

Abstract
Cancer vaccines critically rely on the availability of targetable immunogenic cancer‐specific neoepitopes. However, mutation‐based immunogenic neoantigens are rare or even non‐existent in subgroups of cancer types. To address this issue, we exploited a cancer‐specific aberrant transcription‐induced chimeric RNA, designated A‐Pas chiRNA, as a possible source of clinically relevant and targetable neoantigens. A‐Pas chiRNA encodes a recently discovered cancer‐specific chimeric protein that comprises full‐length astrotactin‐2 (ASTN2) C‐terminally fused in‐frame to the antisense sequence of the 18th intron of pregnancy‐associated plasma protein‐A (PAPPA). We used extracellular vesicles (EVs) from A‐Pas chiRNA‐transfected dendritic cells (DCs) to produce the cell‐free anticancer vaccine DEXA‐P. Treatment of immunocompetent cancer‐bearing mice with DEXA‐P inhibited tumour growth and prolonged animal survival. In summary, we demonstrate for the first time that cancer‐specific transcription‐induced chimeric RNAs can be exploited to produce a cell‐free cancer vaccine that induces potent CD8+ T cell‐mediated anticancer immunity. Our novel approach may be particularly useful for developing cancer vaccines to treat malignancies with low mutational burden or without mutation‐based antigens. Moreover, this cell‐free anticancer vaccine approach may offer several practical advantages over cell‐based vaccines, such as ease of scalability and genetic modifiability as well as enhanced shelf life.
Keywords: EV‐based cancer vaccine, mutation‐independent neoantigen, transcription‐induced chimeric RNA
1. INTRODUCTION
Ideally, a cancer vaccine targets a fully defined immunogenic cancer‐specific neoantigen that induces adequate amounts of potent cancer‐selective T‐cells with no or minimal deleterious activity towards normal cells or tissues (Banchereau & Palucka, 2018; Tran et al., 2019). Recent insights indicate that cancer‐specific aberrant mRNA processing may significantly expand the repertoire of targetable neoantigens for cancer immunotherapy (Frankiw et al., 2019; Yang et al., 2019). Transcription‐induced chimeric RNAs (chiRNAs) are fused mRNA transcripts derived from two (or more) unrelated genes which are generated by aberrant ‘read‐through/splicing’ or ‘trans‐splicing’ (Ke et al., 2020; Lin et al., 2019; Varley et al., 2014; Zhang et al., 2013). Detailed analyses of aberrant mRNA processing events in cancer cells have identified potential new targets for cancer immunotherapy.
In this respect, we recently discovered an aberrant transcription‐induced exon‐intronantisense chimeric RNA ASTN2‐PAPPAantisense (A‐Pas chiRNA) that is selectively present in esophageal cancer (EC) tissue (Zhang et al., 2013). The nucleotide sequence of the cDNA of the A‐Pas chiRNA consists of the full‐length coding sequence of the ASTN2 gene, the subsequent nucleotide sequence corresponds to the antisense strand of part of 18th intron of the PAPPA gene (Wang et al., 2021). Normally, the ASTN2 gene encodes for astrotactin 2, an integral membrane protein involved in neural development (Glessner et al., 2009), whereas PAPPA encodes pregnancy‐associated plasma protein‐A, a secreted metalloproteinase which cleaves insulin‐like growth factor binding proteins (Bonaca et al., 2012; Conover et al., 2016). Given its selective presence in EC tissue (Zhang et al., 2013), we wondered whether A‐Pas chiRNA could be exploited for the development of neoantigen‐based therapeutic vaccine to induce T cells against A‐Pas chiRNA‐positive EC.
Dendritic cells (DCs) are highly effective antigen‐presenting cells with unique capacity to (cross‐) present antigens to naive cognate T cells (Garg et al., 2017). Consequently, DC‐based vaccines are considerably promising for cancer immunotherapy (Banchereau & Palucka, 2018; Garg et al., 2017; Sabado & Bhardwaj, 2015). However, cell‐based DC vaccine approaches are highly complex and require costly good manufacturing practices (GMP) (Garg et al., 2017; Sabado & Bhardwaj, 2015). Extracellular vesicles (EVs) are nanoscale lipid bilayer‐enclosed particles of endocytic origin that are released by numerous cell types, including DCs. DC‐derived EVs (DEXs) harbour many of the key immunostimulatory characteristics of DCs, including exposure of MHC‐I, MHC‐II and various key co‐stimulatory molecules (Pitt et al., 2016). Consequently, DEXs maintain the intrinsic capacity of DCs to present foreign antigens to T cells (Pitt et al., 2016). The lipid bilayer makeup endows DEXs with significant serum stability, long shelf‐life when frozen, and relatively simple GMP handling (El Andaloussi et al., 2013; Jiang & Gao, 2017). In this respect, DEXs can be considered as ‘natural nanodrugs’ of controllable size and composition, and with stable immunomodulatory capacity.
Here, we describe a novel therapeutic cell‐free anticancer vaccine that is based on EVs derived from DCs transduced with A‐Pas chiRNA. To the best of our knowledge, this is the first example of a therapeutic DC‐EV anticancer vaccine based on an immunogenic neoantigen encoded by a cancer‐specific transcription‐induced chiRNA.
2. MATERIALS AND METHODS
2.1. Study design
The purpose of this study was to evaluate the effects of EV‐based therapeutic cancer vaccine in esophageal cancer. We investigated the possibility of exploiting transcription‐induced chimeric RNA as neoantigen and developed a cell‐free EV‐based vaccine containing the neoantigen. The antitumour efficacy was evaluated in mouse model. Sample sizes were chosen on the basis of previously published studies. Mice were randomized into various groups before treatment. Experiments were not performed in a blinded fashion.
2.2. Cell lines
All cell lines (except TE‐1) were provided by the Cell Bank of the Chinese Academy of Medical Sciences and cultured in a humidified atmosphere containing 5% CO2 at 37°C using the indicated culture medium supplemented with 10% FBS, 100 unit/ml penicillin and 100 unit/ml streptomycin, unless indicated otherwise. Human EC cell line TE‐1 (HLA‐A24+), kindly provided by Dr. X.C. Xu (UT M.D. Anderson Cancer Centre, USA) was cultured in Dulbecco's Modified Eagle Medium (DMEM). Human EC cell line KYSE140 (HLA‐A2+) was cultured in RPMI medium. Murine DC cell line DC2.4 (H‐2b), murine EC cell line AKR (H‐2b), and HEK 293T were cultured in DMEM supplemented with 10% FBS. Before EV isolation, the culture medium of DC2.4 cells was replaced by DMEM with 10% of EV‐depleted FBS and cultured for 48 h, after which the conditioned culture medium was collected for subsequent EV isolation.
2.3. Generation of A‐Pas fusion protein‐encoding lentivirus particles
The cDNA encoding the A‐Pas fusion protein (4.2 kb) was fused at its C‐terminal in‐frame to a 3×FLAG detection tag and then cloned into lentivirus expression vector pCDH‐CMV‐puro (System Biosciences, CA, USA). Recombinant A‐Pas‐encoding lentivirus particles (LVA‐P) were produced according to standard procedures using HEK293T cells. Cell lines stably expressing Flag‐tagged A‐Pas were generated by repeated infection with LVA‐P for three times every 18 h and followed by puromycin selection (2 μg/ml). Analogously, empty vector‐LV (LVVEC) particles were generated and used to conduct appropriate control experiments.
2.4. Generation of mAb A‐Pas‐16 against ASTN2‐PAPPAantisense
The mAb A‐Pas‐16 was generated using conventional mouse hybridoma technology (Abmart company). In brief, three peptide sequences derived from the 81 aa of PAPPA in A‐Pas (seven NTRVLIPTVSYSEE 20; 32 QKRMDSPHEPSLSN 45; 62 DEDHVGSMHIQITF 75) were used as immunogens. ELISA was used to screen for A‐Pas‐specific mAbs, which yielded 17 mAbs that were specific for an A‐Pas peptide. From this panel we selected mAb A‐Pas‐16 for further validation by immunoblot analysis.
2.5. MHC‐I peptide prediction
NetMHCpan (www.cbs.dtu.dk/services/NetMHCpan/) was used to predict A‐Pas‐derived peptides for binding to mouse MHC class 1 (H‐2Db, H‐2Kb) and human MHC class 1 (HLA‐A01:01, HLA‐A02:01, HLA‐A03:01, HLA‐A24:02, HLA‐A26:01, HLA‐B07:02, HLA‐B08:01, HLA‐B27:05, HLA‐B39:01, HLA‐B40:01, HLA‐B58:01, HLA‐B15:01) molecules. Subsequently, shared amino acid sequences within each group were selected and use to synthesize as the epitope peptides (Table S1)
2.6. Preparation of cell‐free vaccine DEXA‐P
The cell‐free vaccine DEXA‐P was produced in the absence of exogenous LPS stimulation using EVs derived from the murine DC cell line DC2.4 (H‐2b) transduced with LVA‐P. In a similar manner, control vaccine DEXVEC was produced using DC2.4 cells transduced with LVVEC.
In short, DC2.4 cells transduced with LVA‐P or LVVEC were cultured for 48 h in DMEM supplemented with 10% EV‐depleted FBS; then DEXA‐P and DEXVEC were collected from the respective conditioned culture medium by sequential centrifugation (first at 500 × g for 10 min, followed by 10,000 × g for 30 min; EV‐containing supernatants were filtered through a 0.22‐μm filter; then Evs in the filtrate were pelleted by centrifugation at 100,000 × g for 1 h and resuspended in PBS). The total protein concentration of EV preparations was quantified by Bradford assay (Sangon Biotech, USA).
2.7. Isolation and generation of human DCs
Human DCs were generated from venous blood of healthy volunteers (HLA‐A24+ and/or HLA‐A2+)after informed written consent. Briefly, PBMCs obtained by standard density gradient centrifugation (Lymphoprep) were subjected to magnetic‐activated cell sorting (MACS) using anti‐CD14‐beads and MS columns (Miltenyi Biotec) to isolate monocytes. Immature DCs (iDCs) were generated by treatment with 500 U/ml GM‐CSF and 1000 U/ml IL‐4 for 5 d. On day 6, LVA‐P, empty LV or A‐Pas‐derived peptides were added to the DCs followed by incubated in the presence 500 U/ml GM‐CSF and 1000 U/ml IL‐4 for 24 h. Subsequently, DCs were cultured for 2 days in the presence of 1 μg/ml LPS to generate mature DCs (mDCs). DC phenotype was confirmed by flow cytometric analysis of CD14, CD83, CD86, HLA‐ABC and HLA‐DR expression.
2.8. T cell proliferation assays
For human T cell proliferation assays, antigen‐loaded DCs were pretreated with 10 μg/ml mitomycin C (Sigma, St Louis, MO, USA) to inhibit cell division. Autologous CD3+ T cells were obtained using the human Pan T Cell Isolation Kit (Miltenyi Biotec). These T cells were co‐incubated with antigen‐loaded DCs (10,000 cells/well) at a cell ratio of 10:1 for 72 h. For mouse T cell proliferation assays, T lymphocytes derived from the spleen of C57BL/6 mice were co‐incubated with antigen‐loaded DC2.4 cells at a cell ratio of 10:1, or re‐stimulation with 40 μg DEX for 72 h. Antigen‐loaded DC2.4 cells were pretreated with 10 μg/ml mitomycin C to inhibit cell division. Subsequently, cell viability was determined using CCK‐8 assay (Cell Counting Kit‐8, MedChemExpress).
2.9. Real‐time cytolysis assays using the xCELLigence RTCA
Real‐Time Cell Analysis (RTCA, xCELLigence Roche, Penzberg, Germany) (Wang et al., 2019; Wang et al., 2018; Xiong et al., 2020) was used to monitor T cell‐mediated cell killing. Briefly, target cells (7,000 cells/well for KYSE140, 6,000 cells/well for TE1, and 10,000 cells/well for AKR cells) were seeded in duplicate wells of a 96‐well E‐plate (Roche) and allowed to adhere for 16–25 h. Then 50 μl of the spend culture medium was removed and replaced with 50 μl of fresh medium containing (or not) DC‐ or DEX‐pulsed effector T cells at the indicated E:T cells ratios. Fresh medium containing 1% Triton X‐100 was used as set point for maximum cell killing.
