

ABSTRACT
Inflammatory bowel disease (IBD) is a chronic life-long inflammatory disease affecting almost 2 million Americans. Although new biologic therapies have been developed, the standard medical treatment fails to selectively control the dysregulated immune pathways involved in chronic colonic inflammation. Further, IBD patients with uncontrolled colonic inflammation are at a higher risk for developing colorectal cancer (CRC). Intestinal microbes can impact many immune functions, and here we asked if they could be used to improve intestinal inflammation. By utilizing an intestinal adherent E. coli that we find increases IL-10 producing macrophages, we were able to limit intestinal inflammation and restrict tumor formation. Macrophage IL-10 along with IL-10 signaling to the intestinal epithelium were required for protection in both inflammation and tumor development. Our work highlights that administration of immune modulating microbes can improve intestinal outcomes by altering tissue inflammation.
KEYWORDS: Microbiota, intestinal inflammation, colorectal cancer, colitis-associated cancer, E. coli, IL-10, intestinal macrophages, intestinal epithelium
Introduction
Inflammatory bowel disease (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC), is a chronic inflammatory intestinal disease with no cure1. It affects almost 2 million people in the United States.2 IBD is thought to result from unchecked inflammation against the microbiota in a genetically susceptible individual.3 Although new biologic therapies have been developed, the standard medical treatment fails to selectively control the dysregulated immune pathways involved in chronic colonic inflammation.4 In IBD patients, changes in the microbiota are found, including expanded pro-inflammatory microbes such as Ruminococcus gnavus and proteobacteria, such as Escherichia coli and reduced anti-inflammatory microbes such as Bacteroidetes, Lachnospiraceae, and Faecalibacterium prausnitzii.5–8 This altered microbiota is thought to support disease progression and inflammatory changes.9 In parallel, genetic, and environmental factors can interfere with proper interaction between the host immune system and microbiota leading to enhanced inflammation.10,11
IBD patients with poorly controlled intestinal inflammation are at an elevated risk for CRC development.10–13 Furthermore, chronic intestinal inflammation corresponds with advanced disease stage and decreased survival in CRC.14,15 Animal models support a causative role for inflammation in CRC with inflammation promoting tumorigenesis.14
Numerous groups find the microbiota can both enhance and limit intestinal inflammation. Specifically, we and others find that microbiota-derived signals drive anti-inflammatory processes including macrophage IL-10 production,16 induction of naive and regulatory T cells,17,18 and fortification of the epithelial barrier.19–21 We previously identified that colonization with multiple human E. coli isolates could replace the microbiota in inducing macrophage IL-10 production, limiting pathology in a model of Salmonella infection16 but it remained unclear if such microbes could further enhance IL-10 and anti-inflammatory effects in the context of the normal microbiota.
Here, we find that colonization with the IL-10 inducing E. coli, strain 541–15, increased macrophage IL-10 production but only after intestinal damage. This increased IL-10 production after 541–15 colonization offered protection from intestinal injury as well as limited development of colitis-associated CRC (CAC). These data demonstrate that addition of select microbes is sufficient to shift immune function leading to improved outcomes in models of colitis, and offers insight into the role of gut microbes in intestinal inflammation and cancer.
Results
Intestinal colonization with E. coli 541-15 protects from colitis
As we previously found that colonization with E. coli 541–15 after microbiota depletion with antibiotics was sufficient to induce intestinal CX3CR1+ macrophage production of IL-10 which restricted Salmonella-specific Th1 cells,16 we asked if 541–15 protected from intestinal injury and inflammation in the presence of the steady-state SPF microbiota. We left E. coli-free specific-pathogen-free (SPF) C57BL/6 (B6) mice uncolonized or colonized with E. coli 541–15 2 days before treatment with dextran sodium sulfate (DSS). In this acute intestinal inflammation model, DSS treatment causes epithelial damage and inflammation in the intestine.22 E. coli colonization was confirmed by qPCR using E. coli specific 16s rRNA primers (Figure S1A). E. coli 541–15 colonization did not alter the overall composition of the microbiome (Figures S1B-S1D). We found that colonization with E. coli 541–15 prevented weight loss and colon shortening, and improved histopathology with less incidence of ulcerations and fewer neutrophil clusters in lamina propria and epithelium (Figures 1A-1d). We also found decreased expression of pro-inflammatory markers lipocalin-2 and myeloperoxidase in fecal samples from E. coli 541–15 colonized mice as well as reduced expression of tumor necrosis factor alpha supporting reduced colonic inflammation (Figures 1E, 1f and S1E). As intestinal inflammation is linked with increased gut permeability, we assayed for intestinal permeability after DSS treatment by gavaging mice with fluorescein isothiocyanate (FITC) conjugated dextran (FITC-dextran) and measuring serum FITC-dextran levels.23 DSS-treated mice had increased serum FITC-dextran levels that were reduced in 541–15 colonized mice demonstrating decreased intestinal permeability (Figure 1g). Decreased permeability reflects improved intestinal barrier integrity, likely the consequence of lower intestinal inflammation and improved repair.24–26 We also characterized immune infiltration in DSS-treated E. coli 541–15 colonized mice. In the colon lamina propria, E. coli 541–15-colonized mice had decreased immune infiltration (Figure 1h) with decreased Th1 cells, CTLs, and neutrophils as well as increased Tregs and no change in macrophages or monocytes (Figures 1I-1m). Supporting these findings, in the mesenteric lymph node (MLN), E. coli 541–15-colonized mice had decreased IFNγ-producing CD4+ T cells and ILCs (Figure S1F). Our mice are segmented filamentous bacteria (SFB) free and so we do not see T helper 17 (Th17) cell responses, and these are not altered by 541–15 colonization (Figure S1F). To test if 541–15 enhanced IL-10 production in the presence of the SPF microbiota, we colonized E. coli-free SPF IL-10-GFP reporter mice (Vert-X) with 541–15 and left untreated or treated with 2% DSS for 7 days and used flow cytometry to detect IL-10+ cells.27,28 As previously described27,28 we find CX3CR1+ macrophages to be the main source of IL-10 (Figure S2A). However, 541–15 colonization did not enhance IL-10 production in the presence of the SPF microbiota (Figure S2B). In contrast, after DSS treatment, we find increased IL-10+ cells in the colon of 541–15 colonized mice with increased IL-10 expression by CX3CR1+ macrophages (Figures 2A and 2b).
Figure 1.
