheme the conversion of succinyl-CoA to succinate by succinyl-CoA:acetoacetyl-CoA transferase is dependent on the continuous supply of acetoacetate and degradation of acetoacetyl-CoA (11). Alternatively, a continuous regeneration of acetoacetate from acetoacetyl-CoA may take place, possibly in reactions generating metabolic energy. It is also possible that a succinyl-CoA hydrolase (EC 3.1.2.3), for which no gene is presently known, catalyzes the hydrolysis of succinyl-CoA. As can be seen, the generation of NADH is avoided in central metabolism of H. pylori. For example, the pyruvate and α-ketoglutarate dehydrogenase of H. pylori are flavodoxin and ferredoxin dependent instead of NAD dependent (18). Consequently, the main pyridine nucleotide dehydrogenase activity of H. pylori is an NADPH dehydrogenase instead of an NADH dehydrogenase. The presence of an MQO instead of an NAD-dependent MDH would be in accordance with this fact, whereas an NAD-dependent MDH with a role in oxidative phosphorylation would constitute an exception to this scheme.
ACKNOWLEDGMENTS
Ubiquinone-1 was a kind gift from Hoffman-La Roche.
This research was funded by the German Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie (project 0316712) and the Fonds der Chemischen Industrie.
B.K. and K.S. contributed equally to the paper.
REFERENCES
- 1.Abe S, Takayama K, Kinoshita S. Taxonomical studies on glutamic acid producing organisms. J Gen Appl Microbiol. 1967;13:279–301. [Google Scholar]
- 2.Alm R A, Ling L S, Moir D T, King B L, Brown E D, Doig P C, Smith D R, Noonan B, Guild B C, de Jonge B L, Carmel G, Tummino P J, Caruso A, Uria-Nickelsen M, Mills D M, Ives C, Gibson R, Merberg D, Mills S D, Jiang Q, Taylor D E, Vovis G F, Trust T J. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- 3.Altschul S F, Madden T L, Schaffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casadaban M J. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and mu. J Mol Biol. 1976;104:541–555. doi: 10.1016/0022-2836(76)90119-4. [DOI] [PubMed] [Google Scholar]
- 5.Chang H T, Marcelli S W, Davison A A, Chalk P A, Poole R K, Miles R J. Kinetics of substrate oxidation by whole cells and cell membranes of Helicobacter pylori. FEMS Microbiol Lett. 1995;129:33–38. doi: 10.1016/0378-1097(95)00130-W. [DOI] [PubMed] [Google Scholar]
- 6.Charnock C. Structural studies of malate dehydrogenases (MDHs): MDHs in Brevundimonas species are the first reported MDHs in Proteobacteria which resemble lactate dehydrogenases in primary structure. J Bacteriol. 1997;179:4066–4070. doi: 10.1128/jb.179.12.4066-4070.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen M, Andersen L P, Zhai L, Kharazmi A. Characterization of the respiratory chain of Helicobacter pylori. FEMS Immunol Med Microbiol. 1999;24:169–174. doi: 10.1111/j.1574-695X.1999.tb01278.x. [DOI] [PubMed] [Google Scholar]
- 8.Cohn D V. The enzymatic formation of oxalacetic acid by nonpyridine nucleotide malic dehydrogenase of Micrococcus lysodeikticus. J Biol Chem. 1958;233:299–304. [PubMed] [Google Scholar]
- 9.Collins M D, Pirouz T, Goodfellow M, Minnikin D E. Distribution of menaquinones in actinomycetes and corynebacteria. J Gen Microbiol. 1977;100:221–230. doi: 10.1099/00221287-100-2-221. [DOI] [PubMed] [Google Scholar]
- 10.Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988;16:10881–10890. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corthesy-Theulaz I E, Bergonzelli G E, Henry H, Bachmann D, Schorderet D F, Blum A L, Ornston L N. Cloning and characterization of Helicobacter pylori succinyl CoA:acetoacetate CoA-transferase, a novel prokaryotic member of the CoA-transferase family. J Biol Chem. 1997;272:25659–25667. doi: 10.1074/jbc.272.41.25659. [DOI] [PubMed] [Google Scholar]
- 12.Cover T L, Dooley C P, Blaser M J. Characterization of and human serologic response to proteins in Helicobacter pylori broth culture supernatants with vacuolizing cytotoxin activity. Infect Immun. 1990;58:603–610. doi: 10.1128/iai.58.3.603-610.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davison A A, Kelly D J, White P J, Chalk P A. Citric-acid cycle enzymes and respiratory metabolism in Helicobacter pylori. Acta Gastro-Enterol Belg. 1993;56S:96. [Google Scholar]
