

Abstract
Heart failure (HF) is a leading cause of morbidity and mortality. Studies in animal models and patients with HF revealed a prominent role for CD4+ T cell immune responses in the pathogenesis of HF and highlighted an active crosstalk between cardiac fibroblasts and IFNγ producing CD4+ T cells that results in profibrotic myofibroblast transformation. Whether cardiac fibroblasts concomitantly modulate pathogenic cardiac CD4+ T cell immune responses is unknown. Here we show report that murine cardiac fibroblasts express major histocompatibility complex type II (MHCII) in two different experimental models of cardiac inflammation. We demonstrate that cardiac fibroblasts take up and process antigens for presentation to CD4+ T cells via MHCII induced by IFNγ. Conditional deletion of MhcII in cardiac fibroblasts ameliorates cardiac remodelling and dysfunction induced by cardiac pressure overload. Collectively, we demonstrate that cardiac fibroblasts function as antigen presenting cells (APCs) and contribute to cardiac fibrosis and dysfunction through IFNγ induced MHCII.
Introduction
In the adult murine myocardium, cardiac fibroblasts comprise a large fraction of noncardiomyocytes (<20%),1 and are largely responsible for the synthesis and degradation of the extracellular matrix (ECM) components to maintain tissue homeostasis.2 Under pathological conditions including pressure overload, infection or ischemia, increased fibroblast proliferation and activation is associated with fibrosis.2 Activated fibroblasts deposit excessive ECM components into the cardiac interstitium, resulting in detrimental loss of myocardial compliance and decreased cardiac function. In accordance with this, ablation of activated fibroblasts in mice is associated with decreased fibrosis and improved cardiac function in response to pressure overload and myocardial infarction (MI).3,4 In pressure overload induced HF in mice, the development of cardiac fibrosis coincides with the infiltration of CD4+ T cells into the myocardium and is characterized by the transformation of cardiac fibroblasts to myofibroblasts, which deposit excess collagen.5,6 CD4+ T cell deficient mice are protected from development of cardiac fibrosis, 5,7 and direct interactions between cardiac fibroblasts and IFNγ producing Th1 cells have been shown to promote cardiac fibroblast TGFβ production, resulting in myofibroblast transformation.6 Here, we aim to uncover the bi-directionality of these cellular interactions and determine if cardiac fibroblasts concomitantly promote CD4+ T cell activation, a novel capability that is yet to be established.
CD4+ T cells are classically activated by professional antigen presenting cells (APCs), which efficiently internalize and process antigens that are displayed by MHCII molecules on the cell membrane.8 Additional co-costimulatory signals (CD80 or CD86) expressed by activated APCs are required for full activation of CD4+ T cells. Professional APCs include dendritic cells, macrophages and B cells, which are all present in the heart at lower frequencies relative to cardiac fibroblasts.8 Unlike professional APCs, non-professional APCs do not constitutively express MHCII molecules, but its expression can be induced in neutrophils, mast cells and endothelial cells by various stimuli.9 Additionally, human dermal fibroblasts exposed to IFNγ in vitro express MHCII, and promote recall antigen-dependent T cell responses.10 We hypothesized that the cellular interactions between cardiac fibroblasts and CD4+ T cells in the heart imply a novel role for cardiac fibroblasts as non-professional APCs central to cardiac CD4+ T cell responses and pathology.
Results
IFNγ induces MHCII expression in cardiac fibroblasts
Non-professional APCs do not typically express MHCII unless induced by specific stimuli.9 In conjunction with MHCII, either CD80 or CD86 costimulatory signals are required for full activation of CD4+ T cells upon engagement of the T cell receptor (TCR). We isolated the left ventricle (LV) from adult wt mice and cultured purified CD31-CD45-MEFSK4+cardiac fibroblasts (Fig. 1A). We found that MHCII was not expressed at baseline but was progressively induced over 5 days of IFNγ stimulation in culture (Fig. 1, B-C). Conversely, CD80 was highly expressed at baseline and was refractory to IFNγ stimulation (Fig. 1, D-E). CD86 was neither expressed at baseline, nor induced by IFNγ (Fig. 1, F-G). Expression of MHCII after IFNγ stimulation in vitro was further demonstrated by immunofluorescence microscopy on cardiac fibroblasts (Fig. 1H). In the heart, the majority of resident cardiac fibroblasts express transcription factor 21 (Tcf21).11 To delineate cardiac fibroblast expression of MHCII more specifically, we utilized cardiac fibroblast lineage tracing reporter mice (Tcf21iCre/+; R26eGFP) with a tamoxifen-regulated Cre recombinase knocked into 1 allele of Tcf21 that drives membrane targeted GFP expression from the Rosa26 locus (R26). GFP+ cells from Tcf21iCre/+; R26eGFP mouse hearts were sorted by fluorescent-activated cell sorting (FACS) and cultured for 72hrs in the presence of IFNγ. Similarly, we found that CD80 was highly expressed at baseline, while MHCII was induced after 72hrs of IFNγ stimulation (Fig. 1, I-K). Furthermore, expression of MHCII after IFNγ stimulation in vitro was demonstrated by immunofluorescence microscopy on GFP+ cardiac fibroblasts derived from Tcf21iCre/+; R26eGFP mice (Fig. 1L). We next evaluated in vivo induction of MHCII and costimulatory molecule expression on CD31-CD45-MEFSK4+ cardiac fibroblasts in the myocardial tissue microenvironment by flow cytometry (Fig. 2A). IFNγ was administered daily to wt mice for 5 days (Fig. 2B). In vehicle treated mice, 30–50% of cardiac fibroblasts expressed CD80 on the surface but did not express MHCII or CD86 (Fig. 1, C-H). However, IFNγ treatment induced MHCII surface expression on 28–34% of cardiac fibroblasts, while CD80 and CD86 expression was not altered (Fig. 2, C-H). These data demonstrate that IFNγ induces expression of MHCII in cardiac fibroblasts, and together with the expression of CD80 suggests that cardiac fibroblasts are capable of functioning as non-professional APCs that can induce CD4+ T cell activation.
Figure 1. Cardiac fibroblasts express MHCII in response to IFN𝛾 stimulation in vitro.

(A-H) CD31-CD45-MEFSK4+ primary adult murine cardiac fibroblasts were sorted from heart digests and cultured in the presence of 100U/mL recombinant murine IFN𝛾 for up to 5 days. (A) CD31-CD45-MEFSK4+ cardiac fibroblasts were analysed by flow cytometry (≥ 20,000 target cells acquired) to determine surface expression of (B-C) MHCII as well as (D-E) CD80 and (F-G) CD86 costimulatory molecules. n= 5 (Control and D3), and n=3 (D5) independent experiments and cellular preparations. (H) Immunofluorescence staining for MHCII as well as Vimentin was performed after 3 days of IFN𝛾 stimulation. n= 3 independent experiments and cardiac fibroblasts preparations. (I-L) Sorted GFP+ cardiac fibroblasts from Tcf21iCre/+; R26eGFP lineage tracing reporter mice were cultured in the presence of 100U/mL recombinant murine IFN𝛾 for 72hrs. (I) GFP+ cardiac fibroblasts were analysed by flow cytometry (≥ 5,000 target cells acquired) to determine surface expression of MHCII and CD80 and (J-K) quantified. n=3 (MCHII), and n=5 (CD 80), independent experiments and cellular preparations. (L) Immunofluorescence staining for MHCII on GFP+ cardiac fibroblasts was performed. n=3 independent experiments and cardiac fibroblasts preparations. Scale bars: 100μm. Error bars represent mean ± SD. * p<0.05, *** p<0.001; One-way ANOVA (1C, E and G) and two tailed unpaired T test (1J and K).
Figure 2. Cardiac fibroblasts express MHCII in response to IFN𝛾 stimulation in vivo.

(A) CD31-CD45-MEFSK4+ cardiac fibroblasts were directly analysed by flow cytometry from the LV of (B) wt mice treated with daily intraperitoneal injections of IFN𝛾 (25,000U) or PBS vehicle and harvested on day 5. Surface expression of (C-D) MHCII, (E-F) CD80 and (G-H) CD86 was determined (≥ 5,000 target cells acquired). n= 4 vehicle and n= 5 IFNγ treated mice. Error bars represent mean ± SD. * p=0.0159; two tailed Mann-Whitney test.
