

Radiologists must become familiar with the appearance of amyloid-related imaging abnormalities, how to select an appropriate imaging protocol, and how to report findings in clinical practice.
SUMMARY:
Monoclonal antibodies are emerging disease-modifying therapies for Alzheimer disease that require brain MR imaging for eligibility assessment as well as for monitoring for amyloid-related imaging abnormalities. Amyloid-related imaging abnormalities result from treatment-related loss of vascular integrity and may occur in 2 forms. Amyloid-related imaging abnormalities with edema or effusion are transient, treatment-induced edema or sulcal effusion, identified on T2-FLAIR. Amyloid-related imaging abnormalities with hemorrhage are treatment-induced microhemorrhages or superficial siderosis identified on T2* gradient recalled-echo. As monoclonal antibodies become more widely available, treatment screening and monitoring brain MR imaging examinations may greatly increase neuroradiology practice volumes. Radiologists must become familiar with the imaging appearance of amyloid-related imaging abnormalities, how to select an appropriate imaging protocol, and report findings in clinical practice. On the basis of clinical trial literature and expert experience from clinical trial imaging, we summarize imaging findings of amyloid-related imaging abnormalities, describe potential interpretation pitfalls, and provide recommendations for a standardized imaging protocol and an amyloid-related imaging abnormalities reporting template. Standardized imaging and reporting of these findings are important because an amyloid-related imaging abnormalities severity score, derived from the imaging findings, is used along with clinical status to determine patient management and eligibility for continued monoclonal antibody dosing.
As clinically defined, probable Alzheimer disease (AD) dementia is estimated to affect approximately 11% of Americans aged 65 years and older, and there is a large group of patients who may be potential candidates for emerging disease-modifying therapies.1 Recently, monoclonal antibodies against beta-amyloid have become available (in clinical trials and early clinical practice) for the treatment of AD. These therapies require frequent brain MR imaging examinations to detect contraindications to treatment and to monitor for subclinical or symptomatic adverse events associated with treatment, which are used to guide decisions on dose-adjustment or discontinuation.2 Neuroradiologists will play an important role in diagnostic evaluations, which will include MRIs and either lumbar punctures or amyloid PET scans, and monitoring adverse events associated with treatment along with longer-term structural and functional effects of therapy, analogous to safety monitoring for progressive multifocal leukoencephalopathy in patients undergoing treatment for multiple sclerosis.3,4 Given the large number of AD therapeutic candidates, implementation of treatment and monitoring may greatly increase neuroradiology practice volumes. Radiologists, both neuroradiologists and generalists in private practice and academic institutions, should, therefore, be familiar with the pathophysiology of AD relevant to anti-amyloid therapy and the mechanism and appearance of amyloid-related imaging abnormalities (ARIA) that may result from treatment. In addition, knowledge of the pitfalls in interpretation of these imaging abnormalities, selection of an appropriate imaging protocol, and standardization of imaging and reporting of these findings in clinical practice are important. Use of the recommended standardized imaging protocols and reporting templates will improve ARIA detection and timely communication of findings to referring providers, ensuring optimal patient care and management.
Background: Amyloid Beta, ARIA, and Cerebral Amyloid Angiopathy
Neuropathology and Pathophysiology of Alzheimer Disease and Cerebral Amyloid Angiopathy.
The major proteinopathy that forms amyloid plaques is amyloid-beta (Aβ), specifically the 42 amino acid peptide Aβ42. Amyloid plaques are one of the 2 defining pathologic features of AD, the other being neurofibrillary tangles.5 Aβ42 is derived from proteolytic metabolism of amyloid precursor protein (APP). APP may be cleaved by α-secretase in a nonamyloidogenic pathway or by β-secretase in an amyloidogenic pathway to form α- or β-C terminal fragments that are subsequently cleaved by γ-secretase to form P3 and Aβ peptides, respectively.6 Due to differential cleavage sites, Aβ exists in many isoforms, but the 2 most relevant for this discussion are Aβ42 and Aβ40. Soluble Aβ monomers may undergo clearance via enzymatic degradation, transport across the blood-brain barrier, or efflux out of the brain via perivascular drainage pathways, which include periarterial as well as perivenous or recently detailed glymphatic drainage pathways.7,8 Soluble Aβ monomers may also aggregate into a range of successively larger protein complexes, oligomers, protofibrils, and mature fibrils, that can subsequently deposit in the brain as amyloid plaques (predominantly Aβ42) or in the vessel wall (predominantly Aβ40) and result in AD pathology and cerebral amyloid angiopathy (CAA), respectively.9 Accumulation of Aβ in vessel walls may result from and further contribute to impaired Aβ clearance and loss of vascular integrity. Aβ is, therefore, central to the development of both AD and CAA, which often co-occur.10
Rationale for Beta-Amyloid Removal as Treatment for Alzheimer Disease.
Aβ removal for treatment of AD is based on the amyloid cascade hypothesis.11 This model proposes that amyloid plaque deposition facilitates downstream pathophysiologic events including tau phosphorylation, neurofibrillary tangle formation, microglial activation, and eventually neurodegeneration and progressive cognitive decline.5,12,13 In addition, rare familial (and often young-onset) cases of AD are caused by mutations in genes with central roles in amyloid biology (presenilin 1 and 2, PSEN1 and PSEN2, and APP), further pointing to a central early role of amyloid accumulation in the disease.14 Although tau aggregation has been shown to be more closely related to neuronal loss and cognitive decline both spatially and temporally, it is thought that amyloid plaque deposition is the key initiating step in AD pathophysiology.15-17 Halting β-amyloid formation or facilitating its removal is therefore expected to decrease or halt downstream pathophysiologic processes, tau phosphorylation, tau deposition, neurodegeneration, and cognitive decline. Recent clinical trials of anti-amyloid therapies have indeed demonstrated that removal of amyloid plaques from the brain can result in short-term improvement in downstream biomarkers of tauopathy and neurodegeneration, though longer-term consequences on disease biomarkers and clinical course are not fully understood.18,19
Treatment Strategies and Origin of the Term ARIA.
Therapeutic approaches for reducing amyloid in the brain have included inhibitors of amyloid aggregation, inhibitors of β-secretase, inhibitors of γ-secretase, and immunotherapy to remove amyloid from the brain, with immunotherapy being the most extensively employed mode of action in clinical trials.20 Active and passive immunotherapy approaches have been investigated, though the use of active immunotherapy has been limited due to adverse reactions including meningocephalitis.21 Clinical trials using passive immunotherapy, exogenously administered monoclonal antibodies (mAb) against Aβ, have been ongoing for approximately 2 decades. Bapineuzumab was the first mAb to enter clinical trials. It binds the N-terminus of Aβ and clears both soluble and fibrillary Aβ. In early studies of bapineuzumab, monitoring MR imaging brain examinations showed edema and microhemorrhages in 3/10 participants.22,23 The Alzheimer’s Association Research Roundtable convened a workgroup in 2010 to provide information and recommendations regarding the imaging abnormalities encountered in the anti-amyloid trials. This workgroup termed these amyloid-related imaging abnormalities as ARIA with ARIA-E for edema or effusion and ARIA-H for microhemorrhages and hemosiderosis.24
Multiple monoclonal antibodies with variable targets and incidence of ARIA have since been developed and tested in clinical trials (Table 1).18,19,25-32 Aducanumab recently received accelerated approval by the FDA for potential clinical use in mild, symptomatic AD, based on reduction in amyloid plaque.2 Other agents (donanemab, lecanemab, gantenerumab) are currently in late-phase clinical trials and will undergo similar FDA reviews.18,19,31 The Centers for Medicare and Medicaid Services currently proposes coverage for FDA-approved anti-amyloid mAbs in Centers for Medicare and Medicaid Services–approved randomized control trials. To date, uncertainties remain on several fronts including the presence and extent of insurance coverage, the results of an anticipated Phase IV confirmatory study required by the FDA, multiple stakeholder preparedness across a wide range of clinical practices, and other factors.
Table 1:
Summary of anti-amyloid monoclonal antibodies in clinical trials
| Monoclonal Antibody | Aβ Binding Site | Amyloid Target | Completed Phase III Trials | Main Results | ARIA-E Incidencea | ARIA-H Incidencea | Active Phase III Trials |
|---|---|---|---|---|---|---|---|
| Aducanumab (Salloway et al, 2022) | N-terminus conformational epitope | Aβ oligomers, fibrils, and plaques | EMERGEENGAGE | Reduced decline in cognitive end pointsIncrease in CSF Aβ42Decrease in amyloid PET SUVR and CSF p-tau | 35.2%20.3% NC43.0% C | mH 19.1%12.4% NC22.7% CSS 14.7%6.2% NC19.1% C | EMBARKTRAILBLAZER-ALZ-4 |
| Bapineuzemab (Salloway et al, 2014)28 | N-terminus | Aβ monomers, oligomers, and fibrils | NCT00574132NCT00575055 | No effect on cognitive end pointsDecrease in amyloid PET SUVR and CSF p-tau in APOEε4 carriers | 4.2% NC15.3% C | – | – |
| Crenezumab (Guthrie et al, 2020)29 | Aβ peptides | Aβ oligomers, fibrils, and plaques | CREADCREAD 2 | No effect on cognitive end points, amyloid PET or CSF p-tauIncrease in CSF Aβ42 | 0% | 4.9% | – |
| Donanemab (Mintun et al (2021)18 | Pyroglutamate form of Aβ | Aβ plaques | TRAILBLAZER-ALZ-2 | Reduced decline cognitive end pointsDecrease in amyloid PET SUVR | 27.5%11.4% NC44.0% C | 30.5% | TRAILBLAZER-ALZ-3TRAILBLAZER-ALZ-4 |
| Ponezumab (Landen et al, 2017)30 | C-terminus | Soluble Aβ1-40 | – | No effect on cognitive end points, CSF Aβ42 or amyloid PET | 0.7% | 16.4% | – |
| Gantenerumab (Ostrowitzki et al, 2017)31 | N-terminus and central amino acids | Aβ oligomers, fibrils, and plaques | SCarlet RoADMarguerite RoAD | No effect on cognitive end points or CSF Aβ42Decrease in amyloid PET SUVR and CSF p-tau | 13.5%11.0% NC15.0% C | 16.2%11.0% NC19.4% C | GRADUATE 1GRADUATE 2DIAN-TU |
| Lecanemab (Swanson et al, 2021)19 | Aβ protofibril | Aβ protofibrils | Reduced decline in cognitive end pointsIncrease in CSF Aβ42Decrease in amyloid PET SUVR and CSF p-tau | 9.9%8.0% NC14.3% C | 10.7%4.6% NC13.1% C | CLARITY ADAHEAD 3–45 | |
| Solanezumab (Doody et al, 2014)32 | Mid-domain | Aβ monomers | EXPEDITION 1EXPEDITION 2EXPEDITION 3EXPEDITION PRO | No effect on cognitive end points, amyloid PET SUVR, or CSF p-tauIncrease CSF Aβ42 | 0.9% | 4.9% | A4DIAN-TU |
Note:—mH indicates microhemorrhage; SS, siderosis; NC, APOEε4 noncarrier; C, APOEε4 carrier; SUVR, standardized uptake value ratio; –, none or not reported.
