

Abstract
Glioma is one of the primary malignant brain tumours in adults, with a poor prognosis. Pharmacological reagents targeting glioma are limited to achieve the desired therapeutic effect due to the presence of blood‐brain barrier (BBB). Effectively crossing the BBB and specifically targeting to the brain tumour are the major challenge for the glioma treatments. Here, we demonstrate that the well‐defined small extracellular vesicles (sEVs) with dual‐targeting drug delivery and cell‐penetrating functions, modified by Angiopep‐2 and trans‐activator of transcription peptides, enable efficient and specific chemotherapy for glioma. The high efficiency of engineered sEVs in targeting BBB and glioma was assessed in both monolayer culture cells and BBB model in vitro, respectively. The observed high targeting efficiency was re‐validated in subcutaneous tumour and orthotopic glioma mice models. After loading the doxorubicin into dual‐modified functional sEVs, this specific dual‐targeting delivery system could cross the BBB, reach the glioma, and penetrate the tumour. Such a mode of drug delivery significantly improved more than 2‐fold survival time of glioma mice with very few side effects. In conclusion, utilization of the dual‐modified sEVs represents a unique and efficient strategy for drug delivery, holding great promise for the treatments of central nervous system diseases.
Keywords: Angiopep‐2, blood‐brain barrier, dual‐targeting, glioma, small extracellular vesicles, TAT
1. INTRODUCTION
Glioma is a malignant tumour commonly found in the glial tissue of the central nervous system, accounting for approximately 45% of intracranial tumours with a risk of 30–100 per millions (Jain et al., 2007). The median survival time of patients with glioma is approximately 15 months and < 4% on an overall 5‐year survival, which is accompanied by a huge impact on human health. Due to high invasiveness, rapid growth, and enhanced angiogenesis, glioma cannot be cleanly resected through traditional/routine surgery, resulting in a poor prognosis and short survival time (Cuddapah et al., 2014; Omuro & DeAngelis, 2013). To suppress and eventually eradicate glioma, a chemical drug treatment is considered as one of the most important interventions. However, there is an intrinsic obstacle of glioma cells to chemotherapy and radiotherapy along with the presence of two biological barriers, the blood‐brain barrier (BBB) and the blood‐brain tumour barrier, severely obstructs the entry of almost all macromolecules and 98% of small molecule drug into the tumour regions (Bao et al., 2006; Pardridge, 2007). This leads to tumour recurrence and thus weakens the effectiveness of existing therapies (Liu & Lu, 2012). For example, doxorubicin (Dox), a broad‐spectrum antitumour drug, cannot be delivered efficiently into the glioma by regular intravenous injection (Liu et al., 2014). Worse still, the side effects of cardiotoxicity and toxicity significantly affect the other organs due to unspecific target (Wenningmann et al., 2019). Therefore, reducing cardiotoxicity and targeting the brain tumour regions while maintaining its anticancer efficacy are critical for improving its therapeutic application (Yu et al., 2018). One of the major difficulties faced by conventional synthetic delivery systems is their inability to effectively cross biological barriers, including tissue, cellular, and intracellular barriers. Small extracellular vesicles (sEVs) have recently emerged as a new delivery system that can efficiently cross these biological barriers and be engineered for precision medicine (Alvarez‐Erviti et al., 2011; Tian et al., 2018).
sEVs are a type of extracellular vesicles with a diameter ranging from 30 to 150 nm and secreted by various cells into the extracellular fluid of blood, cerebrospinal fluid, urine, and saliva (Colombo et al., 2014). sEVs originate from the inward budding of multivesicular bodies, releasing intraluminal vesicles into the extracellular space upon fusion with the plasma membrane (Théry et al., 2002). Additionally, they facilitate the transport of specific proteins, lipids, miRNA, and genetic materials from their parental cells to recipient cells and thus plays the important roles in cell‐to‐cell communication (Boorn et al., 2011). Furthermore, sEVs are a kind of natural carrier system with endogenous cytotaxis, avoiding lysosomal degradation by the endosomal pathways, which aid them to bypass phagocytosis process and evade from the immune system (Mehrotra & Tripathi, 2015). So, sEVs can deliver their cargoes directly into the cytoplasm, allowing the better delivery of therapeutic molecules (Rufino‐Ramos et al., 2017). Moreover, as a carrier, sEVs have the advantages of nano size, biodegradation, non‐toxicity, endogeneity, low immunogenicity, strong cargo‐loading capacity and the ability to cross the BBB (Andaloussi et al., 2013; Wang et al., 2021). sEVs have been shown to deliver Dox across the BBB in a xenotransplant brain cancer model of zebrafish embryos, successful distributing the drug in the brain of zebrafish and subsequently exerting the cytotoxic effects (Wang et al., 2021). However, sEVs lack natural targeting ability in the absence of cell source selection and need to be modified to achieve an accurate targeting system for drug delivery (Alvarez‐Erviti et al., 2011; Kojima et al., 2018; Stockinger et al., 2021; Tian et al., 2014).
A great challenge in the glioma treatments is that the chemical drug currently cannot be delivered in a tissue‐ and tumour‐specific manner. Interestingly, it has been reported that low‐density lipoprotein receptor‐related protein‐1(LRP1), which mediates the transcytosis of multiple ligands across the BBB, such as lactoferrin, melanin, transferrin, and receptor‐related proteins (Pan et al., 2004), is extensively over‐expressed not only in brain glioma cells but also in vascular endothelial cells of BBB (Xin et al., 2011). Angiopep‐2(Ang) is a type of active target peptide with a high affinity for LRP1 and has a high brain penetration capability (Xin et al., 2012). The Ang peptide‐modified drug delivery system could enhance the near‐infrared fluorescent probe cross the BBB and deliver genetic drug to the brain (Ke et al., 2009). However, due to the receptor saturation effect, it is difficult to provide adequate doses of drug to glioma patients via LRP/Ang‐based delivery system alone (Zong et al., 2014). Trans‐activator of transcription (TAT) peptide is an efficient cell‐penetrating peptide, which could penetrate the plasma membrane as well as the nuclear envelop of majority of living cells (Zong et al., 2014). Moreover, TAT can easily penetrate biofilm barriers of BBB and penetrate dense tumours tissues by triggering internalization through an unsaturated and receptor/transporter‐independent pathway (Torchilin et al., 2001). Through the synergistic actions of Ang and TAT, the dual peptide‐modified sEVs not only take advantage of the efficient cell membrane penetration mediated by TAT and the targeting ability to glioma mediated by Ang, but also overcome the Ang receptor saturation. So far, the dual peptide‐modified sEVs represent a novelty in the drug delivery systems for brain diseases.
In this study, dual peptide‐modified functional sEVs (Ang/TAT‐sEVs‐Dox) were generated as a high‐efficiency delivery system, which not only targeted the BBB and glioma, but also permeated the BBB and penetrated the tumour. The Ang/TAT‐sEVs were isolated and purified from the engineered cells and characterized by the physical and biological methods. In vitro, the targeted selectivity of Ang/TAT‐sEVs was confirmed by observation of cellular uptake and the ability to permeate BBB was investigated using the BBB model. In addition, a series of in vivo experiments were conducted to further verify the targeting, BBB permeating, glioma penetrating and anti‐tumour activities of Ang/TAT‐sEVs‐Dox (Figure 1).
FIGURE 1.
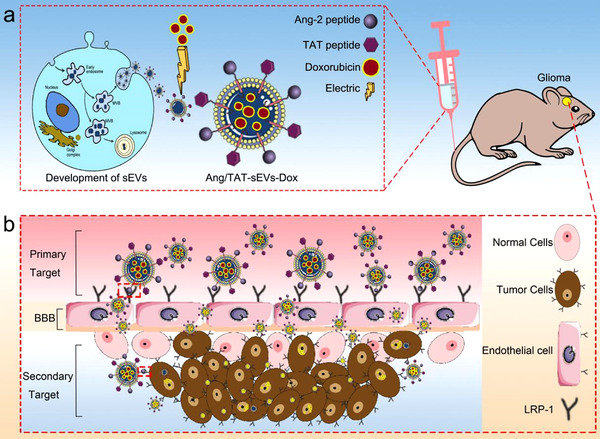
The dual‐modified functional sEVs loaded chemotherapy drug delivery system for the treatment of central nervous system diseases. Angiopep‐2 and TAT peptide are modified to sEVs membrane surface to obtain engineered sEVs with dual targeting and cell‐penetrating functions, which could permeate the BBB, target the glioma, and penetrate the tumour, so as to achieve efficient and specific chemotherapy for glioma
2. MATERIALS AND METHODS
2.1. Materials, cell culture and animal
Doxorubicin hydrochloride (Dox, MedChemExpress LLC, Shanghai, China). BCA protein assay Kit, 3,3′‐dioctadecyloxacarbocyanine perchlorate (DiO), 1,1′‐dioctadecyl‐3,3,3′,3′‐tetramethylindocarbocyanine perchlorateand (DiI), and 1,1′‐dioctadecyl‐3,3,3′,3′‐tetramethyl indotricarbocyanine iodide (DiR) were supplied by Beyotime (Shanghai, China). Lipofectamine 2000 transfection reagent was sourced from Invitrogen (USA). Matrigel was obtained from BD (USA). Creatine kinase MB isoenzyme and aspartate aminotransferase were purchased from Wuhan USCN Business Co. Ltd, China. LRP1, Alix, CD63, and CD9 antibodies were purchased from Abcam (USA), while HA, EGFP, Calnexin and β‐actin antibodies were purchased from Beyotime. All primers were synthesised by GENEWIZ (Suzhou, China). bEnd.3, U87MG, HEK293T, A549, SH‐SY5Y and HepG2 cell lines were purchased from Procell (Wuhan, China) and cultured in DMEM supplemented with 10% foetal bovine serum and 1% penicillin and streptomycin (Gibco, USA) under a 5% CO2 atmosphere at 37 °C. BALB/c nude mice, aged 4–6 weeks, were provided by Cavens (Changzhou, China). All experiments related to animals were performed according to the guidelines estimated and approved by the Ethics Committee of the Suzhou Institute of Nano‐Tech and Nano‐Bionics, Chinese Academy of Sciences (Assigned approval number: SINANO/EC/2021‐065).