MHCII+ cardiac fibroblasts induce CD4+ T cell activation
We next evaluated the ability of cardiac fibroblasts to activate naïve OTII CD4+ T cells, which express a transgenic TCR specific for chicken ovalbumin (OVA) peptide. CD62LhiCD44lo naïve OTII CD4+ T cells were sorted by FACS (Fig. 3A) and cocultured with primary LV cardiac fibroblasts stimulated with IFNγ to induce MHCII expression and loaded with OVA peptide. We identified robust induction of CD4+ T cell proliferation after 72hrs of coculture as determined by CFSE dilution (Fig. 3, B and D); although, not to the same extent as professional antigen presenting bone marrow-derived dendritic cells (BMDCs) (Fig. 3, C-D). These responses were MHCII-restricted as MHCII deficient (MhcII−/−) cardiac fibroblasts did not induce naïve CD4+ T cell proliferation (Fig. 3B). In order to exclude possible contamination with professional APCs in culture, purified GFP+ CD45- cardiac fibroblasts from Tcf21iCre/+; R26eGFP mice were sorted by FACS (Fig. 3E) and stimulated with IFNγ to induce MHCII expression and cocultured with FACS-sorted CD62LhiCD44lo naïve OTII CD4+ T cells in the presence of OVA peptide. Similarly, we identified robust induction of CD4+ T cell proliferation after 72hrs of coculture as determined by CFSE dilution (Fig. 3, F-G). T helper type 1 (Th1) cells are a major cellular source of endogenous IFNγ and establish intimate contact with cardiac fibroblasts upon infiltration into the heart.6 We examined the possibility that antigen presentation to Th1 cells during this contact triggers further IFNγ production to support sustained antigen presentation. We cocultured cardiac fibroblasts and differentiated OTII Th1 cells in the presence of OVA peptide (Fig. 4A). After 24 hours, Th1 cells showed significant induction of IFNγ production determined by intracellular cytokine staining (Fig. 4, B-C). Moreover, 72h cocultures of primary adult cardiac fibroblasts with OTII Th1 cells labelled with CFSE, in the presence of OVA peptide, demonstrated increased MHCII surface expression in cardiac fibroblasts as compared to cardiac fibroblasts alone (Fig. 4, D-E), and resulted in OTII Th1 cell, but not wt Th1 cell proliferation (Fig. 4, F-G). These data demonstrate the ability of cardiac fibroblasts in the presence of IFNγ to present peptide antigens to naïve CD4+ T cells. Additionally, cardiac fibroblasts express MHCII during cardiac fibroblast-Th1 cell contact and promote antigen-specific Th1 cell activation.
Figure 3. Cardiac fibroblasts induce antigen-specific CD4+ T cell proliferation in vitro in the presence of IFNγ.

(A) Naïve CD62LhiCD44lo CD4+ T cells were sorted by FACS from OTII bulk splenocytes and labelled with CFSE (2μM). (B-D) Primary adult murine cardiac fibroblasts and BMDCs derived from wt or MhcII−/− mice were cultured in the presence or absence of IFN𝛾 (100U/mL) for 24hrs and loaded with OVA peptide (4μg/mL) where indicated, 2hrs prior to coculture with OTII naïve CD4+ T cells. After 72hrs CFSE dilution was evaluated by flow cytometry (≥ 10,000 target cells acquired) and (D) quantified. n=3 (CFB OVA), n=5 (CFB+IFNγ+OVA; BMDC+OVA), independent experiments and cell preparations. (E) Sorted GFP+ murine cardiac fibroblasts from Tcf21iCre/+; R26eGFP lineage tracing reporter mice were cultured in the presence or absence of IFN𝛾 (100U/mL) for 24hrs and loaded with OVA peptide (4µg/mL) where indicated, 2hrs prior to coculture with OTII naïve CD4+ T cells. (F) After 72hrs CFSE dilution was evaluated by flow cytometry (≥ 5,000 target cells acquired) and (G) quantified. n=3, independent experiments and cell preparations. Error bars represent mean ± SD. ** p<0.01, *** p<0.001; one-way ANOVA.
Figure 4. IFNγ produced by Th1 cells induce MHCII expression by cardiac fibroblasts and promotes antigen-specific CD4+ T cell activation.

(A) OTII Th1 cells were differentiated in vitro from bulk CD4+ splenocytes, and cocultured with adult cardiac fibroblasts and OVA peptide (4μg/mL). (B-C) After 24hrs of coculture Th1 IFN𝛾 production was determined by intracellular cytokine staining (≥ 15,000 target cells acquired). n=3 control and n=6 OVA, independent experiments and cell preparations. (D-E) After 72hrs of coculture CD31-CD45-MEFSK4+ cardiac fibroblasts were analysed by flow cytometry (≥ 5,000 target cells acquired) for surface expression of MHCII, (n=3, independent experiments and cell preparations) and (F-G) antigen-specific Th1 cell proliferation via CFSE dilution (≥ 8,000 target cells acquired). n=3 independent experiments and cell preparations. Error bars represent mean ± SD. * p<0.05, ** p<0.01, two tailed student unpaired t test (4C and E ) and One-way ANOVA (4G)
Cardiac fibroblasts capture antigens for MHCII presentation
Exogenously delivered OVA peptide can directly bind the peptide groove of cell surface MHCII molecules for presentation to CD4+ T cells (as shown in Fig. 2). However, defining attributes of APCs include the capacity to internalize soluble and particulate antigens, process, load peptides onto MHCII in the cytosol, and translocate MHCII-peptide complexes to the cell surface to activate CD4+ T cells. Soluble OVA protein is taken up via macropinocytosis or mannose receptor mediated endocytosis,12 whereas the uptake of relatively larger particles involves phagocytosis, with phagosomes containing the ingested material maturing into acidified phagolysosomes.13 To track soluble antigen uptake and processing by cardiac fibroblasts we used DQ OVA, a self-quenched conjugate of OVA protein that fluoresces upon proteolytic degradation. Strikingly, cardiac fibroblasts efficiently took up and processed DQ OVA, as demonstrated by flow cytometry and immunofluorescence, and unlike MHC II expression, these functions were independent of IFNγ stimulation (Fig. 5, A-B). Moreover, OVA protein internalized by cardiac fibroblasts, combined with IFNγ stimulation to promote MHCII expression, resulted in naïve OTII CD4+ T cell proliferation (Fig. 5, C-D). We next evaluated phagocytic activity of cardiac fibroblasts using pHrodo green E. coli BioParticles, which become fluorescent in acidified phagolysosomes. A strong fluorescent signal emitted from cardiac fibroblasts was detected by both flow cytometry and immunofluorescence, demonstrating efficient phagocytosis independent of IFNγ stimulation (Fig. 5, E-F). Furthermore, E. coli transformed with a plasmid conferring OVA protein expression were engulfed by cardiac fibroblasts and induced antigen-specific naïve OTII CD4+ T cell proliferation, which was remarkably comparable to that induced by BMDCs (Fig. 5, G-H). These data demonstrate several functions of cardiac fibroblasts as APCs, including efficient uptake and processing of soluble and particulate antigens, and presentation of peptide fragments via MHCII to generate CD4+ T cell immune responses.
Figure 5. Cardiac fibroblasts take up, process, and present antigens to CD4+ T cells in vitro.

