

Abstract
Bile acids (BAs) are the major components of bile and products of cholesterol metabolism. Cholesterol is catalyzed by a variety of enzymes in the liver to form primary BAs, which are excreted into the intestine with bile, and secondary BAs are formed under the modification of the gut microbiota. Most of the BAs return to the liver via the portal vein, completing the process of enterohepatic circulation. BAs have an important role in the development of hepatocellular carcinoma (HCC), which may participate in the progression of HCC by recognizing receptors such as farnesoid X receptor (FXR) and mediating multiple downstream pathways. Certain BAs, such as ursodeoxycholic acid and obeticholic acid, were indicated to be able to delay liver injury and HCC progression. In the present review, the structure and function of BAs were introduced and the metabolism of BAs and the process of enterohepatic circulation were outlined. Furthermore, the mechanisms by which BAs participate in the development of HCC were summarized and possible strategies for targeting BAs and key sites of their metabolic processes to treat HCC were suggested.
Keywords: bile acid, farnesoid x receptor, hepatocellular carcinoma, enterohepatic circulation, ursodeoxycholic acid, obeticholic acid
1. Introduction
Liver cancer is a common malignancy. In recent years, the incidence rate and mortality rate of liver cancer have been rising. Primary liver cancer was the sixth most commonly diagnosed cancer type and the third leading cause of cancer-associated death in the world in 2020 (1). Primary liver cancer includes hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma, as well as other rare types; HCC accounts for 75-85% of cases (1). Risk factors for HCC include chronic hepatitis B and C, alcohol addiction, metabolic liver disease [particularly non-alcoholic fatty liver disease (NAFLD)] and exposure to dietary toxins, such as aflatoxin and carmine acid (2). Liver cancer is an advanced outcome of a range of liver diseases. NAFLD and non-alcoholic steatohepatitis (NASH) are increasingly recognized as important underlying causes of HCC (3). Genetic predisposition, interactions between viral and nonviral risk factors, the cellular microenvironment and various immune cells, as well as the severity of an underlying chronic liver disease, among others, are at the origin of the early steps in the malignant transformation of hepatocytes and development of HCC, whereas an altered microenvironment is a key contributing feature of cancer and is involved in all stages of malignant progression (4).
Bile acids (BAs) are attracting increasing attention from researchers and this field has developed rapidly. Hepatic accumulation of BAs is central to the pathogenesis of cholestasis-induced liver injury and excessive cytotoxic BAs in the liver may lead to liver fibrosis and cirrhosis, and even liver cancer (5). The interest in the role of BAs in HCC has increased and there is increasing evidence that BAs have a role in HCC. In the present review, the biosynthesis, metabolism and transport of BAs are presented and the mechanistic links between BAs and HCC, as well as the opportunities of targeting BAs for the prevention or treatment of HCC are discussed.
2. BAs
The human understanding of BAs dates back to nearly 3,000 years ago, when animal bile was widely used in Traditional Chinese Medicine (6). Since the 19th century, BAs have become the subject of detailed research by scientists (6). BAs, the main lipid components of bile, are a general term for a class of cholanic acids that are converted from cholesterol in hepatocytes through a series of enzymatic reactions (7). In general, BAs have steroid cores and four fused hydrocarbon rings with polar hydroxyl functional groups. Three hydroxyl and carboxyl groups face one side of the carbon skeleton, forming a hydrophilic surface, in contrast to a highly hydrophobic surface (7) (Fig. 1). Thus, BAs are amphipathic molecules with powerful detergent properties.
Figure 1.
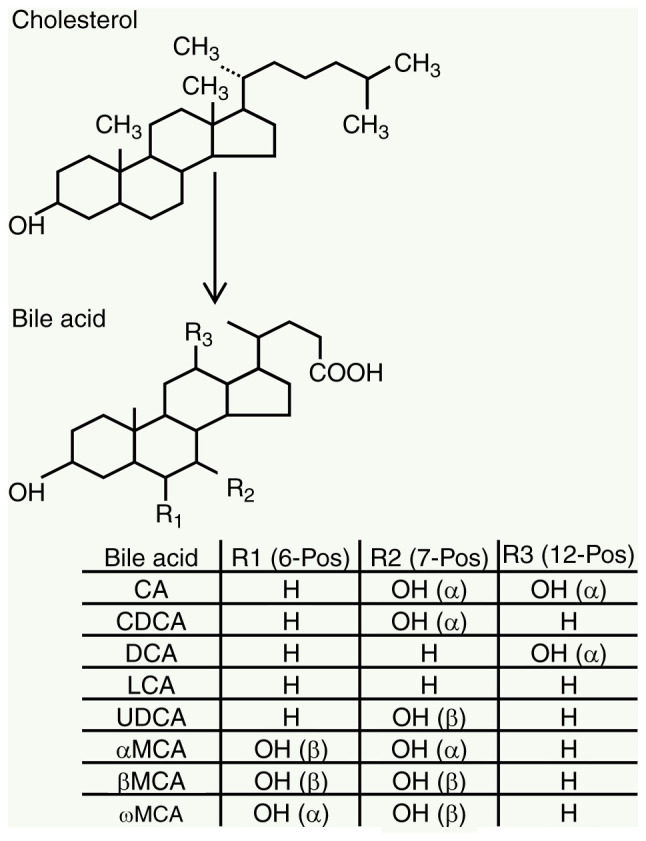
Structure of cholesterol and bile acids [refs. (6-8)]. CA, cholic acid; UDCA, ursodeoxycholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; LCA, lithocholic acid; MCA, muricholic acid.
According to the source of BAs, they may be divided into primary BAs and secondary BAs. In hepatocytes, BAs synthesized directly from cholesterol are called primary BAs, which include cholic acid (CA) and chenodeoxycholic acid (CDCA). After primary BA synthesis, most BAs bind to glycine or taurine, changing from a free form to a binding form; BAs form sodium salts at physiological pH values, which increases their solubility (8). Primary BAs enter the intestine and are converted to secondary BAs, mainly deoxycholic acid (DCA) and trace amounts of lithocholic acid (LCA), through the enzymatic activity of intestinal bacteria (7). These total BAs circulate in the enterohepatic circulation of the human body, including those in the liver (<1%), intestine (85-90%) and gallbladder (10-15%), constituting the BA pool (9). The human BA pool consists of CA (40%), CDCA (40%) and DCA (20%), in which the ratio of glycine (G)-/taurine (T)-conjugated BAs is 3:1, and the BA pool has high hydrophobicity (9). Due to the different kinds of enzymes and metabolic pathways, the BAs in mice have other types in addition to those mentioned above, mainly muricholic acids (MCAs) (9).
BA synthesis
The synthesis and secretion of BAs are the main pathways of cholesterol catabolism in the human body. Cholesterol is eventually converted into water-soluble and easily excreted BAs. BA formation is complex, including several reaction steps catalyzed by at least 17 different enzymes, one or more transporters and multiple cellular compartments, which include the cytosol, endoplasmic reticulum (ER), mitochondria and peroxisomes (6). BA synthesis occurs in the liver, which is the only organ that possesses all the enzymes required for BA synthesis (9). BA synthesis is divided into the classical and the alternative pathway, which are initiated by the microsomal cytochrome P450 (CYP) enzymes cholesterol 7α-hydroxylase (CYP7A1) and mitochondrial sterol 27-hydroxylase (CYP27A1), respectively (Fig. 2). In humans, the classical pathway of BA synthesis accounts for at least 75% of total BA production and is considered to be the main pathway of BA biosynthesis (10). Primary BAs are amino-conjugated at the carboxyl group, with a ratio of glycine to taurine conjugates of approximately 3:1 (11). BAs may also bind to sulfate or glucuronic acid to form fully ionized, negatively charged hydrophilic polar molecules, and these bound forms of BAs subsequently discharge into the intestine with bile. The step of binding BAs to amino acids is markedly efficient and >98% of the BAs secreted into bile are in taurineor glycine-conjugated forms (6). In the intestine, the bound form of primary BAs undergoes dissociation and dehydroxylation to produce secondary BAs. The primary BAs synthesized in the liver of mice also include αMCA and βMCA, and ωMCA may be produced by gut microbial 7α/β-epimerization in the intestine of mice (10).
Figure 2.
