

Abstract
Discovery of small-molecule degraders that activate ubiquitin ligase–mediated ubiquitination and degradation of targeted oncoproteins in cancer cells has been an elusive therapeutic strategy. Here, we report a cancer cell–based drug screen of the NCI drug-like compounds library that enabled identification of small-molecule degraders of the small ubiquitin-related modifier 1 (SUMO1). Structure-activity relationship studies of analogs of the hit compound CPD1 led to identification of a lead compound HB007 with improved properties and anticancer potency in vitro and in vivo. A genome-scale CRISPR-Cas9 knockout screen identified the substrate receptor F-box protein 42 (FBXO42) of cullin 1 (CUL1) E3 ubiquitin ligase as required for HB007 activity. Using HB007 pull-down proteomics assays, we pinpointed HB007’s binding protein as the cytoplasmic activation/proliferation-associated protein 1 (CAPRIN1). Biolayer interferometry and compound competitive immunoblot assays confirmed the selectivity of HB007’s binding to CAPRIN1. When bound to CAPRIN1, HB007 induced the interaction of CAPRIN1 with FBXO42. FBXO42 then recruited SUMO1 to the CAPRIN1-CUL1-FBXO42 ubiquitin ligase complex, where SUMO1 was ubiquitinated in several of human cancer cells. HB007 selectively degraded SUMO1 in patient tumor–derived xenografts implanted into mice. Systemic administration of HB007 inhibited the progression of patient-derived brain, breast, colon, and lung cancers in mice and increased survival of the animals. This cancer cell–based screening approach enabled discovery of a small-molecule degrader of SUMO1 and may be useful for identifying other small-molecule degraders of oncoproteins.
INTRODUCTION
The strategy of targeting protein degradation is shifting drug discovery from functional inhibitors to proteolytic degraders of targeted proteins through activation of the ubiquitin (UB) proteasome system (1, 2). Such targeting approaches include bifunctional molecules such as proteolysis-targeting chimeras and small-molecule degraders such as lenalidomide (3). The anticancer activity of small-molecule degraders has been validated by lenalidomide and its analogs that bind to cereblon, a substrate receptor of cullin 4 (CUL4) E3 UB ligase to recruit, ubiquitinate, and degrade Cys2-His2 zinc finder proteins (4–10). Studies of investigative splicing inhibitor sulfonamides provide the structural basis for the small molecules that act as molecular glues binding the substrate receptor DNA damage-binding protein 1 and CUL4 associated factor 15 (DCAF15) of CUL4 ligase, resulting in ubiquitination and degradation of RNA binding motif protein 39 (11–13). Mammalian cells express 660 E3 UB ligases, but only a few have been explored for targeted protein degradation. This is, in part, due to lack of strategies for discovery of small molecules that initiate specific E3 ligase–mediated degradation of targeted proteins in particular types of diseased cells. Here, we provide a cancer cell–based strategy for the identification of small-molecule degraders of small ubiquitin-related modifier 1 (SUMO1) protein resulting in its ubiquitination and degradation through CUL1 E3 ligase in human brain, breast, colon, and lung cancer cells.
SUMO itself is a UB-related modifier that is conjugated to substrate proteins by the E1 SUMO-activating enzyme 1/2 (SAE1/2), the E2 UB/SUMO-conjugating enzyme-9 (UBC9) and E3 SUMO ligases (14–16). SUMO exists in three conjugated forms SUMO1, 2 and 3, but SUMO2 and SUMO3 are commonly referred to as SUMO2/3 because they share 96% of their amino acid sequences. Whereas all three forms are conjugated by the same enzymes, the mechanisms governing the specificity of each form remain unclear. SUMO was initially linked to cancer through the discovery that promyelocytic leukemia protein (PML), and PML-retinoic acid receptor-α are SUMO substrates (17, 18). More oncoproteins and tumor suppressor proteins including breast cancer type 1 susceptibility protein (BRCA1) have been reported to be SUMO substrates (19). Sumoylation was reported as being involved in oncogenesis and metastasis through the modification of chromatin, extracellular signal–regulated kinase (ERK), and Myc-driven pathways (20–22).
Sumoylation has been implicated in cancer, yet developing targeted therapeutics that block sumoylation has been challenging because of the lack of safe and specific drug targets (23). High-throughput screenings using biochemical sumoylation assays have identified SAE1/2 and UBC9 inhibitors that block global sumoylation, such as SUMO1/3 conjugation of substrates (24). Genomic knockout of UBC9 in mice, however, cripples nuclear functions and is lethal (25, 26). SUMO2-deficient mice die at the embryonic stage, whereas SUMO1-deficient mice are viable (27–29). In contrast, SUMO1 is highly expressed in various types of human cancers, and its knockdown inhibits growth in cancer cell lines and xenografts (30–33). Whereas SUMO1 could be an anticancer target, biochemical sumoylation screens have not yet yielded agents that are selective for SUMO1. Here, using cancer cell–based sumoylation screening, we report the identification of SUMO1 small-molecule degraders that induce ubiquitination and degradation of SUMO1 protein through CUL1 E3 ligase in human brain, breast, colon, and lung cancer cells.
RESULTS
Discovery of small-molecule degraders of SUMO1 protein
We have reported that denatured Western blots can distinguish between SUMO1 and SUMO2/3 conjugation in glioblastoma LN229 cells (33). To selectively target SUMO1 conjugation, we carried out LN229 cell–based denatured Western blot screening of the National Cancer Institute (NCI) diversity set IV library that consists of 1596 compounds selected from >140,000 small molecules based on their structural and pharmacological features. LN229 cells were treated with compound (10 μM each) for 3 days and analyzed by denatured Western blotting for inhibition of the conjugation of SUMO1 but not SUMO2/3 that was used as a selectivity control (Fig. 1A). The compounds that inhibited SUMO1 conjugation were then analyzed by cell viability assays in which LN229 cells were treated with each compound for 5 days. Of 1596 compounds, 11 were effective at inhibiting cell growth and SUMO1 conjugation. The compound D5, referred to as the hit compound (CPD1), was the most effective in blocking SUMO1 conjugation to its substrate proteins and inhibiting cancer cell growth with a concentration of 2.3 μM required to reduce cell viability by 50% [median inhibitory concentration (IC50)]. CPD1 is chemically referred to as 1-(1,3-benzothiazol-2-yl)-3-(4-chloro-3-nitrophenyl)urea and has drug-like properties as indicated by its molecular weight (MW), partition coefficient log P (cLogP), and polar surface area (Fig. 1B). Cancer colony formation assays confirmed the broad activity of this compound against human cancer cell lines derived from brain glioblastoma, breast, colorectal carcinoma, and non–small cell lung carcinoma (NSCLC) (fig. S1A).
Fig. 1. Discovery of the hit compound CPD1 and chemical lead HB007.
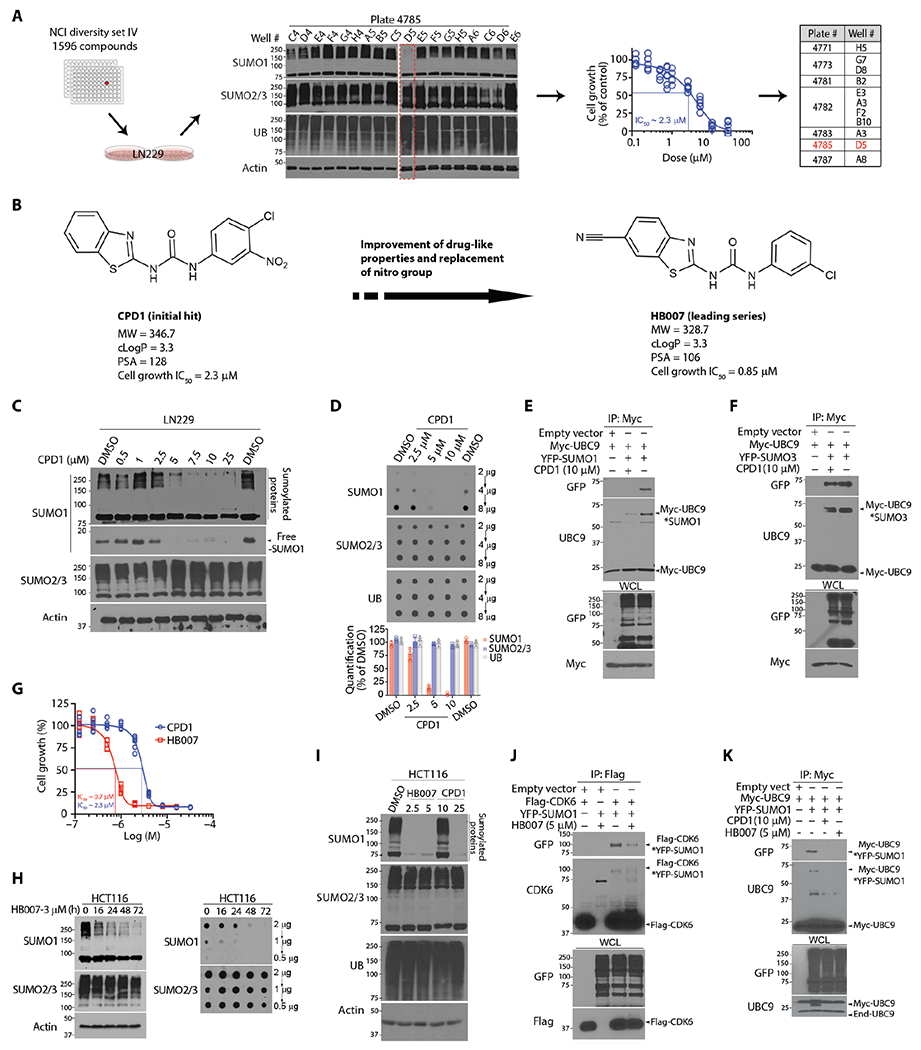
(A) Workflow in LN229 cell–based drug screening of the NCI library through Western blots and cell viability assay with the identification of 11 active compounds with D5 characterized as the hit compound (highlighted in red). (B) The chemical structures and pharmacological properties of the hit compound CPD1 and the lead compound HB007. cLogP, partition coefficient log P; PSA, polar surface area. (C) LN229 cells were treated for 72 hours with indicated doses of CPD1 and analyzed by Western blots for conjugated and unconjugated/free SUMO1 as indicated (right). SUMO2/3 and β-actin were used, respectively, as the selectivity and the loading control. (D) LN229 cells were treated with CDP1 for 72 hours and examined by dot blots for total SUMO1 concentrations with the amounts of loading proteins indicated (right). Dot intensity was evaluated using ImageJ (bottom). (E and F) LN229 cells were cotransfected with Myc-UBC9 and YFP-SUMO1 or YFP-SUMO3 or empty vector as control and subjected to myc IP and Western blotting for UBC9-SUMO1 (E) and UBC9-SUMO3 conjugates (F) as indicated (right) with whole-cell lysate (WCL) as the loading control. (G) LN229 cells were treated with a series of dilutions of CPD1 or HB007 for 5 days and examined by cell viability for cell growth inhibition with the IC50 values indicated (points: n = 6). (H) HCT116 cells were treated with HB007 for the time indicated and analyzed by Western (left) and dot blots (right) for conjugated and total SUMO1 concentrations. (I) HCT116 cells were treated with DMSO (control), CPD1, or HB007 for 72 hours with the indicated doses (micromolar) and analyzed by Western blotting using the indicate antibodies (left). (J) LN229 cells were cotransfected with Flag-CDK6 and YFP-SUMO1, treated with HB007 for 24 hours, and subjected to Flag IP and Western blotting using CDK6 and green fluorescent protein (GFP) antibody (that recognizes YFP) for SUMO1-CDK6 conjugates as indicated (right). (K) Myc IP and Western blotting for SUMO1-UBC9 conjugates as indicated (right) in myc-UBC9– and YFP-SUMO1–transfected LN229 cells after CPD1 and HB007 treatment for 24 hours.
To confirm the selectivity of CPD1, we determined that its treatment reduced the conjugated and unconjugated forms and total amounts of SUMO1 but not SUMO2/3 protein (Fig. 1, C and D). In contrast, CPD1 had no effects on SUMO1 mRNA expression in LN229 cancer cells (fig. S1B). Next, we analyzed the effects of CPD1 on SUMO1 conjugation of the substrate cyclin-dependent kinase 6 (CDK6) using in vivo sumoylation assays (33). Yellow fluorescent protein (YFP)+–SUMO1 and Flag-CDK6 were cotransfected into LN229 cells, and Flag-CDK6 was isolated by Flag immunoprecipitation (IP). Western blotting showed that CPD1 abrogated SUMO1-CDK6 conjugation (fig. S1C). The in vivo sumoylation assay using UBC9 as a SUMO1-3 substrate (34) confirmed that CPD1 selectively reduced SUMO1-UBC9 but not SUMO3-UBC9 conjugation (Fig. 1, E and F). In contrast, the in vitro sumoylation assay using CDK6 as a SUMO1 substrate and RanGap1 as a SUMO1-3 substrate (35) showed no effects of CPD1 on SUMO1-CDK6, SUMO1-RanGAP1, and SUMO3-RanGAP1 conjugation (fig. S1, D to F).