2.2. Construction of Ang/TAT‐Lamp2b plasmid and transfection
Ang peptide (TFFYGGSRGKRNNFKTEEYC) and TAT peptide (YGRKKRRQRRRC) were fused Ang‐Lamp2b‐HA, and TAT‐Lamp2b‐EGFP fragments separately and inserted into the lentiviral vector pLVX‐IRES‐Puro. HEK293T was transfected with the vector expressing Ang ‐Lamp2b‐HA and TAT‐Lamp2b‐EGFP fusion proteins by using Lipofectamine 2000(Invitrogen, USA) transfection reagent with a lentivirus transfection system. The resulting virus particles were then used to infect the HEK293T cells to obtain a new cell line stably expressing Ang‐Lamp2b‐HA and TAT‐EGFP‐Lamp2b fusion proteins.
2.3. Isolation, purification, characterisation and labelling of sEVs
Ultracentrifugation is the most frequently employed method for the isolation of sEVs (Gardiner et al., 2016; Yang et al., 2020). For sEVs isolation, FBS used in the present study was depleted of sEVs through overnight centrifugation at 120,000 g. The cell culture supernatants containing sEVs was harvested after 48 h of culture. Then, the sEVs were purified from culture supernatants via gradient centrifugation. The supernatant obtained was centrifuged at 300 g for 10 min to eliminate the dead cells, then at 2000 g for 10 min to remove cell debris, then at 10,000 g for 30 min to remove large vesicles, and finally at 120,000 g for 90 min to aggregate the sEVs. Then, the sEVs were resuspended in cold PBS, filtrated through a 0.22 μm filter, and re‐ultracentrifuged at 120,000 g for 90 min, after which the pelleted sEVs were resuspended in cold PBS and stored at ‐80°C until further use. The concentration of sEVs was quantitated using the BCA protein assay Kit. The marker proteins CD9, CD63, and Alix of sEVs and the tag proteins HA and EGFP were analysed by Western blotting. sEVs were then applied to a copper grid for 1 min and negatively stained with 1% uranyl nitrate for 30 s. The dried grids were examined using the Tecnai G2 F20 S‐Twin (FEI, USA) transmission electron microscope at 120 kV. The size distribution and the number of sEVs were captured through the NTA (Malvern, UK). The DiO, DiI, and DiR were used to label the sEVs. Purified sEVs were incubated with a fluorescent dye for 20 min at 37°C under dark conditions, after which the labelled sEVs were re‐precipitated through ultracentrifugation at 120,000 g for 90 min to remove the free dye.
2.4. Flow cytometry and confocal imaging
U87MG and bEnd.3 were, respectively, seeded at the density of 5 × 105 cells well–1 in 6‐well plates and cultured for 24 h. Then, the engineered sEVs with DiI‐labelling were added and incubated for 1 h (U87MG) or 2 h (bEnd.3) at 37°C. The cells were rinsed with PBS thrice, digested with 0.25% trypsin, centrifuged at 1000 g for 4 min, resuspended in PBS, and finally subjected to fluorescence intensity examination at an excitation wavelength of 549 nm by flow cytometry (BD, USA). For confocal imaging, U87MG and bEnd.3 were seeded in a confocal dish at the density of 2 × 104 cells and cultured for 24 h. Then, cells were co‐incubated with DiI‐labelling sEVs for 30 min. Subsequently, cells were washed thrice with PBS, fixed with 4% paraformaldehyde and the nucleus was dyed with DAPI. Finally, the cellular uptake of different DiI‐labelled sEVs was captured by confocal laser scanning microscopy.
2.5. Real‐time PCR
Total RNA was isolated from screened stable cell line with TRIzol reagent (Thermo, USA). Then, cDNA was synthesized using the First‐Strand cDNA Synthesized Kits (Thermo, USA), according to the manufacturer instruction. The mRNA expression level was quantified by Applied Biosystems 7500 Real‐time PCR system (Applied Biosystems, USA) with a qPCR Mix kit (Monod, China). GAPDH was used as the internal reference to the uniform standard line. The primer sequences used in this study are as follows: Ang‐Lamp2b‐HA forward primer, 5′‐GCAGCGGCACATTCTTCTAC‐3′ and Ang‐Lamp2b‐HA reverse primer, 5′‐GTAGCGGACGGTGAAGTTCA‐3′; TAT‐Lamp2b‐EGFP forward primer, 5′‐GAAGAAGAGGCAGCGGCTC‐3′ and TAT‐Lamp2b‐EGFP reverse primer, 5′‐GGCGGCCTTTGTAAAGTTGG‐3′; LRP1 forward primer, 5′‐CTATCGACGCCCCTAAGACTT‐3′ and LRP1 reverse primer, 5′‐CATCGCTGGGCCTTACTCT‐3′; GAPDH forward primer, 5′‐AGAAGGCTGGGGCTCATTTG‐3′ and GAPDH reverse primer, 5′‐AGGGGCCATCCACAGTCTTC‐3′.
2.6. Western blotting
The total protein in the cell or sEVs was extracted with the RIPA lysis buffer by using the Protease Inhibitor Cocktail (Transgen, China). The protein concentration was quantified using the BCA protein assay kit, and the protein was subjected to denaturation in boiling water for 10 min. Then, 20 μg of the protein was loaded and separated by 10% SDS‐PAGE and then transferred onto a 0.45‐μm PVDF membrane (Mllipore, USA), followed by blocking in 5% non‐fat milk for 1 h at room temperature. The membrane was incubated with the antibodies to LRP1, Alix, CD63, CD9, HA, EGFP, Calnexin and β‐actin at 4°C overnight. The membrane was washed with TBST thrice, after which the membrane was incubated with HRP‐conjugated secondary antibody at room temperature for 2 h. The protein bands were visualised using the ECL kit(Vazyme, China) and LAS4000 imaging system (Fuji Film, Japan).
2.7. In vitro BBB model
Milli hang culture inserts (Millipore, USA) were added to a 12‐well plate to which bEnd.3 cells were seeded in the upper chamber at the density of 5 × 104 cells in 400 μl DMEM medium with 10% FBS. To maintain the osmotic pressure inside and outside the membrane, the lower chamber was filled with 1950 μl of the same medium. The compactness of the bEnd.3 monolayer was continuously and intermittently monitored by observing the cellular morphology under a phase‐reversal microscope and measuring the TEER value with the TEER equipment (MERCK, Millicell, Germany). Only the TEER value of the tight monolayer cells > 200 Ω·cm2 in the in vitro BBB model was selected for subsequent experiments. U87MG were seeded onto a round glass slide at the bottom of the lower chambers at the density of 5 × 104 cells. Next, the culture medium was replaced with the DMEM basic medium, and the DiI‐labelled sEVs (2 μg ml–1) were added into the upper chamber, after which the culture formula of the lower chamber was kept unchanged. Subsequently, the plates were placed on a mini shaking table and cultured for 24 h. Finally, the U87MG and bEnd.3 were treated as mentioned above and examined with CLSM. The cellular uptake of sEVs by bEnd.3 on the polycarbonate membrane and that of the U87M in the lower chambers were also quantified by flow cytometry. Briefly, U87MG cells were seeded in the lower chambers at the density of 1 × 105 cells. When the TEER value of the bEnd.3 cells reached > 200 Ω·cm2, the culture medium was added with FBS‐free, mixed DiI‐labelled sEVs into the upper chamber. Then, the plates were placed on a mini shaking table and cultured for 24 h. Finally, cells were digested and suspended in a flow cytometry tube, followed by analyses via flow cytometry.
2.8. Therapeutic cargo‐loading into sEVs
The sEVs and Dox (mass ratio of 2:1) were gently mixed in 300 μl electroporation EL transfection buffer at 4°C. After electroporation at 350 V, with pulse duration of 100 μs, pulse separation at 1000 ms, and 10 pulses by X‐Poraot H1(Etta Biotech, China), the mixture was incubated at 37°C with 5% CO2 for 30 min to ensure that the biological membrane of the sEVs was fully recovered. Then, the sEVs were washed with cold PBS and ultracentrifuged at 120,000 g for 90 min to remove the unincorporated free Dox. The Dox concentration encapsulated into sEVs was quantified by detecting the intrinsic ultraviolet absorption of Dox by using an ultraviolet‐visible spectrophotometer (PerkinElmer Lambda25, USA) detecting at 594 nm following excitation at 480 nm.
2.9. Tumour‐bearing nude mouse and orthotopic glioma model
The plasmid of pLVX‐acGFP‐N1‐Flu was purchased from Public Protein/Plasmid Library (PLL, China), and the stable cell line of U87MG‐acGFP‐Flu was screened after lentivirus infection. For tumour‐bearing nude mouse, the reconstructed human brain astroblastoma cells with firefly luciferase reporter gene (U87MG‐acGFP‐Flu, 5 × 106 cells) mixed with Matrigel were transplanted into the back of 6‐week‐old BALB/c nude mice and allowed to grow to a tumour size of approximately 0.2 cm3(volume = ½ × length x width2, as measured with vernier calliper). Finally, the mice were randomly assigned into different experimental groups, as described in the results section. For the orthotopic glioma model, 5 × 105 U87MG‐acGFP‐Flu cells were resuspended in 8 μl of 0.9% NaCl and 10 mg ml–1 of pentobarbital sodium at the dose of 62.5 mg kg–1 to the nude mouse before surgery. The prepared cells were implanted at 0.98‐mm anterior of the bregma, 1.5‐mm laterally from the midline of the skull at an injection depth of 2.5 mm, with the needle moved towards the midline in the coronal plane into the left hemisphere. Then, the burr hole was filled with bone wax (Fine Science Tools, Germany), and the scalp was sutured with needle sutures (Chenghe Microinstrument Factory, China). The growth of the glioma was monitored on the sixth day by bioluminescence using a small animal imaging system (IVIS Lumina XRMS Series III, USA) 10 min after the nude mouse was anaesthetised with pentobarbitone combined with luciferase substrate D‐luciferin potassium (15 mg ml–1 in DPBS, 0.22‐μm membrane filter) at the dose of 75 mg kg–1.