(A) CD31-CD45-MEFSK4+ primary adult murine cardiac fibroblasts were cultured in the presence of DQ OVA (100μg/mL) and IFN𝛾 (100U/mL) overnight to evaluate uptake and intracellular processing of DQ OVA by flow cytometry (≥ 10,000 target cells acquired), and (B) immunofluorescence microscopy in cardiac fibroblasts and BMDCs. (n=3 independent experiments). (C-D) Wt cardiac fibroblasts and BMDCs were treated with purified OVA protein (100μg/mL) overnight and cocultured with CFSE-labelled naïve OTII CD4+ T cells in the presence of IFN𝛾 (100U/mL) for 72 hrs to evaluate CD4+ T cell proliferation by flow cytometry (≥ 5,000 target cells acquired). n=7 and n=5 (CFB untreated and OVA+IFNγ treated) and n=6 and n=7 (BMDC untreated and OVA treated), independent experiments and cell preparations. (E) Wt cardiac fibroblasts were cultured in the presence of pHrodo Green E. coli bioparticles overnight to evaluate phagocytosis by flow cytometry (≥ 7,000 target cells acquired). (F) Intracellular accumulation of bioparticles in acidified phagolysosomes was further evaluated by immunofluorescence microscopy in cardiac fibroblasts and BMDCs. n=3 independent experiments. (G-H) Wt cardiac fibroblasts and BMDCs were cultured overnight in the presence of transformed E. coli particles expressing either chicken ovalbumin or empty vector (EV), and cocultured with CFSE-labelled naïve OTII CD4+ T cells for 72 hrs to evaluate CD4+ T cell proliferation by flow cytometry (≥ 10,000 target cells acquired). n=11 BMDC EV, and n=8 independent experiments for all other conditions. Scale bars: 100μm.. Error bars represent mean ± SD. ** p<0.01, *** p<0.001; one-way ANOVA test.
Cardiac fibroblast MHCII expression modulates pathology
Because myocardial IFNγ is upregulated in response to transverse aortic constriction (TAC), a well-established model of heart failure,6,14 and Chagas disease15,16, we investigated the induction of MHCII on cardiac fibroblasts in vivo, in response to these distinct models of cardiac inflammation. We analysed the surface expression of MHCII and CD80 on CD31-CD45-MEFSK4+ cardiac fibroblasts by flow cytometry (Fig. 6A). 4 weeks of TAC in wt mice resulted in a significant increase in expression of both MHCII and CD80 on cardiac fibroblasts relative to Sham control mice (Fig. 6, B-E). Similarly, wt mice infected with T. cruzi and harvested after 19 days, at the peak of parasitaemia, demonstrated even greater MHCII induction on cardiac fibroblasts relative to mock infected controls. However, CD80 expression did not significantly change (Extended data Fig. 1). Expression of MHCII in vivo by cardiac fibroblasts after TAC in LV tissue sections was further demonstrated by immunofluorescence microscopy on aSMA+ cells in wt mice (Fig. 6F), and in GFP+ cells traced from Tcf21iCre/+; R26eGFP reporter mice (Fig. 6G). Additionally, direct contact can be identified between CD4+ T cells and αSMA+ cardiac fibroblasts cells within LV fibrotic lesions in wt TAC mice (Fig. 6H). We next generated Tcf21iCre/+MhcIIfl/fl mice expressing Tamoxifen-inducible Cre recombinase driven by Tcf21 expression to conditionally delete MhcII in cardiac fibroblasts (Fig. 7A). After Tamoxifen treatment of Tcf21iCre/+MhcIIfl/fl mice, MhcII recombination in sorted CD31-CD45-MEFSK4+ cardiac fibroblasts was detected by PCR, and occurred in a cell-specific manner, as CD31-CD45+ cardiac leukocytes did not undergo MhcII recombination (Fig. 7B, Extended data Fig. 2). Furthermore, CD31-CD45-MEFSK4+ cardiac fibroblasts cultured with 4-Hydroxytamoxifen showed 3.5 fold reduction in MHCII protein expression after 3 days of IFNγ stimulation (Fig. 7, H-I), while CD45+CD11b+CD11c+ BMDCs did not show significant reduction in MHCII in response to 4-Hydroxytamoxifen treatment (Fig. 7, E-F). Tcf21iCre/+MhcIIfl/fl mice were treated with Tamoxifen by intraperitoneal injection prior to TAC surgery and maintained on Tamoxifen chow for 4 weeks until harvest (Fig. 7G). MHCII protein surface expression on myeloid cells in the mediastinal lymph nodes (mLNs) draining the heart (Fig. 7H), as well as in cardiac leukocytes and cardiac endothelial cells, was similar across the treatment groups, with only observed decreased MHCII protein surface expression in cardiac fibroblasts from Tcf21iCre/+MhcIIfl/fl mice treated with Tamoxifen (Extended data Fig. 3). Vehicle and Tamoxifen treated (cardiac fibroblast MHCII deficient) mice showed similar LV CD4+ T cell infiltration (Fig. 7, I-J) and similar CD4+ T cell activation in the mLNs (Extended data Fig. 4), however, only tamoxifen treated mice had decreased cardiac fibroblasts compared to vehicle treated mice in the onset of TAC (Extended data Fig. 5), and a significant amelioration of adverse cardiac remodelling, including perivascular fibrosis (Fig. 7, K-L) and cardiomyocyte hypertrophy (Extended data Fig. 4) compared to the vehicle treatment group. Additionally, cardiac systolic function remained preserved in Tamoxifen treated mice in contrast to the vehicle treatment group as demonstrated by preserved fractional shortening (Fig. 7, M-N, Table 1). These results demonstrate that IFNγ-associated cardiac inflammation promotes MHCII expression in cardiac fibroblasts, and that MHCII in cardiac fibroblasts is central to cardiac remodelling and dysfunction in response to TAC.
Figure 6. Cardiac fibroblasts express MHCII in response to cardiac pressure overload.

(A-E) Transverse aortic constriction (TAC) surgery was performed on mice and harvested after 4 weeks. (A) CD31-CD45-MEFSK4+ cardiac fibroblasts from the LV of wt mice were directly analysed by flow cytometry (≥ 5,000 target cells acquired) to determine surface expression of (B-C) MHCII and (D-E) CD80. n= 3 Sham and TAC treated mice. Immunofluorescence staining of LV tissue sections was performed to detect (F) MHCII on aSMA+ cells in wt mice (white arrows) and (G) MHCII on GFP+ cells in Tcf21iCre/+; R26eGFP lineage tracing reporter mice (indicated with a white arrow) (Scale bars: 100μm). (H) Immunofluorescence staining of wt LV tissue sections was performed to evaluate colocalization of CD4+ cells and aSMA+ cells (Scale bars: 25μm). Images are representative of n=3 mice (F and H) and n=5 mice (G). Error bars represent mean ± SD. (* p<0.05, ** p<0.01; two tailed unpaired T test).
Figure 7. Cardiac fibroblast MHCII expression is required for cardiac dysfunction to develop in response to pressure overload.

(A) Conditional deletion of MHCII in cardiac fibroblasts was induced by administering Tamoxifen to Tcf21iCre/+MhcIIfl/fl mice. (B) CD31-CD45-MEFSK4+ cardiac fibroblasts and CD31-CD45+ leukocytes were FACS sorted from the LV of vehicle and Tamoxifen treated Tcf21iCre/+MhcIIfl/fl mice and genomic DNA was probed for the intact (P1 and P2 primers) and recombined (P1 and P3 primers) MhcII alleles by PCR using specific primers (blue arrows) that detect the recombination (MhcII excision) or the intact-non recombined- in the different conditions. Note that excision is only observed in cardiac fibroblasts from tamoxifen treated mice with the P1-P3 primers. GAPDH is shown as a loading control. Gel represents DNA from cells sorted from n=3 mice (pooled hearts). (C-D) CD31-CD45-MEFSK4+ cardiac fibroblasts from Tcf21iCre/+MhcIIfl/fl mice were cultured overnight in the presence of 4-Hydroxytamoxifen (1μM), then stimulated with IFN𝛾 (100U/mL) for 72 hrs to determine surface expression of MHCII by flow cytometry (≥ 2,000 target cells acquired. n= 4 vehicle and n= 3 for 4HO-TMX, independent experiments and cell preparations. (E-F) CD45+CD11b+CD11c+ BMDCs from Tcf21iCre/+MhcIIfl/fl mice were cultured overnight in the presence of 4-Hydroxytamoxifen (1μM), then stimulated with LPS (10ng/mL) for 72 hrs to determine surface expression of MHCII by flow cytometry (≥ 12,000 target cells acquired) n= 5, independent experiments and cell preparations. (G) Tcf21iCre/+MhcIIfl/fl mice were treated with Tamoxifen (75mg/Kg) by 5 daily I.P injections prior to TAC surgery and maintained on Tamoxifen citrate chow for 4 weeks after surgery to decrease MHCII expression in cardiac fibroblasts, or with vehicle (mice with MHCII sufficient cardiac fibroblasts). (H) MHCII expression on CD11b+ myeloid cells in the mediastinal lymph nodes (mLNs) was determined by flow cytometry. (I-J) IHC of frozen LV sections was used to determine LV CD4+ T cell infiltration, scanning the entire LV section area. Representative images of n=3 hearts (vehicle and TMX Sham), n=4 (TAC vehicle) and n=7 (TAC TMX). (K-L) Picrosirius red staining of fixed LV tissue sections was used to determine perivascular fibrosis. Representative images of n=3 hearts (vehicle and TMX Sham), n=4 (TAC vehicle) and n=7 (TAC TMX). (M-N) Transthoracic echocardiography was used to measure LV fractional shortening. Scale bars: 100μm. Each data points represents individual animal in Fig 7H, 7J, 7L and 7N. Error bars represent mean ± SD. * p<0.05; two tailed unpaired T test (7D) and One-way ANOVA (7H, J, L and N).