To establish CPD1 as a therapeutic, we conducted multiple rounds of structure-activity relationship (SAR) studies to improve potency and pharmacokinetics (PKs). CPD1 consists of the core structural moieties: benzothiazole, urea, nitro, and phenyl (fig. S1G). The nitro moiety forms carcinogenic metabolites through the activity of cytochrome P450 (CYP). Our SAR studies focused on modifications of the core structure and replacement of the nitro (fig. S1H), which led to the identification of several non-nitro analogs with improved biological activity, acceptable MWs, and cLogP values (fig. S1I). Among these analogs, HB007 (Fig. 1B) was more effective in cell growth inhibition assays and SUMO1 degradation assays than the parent compound CPD1 (Fig. 1G and fig. S1, J to N). Western blot and dot blot assays showed that treatment with HB007 reduced the conjugated and total amounts of SUMO1 but not SUMO2/3 protein gradually over about 24 hours in the colon cancer HCT116 cell line (Fig. 1H). Consistently, the treatment of CPD1 and HB007 abrogated SUMO1 conjugation of the substrates CKD6 and UBC9 in HCT116 and LN229 cells (Fig. 1, I to K, and fig. S1O).
CPD1 and HB007 induce the ubiquitination and degradation of SUMO1 protein
To confirm SUMO1 as the target, we generated SUMO1 knockout clones using a lentiCRISPRv2 system in the HCT116 cancer cell line (36). SUMO1 and control single-guide RNAs (sgRNAs) were cloned into Cas9 lentiviral vectors and introduced into HCT116 cells. Single clones were established, and Western blotting confirmed that SUMO1 was deleted (Fig. 2A). SUMO1 knockout drastically inhibited HCT116 cell growth (Fig. 2B) and abrogated the activity of CPD1 and HB007 against cancer cell growth (Fig. 2, C and D). The studies were repeated by transducing two independent SUMO1-targeted short hairpin RNAs into LN229 cancer cells with similar results (fig. S2, A to C), consistent with our previous report (33).
Fig. 2. CPD1 and HB007 induce SUMO1 ubiquitination and degradation.
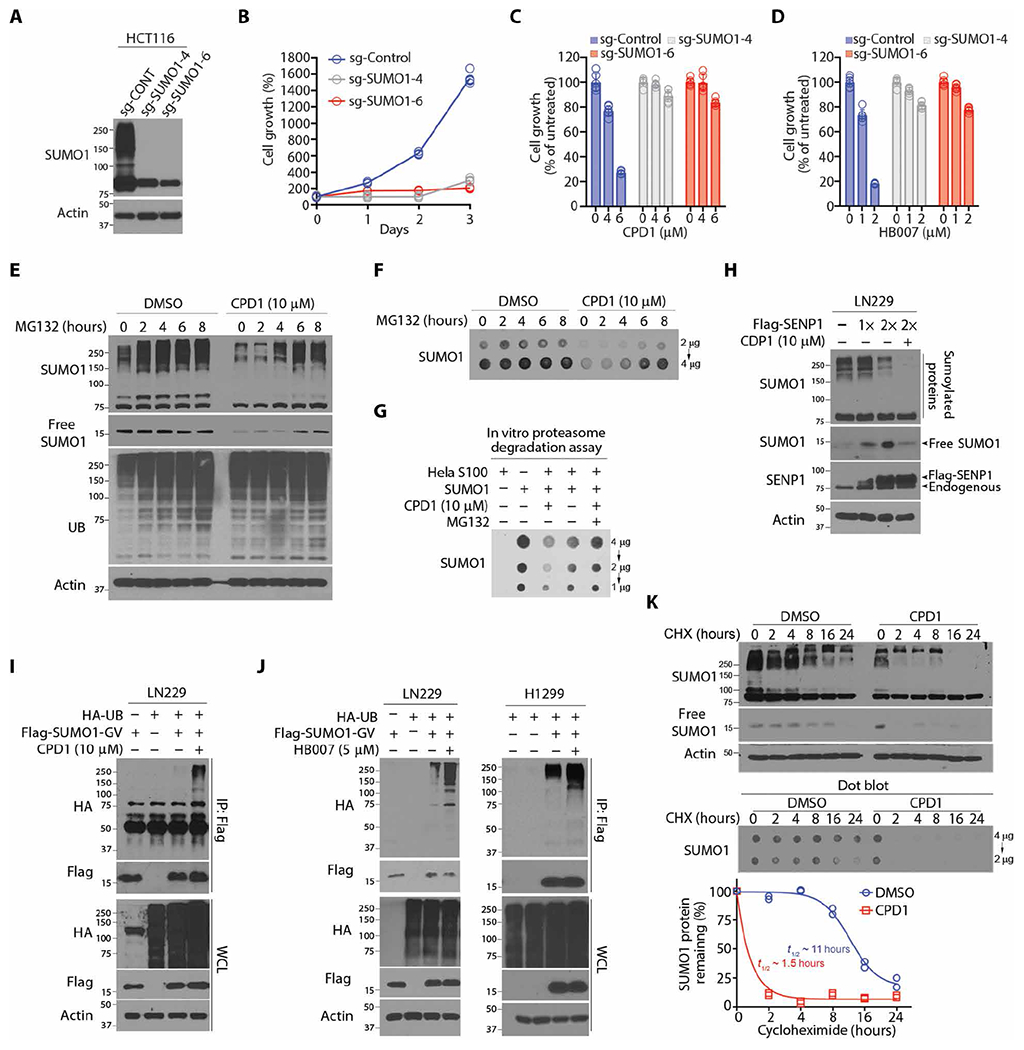
(A) Western blots for SUMO1 protein concentrations in SUMO1 knockout HCT116 sgSUMO1-4, sgSUMO1-6, and control (sg-CONT) clones. (B) Cell growth analysis of SUMO1 knockout HCT116 sgS1-4, sgS1-6, and sg-CONT clones (n = 3). (C and D) Cell viability assay of SUMO1 knockout HCT116 and sg-CONT clones after being treated with CPD1 (C) or HB007 (D) with the indicated doses for 3 days (points: n = 6). (E and F) LN229 cells were treated with CPD1 for 24 hours, followed by MG132 for indicated times (top), and then analyzed by Western blots for conjugated and unconjugated/free forms (E) and dot blots for the total amounts of SUMO1 protein (F). (G) HeLa cell S100 fraction was added with SUMO1, CPD1, and/or MG132 and analyzed by dot blotting for the total amount of SUMO1. (H) Flag-SENP1 was overexpressed in LN229 cells, and the cells were treated with CDP1 for 48 hours and examined by Western blot for conjugated and unconjugated/free SUMO1 as indicated (right). (I) Flag-SUMO1-GV– and HA-UB–transfected LN229 cells were treated or untreated with CDP1 for 24 hours; Flag IP was analyzed by Western blots for SUMO1 polyubiquitination. (J) Flag-SUMO1-GV and HA-UB were cotransfected in LN229 (left) or H1299 (right), and the cells were treated or untreated with HB007 for 48 hours and subjected to Flag IP and Western blots for SUMO1 polyubiquitination. (K) LN229 cells were treated with CPD1 in the presence or absence of CHX for indicated times and examined by Western and dot blots for conjugated and unconjugated/free (top) and total amounts of SUMO1 protein (middle). The total amounts of SUMO1 protein were normalized with actin using the ImageJ and plotted for the half-life of SUMO1 protein (bottom) (n = 2 technical replicates). t1/2, half-time.
To reveal the mechanism of drug action in SUMO1 degradation, we first determined that pretreatment of HCT116 cancer cells with the proteasome inhibitor MG132 blocked the activity of CPD1 and HB007 in SUMO1 degradation (fig. S2D). Next, we confirmed that SUMO1 degradation occurred through the 26S proteasome by showing that posttreatment of cancer cell lines with MG132 restored the conjugated, unconjugated, and total amounts of SUMO1 in several cancer cell lines (Fig. 2, E and F, and fig. S2, E and F). An in vitro proteasome degradation assay using the HeLa cell S100 fraction confirmed that MG132 blocked CPD1-induced degradation of SUMO1 (Fig. 2G). To test the effects of CPD1 on conjugation, we found that the overexpression of SUMO1-specific protease 1 (SENP1) but not SUMO3-specific SENP3 (37) reduced the conjugated form and accumulated the unconjugated form of SUMO1; however, CPD1 treatment abrogated the accumulation of the unconjugated SUMO1 (Fig. 2H and fig. S2G).
To provide supporting evidence that CPD1 and HB007 degraded unconjugated SUMO1 and thereby abrogated its conjugation, we carried out in vivo ubiquitination assays using nonconjugated Flag-SUMO1-Gly-Val (Flag-SUMO1-GV) generated from the conjugated YFP-SUMO1-Gly-Gly (YFP-SUMO1-GG) through Gly to Val mutagenesis (33). YFP-SUMO1-GV and hemagglutinin (HA)–UB were cotransfected in LN229, HCT116, H1299, and A549 cells. The transfected cells were treated with CPD1 and HB007 and lysed in a denatured buffer. Flag IP followed by Western blot revealed the polyubiquitination of Flag-SUMO1-GV in these cancer cells (Fig. 2, I and J, and fig. S2, H and I). Overexpression of HA-UB enhanced CPD1-induced degradation, but overexpression of mutant HA-K0-UB, in which all lysine residues were replaced with arginines, reduced CPD1-induced degradation of SUMO1 but not SUMO2/3 in LN229 cells (fig. S2J). To evaluate the effects of CPD1 and HB007 on SUMO1 half-life, we treated LN229 cells with CPD1 or HB007 in the presence of cycloheximide (CHX), which reduced the half-life of SUMO1 from 11 to 1.5 hours (Fig. 2K and fig. S2K).
A genomic CRISPR-Cas9 screen identifies an E3 UB ligase pathway as the target of HB007
To explore the pathway targeted by HB007, we conducted genome-scale CRISPR-Cas9 knockout screening using a CRISPR-Cas9 knockout library consisting of 123,411 sgRNAs targeting 5′ constitutive exons of 19,050 genes (38, 39). The library was transduced using lentivirus in HCT116 cells. After the completion of the screening treatment phase, which was terminated at 12 population doublings of the vehicle control (fig. S3A), samples were harvested and genomic DNA was extracted for amplicon-based sequencing to quantitatively identify genotype abundance in each sample. Replicates were analyzed side by side and showed high concordance (fig. S3B). Next, we evaluated the performance of the control guide and showed that the control guides in the CRISPR library performed as anticipated with dropout rates for targeting essential genes of up to 128-fold (fig. S3C).
We analyzed the data using the Model-based Analysis of Genomewide CRISPR-Cas9 knockout (MAGeCK) algorithm (40) and identified 28 genes with differing expression between the control and treated groups for each sgRNA using a false discovery rate (FDR) of <0.05 as a cutoff (fig. S3D and data file S1). The prospective HB007-targeted gene list included two genes in the UB proteasome system: FBXO42 and UB-specific protease 14 (USP14). FBXO42 is a substrate receptor of CUL1 E3 UB ligases (41, 42), whereas USP14 is a proteasome-associated protease that removes conjugated poly-UB chains to promote protein degradation through the 26S proteasome (43). To validate the data, we generated FBXO42 and USP14 knockout and sgRNA control clones through transduction of the sgRNAs into HCT116 cells. Single clones were established and analyzed by Western blotting to confirm FBXO42 and USP14 protein deletion. CRISPR-Cas9 knockout of FBXO42 and USP14 diminished the HB007 activity in SUMO1 degradation and cancer cell growth inhibition in HCT116 cells (Fig. 3, A and B, and fig. S3E).
Fig. 3. Discovery of a HB007-targeted E3 ligase pathway using a genome-scale CRISPR-Cas9 knockout screen.