2.10. In vivo imaging
After 6 days of cell implantation, the tumour‐bearing nude mice and orthotopic glioma mice were appraised by injecting the luciferase substrate D‐luciferin potassium and analysed using the small animal imaging system. For the tumour‐bearing nude mouse model, when the volume of the tumour reached approximately 0.2 cm3 after 2 or 3 weeks of implantation, 200 μl of DiR‐labelled sEVs (Ex = 748 nm, Em = 780 nm) were injected via the tail vein. At 8, 24, 48, and 72 h post‐injection, the mice were anaesthetised with pentobarbital sodium (10 mg ml–1 in PBS) at the dose of 62.5 mg kg–1, and relevant fluorescence images were acquired using the fluorescence imaging system to detect sEVs distribution. The organs (the brain, heart, liver, lung, spleen, and kidney) and tumour were separated, and the sEVs distribution was detected using the aforementioned method. For the orthotopic glioma model, after implanting U87MG‐acGFP‐Flu cells for 15 days, we injected the DiR‐labelled or DiO‐labelled sEVs into the mice via the tail vein. At 8, 24, 48, and 72 h post‐injection, we assessed the sEVs accumulation in the brain at different time points by using the fluorescence imaging system. The organs of model mice were collected, and the sEVs distribution was imaged.
2.11. In vivo assessment of antitumour efficacy
The day of cellular implantation was designated as day 0. After 6 days of U87MG‐acGFP‐Flu cellular implantation, the mouse models were treated with PBS, free Dox, Dox‐loaded blank sEVs, and Dox‐loaded Ang/TAT‐sEVs to evaluate the therapeutic efficacy. Then, we selected the successful model mice that were identified by injecting the luciferase substrate D‐luciferin potassium. At day 6, the mice were randomly assigned to four groups (n = 6) and injected with sEVs‐loaded Dox (2 mg kg–1), as described earlier, via the tail vein. The treatment was repeated every 3 days for a total of four doses. The growth volume of the U87MG glioma was monitored using the small animal imaging system, as described previously. On day 21, the brain, heart, lung, liver, spleen, and kidney were separated from each group mouse and fixed with 4% paraformaldehyde and embedded in paraffin. To evaluate the occurrence of any cardiac damage, the sliced tissues were stained with haematoxylin and eosin (H&E), and the serum was collected to determine the activities of creatine kinase MB isoenzyme (CK‐MB, USCN Co. Ltd, China) and AST (USCN Co. Ltd., China).
2.12. Immunohistochemistry
The brain samples were embedded in paraffin and sliced into 4‐μm thick sections. The brain sections were dewaxed to water and placed into a box containing the citric acid antigen repair solution (Servicebio, China) in the microwave for heating repair. Then, the sections were blocked with 3% bovine serum albumin for 30 min at room temperature, followed by incubation first with the primary antibody CD63 at 4°C overnight and then with HRP‐conjugated secondary antibody. The brain sections were stained with DAB, and the nucleus was stained with haematoxylin for 3 min. The images were obtained under an orthographic biological microscope (CIC, XPS‐C204, China).
2.13. Statistical analysis
Statistical analysis of multiple groups of data was performed with one‐way analysis of variance, followed by the least significant difference tests and Student's t test to compare the difference between the two groups. All data are expressed as the mean ± standard deviation (SD). Tumour volumes were compared using the Kruskal‐Wallis test, followed by Mann‐Whitney U test. Survival data significance was calculated using the log‐rank test. Statistical significance was established at *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
3. RESULTS
3.1. Fabrication and characterization of Ang/TAT‐sEVs
To establish the brain tumour‐targeting and cell‐penetrating dual‐functional sEVs simultaneously expressing the Ang peptide and TAT peptide (Figure 2a), we fused the targeting peptides Ang and cell‐penetrating peptides TAT to the extra‐extracellular vesicles N terminus of Lamp2b protein reported to be present abundantly in extracellular vesicles membranes, generating pLVX‐Ang‐Lamp2b‐HA plasmid (Figure 2b) and TAT pLVX‐TAT‐Lamp2b‐EGFP plasmid (Figure 2c). HA and EGFP tags were added to the C terminus of Ang‐Lamp2b or TAT‐Lamp2b, respectively. The HEK293T cells were infected with the lentivirus particles to acquire the stable expressions of Ang‐Lamp2b‐HA and TAT‐Lamp2b‐EGFP fusion proteins (Table S1 and S2), thus generating four stably transfected cell lines: 293T‐Blank, 293T‐Ang, 293T‐TAT and 293T ‐293‐Ang/TAT (Figure S1). The transcriptional expression levels of Ang‐Lamp2b or TAT‐Lamp2b was confirmed by real‐time PCR analysis (Figure 2d, e). Western blotting results indicated that both Ang‐Lamp2b‐HA and TAT‐Lamp2b‐EGFP were highly expressed in the transfected HEK293T (Figure 2f). Blank‐sEVs, Ang‐sEVs, TAT‐sEVs, and Ang/TAT‐sEVs derived from the corresponding transfected HEK293T cells were isolated from the cell culture supernatants derived from the transfected HEK293T through ultracentrifugation. All types of sEVs were expressed with the biomarker proteins of Alix, CD9, CD63, HA tag, EGFP tag and negative marker Calnexin (Figure 2g). Transmission electron microscopy (TEM) and nanoparticle tracking analysis (NTA) revealed that the peptide modifications did not affect the morphology and size distribution of the sEVs. The micro‐images showed all sEVs were in round vesicles, with a lipid bilayer (Figure 2h) and a uniform size distribution of 120 ± 20 nm (Figure 2i and Table S3). Referring to the MISEV2018 guidelines of the international society for extracellular vesicles (Théry et al., 2018), the WB, TEM and NTA results verified that the isolated extracellular vesicles are sEVs.
FIGURE 2.
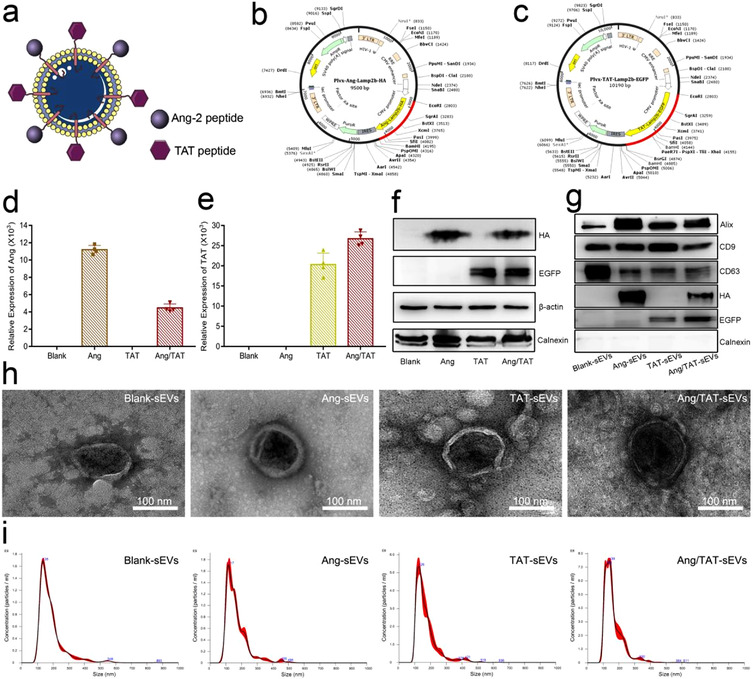
Characterisation of engineered HEK293T cells and peptide‐modified sEVs. (a, b, c) The schematic of Ang/TAT‐sEVs, the plasmid of pLVX‐IRES‐Ang‐Lamp2b‐HA, and the plasmid of pLVX‐IRES‐TAT‐Lamp2b‐EGFP. (d, e) The mRNA expression level of the Ang‐Lamp2b‐HA and TAT‐Lamp2b‐EGFP in HEK293T cells (n = 4). (f) HA and EGFP‐tag protein expression level in HEK293T cells, with β‐actin as the normalisation control. (g) HA and EGFP Tag protein and small extracellular vesicles biomarker such as CD63, CD9, Alix and negative marker (calnexin) were assessed by western blot. (h) The morphology of sEVs detected with TEM. Scale bar = 100 nm. (i) Size distribution of blank‐sEVs, Ang‐sEVs, TAT‐sEVs, and Ang/TAT‐sEVs detected through NTA
3.2. In vitro assessments of targeting efficiency and cellular uptake
The high expression of LRP1 on cells can enhance cells uptake of Ang‐sEVs. Firstly, western blotting results indicated that LRP1 protein was highly expressed in mouse glioblastoma cell line U87MG and immortalised mouse cerebral microvessel endothelial cell line bEnd.3, while lowly expressed in human pulmonary epithelial cell line A549 and the human neuroblastoma cell line SH‐SY5Y (Figure 2a, 2b). Moreover, their expression of LRP1 mRNA was confirmed by real‐time PCR analysis (Figure 2c). Both western blotting and RT‐PCR results indicated a high LRP1 expression for U87MG cells and bEnd.3 cells, while a low LRP1 expression for A549 cells and SH‐SY5Y cells. The Ang‐sEVs were labelled with DiI (red fluorescence) and incubated with the cell lines U87MG or bEnd.3 or A549 or SH‐SY5Y, respectively. The DiI positive percentages for U87MG, bEnd.3, A549 and SH‐SY5Y are 63.74%, 58.13%, 26.55% and 28.5%, respectively (Figure 3d). Flow cytometry demonstrated that both U87MG and bEnd.3 could enhance uptake of the dual peptide‐modified Ang‐sEVs, compared to A549 and SH‐SY5Y. Furthermore, the DiI‐labelled Ang‐sEVs were incubated for 30 min and confocal microscopy indicated that Ang‐sEVs could be highly uptaken in both U87MG and bEnd.3, compared with A549 and SHSY5Y (Figure 3e). These results showed that the LPR1 high expressing in U87MG and bEnd.3 raises cell uptake of Ang‐sEVs.
FIGURE 3.