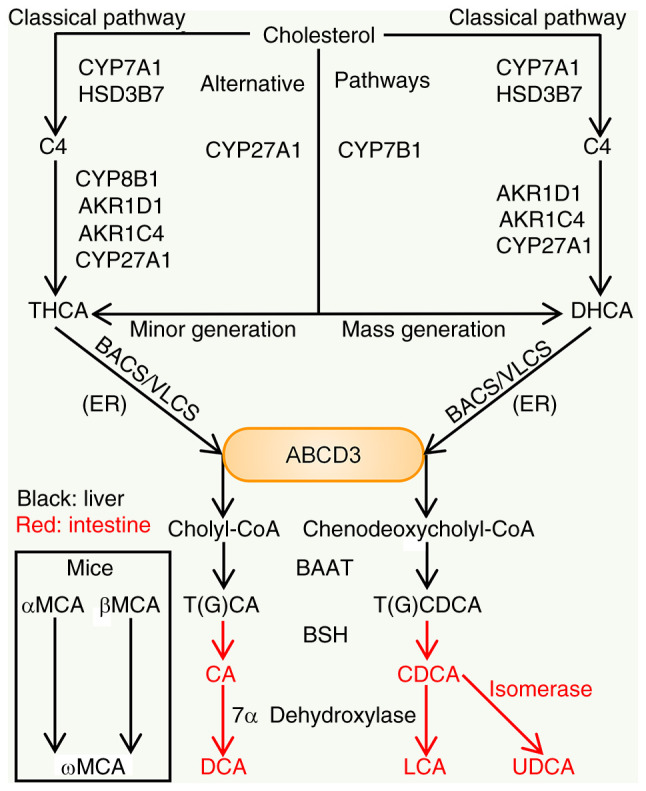
Processes of BA synthesis. In the liver, the classical pathway is initiated by CYP7A1, the rate-limiting enzyme. CYP7A1 and HSD3B7 are able to convert cholesterol to form C4. CYP8B1 performs 12α-hydroxylation of C4. Subsequently, under the catalysis of AKR1D1, AKR1C4 and CYP27A1, THCA is generated. However, without 12α-hydroxylation, C4 is converted to DHCA. BACS or VLCS in the ER then ligate Co-A to the carboxyl groups. After transport by peroxisomal transporter ABCD3 and catalysis by a series of enzymes, THCA and DHCA synthesize cholyl-CoA and chenodeoxycholyl-CoA. These two substances are then conjugated to taurine or glycine by BAAT. The alternative pathway is mainly initiated by CYP27A1. Next, through CYP7B1 and other enzymes that belong to CYP proteins, cholesterol is being subjected to modifications to finally generate CDCA and a small amount of CA. Certain conjugated forms of BAs entering the intestine may be dissociated by BSH and bacterial 7α dehydroxylase may then convert CA and CDCA into DCA and LCA, respectively. CDCA may also be isomerized to UDCA. In mice, the generation of MCA was observed in addition to the above synthetic processes [Refs. (6-8,12)]. BA, bile acid; BSH, BA hydrolase; HSD3B7, 3β-hydroxy-5-C27-steroid dehydrogenase; C4, 7α-hydroxy-4-cholesten-3-one; CYP, cytochrome P450; CYP8B1, sterol 12α-hydroxylase; CYP7B1, nonspecific oxysterol 7α-hydroxylase; AKR1D1, aldo-keto reductase family 1 member D1; BAAT, BA-CoA:amino acid N-acyltransferase; THCA, 3α,7α,12α-trihydroxy-5β cholestanoic acid; DHCA, 3α,7α-dihydroxy-5β cholestanoic acid; BACS, BA-Co-A synthase; VLCS, very long-chain Co-A synthase; ER, endoplasmic reticulum; CA, cholic acid; CDCA, chenodeoxycholic acid; UDCA, ursodeoxycholic acid; DCA, deoxycholic acid; LCA, lithocholic acid; MCA, muricholic acid; ABCD3, ATP binding cassette subfamily D member 3.
Enterohepatic circulation
The enterohepatic circulation of BAs refers to the system in which BAs are synthesized by the liver, discharged into the intestine with bile and then reabsorbed in the intestine and returned to the liver via the portal vein. BAs are synthesized in the liver and secreted by the canalicular membrane transporters, of which the most important is the bile salt export pump [BSEP/ATP binding cassette (ABC) subfamily B member 11] (12,13). In addition, BAs conjugated with sulfate or glucuronic acid become dianionic compounds and may be transported by multidrug resistance-associated protein 2 (MRP2) on the canalicular membrane (14). In addition, MRP3 and MRP4, located on the basolateral side of hepatocytes, are considered compensatory BA efflux transporters and they mainly act when bile excretion by BSEP is impaired (15).
BAs are stored in the gallbladder and released into the intestine after a meal. In the small intestine, a small proportion of free BAs are reabsorbed into intestinal epithelial cells via passive diffusion in the small intestine and colon (16). However, the majority of conjugated BAs can hardly be absorbed in the proximal small intestine, and at the end of the ileum, they are mainly actively and effectively reabsorbed into intestinal epithelial cells by the apical sodium-dependent BA transporter [ASBT/solute carrier family 10 member 2 (SLC10A2)] of the apical membrane (16). Subsequently, BAs bind to intestinal BA binding protein and are transported to the basement membrane for secretion (17). Although the absorption of BAs in the terminal ileum is effective, certain molecules escape and reach the large intestine. BAs entering the colon undergo modifications to produce secondary BAs. BAs in the small and large intestine may be reabsorbed by the heterodimer organic solute transporter α/β at the terminal lumen of the basement membrane and transported back to the liver (18). A small number of BAs that escape absorption may pass into the colon to be eliminated in the feces.
The last step in the enterohepatic circulation is the absorption of BAs from the portal vein by hepatocytes through transporters. Sodium-taurocholate co-transporting polypeptide (NTCP/SLC10A1) and organic anion transporting polypeptides are the main transporters involved in this process (12). Hepatocytes reprocess BAs and secrete them along with the bile to complete enterohepatic circulation. BAs that are not re-ingested by hepatocytes spread to the systemic circulation and may eventually be excreted through the kidney (19) (Fig. 3).
Figure 3.

Enterohepatic circulation of BAs. BA, bile acid; BSEP, bile salt export pump; MRP2, multidrug resistance-associated protein 2; ASBT, apical sodium-dependent BA transporter; IBABP, intestinal BA binding protein; OST, organic solute transporter; NTCP, sodium-taurocholate co-transporting polypeptide; OATP, organic anion transporting polypeptide.
The enterohepatic circulation of BAs occurs 6-8 times a day on average to maintain a constant BA pool size (~3 g) (20,21). The enterohepatic circulation of BAs is highly efficient, with ~95% of the BAs reabsorbed in the ileum and only 5% of the BAs being lost in the feces (22,23). The full elucidation of BA synthesis and transport regulators in the enterohepatic circulation may provide potential targets for drug treatment of cholestatic liver diseases (20).
Functions of BA
BAs have a variety of physiological roles, the most important of which is to promote the digestion and absorption of lipids (11). BA molecules have hydrophilic and hydrophobic sides in their configuration, which endows them with strong interfacial activity to reduce the surface tension between oil and water phases and promote lipid emulsification. At the same time, BAs enlarge the contact surface between lipids and lipase and accelerate the digestion of lipids (11,24). BAs may also inhibit the precipitation of cholesterol in bile and prevent the formation of cholesterol stones. Cholesterol is poorly soluble in water and must be incorporated into lecithin-bile salts to be transported through the biliary tract into the small intestine without precipitation (25).
BAs recognize a variety of receptors and mediate downstream signals. The main BA-mediated nuclear receptors are farnesoid X receptor (FXR), pregnane X receptor and vitamin D receptor. FXR, the first identified BA receptor, is essentially a BA-binding transcription factor that functions by triggering transcriptional changes (26). FXR is mainly expressed in the intestine and liver, and the order of binding potency of BAs to FXR is CDCA>LCA=DCA>CA (27,28). Furthermore, the binding activity is different under different physiological conditions. FXR signaling may have multiple physiological roles, such as negatively feedback-regulating BA synthesis, regulating BA transport and regulating energy metabolism and immune responses (29). In terms of cell membrane receptors, BAs mainly recognize Takeda G protein-coupled receptor 5 (TGR5, also known as GPBAR1) (30). In addition to TGR5, BA-activated G-protein-coupled receptors also include sphingosine-1-phosphate receptor 2 (31). Studies have indicated that TGR5 is highly expressed in liver cells other than hepatocytes, including Kupffer cells and cholangiocytes, and in gallbladder epithelial cells and immune cells (32). TGR5 is being dose-dependently activated by BAs, with the following rank order of potency: LCA≥DCA>CDCA>CA (30). FXR and TGR5 are the two most important receptors for BA mediation, whose signals provide crosstalk between the intestine and the liver. BAs-FXR and BAs-TGR5 signals are widely involved in the pathogenesis of HCC. Therefore, the development of FXR and TGR5 modulators may provide therapeutic interventions for HCC.