2.10. Mouse vaccination and subsequent isolation of murine splenocytes
Female C57BL/6 mice were injected i.v. with DEXA‐P, DEXVEC, DEX or PBS, respectively (40 μg/mouse/week for 3 weeks). Three days after the last vaccination, splenic T cells were harvested from the mice, followed by re‐stimulated with DEXA‐P (40 μg) or DEXVEC (40 μg) for 72 h. Briefly, spleens of vaccinated mice were harvested, crushed and vigorously resuspended to make single‐cell suspensions. To remove aggregates or clumps, the resuspended cells were filtered through a 70 μm cell strainer.
2.11. Esophageal tumour mouse model, T‐cell depletion and vaccination
A total of 5 × 105 AKR cells were resuspended in 100 μl PBS and s.c. inoculated in the flanks of 6‐wk‐old female C57BL/6 mice (H‐2b) and nude athymic mice (strain NU/NU Crl:NUFoxn1nu), respectively. DEXA‐P, DEXVEC and PBS were administered i.v. into 7‐day‐established AKR tumour‐bearing mice every 5 days for three times, respectively. Tumour volumes were measured every 5 days, and tumour mass was calculated using the following formula: volume = 0.5236 × length × width2. Mice were sacrificed by cervical dislocation at desired time‐points, and tumours were excised for paraffin block preservation. For T‐cell depletion studies, antibodies to deplete CD4+ T cells (clone GK1.5; BioXcell) or CD8+ T cells (clone 2.43; BioXcell) were injected i.p. at 100 μg per mouse on day 5, 10, 15 and every other 5 days thereafter until completion of the study. 7‐day‐established AKR tumour‐bearing mice were treated three times with DEXA‐P (40 μg) i.v. and were monitored for tumour growth, body weight and survival. The extent of T cell depletion was determined at the end of the study by flow cytometry of PBL and spleen cell homogenate. Animal experiments were reviewed and approved by the Ethics Committee of Shantou University Medical College.
2.12. Measurement of T lymphocytes in mouse spleen and tumour tissues
Splenocytes were collected from mouse spleen and incubated at 37°C for 6 h in DMEM/F‐12 medium supplemented with 10% FBS. Golgi stop (1.5 μg/ml, BD Biosciences, USA) was added for the final 5 h of incubation to block cytokine secretion. Cells were harvested, washed and blocked with CD16/CD32‐blocking Ab (BD Bioscences, USA) for 5 min at room temperature, and stained with 0.4 μg/ml BV510‐labelled CD45, PerCP‐Cy5.5‐labelled CD3, FITC‐labelled CD8, PE‐Cy7‐labelled CD4, BV421‐labelled CD25 (BD Bioscences, USA) for 15 min at room temperature. Then cells were washed and incubated with permeabilisation solution (BD Bioscences, USA) for another 20 min at 4°C. Finally, cells were further labelled with 0.4 μg/ml Alexa 647‐labeleld IFN‐γ, PE‐labelled FOXP3 (BD Bioscences, USA) for 15 min at room temperature and analysed by flow cytometry. Tumour tissues were minced into small pieces and digested in collagenase type IV suspension (0.05 mg/ml, Worthington Biochem) for 40 min at 37°C. The resulting suspension was filtered through the 70 μm cell strainer. The extract was centrifuged at 500×g for 10 min after which the supernatant was removed. The mixture was re‐suspended in ACK lysis buffer to remove red blood cells and the rest cells were stained with different fluorescently‐labelled antibodies as described above.
2.13. Cytokine release assay
Supernatants from human T cells co‐incubated with antigen‐loaded DCs were collected and secretion of IFN‐γ was quantified using ELISA kit (# 88–7316, Invitrogen, USA). Splenocytes were harvested from DEXA‐P, DEXVEC or PBS treated tumour‐bearing C57BL/6 mice as described above. The supernatant was collected and levels of IFN‐γ (# BMS606, Invitrogen, USA), IL‐2 (# 88‐7024‐88, Invitrogen, USA), IL‐10 (# 88‐7105‐88, Invitrogen, USA) and TGF‐β (# 88‐8350‐88, Invitrogen, USA) in supernatant were quantified using ELISA kits. For measurement of cytokine released in vivo, mouse serum was harvested from tumour‐bearing mice 3 days after the last vaccination and centrifuged at 3000×g for 10 min at room temperature, followed by analysis of IFN‐γ, IL‐2, IL‐10 and TGF‐β content, respectively.
2.14. Flow cytometry analysis
Human DCs were stained with 0.6 μg/ml FITC‐labelled CD14, HLA‐ABC and CD86, PE‐labelled HLA‐DR and CD83 (Thermo Fisher Scientific, MA, USA) mAbs for 45 min at 4°C and analysed by flow cytometry. Murine DCs were stained with FITC‐labelled ICAM1 and MHC‐II, PE‐labelled CD80, CD86 and MHC‐I (Thermo Fisher Scientific, MA, USA) mAbs at the final concentration of 0.6 μg/ml at 4°C for 45 min, respectively, followed by flow cytometry. For DEX assay, 30 μg of EVs (or 30 μg of FCS as negative control) were incubated with 10 μl of 4 μm diameter aldehyde/sulphate latex beads (Interfacial Dynamics, Portland, OR) for 15 min at room temperature in a 30–100 μl final volume, followed by 2 h with gentle shaking in 1 ml PBS. The reaction was stopped by incubation for 30 min in 100 mM glycine. EV‐ or FCS‐coated beads were washed three times PBS with 3% FCS and 0.1% NaN3 and resuspended in 200 μl of the same buffer. 10 μl coated beads were incubated for 1 h at 4°C with each fluorescently‐labelled abs as described above.
2.15. Multiplexed Immunofluorescence (mIF)
Multiplexed Immunofluorescence (mIF) was performed for multiple markers staining using PerkinElmer Opal kit (Perkin‐Elmer, Waltham, MA, USA) (Wang et al., 2020). Briefly, 4 μm sections cut from FFPE mouse tumour tissues were deparaffinized, rehydrated, followed by endogenous peroxidase blockade and antigen retrieval. For the detection of various T cell subpopulations, sections were incubated with Ab against CD4 (Cat. ab183685; Abcam), followed by HRP‐conjugated secondary Ab. A second antigen retrieval was performed to denature any antibodies in these tissues, followed by incubation with Ab against CD8α (Cat. #98941; Cell Signalling Technology) and then incubated with HRP‐conjugated secondary Ab. A third antigen retrieval was conducted in these slides, followed by incubated with Ab against FoxP3 (Cat. ab99964; Abcam) and then incubated with HRP‐conjugated secondary Ab. Between the consecutive antigen retrieval steps the slides were visualized using Opal 650 TSA Plus (1:50). For the detection of macrophage subpopulations, slides were triple stained with F4/80 (Cat. ab240946; Abcam), CD206 (Cat. ab64693; Abcam), along with CD11c (Cat. ab52632; Abcam) were perform according to the sequential staining protocol above. For all sections, nuclei were stained with DAPI. Images were acquired using a Vectra 3 pathology imaging microscope (PerkinElmer, Waltham, MA, USA).
2.16. Histology
For haematoxylin and eosin (H&E) staining, mice were euthanized 3 days after the final vaccination. Tumous, various organs and tissues were fixed in 10% formalin, embedded in paraffin, and stained with haematoxylin and eosin. Histological images were photographed using a microscope.
2.17. RNA analysis
Total RNA was extracted from the cells or clinical specimens using TRIzol (Invitrogen). RNA (4 μg) was reverse‐transcribed using M‐MLV Reverse Transcriptase (Invitrogen) and oligo‐(dT)20 primer (Invitrogen) according to the manufacturer's instructions. Then an equal amount of cDNA was amplified using an SYBR Green PCR amplification kit (Invitrogen) with the Applied Biosystems 7500 Real Time PCR system (Applied Biosystems, Foster City, CA, USA) as described previously (Dong et al., 2017; Feng et al., 2014). PCR product was visualized in a 2% agarose gel. The results were normalized to β‐actin as an internal control. All reactions were run in triplicate. The cDNA was subjected to PCR with the following primers:
A‐Pas forward: 5′‐TGGTGTCAATGGCCCGAAACA‐3′ (designed to hybridize to ASTN2 exon 22)
A‐Pas reverse: 5′‐TCACCAGGGGTGTCTGGTGT‐3′ (designed to hybridize to PAPPA intron 18)
β‐actin forward: 5′‐GAACCCCAAGGCCAACCGCGAGA‐3′
β‐actin reverse: 5′‐TGACCCCGTCACCGGAGTCCATC‐3′
2.18. Quantitative Real‐Time PCR
The amplified PCR product from Real Time PCR analysis was used as standard in quantitative real‐time PCR. The DNA target sequence of A‐Pas was ligated into a pUC57 vector to produce pUC57‐A‐Pas. The concentration of the isolated and purified pDNA was measured with a Cytation 5 (BioTek, USA). The quantification of the pUC57‐A‐Pas in picomoles was performed considering the average weight of a one pair base (660 Da) and the number of base pairs of vector and insert (Nb). The following mathematical formula was applied: pmol of dsDNA = (μg of dsDNA × 106)/(660 × Nb). Avogadro's constant (6.02214086 × 1023 mol–1) was used to estimate the number of pUC57‐A‐Pas molecules. Number of copies was calculated per μ, which was the volume used as template in each quantitative real‐time PCR. Dilutions containing 10–107 copies were used to generate the standard curve. The quantification analysis was carried out in a CFX Connect Real‐Time PCR Detection System (Bio‐Rad, USA) using SYBR Green. Standards and negative control were added to the 96‐well plate in triplicate at each running. Data acquisition, standard curve fittings, and analysis were performed with the CFX Maestro software.
2.19. Western blot
Western blotting was performed as described previously (Gan et al., 2016). Briefly, the cells or tissues were lysed in RIPA buffer, and then equivalent amounts of the protein extracts were separated using 10% SDS/PAGE and then transferred to a polyvinylidene difluoride (PVDF) membrane. The membranes were blocked in 5% skim milk in TBS containing 0.1% Tween‐20 (TBST) buffer and then incubated with the primary abs against A‐Pas‐16, FLAG tag (#F1804, Sigma‐Aldrich, USA), Alix (#2171, Cell Signalling Technology, USA), Calnexin (#2433, Cell Signalling Technology, USA), GAPDH (#5174, Cell Signalling Technology, USA), at 4°C for 16 h. Subsequently, the secondary antibodies were added and incubated at room temperature for 2 h. The immunoreactive bands were visualized with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) using X‐ray film (Eastman Kodak).
2.20. Statistical analysis
All statistical analyses were performed using the SPSS 13.0 statistical software package (SPSS Inc.). Overall survival was estimated using the Kaplan–Meier method, and the difference in survival was evaluated using the log‐rank test. Comparisons between two groups were performed with a Student's t test, or paired t test, and comparisons among more than two groups were performed with one‐way ANOVA with post hoc intergroup comparisons. All bar graphs show the mean ± SEM of at least three independent experiments. A p value of less than 0.05 was considered statistically significant.
3. RESULTS
3.1. A‐Pas chiRNA is selectively transcribed and translated in primary human EC
Transcription read‐through and splicing in EC cells produced a 4296‐nt‐long A‐Pas chiRNA that contained almost the full‐length coding sequence of the ASTN2 gene followed by a 243‐nt‐long antisense sequence of part of 18th intron of the PAPPA gene (Figure 1A) (Wang et al., 2021). The predicted ASTN2‐PAPPAantisense (A‐Pas) fusion protein has the 1339 amino acids from ASTN2 protein at its N‐terminal, and the subsequent 81 new amino acids at the C‐terminal portion are in a novel sequence that does not correspond to any part of the PAPPA protein (Figure 1B). Recombinant lentivirus particles encoding the A‐Pas fusion protein tagged with three FLAG peptide sequences at the C‐terminal (LVA‐P‐FLAG) were produced, as illustrated in Figure 1(B).
FIGURE 1.