Table 1.
Characterization of left ventricular function by echocardiography 4 weeks after surgery in Tcf21iCre/+MhcIIfl/fl
| Sham Vehicle | Sham Tamoxifen | TAC Vehicle | TAC Tamoxifen | |
|---|---|---|---|---|
| Body weight / Tibia length (mg/mm) | 1.55 ± 0.05 | 1.33 ± 0.04* | 1.53 ± 0.09 | 1.21 ± 0.04* |
| LV weight/ Tibia length (mg/mm) | 4.90 ± 0.16 | 4.43 ± 0.14 | 6.82 ± 1.10 | 5.45 ± 0.23* |
| Heart Rate (bpm) | 423 ± 9 | 412 ± 7 | 417 ± 14 | 436 ± 25 |
| Fractional Shortening (%) | 34.9 ± 2.7 | 35.0 ± 2.8 | 21.1 ± 3.0 | 41.2 ± 3.4*** |
| Anterior Wall Thickness (mm) | 1.00 ± 0.10 | 1.04 ± 0.06 | 1.07 ± 0.05 | 0.97 ± 0.04 |
| Posterior Wall Thickness (mm) | 0.79 ± 0.06 | 0.94 ± 0.05 | 0.96 ± 0.05 | 0.96 ± 0.04 |
| End systolic Volume (µL) | 19.3 ± 3.0 | 18.5 ± 2.7 | 44.7 ± 9.0 | 16.4 ± 3.4** |
| End diastolic Volume (µL) | 54.2 ± 2.5 | 51.1 ± 2.3 | 74.1 ± 7.9 | 55.8 ± 3.5* |
| End Diastolic Diameter (mm) | 3.59 ± 0.07 | 3.50 ± 0.06 | 4.07 ± 0.17 | 3.63 ± 0.09* |
| End Systolic Diameter (mm) | 2.34 ± 0.14 | 2.29 ± 0.13 | 3.24 ± 0.26 | 2.14 ± 0.16** |
Values are means ± SEM
p<0.05
p<0.001
p<0.0001
Tamoxifen versus vehicle treatment groups using two Tailed unpaired T test. Vehicle=Cardiac fibroblast sufficient mice; Tamoxifen=Cardiac fibroblast deficient mice.
Discussion
We report for the first time, to our knowledge, that cardiac fibroblasts function as APCs and contribute to cardiac fibrosis and dysfunction through IFNγ induced MHCII. We find that cardiac fibroblasts: express MHCII in vitro and in vivo in response to IFNγ as well as in two different experimental models of cardiac disease; express the requisite costimulatory signal CD80 necessary for CD4+ T cell activation; efficiently capture and process extracellular soluble and particulate antigens for presentation; and induce naïve CD4+ T cell and Th1 cell activation in an MHCII and antigen dependent manner. Moreover, we identify a central role for cardiac fibroblast MHCII expression in cardiac remodelling and dysfunction in an experimental model of HF that is highly dependent on IFNγ+ Th1 cell immune responses.
It has been known for decades that stromal cells, including dermal fibroblasts, can express MHCII in response to IFNγ in vitro17,18, therefore this finding in cardiac fibroblasts could be perhaps unsurprising. The novelty of our study emerges from the relevance of cardiac fibroblasts as APCs in the context of cardiac inflammation and pathology. Our in vitro results demonstrate that MHCII expressed on cardiac fibroblasts, loaded with a peptide antigen, can induce naïve CD4+ T cell proliferation, a capability considered exclusive to mature DCs.8 This serves as decisive validation of the functionality of cardiac fibroblasts as APCs. The quality of TCR interactions with the peptide-MHCII complex is determinant of the strength of CD4+ T cell activation, with higher affinity interactions generating a more potent response.19 In our in vitro studies, we utilize OVA-specific naïve OTII CD4+ T cells against their optimal OVA peptide ligand, thus it is possible that cardiac fibroblasts have a diminished ability to activate CD4+ T cells when presenting suboptimal cognate antigens. However, given the confinement of cardiac fibroblasts to the heart it is unlikely that they establish contact with naïve CD4+ T cells in vivo, but rather with effector CD4+ T cells which are more sensitive to triggering through the TCR. We have reported strong TCR engagement of CD4+ T cells within the murine heart20 and induction of T helper responses in the mLNs during the course of HF development.6,21 Here, we find that IFNγ treatment, and two cardiac pathologies associated with elevated IFNγ show elevated expression of MHCII on cardiac fibroblasts. We demonstrate the ability of cardiac fibroblasts to present peptide antigen to Th1 cells in vitro and propose a role in generating recall responses from Th1 cells infiltrated in the heart through cardiac fibroblasts expressed MHCII and the involvement of this axis in cardiac fibrosis and dysfunction.
We identify baseline expression of the co-stimulatory molecule CD80 in >90% of the cells in vitro, and in approximately 20–50% of cardiac fibroblasts in vivo, and detect increased expression in response to cardiac pressure overload. This variability may arise due to heterogeneity of cardiac fibroblasts in the myocardium at varying stages of maturity,22 in comparison to synchronized cells in culture. A variety of stimuli have also been shown to increase CD80 expression in APCs, including CD40 ligand, IFNα, granulocyte–macrophage colony-stimulating factor (GM-CSF), TLR ligands, and TNFα, which may become expressed in the heart during TAC.23–25 In addition to direct antigen presentation by cardiac fibroblasts, CD80 expression may have further relevance in global enhancement of CD4+ T cell activation in the heart via trans-costimulation, by augmenting costimulatory signals delivered to CD4+ T cells already engaged with an APC.26 CD80 and CD86 are thought to be interchangeable co-stimulators, and we find that cardiac fibroblasts constitutively express CD80 but not CD86. In contrast to cardiac fibroblasts, CD86 is abundantly expressed on DCs and is thought to play a prominent role in initial T cell activation.27 CD80 expression is more slowly induced following DC activation and functional studies suggest that CD80 is a more potent CD28 ligand due its higher relative affinity for CD28.24 Our data demonstrate that in the presence of IFNγ, MHCII and CD80 are available to engage with the TCR and CD28 in CD4+ T cells, respectively, and induce T cell proliferation.