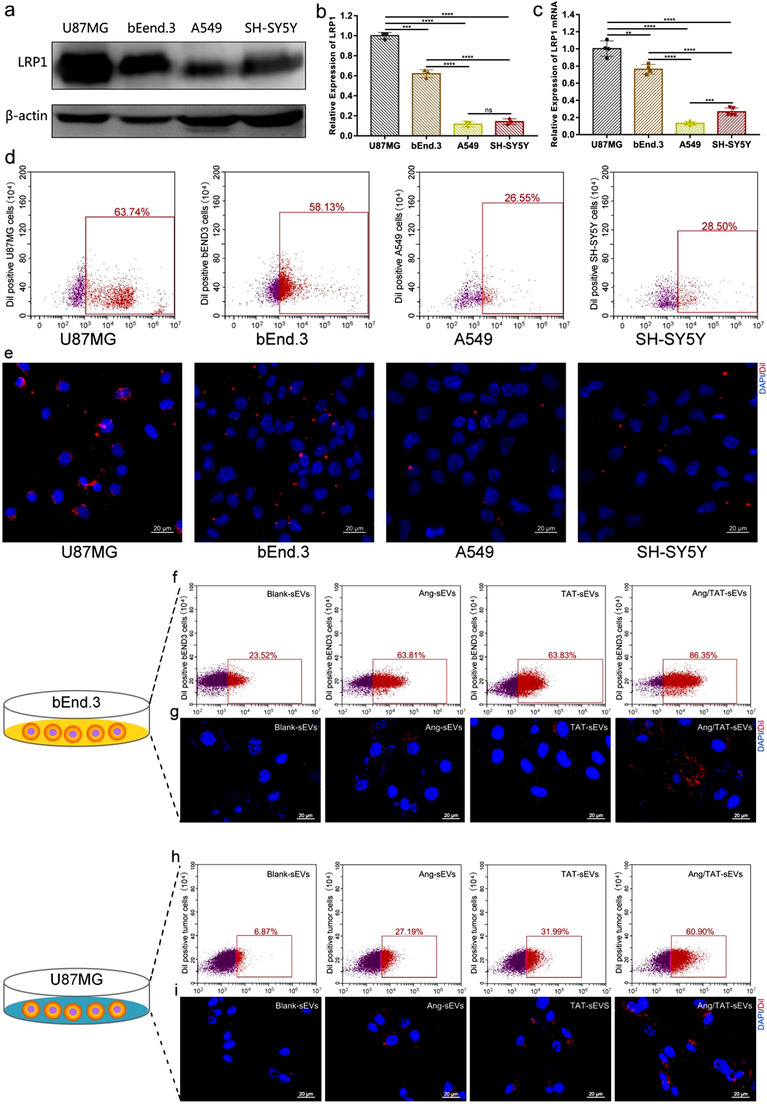
Targeting efficiency and cell uptake of sEVs in the monolayer cell culture. (a and b) Western blot analysis of the LRP1 protein levels in U87MG, bEnd.3, A549 and SH‐SY5Y (n = 3, ***P < 0.001, ****P < 0.0001. ns, not significant). (c) The mRNA expression level of the LRP1 in U87MG, bEnd.3, A549 and SH‐SY5Y (n = 4, **P < 0.01, ***P < 0.001, ****P < 0.0001). (d) Flow cytometry detected DiI‐positive U87MG, bEnd.3, A549 and SH‐SY5Y after incubation with 2 μg ml–1 DiI‐labelled Ang‐sEVs for 2 h. (e) Confocal microscopy images of U87MG, bEnd.3, A549 and SH‐SY5Y uptake of DiI‐labelled Ang‐sEVs after 1 h of incubation. The cell nuclei were stained with DAPI. (f) Flow cytometry detected DiI‐positive bEnd.3 after incubation with DiI‐labelled blank‐sEVs, Ang‐sEVs, TAT‐sEVs, and Ang/TAT‐sEVs for 2 h. (g) Confocal microscopy images of bEnd.3 uptake of DiI‐labelled sEVs after 30 min of incubation. The cell nuclei were stained with DAPI. (h) Flow cytometry detected DiI‐positive U87MG after incubation with DiI‐labelled blank‐sEVs, Ang‐sEVs, TAT‐sEVs, and Ang/TAT‐sEVs for 2 h. (i) Confocal microscopy images of U87MG uptake of DiI‐labelled sEVs after 30 min of incubation. The cell nuclei were stained with DAPI
To investigate the targeting efficiency and cell uptake ability of the peptide‐modified sEVs, Blank‐sEVs, Ang‐sEVs, TAT‐sEVs, and Ang/TAT‐sEVs were labelled with DiI and incubated with the monolayer culture bEnd.3 or U87MG, respectively. Both bEnd.3 and U87MG glioblastoma cells over‐express LRP1, which is a target of Ang‐2. The DiI positive percentages for the bEnd.3 are 23.52%, 63.81%, 63.83%, and 86.35% (Figure 3f), while for the U87MG, 6.87%, 27.19%, 31.99%, and 60.90% (Figure 3h), respectively. Flow cytometry demonstrated that both bEnd.3 and U87MG could efficiently uptake the dual peptide‐modified Ang/TAT‐sEVs as compared to Blank‐sEVs, single peptide‐modified Ang‐sEVs and TAT‐sEVs. To further confirm peptide‐modified sEVs have a high targeting efficiency towards bEnd.3 and U87MG, the cells with DiI‐labelled sEVs were incubated for 30 min. The images indicated that Ang/TAT‐sEVs possess the highest targeting efficiency in both bEnd.3 and U87MG as compared with single peptide‐modified sEVs and blank‐sEVs (Figure 3g, i). Monolayer cell culture experiments with the flow cytometry and confocal microscopy revealed that Ang‐2 targeting peptide‐modified could enhance the efficiency of sEVs uptake by LRP1‐positive cells, and similar enhancement in the efficiency of sEVs uptake was also seen in TAT‐sEVs. Nevertheless, the further improved uptake efficiency of Ang/TAT‐sEVs might be due to the strong penetrating ability of the TAT peptide. Overall, these results showed that Ang/TAT‐sEVs could mediate a higher level of cell‐targeting and cellular uptake capacity, displaying the potential in delivery systems.
3.3. In vitro BBB permeation
To assess the BBB permeation ability of dual peptide‐modified sEVs, an in vitro model was established with transwell inserts placed in 12‐well plates. bEnd.3 were loaded at the membrane insert of the transwell, while U87MG were seeded at the bottom (Figure 4a). bEnd.3 were allowed to form an intact barrier with a TEER of > 200 Ω·cm before the exposure to sEVs (Figure S2). The DiI‐labelled sEVs were added in the apical chamber against bEnd.3 with a minimum shaking for 24 h. The flow cytometry results showed that the percentage of DiI‐positive bEnd.3 were close to 100% in all four groups (Figure 4b), but the DiI‐labelled Ang/TAT‐sEVs presented the highest fluorescence intensity (Figure 4c). Analysis by confocal microscopy demonstrated that the sEVs uptake by bEnd.3 was high (Figure 4d).The sEVs transport across the BBB layer was determined by the uptake of DiI‐labelled sEVs in U87MG in the basolateral chamber. Flow cytometry revealed that the Ang/TAT‐sEVs exhibited the highest efficiency in crossing BBB and binding to U87MG, as compared with the other three groups comprising Ang‐sEVs, TAT‐sEVs, and blank‐sEVs (Figure 4e). Quantitative analysis of the fluorescence intensity also indicated that the U87MG had a remarkable ability to uptake DiI‐labelled sEVs (Figure 4f). Meanwhile, the fluorescence intensity of Ang/TAT‐sEVs group was up‐regulated to nearly 5‐fold relative to blank‐sEVs group, 4‐fold to Ang and 2‐fold to TAT peptide in single‐modified groups, respectively (Figure 4g, h). Interestingly, Ang‐sEVs group was close to blank‐sEVs group and significantly lower than TAT‐sEVs group. Collectively, the quantitative analysis of fluorescence and confocal microscopy imaging demonstrated that Ang/TAT‐sEVs had significantly higher efficiency of BBB permeation and ability to target U87MG cells than Ang‐sEVs, TAT‐sEVs, and blank‐sEVs. In addition, the single TAT peptide‐modified sEVs were found to significantly contribute to high cellular uptake in BBB cells.
FIGURE 4.
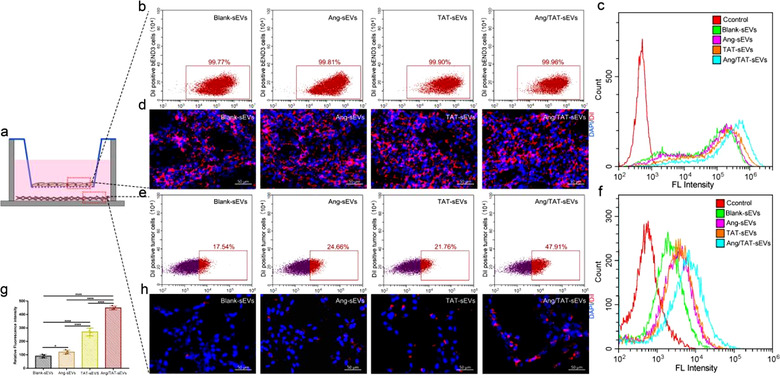
Targeting efficiency and cell uptake of sEVs in the BBB cell model in vitro. (a) bEnd.3 were plated at the membrane insert of the transwell for 5 days, and the U87MG were seeded at the bottom of the plates for 24 h. (b) Flow cytometry detected DiI‐positive bEnd.3 after incubation with blank‐sEVs, Ang‐sEVs, TAT‐sEVs, and Ang/TAT‐sEVs for 24 hou. (c) Flow cytometry evaluated the fluorescence intensity of bEnd.3 with DiI‐labelled sEVs. (d) Confocal microscopy images of bEnd.3 uptake of DiI‐labelled sEVs after 24 h of incubation. The cell nuclei were stained with DAPI. (e) Flow cytometry detected DiI‐positive U87MG seeded at the bottom of the plates. (f) Flow cytometry analysed the fluorescence intensity of U87MG. (g, h) The fluorescence quantitative analysis and confocal microscopy images of U87MG (n = 4, *P < 0.05, ****P < 0.0001). The cell nuclei were stained with DAPI
3.4. In vitro antitumour effect
Given that the intrinsic optical density of pharmaceutical Dox can be detected at 594 nm with an excitation at 480 nm, sEVs could be loaded with Dox by using an optimal condition after electroporation (Figure S3a). Electroporation did not drastically modify the physical properties of sEVs, as demonstrated by NTA and TEM results (Figure S3b, c). Thus, electroporation was found to be an effective method for loading the drug into sEVs.