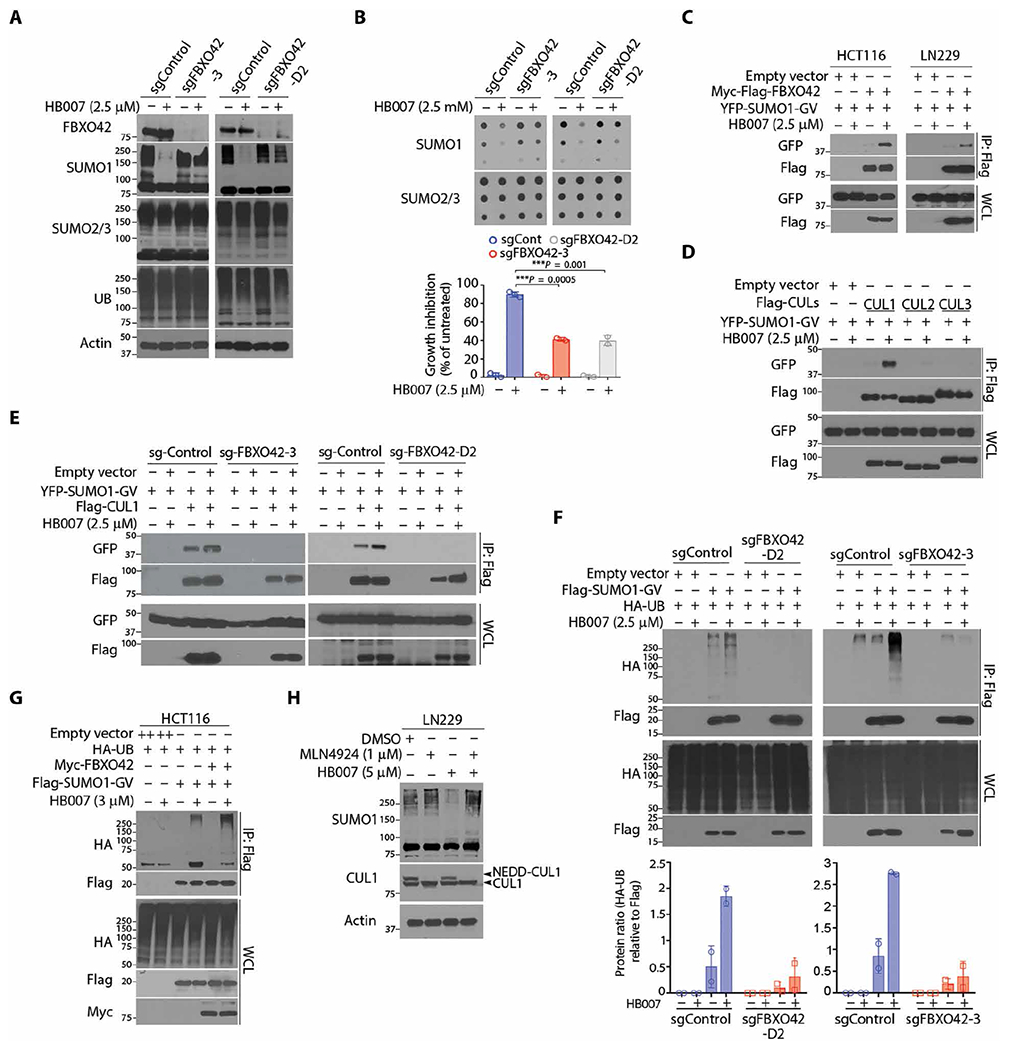
(A and B) CRISPR-Cas9 FBXO42 knockout (sgFBXO42-3 and sgFBXO42-D2) and sgRNA control (sgControl) HCT116 clones were treated with HB007 for 72 hours and analyzed by Western blot for conjugated SUMO1 (A), dot blots for SUMO1 total amounts [(B) top], and cell viability assay for cell growth inhibition [(B) bottom)] (means ± SD; n = 3; ***P < 0.001 by unpaired t test). (C) Flag-FBXO42 and YFP-SUMO1-GV were cotransfected in HCT116 or LN229 cells. After 24 hours of treatment with HB007, the cells were subjected to Flag IP and Western blots using a GFP/YFP antibody for the interaction of FBXO42 and SUMO1. (D) Flag-CUL1, CUL2, or CUL3 was cotransfected with YFP-SUMO1-GV in HCT116 cells, and after treatment with HB007 for 48 hours, the cells were subjected to Flag IP and Western blots using GFP/YFP antibodies for the interaction of SUMO1 and CUL1, CUL2, or CUL3. (E) The FBXO42 knockout sg-FBXO42-3, sg-FBXO42-D2, and sgRNA control HCT116 clone were transfected with YFP-SUMO-GV and Flag-CUL1; treated with HB007 for 24 hours; and subjected to Flag IP and Western blot. (F) The sgRNA control and sg-FBXO42-D2 (left) or sg-FBXO42-3 HCT116 clone (right) was transfected with Flag-SUMO1-GV and HA-UB, treated with HB007 for 24 hours, and subjected to Flag IP and Western blot for SUMO1 polyubiquitination (top) and densitometry analysis of the HA-UB blots for the poly-UB amounts (bottom) (n = 2). (G) HCT116 cells were cotransfected with Flag-SUMO1-GV, Myc-FBXO42, and/or HA-UB; treated or untreated with HB007 for 24 hours; and subjected to Flag IP and Western blotting for SUMO1 polyubiquitination. (H) LN229 cells were treated with MLN4924 for 24 hours, alone or in combination with HB007, and analyzed by Western blots for conjugated SUMO1 and neddylated or unneddylated CUL1 as indicated (right).
HB007 induces SUMO1 ubiquitination and degradation through CUL1-FBXO42 ligase
CUL-based E3 ligases use modular multisubunit organizations consisting of substrate receptors, adaptors such as S-phase kinase-associated protein 1 (SKP1), CUL scaffolds, and E2 enzyme-binding RING-box (RBX) proteins (44). F-box proteins are substrate receptors that recognize and recruit substrates to CUL ligases (45). To define SUMO1 as a substrate of FBXO42, we carried out protein-protein interaction assays. Flag-FBXO42 and YFP-SUMO1-GV were cotransfected in HCT116 and LN229 cancer cells, where Flag IP and Western blot revealed FBXO42 and SUMO1 interaction in HB007-treated but not untreated cells (Fig. 3C). Transfection of Flag-FBXO42 with either YFP-SUMO1-GG or YFP-SUMO1-GV in LN229 cells confirmed that HB007 induced the interaction of FBXO42 with unconjugated SUMO1-GV but not conjugated SUMO1-GG (fig. S3F). To identify the CUL scaffold, YFP-SUMO1-GV was cotransfected with Flag-CUL1, Flag-CUL2 or Flag-CUL3, where Flag IP and Western blot revealed the interaction of SUMO1 with CUL1 and, to a much lesser extent, CUL2 but not CUL3 in HB007-treated but not untreated HCT116 and LN229 cells (Fig. 3D). Next, cotransfection of YFP-SUMO1-GV with Flag-SKP1 or Flag-RBX1 confirmed that HB007 induced SUMO1 recruitment to the CUL1-SKP1-RBX1 ligase complex in HCT116 cells (fig. S3, G and H).
To further define the role of FBXO42 in CUL1 ligase, Flag-CUL1 and YFP-SUMO1-GV were cotransfected in FBXO42 knockout HCT116 clones. FBXO42 knockout blocked the HB007-induced interaction of SUMO1 and CUL1 (Fig. 3E). Next, Flag-SUMO1-GV and HA-UB were cotransfected in FBXO42 knockout and sgRNA control HCT116 clones. HB007 treatment induced SUMO1 polyubiquitination in the control but not FBXO42 knockout clones (Fig. 3F). In contrast, overexpression of FBXO42 enhanced HB007-induced polyubiquitination of SUMO1 in HCT116, LN229, and H1299 cancer cells (Fig. 3G and fig. S3I). Neural precursor cell expressed developmentally down-regulated protein 8 (NEDD8) conjugation to CUL1 backbone induces a conformational change that facilitates RBX1 binding of E2 (46); thus, we determined that the NEDD8-activating enzyme inhibitor MLN4924 markedly reduced HB007-induced SUMO1 degradation in LN229 cells (Fig. 3H). Collectively, these results indicate that HB007 induces FBXO42 recruitment of SUMO1 to CUL1 E3 ligase for its ubiquitination and degradation in cancer cells (fig. S3J).
Identification of the HB007 binding protein CAPRIN1
To identify binding proteins of HB007, we conducted HB007 pull-down assays using streptavidin-coated bead/biotin and ferrite glycidyl methacrylate (FG) beads (47). The compound HB007 was linked to biotin or FG beads using five different linkers to avoid interfering with the HB007 binding site (fig. S4A). HB007-biotin and FG pull-downs of HCT116 cell lysates were analyzed by liquid chromatography–tandem mass spectrometry (LC-MS/MS) (48, 49), which identified a few hundred candidates per pull-down. Venn diagram comparison of HB007 pull-down and genomic CRISPR-Cas9 knockout screen data pinpointed the binding protein to be cytoplasmic activation/ proliferation-associated protein 1 (CAPRIN1) (Fig. 4A and fig. S4B) (50, 51), consistent with the systematic quantitative proteomic finding that CAPRIN1 is one of CUL1 integrating proteins (52).
Fig. 4. Identification of the HB007 binding protein CAPRIN1.
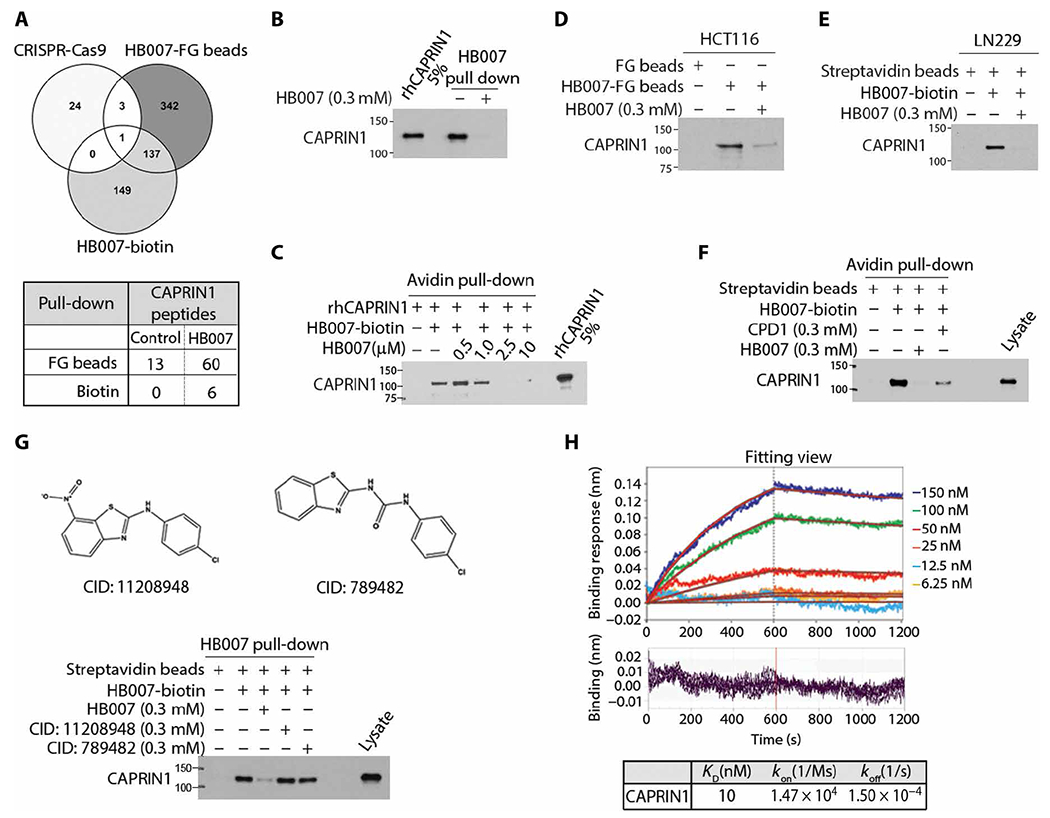
(A) Venn diagram of the data from genome-scale CRISPR-Cas9 knockout screen, HB007-FG bead and HB007-biotin/streptavidin-coated bead pull-down (top), and spectrometric total peptides counts of CAPRIN1 from LC-MS/MS analysis (bottom). (B) HB007-biotin was incubated with rhCAPRIN1 in the presence or absence of free HB007 and pulled down by streptavidin-coated beads and tested by immunoblotting for the binding of rhCAPRIN1 to HB007-biotin, with rhCAPRIN1 (5%) used as the loading control. (C) rhCAPRIN1 was premixed with 1 μM biotin, followed by HB007-biotin/streptavidin-coated bead pull-down in the presence of various doses of HB007. CAPRIN1 binding was identified by immunoblotting using CAPRIN1 antibodies. (D and E) HB007-FG beads and HB007-biotin were incubated with HCT116 lysate (D) and LN229 lysate (E) added or not with free HB007, and the pull-downs were tested by immunoblotting for the binding of cellular CAPRIN1 to HB007. (F) Immunoblot of HB007-biotin/streptavidin pull-down of HCT116 lysate added or not with the indicated concentrations of CPD1 or HB007. (G) The CPD1 similar but inactive compounds CID: 11208948 or CID: 789482 (top) or HB007 was added to HCT116 lysate that was then incubated with HB007-biotin. HB007-biotin/streptavidin pull-down was analyzed by immunoblotting. (H) Representative BLI sensorgrams of the interactions between rhCAPRIN1 and biotinylated HB007. Plots of the binding response during the association (0 to 600 s) and dissociation (600 to 1200 s) periods of the BLI assay at varying concentrations of CAPRIN1 (top) when HB007-biotin–loaded biosensors (quenched with biocytin) were dipped in CAPRIN1 wells. The plots have been processed with the double referencing technique and aligning of x and y axes. The red curves indicate the fit data. Residual binding for each plot at the varying concentrations (bottom). Binding curves were fit globally to a 1:1 binding model to calculate the binding constant (KD) from kinetic analysis as the ratio of the association (koff) and dissociation (kon) rate constants.
Immunoblotting confirmed the binding of recombinant human CAPRIN1 (rhCAPRIN1) to HB007-linked biotin (Fig. 4B). The binding of rhCAPRIN1 to HB007-linked biotin was blocked competitively by free HB007 in a dose-dependent manner (Fig. 4C). HB007 binding of cellular CAPRIN1 was confirmed by HB007-biotin and FG pull-downs of HCT116 and LN229 cell lysates (Fig. 4, D and E), and the binding was competitively blocked by addition of free HB007 and, to a lesser extent, CPD1 to the cell lysates in a dose-dependent manner (Fig. 4F). In contrast, CPD1 similar but inactive compounds (the compound identification number: 11208948 and 789482 in PubChem.com) failed to competitively inhibit the binding of cellular CAPRIN1 to HB007-linked biotin (Fig. 4G).