3. BAs and HCC
Relationship between alterations of BAs and HCC
The delicate connection between BAs and HCC is gradually being confirmed experimentally. In particular, an imbalance between nontoxic hydrophilic BAs and toxic hydrophobic BAs occurs when BA transporter expression is downregulated for various reasons and the accumulation of toxic BAs drives HCC progression (33,34). A retrospective cohort study from 2004 to 2014 including 2,262 patients with chronic hepatitis B on conventional antiviral therapy indicated that persistent elevation of serum total BAs was an independent risk factor for HCC (35). However, the result of another study was that conjugated primary BAs were significantly elevated, whereas the ratios of secondary BAs over primary BAs were significantly lower in HCC cases than in controls (36). The doubling ratio of taurine-over glycine-conjugated CDCA was significantly associated with a 40% increased risk of HCC, whereas the doubling ratio of secondary over primary BAs was associated with a 30-40% reduced risk of HCC (36). In addition, a weighted relative difference accumulation algorithm study suggested that patients with hepatitis and cirrhosis had increased serum levels of G-CDCA, G-CA and T-CA and decreased serum levels of CDCA (37). After 0.2% CA treatment, diethylnitrosamine (DEN)-induced liver tumors in mice increased by three-fold in number and size, and the mRNA levels of TNF-α and IL-1β were significantly increased (38). G-CDCA promotes the survival of HepG2 and QGY-7703 liver cancer cells by activating antiapoptotic genes such as Bcl-2; furthermore, G-CDCA was able to reduce the chemosensitivity of 5-fluorouracil to both cell lines (34). In addition, experiments in which C57BL/6J mice were fed a high-fat diet for 58 weeks demonstrated that long-term high-fat diet feeding induced liver tumors in mice, along with the observation of significantly increased T-CDCA, T-CA and G-CA in plasma and liver (39). T-CDCA treatment of HepG2 cells significantly increased cell proliferation and decreased the expression of CEBPα (CEBPα is a tumor suppressor protein) in HCC, which suggests that BAs alone may have a tumor-promoting effect (39). There is also an experimental finding that metabolites related to BA biosynthesis, such as glycochenodeoxycholic acid 3-sulfate, G-CA, G-DCA, T-CA and T-CDCA, are downregulated in patients with HCC compared to cirrhotic patients (40). All of these studies indicate the possibility that BAs may be dynamically altered all the way to the detriment of the organism in the course of HCC progression.
The effect of BAs on HCC is complex. For instance, in the case of CDCA, a study suggested that it promotes the growth of a variety of tumor cell lines (39). However, another study observed downregulation of CDCA levels in patients with HCC (40). In different experiments, contradictory results regarding the effect of certain BAs on HCC have been obtained, which may be due to the differences in samples, experimental methods and measurements selected by different research institutes. Future studies are required to have a better design, for instance, to control a specific BA as a variable and set up controls. It was indicated that gene knockout of key enzymes during BA synthesis (e.g., CYP7A1, CYP27A1) in mice was able to better qualitatively and quantitatively analyze the role of BAs in the pathogenesis of diseases (41). This method may also be applied to the study of HCC. However, it may be a better way to study the signaling pathways mediated by BAs and their metabolites in HCC, which may contribute to the future targeted treatment of BAs.
Mechanisms by which BAs mediate HCC
FXR. FXR is thought to be the most important receptor for BAs to mediate the development of HCC. The expression of hepatic FXR may inhibit the occurrence of HCC through the following mechanisms: i) FXR maintains the normal liver metabolism of BAs, glucose and lipids; ii) FXR suppresses hepatic inflammation and promotes liver regeneration and repair after injury; iii) FXR protects liver cells from death and enhances cell survival; and iv) FXR may directly increase the expression of certain tumor-suppressor genes and repress the transcription of several oncogenes (42). Decreased FXR signaling leads to decreased liver transporter function, resulting in enhanced hepatic BA sequestration and persistent inflammation, which may promote HCC development. Liver tumors are observed in 90% of global FXR-null mice, but only 20% of liver-specific FXR-null mice develop spontaneous HCC (43). Sirtuin 1 is a transcriptional regulator of FXR, and under pathological conditions of cholestasis, it is downregulated by toxic BAs such as T-DCA, T-CA and DCA, resulting in the inhibition of FXR (44). In the course of liver injury, signals mediated by inflammatory factors such as TNF-α and NF-κB are also involved in the downregulation of FXR (45). FXR was indicated to bind directly to β-catenin, leading to reduced transcriptional activity in HCC (46). However, during HCC progression, Wnt/β-catenin signaling is enhanced and mRNAs of its target gene Myc are mainly found in the liver of FXR-null mice (43). T-CA was able to increase Myc expression in FXR-null hepatocytes and Myc has a crucial role in HCC development due to the induction of cell proliferation and migration (47).
Fibroblast growth factor 15/19 (FGF15/19) is also a target gene of FXR. Activation of the FGF15/19-fibroblast growth factor receptor 4 (FGFR4)-SRY-related high-mobility group box 18 pathway may directly promote epithelial to mesenchymal transition of HCC cells in vitro (29,48). FXR may also directly affect HCC cell proliferation by regulating several tumor suppressors downstream, such as suppressor of cytokine signaling 3 (SOCS3), N-myc downstream-regulated gene 2 and microRNA-122 (miR-122) (29) (Fig. 4). Therefore, when FXR is downregulated, the body's inhibitory effect on HCC is diminished.
Figure 4.

Intracellular signaling in HCC mediated by FXR and TGR5. BA, bile acid; HCC, hepatocellular carcinoma; EMT, epithelial to mesenchymal transition; FXR, FXR, farnesoid X receptor; SOCS3, suppressor of cytokine signaling 3; TGR5, Takeda G protein-coupled receptor 5; miR, microRNA; FGFR, fibroblast growth factor receptor; NDRG2, N-myc downstream-regulated gene 2; α7-nAChR, α7-nicotinic acetylcholine receptor.
TGR5. The secondary BAs DCA and LCA are the most potent natural ligands for TGR5 (49). TGR5 participates in the regulation of nutrient metabolism and energy consumption after activation. The induction of TGR5 in intestinal endocrine cells may promote the release of glucagon-like peptide 1 (GLP-1) (50). Certain studies have also indicated that this process may be initiated by FXR. FXR increases the production of LCA in the intestine, which activates TGR5 to stimulate the secretion of GLP-1 and improve glucose and lipid metabolism. The intestinal 'FXR-gut microbiota-TGR5-GLP-1' axis has a key role in mediating intestinal BA receptor signal transduction and regulating liver metabolism and homeostasis (51). The binding of BAs to TGR5 may also activate the cyclic adenosine monophosphate-protein kinase A signaling pathway and ultimately increase energy metabolism and oxygen consumption (52). TGR5 is also involved in immune regulation and inflammation. The high expression of TGR5 in monocytes and macrophages was indicated to decrease the phagocytic activity of these cells and inhibit the production of numerous proinflammatory cytokines induced by lipopolysaccharide, such as TNF-α, IL-1, IL-6 and IL-8 (53). Most studies point to TGR5-dependent immunosuppression partly due to the suppression of the Toll-like receptor 4/NF-κB pathway (49). Since these inflammatory signals are closely related to HCC, the downregulation of TGR5 may be an important factor in the progression of HCC. A retrospective study indicated that activation of α7-nicotinic acetylcholine receptor in smoking patients with HCC promoted HCC metastasis and recurrence by regulating the JAK/STAT3 axis and TGR5 is down-regulated in this process (54). The abnormality of TGR5 was also indicated to be related to HCC. Another retrospective analysis suggested that hypermethylation of the TGR5 promoter occurred significantly more frequently in patients with HCC (48.13%) than in those with chronic hepatitis B (13.64%) and healthy controls (4.44%) (55). However, there remains a lack of in vivo and in vitro evidence on the direct link between TGR5 and HCC. Revealing the subtle role of TGR5 in the development of HCC may be a promising direction in the future.
BA-mediated inflammation and injury in hepatocytes
BAs mediate a variety of signals that lead to inflammation and hepatocyte injury. Key factors in these pathways include IL-6, STAT3, NF-κB, reactive oxygen species (ROS), MAF bZIP transcription factor G (MAFG), Yes-associated protein (YAP) and PI3K class I isoforms (p110γ).
BAs may directly damage the plasma membrane and cause activation of protein kinase C, which activates the p38 MAPK pathway, leading to the activation of p53 and NF-κB. When this activation is increased, the expression of several inflammatory factors, such as IL-6, is enhanced, ultimately leading to increased apoptosis and inflammation (45). Data from experiments investigating DEN-elicited-CA-induced tumors in mice suggest that BAs may promote liver tumors by increasing inflammatory signaling, ER stress and possibly the selective survival of tumor-initiating stem cells (38). Apoptotic cells, as a result of BAs, may trigger inflammation. IL-6 also activates the signal sensor and activator of the JAK-STAT3 pathway, leading to reduced apoptosis and progression of HCC (56). Another study suggested that the expression level of STAT3 was positively associated with chemoresistance of HCC cells. G-CDCA is able to stimulate the phosphorylation of STAT3 at the Ser727 site and mediate pSer727-STAT3 protein translocation and aggregation in the nucleus, which is important for cell survival (57). The study suggested that G-CDCA may activate STAT3 by phosphorylation at the Ser727 site via the MAPK-ERK1/2 pathway, which may contribute to the progression and chemoresistance of human liver cancer (57). Membrane perturbation by BAs may also activate phospholipase A2, resulting in the release of arachidonic acid from the cell membrane via cyclooxygenase and lipoxygenase, ultimately leading to increased levels of ROS in hepatocytes (45). ROS are substances that jointly act on these several pathways to produce their final effects. ROS may also directly activate NF-κB in a feedback manner, inducing direct DNA damage in cells and the occurrence of HCC (58).