ARIA incidence reported for the highest dose in studies with variable dosing arms. ARIA incidence reported for all participants and separately for APOEε4 noncarriers and carriers when data are available.
ARIA Mechanism.
Amyloid deposition in vessel walls (CAA) may result in loss of vascular integrity and reduced perivascular clearance and may be related to spontaneously occurring microhemorrhages.33 When anti-amyloid monoclonal antibody therapy is initiated, antibody-mediated breakdown of amyloid plaque and mobilization of parenchymal and vascular Aβ increase the load of perivascular drainage.7 The overload of perivascular drainage pathways may transiently increase amyloid deposition in the arterial wall. At the same time, antibody-mediated inflammation and breakdown of amyloid also occur in the vessel wall. These processes cause further loss of vascular integrity and blood-brain barrier breakdown.34 As a result, proteinaceous fluid and/or red blood cells leak into the parenchyma and/or leptomeningeal space and result in edema/effusion (ARIA-E) or microhemorrhages/superficial siderosis (ARIA-H).
ARIA versus CAA-Related Inflammation.
The further loss of vascular integrity and blood-brain barrier breakdown with immunotherapy against Aβ may be thought of as transient exacerbation of the effects of CAA, similar to CAA-related inflammation (CAA-RI). CAA-RI is a spontaneously occurring inflammatory condition that responds to steroid treatment or immunosuppression,35 whereas ARIA occurs secondary to monoclonal antibody therapy and generally resolves spontaneously on interruption or discontinuation of therapy. The presence of spontaneously occurring CSF autoantibodies against Aβ in patients with CAA-RI suggests that CAA and CAA-RI are a natural model for ARIA.36 CAA-RI and ARIA have similar imaging findings of sulcal effusion and edema involving the white and gray matter as well as microhemorrhages and siderosis and are best differentiated by whether or not the patient is undergoing anti-amyloid therapy.
ARIA Risk Factors.
Risk factors for ARIA are drug exposure, APOE-e4 allele carriership, and pretreatment microhemorrhages.24,27,37,38 Regarding drug exposure, the risk of ARIA was found to be greater at higher drug doses and earlier in the treatment course. It is noted that the risk for developing ARIA is reduced if patients are started at a low drug dose and progressively titrated over time to the higher final optimal treatment dose. In theory, this titration phase allows time for the cerebral vasculature to undergo the transient process of loss of structural integrity, due to amyloid removal, over a more prolonged period, thus allowing reconstitution of vascular integrity by the time the patient is titrated to higher treatment doses. As such, dose titration or dose escalation strategies have become commonplace in anti-amyloid mAb treatment studies. For example, in Phase III trials of aducanumab, the rates of ARIA were reduced with dose titration compared with nontitration. Additionally, most ARIA developed within the first 8 doses of the final target dose, during this transient phase of presumed loss of vessel wall integrity.27 APOE-e4 allele carriership remains the greatest risk factor for the development of ARIA, second only to drug dose, and is likely related to higher load of vascular amyloid and poorer vascular integrity pretreatment. In a Phase III trial of gantenerumab, ARIA-E occurred in 10.7% of homozygous APOE-e4 carriers, 5.4% of heterozygous carriers, and 1.8% of noncarriers. Similarly, ARIA-H occurred in 32.0% of homozygous carriers compared with 19.8% in heterozygous carriers and 12.3% in noncarriers.31 The presence of pretreatment hemosiderin products most consistent with CAA, lobar microhemorrhages and superficial siderosis, is a serious imaging risk factor predictive of ARIA with the use of anti-amyloid mAb therapies, particularly in APOE-e4 carriers.39,40 Due to the increased risk of adverse events in homozygous APOE-e4 carriers, APOE-e4 testing could be considered before drug initiation and could be used to help determine the frequency of safety monitoring examinations in future, updated treatment guidelines.
Additionally, in clinical trials, the incidence of ARIA has varied with the Aβ binding site and targeted Aβ structure (Table 1). ARIA incidence was higher with mAbs that bind the N- versus C-terminus and target aggregated-versus-soluble forms of Aβ.
Conversely, risk factors for CAA are AD pathology and genetic factors that promote AD pathology, namely APOE-e4, Down syndrome, PSEN1, PSEN2, and APP mutations.41 Note, vessel-related risk factors for ARIA relate to amyloid deposition and as with CAA are not related to common vascular risk factors such as hypertension, hyperlipidemia, diabetes mellitus, or the severity of atherosclerosis.
ARIA-E
Imaging Appearance.
The E in ARIA-E stands for edema, effusion, and exudate. A leak of proteinaceous fluid into the parenchyma results in edema, with the imaging appearance similar to that of vasogenic edema and best visualized on a T2-FLAIR sequence (Figs 1 and 2). T2-hyperintense signal occurs in the white matter, gray matter, or both. There may be associated local mass effect and gyral swelling. Findings may be differentiated from cytotoxic edema by absent diffusion restriction; intense diffusion restriction associated with an acute infarct is not a characteristic of ARIA. When the leak occurs in the leptomeningeal space, the result is a sulcal effusion or exudate, only appreciated on T2-FLAIR sequences due to T1-shortening related to proteinaceous content (Fig 3). ARIA-E may present as either parenchymal edema or sulcal effusion, or both may occur together; sulcal effusion was the most common manifestation of ARIA-E in some mAb trial analyses, and parenchymal edema, in others.42,43 ARIA-E most commonly affects the occipital lobes followed by the parietal, frontal, and temporal lobes and, least frequently, the cerebellum. The intensity and size of the signal abnormality are variable, from subtle small, 1- to 2-cm zones of cortico-subcortical abnormality to multifocal-to-near hemispheric signal T2-hyperintense signal alterations.24,42 These regions of signal abnormality generally have ill-defined margins, though they may infrequently have circumscribed margins and mimic a neoplastic lesion (Fig 4).
FIG 1.

Dynamic and transient nature of ARIA-E. Axial T2 FLAIR images over 3 sequential time points for a patient undergoing anti-amyloid monoclonal antibody therapy. On the postdosing examination (middle, red circle), there is FLAIR hyperintensity involving the left superior frontal cortex and subcortical white matter measuring <5 cm in transverse dimension (mild ARIA-E) that is new from the baseline examination. On the 1-month postdosing follow-up examination, performed to reassess the ARIA-E, the left frontal FLAIR hyperintensity had resolved, as is typically seen and in keeping with the transient nature of ARIA-E. Images courtesy of Biogen.
FIG 3.