To validate the direct binding of the HB007 compound and CAPRIN1 protein, we applied the biolayer interferometry (BLI) assay using high precision streptavidin biosensors. To block nonspecific binding of CAPRIN1 to the biosensors, biocytin was used to quench the sensors. The data demonstrated that biocytin successfully blocked nonspecific binding of CAPRIN1 protein to the biosensors (fig. S4C). The BLI assay probing the interaction between rhCAPRIN1 protein and biotinylated HB007 compound yielded the binding affinity of 10 nM in 10 mM tris, 1 mM EDTA, and 6% glycerol buffer, and the high binding affinity and slow dissociation rate suggest tight binding of HB007 to CAPRIN1 (Fig. 4H). The nanomolar affinity was determined after considering the nonspecific binding between high precision streptavidin biosensor and CAPRIN1 protein through quenching of the biosensors with biocytin.
HB007 induces CAPRIN1-FBXO42 interaction and SUMO1 recruitment to CUL1 Ligase
To define the role of CAPRIN1 in SUMO1 degradation, we generated CRISPR-Cas9 knockout of CAPRIN1 through introduction of CAPRIN1 and control sgRNA into HCT116 cells (fig. S5A). CAPRIN1 knockout drastically reduced HB007 activity in SUMO1 degradation, cancer cell growth, and colony formation inhibition (Fig. 5, A and B, and fig. S5, B to D). Moreover, myc-FBXO42 and YFP-SUMO1-GV were cotransfected in CAPRIN1 knockout and control clones, and myc IP and Western blots showed that CAPRIN1 knockout blocked HB007-induced interaction of SUMO1 and FBXO42 (Fig. 5C). Consequently, CAPRIN1 knockout abrogated HB007-induced polyubiquitination of SUMO1 protein in Flag-SUMO1-GV and HA-UB–transfected clones (Fig. 5D).
Fig. 5. HB007-induced CAPRIN1-FBXO42 interaction and SUMO1 recruitment to CUL1 E3 Ligase.
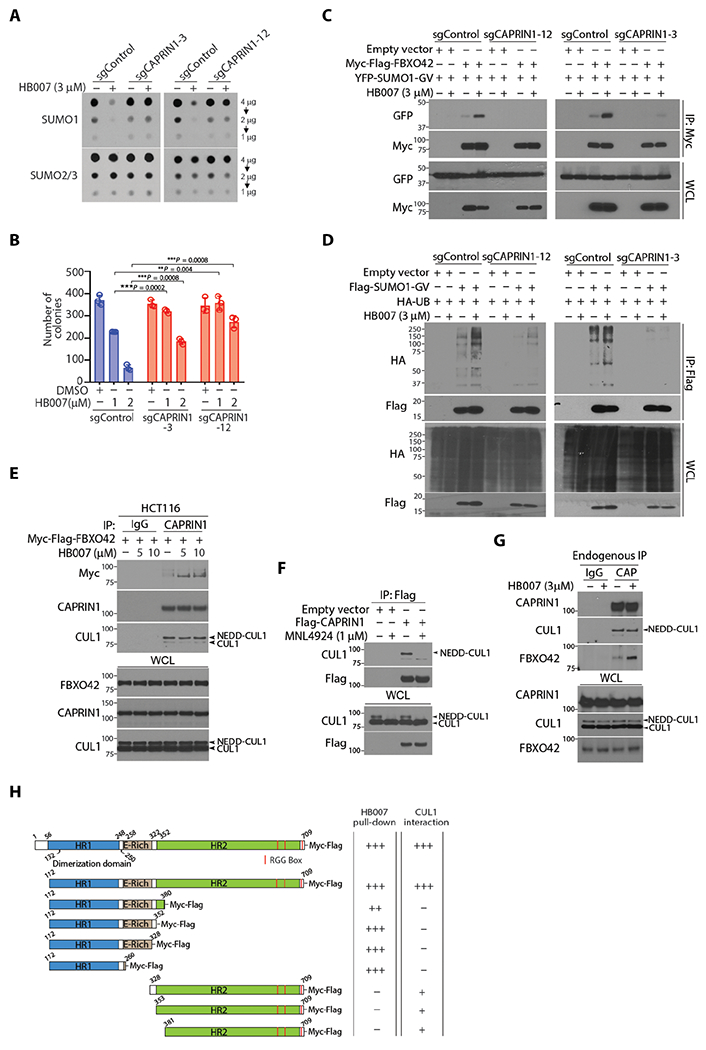
(A) The CAPRIN1 knockout sgCAPRIN1-3, sgCAPRIN1-12, and sgRNA control clones of HCT116 cells were treated or untreated with HB007 (3 μM) for 72 hours and analyzed by dot blotting for total amounts of SUMO1 and SUMO2/3. (B) CAPRIN1 knockout and control HCT116 clones were treated with DMSO or HB007 (1 or 2 μM) for 10 days and tested by colony formation assays with colony numbers calculated and presented (means ± SD; n = 6; **P < 0.01 and ***P < 0.001 by unpaired t test). (C) CAPRIN1 knockout and control HCT116 clones were cotransfected with myc-FBXO42 and YFP-SUMO1-GV, treated or untreated with HB007 for 24 hours, and subjected to myc IP and Western blotting for FBXO42 and SUMO1 interaction. (D) CAPRIN1 knockout HCT116 clones were cotransfected with Flag-SUMO1-GV and HA-UB, treated or untreated with HB007 for 24 hours, and subjected to Flag IP and Western blotting for SUMO1 polyubiquitination. Whole-cell lysate was included as the loading control. (E) HCT116 cells were transfected with Myc-Flag-FBXO42 and treated with HB007 for 8 hours with the doses indicated, followed by endogenous CAPRIN1-IP using CAPRIN1 antibodies. Western blots revealed the interaction of Myc-Flag-FBXO42 and endogenous CUL1. (F) HCT116 cells were transfected with Flag-CAPRIN1, treated with MLN4924 for 4 hours, and submitted to Flag IP and Western blotting for CAPRIN1-CUL1 interaction. (G) HCT116 were treated with HB007 for 8 hours and subjected to endogenous CAPRIN1-IP, followed by Western blot for CUL1 and FBXO42 interaction. Immunoglobulin G (IgG)–IP was used as a negative control. (H) Schematic representation of CAPRIN1 and its deletion mutants (left). Interaction of CAPRIN1 with HB007 and CUL1 was analyzed by HB007-biotin pull-down and Flag IP of CAPRIN1 and its mutants.
To determine whether CAPRIN1 interacts with FBXO42, myc-FBXO42 was transfected in HCT116 and H1299 cells. IP of endogenous CAPRIN1 revealed that HB007 induced the CAPRIN1-FBXO42 interaction (fig. S5E). To link CAPRIN1 to CUL1 E3 ligase complex, we transfected myc-FBXO42 in HCT116 cells and demonstrated the CAPRIN1-FBXO42 interaction in HB007-treated but not untreated cells and noticed CAPRIN1 interacted with the neddylated-CUL1 in both HB007-treated and untreated cells (Fig. 5E). The NEDD8-activating enzyme inhibitor MLN4924 abolished the interaction of Flag-CAPRIN1 and neddylated-CUL1 (Fig. 5F). Last, endogenous IP of CAPRIN1 confirmed the interaction of CAPRIN1 and neddylated-CUL1 in both HB007-treated and HB007-untreated cells, whereas CAPRIN1-FBXO42 interaction was observed only in HB007-treated cells (Fig. 5G). FBXO42 knockout did not interrupt the CAPRIN1-CUL1 interaction (fig. S5F). In contrast, however, CAPRIN1 knockout blocked the FBXO42-CUL1 interaction in HB007-treated and untreated cells (fig. S5G).
To test whether CAPRIN1 is a substrate of SUMO1 and UB, Flag-CAPRIN1 was cotransfected with YFP-SUMO1-GV or YFP-SUMO1-GG in cells. Flag IP and Western blotting revealed no CAPRIN1-SUMO1 conjugates (fig. S5H). Flag-CAPRIN1 and HA-UB were both cotransfected in HCT116 cells, and Flag IP demonstrated the polyubiquitination of CAPRIN1 in both HB007-treated and HB007-untreated cells (fig. S5I). CAPRIN1 consists of two conserved homolog domains HR1 and HR2, an E-rich region, Arg-Gly-Gly rich (RGG) boxes, and three short RG-rich sequences (53). To identify the binding domains of CAPRIN1 to HB007 and CUL1, we generated Flag-CAPRIN1 domain constructs and expressed these in HCT116 cells. HB007-biotin pull-down of cell lysates followed by immunoblot revealed the binding of HB007 to the domain HR1, whereas Flag IP demonstrated the binding of CUL1 to the domain HR2 (Fig. 5H and fig. S5, J and K). HB007 binding of the N-terminal HR1 induces CAPRIN1-FBXO42 interaction and SUMO1 recruitment to the CAPRIN1-CUL1 E3 ligase for SUMO1 ubiquitination and degradation (fig. S5L).
The selective activity of SUMO1 degraders against various types of cancer cells
To evaluate the targeted anticancer activity of SUMO1 degraders, we first showed that SUMO1 and CAPRIN1 protein concentrations were elevated in breast, colorectal carcinoma, and NSCLC patient tissues and established cell lines as compared to matched normal tissues and normal cells (Fig. 6A and fig. S6A). CPD1 and HB007 treatment inhibited the growth of a panel of cancer cell lines with HB007 being more effective at inhibiting growth (IC50 = 0.3 to 1.5 μM) (Fig. 6B). In contrast, HB007 displayed much lower growth inhibition effects on matched normal lung, colon, breast and brain cells with the cell-based IC50 values much higher than that obtained from cancer cell lines (Fig. 6C and fig. S6B). Consistently, Western blots showed that HB007 had no effects on the conjugated and total concentrations of SUMO1 in normal lung epithelial cells as compared to lung cancer cells (Fig. 6, D and E).
Fig. 6. The selective activity of CPD1 and HB007 against different cancer cell types.
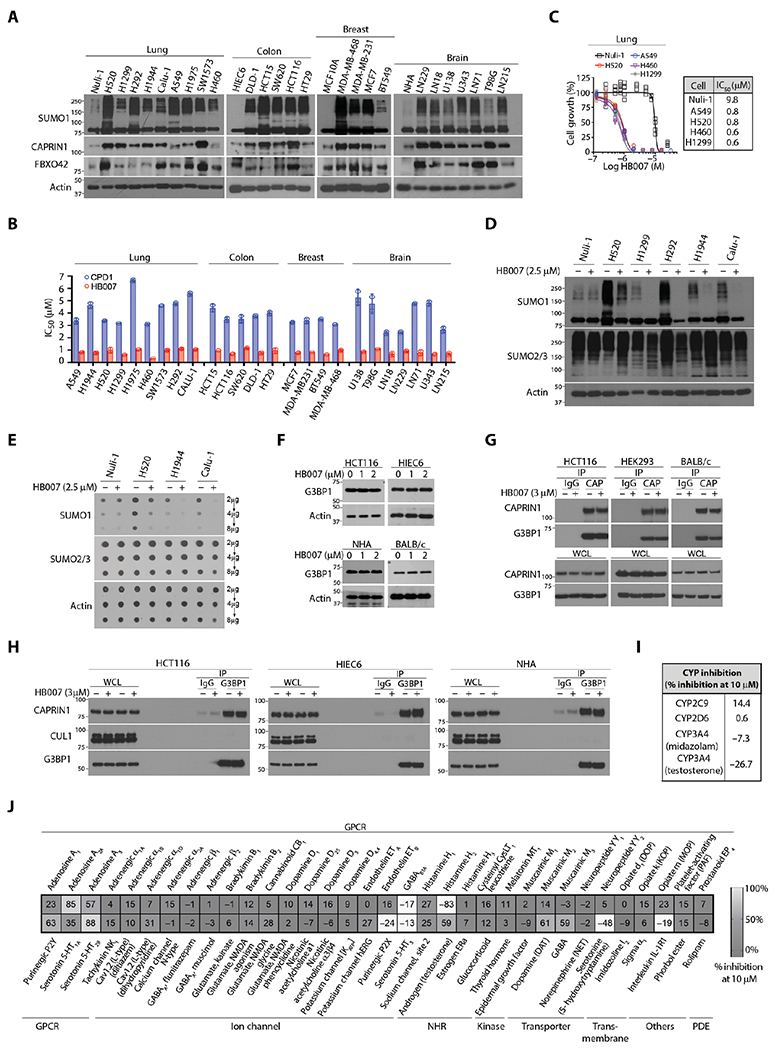
(A) NSCLC (lung), colon, breast carcinoma, and brain glioblastoma cell lines and matched normal lung Nuli-1, colon HIEC6, and breast MCF10A epithelial cells, and brain normal human astrocytes (NHAs) were analyzed by Western blotting for SUMO1 conjugation, CAPRIN1, and FBXO42 expression. (B) The IC50 values were compared between CPD1 and HB007 5 days treatment of 25 cancer cell lines (n = 2 calculated from two independent experiments totaling n = 12). (C to E) The normal lung epithelial cell Nuli-1 and NSCLC cell lines were treated or untreated with HB007 for 72 hours and analyzed by cell viability assay for growth inhibition (n = 6 biological replicate) (C), Western blot for conjugated SUMO1 (D), and dot blot for total SUMO1 amounts (E). SUMO2/3 was used as the selectivity control. (F) The human colon carcinoma HCT116, normal colon epithelial HIEC6, normal human astrocytes NHA, and BALB/c mouse fibroblasts were treated with HB007 for 48 hours and analyzed by Western blot for G3BP1 amounts. (G) HCT116, HEK293, and BALB/c cells were treated with HB007 for 8 hours and subjected to IP by a CAPRIN1 antibody (CAP), followed by Western blot for CAPRIN1 and G3BP1 interaction. IgG was used as the negative control. (H) HCT116, HIEC6, and NHA were treated with HB007 and subjected to IP using a G3BP1 antibody, followed by Western blot for its interaction with CAPRIN1 and CUL1. (I) Enzymatic analysis of the inhibition of CYP enzymes by HB007 at 10 μM in human liver microsomes. (J) The selectivity profile of HB007 (10 μM) against 67 diverse key human proteins as indicated.