We and others have demonstrated intimate contact between cardiac fibroblasts and immune cells, including Th1 cells and macrophages, in the context of cardiac dysfunction due to pressure overload or age.6,28 While these reports focused on the unidirectional signals exerted by immune cells on cardiac fibroblasts and highlighted the requirement for intimate cellular contact to develop cardiac fibrosis, the bidirectionality of this interaction remained unexplored. We now demonstrate that IFNγ secreting Th1 cells induce MHCII on cardiac fibroblasts, that subsequently induce further IFNγ production by Th1 cells, propagating a positive feedback loop that likely exacerbates Th1 cell activation and myofibroblast transformation. The proximity of MHCII+ cardiac fibroblasts and CD4+ T cells in the TAC hearts, together with our findings in mice lacking MHCII in cardiac fibroblasts having decreased fibrosis, are in support of this loop. We demonstrate the ability of cardiac fibroblasts to capture extracellular antigens and process them into peptide antigens loaded onto MHCII to induce CD4+ T cell activation. Cardiac fibroblasts are non-professional phagocytes capable of engulfing apoptotic cells to assist with debris clearance after myocardial ischemia.29,30 Our results demonstrate that cardiac fibroblasts internalize both soluble and particulate material, process and load peptides onto MHCII and induce CD4+ T cell proliferation. We additionally report that Tamoxifen treated (cardiac fibroblast MHCII deficient) Tcf21iCre/+MhcIIfl/fl mice have intact effector CD4+ T cell expansion in the mLNs and CD4+ LV T cell recruitment in response to cardiac pressure overload, as compared to vehicle treated (MHCII sufficient) mice. Yet, the lack of cardiac fibroblast MHCII results in decreased cardiac fibroblasts and is sufficient to prevent adverse cardiac remodelling and dysfunction. These results are in line with a central role for DCs in antigen presentation and Th1 cell expansion in the mLNs in the context of heart inflammation as we reported6,20,31, and as demonstrated by DC ablation being effective in preventing cardiac fibrosis and dysfunction.32 We recently reported that DCs present cardiac neoantigens to CD4+ T cells, and that interrupting this presentation prevents TCR engagement by CD4+ T cells in the heart and ameliorates cardiac dysfunction20. Because cardiac fibroblast MHCII deficient mice have intact MHCII expression in myeloid cells, the observed similar number of effector T cells in the lymph nodes, devoid of cardiac fibroblasts, is not surprising. The similar T cell numbers observed in the heart in the presence or absence of MHCII may suggest continuous cardiotropism that is not altered by cardiac fibroblast MHCII expression, as well as a T cell recall response in situ induced by cardiac DCs expressing MHCII that additionally takes place. It is also possible that DCs and cardiac fibroblasts participate in trans-costimulation augmenting signals delivered to CD4+ T cells already engaged with an APC and that this is altered in the absence of cardiac fibroblast MHCII, something worth addressing in future studies. While our in vitro results demonstrate cardiac fibroblast induction of T cell proliferation and IFNγ production through MHCII, we have not directly tested T cell proliferation in the heart in vivo. Nevertheless, our in vivo data demonstrate that cardiac fibroblast expression of MHCII is required for cardiac fibrosis and systolic dysfunction, which are both T cell dependent 5–7,20. Our new findings endorse the intriguing possibility of treating HF, the leading cause of death in the United States,33 with targeted therapies to the cardiac fibroblast that specifically modulate cardiac CD4+ T cell responses without impairing systemic CD4+ T cell activation by DCs that could have undesired immunosuppressive side effects. A detailed investigation of the dynamics of cell-specific MHCII engagement of the TCR in the mLNs and in the heart will identify when such targeted therapies may be more effective.
We acknowledge some limitations in this study. For instance, while we clearly demonstrate that IFNγ induces MHCII expression of the vast majority of cardiac fibroblasts in vitro, a lower frequency of cardiac fibroblasts express MHCII in vivo in the three experimental models we use (TAC, Chagas, or IFNγ injections) compared to purified cardiac fibroblasts in vitro. It is possible that in vivo in the onset of TAC other factors besides IFNγ contribute to MHCII expression, and that, given the plasticity of cardiac fibroblasts, specific cardiac fibroblast populations are responsible for antigen presentation. Our data showing that conditional deletion of MHCII in cardiac fibroblasts results in decreased cardiac fibroblast presence in TAC suggest an additional role for cardiac fibroblast MHCII in fibroblast proliferation, which is critical for cardiac fibrosis in pathological conditions that include cardiac pressure overload. Future studies will investigate which fibroblast subsets express MHCII and how MHCII regulates fibroblast function and proliferation. These results are in line with recently published single cell RNA sequencing data demonstrating enhanced expression of MHCII in mice 5 weeks after TAC and variable expression in different fibroblast clusters identified in the human cardiac samples obtained from patients with dilated cardiomyopathy34,35. Noteworthy, recent RNAseq data in patients with dilated cardiomyopathy pre and post Left ventricular assist device (LVAD) demonstrate lower expression of cardiac fibroblast HLA-DR in patients post LVAD support, in line with our findings that MHCII expression in cardiac fibroblasts contributes to systolic function 35. It is also plausible that other APCs sequentially dominate this response during the progression of adverse cardiac remodelling post TAC. These APCs may include lymph node fibroblastic reticular cells (FRCs), which have been reported to express MHCII in response to IFNγ36,37. Whether this is the case in mLNs in this context is unknown. For instance, CD4+ T cell engagement of antigen presented by DC or FRC may happen early on in the mLNs, before T cell infiltration in the heart occurs, and continues once T cells Infiltrate the heart, co-existing with cardiac fibroblast presentation to intramyocardial CD4+ T cells and cardiac fibroblast proliferation and transformation. The data presented here is limited to one time point after cardiac pressure overload has been induced and future studies will be necessary to determine the dynamics of the cell specific role of MHCII. This is something worth studying in the immediate future that may have consequences to understand immune tolerance in the heart, that has not been investigated herein. The mechanisms of peptide loading of MHCII in cardiac fibroblasts, and whether these are shared with professional APCs are also an ongoing research direction we are pursuing. Additionally, we have only used male mice in this study, and given that there are sex specific differences in heart failure, it is possible that sex specific differences also exist in the way cardiac fibroblasts sustain cardiac T cell immune responses. Despite these limitations, our study unveils a novel role for cardiac fibroblasts as central contributors to cardiac inflammation and adverse remodelling through MHCII that may have potential implications when targeting inflammation in heart disease (Extended data Fig. 6).
Taken together, our results contribute to the growing field of cardio-immunology and advance the intriguing possibility that diverse subsets of fibroblasts abundantly dispersed within other organs perform similar functions, serving as sentinel cells that sense local insults and directly boost adaptive immune responses.
Methods
Mice.
Mice were bred and maintained under pathogen-free conditions at Tufts University animal facilities and treated in compliance with the Guide for the Care and Use of Laboratory Animals (National Academy of Science). C57BL/6 wild-type (wt), OTII, MhcII-flox mice (all purchased from Jackson Laboratories) and Tcf21-cre mice (provided by Jennifer Davis, University of Washington) were bred and maintained in house and euthanized at 4–14 weeks of age for tissue collection. Tcf21iCre/+; R26eGFP mice were bred and maintained under pathogen-free conditions at the University of Washington, with all animal experimentation was approved by the University of Washington Institutional Animal Care and Use Committee. All animal studies were approved by the Tufts University Institutional Animal Care and Use Committee.
Adult Cardiac Fibroblasts Preparation.
Heart ventricles from adult wt and Tcf21iCre/+; R26eGFP mice (4–8 wk old) were excised, minced, and digested at 37°C for 30 min with agitation in a mixture of 0.25% trypsin and 40 μg/ml liberase TL (Roche). Digested tissue was then centrifuged at 300xg for 5 min and the pellet resuspended in fibroblast growth media (Lonza) and plated on 0.1% gelatin coated plates for 2 h at 37°C with 5% CO2. Unattached cells were discarded, and adherent cells were further cultured. The digested heart cell suspension was filtered through a 40µm cell strainer and stained with BV711-conjugated anti-CD45.2 (104), PE-conjugated anti-CD31 (MEC13.3), APC-conjugated anti-Feeder Cells (mEF-SK4) as outlined below for quantitative flow cytometry and CD31-CD45-MEFSK4+ cells, or GFP+ cells when using Tcf21iCre/+; R26eGFP lineage tracing mice, were sorted on a BD FACSAria and plated on 0.1% gelatin coated plates at 37°C with 5% CO2. Once confluent, cardiac fibroblasts were detached with trypsin and either used directly in experiments or passaged one more time to use in experiments. Confirmation of purity was confirmed using these markers, or GFP, in every experiment.
Dendritic Cell Preparation.
Bone marrow was flushed from femurs and tibias of wt mice. Red blood cells were lysed with Tris ammonium chloride buffer (Roche) and cells were cultured in complete RPMI media with recombinant GM-CSF (15ng/ml) (Peprotech) for 7 days, with additional media added on days 3 and 5. CD11c+ DC purity was >85% by FACS analysis.