It has been experimentally evidenced that MAFG is induced in human HCC and its upregulation correlates with unfavorable prognosis in HCC. It was demonstrated that LCA, through activation of activating protein-1, NF-κB and E-box, induce MAFG expression, and all these enhancer elements are present in the human MAFG promoter. S-adenosylmethionine and UDCA have complementary roles to reduce LCA-mediated changes in the expression and DNA-binding activity of transcription factors that bind to these elements (59).
There are also experimental results demonstrating that BA is an upstream regulator of the Hippo pathway and its target YAP has been identified as a key driver of liver growth and carcinogenesis (60,61). BAs function as a tumor promoter by driving YAP activation and the ability of BAs to activate YAP depends on the concentration. Normal physiological or modestly elevated BA concentrations do not lead to YAP activation; however, chronically elevated pathological concentrations of BAs, which are commonly seen in cholestatic patients, may activate YAP and promote carcinogenesis (62,63).
Another study indicated that p110γ is activated by hydrophobic, but not hydrophilic BAs. BA-induced hepatocyte apoptosis is partly mediated via a PI3K p110γ-dependent signaling pathway (64).
Secondary BA-induced damage to other cells in the liver
Liver sinusoidal endothelial cells (LSECs)-natural killer T (NKT) cells: Primary BAs (β-MCA, CDCA) may increase C-X-C motif chemokine ligand (CXCL)16 expression, whereas secondary BAs (ω-MCA, LCA) had the opposite effect. C-X-C motif chemokine receptor type 6 forms the lining of liver capillaries and the first barrier to blood from the intestine entering the liver, and its expression on LSECs may regulate NKT cell accumulation (65). The increase of NKT cells is beneficial to the protective effect on the liver. Thus, when NKT cells induced by LSECs decrease, HCC progresses. In non-neoplastic liver tissues from patients with primary liver cancer, primary BA CDCA levels were correlated with CXCL16 expression, whereas an inverse correlation was observed with secondary BA G-LCA (65). This suggests that the finding may also apply to humans.
Hepatic stellate cells (HSCs): Blocking DCA production or reducing DCA-producing gut microbes has been indicated to prevent liver cancer development in obese mice. Relevant studies also suggested that enterohepatic circulation of DCA causes a related senescence-associated secretory phenotype (SASP) in HSCs (DCA would cause DNA damage through the generation of ROS, the key trigger of SASP), which in turn secretes various inflammatory and tumor-promoting factors in the liver to promote HCC development in mice after exposure to chemical carcinogens (66,67). Furthermore, DCA-induced senescent HSCs may also contribute to at least certain aspects of obesity-associated HCC development via SASP in humans (68,69) (Fig. 5).
Figure 5.

Mechanisms involved in BA-mediated hepatocarcinogenesis. BA, bile acid; HCC, hepatocellular carcinoma; FXR, FXR, farnesoid X receptor; TGR5, Takeda G protein-coupled receptor 5; ROS, reactive oxygen species; MAFG, MAF bZIP transcription factor G; YAP, Yes-associated protein; p110γ, PI3K class I isoforms γ; PKC, protein kinase C; LSEC, liver sinusoidal endothelial cell; CXCR, C-X-C motif chemokine receptor type; HSC, hepatic stellate cell; NKT, natural killer T; SASP, senescence-associated secretory phenotype.
Emerging strategies for the treatment of HCC utilizing BAs Targeting FXR: Obeticholic acid (OCA)
The semi-synthetic BA analogue 6α-ethyl-chenodeoxycholic acid, more commonly known as OCA, was developed as the first FXR agonist to be used in humans (70). A study investigated the effect of OCA on a NASH-associated HCC animal model induced by diethylnitrosamine and a high-fat choline-deficient diet. The results suggested that FXR activation by OCA is able to alleviate the progression of NASH-associated HCC by regulating the SOCS3/JAK2/STAT3 signaling axis (71). Small heterodimer partner, caspase-3 and p53 were upregulated in this process. Sirtuin-1, a key regulator of FXR that controls liver regenerative response, was also elevated after OCA treatment (71). These findings highlight the potential role of FXR agonists in the effective treatment of NASH-induced HCC. It is worth noting that OCA does not appear to have a simple positive therapeutic effect on HCC, as two studies have obtained contradictory results. An in vitro study indicated that OCA suppresses HCC cell proliferation and metastasis by inhibiting the IL-6/STAT3 signaling pathway, while another study established that OCA promoted HCC cell proliferation in vitro and xenograft tumor growth in vivo (29,59,72). The long-term safety of OCA also requires further evaluation, as the continuous activation of FXR may significantly change the body's energy metabolism. More patients with NAFLD/NASH for clinical trials should also be selected, thereby clarifying the applicable criteria for OCA. The modification of FXR itself may also affect the therapeutic effect. Activated HSCs may mediate hepatic fibrosis. It was reported that activated HSCs have a limited response to OCA and other FXR agonists due to enhanced FXR SUMOylation (73). Therefore, SUMOylation inhibitors rescue FXR signaling, thereby increasing the efficacy of OCA against HSC activation and fibrosis (73). In addition to OCA, other FXR agonists exhibiting therapeutic effects have now been discovered. For instance, GW4064 is able to delay the progression of HCC by blocking the STAT3 pathway through the regulation of the suppressor of cytokine signaling 3 (70). WAY-362450, which reduces inflammation, and INT-767 (a dual FXR and TGR5 agonist), which has lipid-lowering effects, are also being tested in animal experiments (74).
OCA is still the best and most important FXR agonist for treatment. There may be a scope modify OCA to improve its efficacy. An innovative idea is to prepare OCA as nanoparticles, resulting in a stronger agonistic effect on FXR. Studies have indicated that manipulation of FXR by this nanoapproach significantly improves antitumor immune responses in murine HCC (75). This approach undoubtedly markedly increases the efficiency compared with oral administration. Therefore, the improvement of OCA dosage and administration route is a direction worth pursuing in the future. In addition, the rise of nanotechnology will also provide an incentive to develop novel FXR agonists. Perhaps, the replacement of OCA as the main substance in nanoparticles with novel FXR agonists may yield even more surprising results. Exosomes also appear to be a better way to agonize FXR. Studies have indicated that Lactobacillus rhamnosus GG-derived exosome-like nanoparticles may protect against alcohol-associated liver disease through regulation of the FXR signal in mice (76). Perhaps in the future, BAs may be used as components of exosomes to agonize FXR with greater precision. Alternatively, the related genes of FXR may be used as components of exosomes to better delay the progression of HCC.
Targeting BA transporters
Targeting specific sites in the enterohepatic circulation for regulation also appears to be a viable direction for the treatment of HCC. Inhibition of ASBT may reduce intestinal reabsorption of BAs and, at the same time, increase cholesterol catabolism and BA synthesis in the liver (77). Thus, ASBT inhibitors (e.g., SC-435 and 264W94) improve lipid metabolism in obese patients and perhaps also have efficacy against HCC due to NAFLD/NASH (74). BA sequestrants (e.g., cholestyramine, colesevelam), which act similarly to ASBT inhibitors, may also reduce lipids by impairing intestinal reabsorption of BAs, but their effects are more limited (74,77). In addition, studies have demonstrated that DCA has a dual effect in HCC cells and is dependent on the expression of the BA transporter NTCP. DCA may induce apoptosis in NTCP-positive HCC cells, particularly under hypoxic conditions, while in NTCP-negative HCC cells, DCA markedly decreased aggressive cellular behaviors (78). Thus, if it were possible to examine NTCP expression and the apoptotic signaling cascade by immunoblot analysis, hydrophobic BAs may even be a suitable choice for the treatment of NTCP-positive HCC.
Currently, no drugs targeting BA transporters have been approved for clinical use in patients with HCC. Despite the ability of ASBT inhibitors to ameliorate metabolic disorders in patients, the pathogenesis of HCC is complex. The conditions of ASBT inhibitor application must be considered due to their potential to have vastly different treatment outcomes for patients with NAFLD-related HCC and those with non-NAFLD-related HCC. It is also worth noting that blocking any site in the enterohepatic circulation has the potential to create a disorder of BA metabolism. Therefore, the therapeutic effects and adverse effects must be compared and weighed when developing related drugs. Drugs with fewer side effects, such as diarrhea and abdominal pain, would have greater advantages on the basis of guaranteeing therapeutic efficacy (79).