To determine whether Ang/TAT‐sEVs could effectively deliver Dox to LRP1‐positive U87MG in vitro, 2 μM free Dox and an equivalent dose of Dox were loaded into Blank‐sEVs (Blank‐sEVs‐Dox) and Ang/TAT‐sEVs (Ang/TAT‐sEVs‐Dox), followed by an incubation with U87MG. Fluorescence imaging indicated that the delivery of Dox to the U87MG by Ang/TAT‐sEVs was nearly as efficient as the accumulation of free Dox, while Blank‐sEVs showed much lower Dox delivery efficiency (Figure 5a). CCK‐8 assay showed that Ang/TAT‐sEVs‐Dox could inhibit the cell viability with an efficiency comparable to free Dox, while the Blank‐sEVs‐Dox showed a significantly lower inhibition of cell growth than free Dox and Ang/TAT‐sEVs‐Dox (Figure 5b). We also determined the inhibitory effect of Ang/TAT‐sEVs‐Dox on cell viability by using other cell line. Human hepatocellular carcinoma cells (HepG2) also show a high expression of LRP1(Rondón‐Ortiz et al., 2017). High targeting ability and excellent Dox delivery efficiency of Ang/TAT‐sEVs to LRP1‐positive cells were observed in these cells similar to those in U87MG (Figure 5c). Interestingly, although free Dox and Ang/TAT‐sEVs‐Dox exhibited significant inhibition of cells relative to the Blank‐sEVs‐Dox in the A549 cells that has low expression of LRP1(Masaldan & lyer, 2014), the inhibitory effect of Ang/TAT‐sEVs‐Dox was weaker than that of free Dox (Figure 5d). These results indicated that the binding of the receptor to target cell could effectively improve drug delivery. Moreover, nonsignificant inhibition of cell growth was observed in the blank‐sEVs‐ and Ang/TAT‐sEVs‐treated groups, indicating that the sEVs from HEK293T had no or low cytotoxicity.
FIGURE 5.
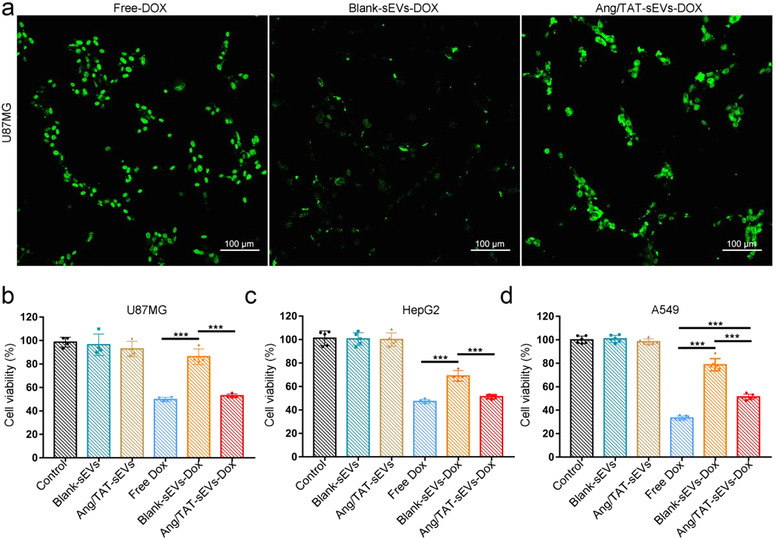
In vitro Dox delivery and antitumour effect. (a) Fluorescence imaging of free Dox, Dox‐loading blank‐sEVs or Ang/TAT‐sEVs incubated with U87MG for 2 h. Scale bars = 100 μm. (b) U87MG, (c) HepG2, (d) A549 viability evaluated by the CCK‐8 assay after incubation with culture medium (control), blank‐sEVs, Ang/TAT‐sEVs, free Dox, blank‐sEVs‐Dox, and Ang/TAT‐sEVs‐Dox for 24 h. Each bar represents the mean ± SD of four replicates, ***P < 0.001
3.5. In vivo targeting, BBB permeating and glioma penetrating
To investigate the tumour‐targeting and cell‐penetrating capabilities of Ang/TAT‐sEVs in vivo, a glioma mouse model was established by subcutaneously injecting U87MG cells into nude mice. To monitor the glioma growth, acGFP and luciferase were transfected in a stable cell line U87MG‐acGFP‐Flu (Figure S4). After implantation of 6 days, the tumour‐bearing nude mouse model was established and assessed by injecting luciferase substrate D‐luciferin potassium (Figure 6a). When the tumour presented a size of approximately 0.2 cm3 on the day 18, the sEVs labelled with DiR dye were injected into the nude mice intravenously through the tail vein. At 24 h after administering injection, DiR fluorescence intensity of tumour was significantly improved up to approximately 4‐fold as compared to the control and 2 to 3‐fold compared to Ang or TAT peptide single‐modified groups, respectively (Figure 6b, d). The fluorescence signal analysis and quantification showed that Ang/TAT‐sEVs had much better targeting ability towards glioma than TAT‐sEVs, Ang‐sEVs, and Blank‐sEVs. To further analyse the targeting of the engineered sEVs in ectopic U87MG xenografts, organs and tumours were harvested. Ex vivo fluorescence imaging and quantifying showed that Ang/TAT‐sEVs had the strongest fluorescence signal in tumour tissue (Figure 6c, e). These results demonstrated that Ang/TAT‐sEVs could effectively target tumour tissue in the subcutaneous glioma model.
FIGURE 6.
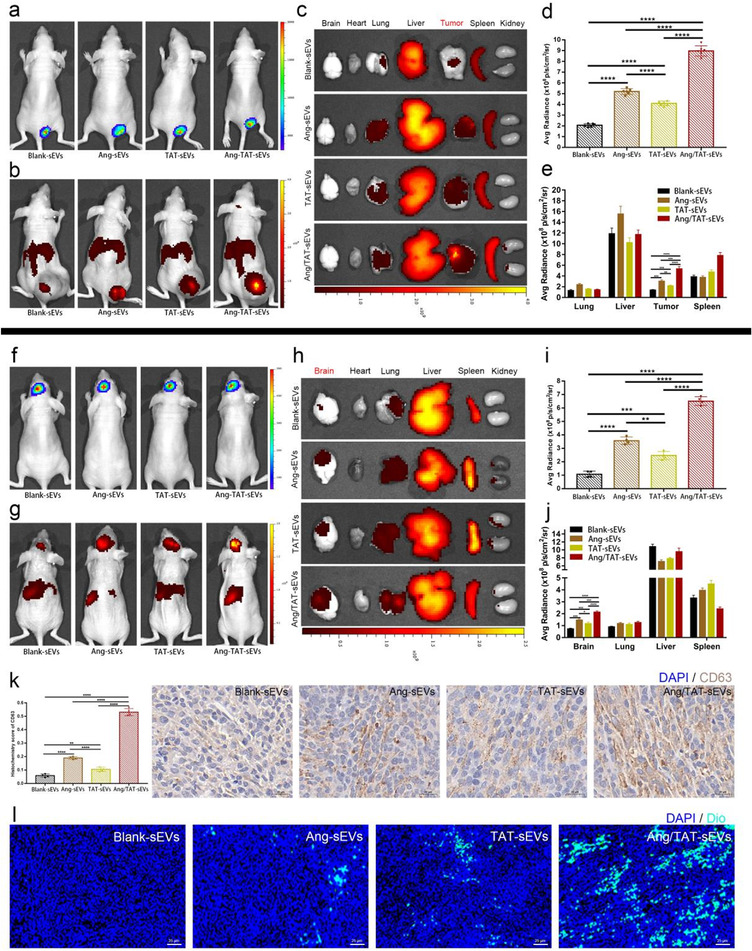
Tumour‐targeting and BBB‐permeating ability of the peptide‐modified sEVs in vivo. (a) In vivo imaging of subcutaneous U87MG glioma identified by injecting luciferase substrate D‐luciferin potassium. (b) In vivo imaging of DiR‐labelled sEVs. (c) Ex vivo fluorescence imaging of organs and tumours from the model mice after DiR‐labelled sEVs intravenous injection. (d) Quantified data of fluorescence signal in (b) (n = 6, ****P < 0.0001). (e) Quantified data of fluorescence signal in (c) (n = 6, **P < 0.01, ***P < 0.001, ****P < 0.0001). (f) In vivo imaging of orthotopic U87MG glioma identified by injecting the luciferase substrate D‐luciferin potassium. (g) In vivo imaging of the DiR‐labelled sEVs. (h) Ex vivo fluorescence imaging of organs and tumour from the model mice after DiR‐labelled sEVs intravenous injection. (i) Quantified data of fluorescence signal in (g) (n = 4, **P < 0.01, ***P < 0.001, ****P < 0.0001). (j) Quantified data of fluorescence signal in (h) (n = 4, *P < 0.05, ***P < 0.001, ****P < 0.0001). (k) Immunohistochemistry images of CD63 expression in the brain tumour (n = 4, **P < 0.01, ****P < 0.0001). (l) Fluorescence images of DiO‐labelled sEVs targeting and penetrating the brain tumour observed through GLSM
To further assess the ability of Ang/TAT‐sEVs to cross BBB and penetrate the glioma, an orthotopic U87MG glioma model was employed (Figure S5). Likewise, the glioma model was established by injecting luciferase substrate D‐luciferin potassium on the day 6 after implantation (Figure 6f). DiR‐labelled sEVs and DiO‐labelled sEVs were injected into the mice through the tail vein. The fluorescence signal was recorded using a live imaging system for 24 h post‐injection. The brain DiR fluorescence intensity of the Ang/TAT‐sEVs group was up‐regulated up to about 6‐fold relative to the controls and 2‐fold or 3‐fold relative to Ang or TAT peptide single‐modified groups, respectively (Figure 6 g, i). The TAT‐sEVs can significantly improve the efficiency of reaching the glioma region, compared with the Blank‐sEVs, it may be that the transmembrane peptide TAT promotes the high BBB permeability and high tumour penetration of sEVs. Fluorescence analysis and quantification showed that the DiR fluorescence of Ang/TAT‐sEVs had a higher accumulation in the brain than other groups of sEVs, indicating that Ang/TAT‐sEVs were highly effective in BBB permeation. Organs and tumours harvested for fluorescence signal analysis and quantification showed similar properties (Figure 6 h, j). These results demonstrated that Ang/TAT‐sEVs could permeate BBB very well and target orthotopic glioma.