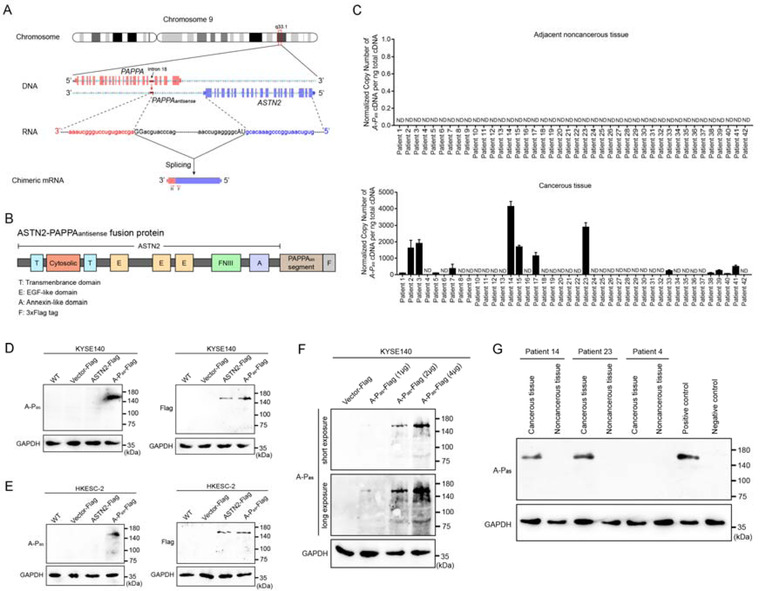
A‐Pas chiRNA is produced by aberrant mRNA processing and is selectively transcribed in EC tissues. (A) Schematic diagram of the A‐Pas chiRNA. Exons are represented by solid blocks connected lines representing introns. Transcription read‐through and splicing produce the A‐Pas chiRNA. ASTN2‐derived sequences are in blue; antisense sequence derived from the 18th intron of the PAPPA gene are in red; RNA splice junctions are shown in capital letters. Positions of the forward (F) and reverse (R) primers for detection A‐Pas chiRNA by RT‐PCR are indicated by red arrows. (B) The predicted structure of A‐Pas fusion protein. (C) The expression levels of A‐Pas chiRNA in EC and paired noncancerous tissues were evaluated by RT‐qPCR. The data were from three independent RT‐PCR reactions. Error bars indicate SEM. (D) Expression of A‐Pas fusion protein in KYSE140 cells transduced with LVvector, LVASTN2‐FLAG and LVA‐P‐FLAG was evaluated by immunoblotting using an A‐Pas‐specific mAb (A‐Pas‐16) and an anti‐FLAG mAb. GAPDH was used as a loading control. (E) Similar results from HKESC‐2 cells are shown. (F) Ectopic expression of A‐Pas fusion protein in KYSE140 cells transduced with different DNA concentrations of LVA‐P‐FLAG was detected by immunoblotting using mAb A‐Pas‐16. (G) Endogenous expression of A‐Pas fusion protein in EC and paired noncancerous tissues were evaluated by immunoblotting using the mAb A‐Pas‐16. A‐Pas chiRNA‐positive cell line HKESC‐3 served as a positive control, A‐Pas chiRNA‐negative cell line KYSE140 served as a negative control. GAPDH was used as a loading control
In 14 out of 42 EC patients, the transcription of A‐Pas chiRNA was restricted to cancerous tissues, and was undetectable in the respective adjacent noncancerous tissues of all 42 patients (Figure 1C, and Figure S1). To examine expression of the A‐Pas fusion protein translated from A‐Pas chiRNA, we generated a series of 17 A‐P as ‐specific monoclonal antibodies (mAbs) using three A‐P as ‐derived peptides. From this series, we selected mAb A‐Pas‐16, which was generated against the 14‐amino acid antigen peptide NTRVLIPTVSYSEE, which is located in the PAPPAas portion. MAb A‐Pas‐16 selectively bound to A‐Pas chiRNA‐negative KYSE140 and HKESC‐2 cells, but only when transduced with LVA‐P‐FLAG. Parental, LVvector and LVASTN2‐FLAG transduced KYSE140 and HKESC‐2 cells were used as negative controls. Immunoblot analysis using mAb A‐Pas‐16 detected a protein band of 160 kDa in LVA‐P‐FLAG‐transduced cells only. This apparent molecular weight (MW) is in good agreement with the calculated molecular weight of 156 kDa for A‐Pas. Immunoblot analysis of LVASTN2‐FLAG and LVA‐P‐FLAG‐transduced cells using an anti‐FLAG mAb revealed the FLAG‐tagged protein bands of expected MW (Figure 1D and E). To further evaluate the sensitivity and specificity of this antibody for A‐Pas, KYSE140 cells were transduced with LVA‐P‐FLAG at increasing DNA concentrations, and immunoblots images were obtained at short and long exposures. Even at short exposure, reasonably specific detection of low level of A‐Pas is demonstrated (Figure 1F). Importantly, in consistent with the RT‐qPCR result, endogenous expression of the A‐Pas fusion protein was detected in cancerous tissues derived from patient 14 and patient 23, but not in that from patient 4 by mAb A‐Pas‐16 (Figure 1G).
Taken together, these results indicated that cancer cells could indeed translate A‐Pas chiRNA into a fusion protein A‐Pas, and that mAb A‐Pas‐16 would be a useful tool to analyse A‐Pas protein expression. Subsequently, we analysed whether A‐Pas might be exploited as an immunogenic neoantigen.
3.2. Ectopic expression of A‐Pas chiRNA in DC promotes maturation status and enhances capacity to prime A‐Pas‐specific anticancer T cells in vitro
Both transcription of A‐Pas chiRNA and subsequent translation into a A‐Pas fusion protein were confirmed in LVA‐P‐FLAG‐transduced DCs (DCA‐P) (Figure 2A and B). In immunoblot analysis, A‐Pas fusion protein could be detected by both mAb A‐Pas‐16 and anti‐FLAG mAb (Figure 2B). Of note, DCA‐P showed downregulation of CD14 expression and upregulation of CD83, CD86, MHC‐I (HLA‐ABC) and MHC‐II (HLA‐DR) expression, collectively indicative of a matured DC phenotype (Prechtel & Steinkasserer, 2007) (Figure 2C).
FIGURE 2.
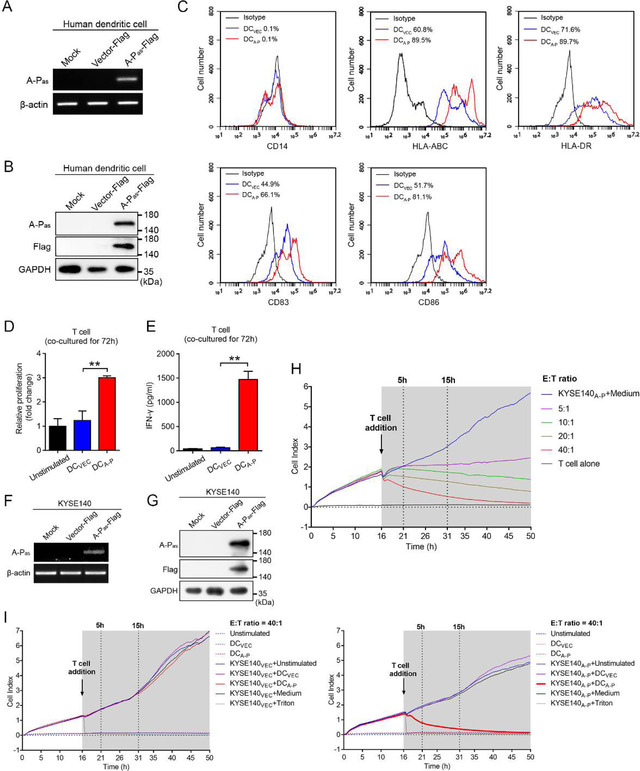
Protein product encoded by A‐Pas chiRNA induces antigen‐specific immune T cell responses against human EC cell. (A‐B) Expression of A‐Pas chiRNA in human DCA‐P and DCVEC was evaluated by RT‐PCR using β‐actin as loading control (A) and by immunoblotting using mAb A‐Pas‐16 and an anti‐FLAG mAb, GAPDH as a loading control (B). (C) Flow cytometric analysis of cell surface protein expression on DCA‐P or DCVEC. (D) The viability of autologous T cells primed with DCA‐P or DCVEC at a ratio of 1:10 (DC:T) for 72 h was evaluated by Cell Counting Kit‐8 (CCK‐8) assay. (E) Measurement of IFN‐γ in supernatants from the co‐culture of autologous T cells with DCA‐P or DCVEC. (F‐G) Expression of A‐Pas chiRNA was evaluated by RT‐PCR (F) and immunoblotting (G) in KYSE140A‐P or KYSE140VEC cells. (H) Monitoring of cytotoxicity mediated by DCA‐P‐activated T cells against KYSE140A‐P cells at the indicated E:T ratios by RTCA using the xCELLigence apparatus. (I) Monitoring of cytotoxicity mediated by DCA‐P or DCVEC‐activated T cells against KYSE140VEC (left panel) and KYSE140A‐P (right panel) at an E:T ratio of 40:1. 1% Triton X‐100 was added instead of T cells as positive control. Samples shown are representative of three independent experiments. Error bars indicate SEM. **p < 0.01 by one‐way ANOVA with post hoc intergroup comparisons in (D‐E)
Upon co‐culturing, DCA‐P exhibited an enhanced potency to promote the capacity of autologous T cells to proliferate and secrete IFN‐γ compared to unstimulated T cells and T cells that were primed by LVvector‐transduced DCs (DCVEC) (Figure 2D and E). Importantly, DCA‐P showed enhanced capacity to prime and activate T cells that selectively targeted and killed HLA‐matched LVA‐P‐FLAG‐transduced KYSE140 EC cells (KYSE140A‐P) (Figure 2F to H), but not empty‐vector transduced cells (KYSE140VEC) (Figure 2I). Moreover, DCA‐P‐primed T‐cells selectively killed HLA‐matched TE1 EC cells that have endogenous A‐Pas expression, but not KYSE140 cells that lack A‐Pas expression (Figure S2). Of note, the observed T cell responses towards A‐Pas transduced cells were not attributable to FLAG‐related immunogenic peptides processed from the FLAG‐tagged A‐Pas fusion protein (Figure S3 and Table S1).
Taken together, these results indicated that ectopic expression of A‐Pas chiRNA promotes maturation and subsequent capacity of DCA‐P to prime and activate A‐Pas‐specific T‐cells with cytolytic activity towards A‐Pas‐positive cancer cells.
3.3. Mouse DC2.4A‐P cells prime A‐Pas‐specific anticancer T cells in vitro
Upon transduction with LVA‐P‐FLAG, murine DC2.4A‐P cells showed a marked upregulation of MHC‐I, CD80, CD86, and ICAM and a moderate upregulation of MHC‐II, collectively indicative for a matured DC phenotype (Figure 3A to C). Importantly, DC2.4A‐P cells showed enhanced capacity to promote proliferation and IFN‐γ secretion by splenic T‐cells derived from H‐2Kb‐matched C57Bl/6 mice (Figure 3D and E). Moreover, DC2.4A‐P‐primed T‐cells selectively killed H‐2b‐matched LVA‐P‐FLAG‐transduced AKR EC cells (AKRA‐P) (Figure 3F to H). Of note, peptides containing potential FLAG tag‐related murine H‐2Kb class‐I epitopes failed to induce proliferation, IFN‐γ secretion and antigen‐specific cytolysis of AKRA‐P cells by CD8+ T cells (Figure S4).
FIGURE 3.