To investigate the mechanism of drug action, we determined whether the compounds either inhibit cell proliferation or promote cell apoptosis. We performed a 5-bromo-2′-deoxyuridine assay, which demonstrated that HB007 treatment inhibited the proliferation of various types of cancer cell lines (fig. S6C). In contrast, the treated cells showed neither caspase-3/7 activation nor caspase-3 cleavage (fig. S6, D and E), and HB007 did not induce apoptotic cell death in these cancer cells. Last, cell viability and Western blot analysis of a large panel of brain glioblastoma, colon, breast carcinoma, and NSCLC cell lines demonstrated the broad activity of HB007 in the inhibition of cancer cell growth and degradation of SUMO1 but not SUMO2/3 protein (fig. S6, F to I).
CAPRIN1 forms a stable complex with Ras guanosine triphosphatase–activating protein binding protein 1 (G3BP1) responsible for normal cell growth and neuronal synaptic formation (53); thus, CAPRIN1 or G3BP1 knockout is lethal to newborn mice due to synaptic and respiratory failure (54, 55). To determine whether HB007 binding of CAPRIN1 alters its normal functions, we treated HCT116 cells, normal colon epithelial HIEC6 cells, normal human astrocytes (NHAs), and BALB/c mouse fibroblasts with HB007 and measured G3BP1 expressions. We determined that HB007 did not affect expression and interaction of CAPRIN1 and G3BP1 in NHAs, colon epithelial HIEC6, embryonic kidney HEK293, and mouse fibroblasts (Fig. 6, F and G). IP of cellular G3BP1 followed by Western blotting in these cells further showed no effect of HB007 on G3BP1-CAPRIN1 interaction in both normal and cancer cells (Fig. 6H).
To evaluate possible off-target effects of the lead compound HB007, we performed enzymatic analyses of the inhibition of CYP enzymes by HB007 in human liver microsomes and found that HB007 did not inhibit CYPs (Fig. 6I). The possible off-target effects of HB007 were analyzed by the LeadProfilingScreen that includes 68 representative key normal human functional proteins including G protein–coupled receptors (GPCRs), kinases, ion channels, transporters, transmembrane, and phosphodiesterase and other enzymes. HB007 selectivity against these human proteins was tested at 10 μM, and no activity against kinases, ion channels, NHRs, enzymes, and human ether-a-go-go–related gene was observed. There was some inhibition of adenosine A2A, histamine H2, and 5-hydroxytryptamine receptor 2B (5-HT2B) but was considered neither confounding off-targets for pharmacology nor threatening off-targets of HB007 as a therapeutic (Fig. 6J) because clinical drugs such as Zantac, Clozapine, and theophylline antagonize these GPCRs.
The PKs and in vivo anticancer activity of CPD1 and HB007
To determine the activity in vivo, we first tested the metabolic stability of the HB007 and CPD1. HB007 was incubated with purified mouse and human liver microsomes, and its degradation was measured by LC-MS. The data showed that HB007 was sufficiently stable in the microsomes (Fig. 7A). To test the PKs of CPD1 and HB007, we analyzed plasma and organ samples over 12 hours from rodents after intraperitoneal injection and found that both CPD1 and HB007 exhibited favorable PK profiles in plasma and quick distribution in organs including brains with the brain-to-plasma partition coefficient 0.96 for CPD1 and 0.57 for HB007 (Fig. 7, B and C, and fig. S7, A and B). In contrast, oral and intravenous administration of HB007 as a suspension resulted in the low plasma concentrations (fig. S7, C and D), but a solution formulation of HB007 improved the oral bioavailability of the compound up to 82% in plasma (Fig. 7D).
Fig. 7. The PKs and in vivo anticancer activity of CPD1 and HB007.
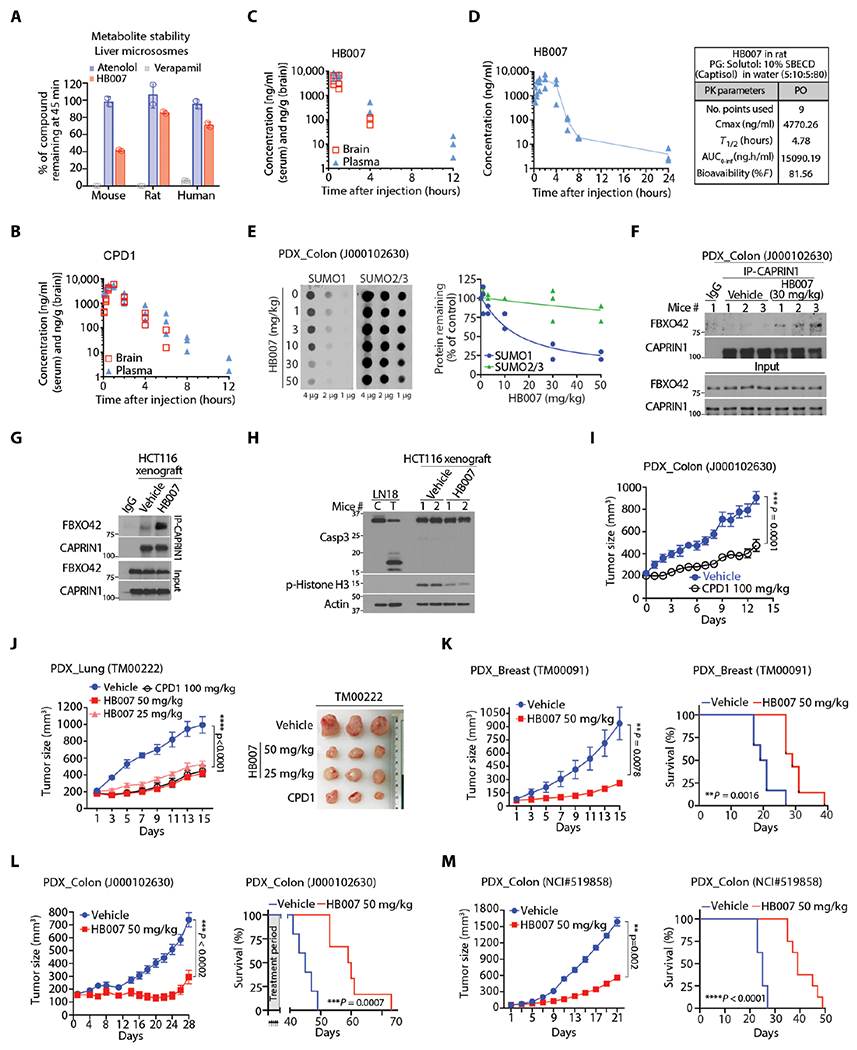
(A) HB007 was incubated for 45 min with mouse, rat, and human microsomes, and the amount of HB007 remaining was quantified by LC-MS (n = 3). Atenolol and verapamil were used as controls. (B and C) The PK assessment of CPD1 (B) and HB007 (C) by analyzing mouse plasma and brain tissue was carried out following intraperitoneal injection of a compound (20 mg/kg) (n = 3 mice per time point) in NOD/SCID mice. (D) The PKs of HB007 were determined by analyzing the compound in rat plasma samples after administrated orally in a solution formulation (n = 3). (E) Colon cancer PDX mice were treated with the indicated doses of HB007 through intraperitoneal injection once per day for 3 days beginning after a week of tumor inoculation; xenograft tissues were analyzed by dot blotting for total amounts of SUMO1 with SUMO2/3 as the control (left) with the densities quantified (right). (F) Colon cancer PDX mice [as in (E)] were subjected to IP by a CAPRIN1 antibody followed by Western blot for FBXO42 and CAPRIN1 interaction. (G) HCT116 xenograft mice were treated with vehicle or HB007 once per day for 3 days beginning after 10 days tumor inoculation; xenograft tissue and subjected to IP using CAPRIN1 antibody followed by Western blot for FBXO42 and CAPRIN1 interaction. (H) HCT116 xenografts [as in (G)] were subjected to Western blotting for caspase-3 cleavage for apoptotic cell death and phosphorylated histone (p-histone) H3 as a proliferation marker. (I) Colon cancer PDX mice were treated with CPD1 (100 mg/kg) for 15 days beginning after a week of tumor inoculation (means ± SEM, n = 10 per group, ***P=0.0001 by Wilcoxon tests). (J and K) PDX mice of lung NSCLC (J) and breast carcinoma (K) were treated with the indicated doses of CPD1 and/or HB007 for 15 days beginning after a week of tumor inoculation, and tumor sizes indicated that the treatment suppressed xenografts. (G) Right: Representative images of lung xenografts at the end of the treatment were presented. Data represent as means ± SEM. PDX mice of breast: n = 6 per group for vehicle and n = 7 per HB007, **P = 0.0078 at day 15 by Wilcoxon tests; PDX mice of lung: n = 7 per vehicle and n = 8 to 9 per treatment group, ****P < 0.0001 by Friedman test. (L and M) Mice bearing primary (L) and metastatic colon PDX xenografts (M) were treated with HB007; effect of the treatment on xenograft growth (left) and survival of mice as indicated by Kaplan-Meier survival curves (right). Data represent means ± SEM. PDX primary colon (J000102630): n = 5 per vehicle, n = 6 per treatment group, ***P < 0.0002 by Wilcoxon test; PDX metastatic colon (NCI #519858): n = 8 per group, **P = 0.002 by Wilcoxon test.
We next tested SUMO1 target engagement with HB007 and CPD1 using patient-derived xenograft (PDX) mouse models. The colon cancer PDXs were established by implanting 3-mm3 pieces of the cancer tissues subcutaneously in mice. When xenografts reached 50 to 70 mm3 in a week, the mice were treated through intraperitoneal injection with HB007 or CPD1 once per day for 3 days. Dot and Western blot analysis of the xenograft tissues showed that the treatment markedly reduced both the total amounts and conjugate SUMO1 but not SUMO2/3 protein in the xenografts in a dose-dependent manner (Fig. 7E and fig. S7E). To reveal the mechanism of HB007 action in SUMO1 degradation in the PDX mouse models, we carried out an IP of endogenous CAPRIN1 using a CAPRIN1 antibody and showed that HB007 induced the interaction of CAPRIN1 and FBXO42 in the xenograft tissues (Fig. 7F). The experiment was repeated using HCT116-derived xenografts, and the data showed a drastic increased interaction of CAPRIN1 and FBXO42 protein in HB007-treated xenografts (Fig. 7G). In contrast, HB007 treatment did not affect the concentration of CAPRIN1 and FBXO42 proteins in the xenografts (fig. S7F). Western blot analysis revealed the decrease of the proliferation marker, phospho-histone H3, but showed no caspase-3 cleavage in HB007-treated xenografts (Fig. 7H).
Next, we examined the anticancer activity of CPD1 and HB007 first by treating cancer cell line-derived xenograft mice. NSCLC A549 and colon cancer HCT116 cells (5 × 106) were injected subcutaneously in right flanks of mice. When xenografts reached 50 to 70 mm3 in 10 days, mice were treated with CPD1 and HB007 via intraperitoneal injection, once per day for 14 days. Xenograft volumes were measured once per 2 days; the data indicated that the treatment suppressed xenograft growth with HB007 being more effective than CPD1 (fig. S7, G and H). In addition, LN229-derived brain xenografts were generated through intracranial injection of the cells (106 cells) in the right striatum of mice. Two weeks after injection, the mice were treated with CPD1 or vehicle control through intraperitoneal injection, once per day for 14 days. Kaplan-Meier analysis revealed that treatment with CPD1 increased the survival of brain xenograft-bearing mice (fig. S7I).
Last, we evaluated the therapeutic efficacy of CPD1 and HB007 in cancer PDX mouse models. PDXs were established by implanting 3-mm3 pieces of cancer tissues subcutaneously in mice. Once xenografts reached 50 to 70 mm3 in a week, the mice were treated with CPD1 or HB007 through intraperitoneal injection, once per day for 14 days. Xenografts were measured once every 2 days. The data showed that treatment with CPD1 or HB007 effectively suppressed the progression of human colon, breast carcinoma, and NSCLC PDXs (Fig. 7, I to K). To evaluate the survival, PDX mice bearing BRCA1 mutant breast carcinoma and primary or metastatic colon carcinoma were treated with HB007. HB007 treatment effectively suppressed all xenograft models and increased the survival of these xenograft mice (Fig. 7, I to M). Across all xenograft mice, the compounds were well tolerated over the treatment period with no clinical signs of lethargy, ataxia, paralysis, seizure, or weight loss observed (fig. S7, J to L). Histological examination of PDX mouse organs showed no tissue damage in the brain, heart, lung, liver, colon, and kidney of treated mice (fig. S7M).