Hydrophilic BA with therapeutic effects
UDCA. UDCA may inhibit cholestasis and has a protective effect on CLDs (80). T-UDCA has been indicated to exert its cytoprotective activity by reducing ER stress and preventing apoptosis. The related mechanisms are dependent on inhibition of the translocation of pro-apoptotic Bax from cytosol to mitochondria, inhibition of cytochrome c release and subsequent suppression of mitochondrial apoptosis and reduction of the expression of cyclin D1 (81,82). In addition, UDCA has been indicated to interfere with the E2F-1/Mdm-2/p53 apoptotic pathway, resulting in subsequent nuclear translocation of the BA-receptor complex and reduction of apoptosis (81,83). UDC-dihydroartemisinin (DHA), which is composed of a mixture of UDCA and DHA, has an inhibitory effect on HepG2 and Huh-7 cells (84). In conclusion, numerous studies suggest that the application of UDCA is an effective strategy for the management of advanced hepatobiliary diseases in the future. Based on the anti-inflammatory, antioxidant and cytoprotective activities, UDCA may be useful to improve treatments of advanced liver diseases, notably in combination with other drugs such as sorafenib, to enhance the therapeutic efficacy of targeting drugs for HCC (49,85,86). Furthermore, based on UDCA, monoclonal antibodies may be designed at the key sites of these inflammatory signaling pathways to achieve the purpose of targeted therapy. Therefore, unraveling the signaling pathways underlying the therapeutic effects of UDCA may provide a more definitive direction for the doses and modalities administered, aiming to improve therapeutic outcomes for patients.
Immunotherapy: Possible utility of BAs
BAs have been indicated to have an immunotherapeutic role in a variety of gastrointestinal and biliary diseases. Immunotherapy for HCC is a more emerging strategy. At present, the commonly used immunotherapy drugs for HCC are antiangiogenic tyrosine kinase inhibitors (such as sorafenib), programmed death ligand 1 (PDL1) blockade with atezolizumab and VEGF blockade with bevacizumab (87). The question arises whether BAs and immunotherapy for HCC have a link. As mentioned above, primary BAs may increase hepatic NKT-cell accumulation by upregulating CXCL16. However, abundant expression of receptors for primary BAs across the gastrointestinal tract overwhelms the possibility of using agonists against these receptors for HCC control. Therefore, one study prepared an OCA-nanoemulsion (OCA-NE) and injected it into mice. The results suggested that OCA-NE significantly suppressed hepatic tumor growth in a murine orthotopic H22 tumor model; furthermore, OCA-NE led to the increase of CXCL16, IFN-γ and the number of NKT cells (88). The study made good use of the intrinsic property of LSECs in capturing circulating nanoparticles and a large amount of injected OCA-NE accumulated in LSECs. This strategy for precise manipulation of LSECs should be extended. For instance, it may be possible to design a nanoparticle of a DCA analogue for targeted manipulation of HSCs and blocking SASP signals inside these cells. The feasibility of this remains to be verified by further experiments.
Immunologic agents that target BA receptors will also be an important direction for treatments in the future. In addition to FXR, TGR5 is also an important receptor for BAs. Studies have indicated that nanoparticles prepared from 5β-CA may act as antagonists of TGR5, thereby exerting a role in recruiting immune cells and suppressing HCC (75). Dual FXR/TGR5 agonist INT-767 was indicated to delay HCC progression and improve liver function in mice (89). Previously, it was mentioned that UDCA is a therapeutic BA widely used in clinics. It may be possible to improve UDCA to prepare specific FXR agonist/TGR5 antagonist, which is an attractive prospect. In addition, another study suggested that UDCA may enhance anti-tumor immunity by promoting the degradation of TGF-β (90). TGF-β is essential for tumor immune evasion. It is also due to its effects that anti-programmed death 1 (PD1) or anti-PDL1 treatments alone do not improve the immunosuppressive tumor microenvironment (90). UDCA is a potential TGF-β inhibitor, which outperforms existing developed TGF-β blocking antibodies in terms of both efficacy and safety (90). UDCA and anti-PD1/anti-PDL1 combination will also be a direction to improve the efficacy of immune checkpoint inhibitor therapy. Combination therapy with anti-PD1 or anti-PDL1 and UDCA may have increased efficacy in patients with HCC.
Other emerging BA-based therapeutic approaches
Functionalized gold nanoparticles (AuNPs) have been widely applied due to their good biocompatibility and long drug half-life. In one study, the researchers synthesized AuNPs capped with ligands that possess polyethylene glycol (PEG) and LCA linked by carboxyl groups (AuNP@MPA-PEG-LCA). The results indicated that AuNP@MPA-PEG-LCA was more effective in promoting programmed cell death of HCC cells due to its better cell selectivity and the related mechanism was the activation of ROS and mediated mitochondrial dysfunction and apoptosis (91).
Probiotics are emerging viable treatments for HCC. Probiotics may affect BA metabolism through the modulation of the gut microbiota. Probiotics modestly regulate the intestinal FXR pathway to promote liver regeneration by affecting the secretion of downstream FGF15 (92). A study indicated that VSL#3 probiotics promote ileal BA deconjugation with subsequent fecal BA excretion and induce hepatic BA neosynthesis via the downregulation of the gut-liver FXR-FGF15 axis (93). VSL#3 increases the excretion of BAs in feces, with distinct alterations in the composition of the fecal microbiota in mice (93,94). Administration of VSL#3 for 21 days resulted in a significantly higher abundance of Firmicutes (51.47%) and Actinobacteria (3.31%) at the expense of Bacteroidetes (44.22%) and Proteobacteria (1%) (93). These findings suggest the possibility that probiotics may be used to treat HCC by affecting the body's BA metabolism.
4. Conclusions
The subtle relationship between BAs and HCC is gradually being explored. FXR signaling is the most important pathway through which BAs mediate HCC development. OCA and several other novel FXR agonists have demonstrated promising therapeutic effects against metabolic liver diseases and even HCC (95). In addition to FXR, BA-mediated HCC development involves TGR5 and multiple inflammatory signals, which implicates BAs in the immunotherapeutic process of HCC. The hydrophilic BA UDCA also exhibits therapeutic effects on HCC by downregulating inflammatory signaling. In addition, targeting BA transporters, such as ASBT inhibitors, also appears to have beneficial effects on HCC, but their efficacy requires further experimental validation. In the future, a deeper understanding of the mechanisms by which BAs mediate HCC is required in order to provide further clinical treatments for HCC and to exploit the therapeutic potential of BAs.
Acknowledgments
Not applicable.
Abbreviations
- HCC
hepatocellular carcinoma
- FXR
farnesoid X receptor
- UDCA
ursodeoxycholic acid
- NAFLD
non-alcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- BAs
bile acids
- CA
cholic acid
- CDCA
chenodeoxycholic acid
- DCA
deoxycholic acid
- LCA
lithocholic acid
- MCA
muricholic acid
- ER
endoplasmic reticulum
- CYP
cytochrome P450
- HSD3B7
3β-hydroxy-5-C27-steroid dehydrogenase
- C4
7α-hydroxy-4-cholesten-3-one
- AKR1D1
aldo-keto reductase family 1 member D1
- THCA
3α,7α,12α-trihydroxy-5β cholestanoic acid
- DHCA
3α,7α-dihydroxy-5β cholestanoic acid
- BACS
BA-Co-A synthase
- VLCS
very long-chain Co-A synthase
- BAAT
bile acid-CoA, amino acid N-acyltransferase
- BSH
bile acid hydrolase
- BSEP
bile salt export pump
- MRP2
multidrug resistance-associated protein 2
- ASBT
apical sodium-dependent bile acid transporter
- IBABP
intestinal bile acid binding protein
- OST
organic solute transporter
- NTCP
sodium-taurocholate co-transporting polypeptide
- OATP
organic anion transporting polypeptide
- TGR5
Takeda G protein-coupled receptor 5
- DEN
diethylnitrosamine
- NF-κB
nuclear factor kappa-B
- FGF
fibroblast growth factor
- FGFR4
fibroblast growth factor receptor 4
- SOX18
SRY-related high-mobility group box 18
- EMT
epithelial to mesenchymal transition
- SOCS3
suppressor of cytokine signaling 3
- NDRG2
N-myc downstream-regulated gene 2
- miR-122
microRNA-122
- GLP-1
glucagon-like peptide 1
- α7-nAChR
α7-nicotinic acetylcholine receptor
- ROS
reactive oxygen species
- MAFG
MAF bZIP transcription factor G
- YAP
Yes-associated protein
- p110γ
PI3K Class I isoforms γ
- PKC
protein kinase C
- COX
cyclooxygenase
- LSECs
liver sinusoidal endothelial cells
- CXCR
C-X-C motif chemokine receptor type
- HSC
hepatic stellate cell
- SASP
senescence-associated secretory phenotype
- OCA
obeticholic acid
- DHA
dihydroartemisinin
- AuNPs
gold nanoparticles
- PEG
polyethylene glycol
Funding Statement
This work was supported by a grant from the Natural Science Foundation of Guangxi (grant no. 2021GXNSFAA325001) and a grant from the Shenyang Science and Technology Plan Fund Project (grant no. 20-205-4-094).