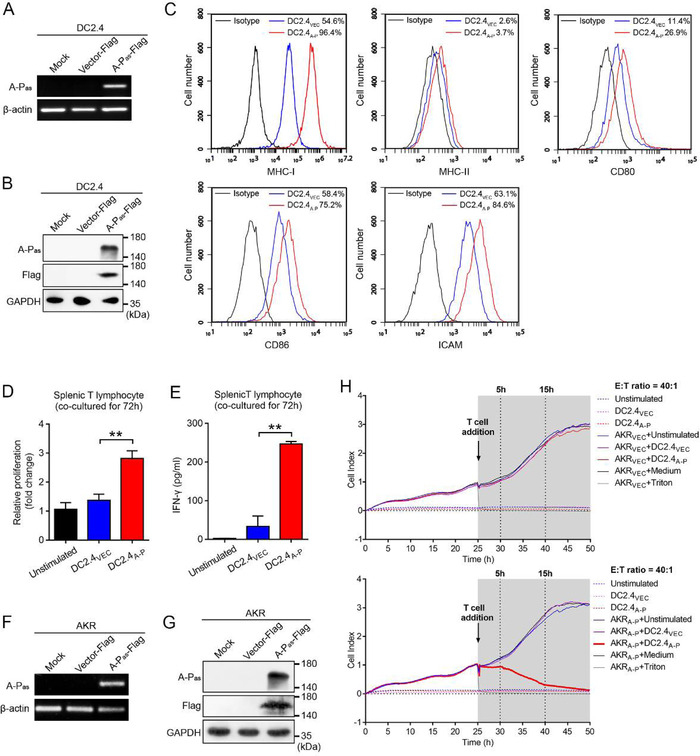
A‐Pas chiRNA induces antigen‐specific immune T cell responses against murine EC cell. (A‐B) Expression of A‐Pas chiRNA was evaluated by RT‐PCR (A) and immunoblotting (B) in murine DC2.4A‐P and DC2.4VEC. (C) Flow cytometric analysis of cell surface protein expression on DC2.4A‐P versus DC2.4VEC. (D) Splenic T cells were harvested from C57Bl/6 mice and primed with DC2.4A‐P or DC2.4VEC at a ratio of 1:10 (DC:T) for 72 h. The viability of splenic T cells was evaluated by CCK‐8 assay. (E) Measurement of IFN‐γ in supernatants from the co‐culture of splenic T cells with DC2.4A‐P versus DC2.4VEC. (F‐G) Expression of A‐Pas chiRNA was evaluated by RT‐PCR (F) and immunoblotting (G) in AKRA‐P and AKRVEC cells. (H) Monitoring of cytotoxicity mediated by DC2.4A‐P‐activated T cells versus DC2.4VEC‐activated T cells against AKRVEC cells (upper panel) or AKRA‐P cells (lower panel) at an E:T ratio of 40:1. 1% Triton X‐100 was added instead of T cells as positive control. Samples shown are representative of three independent experiments. Error bars indicate SEM. **p < 0.01 by one‐way ANOVA with post hoc intergroup comparisons in (D‐E)
3.4. Characterization of DCA‐P‐derived EVs (DEXA‐P)
DC‐derived EVs (DEX) are secreted nanometre‐sized membrane vesicles that maintain various key immunostimulatory characteristics of DCs, including ability to present antigens to T cells (Pitt et al., 2016). A characteristic saucer‐cup shape for DEXA‐P was visualized by transmission electron microscope (TEM) (Figure 4A). An average diameter of 103 nm of DEXA‐P was determined by nanoparticle tracking analysis (Figure 4B).
FIGURE 4.
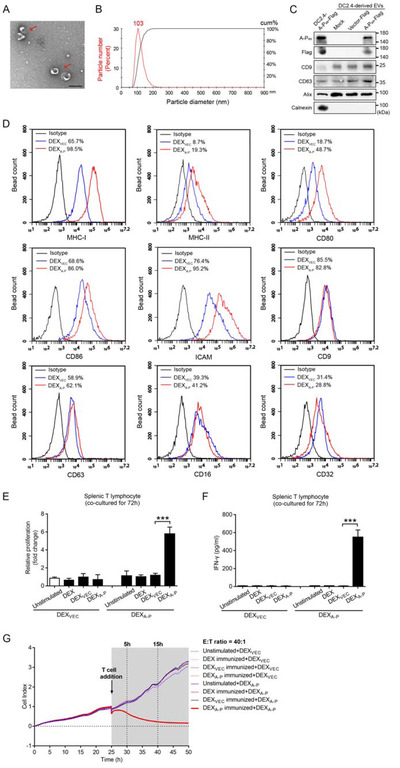
Characterization of DC2.4A‐P‐derived EVs (DEXA‐P) and evaluation of their capacity to induce antigen‐specific immune responses in vitro. (A) Transmission electron microscopy (TEM) of DEXA‐P. Scale bar is 200 nm. (B) NanoSight analysis of DEXA‐P concentration and size distribution. (C) Immunoblotting confirming the presence of fusion protein A‐Pas and EV markers CD9, CD63, Alix and the absence of endoplasmic reticulum protein calnexin in DEX. The A‐Pas fusion protein was detected using mAb A‐Pas‐16 and an anti‐FLAG mAb. (D) Flow cytometric analysis of cell surface protein expression on DEXA‐P versus DEXVEC. (E) Splenic T cells harvested from C57Bl/6 mice vaccinated with DEXA‐P, DEXVEC, DEX or PBS were re‐stimulated with DEXA‐P (40 μg) or DEXVEC (40 μg) for 72 h. The viability of splenic T cells was evaluated by CCK‐8 assay. (F) IFN‐γ production by splenic T cells from DEXA‐P‐, DEXVEC‐, DEX‐ and PBS‐vaccinated mice after re‐stimulation with DEXA‐P versus DEXVEC. (G) Monitoring of cytotoxicity mediated by splenic T cells harvested from DEXA‐P‐, DEXVEC‐, DEX‐ and PBS‐vaccinated mice after re‐stimulation with DEXA‐P or DEXVEC against AKRA‐P cancer cells at an E:T ratio of 40:1. Samples shown are representative of three independent experiments. Error bars indicate SEM. ***p < 0.001 by one‐way ANOVA with post hoc intergroup comparisons in (E‐F)
Next, we investigated whether DEXA‐P derived from DC2.4A‐P contained the A‐Pas neoantigen and displayed enhanced levels of immunostimulatory molecules. Immunoblot analysis demonstrated that purified DEXA‐P was enriched in EV markers CD9, CD63 and Alix as wells as the A‐Pas fusion protein, whereas the cellular endoplasmic reticulum protein calnexin was absent (Figure 4C). Importantly, among the surface proteins that were evaluated, CD80, CD86, ICAM, MHC‐I and MHC‐II were elevated on DEXA‐P, as compared to those on EVs derived from DC2.4VEC (DEXVEC) (Figure 4D). This indicates that the elevated presence of immunostimulatory molecules induced by ectopic expression of A‐Pas in DC2.4A‐P cells is maintained on DEXA‐P.
3.5. DEXA‐P induces A‐Pas‐specific anticancer T‐cells
Next, we assessed in vivo the capacity of DEXA‐P to prime and activate A‐Pas‐specific T‐cells with cytolytic activity towards A‐Pas‐positive EC. To this end, four groups of C57BL/6 mice were vaccinated with DEXA‐P, DEXVEC, DEX or PBS, respectively for 3 weeks. After the final vaccination, splenic T cells were harvested and re‐stimulated with DEXA‐P or DEXVEC for 72 h in vitro, after which proliferation and secretion of IFN‐γ was evaluated. In vitro re‐stimulation of T cells from DEXA‐P‐vaccinated mice with DEXA‐P potently triggered T cell proliferation and IFN‐γ secretion (Figure 4E and F). Importantly, only T cells from DEXA‐P‐vaccinated mice re‐stimulated with DEXA‐P showed potent A‐Pas‐selective cytolytic activity towards AKRA‐P cells. (Figure 4G).
Of note, EVs from DC2.4 cells pulsed with FLAG tag‐associated peptides failed to induce in vitro T‐cell proliferation, IFN‐γ secretion and A‐Pas‐specific cytolytic activity (Figure S5).
Taken together, these results indicated that DEXA‐P may be used as cell‐free vaccine to induce immunity towards A‐Pas‐expressing cancer cells.
3.6. DEXA‐P vaccination prolongs survival of EC‐bearing mice
The capacity of DEXA‐P vaccination to induce rejection of A‐Pas‐expressing tumours was investigated in vivo using a syngeneic immunocompetent mouse tumour model. To this end, C57BL/6 mice bearing 7‐day‐established AKRA‐P tumours were i.v. injected (3 times, once every 5 days) with DEXA‐P, DEXVEC or PBS (Figure 5A). Already after two vaccination rounds with DEXA‐P the tumour volumes were remarkably reduced (Figure 5B). Moreover, a significantly decrease in tumour size and weight was detected in mice that were sacrificed 3 days after the third and final vaccination with DEXA‐P (Figure 5C). The log‐rank test indicated that DEXA‐P vaccination prolonged the survival rate of tumour‐bearing mice (Figure 5D). In addition, there were no differences in terms of tumour growth and survival rate between DEXVEC and DEXA‐P vaccinated AKRVEC tumour‐bearing mice, demonstrating that the antitumour effects of DEXA‐P vaccination are indeed specifically directed towards the A‐Pas antigen (Figure S6). Of note, DEXA‐P vaccination appeared not to induce overt adverse effects, as mice did not show loss of bodyweight (BW) or signs of autoimmunity in vital organs upon histological examination (Figure S7).
FIGURE 5.
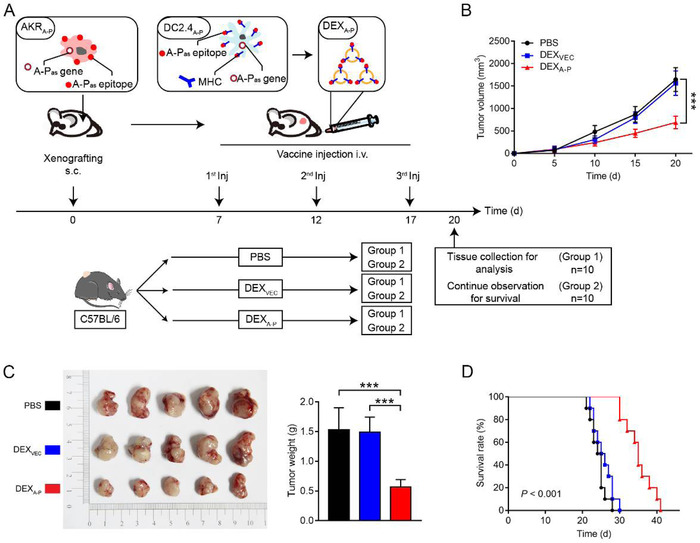
DEXA‐P vaccination results in growth inhibition of syngeneic esophageal carcinoma t u m o u r s in immunocompetent C57BL/6 mice. (A) DEXA‐P were i.v. injected in AKRA‐P tumour‐bearing mice. Treatment scheme indicates the timing of tumour inoculation and repeated vaccination of C57BL/6 mice (n = 10 animals per group). (B) Tumour growth curves after vaccination with DEXA‐P, DEXVEC or PBS. (C) Representative images (left panel) and average weights (right panel) of tumours harvested on 20 d after AKRA‐P cancer cell inoculation. (D) Kaplan‐Meier survival curves of tumour‐bearing mice vaccinated with DEXA‐P, DEXVEC or PBS. Error bars indicate SEM. ***p < 0.001 by one‐way ANOVA with post hoc intergroup comparisons in (B‐C). Log‐rank test was used in (D)
3.7. DEXA‐P vaccination modulates the tumour immune microenvironment as well as systemic immune responses
Compared to controls, mice vaccinated with DEXA‐P showed increased IFN‐γ and IL‐2 and reduced TGF‐β and IL‐10 serum levels, respectively (Figure 6A and B). This suggests that DEXA‐P vaccination appears to skew the tumour immune milieu from an inhibitory ‘cold’ to immune‐stimulatory ‘hot’ one.
FIGURE 6.