To investigate glioma penetrating properties of dual peptide‐modified sEVs, paraffin sections of the brain tissue were prepared. CD63, a marker protein of sEVs in the intratumour region of brain glioma, was analysed and quantified through immunohistochemistry. The Ang/TAT‐sEVs group showed significantly higher accumulation in the intratumour than other groups (Figure 6k and Figure S6a). Furthermore, we analysed the CD63 of the anti‐tumour therapeutic groups, which showed similar properties (Figure S6b). In order to confirm that the sEVs enriched in the intratumour domain were mainly from the delivered engineered sEVs, instead of the sEVs secreted by the brain tissue. DiO‐labelled Ang/TAT‐sEVs were injected into the mice and the fluorescence signals from frozen sections of brain tissue were detected. Confocal imaging demonstrated that the accumulation of Ang/TAT‐sEVs was significantly higher inside the glioma, indicating Ang/TAT‐sEVs had a higher capacity of penetrating the glioma (Figure 6l and Figure S6c). These results indicated that the Ang/TAT‐sEVs possesses a superior targeting ability towards BBB and glioma and can efficiently permeate the BBB and glioma.
3.6. In vivo anti‐tumour efficacy
The anti‐glioma efficacy of Dox‐loaded dual peptide‐modified sEVs was assessed in nude mice with orthotopic glioma. The growth of glioma was monitored by the bioluminescence emitted in a given time period. The suppression of glioma growth was observed in the mice treated with Blank‐sEVs‐Dox, however, to a lesser extent than that in Ang/TAT‐sEVs‐Dox. By contrast, mice treated with free Dox and PBS showed faster tumour growth (Figure 7a). At day 21, the Ang/TAT‐sEVs‐Dox group of the fluorescence intensity decreased approximately 6‐fold relative to free Dox group and 2‐fold to Blank‐sEVs‐Dox group (Figure 7b), indicating that Ang/TAT‐sEVs‐Dox was significantly more efficient in suppressing glioma growth than Blank‐sEVs‐Dox and free Dox, probably due to the efficient targeting of glioma, effective BBB permeation, and deeper glioma penetration. Interestingly, the body weight of mice treated with Ang/TAT‐sEVs‐Dox did not decrease until the day 21, however, the Blank‐sEVs‐Dox group showed a decreasing trend of the same. Conversely, the free Dox group showed a significant decrease in body weight, probably due to the cytotoxicity of free Dox and brain malfunction because of tumour invasion (Figure 7c). The Kaplan‐Meier survival curves indicated that the median survival in orthotopic brain tumour‐bearing mice treated with Ang/TAT‐sEVs‐Dox was much longer than that in the other groups. The median survival time was 24, 32, 40, and 52 days for the groups treated with PBS, free Dox, Blank‐sEVs‐Dox, and Ang/TAT‐sEVs‐Dox (Figure 7d), respectively. Meanwhile, H&E staining analysis of the brain tissue indicated that the Ang/TAT‐sEVs‐Dox treated group had significantly inhibited the growth of tumours as compared with other groups (Figure 7e). These results suggested that the Ang/TAT‐sEVs‐Dox exhibited an effective and targeted therapy for glioma as against the use of free Dox and Blank‐sEVs‐Dox.
FIGURE 7.
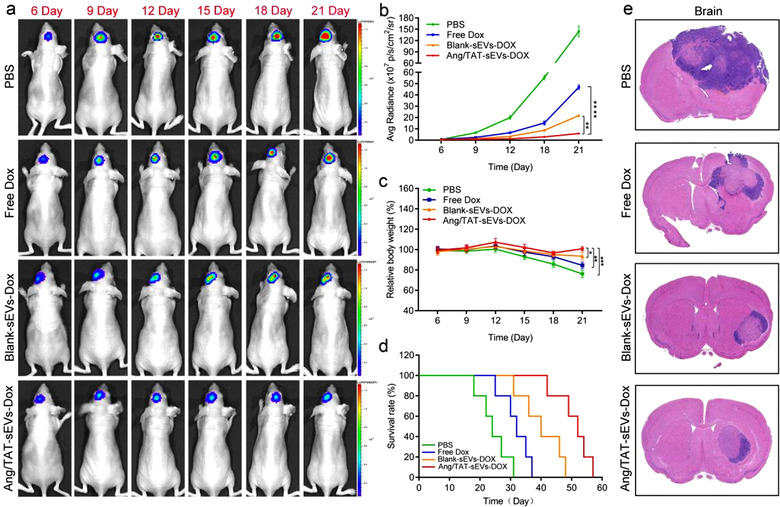
Targeted therapy of Ang/TAT‐sEVs‐Dox in the orthotopic U87MG glioma model mice. (a) Tumour growth was monitored by bioluminescence imaging at 6, 9, 12, 15, 18, and 21 days. (b) Bioluminescence intensity analysis of the change in U87MG glioma in nude mice at different time points (n = 6, **P < 0.01, ****P < 0.0001). (c) Body weight changes of the orthotopic U87MG glioma model mice in different treatment groups (n = 6, *P < 0.05, **P < 0.01, ***P < 0.001). (d) Kaplan–Meier survival curves of the orthotopic U87MG glioma model mice in different treatment groups. Statistical analysis: Ang/TAT‐sEVs‐Dox versus blank‐sEVs‐Dox, P < 0.01; blank‐sEVs‐Dox versus free Dox, P < 0.01 (n = 6, Kaplan–Meier, log‐rank test). (e) H&E staining images of U87MG glioma in the brain
The biosafety of dual peptide‐modified sEVs was also evaluated in vivo. The heart, liver, spleen, and lungs were obtained from the orthotopic glioma mouse model and stained with H&E. No evidence of tissue damage was observed in the heart and lungs, but the liver tissue of the free Dox group exhibited slight tissue damage compared with that of the other groups, indicating that free Dox showed little cytotoxicity (Figure S7a). After 21 days of the treatment, the serum from the mice was collected for measuring the activities of creatine kinase MB isoenzyme (Figure S7b) and AST (Figure S7c), which are commonly used as markers to evaluate cardiac damage. The activities of CK‐MB and AST were significantly lower in the serum of mice treated with Ang/TAT‐sEVs‐Dox and Blank‐sEVs‐Dox than in the serum of free Dox treated mice and were comparable to those of the PBS treated group, indicating that Dox‐loaded dual peptide‐modified sEVs are less cardiotoxic than free Dox. These results demonstrated that Ang/TAT‐sEVs‐Dox combining the sEVs engineering with therapeutic drug loading possessed the ability to cross the BBB, target the glioma and improve the survival of glioma‐bearing mice with minimal side effects, representing a safe and effective approach to deliver therapeutic drugs for glioma treatment.
4. DISCUSSION
The main challenge in treating glioma is BBB permeation and delivery of anticancer drug at the therapeutic dose to the tumour (Islam et al., 2021; Pardridge, 2007). Nonspecific cytotoxicity, poor biocompatibility and low delivery efficiency have been reported in the traditional nano‐delivery systems (Thomas et al., 2003). sEVs are one of the endogenous carriers with an inherently small size, low immunogenicity, strong cargo‐carrying capacity and the capacity to penetrate the BBB, rendering them the potential to be a potent tool for brain cancer therapy (Zhang et al., 2021). However, the molecular mechanism in BBB crossing of sEVs remains unclear. Mannose 6‐phosphate receptor has been reported as one kind of transporter for sEVs to cross the BBB by adsorptive transcytosis (William et al., 2020), and specific miRNA (such as miRNA‐181c) could promote the destruction of BBB through the abnormal localization of actin via the downregulation of its target gene (Naoomi et al., 2015). An efficient tissue‐targeting delivery system, the neuron‐specific rabies viral glycoprotein (RVG) peptide‐modified sEVs, has been reported for treating Alzheimer's disease and glioblastoma (Alvarez‐Erviti et al., 2011; Zhu et al., 2019). However, sEVs lack the reliable targeting capacities to treat brain tumours, which probably resulted into unexpected side effects to unintended organ or tissues, thus severely impeding their application in tumour therapeutic treatment.
In this work, Ang peptide‐modified functional sEVs could efficiently cross the BBB and target the brain glioma. Meanwhile, the TAT peptide‐modified sEVs can move across the cell membrane and overcome the Ang receptor saturation on the cell membrane, which can improve BBB permeation and promote tumour penetration. To load the drug into cells, sEVs need to fuse with the membrane, which may fuse directly with the plasma membrane or undergo endocytosis with the endosomal membrane. Endocytosis is believed to be responsible for en entry of the sEVs (Mulcahy et al., 2014). The sEVs are taken up by cells through a wide range of endocytosis pathways of clathrin‐dependent endocytosis and clathrin‐independent endocytosis, including phagocytosis, micropinocytosis, caveolin‐mediated, and lipid raft‐mediated uptake (Feng et al., 2010; Fitzner et al., 2011). However, whether or not the tailored/modified sEVs comply with the similar mechanism needs further investigation.
Moreover, facilitating the entry of sEVs that exhibit non‐specific biodistribution into a specific organ (liver, kidney, lungs, and spleen) is an essence for sEVs‐based drug delivery applications (Li et al., 2020; Munagala et al., 2016). The observation of < 5% of the initial dose of sEVs accumulated in the brain via intravenous injection can be ascribed to the existence of the BBB and the non‐specific targeting of the sEVs (Banerjee et al., 2019; Lai et al., 2014), which also required the specific tumour‐targeting capacity for engineered sEVs. Although the functional tumour‐targeting sEVs in this study could be enriched in the BBB and tumour region, the liver showed a strong interception effect for customised/modified sEVs. Therefore, it is believed to develop engineered sEVs that can avoid the interception and clearance by the organs, such as liver (Wiklander et al., 2015).