Availability of data and materials
Data sharing not applicable to this article, as no datasets were generated or analyzed during the current study.
Authors' contributions
WL and SG drafted the manuscript, performed the selection and organization of the literature and prepared the figures. BW, YZ, JWZ, MW, JFZ and LS revised the manuscript. BC carried out the design of this review and revised the manuscript. All authors contributed to this manuscript and read and approved the final manuscript. Data authentication is not applicable.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer. J Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16:589–604. doi: 10.1038/s41575-019-0186-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foerster F, Gairing SJ, Müller L, Galle PR. NAFLD-driven HCC: Safety and efficacy of current and emerging treatment options. J Hepatol. 2022;76:446–457. doi: 10.1016/j.jhep.2021.09.007. [DOI] [PubMed] [Google Scholar]
- 4.Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Zucman-Rossi J, Finn RS. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7:6. doi: 10.1038/s41572-020-00240-3. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Chen K, Li F, Gu Z, Liu Q, He L, Shao T, Song Q, Zhu F, Zhang L, et al. Probiotic Lactobacillus rhamnosus GG prevents liver fibrosis through inhibiting hepatic bile acid synthesis and enhancing bile acid excretion in mice. Hepatology. 2020;71:2050–2066. doi: 10.1002/hep.30975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J, Dawson PA. Animal models to study bile acid metabolism. Biochim Biophys Acta Mol Basis Dis. 2019;1865:895–911. doi: 10.1016/j.bbadis.2018.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Ciaula A, Garruti G, Lunardi Baccetto R, Molina-Molina E, Bonfrate L, Wang DQ, Portincasa P. Bile acid physiology. Ann Hepatol. 2017;16(Suppl 1):s4–s14. doi: 10.5604/01.3001.0010.5493. [DOI] [PubMed] [Google Scholar]
- 8.Shulpekova Y, Shirokova E, Zharkova M, Tkachenko P, Tikhonov I, Stepanov A, Sinitsyna A, Izotov A, Butkova T, Shulpekova N, et al. A recent ten-year perspective: Bile acid metabolism and signaling. Molecules. 2022;27:1983. doi: 10.3390/molecules27061983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiang JYL, Ferrell JM. Bile acid metabolism in liver pathobiology. Gene Expr. 2018;18:71–87. doi: 10.3727/105221618X15156018385515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wahlström A, Sayin SI, Marschall HU, Bäckhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016;24:41–50. doi: 10.1016/j.cmet.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 11.Zhang B, Kuipers F, de Boer JF, Kuivenhoven JA. Modulation of bile acid metabolism to improve plasma lipid and lipoprotein profiles. J Clin Med. 2021;11:4. doi: 10.3390/jcm11010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perino A, Demagny H, Velazquez-Villegas L, Schoonjans K. Molecular physiology of bile acid signaling in health, disease, and aging. Physiol Rev. 2021;101:683–731. doi: 10.1152/physrev.00049.2019. [DOI] [PubMed] [Google Scholar]
- 13.Sohail MI, Dönmez-Cakil Y, Szöllősi D, Stockner T, Chiba P. The bile salt export pump: Molecular structure, study models and small-molecule drugs for the treatment of inherited BSEP deficiencies. Int J Mol Sci. 2021;22:784. doi: 10.3390/ijms22020784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jetter A, Kullak-Ublick GA. Pharmacol Res. 2020;154:104234. doi: 10.1016/j.phrs.2019.04.018. [DOI] [PubMed] [Google Scholar]
- 15.Köck K, Ferslew BC, Netterberg I, Yang K, Urban TJ, Swaan PW, Stewart PW, Brouwer KL. Risk factors for development of cholestatic drug-induced liver injury: Inhibition of hepatic basolateral bile acid transporters multidrug resistance-associated proteins 3-4. Drug Metab Dispos. 2014;42:665–674. doi: 10.1124/dmd.113.054304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao L, Pan G. An important intestinal transporter that regulates the enterohepatic circulation of bile acids and cholesterol homeostasis: The apical sodium-dependent bile acid transporter (SLC10A2/ASBT) Clin Res Hepatol Gastroenterol. 2017;41:509–515. doi: 10.1016/j.clinre.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 17.Deng F, Bae YH. Bile acid transporter-mediated oral drug delivery. J Control Release. 2020;327:100–116. doi: 10.1016/j.jconrel.2020.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suga T, Yamaguchi H, Ogura J, Mano N. Characterization of conjugated and unconjugated bile acid transport via human organic solute transporter α/β. Biochim Biophys Acta Biomembr. 2019;1861:1023–1029. doi: 10.1016/j.bbamem.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 19.Vaz FM, Ferdinandusse S. Bile acid analysis in human disorders of bile acid biosynthesis. Mol Aspects Med. 2017;56:10–24. doi: 10.1016/j.mam.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Trauner M, Fuchs CD, Halilbasic E, Paumgartner G. New therapeutic concepts in bile acid transport and signaling for management of cholestasis. Hepatology. 2017;65:1393–1404. doi: 10.1002/hep.28991. [DOI] [PubMed] [Google Scholar]
- 21.Li T, Chiang JY. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev. 2014;66:948–983. doi: 10.1124/pr.113.008201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daruich A, Picard E, Boatright JH, Behar-Cohen F. Review: The bile acids ursoand tauroursodeoxycholic acid as neuroprotective therapies in retinal disease. Mol Vis. 2019;25:610–624. [PMC free article] [PubMed] [Google Scholar]
- 23.Sato R. Recent advances in regulating cholesterol and bile acid metabolism. Biosci Biotechnol Biochem. 2020;84:2185–2192. doi: 10.1080/09168451.2020.1793658. [DOI] [PubMed] [Google Scholar]
- 24.Ko CW, Qu J, Black DD, Tso P. Regulation of intestinal lipid metabolism: Current concepts and relevance to disease. Nat Rev Gastroenterol Hepatol. 2020;17:169–183. doi: 10.1038/s41575-019-0250-7. [DOI] [PubMed] [Google Scholar]
- 25.Blanchet M, Brunel JM. Bile acid derivatives: From old molecules to a new potent therapeutic use: An overview. Curr Med Chem. 2018;25:3613–3636. doi: 10.2174/0929867325666180309113737. [DOI] [PubMed] [Google Scholar]
- 26.Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, Noonan DJ, Burka LT, McMorris T, Lamph WW, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81:687–693. doi: 10.1016/0092-8674(95)90530-8. [DOI] [PubMed] [Google Scholar]
- 27.Massafra V, Pellicciari R, Gioiello A, van Mil SWC. Progress and challenges of selective farnesoid X receptor modulation. Pharmacol Ther. 2018;191:162–177. doi: 10.1016/j.pharmthera.2018.06.009. [DOI] [PubMed] [Google Scholar]
- 28.Schubert K, Olde Damink SWM, von Bergen M, Schaap FG. Interactions between bile salts, gut microbiota, and hepatic innate immunity. Immunol Rev. 2017;279:23–35. doi: 10.1111/imr.12579. [DOI] [PubMed] [Google Scholar]
- 29.Sun L, Cai J, Gonzalez FJ. The role of farnesoid X receptor in metabolic diseases, and gastrointestinal and liver cancer. Nat Rev Gastroenterol Hepatol. 2021;18:335–347. doi: 10.1038/s41575-020-00404-2. [DOI] [PubMed] [Google Scholar]
- 30.Duboc H, Taché Y, Hofmann AF. The bile acid TGR5 membrane receptor: From basic research to clinical application. Dig Liver Dis. 2014;46:302–312. doi: 10.1016/j.dld.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ticho AL, Malhotra P, Dudeja PK, Gill RK, Alrefai WA. Bile acid receptors and gastrointestinal functions. Liver Res. 2019;3:31–39. doi: 10.1016/j.livres.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Portincasa P, Di Ciaula A, Garruti G, Vacca M, De Angelis M, Wang DQ. Bile acids and GPBAR-1: Dynamic interaction involving genes, environment and gut microbiome. Nutrients. 2020;12:3709. doi: 10.3390/nu12123709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang R, Sheps JA, Ling V. ABC transporters, bile acids, and inflammatory stress in liver cancer. Curr Pharm Biotechnol. 2011;12:636–646. doi: 10.2174/138920111795163986. [DOI] [PubMed] [Google Scholar]