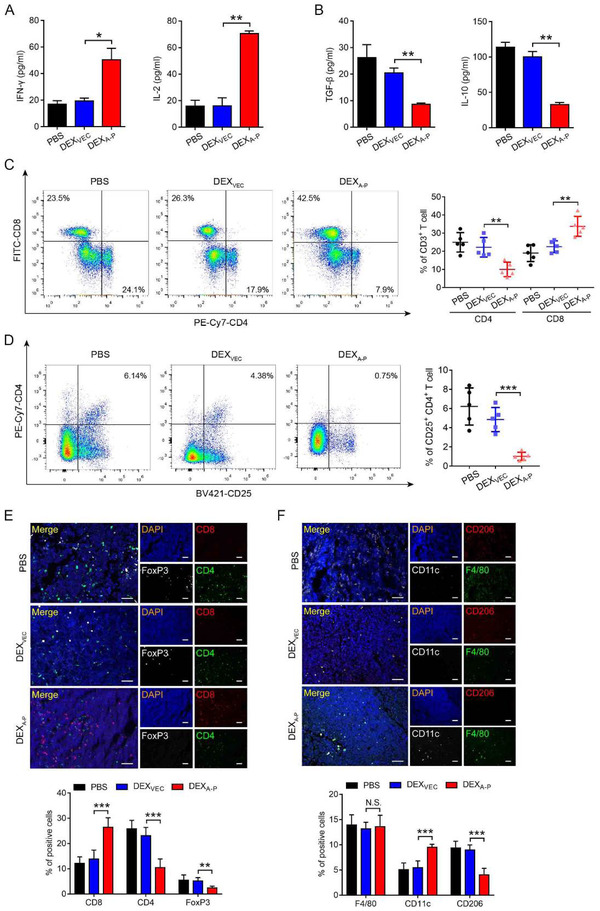
DEXA‐P vaccination modulates the t u m o ur immune microenvironment in t u m o u r ‐bearing mice. (A‐B) Measurement of IFN‐γ and IL‐2 (A), TGF‐β and IL‐10 (B) in serum from DEXA‐P‐, DEXVEC‐, and PBS‐vaccinated mice 20 d after AKRA‐P tumour cell inoculation. (C‐D) Flow cytometry analysis of CD8+CD3+, CD4+CD3+ T cells (C) and CD25+CD4+ T cells (D) present in vaccinated mice 20 d after AKRA‐P cancer cell inoculation (left panels). Quantification results are plotted in right panels. (E‐F) Representative images of multiplexed ImmunoFluorescence (mIF) staining for CD8, CD4, and FoxP3 (E, upper panel) or F4/80, CD11c, and CD206 (F, upper panel) in tumour sections derived from vaccinated mice 20 d after AKRA‐P cancer cell inoculation. Quantification results are plotted in lower panels. Scale bar is 50 μm. (n = 5 animals per group). Error bars indicate SEM. N.S. not significant, *p < 0.05, **p < 0.01, **p < 0.001 by one‐way ANOVA with post hoc intergroup comparisons in (A‐ F)
Furthermore, an enhanced CD8+ to CD4+ T cell ratio was detected in tumour tissues from DEXA‐P‐treated mice compared to controls (Figure 6C, and Figure S8A). Importantly, immunofluorescence showed that, compared to controls, DEXA‐P vaccination resulted in a decreased presence of tumour‐infiltrating CD25+/CD4+ Tregs and an increased presence of CD8+ T cells at the tumour site (Figure 6D and E). Additionally, compared to controls, DEXA‐P vaccination resulted in an increased presence of CD11c+ cells (M1 macrophages) and a decreased presence of CD206+ cells (M2 macrophages) in tumour tissues (Figure 6F). The latter suggests that the anticancer activity of DEXA‐P vaccination promotes innate immunity. Taken together, vaccination with DEXA‐P appears to stimulate both innate and acquired immunity which may reprogram the tumour immune microenvironment to an activated status.
To further evaluate the effect of DEXA‐P vaccination on systemic immune responses, the spleens of immunocompetent mice were analysed. DEXA‐P vaccination resulted in an increase in CD8+ ‐ and a decrease CD4+ T cell number in the spleen (Figure 7A). In particular, the number of IFN‐γ‐expressing CD8+ CTLs was significantly increased (Figure 7B, and Figure S8B) which was accompanied by decreased number of CD4+FoxP3+CD25+ Tregs (Figure 7C). In line with this, DEXA‐P vaccination enhanced the production of IFN‐γ and IL‐2 (Figure 7D) and reduced production of TGF‐β and IL‐10 by splenic T cells (Figure 7E). This suggests that DEXA‐P vaccination also systemically modulates immune responses in immunocompetent mice.
FIGURE 7.
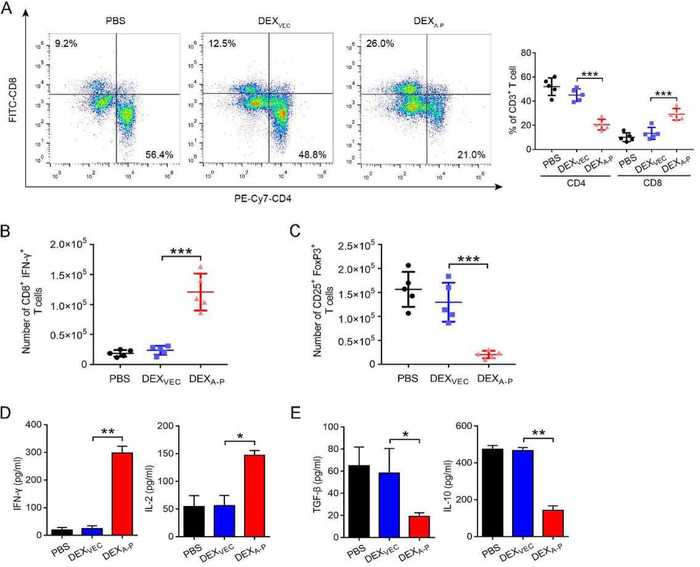
DEXA‐P vaccination promotes systemic immune responses in the spleen. (A) Flow cytometric analysis of CD8+CD3+, CD4+CD3+ T cells from the spleens of DEXA‐P‐, DEXVEC‐, and PBS‐vaccinated mice 20 d after AKRA‐P cancer cell inoculation (left panel). Quantification results are plotted in the right panel. (B‐C) Flow cytometric analysis of CD8+IFN‐γ+ (B) and CD25+FoxP3+ (C) T cells derived from spleens of vaccinated mice 20 d after AKRA‐P cancer cell inoculation. (D‐E) Measurement of IFN‐γ and IL‐2 (D), TGF‐β and IL‐10 (E) released from splenic T cells harvested from DEXA‐P‐vaccinated mice 20 d after AKRA‐P cancer cell inoculation after re‐stimulation as indicated with PBS, DEXVEC, or DEXA‐P (n = 5 animals per group). Error bars indicate SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by one‐way ANOVA with post hoc intergroup comparisons in (A‐E)
3.8. Antitumour effect of DEXA‐P vaccination is CD8+ T cell‐dependent
We then explicated whether T cells mediated antitumour effect of DEXA‐P in vivo. DEXA‐P vaccination delayed tumour growth in tumour‐bearing immunocompetent mice, but not in tumour‐bearing immunocompromised nude mice (Figure 8A to D) (Van Montfoort et al., 2018). This indicated that the antitumour effect of DEXA‐P vaccination critically relies on the induction and activation of anticancer T cells. To further examine which subset of T cells is responsible for the anti‐tumour immunity of DEXA‐P, we treated tumour‐bearing immunocompetent mice with DEXA‐P in the presence of CD8‐ and/or CD4‐depleting antibodies (Figure 8E, and Figure S9). Depletion of CD4+ T cells alone had only a marginal effect on therapeutic efficacy of DEXA‐P vaccination to control tumour growth (Figure 8F and G). In contrast, depletion of CD8+ T cells, alone or combined with depletion of CD4+ T cells, resulted in a marked reduction of the efficacy of DEXA‐P vaccination to control tumour growth (Figure 8F and G) and abrogated prolonged overall survival of DEXA‐P‐vaccinated mice (Figure 8H). These results indicated that antitumour effect DEXA‐P vaccination is critically dependent on CD8+ T cells.
FIGURE 8.
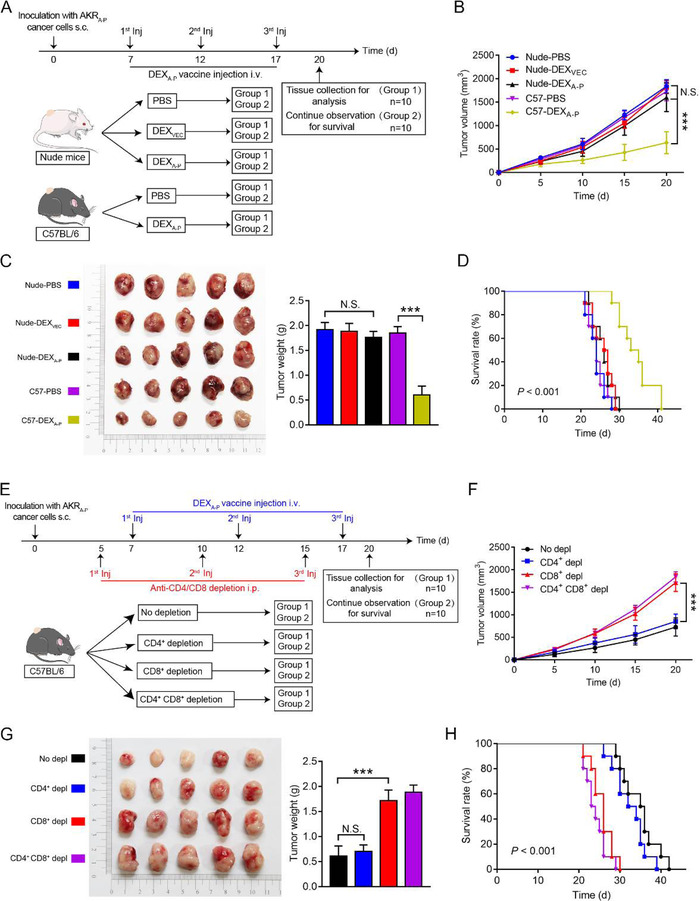
In vivo assessment of antitumour activity of DEXA‐P vaccination. (A) On day 0, nude athymic mice (strain NU/NU) or immunocompetent C57BL/6 mice (n = 10 animals per group) were s.c. inoculated with AKRA‐P cancer cells and then vaccinated with 3 consecutive injections (day 7, 12, and 17) with DEXA‐P, DEXVEC or PBS, respectively. (B) Tumour growth curves in tumour‐bearing nude mice vaccinated with DEXA‐P, DEXVEC or PBS, respectively and C57BL/6 mice vaccinated with DEXA‐P or PBS, respectively. (C) Representative images (left panel) and average weights (right panel) of tumours harvested on 20 d after AKRA‐P cancer cell inoculation. (D) Kaplan‐Meier survival curves of tumour‐bearing nude mice vaccinated with DEXA‐P, DEXVEC or PBS, respectively and C57BL/6 mice vaccinated with DEXA‐P or PBS, respectively. (E) At day 0, C57BL/6 mice (n = 10 animals per group) were s.c. inoculated with AKRA‐P cells and then injected at day 5, 10, and 15 with a CD8‐depleting and/or CD4‐depleting Abs vaccinated with 3 consecutive injections at day 7, 12, and 17 of DEXA‐P (F) Tumour growth curves of DEXA‐P‐ vaccinated tumour‐bearing mice after CD8+‐ and/or CD4+ T cells depletion (Depl). (G) Representative images (left panel) and average weights (right panel) of tumours harvested on 20 d after AKRA‐P cancer cell inoculation. (H) Kaplan‐Meier survival curves of DEXA‐P vaccinated tumour‐bearing mice after CD8+‐ and/or CD4+ T cells Depl. Error bars indicate SEM. N.S. not significant, ***p < 0.001 by one‐way ANOVA with post hoc intergroup comparisons in (B‐C, E‐F). Log‐rank test was used in (D and H)
4. DISCUSSION
EV‐based therapies are increasingly being explored in various diseases, such as bronchopulmonary dysplasia, neonatal lung injury, osteoarthritis and liver necrosis (Lee et al., 2021; Willis et al., 2018; Willis et al., 2021; Yin et al., 2022). Previously, several DC‐derived EVs (DEXs) approaches have been evaluated in mouse tumour models for melanoma, non‐small cell lung cancer (NSCLC), breast cancer, and liver cancer (André et al., 2004; Chaput et al., 2004; Lu et al., 2017; Nikfarjam et al., 2020; Pitt et al., 2016; Taieb et al., 2006; Viaud et al., 2011; Viaud et al., 2010; Zitvogel et al., 1998). Moreover, DEXs were shown to induce promising immunogenic responses in several phase I/II clinical trials in patients with NSCLC (Besse et al., 2016; Morse et al., 2005) and in patients with advanced melanoma (Escudier et al., 2005; Viaud et al., 2009). Collectively, these clinical trials have highlighted feasibility and safety of the use of DEXs in a clinical setting. Of note, most reports on treating cancer with DEXs focus on DEX vaccines pulsed with cancer testis antigens, such as MAGE‐1, MAGE‐3, NY‐ESO‐1 (Besse et al., 2016; Morse et al., 2005), or onco‐fetal antigens like alpha‐fetoprotein (AFP) (Lu et al., 2017). In the current study, we describe the construction of the cell‐free vaccine DEX‐A‐P that is based on A‐PaschiRNA‐loaded DEXs, and its preclinically evaluation for applicability to treat esophageal cancer (EC), a highly refractory and lethal malignancy. Our results demonstrated promising therapeutic effects of DEX‐A‐Ptowards EC, both in vitro and in immunocompetent mouse tumour model. Intriguingly, DEX‐A‐Pdisplayed high levels of MHC‐I, MHC‐II, ICAM, CD80, and CD86, indicating that A‐PaschiRNA transduction promotes antigen cross‐presentation. Moreover, vaccination of immunocompetent mice bearing syngeneic A‐PaschiRNA‐expressing esophageal cancer with DEX‐A‐Ppotently inhibited tumour growth and prolonged survival.