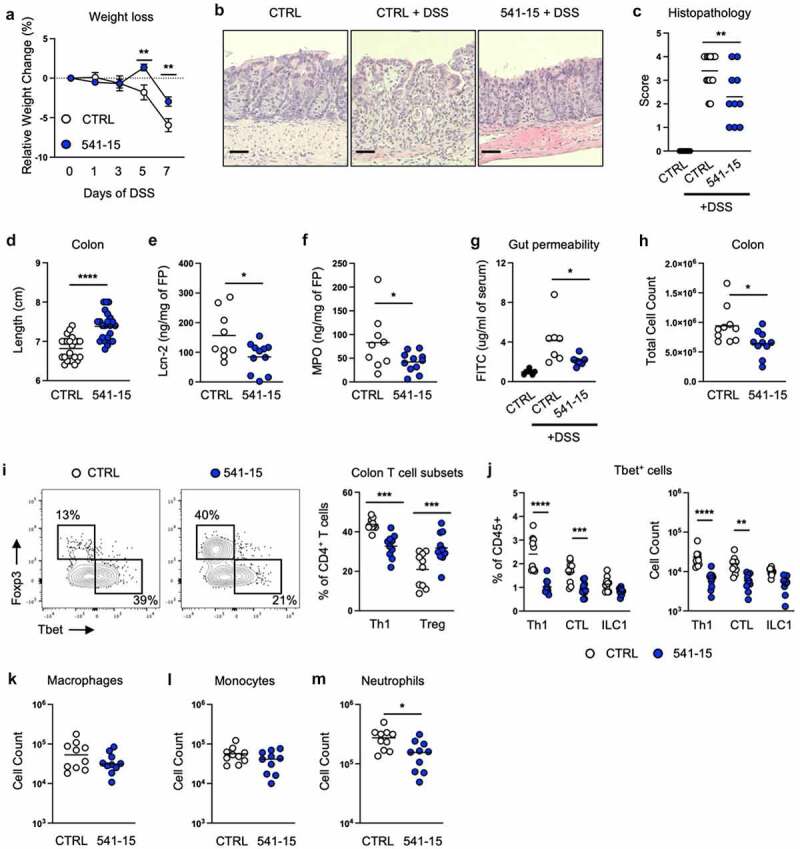
Intestinal colonization with E. coli 541–15 protects from colitis. Wildtype mice were colonized with E. coli 541–15 or gavaged with LB broth (CTRL) and 3 days later 2% DSS was provided in drinking water. At day 7 of DSS treatment, samples were collected. (a) Relative weight change. (b) Representative H&E staining of distal colons. Scale bars represent 50 μm. (c) Histopathology scores. (d) Colon lengths. (e) Lipocalin-2 (Lcn-2) and (f) Myeloperoxidase (MPO) concentration in fecal pellets. At day 7 mice were gavaged with FITC-dextran and 3 hours later blood was collected. (g) FITC concentration in serum. Colon lamina propria cells were isolated. (h) Number of immune cells. (i) Flow cytometry and frequencies of Th1 and Tregs. (j) Frequencies and counts of total Th1 cells, CTLs, and ILC1s. Counts of (k) Macrophages, (l) Monocytes, and (m) Neutrophils. Each replicate is a biologically independent sample. Individual dots represent samples from individual mice. Data are shown as individual values and mean, compared by two-tailed unpaired t-test or two-way ANOVA with Fisher’s LSD post hoc test. The results are representative of at least two independent experiments. *P < .05 was considered statistically significant; **P <.01; ***P < .001; ****P < .0001. See also Figure S1.
Figure 2.
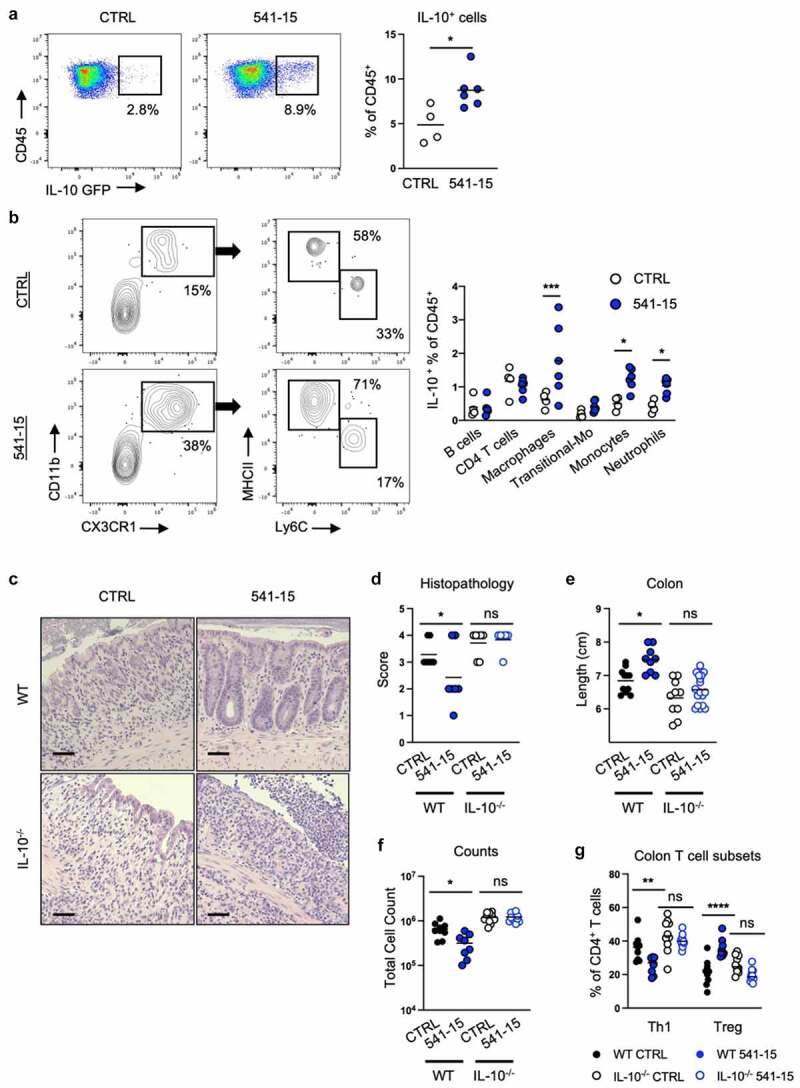
Intestinal colonization with E. coli 541–15 prevents colitis through IL-10 induction. Vert-X mice were colonized with E. coli 541–15 or gavaged with LB broth (CTRL) and 3 days later 2% DSS was provided. At day 7 of DSS treatment, samples were collected. Flow cytometry and frequencies of (a) total IL-10+ immune cells from the colonic lamina propria and (b) identification of IL-10+ immune cells. IL-10 deficient (IL-10-/-) and wildtype (WT) littermate mice were colonized with E. coli 541–15 or gavaged with LB broth (CTRL) and 3 days later 2% DSS was provided. At day 7 of DSS treatment, samples were collected. (c) Representative H&E staining of distal colons. Scale bars represent 50 μm. (d) Histopathology scores. (e) Colon lengths. Colon lamina propria cells were isolated. (f) Number of immune cells. (g) Frequencies of Th1 and Tregs. Each replicate is a biologically independent sample. Individual dots represent samples from individual mice. Data are shown as individual values and mean, compared by two-tailed unpaired t-test or two-way ANOVA with Fisher’s LSD post hoc test. The results are representative of at least two independent experiments. *P < .05 was considered statistically significant; **P <.01; ***P < .001; ****P < .0001. See also Figure S2.
Intestinal colonization with E. coli 541–15 prevents colitis by inducing IL-10 production by CX3CR1+ macrophages which targets the intestinal epithelium
IL-10 deficient mice develop microbiota-driven spontaneous colitis and also show increased sensitivity to DSS.29 To assess the requirement for IL-10 in colitis protection downstream of 541–15 colonization, we left IL-10 wildtype and knockout (IL-10−/−) littermate mice uncolonized or colonized with E. coli 541–15 before DSS treatment. As above, 541–15 colonization improved colitis in wildtype mice (Figures 2C-2g and S2C-S2H). However, in IL-10 knockout mice 541–15 colonization did not improve colitis outcomes including no effect on colon length, histopathology scores, or immune infiltration (Figures 2C-2g and S2C-S2H), supporting the requirement of IL-10 in protection after 541–15 colonization.