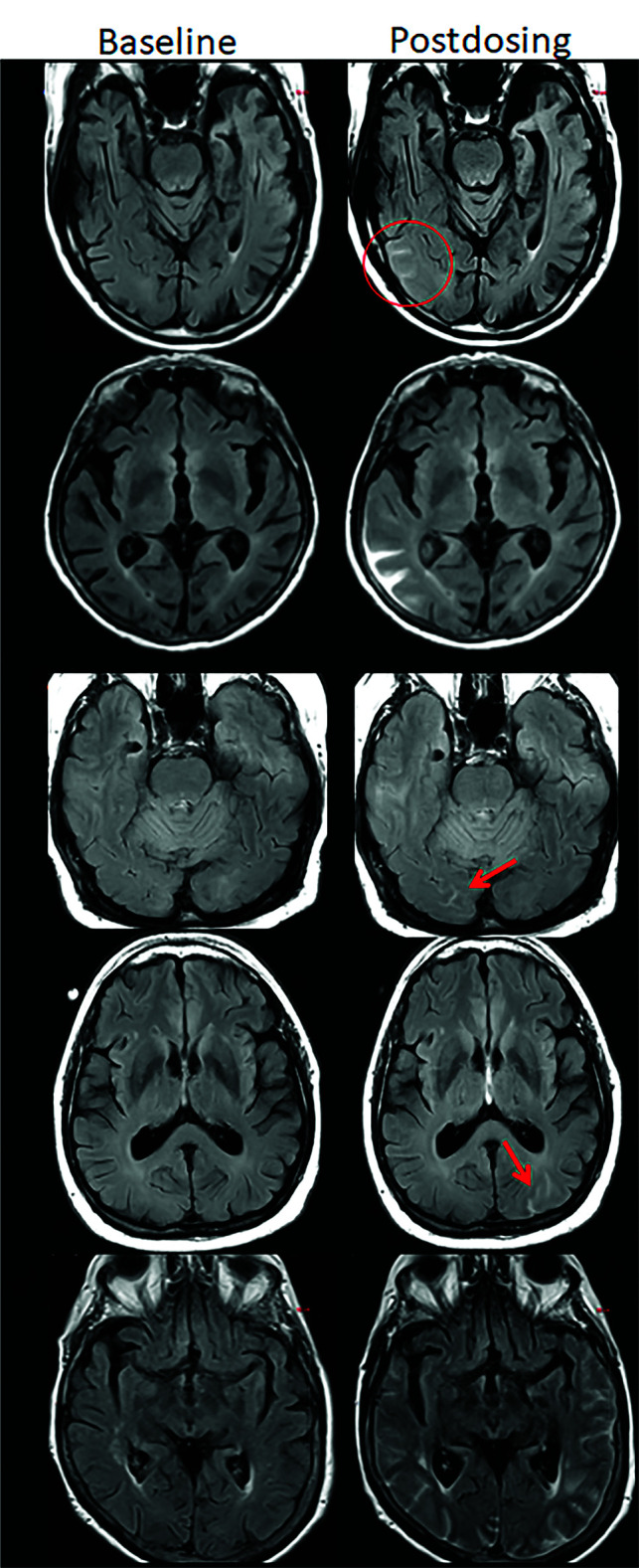
ARIA-E, sulcal effusion. Axial T2-FLAIR images from 4 separate patients at pretreatment baseline (left) and on a monitoring examination following initiation of anti-amyloid monoclonal antibody therapy (postdosing, right). A, Compared with the baseline examination, new sulcal T2-FLAIR hyperintense signal in the right temporal-occipital lobe measuring <5 cm in transverse dimensions (mild ARIA-E, red circle). B, Postdosing, new T2-FLAIR sulcal effusion involving the right posterior temporal and parietal lobes measuring 5–10 cm (moderate ARIA-E). C, Subtle multifocal, bi-occipital, sulcal effusion on the postdosing examination, each region measuring <5 cm (moderate ARIA-E, red arrows). A single region of ARIA-E measuring <5 cm would be classified as mild, but >1 cm yields a classification of moderate. Multiplicity of ARIA-E involvement yields a classification of moderate, as long as each region is <10 cm in diameter. Identification of these subtle abnormalities requires careful comparison with prior monitoring and/or baseline examination. D, Postdosing, extensive T2-FLAIR sulcal effusion involving the bilateral temporal and occipital lobes measuring ≥10 cm in extent (severe ARIA-E). Images courtesy of Biogen and the Dominantly Inherited Alzheimer Network.
FIG 4.
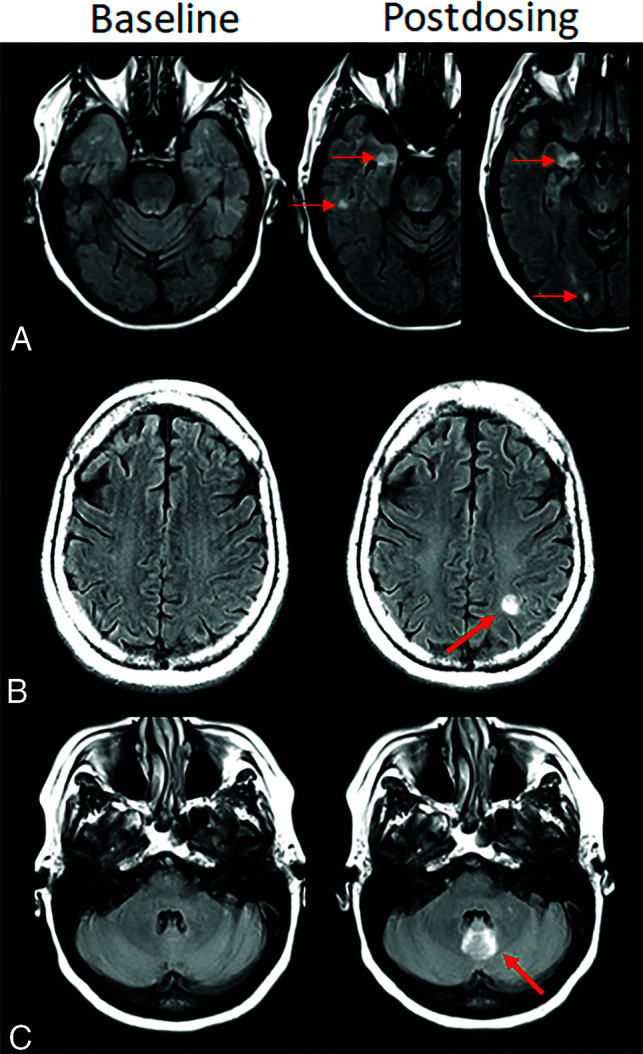
Atypical ARIA-E, parenchymal edema. Axial T2-FLAIR images from 3 separate patients at pretreatment baseline (left) and on a monitoring examination following initiation of anti-amyloid monoclonal antibody therapy (postdosing, right). A, Adjacent slices on postdosing T2-FLAIR show development of multiple nodular areas of ARIA-E (red arrows). This nodular presentation is less commonly encountered in contrast to the typical ARIA-E, which has an amorphous parenchymal pattern as expected for vasogenic edema. In this case, although each area of ARIA-E is small (<5 cm), the multiplicity of lesions yields a classification of moderate ARIA-E. B, Atypical ARIA-E as a rounded focus of T2-FLAIR hyperintense signal in the left parietal white matter (left, red arrow) that may be mistaken for neoplastic process and differentiated by the time course of appearance coinciding with monoclonal antibody dosing and subsequent resolution. C, Atypical ARIA-E in the cerebellar vermis. Postdosing, new T2-FLAIR hyperintense signal in the cerebellar vermis (red arrow), a less common location for ARIA-E relative to the cerebral hemispheres. Although ARIA-E has a slight predilection for the parieto-occipital lobes, similar to posterior reversible encephalopathy, any part of the brain may be affected. Images courtesy of Biogen and the Dominantly Inherited Alzheimer Network.
Both the edema and effusion/exudate of ARIA-E are transient and typically resolve over time upon interruption or discontinuation of anti-amyloid therapy (and have even been observed to resolve under continued dosing).44
Interpretation Pitfalls.
Imaging experience in clinical trials has provided insight into potential interpretation pitfalls in the assessment of ARIA-E. Any condition that results in T2-FLAIR hyperintensity, such as incomplete water suppression, susceptibility artifact, etc, may serve as an ARIA-E mimic (Fig 5). Shading artifacts and scanner or sequence variability may make identification and interpretation of ARIA-E-versus-artifacts difficult. Shading artifacts may occur when prescan normalization is inadvertently turned off or the patient is not centered in the receive coil, resulting in artifactually bright regions. When this occurs focally on the T2-FLAIR sequence, the artifacts may simulate ARIA-E, particularly when occurring in the occipital lobes where ARIA-E is most common. The occipital white matter signal may also vary with MR imaging scan vendor or field strength.45 If a patient is imaged on different scanners, it may be difficult to distinguish true ARIA-E versus technical variation (Fig 5). Similarly, white matter signal may differ with scan technique, such as the use of 3D-versus-2D FLAIR.46 CSF suppression may be suboptimal, and CSF may remain very high in signal in the presence of large susceptibility or due to inflow phenomena.47
FIG 5.

ARIA-E interpretation pitfalls and mimics. A, Shading artifacts with prescan normalize inadvertently turned off, results in T2-FLAIR hyperintense signal in the bilateral occipital white matter (red arrows) that mimics ARIA-E (edema). B, Axial T2-FLAIR images from 2 time points with the 2 scans performed on different vendors. T2-FLAIR hyperintense signal in the bilateral occipital white matter on vendor 2 (red arrows) appears to be new from the prior examination on vendor 1 and may be mistaken for subtle ARIA-E. The participant returned for repeat imaging on vendor 1, and the apparent abnormality was resolved (not shown). C, Diffuse sulcal T2-FLAIR hyperintense signal with administration of supplemental oxygen mimics ARIA-E effusion. The abnormality resolved on repeat imaging without supplemental oxygen. D, Poor CSF suppression results in artifactual sulcal T2-FLAIR hyperintense signal mimicking ARIA-E (red arrow) and was confirmed to be artifacts by resolution on immediate repeat imaging with optimized parameters. E, Susceptibility artifacts from a hearing aid results in apparent T2-FLAIR hyperintense signal (red arrow), new from the prior examination on which the patient’s hearing aid was not in place. The third image in this set is the GRE scan showing the marked signal void artifacts from the hearing aid. The resulting susceptibility effect results in incomplete water suppression on T2-FLAIR, and the resulting artifacts mimic ARIA-E. F, Patient with left occipital subcortical T2-FLAIR hyperintense signal on the postdosing monitoring examination (mild ARIA-E, circle), which resolved on the follow-up monitoring scan. Separate subcentimeter focus of periventricular T2 signal with associated diffusion restriction (arrow, left) was consistent with an incidental acute/subacute infarct that showed expected evolution on the postdosing follow-up examination (right). Images courtesy of Biogen and the Dominantly Inherited Alzheimer Network. Supp indicates supplemental.