Somatic mutations are important sources of cancer‐specific neoepitopes to generate cancer‐selective T‐cells (Ott et al., 2017; Sahin & Türeci, 2018; Sahin et al., 2017). However, mutation‐based immunogenic neoantigens are rare or even non‐existent in subgroups of malignancies (Chan et al., 2019; Martin et al., 2016; Schumacher et al., 2019; Wang et al., 2021). Recent studies revealed that somatic mutations in cancer‐related genes commonly occur in many normal tissues and are caused mainly by intrinsic mutational processes. Interestingly, phenotypically normal cells carrying somatic mutations in cancer‐related genes appear to colonize the normal human esophagus with age. In fact, in middle‐aged and elderly subjects, clones with cancer‐associated mutations covered much of the epithelium. Counterintuitively, the prevalence of NOTCH1 mutations in phenotypically normal esophagus was several times higher than that in esophageal cancers (Chanock, 2018; Dart, 2018; Martincorena et al., 2018; Tomasetti, 2019; Yizhak et al., 2019; Yokoyama et al., 2019). Consequently, conventional somatic mutations in cancer‐related genes may not yield discriminatory esophagus cancer‐selective neoantigens suitable for cancer immunotherapy. In contrast, our study indicates that the esophageal cancer‐specific transcription‐induced chiRNAs (i.e., encoded proteins) may serve as an alternate and potentially more suitable source of immunogenic neoantigens for the development of anticancer vaccines. In particular, cancer‐specific transcription‐induced chiRNAs may be particularly useful for cancer types known to have low mutation burden and in which mutation‐based immunogenic neoantigens are rare or even non‐existent (Chan et al., 2019; Martin et al., 2016; Schumacher et al., 2019; Wang et al., 2021).
Previously, we identified that EC tissue shows aberrant mRNA transcription resulting in a chimeric RNA in which the coding sequence with the full‐length coding sequence of the ASTN2 gene is fused in‐frame to the antisense strand of part of 18th intron of the PAPPA gene (A‐Pas chiRNA) (Zhang et al., 2013). We demonstrate here that A‐Pas chiRNA is selectively transcribed and translated in patient‐derived EC tissue but not in adjacent non‐cancerous tissue. In contrast to most reports that aberrant transcription‐induced chimeric RNAs derived from structural variations of exons only (Kannan et al., 2011; Kovar et al., 1999), we identified A‐Pas chiRNA derived from the coding regions of ASTN2 fused to the antisense strand of an intron of PAPPA, two adjacent genes in a tail‐tail orientation. ASTN2 is an integral membrane protein, with two transmembrane (TM) domains projecting a large C‐terminal domain into endosomal vesicle lumen, while exposing cytosolic domains on the other side of membrane (Wilson et al., 2010). Normally, ASTN2 is expressed in the brain where it is involved in neuronal migration. PAPPA is a secreted metalloproteinase that plays a role in bone formation, inflammation, wound healing, and female fertility (Conover, 2012). The encoded A‐Pas fusion protein has the N‐terminal portion of the ASTN2 protein, and the subsequent 81 new amino acids at the C‐terminal portion are in a novel sequence encoded by the antisense strand of part of 18th intron of the PAPPA gene. Importantly, immunoblot analysis using A‐Pas‐specific mAb confirmed the presence of endogenous A‐Pas fusion protein in EC tissues.
Intriguingly, LVA‐P‐FLAG transduction of DCs (DCA‐P) appeared to promote DC maturation status. We have no explanation for this unique activity of the A‐Pas fusion protein, but it is tentative to speculate that this somehow related to ASTN2‐mediated activities. Next, we investigated whether the A‐Pas fusion protein can act as an immunogenic neoantigen. Indeed, DCA‐P show enhanced capacity to prime A‐Pas‐specific anticancer T cells in vitro. Analogously, LVA‐P‐FLAG‐transduced mouse DC2.4A‐P cells showed capacity to prime A‐Pas‐specific mouse T cells in vitro.
In our study, DEXA‐P vaccination increased serum levels of IFN‐γ and IL‐2, indicating the induction of Th1‐type immune response in vivo. A recent study, using a cancer vaccine that consisted of irradiated murine cancer cells engineered to co‐express an anti‐PD‐1 mAb and GM‐CSF (Tian et al., 2016), reported a similar increase of IFN‐γ and IL‐2. This Th1‐type immune response was reported to be predominantly mediated by CD4+ T cells (Tian et al., 2016). In contrast, our study indicated that depletion of CD4+ T cells only marginally reduced the capacity of DEXA‐P vaccination to control tumour growth. Therefore, we assume that in our mouse tumour model the increase of IFN‐γ and IL‐2 serum levels after DEXA‐P vaccination is most likely mediated by CD8+ T cells.
Of note, DEXA‐P displayed high levels of MHC‐I, MHC‐II, ICAM, CD80, and CD86 indicating that A‐Pas chiRNA transduction of DCs promotes antigen cross‐presentation capacity which endows them with enhanced capacity to prime A‐Pas‐specific anticancer T cell responses. Importantly, vaccination of immunocompetent mice bearing syngeneic A‐Pas chiRNA‐expressing esophageal tumours with DEXA‐P potently inhibited tumour growth and prolonged survival. Animals vaccinated with 40 μg DEXA‐P showed a prolonged survival rate at 30 days after tumour challenge of up to 100%. Intriguingly, DEXA‐P treatment appeared to activate CD8+ T cells and simultaneously suppress Tregs. Furthermore, DEXA‐P vaccination was accompanied by a marked increase in number of M1 (CD11c+) macrophages and a decrease in number of M2 (CD206+) macrophages, indicating that vaccination of DEXA‐P appeared to remodel the immune milieu of the tumour microenvironment, that is, turning a ‘cold’ tumour into a ‘hot’ one (Galon & Bruni, 2019).
Importantly, we detected no overt signs of immune‐related adverse events (irAEs) in mice treated with the DEXA‐P vaccination protocol, suggesting that this approach induces no or only minimal systemic toxicity (Postow et al., 2018). Complete tumour eradication upon DEXA‐P vaccination was not observed in the present study. This is in line with most clinical studies in which anticancer vaccines were used as a monotherapeutic agent (Levy et al., 2014; Schadendorf et al., 2006; Vansteenkiste et al., 2016). This is probably attributable to one or more immunosuppressive checkpoints, such as CTLA4, PD‐1/PD‐L1, and CD47 that are known to blunt the anticancer activity of vaccine‐induced CD8+ T cells and M1 macrophages (Soares et al., 2015). Therefore, we speculate that a combination of DEX‐based vaccination and appropriate checkpoint inhibition may be particularly advantageous to enhance clinical efficacy of difficult‐to‐treat ‘cold’ solid cancers like EC (Munn & Bronte, 2016; Soares et al., 2015).
Our in vitro results indicate that DEXA‐P activates T cells to mount A‐Pas‐directed immune responses. It is likely that the recognized immunogenic TCR epitopes reside in the 81 aa stretch derived from the antisense strand of part of 18th intron of the PAPPA gene or proximal to the junction sequence at the C‐terminus of the ASTN2 domain of the A‐Pas fusion protein. We are currently in the process of designing and producing appropriate tetramers to further analyse A‐Pas‐responsive T cell populations and characterize their TCRs.
Taken together, we developed a novel EV‐based anticancer vaccine. Our work demonstrates for the first time the principle that transcription‐induced chiRNAs have potential to serve as a source of cancer‐specific mutation‐independent neoantigens for inducing therapeutic T cell‐mediated anticancer immunity.
AUTHOR CONTRIBUTIONS
Research design: Hao Zhang; DEXA‐P vaccine production: Xiao Xiong, Xiurong Ke, Yusheng Lin and Lu Wang; Mouse model constructions and mouse experiments: Xiao Xiong, Zhimeng Yao and Kai Li; Data analyses and interpretation: Hao Zhang, Xiurong Ke, Xiao Xiong, Yusheng Lin, Fan Liu, Yunlong Pan, Sai‐Ching J. Yeung and Wijnand Helfrich; Project supervision: Hao Zhang; Manuscript writing and reviewing: Hao Zhang, Xiurong Ke, Wijnand Helfrich and Sai‐Ching J. Yeung.
CONFLICT OF INTERESTS
Authors declare that they have no competing interests.
Supporting information
Supporting information.
ACKNOWLEDGEMENTS
Authors thank Professor Guobin Chen for the discussion and comment. We acknowledge members of H. Zhang's laboratory for the technical help and discussion. The work was supported by a grant in part by the National Natural Science Foundation of China (82072683, 81773087, 81071736, 81572876 and 30973508 to H.Z.); the Natural Science Foundation of Guangdong Province of China (2022A1515010925, 2021A1515012522 and 9151018004000000 to H.Z.); the Science and Technology Planning Project of Guangdong Province of China (2019A030317024 to H.Z.); Special Project on the Integration of Industry, Education and Research of Guangdong Province (2011A090100024 to H.Z.); Flagship specialty construction project General surgery (711003 to H.Z. and Y.P.). Abel Tasman Talent Program (ATTP) Sandwich PhD Scholarship supported by the Graduate School of Medical Sciences (GSMS) of the University of Groningen (to X. K).
Xiong, X. , Ke, X. , Wang, L. , Lin, Y. , Wang, S. , Yao, Z. , Li, K. , Luo, Y. , Liu, F. , Pan, Y. , Yeung, S. ‐ C. J. , Helfrich, W. , & Zhang, H. (2022). Neoantigen‐based cancer vaccination using chimeric RNA‐loaded dendritic cell‐derived extracellular vesicles. Journal of Extracellular Vesicles, 11, e12243. 10.1002/jev2.12243
Xiao Xiong and Xiurong Ke contributed equally to this work.
One Sentence Summary: Development and preclinical evaluation of an extracellular vesicles‐based therapeutic cancer vaccine exploiting an immunogenic neoantigen encoded by an aberrant transcription‐induced exon‐intronantisense chimeric RNA.