To assess the role of CX3CR1+ macrophages in 541–15 protection from colitis, we used mice where the diphtheria toxin receptor (DTR) is expressed from the Cx3cr1 locus (CX3-DTR mice).30 In these mice, all CX3CR1+ cells are depleted after diphtheria toxin (DT) injection. Colonization of these mice with 541–15 before DSS treatment did not induce changes in colon length or infiltrating immune cells (Figures S3A-S3D). This is likely because DT treatment depleted both CX3CR1+ monocytes that drive pathology in colitis and CX3CR1+ macrophages that are important for barrier repair.31 Therefore, we used mice where a floxed-stop cassette upstream of the DTR was inserted into the Cx3cr1 locus (CX3-STOP-DTR). Crossing these mice to CD11c-Cre allows for depletion of CX3CR1+ macrophages and dendritic cells (DCs) but not monocytes or other CX3CR1-expressing cells after DT injection.32 After 541–15 colonization and DSS treatment, mice lacking CX3CR1+ macrophages and DCs had shorter colons with increased immune infiltration including more Th1 cells, CTLs, ILC1s, and fewer Tregs as compared to littermate controls (Figures S3E-S3H).
We next used mice lacking IL-10 production by CX3CR1+ cells to test if CX3CR1+ cell IL-10 was required for colitis protection after E. coli 541–15 colonization. For these experiments, mice with a conditional allele for IL-10 were crossed to the tamoxifen-inducible CX3CR1-CreERT2.33 Tamoxifen treatment results in loss of IL-10 production by CX3CR1+ cells in IL-10flox/- compared to IL-10flox/+ littermate control CX3CR1-CreERT2 mice. As compared to controls, in DSS treated 541–15 colonized mice whose CX3CR1+ cells cannot produce IL-10, we found loss of protection by E. coli 541–15 colonization with increased weight loss, shorter colon length, and increased pathology characterized by significant inflammatory infiltrate and ulcerations (Figures 3A-3c and S3I). We also found increased immune infiltration with increased Th1 cells, ILC1s, and neutrophils (Figures 3D, 3e, and S3J-S3L). In contrast, we found T cell IL-10 was dispensable as we found complete protection after 541–15 colonization of mice where T cells could not produce IL-10, with improved colon length and histopathology after 541–15 colonization (Figures S3M-S3R).
Figure 3.
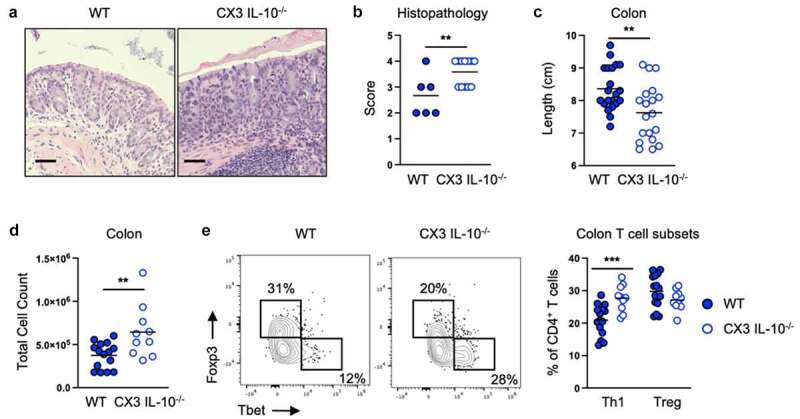
Intestinal colonization with E. coli 541–15 prevents colitis by inducing IL-10 production by CX3CR1+ macrophages. IL-10flox/- (CX3 IL-10-/-) and IL-10flox/+ (WT) littermate CX3CR1-CreERT2 mice were colonized with E. coli 541–15 and 3 days later 2% DSS was provided. At day 7 of DSS treatment, samples were collected. 3 days before colonization and every 2 days throughout the experiment 4OHT was injected to both groups. (a) Representative H&E staining of distal colons. Scale bars represent 50 μm. (b) Histopathology scores. (c) Colon lengths. Colon lamina propria cells were isolated. (d) Number of immune cells. (e) Flow cytometry and frequencies of Th1 and Tregs. Each replicate is a biologically independent sample. Individual dots represent samples from individual mice. Data are shown as individual values and mean, compared by two-tailed unpaired t-test or two-way ANOVA with Fisher’s LSD post hoc test. The results are representative of at least two independent experiments. *P < .05 was considered statistically significant; **P <.01; ***P < .001. See also Figure S3.
To identify the cellular target of CX3CR1+ macrophage IL-10 after E. coli 541–15 colonization we next analyzed mice in which epithelial cells, CX3CR1+ cells, or T cells lack expression of the IL-10 receptor α (IL-10 Rα). Suppression of colitis by E. coli 541–15 was intact when mice lacked IL-10Rα expression in T cells or CX3CR1+ cells (Figures S4A-S4L). In contrast, we found epithelial IL-10Rα is required as 541–15 was no longer protective after DSS treatment in LGR5-CreERT2+ IL-10Rαflox/flox mice as compared to littermates without LGR5-CreERT2, as seen by increased weight loss, shorter colon length, increased histopathology scores, and increased epithelial permeability (Figures 4A-4c, S4M, and S4N). In these mice we found increased immune infiltration with increased Th1 cells (Figures 4D, 4e, and S4O-S4Q). In addition, fecal lipocalin-2 and myeloperoxidase were increased in DSS-treated 541–15 colonized mice when epithelial cells lack IL-10Rα (Figures S4R and S4S). In epithelial cells, IL-10 triggers repair pathways including WNT signaling that leads to downstream expression of the pluripotency gene nanog which supports cell proliferation and epithelial barrier repair.34,35 Confirming the role of IL-10 in epithelial cells, we find that E. coli 541–15 colonization increased nanog expression in the colon of DSS-treated wild-type mice (Figure S4T).
Figure 4.
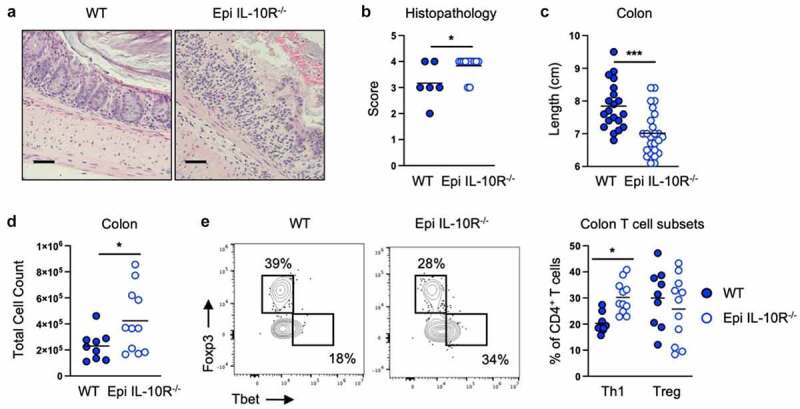
Intestinal colonization with E. coli 541–15 prevents colitis by modulating IL-10 signaling to intestinal epithelium. LGR5-CreERT2+ (Epi IL-10R-/-) and LGR5- CreERT2- (WT) littermate IL-10Raflox/flox mice were colonized with E. coli 541–15 and 3 days later 2% DSS was provided. At day 7 of DSS treatment, samples were collected. 7 days before colonization 4OHT was injected on two consecutive days to both groups. (a) Representative H&E staining of distal colons. Scale bars represent 50 μm. (b) Histopathology scores. (c) Colon lengths. Colon lamina propria cells were isolated. (d) Number of immune cells. (e) Flow cytometry and frequencies of Th1 and Tregs. Each replicate is a biologically independent sample. Individual dots represent samples from individual mice. Data are shown as individual values and mean, compared by two-tailed unpaired t-test or two-way ANOVA with Fisher’s LSD post hoc test. The results are representative of at least two independent experiments. *P < .05 was considered statistically significant; ***P < .001. See also Figure S4.