DISCUSSION
Small-molecule drug discovery has focused on binding site occupation and functional inhibition of targeted proteins, yet 80% of the human proteome is undruggable due to lack of active sites or binding pockets (56). Taking advantage of targeted protein degradation of undruggable proteins (2), we designed and carried out a cancer cell–based drug screen and identified small-molecule degraders of SUMO1 previously considered an undruggable protein. Our cancer cell–based drug screening identified the compound CPD1, and subsequent SAR studies optimized the chemical lead compound HB007, which had improved drug-like properties and potent anticancer activity. Using SUMO2/3 in parallel through the drug screening and SAR studies, we were able to establish the selectivity of SUMO1 degraders. The small-molecule degraders showed high selectivity for SUMO1; the compounds exhibited no harmful off-target effects and reduced cancer cell lines and PDXs in mice without causing damage to normal tissues. The finding that HB007 was efficacious as a single agent across various cancer models highlights the importance of SUMO1 in cancer progression and its pharmacological degradation as a broadly effective cancer therapy in preclinical models.
SUMO1 itself is a UB-like protein, and here, we show that it is also a UB substrate. SUMO1 degraders induced the ubiquitination of unconjugated SUMO1 and ablated its conjugation in cancer cells. Using a genomic CRISPR-Cas9 knockout screen, we identified FBXO42 as the SUMO1 substrate receptor of CUL1 ligase. SUMO1 degraders induced FBXO42 recruitment of SUMO1 to CUL1 ligase for SUMO1 ubiquitination and degradation. CUL1 ligases are bound to the 19S regulatory core (57), where the proteasome-associated protease USP14 trims poly-UB chains and promotes the translation of the protein from the 19S to the 20S core for degradation (43). The discovery of CAPRIN1 as the HB007 binding partner further revealed the mechanism of drug action. The finding that CAPRIN1 constitutes CUL1 E3 ligase suggests that CAPRIN1-CUL1 is a E3 ligase. Using its C terminus, CAPRIN1 binds to neddylated-CUL1 ligase. HB007 binding to the N terminus of CAPRIN1 induced the interaction of CAPRIN1 with FBXO42 and recruitment of SUMO1 to the CAPRIN1-CUL1 E3 ligase. These studies provide the CAPRIN1-CUL1 E3 ligase complex as a targeted protein for degradation drug discovery. Further studies will identify CAPRIN1-CUL1-FBXO42 modulators and substrate protein degraders.
There are some limitations to the study. The degrader HB007 may act as a molecular glue between CAPRIN1 and FBXO42 (3). However, the crystal structure of CAPRIN1 is available only for the N-terminal residues 132 to 251 (58), and further study of the full-length CAPRIN1 structure and its complexes with HB007 and FBXO42 is needed to determine whether the degrader acts as a glue for CAPRIN1 and FBXO42. It is conceivable that CAPRIN1-CUL1-FBXO42 might have other substrates, collectively referred to as a degrome, as has been elucidated for thalidomide (8). However, it remains challenging to identify the degrome of SUMO1 degraders because SUMO1 is a modifier where its conjugation controls the ubiquitination and degradation of many of its substrate proteins (33). Further investigation is necessary to distinguish between the subset of degraded proteins and SUMO1 substrate proteins that are decreased because of the removal of SUMO1 conjugation.
Genomic CRISPR-Cas9 screens also identified HB007-targeted genes in multiple cellular processes including chromatin modeling and DNA repair (CHD8, PAXIP1, TAF5L, H2AFZ, TEN1, G3BP1, and OLA1) (59), gene transcription (KAT2A, METTL23, EED, MED23, and CBX4), protein translation (NOC4L, DNAJC24, CAPRIN1, and EIF3H), Ras/Raf-ERK axis (CAD, PDCD10, NPRL2, MAPK1, WDR83, and NPRL2), and metabolism (STARD7, G6PD, and SLC7A1). Seven genes were directly linked to sumoylation: CBX4 encodes a SUMO E3 ligase (60); the proteins encoded by G6PD, CHD8, USP14, CAD, H2AFZ, and OLA1 are SUMO1 substrates (61). Further investigation will determine whether HB007’s anticancer activity occurs through degradation or removal of SUMO1 conjugation of the substrate proteins in cancer cells.
Our work provides a cancer cell–based approach of targeted protein degradation for the discovery of small-molecule degraders of targeted cellular proteins. As therapeutics, both HB007 and CPD1 were able to degrade SUMO1 protein and ablate its conjugation in cancer cells and PDXs in mice, and these compounds displayed anticancer activity against various types of human cancers. This cancer cell–based approach provides an alternative strategy of targeted protein degradation that can be applied to the discovery of other small-molecule degraders of other cellular proteins in cancer cells.
MATERIALS AND METHODS
Study design
The objective of this study was to identify and develop SUMO1 small-molecule degraders as anticancer drugs. Using denatured Western blots to distinguish between SUMO1 and SUMO2/3, we carried out cancer cell–based drug screening and identified the hit compound CPD1 from the NCI drug-like compounds library as a SUMO1 degrader. A series of SAR studies of CPD1 analogs optimized the chemical lead HB007. To reveal the compound-targeted pathway, we applied a genome-scale CRISPR-Cas9 knockout screen and HB007 pull-down proteomics and identified CAPRIN1 as the target protein of these compounds. Various molecular and cellular assays revealed the CAPRIN1-CUL1-FBXO42 ligase as a target of HB007 and demonstrated that the compound induced the ubiquitination and degradation of SUMO1 through the E3 ligase in cancer cell lines in vitro. A panel of cancer cell lines and matched normal cells was used to evaluate compound anticancer activity and toxicity. All molecular and cellular assays were performed with at least duplicate samples, and each experiment was repeated at least three times. The therapeutic efficacy of these compounds was evaluated in PDXs in mice. All animals were randomly allocated to the treatment groups. Tumor measurement and treatment were not blinded. Sample sizes were determined on the basis of statistical power analysis and ranged from 6 to 10 mice per group. No samples were excluded from the study. All mouse studies were carried out under the protocols approved by the Institutional Animal Care and Use Committee.
Two-dimensional colony formation assay
Cells were grown in triplicate at a low density in six-well plates and treated with CDP1 or HB007 at the concentrations as indicated. The culture medium was replaced every other day with dimethyl sulfoxide (DMSO) or drug treatment. After 15 days, the cells were fixed and stained with 0.5% crystal violet solution in methanol. The plates were air-dried and scanned at 600 dpi, and colonies were counted using OpenCFU colony counting software (http://opencfu.sourceforge.net).
In vivo ubiquitination
Cells were cotransfected with HA-UB and Flag-SUMO1-GV for 24 hours, followed by CPD1 or HB007 treatment for 48 hours. Cell were then harvested in cold phosphate-buffered saline (PBS) and lysed in a denaturing buffer. Lysates were diluted to 0.1% SDS in radioimmunoprecipitation assay (RIPA) buffer and immunoprecipitated overnight with anti-Flag M2 agarose beads. The cells were extensively washed five times with RIPA buffer. Proteins were eluted with 2× Laemmli buffer and examined by Western blotting using HA and Flag antibodies.
In vitro sumoylation
In vitro sumoylation assay was carried out in 10 μl of reaction volume containing 100 ng of CDK6, 1.5 μg of SUMO-1, 500 ng of Ubc9, and 50 ng of SEA1/2 in a buffer consisting 20 mM Hepes (pH 7.3), 110 mM KOAc, 2 mM Mg[OAc]2, 1 mM EGTA, 1 mM dithiothreitol (DTT), 0.05% Tween 20, 0.2 mg of ovalbumin, and 10 mM adenosine 5′-triphosphate. Protease inhibitors were added to the reaction together with CDK6 protein. After 90 min at 30°C, reactions were stopped by addition of 10 μl of 2× Laemmli buffer and analyzed by Western blotting with a CDK6 antibody.
In vivo sumoylation
Flag-CDK6 or Myc-UBC9 vector was transfected in a cell line together with YFP-SUMO1-GG or YFP-SUMO3-GG. After 24 hours of transfection, cells were treated with CPD1 or HB007 for 48 hours and then lysed in denaturing buffer, diluted to 0.1% SDS in RIPA buffer, and immunoprecipitated with anti-tag agarose beads. After extensive washes, proteins were eluted and sumoylation was detected by Western blotting using anti-YFP and CDK6 or UBC9 antibodies.
Protein half-life assay
The cells were treated with CPD1 or HB007 for 24 hours followed by CHX (50 μg/ml) for indicated times in Results. Cell extracts from each time points were resolved by SDS–polyacrylamide gel electrophoresis followed by Western and dot blotting using SUMO1, SUMO2/3, and β-actin antibodies. The β-actin protein amounts were used as protein loading control and normalized for the degradation rate of SUMO1 in quantification graph generated by ImageJ software. The β-actin–normalized SUMO1 protein amounts at 0 hours were defined as 100 for each panel.
S100 fraction degradation
This assay was carried out using an S100 Fraction Degradation kit from Boston Biochem according to its protocol. SUMO1 protein (20 ng) was added to Hela S100 Fraction with or without CPD1 at 10 μM for 4 hours at 37°C. In one sample, degradation was inhibited by the co-addition of MG132. After 4 hours, SUMO1 amount was evaluated by dot blot using a SUMO1 antibody.
MG132 treatment protocols
Cells were either pretreated with MG132 for 4 hours at 1 μM followed by 24 hours of treatment with HB007 or CPD1 or first treated with CPD1/HB007 for 24 hours followed by treatment with MG132. At the end of the treatment, cells were harvested and lysed in denaturing condition. Western blots using antibodies against SUMO1 and UB were used to test SUMO1 amount.
HB007 pull-down assay
Preparation of HB007-immobilized beads and HB007-biotin is described in the chemical synthesis section in Supplementary Materials. HB007-immobilized FG magnetic beads (0.5 to 1 mg) were equilibrated by three washes in binding buffer [0.1% NP-40, 50 mM tris-HCl (pH 8.0), 100 mM KCl, 1 mM MgCl2, 0.2 mM CaCl2, 0.2 mM EDTA, 10% glycerol, 150 mM NaCl, 1 mM DTT, and 0.2 mM phenylmethylsulfonyl fluoride, 1× protease inhibitor]. Cell extracts (1 mg) prepared from HCT116, human embryonic kidney (HEK) 293, and LN229 lysed in binding buffer were incubated with the HB007-FG beads for 2 hours at 4°C. Beads were washed three times with binding buffer. Bound proteins were eluted on ice for 20 min with elution buffer [0.0625 M tris-HCl (pH 6.8), 0.005% bromophenol blue, 2% SDS, 10% glycerol, and 5% 2-mercaptoethanol].
For HB007-biotin pull-down, streptavidin magnetic beads (Thermo Fisher Scientific) were mixed with HB007-biotin for 1 hour at 4°C, washed three times in binding buffer, followed by 5-min incubation with 0.001% biotin in binding buffer at room temperature to block free streptavidin sites on the magnetic beads. The beads were then washed three times and incubated with cell lysate (1 mg) for 1 to 2 hours at 4°C. Bound proteins were eluted as described above. In experiments where compounds were added as competitors, each compound (0.3 mM) or HB007 at different concentrations was added together with the cell extract for 1 hour. For the competition assay using rhCAPRIN1 protein, 200 ng of rhCAPRIN1 was premixed with 1 μM biotin for 30 min at 4°C to block biotin binding site of CAPRIN1, followed by the addition of free HB007 at different concentrations (from 500 nM to 10 μM). The protein was then incubated with the HB007-biotin following the protocol described above.
CRISPR/Cas9-mediated genome-editing in cell lines
The lentiCRISPRv2 expression system was used to construct lentiviral CRISPR for FBOX42 and USP14 as described previously (36). Briefly, gRNA sequence was cloned into lentiCRISPR v2 (Addgene, #52961) according to the protocol. The sequences for FBXO42 CRISPR were as follows: FBXO42 Oligo1 (5′-CACCGGTCCTTTCTCTCACCGTATC-3′) and FBXO42 Oligo2 (5′-AAACGATACGGTGAGAGAAAGGACC-3′). The sequences for USP14 CRISPR were as follows: USP14 Oligo1 (5′-CACCGTGAGCCTTGAATACCATTGG-3′) and USP14 Oligo2 (5′-AAACCCAATGGTATTCAAGGCTCAC-3′). The sequences of CAPRIN1 CRISPR were as follows: CAPRIN1 Oligo1 (5′-CACCGCGACAAGAAACTTCGGAACC-3′) and CAPRIN1 Oligo2 (5′-AAACGGTTCCGAAGTTTCTTGTCG C-3’). The sequence for SUMO1 CRISPR Oligo is 5′-GAAGTTTATCAGGAACAAAC-3′. The sequences for nontargeting control CRISPR (Addgene, #80248) were as follows: Oligo1 (5′- CACCGGTATTACTGATATTGGTGGG-3′) and Oligo2 (5′-AAACCCCACCAATATCAGTAATACC-3′). HCT116 cells were infected with the same virus titer for the control CRIPSR, FBXO42 CRISPR, and USP14 CRIPSR. After 24 hours of infection, puromycin (1 μg/ml) was added to enrich positively infected cells for 2 days when mock-transfected control cells were completely eliminated by puromycin. Single-cell cloning was performed by serial dilution in 96-well plates. After 6 to 8 weeks, the clones were picked for knockout verification by Western blots.