Other entities may simulate ARIA. Posterior reversible encephalopathy syndrome may similarly have T2-hyperintense signal involving the white and gray matter, co-occurring hemorrhage, and a predilection for the occipital lobes, though often with a more near-symmetric parasagittal distribution.48 A subacute infarct that no longer demonstrates diffusion restriction may be difficult to differentiate from the parenchymal edema of ARIA-E in the absence of prior imaging from the acute stage or a history of focal neurologic deficit. Incomplete water suppression on FLAIR, oxygen supplementation, subarachnoid hemorrhage, and other entities that cause FLAIR hyperintense sulcal signal may mimic ARIA-E sulcal effusion.49 Although these entities have overlapping radiographic features, when clinical history is available, they may be differentiated from ARIA by the absence of prior anti-amyloid therapy and, in some cases, the presence of clinical symptoms.
Comparison with the baseline, pretreatment T2-FLAIR study is important in ARIA-E detection. ARIA-E that is subtle or that occurs in the setting of extensive small-vessel disease, particularly in a peripheral pattern, may only be appreciated when a careful comparison is made with the baseline T2-FLAIR study. In other cases, the normal compact configuration of sulci with faint cortical/leptomeningeal hyperintensity may mimic an area of ARIA-E, when this is simply normal anatomy confirmed as present at the baseline scan. Subtraction imaging may help detect subtle ARIA-E cases.50
Spontaneous Edema/Effusion versus Treatment-Emergent ARIA-E.
ARIA-E is not uncommon in clinical trials of mAbs. For example, this was identified in 17% of participants in the bapineuzumab treatment arm and 35% of the aducanumab treatment arm, with incidence varying with treatment and patient-related factors described above.2,23 On the other hand, spontaneous, transient edema is rare and was reported in 0.05% of baseline examinations for early anti-amyloid trials and 3% of the aducanumab placebo arm.2,43
Clinical Consequences.
On the basis of available clinical trial data and experience, ARIA-E is most frequently detected on routine, protocol-specified, surveillance MRIs in patients who are clinically asymptomatic. When ARIA-E is symptomatic, the symptoms are most commonly nonlocalizing, such as headache or confusion, but can additionally include visual disturbances, visuospatial impairment, or praxis difficulties in view of the relative predilection for posterior involvement of ARIA-E.28 Like the imaging findings, the clinical symptoms are generally expected to resolve over time with treatment pause or withdrawal. In the rare event of symptomatic ARIA-E or in cases with asymptomatic radiographically severe ARIA-E, treatment with intravenous methylprednisolone and possibly other therapies (eg, antihypertensives, anti-seizure medications) may be indicated on the basis of anecdotal reports.51
ARIA-H
Imaging Appearance.
ARIA-H, hemorrhage, includes microhemorrhages and superficial siderosis. When a leakage of heme products occurs in the parenchyma, microhemorrhages develop. Microhemorrhages are punctate, rounded, and markedly hypointense foci in the brain parenchyma on T2* sequences, measuring <10 mm in diameter52 (Fig 6). A leak of heme products into the leptomeningeal or subpial space results in superficial siderosis, which manifests as curvilinear hypointensity along the brain surface (Fig 7). Lobar macrohemorrhage (focus of hemorrhage identifiable on T1- or T2-weighted imaging, and usually >10 mm in diameter on gradient recalled-echo [GRE]) rarely occurs with anti-amyloid agents, and when it does, it may be the result of an underlying disease process such as CAA.
FIG 6.

ARIA-H, microhemorrhage. Baseline and postdosing GRE (left, A–C) and T2-FLAIR (right, A–C) for 3 patients. A, Postdosing, few (<5) new peripheral left frontal microhemorrhages (mild ARIA-H, red circle) that occur with new patchy T2-FLAIR hyperintense signal in that region (mild ARIA-E, red circle). B, Postdosing, 5 treatment-emergent microhemorrhages (moderate ARIA, red circle) that occurred with regional mild ARIA-E edema (red circle). C, Postdosing, ≥10 new microhemorrhages (severe ARIA-H, red circle). Associated extensive right cerebral hemisphere T2-FLAIR hyperintense signal involving the cortex and subcortical white matter with mass effect and midline shift (severe ARIA-E). D, At least 12 treatment-emergent cerebral microhemorrhages (severe ARIA-H, red arrows) without ARIA-E. In comparison with case C, these microhemorrhages are scattered, rather than clustered. Regional distribution of microhemorrhages may vary, and both cases C and D are severe ARIA-H and would prompt discontinuation of anti-amyloid therapy per current guidelines. Images courtesy of Biogen and the Dominantly Inherited Alzheimer Network.
FIG 7.
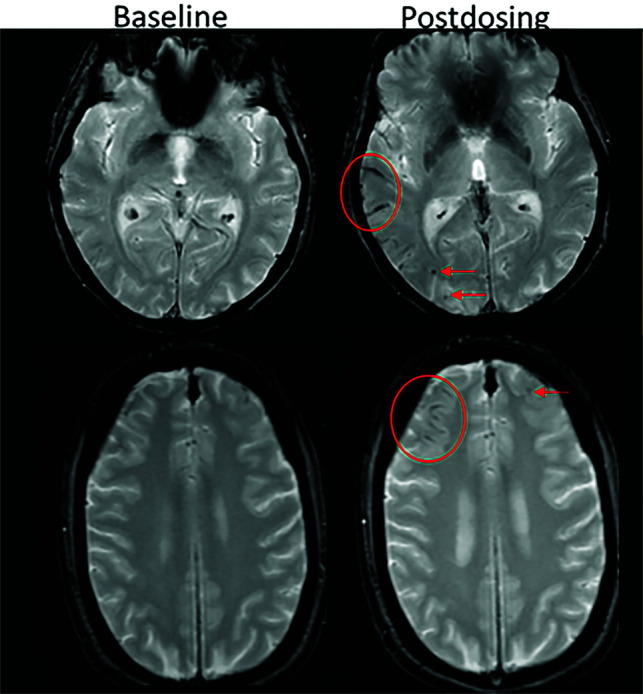
ARIA-H, superficial siderosis. Axial T2*-GRE imaging from 2 patients at baseline and postdosing. A, Postdosing, new right temporal superficial siderosis, which involves contiguous sulci when viewed over multiple slices (mild ARIA-H, siderosis, red circle). This patient also had 2 treatment-emergent microhemorrhages in the right occipital lobe (mild ARIA-H, microhemorrhage, red arrows). B, Two regions of treatment-emergent superficial siderosis in the right greater-than-left frontal lobes (moderate ARIA-H, red circle and arrow). Images courtesy of Biogen.
ARIA-H is detected on heme-sensitive sequences, ie, T2* GRE and SWI. SWI achieves increased sensitivity for microhemorrhage detection by generating both magnitude and phase images and multiplying the magnitude image by the phase image.52 Sensitivity to the detection of ARIA-H is also increased by higher field strength, longer TEs, and lower readout bandwidth. Improved spatial resolution on SWI sequences compared with 2D GRE decreases partial volume effects, which may obscure microhemorrhages, but also results in a reduction in the SNR. The imaging sequence, sequence parameters, scanner, and field strength all affect the sensitivity for detection of microhemorrhages. As discussed below, counting the number of microhemorrhages is a key component of ARIA assessment; therefore, a standardized imaging protocol is needed.
Interpretation Pitfalls.
There are both patient-related and acquisition-related interpretation pitfalls or difficulties with ARIA-H and, in particular, microhemorrhage detection (Fig 8). Blurring due to patient motion may impair detection of small microhemorrhages. Areas of prominent air-tissue susceptibility effects may induce punctate artifacts that look similar to microhemorrhages, especially near the frontal sinuses, mastoid air cells, and skull base. Susceptibility-related signal loss from physiologic mineralization in the basal ganglia may be misinterpreted as microhemorrhages and should not be incorporated into the overall microhemorrhage count. Bulk susceptibility effects that produce signal loss may preclude evaluation of the inferior temporal and anterior frontal lobes. Partial volume effects may cause a small microhemorrhage to be poorly seen or to have a variable appearance on serial examinations. Thick-section acquisitions may also make it difficult to distinguish a microhemorrhage from a vessel flow void.53 A flow void should be able to be tracked across multiple contiguous slices, though a vessel image in profile may mimic microhemorrhage on a single section. T2-weighted images can be useful for comparisons, as flow voids in vessels do not have a blooming effect. Reader biases may also affect interpretation, as readers have been found to undercall possible microhemorrhages in a patient without other microhemorrhages and to overcall in patients with many.
FIG 8.

ARIA-H interpretation pitfalls. A, Motion and partial volume effects (right) result in poor visualization of a previously documented right frontal microhemorrhage (left, red arrow). B, Vessel in profile mimics a microhemorrhage on a single section (left, red arrow) but can be traced as a vessel flow void on adjacent slices (right, arrow). C, Deep gray mineralization is often confluent and ill-defined (left) and may be clearly differentiated from a microhemorrhage. However, when punctate, senescent mineralization may mimic a microhemorrhage (right, arrow). D, Bulk susceptibility effects preclude evaluation of the inferior temporal lobes adjacent to the mastoids and sinuses (arrows). E, Susceptibility artifacts may appear as punctate foci (arrows) adjacent to obvious susceptibility areas. Therefore, punctate foci of susceptibly in these regions should be interpreted with caution and in correlation with prior examinations. F, Phase artifacts, especially about the torcula (red arrows), can mimic microhemorrhages/siderosis and may be differentiated by recognition of the shape of the torcula repeating in the phase direction. Images courtesy of Biogen.
Microhemorrhages: Spontaneous versus Treatment-Emergent versus Cerebrovascular Disease.