Intestinal colonization with E. coli 541-15 protects from colitis-associated colorectal cancer
As intestinal inflammation is a risk for CAC development and dysbiosis characterized by enriched mucosa adherent bacteria such as E. coli is seen during inflammatory conditions in human IBD patients, in mouse IBD models, and in colon cancer,10,11 we asked if colonization with E. coli 541–15 modulated an inflammation-driven mouse model of CAC. 6-week-old E. coli-free SPF B6 mice were subjected to a chemically induced colitis associated carcinoma model. In this colon tumor model, mice are treated with azoxymethane (AOM), a mutagen, followed by three cycles of 2% DSS.36 Under the AOM/DSS model, mice develop tumors in the distal colon as early as 1 week after the second cycle of DSS.36 We colonized mice with E. coli 541–15 before the first DSS administration and assessed tumor formation 4 weeks after the last DSS cycle (Figure S5A). As predicted from its anti-inflammatory effect,16 we found E. coli 541–15 was protective with reduced number and size of tubular adenomas (Figures 5A and 5b). Spleen size was also reduced indicating reduced systemic inflammation (Figure S5B).
Figure 5.
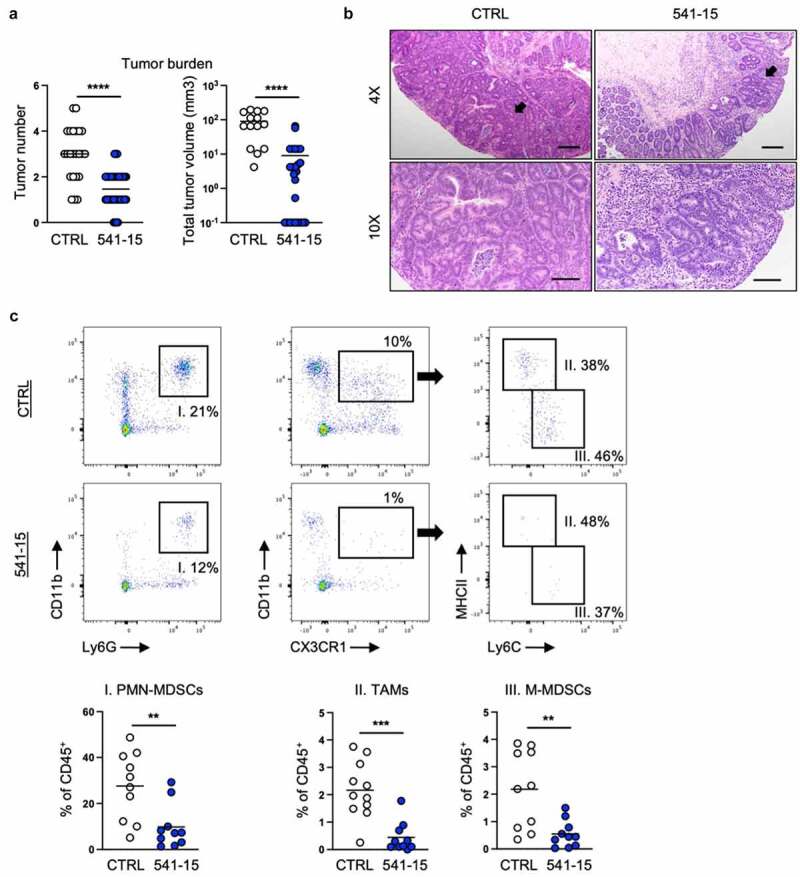
Intestinal colonization with E. coli 541–15 protects from colitis-associated colorectal cancer. Wildtype mice were administered AOM followed by 3 cycles of 2% DSS. Before administration of DSS mice were colonized with E. coli 541–15 or gavaged with LB broth (CTRL) and samples were collected as indicated in Figure S1A. (a) Number and size of colonic tumors. (b) Representative H&E staining of distal colons. Black arrow shows a tubular adenoma. Scale bars represent 4x = 200 μm and 10x = 100 μm. Tumors were individually collected, digested, and the tumor microenvironment was analyzed by flow cytometry. (c) Flow cytometry and frequencies of PMN-MDSCs, TAMs, and MMDSCs. Each replicate is a biologically independent sample. Individual dots represent samples from individual mice. Data are shown as individual values and mean, compared by two-tailed unpaired t-test. The results are representative of at least two independent experiments. *P < .05 was considered statistically significant; **P <.01; ***P < .001; ****P < .0001. See also Figures S5 and S6.
We further assessed how 541–15 colonization affected the immune microenvironment within the tumors (tumor microenvironment, TME) and surrounding tissue. Using flow cytometry, we found decreased infiltration of myeloid cells including tumor associated macrophages (TAMs), mononuclear myeloid-derived suppressor cells (M-MDSCs), and polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) in tumors of mice colonized with 541–15 (Figures 5C and S5C). TAMs both inhibit anti-tumor T cell responses and directly promote tumor growth and angiogenesis.37 M-MDSCs resemble monocytes while PMN-MDSCs are similar to neutrophils and both inhibit T cell recruitment and suppress T cell responses within the tumor.38,39 We also found changes in recruitment of lymphoid cells to tumors in 541–15 colonized mice. As compared to control mice, mice colonized with E. coli 541–15 had increased T helper 1 (Th1) cells, cytotoxic T lymphocytes (CTLs), and type 1 innate lymphoid cells (ILC1s), and decreased regulatory T cells (Tregs) within tumors (Figures S5D and S5E). This shift in immune infiltration supports that 541–15 is restricting tumor development as smaller, better controlled tumors have increased T effector cells as compared to Tregs and MDSCs.37 In contrast to tumor tissue, in tumor adjacent colonic tissue from 541 to 15 colonized mice we found the opposite effects with decreased Th1 cells and increased Tregs as compared to uncolonized mice (Figure S5F). This indicated 541–15 induced tumor-specific changes in infiltrating immune populations. Interestingly, as in DSS-induced colitis, E. coli 541–15 colonization did not alter microbiota composition in mice under the CAC model (Figures S6A and S6B). This indicated that immune changes leading to protection against CAC were specific for 541–15 and is not attributed to changes in other microbial populations.