PK studies
Intraperitoneal PK studies were performed by the Pharmacology Core at the Karmanos Cancer Institute and Syngene international using 6- to 8-week-old nonobese (NOD)/severe combined immunodeficient (SCID) mice (Taconic, Albany, NY). To determine the plasma and tissue concentration, mice (three mice per time point) were treated with a single intraperitoneal dose of CPD1 or HB007 (20 mg/kg). Blood samples were collected at 0.25, 0.5, 1, 2, 4, 6, 10, and 24 hours for CPD1 and 0, 0.5, 1, 2, and 4 hours for HB007 after the injection. Blood was collected into K2 EDTA–containing tubes, and plasma was obtained by centrifugation at 2000g for 5 min. Compound concentration in plasma was quantified by LC-MS/MS. PK parameters were estimated using Phoenix WinNonlin software (version 6.2; Certara USA, Princeton, NJ) from mean plasma concentration time profiles. The area under the curve was calculated using trapezoidal method.
Intravenous (IV) and oral (PO) PK in rodents were processed by Syngene International using 8- to 10-week-old CD1 male mice and 8-to 10-week-old Sprague Dawley male rats. Three mice or rats per group received a single administration of 1 mg/kg diluted in 20% Captisol in Milli-Q water (IV) or 3 mg/kg in 1% hydroxyethyl cellulose in Milli-Q water (PO). An improved formulation for HB007 for PO consisting of propylene glycol, Solutol, 10% sulfobutylether-β-cyclodextrin (SBECD, Captisol) in water (5:10:5:80) was also used for rat PK. Blood samples (~25 μl per each time point from saphenous vein of mice or jugular vein of rats) were collected in K2 EDTA–containing tubes at 0.083, 0.25, 0.5, 1, 2, 4, 6, 8, and 24 hours for IV and 0.25, 0.5, 1, 2, 4, 6, 8, 24 hours for PO. Blood samples were then centrifuged at 13,000 rpm for 10 min at 4°C and stored at −80°C. Samples were analyzed by LC-MS/MS.
Selectivity profiling
The selectivity profiling of HB007 against a selected panel of 68 diverse targets was performed by Eurofins Discovery at the compound test concentration of 10 μM. In the assay, results from primary screen binding interactions were reported as percent of inhibition of specific binding, where higher numbers indicated HB007 binding.
CYP inhibition
CYP inhibition on three cytochrome P450s (2C9, 2D6, and 3A4 at 10 μM) in human liver microsomes was performed by Eurofins Discovery at a compound test concentration of 10 μM.
Microsomal stability
Metabolic stability analysis measured using liver microsomes from mouse, rat, and human was performed by Syngene International. Briefly, 1 μM HB007 was added to a solution of 100 mM potassium phosphate buffer at pH 7.4 with 1 mM nicotinamide adenine dinucleotide phosphate (NADPH). Microsomes from mouse, rat, or human were added and incubated for 45 min at 37°C. The percentage of the compound remaining at 45 min time point was analyzed by LC-MS/MS. Verapamil (high clearance) and atenolol (low clearance) were added as quality control (QC).
Tumor cell line–derived xenografts and PDXs in mice
The animal studies were approved by the Institutional Animal Care and Use Committee of the Indiana University School of Medicine. CPD1 or HB007 was dissolved in 25% DMSO, 25% Kolliphor EL, and 50% PBS for intraperitoneal injection. For cell line–derived xenografts, female 6- to 8-week-old athymic BALB/c mice (Taconic, Albany, NY) were injected subcutaneously with LN229, HCT116, or A549 cells (5 × 106) in 100 μl of PBS. When the tumor reached 50 to 70 mm3, mice were randomized into groups and treated daily with CPD1, HB007, or vehicle once per day for 14 days. Tumor size was measured using digital calipers every 2 days. Tumor volume was determined by calculating (length × width2)/2.
For mouse intracranial xenografts, LN229 cells (106 cells) were injected in the right striatum of NOD/SCID mice (Taconic). Two weeks after injection, the mice were treated daily with CPD1 or vehicle control. The mice were followed up and euthanized until development of the signs (lethargy, ataxia, paralysis, or seizure). Growth curves and survival curves were generated using GraphPad Prism v8.
NOD/SCID gamma (NSG) mice bearing PDXs from the Jackson Laboratory were harvested, processed into 3-mm3 fragments, and stored in liquid nitrogen. PDX fragments in frozen were also obtained from the NCI Patient-Derived Models Repository. The fragments of colon (J000102630, NCI519858), lung (TM00222), and breast cancer (TM00099) PDXs were implanted subcutaneously in right flanks of NSG mice (Taconic). When tumor reached 50 to 70 mm3, mice were randomized into groups and treated with CPD1, HB007, or vehicle once per day for 14 days. Tumor size was measured using digital calipers once per 2 days. Tumor volume was determined by calculating (length × width2)/2. For survival study, animals were monitored for tumor volumes every 3 days until tumor volume exceeded 1500 mm3 or mice died.
Genome-scale CRISPR-Cas9 knockout screening
Library generation
A whole-genome library was developed that exploited informatically optimized guides (36) expressed in tandem with a modified tracrRNA sequence (5′-GTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTT-3′) (39). An all-in-one lentivirus plasmid vector was built comprising a selection marker (puromycin resistance), the expression cassette for Cas9, and sgRNA sequence and cloned by Gibson Assembly [New England BioLabs, #E2611S/L] in accordance with the manufacturer’s instructions. Library plasmids were purified using a QIAGEN Plasmid Plus Purification system based on the manufacturer’s instructions.
Lentivirus production
HEK293T cells (American Type Culture Collection, USA) grown in Dulbecco’s modified Eagle’s medium (DMEM) and 10% fetal bovine serum (FBS) (Gibco, UK) were transfected with the library plasmids using Lipofectamine 3000 (Invitrogen, USA) and ViraPower packaging virus (Life Technologies, UK) according to the manufacturer’s instructions. After 48 hours, the medium was removed and centrifuged at 500g for 10 min at 4°C. The virus was concentrated using Lenti-X concentrator (Clontech, #631232). The viral supernatant was aliquoted and stored at −80°C in DMEM with 10% FBS and 1% bovine serum albumin.
Cell transduction, staining, and screening protocol
HCT116 cells were seeded in complete medium supplemented with polybrene (8 μg/ml; Sigma-Aldrich) and seeded into 12-well dishes at 2 × 106 cells per well and spinfected for 2 hours at 2000 rpm at 37°C using virus diluted to achieve a multiplicity of infection of 0.3. At least 1 × 108 cells were transduced, transferred to a 50-ml falcon, and centrifuged at 1000 rpm for 5 min. The cells were resuspended in 50 ml of fresh media (without polybrene). After 48 hours, cells were treated with puromycin (1 μg/ml) and then separated into replicates and treatment groups of at least 3.6 × 107 cell per condition and grown in continuous culture with the treatment of HB007 and DMSO as indicated for 21 days, during which the control-treated population went through 14 population doublings. Cell pellets were collected and stored at −80°C. All samples were thawed, and guide DNA extracted using the QIAGEN Blood Maxi Kit. DNA concentration was determined using a Nanodrop spectrophotometer, and at least 230 μg of genomic DNA for each sample was amplified with polymerase chain reaction to generate amplicons of the sgRNA cassette using a forward primer (TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGU-[Variable]-TGTGGAAAGGACGAAACACC) and a reverse primer (GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGATCAATTGCCGACCCCTCC). These amplicon samples were purified using Agencourt beads (Beckman) and deep-sequenced on an Illumina NextSeq platform/system (Microsynth AG, Switzerland).
QC analysis
After the deep sequencing, QC analyses of the control group behavior were performed to evaluate how essential genes responded to CRISPR-Cas9 knockout and if the samples were deemed suitable for further analysis and hit calling. First, the sample quality was determined by distribution analysis using a probability density function and evaluation of concordance between sample replicates using Pearson’s correlation test. Second, after mapping of samples to the library sgRNA sequences before treatment and their quantitation, the performance of control guide RNAs was evaluated. This QC metric is important to establish the efficiency of the CRISPR-Cas9 process. Control guides included 2000 nontargeting guides that did not bind anywhere in the genome, 612 negative control guides targeting 102 genes, which are guides designed against putative neutral genes, and, lastly, 294 positive control guides targeting 49 genes, which are guides designed against putative essential genes because the knockout of essential genes should negatively affect cell survival.
The results showed that the control guides performed as expected, with dropout rates for guides targeting essential genes of up to 128-fold. Nontargeting guides showed a modest enrichment probably because these guides do not induce any DNA damage, which resulted in a competitive advantage. On the basis of the QC conducted, all samples were deemed suitable for further analysis, and NSG datasets—e.g., sgRNA abundance—were then analyzed using scripts based on the MAGeCK algorithm (40). The hit genes were called by comparing the difference in abundance between HB007- and DMSO-treated samples for each sgRNA. Using an FDR of <0.05 as a cutoff, 28 genes were identified that rendered cells resistant to HB007 treatment.
Data analysis
Raw NSG libraries were evaluated for quality using FASTQC version 0.11.5. (Babraham Institute, Cambridge, UK). Guide counts were obtained using an in-house customized version of the MAGeCK workflow version 0.5.56, which took into account guide staggering from the experimental protocol. Briefly, guides were trimmed and mapped with exact string counts from each file to provide raw counts for each guide found in the library. Guide counts were normalized within each group (median based), and log2 fold change was calculated to determine the change in abundance of each guide in each sample. Robust rank aggregation (RRA) values (P values) were determined using the MAGeCK algorithm (version 0.5.56) (40).
Statistical analysis
Statistical analyses were performed using GraphPad Prism v8 (GraphPad Software). Statistical significance was determined as indicated in the figure legends and represented as *P < 0.05, **P < 0.01, and ***P < 0.001. The unpaired t test was used for comparisons between two-group means under the assumption of normality. The nonparametric Friedman’s test was used for comparison between three groups. The nonparametric Wilcoxon test was used to compare the means rank between two groups. Kaplan-Meier survival analysis was carried out using GraphPad Prism and statistically analyzed with log-rank test. Sample sizes (n) were incorporated into the figure legends, and individual data points from each replicate were depicted as small circles in all figures. Data analysis for BLI assay was performed with Data Analysis 9.0 (Legacy version). After the experimental data folder was opened in the software, the reference well (with no CAPRIN1) and the reference sensors (with no loading of HB007) were assigned. Double reference method was used to subtract the reference well and sensors. The full baseline before the association step was aligned, and interstep correction was performed. After processing the data, the kinetic fit was analyzed with global fitting and 1:1 modeling.
Supplementary Material
Acknowledgments:
We thank F. Melchior, R. T. Hay, and Z. J. Chen for providing the plasmid YFP-SUMO1ΔC4 (1-97), YFP-SUMO3ΔC11, pcDNA3 6xHis/SUMO1, pCMV-myc-UBC9, pEF-IRES-P-HA-Ub-K48, pEF-IRES-P-HA-Ub-K0, pEF-IRES-P-HA-Ub-WT, and pEF-IRES-P-HA-R48. We also thank the Bindley Biophysical Analysis Facility at Purdue University for assistance with data collection and D. Liu (Sartorius BioAnalytical Instruments Inc.) for technical assistance.
Funding:
This work was supported in part by the National Institutes of Health R01CA203893 (to C. Hao) and R43CA224461 (to A.C.B.), the Indiana University School of Medicine Physician Scientist Initiative funded by Lilly Endowment Inc. (to C. Hao), the start-up fund from the Indiana University School of Medicine Department of Pathology and Laboratory Medicine (A.C.B; C.Hao.) and Indiana University Simon Comprehensive Cancer Center 100 Voices of Hope Fund (to A.C.B. and C. Hao). R.K.H is the 2020 Vera Bradley Foundation Scholar. C. Hao has been supported by the endowment fund of the Bicentennial Chair of the Indiana University School of Medicine.
Footnotes
Competing interests: A.C.B. and C. Hao are the cofounders of HB Therapeutics Inc. H.-Y.L. is an employee of Synovel Laboratory LLC. M.B., C.l.S, and B.C.S.C. are the employees of Horizon Discovery. A.S. is a cofounder of Anagin Corp. A.C.B., C. Hao, and H.-Y.L. are co-inventors on the U.S. provisional patent application 62/669,640 and the PCT/US2019/031245 patent application entitled “Compositions and methods for treating cancer.” The other authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. MS proteomics data have been deposited to the ProteomeXchange Consortium through the PRIDE partner repository with the dataset identifier PXD022399 and 10.6019/PXD022399. CRISPR-Cas9 screen data have been deposited on BioGRID Open Repository of CRISPR Screens (ORCS) with accession bellail2021. The compound CPD1 is available from the NCI drug-like compounds library upon request (https://dtp.cancer.gov/RequestCompounds/). The lead compound HB007 will be made available to academic researchers through a material transfer agreement upon request to A.C.B.