The treatment-emergent microhemorrhages of ARIA-H have a peripheral (lobar) predilection and most commonly occur in the cortex and gray-white matter junction as well as the cerebellum, similar to where ARIA-E may occur. These microhemorrhages have the same morphologic appearance and distribution as those that occur spontaneously and characterize and define CAA. Importantly, these areas of hemosiderin deposition typically occur in different brain areas from those related to vascular risk factors, namely hypertension, which most commonly occur in the deep white matter, deep gray matter, and brainstem.54,55 In the current clinical trials and the aducanumab FDA label ARIA severity grading, all microhemorrhages, regardless of location and suspected etiology, were included in the count used to determine eligibility and continuation of anti-amyloid therapy.2 Of note, although discrete microhemorrhages in the deep gray structures are included in the ARIA assessment, ill-defined susceptibility in the basal ganglia related to senescent change or mineralization (Fig 8) is not. In future clinical trials and clinical practice, we recommend that the inclusion of microhemorrhages in the deep gray matter and brainstem in the count used to determine treatment eligibility be reassessed, as these are most likely secondary to vascular risk factors and not related to amyloidosis. The differentiation of microhemorrhages most likely secondary to vascular risk factors from those related to amyloidosis and an increased risk of adverse events with mAbs may be particularly important as mAb use expands to clinical practice and inclusion of patient populations with higher prevalence of vascular risk factors than clinical trial populations.56
Spontaneous microhemorrhages are relatively common in elderly persons, with a prevalence of up to 15%–30% in memory clinic and AD cohorts and a similar prevalence in baseline MR imaging examinations for mAb clinical trial enrollment.43,57,58 The prevalence of spontaneous superficial siderosis is considerably lower (prevalence 0.4% versus 12.8% for superficial siderosis versus lobar microhemorrhages in the Framingham and Rotterdam cohorts), though it may be as high as 5% in AD cohorts.59,60 The incidence of mAb treatment-related microhemorrhages, ARIA-H, has been variable in clinical trials, for example 4.9% in a trial of solanezumab and 30.5% for donanemab.18,32
Clinical Consequences.
ARIA-H is generally not associated with clinical sequelae, similar to microhemorrhages and superficial siderosis when occurring independent from anti-amyloid therapy. This is in contrast to macrohemorrhages, which are associated with brain tissue injury and potential clinical consequences. In the general community population, the presence of microhemorrhages and superficial siderosis has been shown to be associated with a slightly increased risk of stroke and macrohemorrhage.57,60 In clinical trials, the presence of these hemosiderin products at baseline conferred an increased risk for adverse events with the use of anti-amyloid mAb therapies.39 Although data currently available from clinical trials do not support a significantly higher frequency of lobar macrohemorrhage with anti-amyloid therapy, a slightly elevated risk of lobar macrohemorrhage during treatment amid the wider population with AD may need to be taken into consideration when judging treatment candidacy amid other factors.
Co-Occurrence of ARIA-E and -H.
ARIA-E and ARIA-H may be temporally or spatially associated (Fig 9), or they may be detected independently. For example, in the bapineuzumab trial, ARIA-H occurred in approximately 50% of participants with ARIA-E though often not simultaneously and either before or after ARIA-E.36
FIG 9.

ARIA-E and -H. Patient on anti-amyloid therapy who developed ARIA-E (T2-FLAIR, A–D) and ARIA-H microhemorrhages (T2*-GRE, E–H). On postdosing T2-FLAIR (B), new left parietal T2-FLAIR hyperintense signal involving the cortex and subcortical white matter with associated local mass effect, consisting of sulcal effacement and gyral expansion. Despite suspension of dosing, the extent of T2-FLAIR abnormality and mass effect progressed at the postdosing follow-up No. 1 (1 month). ARIA-E resolved by postdosing follow-up No. 2 (2 months). On postdosing T2*-GRE, 2 new, treatment-emergent microhemorrhages (mild ARIA-H, F, red arrows) that increased in number over postdosing follow-up examinations with severe ARIA-H at postdosing follow-up No. 2 (H, red circle).
As discussed above, both ARIA-E and H share a common mechanism relating to increased vascular permeability, but MR imaging appearance depends on the composition of the leakage products. It is thought that proteinaceous fluid also leaks any time there is leakage of red blood cells or that ARIA-E occurs, to some extent, with any ARIA-H. However, isolated ARIA-H may be detected as ARIA-E is transient and resolves over the course of weeks to months, while the hemosiderin deposition of ARIA-H typically does not resolve.27 In the authors’ experience, the detection of isolated ARIA-H should prompt the imager to pay particular attention to the T2-FLAIR sequence in this area for subtle ARIA-E; treatment-emergent ARIA-H will often be the harbinger of subtle ARIA-E, which will only be appreciated with directed focus to this area. Note, although microhemorrhages are thought to not resolve, when incident ARIA-H is detected in the acute phase, these fresh blood-degeneration products may become less apparent on subsequent imaging, due to some degree of resorption.
In contrast, ARIA-E may occur without ARIA-H if not enough red blood cells have leaked into the extracellular or subarachnoid space or if not enough time has passed to allow heme product degradation to affect T2* signal. In summary, the recognition of ARIA-H or ARIA-E independently is likely due to the timing of imaging relative to the time of the vascular leak.
ARIA Imaging in Clinical Trials
Clinical Trial Imaging Protocols.
Standardization of imaging protocols is employed in clinical imaging trials to obtain consistent ascertainment sensitivity and performance across sites and serial participant visits. As recommended by Sperling et al,24 mAb clinical trials have used axial T2-FLAIR for detection of ARIA-E and axial T2* GRE for detection of ARIA-H.
Baseline/Screening Imaging.
Before therapy initiation, brain MR imaging is required to evaluate for pre-existing hemosiderin deposition associated with increased risk of adverse treatment events and provide baseline for comparison on the subsequent safety-monitoring examinations. In clinical trials, common exclusion criteria are ≥5 microhemorrhages or any superficial siderosis on the baseline MR imaging. The recent FDA guidance for clinical use of aducanumab lists exclusion criteria as ≥10 microhemorrhages, any superficial siderosis, or parenchymal hemorrhages measuring >1 cm in the prior year.2 The baseline imaging should be performed using the same MR imaging protocol as used for the subsequent safety-monitoring examinations.
ARIA Severity Grading on Monitoring Examinations.
Different ARIA grading schemes have been suggested.42,61 However, a specific scheme was included in the recent FDA guidance for clinical use of aducanumab.2 Given the FDA endorsement, this scheme is likely to become a default standard at least as a starting point. In this framework, ARIA-E, ARIA-H microhemorrhage, and ARIA-H superficial siderosis are each categorized by radiographic severity (Table 2 and Figs 2, 3, 6, and 7). ARIA-E scoring was defined as mild: 1 site of sulcal or cortical/subcortical FLAIR hyperintense signal measuring <5 cm; moderate: 1 site measuring 5–10 cm or >1 site each measuring <10 cm; and severe: 1 or more sites measuring >10 cm.
Table 2:
ARIA severity gradinga
| Radiographic Severity |
|||
|---|---|---|---|
| Mild | Moderate | Severe | |
| ARIA-E(sulcal and/or cortical/subcortical FLAIR hyperintensity) | 1 Location < 5 cm | 1 Location 5–10 cm OR>1 Location each <10 cm | 1 more location > 10 cm |
| ARIA-H (microhemorrhage) | ≤4 | 5–9 | ≥10 |
| ARIA-H (superficial siderosis) | 1 Focal area | 2 Focal areas | >2 Focal areas |
ARIA is graded on the basis of treatment-emergent events. For ARIA-H, this count includes cumulative new microhemorrhages or regions of siderosis compared with the baseline, pretreatment examination.
FIG 2.
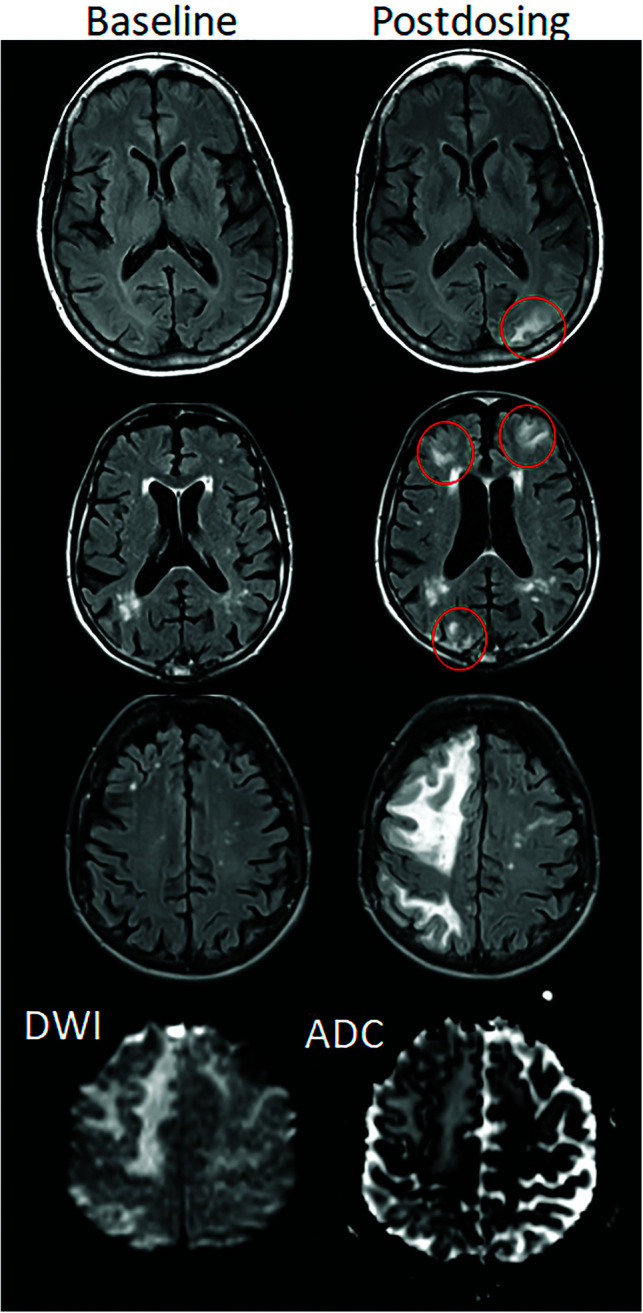
ARIA-E, parenchymal edema. Axial T2-FLAIR images from 3 separate patients at the time of the pretreatment baseline (left) and on a monitoring examination following initiation of anti-amyloid monoclonal antibody therapy (postdosing, right). A, On the postdosing examination, new T2-FLAIR hyperintense signal in the left parieto-occipital subcortical white matter with mild local mass effect and sulcal effacement measuring <5 cm the transverse dimension (mild ARIA-E, red circle). B, New multifocal, patchy T2-FLAIR hyperintense signal in the bifrontal and right occipital subcortical white matter on the postdosing examination, each region measuring <5 cm (red circles). A single region measuring <5 cm would be classified as mild, but >1 yields a classification of moderate ARIA-E. Multiplicity of ARIA-E involvement yields a classification of moderate, as long as each region is <10 cm in diameter. In some regions, there is involvement of the cortex, mild local mass effect, and gyral swelling. C, On the postdosing examination, development of extensive T2-FLAIR hyperintense signal throughout the right frontal and parietal lobes measuring >10 cm (severe ARIA-E). Associated mass effect and sulcal effacement throughout much of the right cerebral hemisphere. Hyperintense signal on DWI (lower left) is confirmed to be T2 shinethrough on the ADC map (lower right), differentiating ARIA-E from acute ischemia or other cause of cytotoxic edema. Images courtesy of Biogen and the Dominantly Inherited Alzheimer Network.