- 14.Doig P, de Jonge B L, Alm R A, Brown E D, Uria-Nickelsen M, Noonan B, Mills S D, Tummino P, Carmel G, Guild B C, Moir D T, Vovis G F, Trust T J. Helicobacter pylori physiology predicted from genomic comparison of two strains. Microbiol Mol Biol Rev. 1999;63:675–707. doi: 10.1128/mmbr.63.3.675-707.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eikmanns B J, Kleinertz E, Liebl W, Sahm H. A family of Corynebacterium glutamicum/Escherichia coli shuttle vectors for cloning, controlled gene expression, and promoter probing. Gene. 1991;102:93–98. doi: 10.1016/0378-1119(91)90545-m. [DOI] [PubMed] [Google Scholar]
- 16.Hoffman P S, Goodman T G. Respiratory physiology and energy conservation efficiency of Campylobacter jejuni. J Bacteriol. 1982;150:319–326. doi: 10.1128/jb.150.1.319-326.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffman P S, Goodwin A, Johnsen J, Magee K, Veldhuizen van Zanten S J O. Metabolic activities of metronidazole-sensitive and -resistant strains of Helicobacter pylori: repression of pyruvate oxidoreductase and expression of isocitrate lyase activity correlate with resistance. J Bacteriol. 1996;178:4822–4829. doi: 10.1128/jb.178.16.4822-4829.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hughes N J, Clayton C L, Chalk P A, Kelly D J. Helicobacter pylori porCDAB and oorDABC genes encode distinct pyruvate:flavodoxin and 2-oxoglutarate:acceptor oxidoreductases which mediate electron transport to NADP. J Bacteriol. 1998;180:1119–1128. doi: 10.1128/jb.180.5.1119-1128.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huynen M A, Dandekar T, Bork P. Variation and evolution of the citric acid cycle: a genomic perspective. Trends Microbiol. 1999;7:281–291. doi: 10.1016/s0966-842x(99)01539-5. [DOI] [PubMed] [Google Scholar]
- 20.Kelly D J. The physiology and metabolism of the human gastric pathogen Helicobacter pylori. Adv Microb Physiol. 1998;40:137–189. doi: 10.1016/s0065-2911(08)60131-9. [DOI] [PubMed] [Google Scholar]
- 21.Molenaar D, van der Rest M E, Frank C, Yücel R, Petrović S. Malate:quinone oxidoreductase. A membrane-associated malate dehydrogenase. Biochim Biophys Acta EBEC Rep. 1998;10:93. [Google Scholar]
- 22.Molenaar D, van der Rest M E, Petrović S. Biochemical and genetic characterization of the membrane-associated malate dehydrogenase (acceptor) (EC 1.1.99.16) from Corynebacterium glutamicum. Eur J Biochem. 1998;254:395–403. doi: 10.1046/j.1432-1327.1998.2540395.x. [DOI] [PubMed] [Google Scholar]
- 23.Möllering H. l-(−)-Malate. Determination with malate dehydrogenase and aspartate transaminase. In: Bergmeyer H U, Bergmeyer J, Grassl M, editors. Methods of enzymatic analysis. Weinheim, Germany: VCH Verlagsgesellschaft; 1985. pp. 39–47. [Google Scholar]
- 24.Pitson S M, Mendz G L, Srinivasan S, Hazell S L. The tricarboxylic acid cycle of Helicobacter pylori. Eur J Biochem. 1999;260:258–267. doi: 10.1046/j.1432-1327.1999.00153.x. [DOI] [PubMed] [Google Scholar]
- 25.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 26.Short J M, Fernandez J M, Sorge J A, Huse W D. Lambda ZAP: a bacteriophage lambda expression vector with in vivo excision properties. Nucleic Acids Res. 1988;16:7583–7600. doi: 10.1093/nar/16.15.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith P K, Krohn R I, Hermanson G T, Mallia A K, Gartner F H, Provenzano M D, Fujimoto E K, Goeke N M, Olsen B J, Klenk D C. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 28.Thauer R K, Jungermann K, Decker K. Energy conservation in chemotrophic anaerobic bacteria. Bacteriol Rev. 1977;41:100–180. doi: 10.1128/br.41.1.100-180.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomb J F, White O, Kerlavage A R, Clayton R A, Sutton G G, Fleischmann R D, Ketchum K A, Klenk H P, Gill S, Dougherty B A, Nelson K, Quackenbush J, Zhou L, Kirkness E F, Peterson S, Loftus B, Richardson D, Dodson R, Khalak H G, Glodek A, McKenney K, Fitzegerald L M, Lee N, Adams M D, Hickey E K, Berg D E, Gocayne J D, Utterback T R, Peterson J D, Kelley J M, Cotton M D, Weidman J M, Fuji C, Bowman C, Watthey L, Wallin E, Hayes W S, Borodovsky M, Karp P D, Smith H O, Fraser C M, Venter J C. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 30.van der Rest M E, Lange C, Molenaar D. A heat shock following electroporation induces highly efficient transformation of Corynebacterium glutamicum with xenogeneic plasmid DNA. Appl Microbiol Biotechnol. 1999;52:541–545. doi: 10.1007/s002530051557. [DOI] [PubMed] [Google Scholar]
- 31.Wierenga R K, Terpstra P, Hol W G J. Prediction of the occurrence of the ADP-binding beta-alpha-beta-fold in proteins, using an amino acid sequence fingerprint. J Mol Biol. 1986;187:101–107. doi: 10.1016/0022-2836(86)90409-2. [DOI] [PubMed] [Google Scholar]