E. coli 541–15 modulates tumorigenesis through intestinal epithelium responses to IL-10 production from CX3CR1+ macrophages
We next assessed if colonization with E. coli 541–15 prevented CAC through the same pathways through which it protected from intestinal inflammation. IL-10flox/+ and IL-10flox/- CX3CR1-CreERT2 mice under the AOM/DSS model were colonized with 541–15 and concomitantly treated with tamoxifen (Figure S6C). Loss of CX3CR1+ cell IL-10 during DSS treatment in the presence of E. coli 541–15 led to increased tumor number and size likely due to increased inflammation as described above (Figure S6E). No change in tumor number or size was observed in mice lacking IL-10 production by T cells colonized with 541–15 (Figure S6E).
To assess the role of epithelial IL-10 signaling in E. coli 541–15 modulation of tumorigenesis, we used mice in which epithelial cells are deficient for IL-10Rα after treatment with tamoxifen. IL-10Rαflox/flox mice with and without LGR5-CreERT2 were colonized with 541–15 and simultaneously treated with tamoxifen followed by treatment with AOM/DSS (Figure S6D). Similar to mice in which CX3CR1+ cells do not produce IL-10, 541–15 colonized mice lacking epithelial IL-10Rα had more frequent, larger tumors (Figure S6E). We saw no change in tumor number or size when CX3CR1+ or T cells lacked IL-10Rα (Figure S6E).
These results identify that microbiota modulation of macrophage anti-inflammatory function has direct outcomes on inflammation and CAC development. IL-10 produced primarily by CX3CR1+ macrophages is sensed by epithelial cells potentially triggering epithelial repair processes helping in the resolution of inflammation and protecting from tumor development34 (Figure S7A).
Discussion
Understanding the role of tissue inflammation is crucial to preventing and treating IBD, a chronic disease linked with changes in the gut microbial landscape that are thought to drive pathogenesis.5–9 Here, we find that colonization with a specific intestinal microbe restricts inflammation in a macrophage-dependent manner.
We and others previously demonstrated that the intestinal microbiota is a key regulator of intestinal macrophage inflammatory potential with the microbiota required for IL-10 production by intestinal macrophages.16 Particularly, we demonstrated that colonization with E. coli 541–15 was sufficient to restore CX3CR1+ macrophage IL-10 production in microbiota depleted mice.16 Here, we find that 541–15 reduces intestinal pathology after DSS treatment. 541–15 colonization followed by DSS treatment results in enhanced macrophage IL-10 in the presence of the normal microbiota. Macrophage IL-10 production is required for protection as 541–15 is unable to protect from pathology in IL-10 deficient mice or in mice lacking IL-10 production by CX3CR1+ macrophages. This confirms a critical role for microbial regulation of macrophage IL-10 in controlling intestinal inflammation.
We also find that intestinal epithelial cell response to IL-10 was required downstream of 541–15 colonization. In epithelial cells, IL-10 triggers repair pathways including WNT signaling pathways that support epithelial barrier repair further limiting tissue inflammation.34,35 Together, colonization with 541–15 protected from colitis only when intestinal epithelial cells were able to sense IL-10, highlighting the importance of crosstalk between macrophages and epithelial cells to support recovery from epithelial injury.
We next asked how this shift toward barrier support by 541–15 colonization regulated tumor development in a CAC model. In CRC, evidence supports that increased intestinal inflammation enhances intestinal tumors,40,41 likely driven by environmental and lifestyle changes that can interfere with proper host–microbiota interactions, leading to unresolved damage and unchecked inflammation that promotes CRC.13 In inflammatory disease, changes in microbiota composition can lead to lost regulatory capacity including decreased IL-10 production, that promotes or sustains inflammation.16,42,43 Recent studies identify select microbes as increased in CRC.10,11 One bacterium associated with worse CRC outcome is the attaching bacterium Fusobacterium nucleatum,10 which induces cell proliferation and modulation of the tumor microenvironment without altering inflammation.44 We find that enrichment of E. coli 541–15, an adherent E. coli isolated from the intestine of an IBD patient,45,46 also affects CRC. However, contrary to F. nucleatum, 541–15 limits tumorigenesis by engaging IL-10 signaling pathways that suppress inflammation. Macrophages are known to produce additional factors that can promote tumor development. This includes transforming growth factor β1 (TGF-β1), insulin-like growth factor 1 (IGF-1), and vascular endothelial growth factor α (VEGF-α), which promote cellular proliferation and blood-vessel development.47 It remains to be seen if 541–15 activates these pathways in addition to IL-10. As predicted by its ability to promote macrophage IL-10, colonization with 541–15 before induction of colitis in the mouse model of CAC led to reduced tumor incidence with fewer, smaller tumors in colonized mice. As in colitis, decreased tumorigenesis depended on intestinal macrophage IL-10 production and IL-10 sensing by the intestinal epithelium.34,35
Our findings suggest that modulating the function of CX3CR1+ macrophages represents a therapeutic avenue to decrease inflammation that can lead to CAC in IBD patients.48,49 Although targeting specific microbes may offer therapeutic advantages, much work remains to be done to better characterize IBD-associated microbes and the downstream pathways they activate. Further studies should assess if using probiotics with immunomodulatory properties have therapeutic potential that provides protection against IBD and IBD-related cancer.
Materials and methods
Mice
All mice were bred in house at the animal facility of Baylor College of Medicine or Memorial Sloan Kettering Cancer Center and maintained as E. coli free. The colony is routinely tested by qPCR and immunophenotyping to confirm lack of E. coli colonization. C57BL/6 J (Jax #000664), IL10-GFP (Vert-X) (JAX #014530), IL-10KO (JAX #003968), CD11c-Cre (JAX #008068), LRG5-CreERT2 (JAX# 008875), CX3CR1-CreERT2 (JAX #021160),33 and CD4-Cre (JAX #017336), mice were originally purchased from Jackson laboratory before being bred in house. CX3CR1-STOP-DTR (JAX #025629),32 CX3CR1-DTR,30 IL-10 flox,50 and IL-10Rα flox51 were previously described. See also Table S1. All mice were crossed at least 12 generations to the C57BL/6 J background. All mouse experiments were performed with mice between 6–20 weeks of age with males and females at similar ratios, unless otherwise specified. Littermate controls were used for each experiment, and mice were randomly assigned to experimental groups. All experiments were performed in accordance with approved protocols by the Institutional Animal Care and Usage Committee at Baylor College of Medicine and Memorial Sloan Kettering Cancer Center.
Induction of colitis and colonic tumors
Induction of colitis was performed by administering dextran sulfate sodium salt (DSS, mol wt 40 kDa; Alfa Aesar).22 Mice were administered 2% DSS in their drinking water for 7 days. Mice were weighed daily and analyzed after 7 days of DSS. For induction of colonic tumors, mice received a single injection of azoxymethane (AOM, Sigma) at 10 mg/kg body weight intraperitoneally. One week later, mice were treated with 3 cycles of 2% DSS (7 days) followed by 7 days of regular water. 4–5 weeks after the last cycle of DSS mice were analyzed.36
Colonization of mice with E. coli
E. coli (EC) 541–15 was cultured overnight in LB media. Mice with a SPF microbiota were colonized with a single gavage of 108 CFU of mouse commensal E. coli. Colonization was confirmed by qPCR with E. coli-specific primers using 16S (for fecal pellets) as housekeeping gene. Primers used were: 16S F: CGGTGAATACGTYCGG, 16S R: GGWTACCTTGTTACGACTT,52 E. coli F: GGTAGAGCACTGTTTTGGCA, E. coli R: TGTCTCCCGTGATAACTTTCT.53
Diphtheria toxin (DT) and 4-hydroxytamoxifen (4OHT) administration
Mice were injected every other day with 200ng diphtheria toxin (DT, Sigma) in PBS. (Z)-4-Hydroxytamoxifen, 98% Z isomer (4OHT, Sigma) was resuspended to 20 mg/ml in ethanol by heating to 37°C. 4OHT was then diluted in corn oil (Sigma) to 2 mg/ml and mice were injected every 3 days with 100ul. All injections were intraperitoneal.