- 34.Wang C, Yang M, Zhao J, Li X, Xiao X, Zhang Y, Jin X, Liao M. Bile salt (glycochenodeoxycholate acid) induces cell survival and chemoresistance in hepatocellular carcinoma. J Cell Physiol. 2019;234:10899–10906. doi: 10.1002/jcp.27905. [DOI] [PubMed] [Google Scholar]
- 35.Wang H, Shang X, Wan X, Xiang X, Mao Q, Deng G, Wu Y. Increased hepatocellular carcinoma risk in chronic hepatitis B patients with persistently elevated serum total bile acid: A retrospective cohort study. Sci Rep. 2016;6:38180. doi: 10.1038/srep38180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas CE, Luu HN, Wang R, Xie G, Adams-Haduch J, Jin A, Koh WP, Jia W, Behari J, Yuan JM. Association between pre-diagnostic serum bile acids and hepatocellular carcinoma: The singapore Chinese health study. Cancers (Basel) 2021;13:2648. doi: 10.3390/cancers13112648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang W, Zhou L, Yin P, Wang J, Lu X, Wang X, Chen J, Lin X, Xu G. A weighted relative difference accumulation algorithm for dynamic metabolomics data: Long-term elevated bile acids are risk factors for hepatocellular carcinoma. Sci Rep. 2015;5:8984. doi: 10.1038/srep08984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun L, Beggs K, Borude P, Edwards G, Bhushan B, Walesky C, Roy N, Manley MW, Jr, Gunewardena S, O'Neil M, et al. Bile acids promote diethylnitrosamine-induced hepatocellular carcinoma via increased inflammatory signaling. Am J Physiol Gastrointest Liver Physiol. 2016;311:G91–G104. doi: 10.1152/ajpgi.00027.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie G, Wang X, Huang F, Zhao A, Chen W, Yan J, Zhang Y, Lei S, Ge K, Zheng X, et al. Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis. Int J Cancer. 2016;139:1764–1775. doi: 10.1002/ijc.30219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ressom HW, Xiao JF, Tuli L, Varghese RS, Zhou B, Tsai TH, Ranjbar MR, Zhao Y, Wang J, Di Poto C, et al. Utilization of metabolomics to identify serum biomarkers for hepatocellular carcinoma in patients with liver cirrhosis. Anal Chim Acta. 2012;743:90–100. doi: 10.1016/j.aca.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rizzolo D, Buckley K, Kong B, Zhan L, Shen J, Stofan M, Brinker A, Goedken M, Buckley B, Guo GL. Bile acid homeostasis in a cholesterol 7α-hydroxylase and sterol 27-hydroxylase double knockout mouse model. Hepatology. 2019;70:389–402. doi: 10.1002/hep.30612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang XF, Zhao WY, Huang WD. FXR and liver carcinogenesis. Acta Pharmacol Sin. 2015;36:37–43. doi: 10.1038/aps.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takahashi S, Tanaka N, Fukami T, Xie C, Yagai T, Kim D, Velenosi TJ, Yan T, Krausz KW, Levi M, Gonzalez FJ. Role of farnesoid X receptor and bile acids in hepatic tumor development. Hepatol Commun. 2018;2:1567–1582. doi: 10.1002/hep4.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao Q, Liu F, Cheng Y, Xiao XR, Hu DD, Tang YM, Bao WM, Yang JH, Jiang T, Hu JP, et al. Celastrol protects from cholestatic liver injury through modulation of SIRT1-FXR signaling. Mol Cell Proteomics. 2019;18:520–533. doi: 10.1074/mcp.RA118.000817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jia W, Xie G, Jia W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Rev Gastroenterol Hepatol. 2018;15:111–128. doi: 10.1038/nrgastro.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu X, Zhang X, Ji L, Gu J, Zhou M, Chen S. Farnesoid X receptor associates with β-catenin and inhibits its activity in hepatocellular carcinoma. Oncotarget. 2015;6:4226–4238. doi: 10.18632/oncotarget.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qu A, Jiang C, Cai Y, Kim JH, Tanaka N, Ward JM, Shah YM, Gonzalez FJ. Role of Myc in hepatocellular proliferation and hepatocarcinogenesis. J Hepatol. 2014;60:331–338. doi: 10.1016/j.jhep.2013.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen J, Du F, Dang Y, Li X, Qian M, Feng W, Qiao C, Fan D, Nie Y, Wu K, Xia L. Fibroblast growth factor 19-mediated up-regulation of SYR-related high-mobility group box 18 promotes hepatocellular carcinoma metastasis by transactivating fibroblast growth factor receptor 4 and fms-related tyrosine kinase 4. Hepatology. 2020;71:1712–1731. doi: 10.1002/hep.30951. [DOI] [PubMed] [Google Scholar]
- 49.Režen T, Rozman D, Kovács T, Kovács P, Sipos A, Bai P, Mikó E. The role of bile acids in carcinogenesis. Cell Mol Life Sci. 2022;79:243. doi: 10.1007/s00018-022-04278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Nierop FS, Scheltema MJ, Eggink HM, Pols TW, Sonne DP, Knop FK, Soeters MR. Clinical relevance of the bile acid receptor TGR5 in metabolism. Lancet Diabetes Endocrinol. 2017;5:224–233. doi: 10.1016/S2213-8587(16)30155-3. [DOI] [PubMed] [Google Scholar]
- 51.Pathak P, Xie C, Nichols RG, Ferrell JM, Boehme S, Krausz KW, Patterson AD, Gonzalez FJ, Chiang JYL. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology. 2018;68:1574–1588. doi: 10.1002/hep.29857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fuchs CD, Trauner M. Role of bile acids and their receptors in gastrointestinal and hepatic pathophysiology. Nat Rev Gastroenterol Hepatol. 2022;19:432–450. doi: 10.1038/s41575-021-00566-7. [DOI] [PubMed] [Google Scholar]
- 53.Pols TW, Noriega LG, Nomura M, Auwerx J, Schoonjans K. The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J Hepatol. 2011;54:1263–1272. doi: 10.1016/j.jhep.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li CL, Lin YK, Chen HA, Huang CY, Huang MT, Chang YJ. Smoking as an independent risk factor for hepatocellular carcinoma due to the α7-nachr modulating the JAK2/STAT3 signaling axis. J Clin Med. 2019;8:1391. doi: 10.3390/jcm8091391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Han LY, Fan YC, Mu NN, Gao S, Li F, Ji XF, Dou CY, Wang K. Aberrant DNA methylation of G-protein-coupled bile acid receptor Gpbar1 (TGR5) is a potential biomarker for hepatitis B virus associated hepatocellular carcinoma. Int J Med Sci. 2014;11:164–171. doi: 10.7150/ijms.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu J, Lin H, Wu G, Zhu M, Li M. IL-6/STAT3 is a promising therapeutic target for hepatocellular carcinoma. Front Oncol. 2021;11:760971. doi: 10.3389/fonc.2021.760971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang J, Zhou M, Jin X, Li B, Wang C, Zhang Q, Liao M, Hu X, Yang M. Glycochenodeoxycholate induces cell survival and chemoresistance via phosphorylation of STAT3 at Ser727 site in HCC. J Cell Physiol. 2020;235:2557–2568. doi: 10.1002/jcp.29159. [DOI] [PubMed] [Google Scholar]
- 58.Zhang WJ, Chen SJ, Zhou SC, Wu SZ, Wang H. Inflammasomes and fibrosis. Front Immunol. 2021;12:643149. doi: 10.3389/fimmu.2021.643149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu T, Yang H, Fan W, Tu J, Li TWH, Wang J, Shen H, Yang J, Xiong T, Steggerda J, et al. Mechanisms of MAFG dysregulation in cholestatic liver injury and development of liver cancer. Gastroenterology. 2018;155:557–571.e14. doi: 10.1053/j.gastro.2018.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cai J, Zhang N, Zheng Y, de Wilde RF, Maitra A, Pan D. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev. 2010;24:2383–2388. doi: 10.1101/gad.1978810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang S, Zhou D. Role of the transcriptional coactivators YAP/TAZ in liver cancer. Curr Opin Cell Biol. 2019;61:64–71. doi: 10.1016/j.ceb.2019.07.006. [DOI] [PubMed] [Google Scholar]