REFERENCES
- André, F. , Chaput, N. , Schartz, N. E. C. , Flament, C. , Aubert, N. , Bernard, J. , Lemonnier, F. , Raposo, G. , Escudier, B. , Hsu, D.‐H. , Tursz, T. , Amigorena, S. , Angevin, E. , & Zitvogel, L. (2004). Exosomes as potent cell‐free peptide‐based vaccine. I. Dendritic cell‐derived exosomes transfer functional MHC class I/peptide complexes to dendritic cells. Journal of Immunology, 172(4), 2126–2136. [DOI] [PubMed] [Google Scholar]
- Banchereau, J. , & Palucka, K. (2018). Immunotherapy: Cancer vaccines on the move. Nature Reviews Clinical Oncology, 15(1), 9–10. [DOI] [PubMed] [Google Scholar]
- Besse, B. , Charrier, M. , Lapierre, V. , Dansin, E. , Lantz, O. , Planchard, D. , Le Chevalier, T. , Livartoski, A. , Barlesi, F. , Laplanche, A. , Ploix, S. , Vimond, N. , Peguillet, I. , Théry, C. , Lacroix, L. , Zoernig, I. , Dhodapkar, K. , Dhodapkar, M. , Viaud, S. , …, Chaput, N. (2016). Dendritic cell‐derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology, 5(4), e1071008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaca, M. P. , Scirica, B. M. , Sabatine, M. S. , Jarolim, P. , Murphy, S. A. , Chamberlin, J. S. , Rhodes, D. W. , Southwick, P. C. , Braunwald, E. , & Morrow, D. A. (2012). Prospective evaluation of pregnancy‐associated plasma protein‐a and outcomes in patients with acute coronary syndromes. Journal of the American College of Cardiology, 60(4), 332–338. [DOI] [PubMed] [Google Scholar]
- Chan, T. A. , Yarchoan, M. , Jaffee, E. , Swanton, C. , Quezada, S. A. , Stenzinger, A. , & Peters, S. (2019). Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Annals of Oncology, 30(1), 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanock, S. J. (2018). The paradox of mutations and cancer. Science, 362(6417), 893–894. [DOI] [PubMed] [Google Scholar]
- Chaput, N. , Schartz, N. E. C. , André, F. , Taïeb, J. , Novault, S. , Bonnaventure, P. , Aubert, N. , Bernard, J. , Lemonnier, F. , Merad, M. , Adema, G. , Adams, M. , Ferrantini, M. , Carpentier, A. F. , Escudier, B. , Tursz, T. , Angevin, E. , & Zitvogel, L. (2004). Exosomes as potent cell‐free peptide‐based vaccine. II. Exosomes in CpG adjuvants efficiently prime naive Tc1 lymphocytes leading to tumor rejection. Journal of Immunology, 172(4), 2137–2146. [DOI] [PubMed] [Google Scholar]
- Conover, C. A. (2012). Key questions and answers about pregnancy‐associated plasma protein‐A. Trends in Endocrinology and Metabolism, 23(5), 242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover, C. A. , Bale, L. K. , & Oxvig, C. (2016). Targeted inhibition of pregnancy‐associated plasma protein‐A activity reduces atherosclerotic plaque burden in mice. Journal of Cardiovascular Translational Research, 9(1), 77–79. [DOI] [PubMed] [Google Scholar]
- Dart, A. (2018). Colony takeover. Nature Reviews Cancer, 18(12), 725. [DOI] [PubMed] [Google Scholar]
- Dong, H. , Xu, J. , Li, W. , Gan, J. , Lin, W. , Ke, J. , Jiang, J. , Du, L. , Chen, Y. , Zhong, X. , Zhang, D. , Yeung, S. ‐ C. J. , Li, X. , & Zhang, H. (2017). Reciprocal androgen receptor/interleukin‐6 crosstalk drives oesophageal carcinoma progression and contributes to patient prognosis. Journal of Pathology, 241(4), 448–462. [DOI] [PubMed] [Google Scholar]
- El Andaloussi, S. , Lakhal, S. , Mager, I. , & Wood, M. J. (2013). Exosomes for targeted siRNA delivery across biological barriers. Advanced Drug Delivery Reviews, 65(3), 391–397. [DOI] [PubMed] [Google Scholar]
- Escudier, B. , Dorval, T. , Chaput, N. , André, F. , Caby, M.‐P. , Novault, S. , Flament, C. , Leboulaire, C. , Borg, C. , Amigorena, S. , Boccaccio, C. , Bonnerot, C. , Dhellin, O. , Movassagh, M. , Piperno, S. , Robert, C. , Serra, V. , Valente, N. , Le Pecq, J.‐B. , …, Zitvogel, L. (2005). Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived‐exosomes: Results of thefirst phase I clinical trial. Journal of Translational Medicine, 3(1), 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Y. , Ke, C. , Tang, Q. , Dong, H. , Zheng, X. , Lin, W. , Ke, J. , Huang, J. , Yeung, S.‐C. , & Zhang, H. (2014). Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Death & Disease, 5, e1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankiw, L. , Baltimore, D. , & Li, G. (2019). Alternative mRNA splicing in cancer immunotherapy. Nature Reviews Immunology, 19(11), 675–687. [DOI] [PubMed] [Google Scholar]
- Galon, J. , & Bruni, D. (2019). Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nature Reviews Drug Discovery, 18(3), 197–218. [DOI] [PubMed] [Google Scholar]
- Gan, J. , Ke, X. , Jiang, J. , Dong, H. , Yao, Z. , Lin, Y. , Lin, W. , Wu, X. , Yan, S. , Zhuang, Y. , Chu, W. K. , Cai, R. , Zhang, X. , Cheung, H. S. , Block, N. L. , Pang, C. P. , Schally, A. V. , & Zhang, H. (2016). Growth hormone‐releasing hormone receptor antagonists inhibit human gastric cancer through downregulation of PAK1‐STAT3/NF‐kappa B signaling. Proceedings of the National Academy of Sciences of the United States of America, 113(51), 14745–14750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg, A. D. , Coulie, P. G. , Van Den Eynde, B. J. , & Agostinis, P. (2017). Integrating next‐generation dendritic cell vaccines into the current cancer immunotherapy landscape. Trends in Immunology, 38(8), 577–593. [DOI] [PubMed] [Google Scholar]
- Glessner, J. T. , Wang, K. , Cai, G. , Korvatska, O. , Kim, C. E. , Wood, S. , Zhang, H. , Estes, A. , Brune, C. W. , Bradfield, J. P. , Imielinski, M. , Frackelton, E. C. , Reichert, J. , Crawford, E. L. , Munson, J. , Sleiman, P. M. A. , Chiavacci, R. , Annaiah, K. , Thomas, K. , …, Hakonarson, H. (2009). Autism genome‐wide copy number variation reveals ubiquitin and neuronal genes. Nature, 459(7246), 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, X.‐C. , & Gao, J.‐Q. (2017). Exosomes as novel bio‐carriers for gene and drug delivery. International Journal of Pharmaceutics, 521(1‐2), 167–175. [DOI] [PubMed] [Google Scholar]
- Kannan, K. , Wang, L. , Wang, J. , Ittmann, M. M. , Li, W. , & Yen, L. (2011). Recurrent chimeric RNAs enriched in human prostate cancer identified by deep sequencing. Proceedings of the National Academy of Sciences of the United States of America, 108(22), 9172–9177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke, X. , Xiong, X. , Lin, Y. , & Zhang, H. (2020). Chimeric RNA and exosomes‐based liquid biopsy. Methods in Molecular Biology, 2079, 211–218. [DOI] [PubMed] [Google Scholar]
- Kovar, H. , Jugovic, D. , Melot, T. , Zoubek, A. , Lenoir, G. , Fink, F.‐M. , Philip, I. , Turc‐Carel, C. , Thomas, G. , & Zucman‐Rossi, J. (1999). Cryptic exons as a source of increased diversity of Ewing tumor‐associated EWS‐FLI1 chimeric products. Genomics, 60(3), 371–374. [DOI] [PubMed] [Google Scholar]
- Lee, J. , Kim, S. R. , Lee, C. , Jun, Y. I. , Bae, S. , Yoon, Y. J. , Kim, O. Y. , & Gho, Y. S. (2021). Extracellular vesicles from in vivo liver tissue accelerate recovery of liver necrosis induced by carbon tetrachloride. Journal of Extracellular Vesicles, 10(10), e12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy, R. , Ganjoo, K. N. , Leonard, J. P. , Vose, J. M. , Flinn, I. W. , Ambinder, R. F. , Connors, J. M. , Berinstein, N. L. , Belch, A. R. , Bartlett, N. L. , Nichols, C. , Emmanouilides, C. E. , Timmerman, J. M. , Gregory, S. A. , Link, B. K. , Inwards, D. J. , Freedman, A. S. , Matous, J. V. , Robertson, M. J. , …, Denney, D. W. (2014). Active idiotypic vaccination versus control immunotherapy for follicular lymphoma. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 32(17), 1797–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. , Dong, H. , Deng, W. , Lin, W. , Li, K. , Xiong, X. , Guo, Y. , Zhou, F. , Ma, C. , Chen, Y. , Ren, H. , Yang, H. , Dai, N. , Ma, L. , Meltzer, S. J. , Yeung, S‐C. J. , & Zhang, H. (2019). Evaluation of salivary exosomal chimeric GOLM1‐NAA35 RNA as a potential biomarker in esophageal carcinoma. Clinical Cancer Research, 25(10), 3035–3045. [DOI] [PubMed] [Google Scholar]
- Lu, Z. , Zuo, B. , Jing, R. , Gao, X. , Rao, Q. , Liu, Z. , Qi, H. , Guo, H. , & Yin, H. (2017). Dendritic cell‐derived exosomes elicit tumor regression in autochthonous hepatocellular carcinoma mouse models. Journal of Hepatology, 67(4), 739–748. [DOI] [PubMed] [Google Scholar]
- Martin, S. D. , Brown, S. D. , Wick, D. A. , Nielsen, J. S. , Kroeger, D. R. , Twumasi‐Boateng, K. , Holt, R. A. , & Nelson, B. H. (2016). Low mutation burden in ovarian cancer may limit the utility of neoantigen‐targeted vaccines. Plos One, 11(5), e0155189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena, I. , Fowler, J. C. , Wabik, A. , Lawson, A. R. J. , Abascal, F. , Hall, M. W. J. , Cagan, A. , Murai, K. , Mahbubani, K. , Stratton, M. R. , Fitzgerald, R. C. , Handford, P. A. , Campbell, P. J. , Saeb‐Parsy, K. , & Jones, P. H. (2018). Somatic mutant clones colonize the human esophagus with age. Science, 362(6417), 911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse, M. A. , Garst, J. , Osada, T. , Khan, S. , Hobeika, A. , Clay, T. M. , Valente, N. , Shreeniwas, R. , Sutton, M. , Delcayre, A. , Hsu, D.‐H. , Le Pecq, J.‐B. , & Lyerly, H. K. (2005). A phase I study of dexosome immunotherapy in patients with advanced non‐small cell lung cancer. Journal of translational medicine, 3(1), 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn, D. H. , & Bronte, V. (2016). Immune suppressive mechanisms in the tumor microenvironment. Current Opinion in Immunology, 39, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikfarjam, S. , Rezaie, J. , Kashanchi, F. , & Jafari, R. (2020). Dexosomes as a cell‐free vaccine for cancer immunotherapy. Journal of Experimental & Clinical Cancer Research, 39(1), 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott, P. A. , Hu, Z. , Keskin, D. B. , Shukla, S. A. , Sun, J. , Bozym, D. J. , Zhang, W. , Luoma, A. , Giobbie‐Hurder, A. , Peter, L. , Chen, C. , Olive, O. , Carter, T. A. , Li, S. , Lieb, D. J. , Eisenhaure, T. , Gjini, E. , Stevens, J. , Lane, W. J. , …, Wu, C. J. (2017). An immunogenic personal neoantigen vaccine for patients with melanoma. Nature, 547(7662), 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt, J. M. , André, F. , Amigorena, S. , Soria, J.‐C. , Eggermont, A. , Kroemer, G. , & Zitvogel, L. (2016). Dendritic cell‐derived exosomes for cancer therapy. Journal of Clinical Investigation, 126(4), 1224–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow, M. A. , Sidlow, R. , & Hellmann, M. D. (2018). Immune‐related adverse events associated with immune checkpoint blockade. The New England Journal of Medicine, 378(2), 158–168. [DOI] [PubMed] [Google Scholar]
- Prechtel, A. T. , & Steinkasserer, A. (2007). CD83: An update on functions and prospects of the maturation marker of dendritic cells. Archives of Dermatological Research, 299(2), 59–69. [DOI] [PubMed] [Google Scholar]