In vivo gut permeability assay with Fluorescein isothiocyanate (FITC)-dextran
Mice were weighed and fasted for 3 hours prior to oral FITC-dextran (4 kDa, Sigma) administration. A total concentration of 250 mg/kg body weight was administered through gavage. 3 hours after administration, blood samples were collected through retro-orbital bleeding and 50 ul of serum was placed in a fluorescence plate reader to determine the concentration by fluorescence excitation at 495 nm/519 nm reference.
DNA isolation and real-Time quantitative PCR (qPCR) analysis
For bacterial DNA identification, fecal pellets were collected and stored at −80°C. DNA was isolated using the DNeasy PowerSoil Kit according to the manufacturer’s protocol (Qiagen). Real-time qPCR was performed using SYBR Green Supermix (Bio-Rad Laboratories) in the Bio-Rad thermocycler 384 well plates. Thermocycling program was 40 cycles at 95°C for 15s, 60°C for 30s, and 72°C for 30s, with an initial cycle of 95°C for 120s. Primers used were described above. Relative expression of target gene was determined using a standard curve to calculate the number of nucleotides per sample and the ΔΔCT method.
Cell isolation
Intestinal lamina propria cells were isolated as previously described.16,32,54 Briefly, mouse large intestines were washed in PBS, once with freshly prepared 30 mM EDTA and 1 mM DTT and once with 30 mM EDTA (both at 37°C), and then digested at 37°C in collagenase 8 (100 U/ml, Sigma) and DNase-containing (150 µg/ml, Sigma) media with 10% fetal bovine serum. Digested material was separated on a 40%/80% Percoll (GE Health) gradient. Tumors were digested as above, washed in PBS, and passed through a 40um strainer.
Antibodies, cell staining, and flow cytometry
Cells isolated from colon lamina propria and tumors were blocked with CD16/32 (Fc receptor) to prevent nonspecific antibody binding. Then, cells were surface-stained in FACS buffer (PBS, 2 mM EDTA, 2% fetal bovine serum) for 30 minutes in the dark at 4°C with fluorescently conjugated antibodies from BD, eBiosciences, or BioLegend specific to CD45 (30-F11), TCR β (H57-597), CD11b (M1/70), CD11c (N418), MHCII (M5/114.15.2), Ly6C (AL-21), CX3CR1 (SA011 F11), Ly6G (1A8), B220 (RA3-6B2), CD3 (145–2C11), CD90.2 (53–2.1), CD4 (GK1.5), and CD8 (53–6.7). For T cell subsets identification, after staining for surface antigens, cells were fixed and permeabilized using a Fixation/Permeabilization Solution Kit (ThermoFisher) overnight at 4°C and stained with antibodies specific to mouse FOXP3 (FJK-16s) or T-bet (4B10) for 45 minutes at room temperature. To assess cytokine production after ex vivo restimulation, single-cell suspensions from mesenteric lymph nodes were incubated for 4 hours at 37°C with 5% CO2 in the presence of 50 ng/mL PMA and 500 ng/mL ionomycin with 2 μM monensin (BD Biosciences). Flow cytometric analysis was performed on an LSR II (BD Biosciences) or Aurora (Cytek) and analyzed using FlowJo software (Tree Star Inc.). DAPI or live/dead fixable blue dead cell stain kit (ThermoFisher) was used to exclude dead cells. Total cell counts were determined with Precision Count Beads (BioLegend). Gating strategy is shown in Figure S5C. PMN-MDSCs were CD11b+Ly6G+, TAMs were CD11b+CX3CR1+MHCII+Ly6C−, M-MDSCs were CD11b+CX3CR1+MHCII−Ly6C+, Th1 cells were CD3+TCRβ+CD4+CD8−Tbet+, CTLs were CD3+TCRβ+CD4−CD8+Tbet+, ILC1s were CD3−TCRβ−CD90.2+Tbet+, and Tregs were CD3+TCRβ+CD4+CD8−Foxp3+. All antibody information is listed below:
| Antibody, Fluorochrome, Clone | Source | Catalog number |
|---|---|---|
| Anti-mouse MHCII (I-A/I-E), Pacific blue, clone M5/114.15.2 | BioLegend | Cat# 107,620 |
| Anti-mouse CD11c, PEcy7, clone N418 | BioLegend | Cat# 117,317 |
| Anti-mouse/human CD11b, APC/Cy7, clone M1/70 | BioLegend | Cat# 101,226 |
| Anti-mouse CX3CR1, FITC, clone SA011 F11 | BioLegend | Cat# 149,020 |
| Anti-mouse Ly6C, Alexa Fluor 700, clone AL-21 | BD | Cat# 561,237 |
| Anti-mouse Ly6G, PE, clone 1A8 | BioLegend | Cat# 127,608 |
| Anti-mouse CD4, Brilliant Violet 510, clone GK1.5 | BioLegend | Cat# 100,449 |
| Anti-human/mouse T-bet, PEcy7, clone 4B10 | eBiosciences | Cat# 25–5825-82 |
| Anti-mouse CD3, APC/Cy7, clone 145–2C11 | BioLegend | Cat# 100,330 |
| Anti-mouse TCRβ, Percp/Cy5.5, clone H57-597 | BioLegend | Cat# 109,228 |
| Anti-mouse/rat Foxp3, eFluor660, clone FJK-16s | eBiosciences | Cat# 50–5773-82 |
| Anti-mouse CD45, Brilliant Violet 785, clone 30-F11 | BioLegend | Cat# 103,149 |
| Anti-human/mouse B220, Brilliant Violet 650, clone RA3-6B2 | BioLegend | Cat# 103,241 |
| Anti-mouse CD90.2, BUV395, clone 53–2.1 | BD | Cat# 565,257 |
| Anti-mouse CD8, APC, clone 53–6.7 | BioLegend | Cat# 100,712 |
| Anti-mouse IFNg, FITC, clone XMG1.2 | BioLegend | Cat# 505,806 |
| Anti-mouse IL-17A, BV421, clone TC11-18H10.1 | BioLegend | Cat# 506,926 |
Enzyme-linked immunosorbent assays (ELISAs)
Fecal lipocalin-2 (Lcn-2) and myeloperoxidase (MPO) were determined by ELISA (R&D Systems). Fecal samples were solubilized in PBS with protease inhibitors at 10 mg/100 µl using a Mixer Mill.