To assess ARIA-E extent, a measure is made along the single greatest dimension of the lesion. This measurement should encompass both parenchymal and sulcal T2-FLAIR hyperintensities as well as any related gyral swelling and sulcal effacement. Measuring in the plane of acquisition should be relevant in most situations. For lesions that have a greater through-plane extent, ie, craniocaudal extent, one may determine lesion size by reformatting data into another plane or estimating extent on the basis of the number of slices on which the abnormality is identified. Each lesion, which is separate and distinct (eg, separated by normal brain tissue, sulci, and gyri), should be measured separately. In other words, reporting the number of sites involved does not need to account for anatomic locations but should only rely on the presence or absence of physically separated lesions. If the lesion spans multiple contiguous brain lobes, it should still be counted as a single location. If there are lesions in both hemispheres, they should be reported as separate locations, as measurements would not cross the midline.
Microhemorrhages were scored using the cumulative number of treatment-emergent microhemorrhages: mild: ≤ 4; moderate: 5–9; and severe: ≥ 10 new microhemorrhages since the baseline examination. Superficial siderosis was scored as cumulative regions of treatment-emergent regions of siderosis: mild: 1; moderate: 2; severe: > 2 new areas of superficial siderosis since the baseline, pretreatment examination. As the presence of any siderosis should exclude a patient from treatment, cumulative treatment-emergent regions of siderosis should correspond with total regions of siderosis present. On the other hand, a patient may have up to 4 or 9 microhemorrhages before treatment, depending on the exclusion criteria implemented, and these would not be included in the count for ARIA-H severity grading. As with ARIA-E, the number of involved regions of superficial siderosis is important, and each involved site, region, or area is similarly defined as a physically separate region of contiguous sulcal signal abnormality.
Clinical Management Based on ARIA Findings.
In deciding to continue with treatment dose and dose escalations, 2 metrics were employed in the clinical trials and are described in the aducanumab FDA label (Fig 10).2 The patient had to be asymptomatic, and the following imaging criteria had to be met to continue with dosing. For asymptomatic patients, dosing was continued with mild ARIA-E and/or mild ARIA-H, but dosing was suspended in patients with moderate ARIA-E and/or moderate ARIA-H. Once dosing was suspended due to imaging findings, serial imaging was performed monthly, and dosing was resumed following the resolution of ARIA-E and stabilization of ARIA-H. If ARIA was associated with the symptoms, dosing resumed only after both the resolution of clinical symptoms and the resolution of ARIA-E and stabilization of ARIA-H. Dosing was permanently discontinued in participants with severe ARIA-H (≥10 treatment-emergent microhemorrhages or >2 areas of treatment-emergent superficial siderosis) or a macrohemorrhage (>10 mm in diameter). Real-world clinical practice guidelines may require adjustments to these protocols on the basis of further experience and depending on the extent to which the population of individuals treated with mAbs in wider practice resembles the target population in the trials.
FIG 10.

Patient management based on ARIA severity and clinical symptoms. ARIA-H management rules are the same for each severity of microhemorrhages and superficial siderosis. C (green) indicates continue dosing at current dose and schedule; S (yellow), suspend dosing; resume dosing at same dose once ARIA-E resolved or ARIA-H stable and clinical symptoms resolve; D (red), discontinue dosing; Serious (other), medical event unrelated to anti-amyloid therapy.
Considerations for Neurology Practice
Developments in anti-amyloid mAbs for AD herald a potential shift in management of an exceptionally common and devastating disease. These new anti-amyloid agents are the first potentially disease-modifying agents for AD. In contrast, current mainstays of therapy for AD are not disease-modifying but, instead, are simply symptomatic and supportive in nature, predominantly related to lifestyle and environmental modifications and use of pharmacotherapies (eg, cholinesterase inhibitors), which target symptoms but do not alter the disease itself (which continues to progress). As a result, current neurology practice operations and infrastructure may need to rapidly evolve to accommodate emerging therapeutics that have very different profiles (relating to adverse effects, financial and nonfinancial costs, test and visit types, and frequencies for treatment initiation and monitoring) from existing options.
Several points of interface between neurology and radiology are relevant in this space. There is broad agreement among cognitive/behavioral neurology subspecialists that appropriate application of anti-amyloid therapies would, at minimum, require biomarker evidence that AD is the etiology for a patient’s cognitive impairment.62,63 As a result, sites may need to plan for increased volumes of lumbar punctures (for CSF biomarker studies) and/or amyloid PET scans as part of initial evaluations. This context could impact logistics and personnel (to ensure appropriate expertise) around fluoroscopically guided lumbar punctures and PET tracer and scanner access. Multidisciplinary input from experts at local sites could also help to inform appropriate candidacy.
In addition, given the need for regular monitoring for ARIA, practices will also benefit from anticipating an increase in MR imaging examinations to prevent delays for patients receiving treatment, including patients in whom symptoms potentially necessitate more urgent evaluation. As part of this, clinical practices will need to decide on the most appropriate timeline for scheduled safety MR imaging scans, with expert recommendations currently favoring more frequent scanning (particularly early in the course of treatment) in line with protocols from existing Phase III trials. Currently, in most clinical trials, anti-amyloid dosing occurs on a monthly basis. MR imaging is performed at baseline, and at weeks 14 and 22 as the patients are being titrated to the maximal drug dose. Subsequently, monitoring MR images are taken a month after receiving the maximal maintenance dose and every 3 months thereafter while patients are on this maintenance dose.
In rare cases in which hospitalization for ARIA may be required, access and education will be crucial, particularly given the subtleties of ARIA detection and distinctions with management (principally related to drug discontinuation and therapeutic steroids) in comparison with other mimics.
More broadly, it is crucial to remember that the collective clinical experience with anti-amyloid mAbs is in its relative infancy and thus far is mostly restricted to clinical trial populations, which may not fully reflect real world practice within the broader population having symptomatic AD. In addition, there are ongoing trials testing an anti-amyloid mAb in asymptomatic individuals with intermediate (A3 trial) and elevated (A45 trial) brain amyloid loads,64 and results from these trials may further influence the timing and target population for therapy. As such, neurology and radiology providers will benefit from being collaborative and nimble over the coming period.
ARIA Imaging in Clinical Practice
Role of the Radiologist in Clinical Management.
Detailed description and quantification of ARIA findings will be used in guiding patient therapeutic dosing, and changes over time must be accurately assessed. To obtain accurate longitudinal assessment of findings, standardized methods are needed for image acquisition and reporting. The minimum sequences and reporting guidelines should be standardized within an institution or practice and ideally across institutions. To promote such efforts, recommendations for an imaging protocol and reporting template are provided below.
Recommendations for Clinical Imaging Protocol
Minimal Required Sequences.
The minimal recommended sequences for ARIA ascertainment are T2-FLAIR, T2* GRE, and DWI (Table 3). T2-FLAIR imaging is necessary for detection of ARIA-E. In clinical trials, 2D axial T2-FLAIR imaging was performed. Over the past decade, 3D FLAIR imaging has become more widely employed and has some advantages relative to 2D FLAIR, including improved CSF suppression and increased sensitivity for parenchymal edema.47,65 Therefore, 3D FLAIR may be preferred if it can be performed routinely with high quality and in a standardized fashion. However, if that is not feasible in your practice at the current time, 2D FLAIR should be performed. In summary, T2 FLAIR imaging is required for ARIA-E detection and should be performed using whichever technique is reliable, reproducible, and works for your practice.