- 62.Anakk S, Bhosale M, Schmidt VA, Johnson RL, Finegold MJ, Moore DD. Bile acids activate YAP to promote liver carcinogenesis. Cell Rep. 2013;5:1060–1069. doi: 10.1016/j.celrep.2013.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Russell JO, Camargo FD. Hippo signalling in the liver: Role in development, regeneration and disease. Nat Rev Gastroenterol Hepatol. 2022;19:297–312. doi: 10.1038/s41575-021-00571-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hohenester S, Gates A, Wimmer R, Beuers U, Anwer MS, Rust C, Webster CR. Phosphatidylinositol-3-kinase p110γ contributes to bile salt-induced apoptosis in primary rat hepatocytes and human hepatoma cells. J Hepatol. 2010;53:918–926. doi: 10.1016/j.jhep.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, Agdashian D, Terabe M, Berzofsky JA, Fako V, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360:eaan5931. doi: 10.1126/science.aan5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Friedman SL. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matsuda M, Seki E. Hepatic stellate cell-macrophage crosstalk in liver fibrosis and carcinogenesis. Semin Liver Dis. 2020;40:307–320. doi: 10.1055/s-0040-1708876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 69.Ohtani N. The roles and mechanisms of senescence-associated secretory phenotype (SASP): Can it be controlled by senolysis? Inflamm Regen. 2022;42:11. doi: 10.1186/s41232-022-00197-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Orabi D, Berger NA, Brown JM. Abnormal metabolism in the progression of nonalcoholic fatty liver disease to hepatocellular carcinoma: Mechanistic insights to chemoprevention. Cancers (Basel) 2021;13:3473. doi: 10.3390/cancers13143473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Attia YM, Tawfiq RA, Gibriel AA, Ali AA, Kassem DH, Hammam OA, Elmazar MM. Activation of FXR modulates SOCS3/Jak2/STAT3 signaling axis in a NASH-dependent hepatocellular carcinoma animal model. Biochem Pharmacol. 2021;186:114497. doi: 10.1016/j.bcp.2021.114497. [DOI] [PubMed] [Google Scholar]
- 72.Attia YM, Tawfiq RA, Ali AA, Elmazar MM. The FXR agonist, obeticholic acid, suppresses HCC proliferation & metastasis: Role of IL-6/STAT3 signalling pathway. Sci Rep. 2017;7:12502. doi: 10.1038/s41598-017-12629-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou J, Cui S, He Q, Guo Y, Pan X, Zhang P, Huang N, Ge C, Wang G, Gonzalez FJ, et al. SUMOylation inhibitors synergize with FXR agonists in combating liver fibrosis. Nat Commun. 2020;11:240. doi: 10.1038/s41467-019-14138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chow MD, Lee YH, Guo GL. The role of bile acids in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Mol Aspects Med. 2017;56:34–44. doi: 10.1016/j.mam.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ji G, Si X, Dong S, Xu Y, Li M, Yang B, Tang Z, Fang X, Huang L, Song W, Chen X. Manipulating liver bile acid signaling by nanodelivery of bile acid receptor modulators for liver cancer immunotherapy. Nano Lett. 2021;21:6781–6791. doi: 10.1021/acs.nanolett.1c01360. [DOI] [PubMed] [Google Scholar]
- 76.Jiang M, Li F, Liu Y, Gu Z, Zhang L, Lee J, He L, Vatsalya V, Zhang HG, Deng Z, et al. Probiotic-derived nanoparticles inhibit ALD through intestinal miR194 suppression and subsequent FXR activation. Hepatology. 2022 Jun;:11. doi: 10.1002/hep.32608. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van de Peppel IP, Verkade HJ, Jonker JW. Metabolic consequences of ileal interruption of the enterohepatic circulation of bile acids. Am J Physiol Gastrointest Liver Physiol. 2020;319:G619–G625. doi: 10.1152/ajpgi.00308.2020. [DOI] [PubMed] [Google Scholar]
- 78.Jang ES, Yoon JH, Lee SH, Lee SM, Lee JH, Yu SJ, Kim YJ, Lee HS, Kim CY. Sodium taurocholate cotransporting polypeptide mediates dual actions of deoxycholic acid in human hepatocellular carcinoma cells: Enhanced apoptosis versus growth stimulation. J Cancer Res Clin Oncol. 2014;140:133–144. doi: 10.1007/s00432-013-1554-6. [DOI] [PubMed] [Google Scholar]
- 79.Yang N, Dong YQ, Jia GX, Fan SM, Li SZ, Yang SS, Li YB. ASBT(SLC10A2): A promising target for treatment of diseases and drug discovery. Biomed Pharmacother. 2020;132:110835. doi: 10.1016/j.biopha.2020.110835. [DOI] [PubMed] [Google Scholar]
- 80.Cabrera D, Arab JP, Arrese M. UDCA, NorUDCA, and TUDCA in liver diseases: A review of their mechanisms of action and clinical applications. Handb Exp Pharmacol. 2019;256:237–264. doi: 10.1007/164_2019_241. [DOI] [PubMed] [Google Scholar]
- 81.Kusaczuk M. Tauroursodeoxycholate-bile acid with chaperoning activity: Molecular and cellular effects and therapeutic perspectives. Cells. 2019;8:1471. doi: 10.3390/cells8121471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Castro RE, Solá S, Ma X, Ramalho RM, Kren BT, Steer CJ, Rodrigues CM. A distinct microarray gene expression profile in primary rat hepatocytes incubated with ursodeoxycholic acid. J Hepatol. 2005;42:897–906. doi: 10.1016/j.jhep.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 83.Solá S, Amaral JD, Castro RE, Ramalho RM, Borralho PM, Kren BT, Tanaka H, Steer CJ, Rodrigues CM. Nuclear translocation of UDCA by the glucocorticoid receptor is required to reduce TGF-beta1-induced apoptosis in rat hepatocytes. Hepatology. 2005;42:925–934. doi: 10.1002/hep.20870. [DOI] [PubMed] [Google Scholar]
- 84.Huang TE, Deng YN, Hsu JL, Leu WJ, Marchesi E, Capobianco ML, Marchetti P, Navacchia ML, Guh JH, Perrone D, Hsu LC. Evaluation of the anticancer activity of a bile acid-dihydroartemisinin hybrid ursodeoxycholic-dihydroartemisinin in hepatocellular carcinoma cells. Front Pharmacol. 2020;11:599067. doi: 10.3389/fphar.2020.599067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Goossens JF, Bailly C. Ursodeoxycholic acid and cancer: From chemoprevention to chemotherapy. Pharmacol Ther. 2019;203:107396. doi: 10.1016/j.pharmthera.2019.107396. [DOI] [PubMed] [Google Scholar]
- 86.Lee S, Cho YY, Cho EJ, Yu SJ, Lee JH, Yoon JH, Kim YJ. Synergistic effect of ursodeoxycholic acid on the antitumor activity of sorafenib in hepatocellular carcinoma cells via modulation of STAT3 and ERK. Int J Mol Med. 2018;42:2551–2559. doi: 10.3892/ijmm.2018.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sangro B, Sarobe P, Hervás-Stubbs S, Melero I. Advances in immunotherapy for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2021;18:525–543. doi: 10.1038/s41575-021-00438-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ji G, Ma L, Yao H, Ma S, Si X, Wang Y, Bao X, Ma L, Chen F, Ma C, et al. Precise delivery of obeticholic acid via nanoapproach for triggering natural killer T cell-mediated liver cancer immunotherapy. Acta Pharm Sin B. 2020;10:2171–2182. doi: 10.1016/j.apsb.2020.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cariello M, Peres C, Zerlotin R, Porru E, Sabbà C, Roda A, Moschetta A. Long-term administration of nuclear bile acid receptor FXR agonist prevents spontaneous hepatocarcinogenesis in Abcb4-/mice. Sci Rep. 2017;7:11203. doi: 10.1038/s41598-017-11549-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shen Y, Lu C, Song Z, Qiao C, Wang J, Chen J, Zhang C, Zeng Z, Ma Z, Chen J, et al. Ursodeoxycholic acid reduces antitumor immunosuppression by inducing CHIP-mediated TGF-β degradation. Nat Commun. 2022;13:3419. doi: 10.1038/s41467-022-31141-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhao MX, Cai ZC, Zhu BJ, Zhang ZQ. The apoptosis effect on liver cancer cells of gold nanoparticles modified with lithocholic acid. Nanoscale Res Lett. 2018;13:304. doi: 10.1186/s11671-018-2653-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu T, Song X, Khan S, Li Y, Guo Z, Li C, Wang S, Dong W, Liu W, Wang B, Cao H. The gut microbiota at the intersection of bile acids and intestinal carcinogenesis: An old story, yet mesmerizing. Int J Cancer. 2020;146:1780–1790. doi: 10.1002/ijc.32563. [DOI] [PubMed] [Google Scholar]
- 93.Degirolamo C, Rainaldi S, Bovenga F, Murzilli S, Moschetta A. Microbiota modification with probiotics induces hepatic bile acid synthesis via downregulation of the Fxr-Fgf15 axis in mice. Cell Rep. 2014;7:12–18. doi: 10.1016/j.celrep.2014.02.032. [DOI] [PubMed] [Google Scholar]
- 94.Jones ML, Tomaro-Duchesneau C, Prakash S. The gut microbiome, probiotics, bile acids axis, and human health. Trends Microbiol. 2014;22:306–308. doi: 10.1016/j.tim.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 95.Polyzos SA, Kountouras J, Mantzoros CS. Obeticholic acid for the treatment of nonalcoholic steatohepatitis: Expectations and concerns. Metabolism. 2020;104:154144. doi: 10.1016/j.metabol.2020.154144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article, as no datasets were generated or analyzed during the current study.