Histology
Distal colons were fixed in 10% neutral buffered formalin, routinely processed, sectioned at 6 mm, and stained with hematoxylin and eosin (H&E) for light microscopic examination. Images were taken at 40X or 100X magnification using a ZEISS Axio Observer 2. Samples were assessed in a blinded fashion and scored 0 to 4 based on the criteria previously described:55 (grade 0) Minimal chronic inflammatory infiltrate; (grade 1) Chronic inflammatory infiltrate; (grade 2) Few or rare neutrophils in lamina propria or epithelium; (grade 3) Multiple clusters of neutrophils in lamina propria and/or epithelium; (grade 4) Ulceration.
16S rDNA high-Throughput sequencing
Mouse fecal samples were collected sterilely and stored at −80 C. DNA was extracted utilizing Promega Max Prep & Promega Maxwell RSC 48 Instrument. The V4 and V5 regions of the 16S rDNA were PCR amplified, normalized, pooled, and sequenced using the Illumina MiSeq platform with 2 × 300 bp paired end reads. Analysis of 16S rDNA sequencing reads was performed using the software USEARCH (version 11.0.667) assigning taxonomy using the RDP 16S training set (version 16) as reference database. Merge reads with an expected error number over 1.0 were filtered. Filtered operational taxonomic units (OTUs) were rarefied to a depth of 17,481 reads per sample. Extraction, sequencing, and data processing were performed by the Microbiome Core Lab of Weill Cornell Medicine.
Statistics and reproducibility
Statistical analysis was performed using GraphPad Prism. All data are presented as individual values and mean. Two-tailed unpaired Student’s t-test using a 95% confidence interval was used to evaluate the difference between two groups. For more than two groups, One-way ANOVA was used. For more than two groups under different conditions Two-way ANOVA was used. A probability value of p < .05 was considered significant. Statistical significance is indicated in each figure. Each experiment was repeated independently at least 2 times with similar results. Representative flow plots and micrographs were selected from the pool of biological replicates indicated in their respective quantifications.
Supplementary Material
Acknowledgments
We thank all members of the Diehl and Longman labs for their critical input on the manuscript.
Funding Statement
This work was supported by the NIH AI125264 (G.E.D.), Rainin Foundation (G.E.D.), Ludwig Center at Memorial Sloan Kettering (D.F.Z.), AAI Careers in Immunology Fellowship (M.K.), MSK Cancer Center Core Grant (P30 CA 008748), the Cytometry and Cell Sorting Core at Baylor College of Medicine (P30 AI036211, P30 CA125123, and S10 RR024574), and the Microbiome Core Lab of Weill Cornell Medicine; Ludwig Family Foundation;
Disclosure statement
The authors declare no conflict of interests.
Contributions
D.F. Zegarra-Ruiz, R.S. Longman, and G.E. Diehl designed the experiments and analyzed data. D.F. Zegarra-Ruiz and G.E. Diehl wrote the manuscript with input from all co-authors. D.F. Zegarra-Ruiz, D.V. Kim, K. Norwood, F.B. Saldana-Morales, M. Kim, C. Ng, R. Callaghan, M. Uddin, L. Chang, and G.E. Diehl conducted experiments and analyzed data.
Data availability
The authors confirm that the data supporting the findings of this study are available within the
article and/or its supplementary materials. 16S sequencing data are openly available in NCBI under BioProject ID PRJNA845002.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2022.2119054
References
- 1.Baumgart DC, Carding SR.. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369(9573):1627–15. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 2.Alatab S, Sepanlou SG, Ikuta K, Vahedi H, Bisignano C, Safiri S, Sadeghi A, Nixon MR, Abdoli A, Abolhassani H, et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: a systematic analysis for the global burden of disease study 2017. Lancet Gastroenterology Hepatology. 2020;5:17–30. doi: 10.1016/S2468-1253(19)30333-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caruso R, Lo BC, Núñez G. Host–microbiota interactions in inflammatory bowel disease. Nat Rev Immunol. 2020;20:411–426. doi: 10.1038/s41577-019-0268-7. [DOI] [PubMed] [Google Scholar]
- 4.Pithadia AB, Jain S. Treatment of inflammatory bowel disease (IBD). Pharmacol Rep. 2011;63:629–642. doi: 10.1016/S1734-1140(11)70575-8. [DOI] [PubMed] [Google Scholar]
- 5.Dalal SR, Chang EB. The microbial basis of inflammatory bowel diseases. J Clin Investigation. 2014;124:4190–4196. doi: 10.1172/JCI72330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frank DN, Amand ALS, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc National Acad Sci. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peterson DA, Frank DN, Pace NR, Gordon JI. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe. 2008;3:417–427. doi: 10.1016/j.chom.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu W-JH, Zegarra-Ruiz DF, Diehl GE. Intestinal microbes in autoimmune and inflammatory disease. Front Immunol. 2020;11:597966. doi: 10.3389/fimmu.2020.597966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elson CO, Cong Y. Host-microbiota interactions in inflammatory bowel disease. Gut Microbes. 2012;3:332–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host Microbe. 2014;15(3):317–328. doi: 10.1016/j.chom.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Pitmon E, Wang K. Microbiome, inflammation and colorectal cancer. Semin Immunol. 2017;32:43–53. doi: 10.1016/j.smim.2017.09.006. [DOI] [PubMed] [Google Scholar]
- 12.Longo DL, Beaugerie L, Itzkowitz SH, Longo DL. Cancers complicating inflammatory bowel disease. New Engl J Medicine. 2015;372:1441–1452. doi: 10.1056/NEJMra1403718. [DOI] [PubMed] [Google Scholar]
- 13.Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13:759–771. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- 14.Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation, and Cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siegel RL, Miller KD, Sauer AG, Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA, Jemal A. Colorectal cancer statistics, 2020. Ca Cancer J Clin. 2020;70:145–164. doi: 10.3322/caac.21601. [DOI] [PubMed] [Google Scholar]
- 16.Kim M, Galan C, Hill AA, W-J W, Fehlner-Peach H, Song HW, Schady D, Bettini ML, Simpson KW, Longman RS, et al. Critical role for the microbiota in CX3CR1+ intestinal mononuclear phagocyte regulation of intestinal T cell responses. Immunity [Internet]. Available from. 2018;49:151–163.e5. https://linkinghub.elsevier.com/retrieve/pii/S1074761318302450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zegarra-Ruiz DF, Kim DV, Norwood K, Kim M, W-JH W, Saldana-Morales FB, Hill AA, Majumdar S, Orozco S, Bell R, et al. Thymic development of gut-microbiota-specific T cells. Nature. 2021;594:413–417. doi: 10.1038/s41586-021-03531-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, et al. Induction of colonic regulatory T Cells by indigenous clostridium species. Science. 2011;331:337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamada N, Seo S-U, Chen GY, Núñez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13:321–335. doi: 10.1038/nri3430. [DOI] [PubMed] [Google Scholar]