CD4+ T cell Preparation.
CD4+ cells were isolated from spleen cell suspensions of OTII mice by positive selection with CD4 microbeads (Miltenyi Biotec) and further FACS sorted based on the expression of CD62L and CD44. Naïve CD4+ T cells were differentiated into Th1 cells by stimulation with plate-bound anti-CD3 (5µg/mL) and soluble anti-CD28 (1µg/mL) in the presence of IL-12 (0.01µg/mL), IL-2 (25U/mL) and anti-IL-4 (0.5µg/mL). On day 3 of stimulation, Th1 cultures were split 1:1 with fresh medium containing IL-2 (25U/mL). All cytokines and blocking antibodies were purchased from Peprotech. Differentiated T cells were harvested a day later and immediately used for experiments. Th1 cells generation was confirmed by IFNγ production upon PMA/ionomycin stimulation. Cells were labelled with 2µM CFDA-SE (ThermoFisher) according to manufacturer specifications.
Proliferation Assay.
In a 24 well plate, cardiac fibroblasts or BMDCs (100,000 cells/ well) were allowed to adhere and either pulsed for 4 hours with 4 µg/mL OVA 323–339 (AnaSpec), or incubated overnight with 100μg/mL Ovalbumin protein (Sigma), 100μg/mL DQ OVA (ThermoFisher) or OVA transformed E. coli (MOI 10). Cells were washed twice with PBS and cocultured for 3 days with 1 million CFDA-SE labelled OTII CD4+ T cells. For FACS sorting of purified CD44loCD62Lhi naïve CD4+ T cells, bulk splenocytes from OTII mice were stained with FITC-conjugated anti-CD4 (GK1.5), PE-conjugated anti-CD62L (MEL-14), and APC-conjugated anti-CD44 (IM7) as outlined below for FACS sorting on a BD FACSAria (BD Biosciences).
Quantitative flow cytometry.
Flow cytometry was performed to analyse cell surface protein expression. The data was acquired on a BD LSRII flow cytometer (BD Biosciences) and analysed using FlowJo software. LV digestion was performed by collagenase type II (0.895mg/mL) for 30minutes at 37C. Cells were then stained with the following antibodies: FITC-conjugated anti-CD4 (GK1.5), APC-conjugated anti-CD4 (RM4–5), BV711-conjugated anti-CD45.2 (104), APC-conjugated anti-CD44 (IM7), PE-conjugated anti-CD62L (MEL-14), AlexaFluor 488 -conjugated anti-I-A/I-E, BV421-conjugated anti-CD80, PerCP/Cy5.5-conjugated anti-CD86, and PE-conjugated anti-CD31 (MEC13.3). All antibodies were purchased from BioLegend (San Diego, CA). APC-conjugated anti-Feeder Cells (mEF-SK4) was purchased from Miltenyi Biotec. Cells were surface stained by incubation with the relevant antibodies diluted in phosphate buffered saline (PBS) + 2% FBS for 20 minutes at 4C, followed by two washes with PBS 2% FBS. When intracellular staining of signature cytokines was performed, cell suspensions were incubated overnight at 37C in the presence of 0.1% brefeldin A (BioLegend) and 0.1% monensin (BioLegend). After the incubation, surface staining was performed as indicated above, followed by cell fixation for 20 minutes at RT with Fixation buffer (Biolegend). Upon fixation and washing with PBS 2% FBS, cell suspension was permeabilized with 1X Perm Wash buffer (Biolegend), and stained intracellularly for 30 minutes at RT in the dark. Absolute cell numbers were quantified using Precision Count Beads (BioLegend).
Immunofluorescence.
Cardiac fibroblasts were cultured on 0.1% gelatin-coated glass coverslips in a 6-well plate until confluent and treated daily with 100U/mL murine IFNγ (Peprotech) for 3 days. At the end of each treatment, cell layers were washed twice with PBS and fixed in 3% PFA. Nonspecific binding was prevented by incubation with PBS containing 10% goat serum (Jackson ImmunoResearch) for 1 h. Cells were incubated with primary antibodies against Vimentin (Abcam Cat# ab20346) and MHCII (Invitrogen Cat #14–5321-82) at 1:250 dilution. The cells were then incubated at 4°C overnight and washed three times with PBS. As controls, parallel coverslips were incubated with no primary antibody. Alexa Fluor 568–conjugated goat anti–mouse (Invitrogen; Cat # A-11004) and Alexa Fluor 488-conjugated goat anti-rat ( Invitrogen; Cat # A48262) at 1:500 dilution were used as secondary antibodies. Visualization was performed with a Nikon Ti inverted fluorescent microscope.
Acute T. cruzi Infection.
8 week old wt mice were infected with 20,000 T. cruzi trypomastigotes (Colombiana strain, discrete typing unit (DTU) TcI, the most widely distributed in Colombia). Trypomastigotes were harvested from infected Vero cells (ATCC, Manassas, Va), collected from culture supernatant, purified by differential centrifugation, and resuspended in phosphate-buffered saline (PBS) before intraperitoneal injection in mice. Mice were euthanized at the peak of parasitemia, at 19 days post infection.
Mouse model of Transverse Aortic Constriction (TAC).
Pressure overload was induced by minimally invasive TAC surgery to constrict the transverse aorta (26G, 0.51mm outer diameter) of randomized 8–10 week old male mice to induce HF as previously described.38 Sham operated mice underwent the same procedure but without aortic constriction. 4 weeks after surgery, mice were euthanized, and tissue was harvested for further analysis.
Tamoxifen Treatment.
To induce Cre recombinase activity, Tcf21iCre/+MhcIIfl/fl mice were treated with Tamoxifen (20mg/mL) (Millipore Sigma, T5648) dissolved in sunflower oil 10% ethanol for 5 consecutive daily i.p. injections at 75 mg/kg body weight. After TAC surgery, mice were maintained on a diet containing 400 mg/kg tamoxifen citrate (Envigo, TD.55125) for 4 weeks until harvest.
In vivo echocardiography.
In vivo transthoracic echocardiography was assessed one day prior to tissue harvest. Mice were lightly sedated at 1–2% isoflurane in medical oxygen (0.7 L/minute), on a heated stage in the supine position as previously described.38 Heart rates and respiratory rates were continuously monitored via the stage electrodes. Depilatory cream (Nair, USA) was applied to the chest to remove fur and ultrasonic gel was applied to the 22–55 MHz echocardiography transducer (MS550D; Vevo 2100, FUJIFILM VisualSonics, Canada) to obtain parasternal short axis views of the LV in M-mode with a target heart rate of 400–500 bpm. LV parameters and heart rate (Table 1) were measured by averaging values obtained from 5 cardiac cycles. All analyses were performed blindly using Vevo 2100 software (v3.1.1, FUJIFILM Visual Sonics, Canada).
Histology.
Immunohistochemistry, and immunofluorescence of cardiac tissue were performed in midpapillary LV tissue, cut into 5μm sections. Frozen LV sections were incubated with primary antibody against murine CD4 (Biolegend, clone GK1.5) for 1 h (1:500 dilution) followed by incubation with goat anti-rat biotinylated secondary antibody (1:300 dilution) (Jackson ImmunoResearch, 112–065-062). Sections were incubated with Streptavidin-HRP (DAKO, K0675) and visualized using AEC Substrate-Chromogen. CD4+ T cells were quantified by manually scanning the entire LV tissue section and counting stained cells per section. LV sections from tissue fixed in 10% formalin and embedded in paraffin were stained with FITC-conjugated WGA (Millipore Sigma, Cat #L4895) at 5 μg/ml to determine cardiomyocyte cross-sectional area by tracing the outline of at least 25 myocytes per section with NIS-Elements software. Antibodies to α-Smooth Muscle Actin (Sigma, Cat #A2547), 1:250 dilution, and to CD4 (Bio legend, Cat#100401), 1:25 dilution, and MHCII (Invitrogen Cat #14–5321-82), 1:250 dilution, were used in immunofluorescence staining of LV tissue of WT and Tcf21iCre/+; R26eGFP mice subjected to Sham and TAC. Fixed sections were stained with Picrosirius red to determine percent fibrotic area quantified using NIH ImageJ software. All analyses were performed blindly.