Table 3:
Recommended imaging sequences for baseline imaging and ARIA monitoring examinations based on clinical trial experience and current guidelinesa
| Minimal | Recommended | Notes | |
|---|---|---|---|
| Field strength | 1.5T | 3T | Use of a consistent field strength for serial imaging of a given patient is important; imaging may be performed at 1.5T if a patient is not a candidate for imaging at 3T or 3T scanners are not available at a site |
| ARIA-E detection | 2D-FLAIR | 2D- or 3D-FLAIR | Either 2D or 3D is acceptable, whichever can be performed with consistent quality and optimal CSF suppression |
| ARIA-H detection | T2*-GRE | T2*-GRE (±SWI) | Recommendations for enrollment and dose suspension are based on T2*-GRE detection of blood products; SWI may also be performed for confirmation and may be of value to gather data going forward |
| Infarct assessment | DWI | DWI | DWI required to differentiate ARIA from acute/subsacute infarct and identification of incidental infarcts |
Additional optional sequences may be added per individual site preference.
T2* sequences are required for ARIA-H assessment, and in clinical trials, the GRE sequence was used. SWI may provide improved sensitivity for microhemorrhage detection52 but is less widely available and has more variability among MR image vendors compared with the GRE sequence. Also, current rules for dose-adjustment are based on 2D-GRE data. In other words, the increased sensitivity of SWI in the detection of microhemorrhages may cause a patient to enter the category of dose suspension due to too many microhemorrhages detected, whereas on the basis of the GRE scan, the lower microhemorrhage count would allow for continued treatment. Therefore, 2D axial GRE is recommended for ARIA-H assessment in clinical practice and should use parameters similar to those employed in clinical trials (non-EPI technique: 3T TE, ∼20 ms; maximum section thickness, 4 mm; acquisition time, ∼4–5 minutes).
Whenever a new signal abnormality is noted on a monitoring MR imaging study, the DWI sequence plays an important role in helping to differentiate ARIA-E from potential cytotoxic edema as may be noted with an incidental acute-to-subacute infarct. As such, it is recommended that a standard clinical axial 2D trace DWI sequence be included as routine protocol.
Standardized versions of these 3 sequences are required at the enrollment (pretreatment screeening/baseline) examination and each treatment monitoring examination. In regard to ARIA-E, standardized imaging will allow the detection of subtle T2-FLAIR changes by comparison with baseline. In regard to ARIA-H, baseline imaging is essential for patient selection to exclude those patients with pre-existing siderosis or too many microhemorrhages, for whom there is a higher rate of adverse events. As such, baseline imaging is critical to ensure proper patient selection, image interpretation, and patient management.
Considerations for Additional Optional Sequences, Enrollment, and Monitoring Protocols.
Additional sequences may be performed at individual sites on the basis of their standard protocols and procedures, interests, and available scan time, and these additional sequences could be tailored to separate enrollment and monitoring protocols. For example, in many practices, MPRAGE and volumetric analyses are performed on all MR imaging scans for the evaluation of dementia, and the enrollment MR imaging scan would fall under this category. However, such volumetric assessment is not needed for safety monitoring during treatment. Therefore, the MPRAGE sequence could be included in a dedicated enrollment protocol. T2 TSE/FSE may be included to help resolve ambiguous T2* findings, such as differentiating a microhemorrhage from a vessel flow void. SWI could be added (but not replace GRE) to either the enrollment or monitoring protocols if there is interest in more sensitive microhemorrhage detection or academic comparison with GRE. Postcontrast imaging is not recommended unless there is a diagnostic dilemma or incidental finding requiring further evaluation.
Importance of Consistent Serial Imaging: Field Strength and Vendor.
In addition to a standardized imaging protocol, patients would also ideally be imaged on the same field strength and vendor on sequential visits due to scanner-related differences (see “ARIA-E” “Interpretation Pitfalls”). As with recent clinical trials, we recommend that imaging be performed at 3T rather than 1.5T due to improved SNR with higher field strengths, which may allow improved detection of small microhemorrhages and subtle ARIA-E.53,65 However, if 3T imaging is not available or the patient is not a candidate for 3T imaging (eg, devices), imaging may be performed on 1.5T. Whichever field strength and vendor is used for imaging a patient, effort should be made to be consistent from scan to scan whenever possible. Patients may be encouraged to visit a consistent imaging facility for their monitoring scans. Within an institution, AD therapeutic imaging could be triaged to a certain group of scanners as scheduling allows.
Need for Routine MR Imaging: Consideration for AD Therapeutic Enrollment.
Patient factors that would preclude routine MR imaging should be considered at the time of enrollment. Patients with MR imaging–unsafe devices or other MR imaging contraindication (eg, metallic foreign body in the eye) would not be able to be imaged. Patients with MR imaging–conditional devices requiring scanning at specific institutions and imaging slots for which scanning may be monitored by an MR imaging physicist may be ineligible. Additionally, patients with claustrophobia, unable to undergo MR imaging without anesthesia, may be considered ineligible for treatment.
Online Resources.
If in the future, vendor-specific protocols are made available for ARIA monitoring, these will be posted on the American Society of Neuroradiology website. As additional mAbs obtain FDA approval and guidelines are modified, the recommended imaging sequences and protocols may evolve.
Recommendations for Reporting/Communication
Clinical History.
Relevant clinical history should be available at the time of image review and include the drug the patient is receiving, drug dose, duration of treatment, date of last dose, and whether the patient has experienced previous episodes of ARIA.
Reporting of Baseline/Enrollment MR Imaging Examination.
The number and location of existing microhemorrhages and superficial siderosis must be tabulated on the baseline examination. In line with exclusion criteria, microhemorrhage number should be summarized as 0–4, 5–9, and ≥10 and superficial siderosis as present or absent. Any significant incidental and acute findings, such as acute or chronic infarctions, also should be documented in the report narrative.62
Reporting of Monitoring MR Imaging Examinations.
The radiology report must allow the patient to be given an ARIA severity score for each ARIA-E, ARIA-H microhemorrhage, and ARIA-H superficial siderosis, which will be used along with the clinical symptom score to determine continued dosing. The quantitative nature of the ARIA severity scoring lends itself to a templated report including the following sections. ARIA-E must be noted as absent or present. If present, the location and maximal transverse diameter of each noncontiguous, involved site must be reported. When following a patient with ARIA-E at the prior time point, one must describe interval change and resolution. For microhemorrhages, the number and location of prior and new microhemorrhages must be reported, and the number of cumulative treatment-emergent microhemorrhages may be reported in relevant categories of 0–4, 5–9, and ≥10. The description of superficial siderosis should include the number and location of each prior and new noncontiguous site. Incidental and acute findings such as acute or interval (nonacute) infarct should be included in the report narrative.
Considerations for ARIA Reporting.
As the presence of ARIA-E and siderosis, the number of microhemorrhages, and changes over time directly determine patient management, radiologists must carefully consider their level of diagnostic uncertainty and level of sensitivity versus specificity for ARIA detection. In clinical trial interpretations, the practice at some central reading sites is that each finding is marked as possible or definite and only definite findings are counted toward ARIA severity scoring and exclusion or dosing-discontinuation criteria. Possible findings may include a subtle abnormality (small, faint possible microhemorrhage or subtle increased extent of occipital white matter hyperintensity) or be related to image acquisition (motion-degraded examination, poor CSF suppression, or change in imaging technique). While only definite findings are counted toward ARIA severity scoring, participants may be asked to return for repeat imaging in the case of possible ARIA-E, as its presence would affect continued drug dosing. Although exact cut-points for microhemorrhages are used in severity scoring, the presence of a possible microhemorrhage is less likely to affect the treatment course.
In clinical ARIA reporting, we recommend that only definite microhemorrhages be included in the quantitative ARIA template; possible microhemorrhages should be described in the report narrative as a pointer to direct the next radiologist interpreting a scan for this patient. For ARIA-E, we recommend both definite and possible ARIA-E (parenchymal edema or effusion) be included in the templated report. Finally, we recommend including whether the image quality is adequate for ARIA assessment or inadequate, such as due to motion or poor CSF suppression, requiring the patient to return for repeat imaging.
Recommended Reporting Template.
A recommended reporting template is posted on the American Society of Neuroradiology website at https://www.asnr.org/alzheimers-webinar-series/. As with the imaging protocol, the recommended reporting template may evolve and will be updated as guidelines are modified on the basis of growing experience in clinical trials and clinical practice.
Communication with Referring Physicians.
Use of the recommended ARIA reporting template will allow clear communication of relevant ARIA findings that will, in part, determine eligibility for treatment and further drug dosing. All ARIA reports should be generated in a timely manner, as each dosing session is typically preceded by imaging and dose administration is dependent on a satisfactory radiographic report. Findings of severe ARIA should be communicated in an urgent manner as they may affect dosing and patient management.
CONCLUSIONS
The emerging monoclonal antibody therapies for AD require both baseline pretreatment brain MR imaging as well as frequent monitoring MR imaging examinations for the detection of potential subclinical adverse events that may require dose adjustment. As these therapies begin to be implemented in clinical practice, treatment enrollment and monitoring brain MR imaging examinations may greatly increase neuroradiology practice volumes and will introduce a new imaging entity, ARIA, requiring awareness by all radiologists. ARIA-E is transient, treatment-induced edema or sulcal effusion, identified on T2-FLAIR that must be differentiated from an acute infarct or other entities causing hyperintense T2 signal. ARIA-H is treatment-induced microhemorrhages or superficial siderosis identified on T2* GRE, qualitatively similar to spontaneous hemosiderin deposition in CAA. Use of the recommended standardized imaging protocols and reporting templates will improve ARIA detection and timely communication of findings to referring providers, ensuring optimal patient care and management.
Acknowledgments
We thank Biogen and the Dominantly Inherited Alzheimer Network for images and the American Society of Neuroradiology for support in development of this manuscript.