- 20.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr Protoc Immunol. 2014;104:15.25.1–15.25.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.B-R L, Wu J, H-S L, Jiang Z-H, Zhou X-M, C-H X, Ding N, Zha J-M, W-Q H. In vitro and in vivo approaches to determine intestinal epithelial cell permeability. J Vis Exp. 2018. doi: 10.3791/57032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zegarra-Ruiz DF, Beidaq AE, Iñiguez AJ, Ricco MLD, Vieira SM, Ruff WE, Mubiru D, Fine RL, Sterpka J, Greiling TM, et al. A diet-sensitive commensal lactobacillus strain mediates TLR7-dependent systemic autoimmunity. Cell Host Microbe. 2019;25:113–127.e6. doi: 10.1016/j.chom.2018.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vieira SM, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, Costa FRC, Tiniakou E, Greiling T, Ruff W, et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science. 2018;359:1156–1161. doi: 10.1126/science.aar7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, Loukov D, Schenck LP, Jury J, Foley KP, et al. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe. 2017;21:455–466.e4. doi: 10.1016/j.chom.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Medina-Contreras O, Geem D, Laur O, Williams IR, Lira SA, Nusrat A, Parkos CA, Denning TL. CX3CR1 regulates intestinal macrophage homeostasis, bacterial translocation, and colitogenic Th17 responses in mice. J Clin Investigation. 2011;121:4787–4795. doi: 10.1172/JCI59150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zigmond E, Bernshtein B, Friedlander G, Walker CR, Yona S, Kim K-W, Brenner O, Krauthgamer R, Varol C, Müller W, et al. Macrophage-restricted interleukin-10 receptor deficiency, but not il-10 deficiency, causes severe spontaneous colitis. Immunity. 2014;40:720–733. doi: 10.1016/j.immuni.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 29.Kühn R, Löhler J, Rennick D, Rajewsky K, Müller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-P. [DOI] [PubMed] [Google Scholar]
- 30.Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, Swaminath A, Bonneau R, Scherl EJ, Littman DR. CX₃CR1+ mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Medicine [Internet]. 2014;211:1571–1583. Available from http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=25024136&retmode=ref&cmd=prlinks. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol [Internet]. 2000;20:4106–4114. Available from http://mcb.asm.org/cgi/do/ 10.1128/MCB.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diehl GE, Longman RS, Zhang J-X, Breart B, Galan C, Cuesta A, Schwab SR, Littman DR. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX3CR1hi cells. Nature [Internet]. 2013;494:116–120. Available from http://www.nature.com/doifinder/ 10.1038/nature11809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, Lafaille JJ, Hempstead BL, Littman DR, Gan W-B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell [Internet]. 2013;155:1596–1609. Available from. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quiros M, Nishio H, Neumann PA, Siuda D, Brazil JC, Azcutia V, Hilgarth R, O’Leary MN, Garcia-Hernandez V, Leoni G, et al. Macrophage-derived IL-10 mediates mucosal repair by epithelial WISP-1 signaling. J Clin Investigation. 2017;127:3510–3520. doi: 10.1172/JCI90229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Werner T, Shkoda A, Haller D. Intestinal epithelial cell proteome in IL-10 deficient mice and il-10 receptor reconstituted epithelial cells: impact on chronic inflammation. J Proteome Res. 2007;6:3691–3704. doi: 10.1021/pr070222x. [DOI] [PubMed] [Google Scholar]
- 36.Parang B, Barrett CW, Williams CS. Gastrointestinal Physiology and Diseases. Methods Mol Biology. 2016;1422:297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Gene Dev. 2018;32:1267–1284. doi: 10.1101/gad.314617.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gabrilovich DI. Myeloid-Derived suppressor cells. Cancer Immunol Res. 2017;5:3–8. doi: 10.1158/2326-6066.CIR-16-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ostrand-Rosenberg S, Fenselau C. Myeloid-Derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol Baltim Md 1950. 2018;200:422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, G-Y Y, Osterreicher CH, Hung KE, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491:254–258. doi: 10.1038/nature11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 42.Zigmond E, Varol C, Farache J, Elmaliah E, Satpathy AT, Friedlander G, Mack M, Shpigel N, Boneca IG, Murphy KM, et al. Ly6Chi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity. 2012;37:1076–1090. doi: 10.1016/j.immuni.2012.08.026. [DOI] [PubMed] [Google Scholar]
- 43.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold GL, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14:207–215. doi: 10.1016/j.chom.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dogan B, Suzuki H, Herlekar D, Sartor RB, Campbell BJ, Roberts CL, Stewart K, Scherl EJ, Araz Y, Bitar PP, et al. Inflammation-associated adherent-invasive Escherichia coli are enriched in pathways for use of propanediol and iron and M-cell translocation. Inflamm Bowel Dis. 2014;20:1919–1932. doi: 10.1097/MIB.0000000000000183. [DOI] [PubMed] [Google Scholar]
- 46.Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG, Woo V, Teng F, Tran NL, Sczesnak A, et al. IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH17-dependent inflammation. Sci Transl Med. 2017;9:eaaf9655. doi: 10.1126/scitranslmed.aaf9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–462. doi: 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Hara RJ, Greenman J, MacDonald AW, Gaskell KM, Topping KP, Duthie GS, Kerin MJ, Lee PW, Monson JR. Advanced colorectal cancer is associated with impaired interleukin 12 and enhanced interleukin 10 production. Clin Cancer Res Official J Am Assoc Cancer Res. 1998;4:1943–1948. [PubMed] [Google Scholar]
- 49.M-C L, S-H H. IL-10 and its related cytokines for treatment of inflammatory bowel disease. World J Gastroentero. 2004;10:620–625. doi: 10.3748/wjg.v10.i5.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roers A, Siewe L, Strittmatter E, Deckert M, Schlüter D, Stenzel W, Gruber AD, Krieg T, Rajewsky K, Müller W. T cell-specific inactivation of the interleukin 10 gene in mice results in enhanced T cell responses but normal innate responses to lipopolysaccharide or skin irritation. J Exp Medicine. 2004;200:1289–1297. doi: 10.1084/jem.20041789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pils MC, Pisano F, Fasnacht N, Heinrich J-M, Groebe L, Schippers A, Rozell B, Jack RS, Müller W. Monocytes/macrophages and/or neutrophils are the target of IL-10 in the LPS endotoxemia model. Eur J Immunol. 2010;40:443–448. [DOI] [PubMed] [Google Scholar]
- 52.Buchan A, Hadden M, Suzuki MT. Development and application of quantitative-PCR tools for subgroups of the Roseobacter clade. Appl Environ Microb [Internet]. 2009;75:7542–7547. Available from http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=19801463&retmode=ref&cmd=prlinks. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chern EC, Siefring S, Paar J, Doolittle M, Haugland RA. Comparison of quantitative PCR assays for Escherichia coli targeting ribosomal RNA and single copy genes. Lett Appl Microbiol [Internet]. 2011;52:298–306. Available from http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=21204885&retmode=ref&cmd=prlinks. [DOI] [PubMed] [Google Scholar]
- 54.Valdez Y, Diehl GE, Vallance BA, Grassl GA, Guttman JA, Brown NF, Rosenberger CM, Littman DR, Gros P, Finlay BB. Nramp1 expression by dendritic cells modulates inflammatory responses during SalmonellaTyphimurium infection. Cell Microbiol [Internet]. 2008;10:1646–1661. Available from 10.1111/j.1462-5822.2008.01155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marchal-Bressenot A, Salleron J, Boulagnon-Rombi C, Bastien C, Cahn V, Cadiot G, Diebold M-D, Danese S, Reinisch W, Schreiber S, et al. Development and validation of the Nancy histological index for UC. Gut. 2015;66:43–49. doi: 10.1136/gutjnl-2015-310187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the
article and/or its supplementary materials. 16S sequencing data are openly available in NCBI under BioProject ID PRJNA845002.