Statistics.
Data are presented as the mean ± SD. Statistical analyses were done by student unpaired t test (2-tailed) or nonparametric Mann-Whitney test (2-tailed) to adjust for nonequal Gaussian distribution when comparing two groups. Multiple group comparisons were performed by one way ANOVA and Turkey’s post-test, where indicated, using GraphPad Prism software (GraphPad). Differences were considered statistically significant at p≤ 0.05. Exact p value are added to source data.
Data Availability.
All data used to generate manuscript figures are included in the manuscript files.
Extended Data
Extended Data Fig. 1. Cardiac fibroblasts express MHCII in vivo in response to acute T. cruzi infection.

(A) Wt mice were inoculated with 20,000 T. cruzi parasites by intraperitoneal injection and the hearts were harvested at 19 days post-infection. (B-E) Ventricular CD31-CD45-MEFSK4+ cardiac fibroblasts were analysed by flow cytometry (≥ 5,000 target cells acquired) to determine surface expression of (B-C) MHCII and (D-E) CD80. n= 4 (mock) and n=3 (Chagas) mice. Error bars represent mean ± SD. (* p≤0.05; *** p<0.001; Mann-Whitney test, two tailed).
Extended Data Fig. 2. MhcII recombination in cardiac fibroblasts occurred in a cell-specific manner.

Detection of Intact allele (310bp), recombined alleles (475 bp) and GAPDH( 156 bp) in sorted CD31-CD45-MEFSK4+ Fibroblasts and leukocytes from Tcf21iCre/+MhcIIfl/fl mice treated with vehicle or Tamoxifen. GAPDH was used as loading control.
Extended Data Fig. 3. Tamoxifen treatment of Tcf21iCre/+/ MhcIIflox/flox mice reduces MHCII surface protein expression specifically in cardiac fibroblasts.
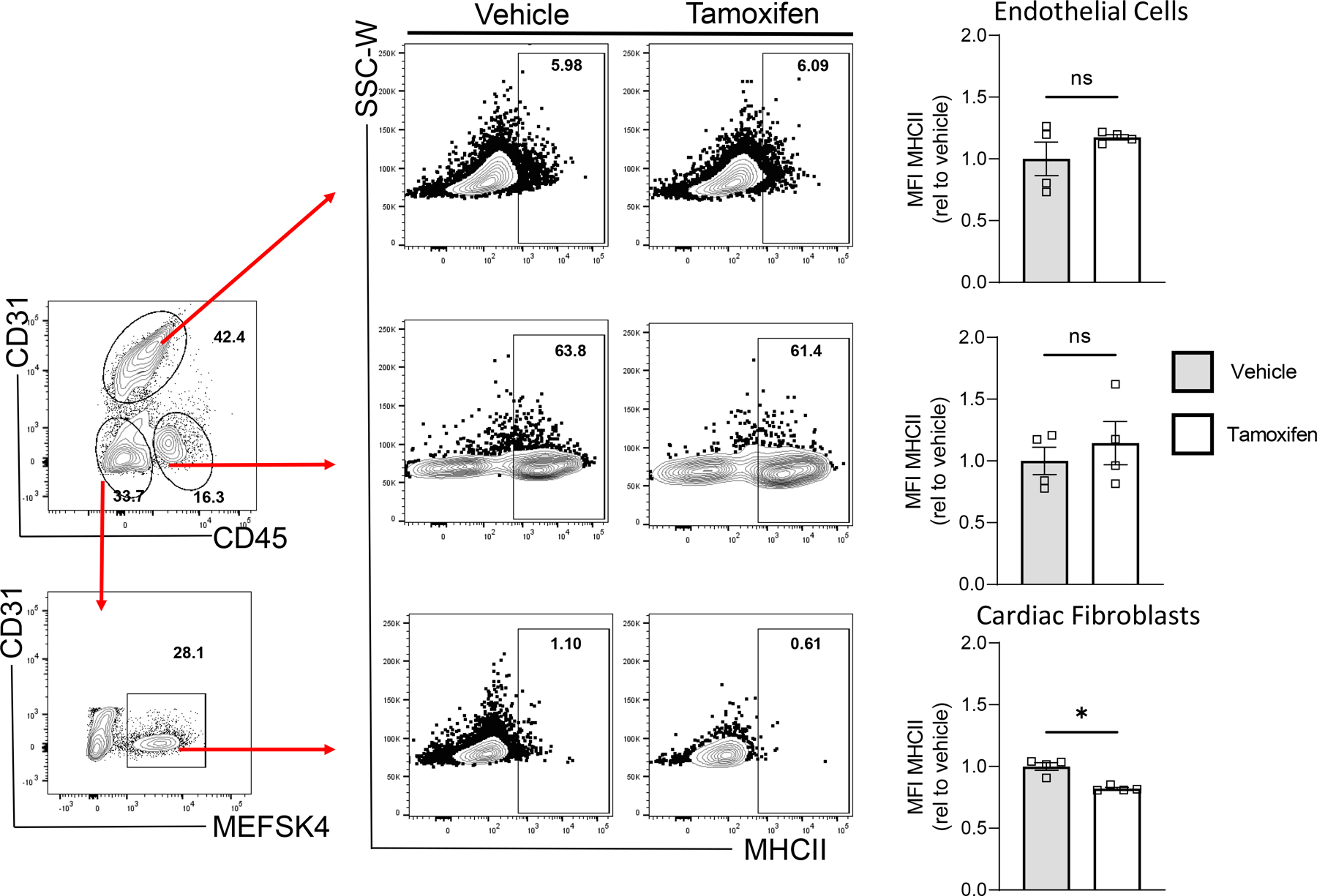
Transverse aortic constriction (TAC) surgery was performed on vehicle and Tamoxifen treated Tcf21iCre/+MhcIIfl/fl mice and the left ventricle was harvested after 4 weeks, digested, stained and analysed by flow cytometry. (A) Representative FACS plot showing gating of CD31+CD45-MESFK4- endothelial cells, CD31-CD45+MESFK4- leukocytes and CD31-CD45-MESFK4+ cardiac fibroblasts. (B-C) Representative FACS plots (B) and quantification of mean fluorescence intensity (MFI) (C) of MHC-II expression in endothelial cells (top), leukocytes (middle) and cardiac fibroblasts (bottom) in the hearts of vehicle and tamoxifen treated mice. n=4 mice/group. Error bars represent mean ± SE. (* p<0.05; Mann-Whitney test, two tailed. C: p value=0.0286).
Extended Data Fig. 4. Cardiac fibroblast MHCII expression does not affect CD4+ T cell activation in the mLNs in response to TAC.

Extended Data Fig. 5. Cardiac fibroblast MHCII expression contributes to cardiomyocyte hypertrophy and to cardiac fibroblast abundance in response to pressure overload.

Transverse aortic constriction (TAC) was performed on vehicle and Tamoxifen treated Tcf21iCre/+MhcIIfl/fl mice and harvested after 4 weeks. Conditional deletion of MhcII on cardiac fibroblasts was induced by administering Tamoxifen via intraperitoneal injections prior to TAC surgery, followed by Tamoxifen citrate chow for 4 weeks. (A) Wheat germ agglutinin (WGA) staining of frozen LV tissue sections was performed and used to calculate (B) mean cardiomyocyte area. Representative images of n=3 hearts (Sham), n=4 (TAC vehicle) and n=7 (TAC TMX). (C) Gross LV mass was acquired and is shown normalized to tibia length. (D-E) LV tissue was digested, stained for CD31 and MESFK4 and analysed by flow cytometry. Representative FACS plot (D) and quantification (E) of vehicle and tamoxifen treated mice hearts (n=4 mice/group). Error bars represent mean ± SE. (* p<0.01; Mann-Whitney test, two tailed: E: p=0286).
Extended Data Fig. 6. Schematic Diagram showing bidirectionality of cardiac fibroblast and Th1 cell interactions.