In this study, the engineered sEVs were derived from HEK293T cells because of their high sEVs production capacity and the ease to handle and transfect. Moreover, the CCK8 cell activity test results of our work, as well as those of other researches showed that the sEVs derived from HEK293T cells exhibit no or few significant cytotoxicity (Liu et al., 2019). However, because lentivirus was used in the construction of stably expressed cell line, its clinical application requires further validation, despite no relevant safety risks observed in the experiment. Further research on safer ways to promote the clinical application of this drug delivery system is required. Furthermore, although HEK293T cells have superior sEVs production to other cell lines, the production efficiency is still required to improve for clinical application, thus limiting the potential of sEVs for personalised drug delivery and treatment (Antimisiaris et al., 2018). Prolonging incubation times, applying higher initial sample density and changing the composition of the culture medium were the potential methods to increase the overall production yield (Yang et al., 2015). However, none of these methods resulted in any significant increase in the yield or purity of sEVs production. Thus, it is essential to develop a new strategy to remarkably increase the yield of sEVs in the future.
In summary, Ang and TAT dual peptide‐modified sEVs are a simple and natural nanoscale drug delivery platform that is highly effective in targeting chemotherapeutic drug to a mouse brain and therefore represent a promising approach for human cancer treatment. It is noteworthy that the sEVs not only show glioma cell and BBB selectivity but also can significantly enhance BBB permeation and glioma tumour penetration in the brain. Moreover, sEVs loaded with Dox can treat glioma well with very few side effects. This kind of unique engineered sEVs possess great potential in the treatment of glioma and promising strategy for future treatment of diseases of the central nervous system.
AUTHOR CONTRIBUTIONS
Lingyan Yang and Guosheng Cheng conceived and designed the study. Zhanchi Zhu, Yuanxin Zhai, Quanwei Wang, Fang Han, Wenlong Zheng, Leisha Cui and Jing Hong performed the experiments. Zhanchi Zhu, Yuanxin Zhai, Ying Hao, and Quanwei Wang analysed the results. Lingyan Yang and Guosheng Cheng provided suggestions on the project design and data presentation. Sancheng Ma and Wei Jin provided resources. Zhanchi Zhu wrote the original draft manuscript. Ying Hao, Lingyan Yang and Guosheng Cheng supervised all experiments and revised the manuscript. All authors critically discussed the results and reviewed and approved the manuscript before submission.
CONFLICT OF INTEREST
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supporting information
Supporting information.
ACKNOWLDGEMENTS
The authors thank Prof. Hua Jinlian from Northwest A&F University for providing pLVX, VSV‐G and psPAX2 plasmids, Prof. Zhiyuan Zhong from Soochow University for providing the trans‐endothelial electrical resistance (TEER) equipment. This work was supported by grants from Guangdong Basic and Applied Basic Research Foundation (2019B1515120090), National Natural Science Foundation of China (51903246, 31700831), and Natural Science Foundation of Jiangsu Province (BK20201196).
Zhu, Z. , Zhai, Y. , Hao, Y. , Wang, Q. , Han, F. , Zheng, W. , Hong, J. , Cui, L. , Jin, W. , Ma, S. , Yang, L. , & Cheng, G. (2022). Specific anti‐glioma targeted‐delivery strategy of engineered small extracellular vesicles dual‐functionalised by Angiopep‐2 and TAT peptides. Journal of Extracellular Vesicles, 11, e12255. 10.1002/jev2.12255
Contributor Information
Sancheng Ma, Email: sanchengma@hotmail.com.
Lingyan Yang, Email: lyyang2013@sinano.ac.cn.
Guosheng Cheng, Email: gscheng2006@sinano.ac.cn.
REFERENCES
- Alvarez‐Erviti, L. , Seow, Y. , Yin, H. , Betts, C. , & Lakhal, S. (2011). Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nature Biotechnology, 29, 341–345. 10.1038/nbt.1807 [DOI] [PubMed] [Google Scholar]
- Andaloussi, S. E. , Lakhal, S. , Ger, I. M. , & Wood, M. J. A. (2013). Exosomes for targeted siRNA delivery across biological barriers. Advanced Drug Delivery Reviews, 65, 391–397. 10.1016/j.addr [DOI] [PubMed] [Google Scholar]
- Antimisiaris, S. G. , Mourtas, S. , & Marazioti, A. (2018). Exosomes and exosome‐inspired vesicles for targeted drug delivery. Pharmaceutics, 10, 218. 10.3390/pharmaceutics10040218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee, A. , Alves, V. , Rondo, T. , Sereno, J. , Neves, N. , Lino, M. , Ribeiro, A. , Abrunhosa, A. J. , & Ferreira, L. S. (2019). A positron‐emission tomography (PET)/magnetic resonance imaging (MRI) platform to track in vivo small extracellular vesicles. Nanoscale, 11, 13243–13248. 10.1039/c9nr02512j [DOI] [PubMed] [Google Scholar]
- Bao, S. , Wu, Q. , Mclendon, R. E. , Hao, Y. , Shi, Q. , Hjelmeland, A. B. , Dewhirst, M. W. , Bigner, D. D. , & Rich, J. N. (2006). Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature, 7120, 756–760. 10.1038/nature05236 [DOI] [PubMed] [Google Scholar]
- Boorn, V. D. , Jasper, G. , Martin, S. , Christoph, C. , & Gunther, H. (2011). SiRNA delivery with exosome nanoparticles. Nature Biotechnology, 4, 325–326. 10.1038/nbt.1830 [DOI] [PubMed] [Google Scholar]
- Colombo, M. , Raposo, G. , & Thery, C. (2014). Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annual Review of Cell and Developmental Biology, 30, 255–289. 10.1146/annurev-cellbio-101512-122326 [DOI] [PubMed] [Google Scholar]
- Cuddapah, V. A. , Robel, S. , Watkins, S. , & Sontheimer, H. (2014). A neurocentric perspective on glioma invasion. Nature Reviews Neuroscience, 7, 455–6515. 10.1038/nrn3765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, D. , Zhao, W. , Ye, Y. , Bai, X. , Liu, R. , Chang, L. , Qiang, Z. , & Sui, S. (2010). Cellular internalization of exosomes occurs through phagocytosis. Traffic, 11, 675–687. 10.1111/j.1600-0854.2010.01041.x [DOI] [PubMed] [Google Scholar]
- Fitzner, D. , Schnaars, M. , Rossum, D. V. , Krishnamoorthy, G. , Dibaj, P. , Bakhti, M. , Regen, T. , Hanisch, U. K. , & Simons, M. (2011). Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. Journal of Cell Science, 124, 447–458. 10.1242/jcs.074088 [DOI] [PubMed] [Google Scholar]
- Gardiner, C. , Vizio, D. D. , Sahoo, S. , Théry, C. , Witwer, K. W. , Wauben, M. , & Hill, A. F. (2016). Techniques used for the isolation and characterization of extracellular vesicles: Results of a worldwide survey. Journal of Extracellular Vesicles, 5, 32945. 10.3402/jev.v5.32945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam, Y. , Leach, A. G. , Smith, J. , Pluchino, S. , Coxon, C. R. , Sivakumaran, M. , Downing, J. , Fatokun, A. A. , Teixidò, M. , & Ehtezazi, T. (2021). Physiological and pathological factors affecting drug delivery to the brain by nanoparticles. Advanced Science, 8, 2002085. 10.1002/advs.202002085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, R. K. , Tomaso, E. d. , Duda, D. G. , Loeffler, J. S. , Sorensen, A. G. , & Batchelor, T. T. (2007). Angiogenesis in brain tumours. Nature Reviews Neuroscience, 8, 610–622. 10.1038/nrn2175 [DOI] [PubMed] [Google Scholar]
- Ke, W. , Shao, K. , Huang, R. , Liang, H. , & Chen, J. (2009). Gene delivery targeted to the brain using an Angiopep‐conjugated polyethyleneglycol‐modified polyamidoamine dendrimer. Biomaterials, 30, 6976–6985. 10.1016/j.biomaterials.2009.08.049 [DOI] [PubMed] [Google Scholar]
- Kojima, R. , Bojar, D. , Rizzi, G. , Hamri, C. E. , El‐Baba, M. D. , Saxena, P. , Nder, S. A. , Tan, K. R. , & Fussenegger, M. (2018). Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson's disease treatment. Nature Communications, 9, 1305. 10.1038/s41467-018-03733-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, C. P. , Mardini, O. , Ericsson, M. , Prabhakar, S. , Maguire, C. A. , Chen, J. W. , Tannous, B. A. , & Breakefield, X. O. (2014). Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano, 8, 483–494. 10.1021/nn404945r [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Wu, J. , Wang, J. , Hu, X. , Cai, J. , & Xiang, D. (2020). Gemcitabine loaded autologous exosomes for effective and safe chemotherapy of pancreatic cancer. Acta Biomaterialia, 101, 519–530. 10.1016/j.actbio.2019.10.022 [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Fang, J. , Wang, M. , & Wang, P. (2014). Codelivery of doxorubicin and paclitaxel by cross‐linked multilamellar liposome enables synergistic antitumor activity. Molecular Pharmacology, 5, 1651–1661. 10.1021/mp5000373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , & Lu, W. (2012). Recent advances in brain tumor‐targeted nano‐drug delivery systems. Drug Delivery, 9, 671–686. 10.1517/17425247.2012.682726 [DOI] [PubMed] [Google Scholar]
- Liu, J. , Ye, Z. , Xiang, M. , Chang, B. , & Wang, Z. (2019). Functional extracellular vesicles engineered with lipid‐grafted hyaluronic acid effectively reverse cancer drug resistance. Biomaterials, 223, 119475. 10.1016/j.biomaterials.2019.119475 [DOI] [PubMed] [Google Scholar]