REFERENCES AND NOTES
- 1.Salami J, Crews CM, Waste disposal-An attractive strategy for cancer therapy. Science 355, 1163–1167 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Schapira M, Calabrese MF, Bullock AN, Crews CM, Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug Discov 18, 949–963 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Baek K, Schulman BA, Molecular glue concept solidifies. Nat. Chem. Biol 16, 2–3 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, Wong KK, Bradner JE, Kaelin WG Jr., The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, Ciarlo C, Hartman E, Munshi N, Schenone M, Schreiber SL, Carr SA, Ebert BL, Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kronke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, Chamberlain PP, Mani DR, Man HW, Gandhi AK, Svinkina T, Schneider RK, McConkey M, Jaras M, Griffiths E, Wetzler M, Bullinger L, Cathers BE, Carr SA, Chopra R, Ebert BL, Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 523, 183–188 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu C-C, Miller K, Fang W, Wang N-Y, Nguyen D, Houston J, Carmel G, Tran T, Riley M, Nosaka L, Lander GC, Gaidarova S, Xu S, Ruchelman AL, Handa H, Carmichael J, Daniel TO, Cathers BE, Lopez-Girona A, Chamberlain PP, A novel cereblon modulator recruits GSPT1 to the CRL4CRBN ubiquitin ligase. Nature 535, 252–257 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Sievers QL, Petzold G, Bunker RD, Renneville A, Slabicki M, Liddicoat BJ, Abdulrahman W, Mikkelsen T, Ebert BL, Thoma NH, Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 362, eaat0572 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petzold G, Fischer ES, Thoma NH, Structural basis of lenalidomide-induced CK1α degradation by the CRL4CRBN ubiquitin ligase. Nature 532, 127–130 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM, Tichkule RB, Schebesta M, Forrester WC, Schirle M, Hassiepen U, Ottl J, Hild M, Beckwith RE, Harper JW, Jenkins JL, Thomä NH, Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han T, Goralski M, Gaskill N, Capota E, Kim J, Ting TC, Xie Y, Williams NS, Nijhawan D, Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 356, eaal3755 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Du X, Volkov OA, Czerwinski RM, Tan H, Huerta C, Morton ER, Rizzi JP, Wehn PM, Xu R, Nijhawan D, Wallace EM, Structural basis and kinetic pathway of RBM39 recruitment to DCAF15 by a sulfonamide molecular glue E7820. Structure 27, 1625–1633.e3 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Bussiere DE, Xie L, Srinivas H, Shu W, Burke A, Be C, Zhao J, Godbole A, King D, Karki RG, Hornak V, Xu F, Cobb J, Carte N, Frank AO, Frommlet A, Graff P, Knapp M, Fazal A, Okram B, Jiang S, Michellys PY, Beckwith R, Voshol H, Wiesmann C, Solomon JM, Paulk J, Structural basis of indisulam-mediated RBM39 recruitment to DCAF15 E3 ligase complex. Nat. Chem. Biol 16, 15–23 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Flotho A, Melchior F, Sumoylation: A regulatory protein modification in health and disease. Annu. Rev. Biochem 82, 357–385 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Hay RT, SUMO: A history of modification. Mol. Cell 18, 1–12 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Gareau JR, Lima CD, The SUMO pathway: Emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol 11, 861–871 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matunis MJ, Coutavas E, Blobel G, A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol 135, 1457–1470 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahajan R, Delphin C, Guan T, Gerace L, Melchior F, A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell 88, 97–107 (1997). [DOI] [PubMed] [Google Scholar]
- 19.Morris JR, Boutell C, Keppler M, Densham R, Weekes D, Alamshah A, Butler L, Galanty Y, Pangon L, Kiuchi T, Ng T, Solomon E, The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature 462, 886–890 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Kubota Y, O’Grady P, Saito H, Takekawa M, Oncogenic Ras abrogates MEK SUMOylation that suppresses the ERK pathway and cell transformation. Nat. Cell Biol 13, 282–291 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, Schmitt EM, Skinner SO, Xu Q, Li MZ, Hartman ZC, Rao M, Yu P, Dominguez-Vidana R, Liang AC, Solimini NL, Bernardi RJ, Yu B, Hsu T, Golding I, Luo J, Osborne CK, Creighton CJ, Hilsenbeck SG, Schiff R, Shaw CA, Elledge SJ, Westbrook TF, A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 335, 348–353 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim JH, Choi HJ, Kim B, Kim MH, Lee JM, Kim IS, Lee MH, Choi SJ, Kim KI, Kim SI, Chung CH, Baek SH, Roles of sumoylation of a reptin chromatin-remodelling complex in cancer metastasis. Nat. Cell Biol 8, 631–639 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Seeler JS, Dejean A, SUMO and the robustness of cancer. Nat. Rev. Cancer 17, 184–197 (2017). [DOI] [PubMed] [Google Scholar]
- 24.He X, Riceberg J, Soucy T, Koenig E, Minissale J, Gallery M, Bernard H, Yang X, Liao H, Rabino C, Shah P, Xega K, Yan ZH, Sintchak M, Bradley J, Xu H, Duffey M, England D, Mizutani H, Hu Z, Guo J, Chau R, Dick LR, Brownell JE, Newcomb J, Langston S, Lightcap ES, Bence N, Pulukuri SM, Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat. Chem. Biol 13, 1164–1171 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Nacerddine K, Lehembre F, Bhaumik M, Artus J, Cohen-Tannoudji M, Babinet C, Pandolfi PP, Dejean A, The SUMO pathway is essential for nuclear integrity and chromosome segregation in mice. Dev. Cell 9, 769–779 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Demarque MD, Nacerddine K, Neyret-Kahn H, Andrieux A, Danenberg E, Jouvion G, Bomme P, Hamard G, Romagnolo B, Terris B, Cumano A, Barker N, Clevers H, Dejean A, Sumoylation by Ubc9 regulates the stem cell compartment and structure and function of the intestinal epithelium in mice. Gastroenterology 140, 286–296 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Evdokimov E, Sharma P, Lockett SJ, Lualdi M, Kuehn MR, Loss of SUMO1 in mice affects RanGAP1 localization and formation of PML nuclear bodies, but is not lethal as it can be compensated by SUMO2 or SUMO3. J. Cell Sci 121,4106–4113 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Zhang FP, Mikkonen L, Toppari J, Palvimo JJ, Thesleff I, Janne OA, Sumo-1 function is dispensable in normal mouse development. Mol. Cell. Biol 28, 5381–5390 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L, Wansleeben C, Zhao S, Miao P, Paschen W, Yang W, SUMO2 is essential while SUMO3 is dispensable for mouse embryonic development. EMBO Rep. 15, 878–885 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo WH, Yuan LH, Xiao ZH, Liu D, Zhang JX, Overexpression of SUMO-1 in hepatocellular carcinoma: A latent target for diagnosis and therapy of hepatoma. J. Cancer Res. Clin. Oncol 137, 533–541 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Zhang H, Kuai X, Ji Z, Li Z, Shi R, Over-expression of small ubiquitin-related modifier-1 and sumoylated p53 in colon cancer. Cell Biochem. Biophys 67, 1081–1087 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Oliveira Alves MG, da Mota Delgado A, Balducci I, Carvalho YR, Cavalcante AS, Almeida JD, Study of MDM2 and SUMO-1 expression in actinic cheilitis and lip cancer. Arch. Dermatol. Res 306, 837–841 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Bellail AC, Olson JJ, Hao C, SUMO1 modification stabilizes CDK6 protein and drives the cell cycle and glioblastoma progression. Nat. Commun 5, 4234 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knipscheer P, Flotho A, Klug H, Olsen JV, van Dijk WJ, Fish A, Johnson ES, Mann M, Sixma TK, Pichler A, Ubc9 sumoylation regulates SUMO target discrimination. Mol. Cell 31 , 371–382 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Pichler A, Gast A, Seeler JS, Dejean A, Melchior F, The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 108, 109–120 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Sanjana NE, Shalem O, Zhang F, Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drag M, Salvesen GS, DeSUMOylating enzymes-SENPs. IUBMB Life 60, 734–742 (2008). [DOI] [PubMed] [Google Scholar]
- 38.le Sage C, Lawo S, Panicker P, Scales TME, Rahman SA, Little AS, McCarthy NJ, Moore JD, Cross BCS, Dual direction CRISPR transcriptional regulation screening uncovers gene networks driving drug resistance. Sci. Rep 7, 17693 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cross BC, Lawo S, Archer CR, Hunt JR, Yarker JL, Riccombeni A, Little AS, McCarthy NJ, Moore JD, Increasing the performance of pooled CRISPR–Cas9 drop-out screening. Sci. Rep 6, 31782 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, Liu XS, MAGeCK enables robust identification of essential genes from genome-scale CRISPR/ Cas9 knockout screens. Genome Biol. 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun L, Shi L, Li W, Yu W, Liang J, Zhang H, Yang X, Wang Y, Li R, Yao X, Yi X, Shang Y, JFK, a Kelch domain-containing F-box protein, links the SCF complex to p53 regulation. Proc. Natl. Acad. Sci. U.S.A 106, 10195–10200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan R, He L, Li Z, Han X, Liang J, Si W, Chen Z, Li L, Xie G, Li W, Wang P, Lei L, Zhang H, Pei F, Cao D, Sun L, Shang Y, SCF(JFK) is a bona fide E3 ligase for ING4 and a potent promoter of the angiogenesis and metastasis of breast cancer. Genes Dev. 29, 672–685 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Collins GA, Goldberg AL, The Logic of the 26S Proteasome. Cell 169, 792–806 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lydeard JR, Schulman BA, Harper JW, Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 14, 1050–1061 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skaar JR, Pagan JK, Pagano M, Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. Mol. Cell Biol 14, 369–381 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skaar JR, Pagan JK, Pagano M, SCF ubiquitin ligase-targeted therapies. Nat. Rev. Drug Discov 13, 889–903 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y, Handa H, Identification of a primary target of thalidomide teratogenicity. Science 327, 1345–1350 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y, Wen Z, Washburn MP, Florens L, Refinements to label free proteome quantitation: How to deal with peptides shared by multiple proteins. Anal. Chem 82, 2272–2281 (2010). [DOI] [PubMed] [Google Scholar]
- 49.Mosley AL, Sardiu ME, Pattenden SG, Workman JL, Florens L, Washburn MP, Highly reproducible label free quantitative proteomic analysis of RNA polymerase complexes. Mol. Cell. Proteomics 10, M110 000687 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grill B, Wilson GM, Zhang KX, Wang B, Doyonnas R, Quadroni M, Schrader JW, Activation/division of lymphocytes results in increased levels of cytoplasmic activation/ proliferation-associated protein-1: Prototype of a new family of proteins. J. Immunol 172, 2389–2400 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Wang B, David MD, Schrader JW, Absence of caprin-1 results in defects in cellular proliferation. J. Immunol 175, 4274–4282 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Bennett EJ, Rush J, Gygi SP, Harper JW, Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics. Cell 143, 951–965 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Solomon S, Xu Y, Wang B, David MD, Schubert P, Kennedy D, Schrader JW, Distinct structural features of caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2alpha, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Mol. Cell. Biol 27, 2324–2342 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shiina N, Yamaguchi K, Tokunaga M, RNG105 deficiency impairs the dendritic localization of mRNAs for Na+/K+ ATPase subunit isoforms and leads to the degeneration of neuronal networks. J. Neurosci 30, 12816–12830 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martin S, Zekri L, Metz A, Maurice T, Chebli K, Vignes M, Tazi J, Deficiency of G3BP1, the stress granules assembly factor, results in abnormal synaptic plasticity and calcium homeostasis in neurons. J. Neurochem 125, 175–184 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Hopkins AL, Groom CR, The druggable genome. Nat. Rev. Drug Discov 1, 727–730 (2002). [DOI] [PubMed] [Google Scholar]
- 57.Bloom J, Peschiaroli A, Demartino G, Pagano M, Modification of Cul1 regulates its association with proteasomal subunits. Cell Div. 1, 5 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu Y, Zhu J, Huang X, Du Z, Crystal structure of a dimerization domain of human Caprin-1: Insights into the assembly of an evolutionarily conserved ribonucleoprotein complex consisting of Caprin-1, FMRP and G3BP1. Acta Crystallogr. D Struct. Biol 72, 718–727 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cubenas-Potts C, Matunis MJ, SUMO: A multifaceted modifier of chromatin structure and function. Dev. Cell 24, 1–12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li J, Xu Y, Long XD, Wang W, Jiao HK, Mei Z, Yin QQ, Ma LN, Zhou AW, Wang LS, Yao M, Xia Q, Chen GQ, Cbx4 governs HIF-1α to potentiate angiogenesis of hepatocellular carcinoma by its SUMO E3 ligase activity. Cancer Cell 25, 118–131 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Becker J, Barysch SV, Karaca S, Dittner C, Hsiao H-H, Berriel Diaz M, Herzig S, Urlaub H, Melchior F, Detecting endogenous SUMO targets in mammalian cells and tissues. Nat. Struct. Mol. Biol 20, 525–531 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. MS proteomics data have been deposited to the ProteomeXchange Consortium through the PRIDE partner repository with the dataset identifier PXD022399 and 10.6019/PXD022399. CRISPR-Cas9 screen data have been deposited on BioGRID Open Repository of CRISPR Screens (ORCS) with accession bellail2021. The compound CPD1 is available from the NCI drug-like compounds library upon request (https://dtp.cancer.gov/RequestCompounds/). The lead compound HB007 will be made available to academic researchers through a material transfer agreement upon request to A.C.B.