Cardiac fibroblasts express the costimulatory molecule CD80 and efficiently capture and process extracellular antigens into small peptides that are uploaded into MHC-II molecules. In response to IFNγ stimulation, cardiac fibroblasts express MHCII and present peptide antigens that induce IFNγ + Th1 cell immune responses and promote cardiac remodelling and dysfunction.
Supplementary Material
Acknowledgements
These studies were supported by NIH R01 HL123658 and R01 HL144477 (PA), NIH T32 HL 69770, NIH T32AI007077–34, NIH F31HL140883 and the American Heart Association 18PRE34020084 (NN), NIH R01 HL141187 and HL142624 (JD), and NIH T32AG066574 (DB).
Footnotes
Competing Interests
The authors declare no competing financial interests.
References
- 1.Pinto AR et al. Revisiting Cardiac Cellular Composition. Circ Res 118, 400–409, doi: 10.1161/CIRCRESAHA.115.307778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prabhu SD & Frangogiannis NG The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ Res 119, 91–112, doi: 10.1161/CIRCRESAHA.116.303577 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaur H et al. Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice. Circ Res 118, 1906–1917, doi: 10.1161/CIRCRESAHA.116.308643 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Aghajanian H et al. Targeting cardiac fibrosis with engineered T cells. Nature 573, 430–433, doi: 10.1038/s41586-019-1546-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nevers T et al. Left Ventricular T-Cell Recruitment Contributes to the Pathogenesis of Heart Failure. Circ Heart Fail 8, 776–787, doi: 10.1161/CIRCHEARTFAILURE.115.002225 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nevers T et al. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J Exp Med 214, 3311–3329, doi: 10.1084/jem.20161791 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laroumanie F et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation 129, 2111–2124, doi: 10.1161/CIRCULATIONAHA.113.007101 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Epelman S, Liu PP & Mann DL Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol 15, 117–129, doi: 10.1038/nri3800 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kambayashi T & Laufer TM Atypical MHC class II-expressing antigen-presenting cells: can anything replace a dendritic cell? Nat Rev Immunol 14, 719–730, doi: 10.1038/nri3754 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Umetsu DT, Katzen D, Jabara HH & Geha RS Antigen presentation by human dermal fibroblasts: activation of resting T lymphocytes. J Immunol 136, 440–445 (1986). [PubMed] [Google Scholar]
- 11.Kanisicak O et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun 7, 12260, doi: 10.1038/ncomms12260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burgdorf S, Kautz A, Bohnert V, Knolle PA & Kurts C Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 316, 612–616, doi: 10.1126/science.1137971 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Ramachandra L, Noss E, Boom WH & Harding CV Phagocytic processing of antigens for presentation by class II major histocompatibility complex molecules. Cell Microbiol 1, 205–214, doi: 10.1046/j.1462-5822.1999.00026.x (1999). [DOI] [PubMed] [Google Scholar]
- 14.Ngwenyama N et al. CXCR3 regulates CD4+ T cell cardiotropism in pressure overload-induced cardiac dysfunction. JCI Insight 4, doi: 10.1172/jci.insight.125527 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michailowsky V et al. Intercellular adhesion molecule 1 deficiency leads to impaired recruitment of T lymphocytes and enhanced host susceptibility to infection with Trypanosoma cruzi. J Immunol 173, 463–470, doi: 10.4049/jimmunol.173.1.463 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Cardillo F, Voltarelli JC, Reed SG & Silva JS Regulation of Trypanosoma cruzi infection in mice by gamma interferon and interleukin 10: role of NK cells. Infect Immun 64, 128–134, doi: 10.1128/IAI.64.1.128-134.1996 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haverson K, Singha S, Stokes CR & Bailey M Professional and non-professional antigen-presenting cells in the porcine small intestine. Immunology 101, 492–500, doi: 10.1046/j.1365-2567.2000.00128.x (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geppert TD & Lipsky PE Antigen presentation by interferon-gamma-treated endothelial cells and fibroblasts: differential ability to function as antigen-presenting cells despite comparable Ia expression. J Immunol 135, 3750–3762 (1985). [PubMed] [Google Scholar]
- 19.Spear TT, Evavold BD, Baker BM & Nishimura MI Understanding TCR affinity, antigen specificity, and cross-reactivity to improve TCR gene-modified T cells for cancer immunotherapy. Cancer Immunol Immunother 68, 1881–1889, doi: 10.1007/s00262-019-02401-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ngwenyama N et al. Isolevuglandin-Modified Cardiac Proteins Drive CD4+ T Cell Activation in the Heart and Promote Cardiac Dysfunction. Circulation, doi: 10.1161/CIRCULATIONAHA.120.051889 (2021). [DOI] [PMC free article] [PubMed]
- 21.Rieckmann M et al. Myocardial infarction triggers cardioprotective antigen-specific T helper cell responses. J Clin Invest 129, 4922–4936, doi: 10.1172/JCI123859 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tallquist MD & Molkentin JD Redefining the identity of cardiac fibroblasts. Nat Rev Cardiol 14, 484–491, doi: 10.1038/nrcardio.2017.57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kong P, Christia P & Frangogiannis NG The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 71, 549–574, doi: 10.1007/s00018-013-1349-6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fields PE et al. B7.1 is a quantitatively stronger costimulus than B7.2 in the activation of naive CD8+ TCR-transgenic T cells. J Immunol 161, 5268–5275 (1998). [PubMed] [Google Scholar]
- 25.Pechhold K et al. Inflammatory cytokines IFN-gamma plus TNF-alpha induce regulated expression of CD80 (B7–1) but not CD86 (B7–2) on murine fibroblasts. J Immunol 158, 4921–4929 (1997). [PubMed] [Google Scholar]
- 26.Smythe JA et al. Human fibroblasts transduced with CD80 or CD86 efficiently trans-costimulate CD4+ and CD8+ T lymphocytes in HLA-restricted reactions: implications for immune augmentation cancer therapy and autoimmunity. J Immunol 163, 3239–3249 (1999). [PubMed] [Google Scholar]
- 27.Sansom DM CD28, CTLA-4 and their ligands: who does what and to whom? Immunology 101, 169–177, doi: 10.1046/j.1365-2567.2000.00121.x (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hulsmans M et al. Cardiac macrophages promote diastolic dysfunction. J Exp Med 215, 423–440, doi: 10.1084/jem.20171274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seeberg JC et al. Non-professional phagocytosis: a general feature of normal tissue cells. Sci Rep 9, 11875, doi: 10.1038/s41598-019-48370-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakaya M et al. Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J Clin Invest 127, 383–401, doi: 10.1172/JCI83822 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salvador AM et al. Intercellular Adhesion Molecule 1 Regulates Left Ventricular Leukocyte Infiltration, Cardiac Remodeling, and Function in Pressure Overload-Induced Heart Failure. J Am Heart Assoc 5, e003126, doi: 10.1161/jaha.115.003126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H et al. Role of bone marrow-derived CD11c(+) dendritic cells in systolic overload-induced left ventricular inflammation, fibrosis and hypertrophy. Basic Res Cardiol 112, 25, doi: 10.1007/s00395-017-0615-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Virani SS et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 141, e139–e596, doi: 10.1161/CIR.0000000000000757 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Ren Z et al. Single-Cell Reconstruction of Progression Trajectory Reveals Intervention Principles in Pathological Cardiac Hypertrophy. Circulation 141, 1704–1719, doi: 10.1161/circulationaha.119.043053 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Wang L et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat Cell Biol 22, 108–119, doi: 10.1038/s41556-019-0446-7 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Dubrot J et al. Lymph node stromal cells acquire peptide-MHCII complexes from dendritic cells and induce antigen-specific CD4⁺ T cell tolerance. J Exp Med 211, 1153–1166, doi: 10.1084/jem.20132000 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perez-Shibayama C et al. Type I interferon signaling in fibroblastic reticular cells prevents exhaustive activation of antiviral CD8(+) T cells. Sci Immunol 5, doi: 10.1126/sciimmunol.abb7066 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Richards DA et al. Distinct Phenotypes Induced by Three Degrees of Transverse Aortic Constriction in Mice. Sci Rep 9, 5844, doi: 10.1038/s41598-019-42209-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used to generate manuscript figures are included in the manuscript files.