- Masaldan, S. , & Iyer, V. V. (2014). Exploration of effects of emodin in selected cancer cell lines: Enhanced growth inhibition by ascorbic acid and regulation of LRP1 and AR under hypoxia‐like conditions. Journal of Applied Toxicology, 34, 95–104. 10.1002/jat.2838 [DOI] [PubMed] [Google Scholar]
- Mehrotra, N. , & Tripathi, R. M. (2015). Short interfering RNA therapeutics: Nanocarriers, prospects and limitations. IET Nanobiotechnology, 9, 386–395. 10.1049/iet-nbt.2015.0018 [DOI] [PubMed] [Google Scholar]
- Mulcahy, L. A. , Pink, R. C. , & Carter, D. R. (2014). Routes and mechanisms of extracellular vesicle uptake. Journal of Extracellular Vesicles, 3, 24641. 10.3402/jev.v3.24641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munagala, R. , Aqil, F. , Jeyabalan, J. , & Gupta, R. C. (2016). Bovine milk‐derived exosomes for drug delivery. Cancer Letters, 371, 48–61. 10.1016/j.canlet.2015.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naoomi, T. , Nobuyoshi, K. , Makiko, O. , Takeshi, K. , Yusuke, Y. , Kenji, T. , Jan, L. , Hitoshi, N. , & Takahiro, O. (2015). Brain metastatic cancer cells release microRNA‐181c‐containing extracellular vesicles capable of destructing blood‐brain barrier. Nature Communications, 6, 6716. 10.1038/ncomms7716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omuro, A. , & DeAngelis, L. M. (2013). Glioblastoma and other malignant gliomas: A clinical review. Journal of the American Medical Association, 17, 1842–1850. 10.1001/jama.2013.280319 [DOI] [PubMed] [Google Scholar]
- Pan, W. , Kastin, A. J. , Zankel, T. C. , Kerkhof, P. V. , Terasaki, T. , & Bu, G. (2004). Efficient transfer of receptor‐associated protein (RAP) across the blood‐brain barrier. Journal of Cell Science, 117, 5071–5078. 10.1242/jcs.01381 [DOI] [PubMed] [Google Scholar]
- Pardridge, W. M. (2007). Blood‐brain barrier deliver. Drug Discovery Today, 12, 54–61. 10.1016/j.drudis.2006.10.013 [DOI] [PubMed] [Google Scholar]
- Rondón‐Ortiz, A. N. , Lino, C. C. L. , Jimena, M. M. , Gonzales‐Urday, A. L. , Gugnani, K. S. , Mark, B. , Maher, T. J. , & Pino‐Figueroa, A. J. (2017). High concentrations of rosiglitazone reduce mRNA and protein levels of LRP1 in HepG2 Cells. Frontiers Pharmacology, 8, 772. 10.3389/fphar.2017.00772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufino‐Ramos, D. , Albuquerque, P. R. , Carmona, V. , Perfeito, R. , & Nobre, R. J. (2017). Extracellular vesicles: Novel promising delivery systems for therapy of brain diseases. Journal of Controlled Release, 28, 247–258. 10.1016/j.jconrel.2017.07.001 [DOI] [PubMed] [Google Scholar]
- Stockinger, T. , Wirthl, D. , Mao, G. , Drack, M. , Pruckner, R. , Demchyshyn, S. , Steiner, M. , Egger, F. , Müller, U. , Schwödiauer, R. , Bauer, S. , Arnold, N. , & Kaltenbrunner, M. (2021). iSens: A fiber‐based, highly permeable and imperceptible sensor design. Advanced Materials, 33, 2102736. 10.1002/adma.202102736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry, C. , Zitvogel, L. , & Amigorena, S. (2002). Exosomes: Composition, biogenesis and function. Nature Reviews Immunology, 2, 569–579. 10.1038/nri855 [DOI] [PubMed] [Google Scholar]
- Théry, C. , Witwer, K. W. , Aikawa, E. , Alcaraz, M. , Anderson, J. D. , Andriantsitohaina, R. , Antoniou, A. , Arab, T. , Archer, F. , Atkin‐Smith, G. K. , Craig Ayre, D. , Bach, J. ‐ M. , Bachurski, D. , Baharvand, H. , Balaj, L. , Baldacchino, S. , Bauer, N. N. , Baxter, A. A. , Bebawy, M. , … Zuba‐Surma, E. K. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal Extracellular Vesicles, 7(1), 1535750. 10.1080/20013078.2018.1535750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, C. E. , Ehrhardt, A. , & Kay, M. A. (2003). Progress and problems with the use of viral vectors for gene therapy. Nature Reviews Genetics, 4, 346–358. 10.1038/nrg1066 [DOI] [PubMed] [Google Scholar]
- Tian, T. , Zhang, H. , He, C. , Fan, S. , Zhu, Y. , Qi, C. , Huang, N. , Xiao, Z. , Lu, Z. , Tannous, B. A. , & Gao, J. (2018). Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials, 150, 137–149. 10.1016/j.biomaterials.2017.10.012 [DOI] [PubMed] [Google Scholar]
- Tian, Y. , Li, S. , Song, J. , Ji, T. , Zhu, M. , Anderson, G. J. , Wei, J. , & Nie, G. (2014). A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials, 35, 2383–2390. 10.1016/j.biomaterials.2013.11.083 [DOI] [PubMed] [Google Scholar]
- Torchilin, V. P. , Rammohan, R. , Weissig, V. , & Levchenko, T. S. (2001). TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proceedings of the National Academy of Sciences of the United States of America, 98, 8786–8791. 10.1073/pnas.151247498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Tang, W. , Yang, M. , Yin, Y. , Li, H. , Hu, F. , Tang, L. , Ma, X. , Zhang, Y. , & Wang, Y. (2021). Inflammatory tumor microenvironment responsive neutrophil exosomes‐based drug delivery system for targeted glioma therapy. Biomaterials, 273, 120784. 10.1016/j.biomaterials.2021.120784 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Pang, J. , Wang, Q. , Yan, L. , Wang, L. , Xing, Z. , Wang, C. , Zhang, J. , & Dong, L. (2021). Delivering antisense oligonucleotides across the blood‐brain barrier by tumor cell‐derived small apoptotic bodies. Advanced Science, 8, 2004929. 10.1002/advs.202004929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenningmann, N. , Knapp, M. , Ande, A. , Vaidya, T. R. , & Ait‐Oudhia, S. (2019). Insights into doxorubicin‐induced cardiotoxicity: Molecular mechanisms, preventive strategies, and early monitoring. Molecular Pharmacology, 2, 219–232. 10.1124/mol.119.115725 [DOI] [PubMed] [Google Scholar]
- Wiklander, O. P. B. , Nordin, J. Z. , Loughlin, A. O. , Gustafsson, Y. , Corso, G. , Mäger, I. , Vader, P. , Lee, Y. , Sork, H. , Seow, Y. , Heldring, N. , Alvarez‐Erviti, L. , Smith, C. E. , Blanc, K. L. , Macchiarini, P. , Jungebluth, P. , Wood, M. J. A. , & Andaloussi, S. E. (2015). Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. Journal of Extracellular Vesicles, 4, 26316. 10.3402/jev.v4.26316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- William, A. , Priyanka, S. , Kristin, M. , Kim, M. , Nils, L. , & Theresa, L. (2020). Transport of extracellular vesicles across the blood‐brain barrier: Brain pharmacokinetics and effects of inflammation. International Journal of Molecular Sciences, 21, 4407. 10.3390/ijms21124407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin, H. , Jiang, X. , Gu, J. , Sha, X. , Chen, L. , Law, K. , Chen, Y. , Wang, X. , Jiang, Y. , & Fang, X. (2011). Angiopep‐conjugated poly(ethylene glycol)‐co‐poly(ε‐caprolactone) nanoparticles as dual‐targeting drug delivery system for brain glioma. Biomaterials, 32, 4293–4305. 10.1016/j.biomaterials.2011.02.044 [DOI] [PubMed] [Google Scholar]
- Xin, H. , Sha, X. , Jiang, X. , Zhang, W. , Chen, L. , & Fang, X. (2012). Anti‐glioblastoma efficacy and safety of paclitaxel‐loading Angiopep‐conjugated dual targeting PEG‐PCL nanoparticles. Biomaterials, 33, 8167–8176. 10.1016/j.biomaterials.2012.07.046 [DOI] [PubMed] [Google Scholar]
- Yang, L. , Zhai, Y. , Hao, Y. , Zhu, Z. , & Cheng, G. (2020). The regulatory functionality of exosomes derived from hUMSCs in 3D culture for Alzheimer's disease therapy. Small, 16, 1906273. 10.1002/smll.201906273 [DOI] [PubMed] [Google Scholar]
- Yang, Y. , Chen, Y. , Zhang, F. , Zhao, Q. , & Zhong, H. (2015). Increased anti‐tumour activity by exosomes derived from doxorubicin‐treated tumour cells via heat stress. International Journal of Hyperthermia, 31, 498–506. 10.3109/02656736.2015.1036384 [DOI] [PubMed] [Google Scholar]
- Yu, J. , Wang, C. , Kong, Q. , Wu, X. , Lu, J. J. , & Chen, X. (2018). Recent progress in doxorubicin‐induced cardiotoxicity and protective potential of natural products. Phytomedicine, 1, 125–139. 10.1016/j.phymed.2018.01.009 [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Zhang, H. , Gu, J. , Zhang, J. , Shi, H. , Qian, H. , Wang, D. , Xu, W. , Pan, J. , & Santos, H. A. (2021). Engineered extracellular vesicles for cancer therapy. Advanced Materials, 33, 2005709. 10.1002/adma.202005709 [DOI] [PubMed] [Google Scholar]
- Zhu, Q. , Ling, X. , Yang, Y. , Zhang, J. , Li, Q. , Niu, X. , Hu, G. , Chen, B. , Li, H. , Wang, Y. , & Deng, Z. (2019). Embryonic stem cells‐derived exosomes endowed with targeting properties as chemotherapeutics delivery vehicles for glioblastoma therapy. Advanced Science, 6, 1801899. 10.1002/advs.201801899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong, T. , Mei, L. , Gao, H. , Cai, W. , Zhu, P. , Shi, K. , Chen, J. , Wang, Y. , Gao, F. , & He, Q. (2014). Synergistic dual‐ligand doxorubicin liposomes improve targeting and therapeutic efficacy of brain glioma in animals. Molecular Pharmacology, 11, 2346–2357. 10.1021/mp500057n [DOI] [PubMed] [Google Scholar]
- Zong, T. , Mei, L. , Gao, H. , Shi, K. , & Chen, J. (2014). Enhanced glioma targeting and penetration by dual‐targeting liposome co‐modified with T7 and TAT. Journal of Pharmaceutical Science, 12, 3891–3901. 10.1002/jps.24186 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.