- Sabado, R. L. , & Bhardwaj, N. (2015). Cancer immunotherapy: Dendritic‐cell vaccines on the move. Nature, 519(7543), 300–301. [DOI] [PubMed] [Google Scholar]
- Sahin, U. , Derhovanessian, E. , Miller, M. , Kloke, B.‐P. , Simon, P. , Löwer, M. , Bukur, V. , Tadmor, A. D. , Luxemburger, U. , Schrörs, B. , Omokoko, T. , Vormehr, M. , Albrecht, C. , Paruzynski, A. , Kuhn, A. N. , Buck, J. , Heesch, S. , Schreeb, K. H. , Müller, F. , …, Türeci, Ö. (2017). Personalized RNA mutanome vaccines mobilize poly‐specific therapeutic immunity against cancer. Nature, 547(7662), 222–226. [DOI] [PubMed] [Google Scholar]
- Sahin, U. , & Türeci, Ö. (2018). Personalized vaccines for cancer immunotherapy. Science, 359(6382), 1355–1360. [DOI] [PubMed] [Google Scholar]
- Schadendorf, D. , Ugurel, S. , Schuler‐Thurner, B. , Nestle, F. O. , Enk, A. , Bröcker, E.‐B. , Grabbe, S. , Rittgen, W. , Edler, L. , Sucker, A. , Zimpfer‐Rechner, C. , Berger, T. , Kamarashev, J. , Burg, G. , Jonuleit, H. , Tüttenberg, A. , Becker, J. C. , Keikavoussi, P. , Kämpgen, E. , & Schuler, G. (2006). Dacarbazine (DTIC) versus vaccination with autologous peptide‐pulsed dendritic cells (DC) in first‐line treatment of patients with metastatic melanoma: A randomized phase III trial of the DC study group of the DeCOG. Annals of Oncology : official Journal of the European Society for Medical Oncology, 17(4), 563–570. [DOI] [PubMed] [Google Scholar]
- Schumacher, T. N. , Scheper, W. , & Kvistborg, P. (2019). Cancer Neoantigens. Annual Review of Immunology, 37, 173–200. [DOI] [PubMed] [Google Scholar]
- Soares, K. C. , Rucki, A. A. , Wu, A. A. , Olino, K. , Xiao, Q. , Chai, Y. , Wamwea, A. , Bigelow, E. , Lutz, E. , Liu, L. , Yao, S. , Anders, R. A. , Laheru, D. , Wolfgang, C. L. , Edil, B. H. , Schulick, R. D. , Jaffee, E. M. , & Zheng, L. (2015). PD‐1/PD‐L1 blockade together with vaccine therapy facilitates effector T‐cell infiltration into pancreatic tumors. Journal of Immunotherapy, 38(1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taieb, J. , Chaput, N. , Schartz, N. , Roux, S. , Novault, S. , Ménard, C. , Ghiringhelli, F. , Terme, M. , Carpentier, A. F. , Darrasse‐Jèse, G. , Lemonnier, F. , & Zitvogel, L. (2006). Chemoimmunotherapy of tumors: cyclophosphamide synergizes with exosome based vaccines. Journal of Immunology, 176(5), 2722–2729. [DOI] [PubMed] [Google Scholar]
- Tian, H. , Shi, G. , Wang, Q. , Li, Y. , Yang, Q. , Li, C. , Yang, G. , Wu, M. , Xie, Q. , Zhang, S. , Yang, Y. , Xiang, R. , Yu, D. , Wei, Y. , & Deng, H. (2016). A novel cancer vaccine with the ability to simultaneously produce anti‐PD‐1 antibody and GM‐CSF in cancer cells and enhance Th1‐biased antitumor immunity. Signal Transduction and Targeted Therapy, 1, 16025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasetti, C. (2019). Mutated clones are the new normal. Science, 364(6444), 938–939. [DOI] [PubMed] [Google Scholar]
- Tran, T. , Blanc, C. , Granier, C. , Saldmann, A. , Tanchot, C. , & Tartour, E. (2019). Therapeutic cancer vaccine: Building the future from lessons of the past. Seminars in Immunopathology, 41(1), 69–85. [DOI] [PubMed] [Google Scholar]
- Van Montfoort, N. , Borst, L. , Korrer, M. J. , Sluijter, M. , Marijt, K. A. , Santegoets, S. J. , Van Ham, V. J. , Ehsan, I. , Charoentong, P. , André, P. , Wagtmann, N. , Welters, M. J. P. , Kim, Y. J. , Piersma, S. J. , Van Der Burg, S. H. , & Van Hall, T. (2018). NKG2A blockade potentiates CD8 T cell immunity induced by cancer vaccines. Cell, 175(7), 1744–1755.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vansteenkiste, J. F. , Cho, B. C. , Vanakesa, T. , De Pas, T. , Zielinski, M. , Kim, M. S. , Jassem, J. , Yoshimura, M. , Dahabreh, J. , Nakayama, H. , Havel, L. , Kondo, H. , Mitsudomi, T. , Zarogoulidis, K. , Gladkov, O. A. , Udud, K. , Tada, H. , Hoffman, H. , Bugge, A. , …, Altorki, N. (2016). Efficacy of the MAGE‐A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE‐A3‐positive non‐small‐cell lung cancer (MAGRIT): A randomised, double‐blind, placebo‐controlled, phase 3 trial. The Lancet Oncology, 17(6), 822–835. [DOI] [PubMed] [Google Scholar]
- Varley, K. E. , Gertz, J. , Roberts, B. S. , Davis, N. S. , Bowling, K. M. , Kirby, M. K. , Nesmith, A. S. , Oliver, P. G. , Grizzle, W. E. , Forero, A. , Buchsbaum, D. J. , Lobuglio, A. F. , & Myers, R. M. (2014). Recurrent read‐through fusion transcripts in breast cancer. Breast Cancer Research and Treatment, 146(2), 287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viaud, S. , Ploix, S. , Lapierre, V. , Théry, C. , Commere, P.‐H. , Tramalloni, D. , Gorrichon, K. , Virault‐Rocroy, P. , Tursz, T. , Lantz, O. , Zitvogel, L. , & Chaput, N. (2011). Updated technology to produce highly immunogenic dendritic cell‐derived exosomes of clinical grade: A critical role of interferon‐gamma. Journal of Immunotherapy, 34(1), 65–75. [DOI] [PubMed] [Google Scholar]
- Viaud, S. , Terme, M. , Flament, C. , Taieb, J. , André, F. , Novault, S. , Escudier, B. , Robert, C. , Caillat‐Zucman, S. , Tursz, T. , Zitvogel, L. , & Chaput, N. (2009). Dendritic cell‐derived exosomes promote natural killer cell activation and proliferation: A role for NKG2D ligands and IL‐15Ralpha. Plos One, 4(3), e4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viaud, S. , Théry, C. , Ploix, S. , Tursz, T. , Lapierre, V. , Lantz, O. , Zitvogel, L. , & Chaput, N. (2010). Dendritic cell‐derived exosomes for cancer immunotherapy: What's next? Cancer Research, 70(4), 1281–1285. [DOI] [PubMed] [Google Scholar]
- Wang, L. , Li, K. , Lin, X. , Yao, Z. , Wang, S. , Xiong, X. , Ning, Z. , Wang, J. , Xu, X. , Jiang, Y. , Liu, D. , Chen, Y. , Zhang, D. , & Zhang, H. (2019). Metformin induces human esophageal carcinoma cell pyroptosis by targeting the miR‐497/PELP1 axis. Cancer Letters, 450, 22–31. [DOI] [PubMed] [Google Scholar]
- Wang, L. , Li, W. , Li, K. , Guo, Y. , Liu, D. , Yao, Z. , Lin, X. , Li, S. , Jiang, Z. , Liu, Q. , Jiang, Y. , Zhang, B. , Chen, L. , Zhou, F. , Ren, H. , Lin, D. , Zhang, D. , Yeung, S. ‐ C. J. , & Zhang, H. (2018). The oncogenic roles of nuclear receptor coactivator 1 in human esophageal carcinoma. Cancer medicine, 7(10), 5205–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Xiong, X. , Yao, Z. , Zhu, J. , Lin, Y. , Lin, W. , Li, K. , Xu, X. , Guo, Y. , Chen, Y. , Pan, Y. , Zhou, F. , Fan, J. , Chen, Y. , Gao, S. , Jim Yeung, S.‐C. , & Zhang, H. (2021). Chimeric RNA ASTN2‐PAPPAas aggravates tumor progression and metastasis in human esophageal cancer. Cancer Letters, 501, 1–11. [DOI] [PubMed] [Google Scholar]
- Wang, P. , Chen, Y. , & Wang, C. (2021). Beyond tumor mutation burden: Tumor neoantigen burden as a biomarker for immunotherapy and other types of therapy. Frontiers in Oncology, 11, 672677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Lin, Y. , Xiong, X. , Wang, L. , Guo, Y. , & Chen, Y. , Chen, S. , Wang, G. , Lin, P. , Chen, H. , Yeung, S. J. , Bremer, E. , & Zhang, H. (2020). Low‐dose metformin reprograms the tumor immune microenvironment in human esophageal cancer: results of a phase II clinical trial. Clinical Cancer Research, 2020;26(18), 4921–32. [DOI] [PubMed] [Google Scholar]
- Willis, G. R. , Fernandez‐Gonzalez, A. , Anastas, J. , Vitali, S. H. , Liu, X. , Ericsson, M. , Kwong, A. , Mitsialis, S. A. , & Kourembanas, S. (2018). Mesenchymal stromal cell exosomes ameliorate experimental bronchopulmonary dysplasia and restore lung function through macrophage immunomodulation. American Journal of Respiratory and Critical Care Medicine, 197(1), 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis, G. R. , Reis, M. , Gheinani, A. H. , Fernandez‐Gonzalez, A. , Taglauer, E. S. , Yeung, V. , Liu, X. , Ericsson, M. , Haas, E. , Mitsialis, S. A. , & Kourembanas, S. (2021). Extracellular vesicles protect the neonatal lung from hyperoxic injury through the epigenetic and transcriptomic reprogramming of myeloid cells. American Journal of Respiratory and Critical Care Medicine, 204(12), 1418–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, P. M. , Fryer, R. H. , Fang, Y. , & Hatten, M. E. (2010). Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial‐guided neuronal migration. Journal of Neuroscience, 30(25), 8529–8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, X. , Ke, X. , Wang, L. , Yao, Z. , Guo, Y. , Zhang, X. , Chen, Y. , Pang, C. P. , Schally, A. V. , & Zhang, H. (2020). Splice variant of growth hormone‐releasing hormone receptor drives esophageal squamous cell carcinoma conferring a therapeutic target. Proceedings of the National Academy of Sciences of the United States of America, 117(12), 6726–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, W. , Lee, K.‐W. , Srivastava, R. M. , Kuo, F. , Krishna, C. , Chowell, D. , Makarov, V. , Hoen, D. , Dalin, M. G. , Wexler, L. , Ghossein, R. , Katabi, N. , Nadeem, Z. , Cohen, M. A. , Tian, S. K. , Robine, N. , Arora, K. , Geiger, H. , Agius, P. , …, Morris, L. G. T. (2019). Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nature Medicine, 25(5), 767–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, B. , Ni, J. , Witherel, C. E. , Yang, M. , Burdick, J. A. , Wen, C. , & Wong, S. H. D. (2022). Harnessing tissue‐derived extracellular vesicles for osteoarthritis theranostics. Theranostics, 12(1), 207–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhak, K. , Aguet, F. , Kim, J. , Hess, J. M. , Kübler, K. , Grimsby, J. , Frazer, R. , Zhang, H. , Haradhvala, N. J. , Rosebrock, D. , Livitz, D. , Li, X. , Arich‐Landkof, E. , Shoresh, N. , Stewart, C. , Segrè, A. V. , Branton, P. A. , Polak, P. , Ardlie, K. G. , & Getz, G. (2019). RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science, 364(6444), eaaw0726. 10.1126/science.aaw0726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama, A. , Kakiuchi, N. , Yoshizato, T. , Nannya, Y. , Suzuki, H. , Takeuchi, Y. , Shiozawa, Y. , Sato, Y. , Aoki, K. , Kim, S. K. , Fujii, Y. , Yoshida, K. , Kataoka, K. , Nakagawa, M. M. , Inoue, Y. , Hirano, T. , Shiraishi, Y. , Chiba, K. , Tanaka, H. , …, Ogawa, S. (2019). Age‐related remodelling of oesophageal epithelia by mutated cancer drivers. Nature, 565(7739), 312–317. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Lin, W. , Kannan, K. , Luo, L. , Li, J. , Chao, P.‐W. , Wang, Y. , Chen, Y.‐P. , Gu, J. , & Yen, L. (2013). Aberrant chimeric RNA GOLM1‐MAK10 encoding a secreted fusion protein as a molecular signature for human esophageal squamous cell carcinoma. Oncotarget, 4(11), 2135–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel, L. , Regnault, A. , Lozier, A. , Wolfers, J. , Flament, C. , Tenza, D. , Ricciardi‐Castagnoli, P. , Raposo, G. , & Amigorena, S. (1998). Eradication of established murine tumors using a novel cell‐free vaccine: Dendritic cell‐derived exosomes. Nature Medicine, 4(5), 594–600. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.