ABBREVIATIONS:
- Aβ
amyloid-beta
- AD
Alzheimer disease
- APP
amyloid precursor protein
- ARIA
amyloid-related imaging abnormalities
- ARIA-E
amyloid-related imaging abnormalities with edema or effusion
- ARIA-H
amyloid-related imaging abnormalities with hemorrhage
- CAA
cerebral amyloid angiopathy
- CAA-RI
CAA-related inflammation
- GRE
gradient recalled-echo
- mAb
monoclonal antibody
References
- 1.Rajan KB, Weuve J, Barnes LL, et al. Population estimate of people with clinical Alzheimer’s disease and mild cognitive impairment in the United States (2020-2060). Alzheimers Dement 2021;17:1966–75 10.1002/alz.12362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.ADUHELM®. (aducanumab-avwa) is FDA Approved for Healthcare Professionals. https://biogencdn.com/us/aduhelm-pi.pdf. Accessed June 7, 2021
- 3.Honce JM, Nagae L, Nyberg E. Neuroimaging of natalizumab complications in multiple sclerosis: PML and other associated entities. Mult Scler Int 2015;2015:e80925 10.1155/2015/809252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wattjes MP, Vennegoor A, Steenwijk MD, et al. MRI pattern in asymptomatic natalizumab-associated PML. J Neurol Neurosurg Psychiatry 2015;86:793–98 10.1136/jnnp-2014-308630 [DOI] [PubMed] [Google Scholar]
- 5.Jack CR, Bennett DA, Blennow K, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018;14:535–62 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen G, Xu T, Yan Y, et al. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin 2017;38:1205–35 10.1038/aps.2017.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, et al. Cerebral amyloid angiopathy and Alzheimer disease: one peptide, two pathways. Nat Rev Neurol 2020;16:30–42 10.1038/s41582-019-0281-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tarasoff-Conway JM, Carare RO, Osorio RS, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol 2015;11:457–70 10.1038/nrneurol.2015.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller DL, Papayannopoulos IA, Styles J, et al. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Arch Biochem Biophys 1993;301:41–52 10.1006/abbi.1993.1112 [DOI] [PubMed] [Google Scholar]
- 10.Corriveau RA, Bosetti F, Emr M, et al. The Science of Vascular Contributions to Cognitive Impairment and Dementia (VCID): a framework for advancing research priorities in the cerebrovascular biology of cognitive decline. Cell Mol Neurobiol 2016;36:281–88 10.1007/s10571-016-0334-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karran E, Mercken M, Strooper BD. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 2011;10:698–712 10.1038/nrd3505 [DOI] [PubMed] [Google Scholar]
- 12.Jack CR, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 2010;9:119–28 10.1016/S1474-4422(09)70299-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jack CR, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013;12:207–16 10.1016/S1474-4422(12)70291-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med 2012;2:a006296 10.1101/cshperspect.a006296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brier MR, Gordon B, Friedrichsen K, et al. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci Transl Med 2016;8:338ra66 10.1126/scitranslmed.aaf2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson KA, Schultz A, Betensky RA, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol 2016;79:110–19 10.1002/ana.24546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jack CR, Therneau TM, Weigand SD, et al. Prevalence of biologically vs clinically defined Alzheimer spectrum entities using the National Institute on Aging-Alzheimer’s Association Research Framework. JAMA Neurol 2019;76:1174 10.1001/jamaneurol.2019.1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer’s disease. N Engl J Med 2021;384:1691–704 10.1056/NEJMoa2100708 [DOI] [PubMed] [Google Scholar]
- 19.Swanson CJ, Zhang Y, Dhadda S, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res Ther 2021;13:80 10.1186/s13195-021-00813-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies P, Koppel J. Mechanism-based treatments for Alzheimer’s disease. Dialogues Clin Neurosci 2009;11:159–69 10.31887/DCNS.2009.11.2/pdavies [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilman S, Koller M, Black RS, et al. ; for the AN1792(QS-21)-201 Study Team. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005;64:1553–62 10.1212/01.WNL.0000159740.16984.3C [DOI] [PubMed] [Google Scholar]
- 22.Salloway S, Sperling R, Gilman S, et al. ; Bapineuzumab 201 Clinical Trial Investigators. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 2009;73:2061–70 10.1212/WNL.0b013e3181c67808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Black RS, Sperling RA, Safirstein B, et al. A single ascending dose study of bapineuzumab in patients with Alzheimer disease. Alzheimer Dis Assoc Disord 2010;24:198–203 10.1097/WAD.0b013e3181c53b00 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sperling RA, Jack CR, Black SE, et al. Amyloid Related Imaging Abnormalities (ARIA) in amyloid modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement 2011;7:367–85 10.1016/j.jalz.2011.05.2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cummings J, Lee G, Zhong K, et al. Alzheimer’s disease drug development pipeline: 2021. Alzheimers Dement (N Y) 2021;7:7:e12179 10.1002/trc2.12179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Dyck CH. Anti-Amyloid-β monoclonal antibodies for Alzheimer’s disease: pitfalls and promise. Biol Psychiatry 2018;83:311–19 10.1016/j.biopsych.2017.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salloway S, Chalkias S, Barkhof F, et al. Amyloid-related imaging abnormalities in 2 Phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol 2022;79:13–21 10.1001/jamaneurol.2021.4161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salloway S, Sperling R, Fox NC, et al. ; Bapineuzumab 301 and 302 Clinical Trial Investigators. Two Phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 2014;370:322–33 10.1056/NEJMoa1304839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guthrie H, Honig LS, Lin H, et al. Safety, tolerability, and pharmacokinetics of crenezumab in patients with mild-to-moderate Alzheimer’s disease treated with escalating doses for up to 133 weeks. J Alzheimers Dis 2020;76:967–79 10.3233/JAD-200134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Landen JW, Cohen S, Billing CB, et al. Multiple-dose ponezumab for mild-to-moderate Alzheimer’s disease: safety and efficacy. Alzheimers Dement (N Y) 2017;3:339–47 10.1016/j.trci.2017.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ostrowitzki S, Lasser RA, Dorflinger E, et al. ; SCarlet RoAD Investigators. A Phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther 2017;9:95 10.1186/s13195-017-0318-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doody RS, Thomas RG, Farlow M, et al. ; Solanezumab Study Group. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med 2014;370:311–21 10.1056/NEJMoa1312889 [DOI] [PubMed] [Google Scholar]
- 33.Kim SH, Ahn JH, Yang H, et al. Cerebral amyloid angiopathy aggravates perivascular clearance impairment in an Alzheimer’s disease mouse model. Acta Neuropathol Commun 2020;8:181 10.1186/s40478-020-01042-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zago W, Schroeter S, Guido T, et al. Vascular alterations in PDAPP mice after anti-Aβ immunotherapy: implications for amyloid-related imaging abnormalities. Alzheimers Dement 2013;9:S105–15 10.1016/j.jalz.2012.11.010 [DOI] [PubMed] [Google Scholar]
- 35.Antolini L, DiFrancesco JC, Zedde M, et al. Spontaneous ARIA-like events in cerebral amyloid angiopathy–related inflammation: a multicenter prospective longitudinal cohort study. Neurology 2021;97:e1809–22 10.1212/WNL.0000000000012778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eng JA, Frosch MP, Choi K, et al. Clinical manifestations of cerebral amyloid angiopathy-related inflammation. Ann Neurol 2004;55:250–56 10.1002/ana.10810 [DOI] [PubMed] [Google Scholar]
- 37.Bohrmann B, Baumann K, Benz J, et al. Gantenerumab: a novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J Alzheimers Dis 2012;28:49–69 10.3233/JAD-2011-110977 [DOI] [PubMed] [Google Scholar]
- 38.Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016;537:50–56 10.1038/nature19323 [DOI] [PubMed] [Google Scholar]
- 39.Brashear HR, Ketter N, Bogert J, et al. Clinical evaluation of amyloid-related imaging abnormalities in bapineuzumab Phase III studies. J Alzheimers Dis 2018;66:1409–24 10.3233/JAD-180675 [DOI] [PubMed] [Google Scholar]
- 40.Barakos J, Purcell D, Suhy J, et al. Detection and management of amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with anti-amyloid beta therapy. J Prev Alzheimers Dis 2022;9:211–20 10.14283/jpad.2022.21 [DOI] [PubMed] [Google Scholar]
- 41.Yamada M. Cerebral amyloid angiopathy and gene polymorphisms. J Neurol Sci 2004;226:41–44 10.1016/j.jns.2004.09.009 [DOI] [PubMed] [Google Scholar]
- 42.Barakos J, Sperling R, Salloway S, et al. MR imaging features of amyloid-related imaging abnormalities. AJNR Am J Neuroradiol 2013;34:1958–65 10.3174/ajnr.A3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carlson C, Siemers E, Hake A, et al. Amyloid-related imaging abnormalities from trials of solanezumab for Alzheimer’s disease. Alzheimers Dement (Amst) 2016;2:75–85 10.1016/j.dadm.2016.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sperling R, Salloway S, Brooks DJ, et al. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol 2012;11:241–49 10.1016/S1474-4422(12)70015-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neema M, Guss ZD, Stankiewicz JM, et al. Normal findings on brain fluid-attenuated inversion recovery MR images at 3T. AJNR Am J Neuroradiol 2009;30:911–16 10.3174/ajnr.A1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kakeda S, Korogi Y, Hiai Y, et al. Pitfalls of 3D FLAIR brain imaging: a prospective comparison with 2D FLAIR. Acad Radiol 2012;19:1225–32 10.1016/j.acra.2012.04.017 [DOI] [PubMed] [Google Scholar]
- 47.Kallmes DF, Hui FK, Mugler JP. Suppression of cerebrospinal fluid and blood flow artifacts in FLAIR MR imaging with a single-slab three-dimensional pulse sequence: initial experience. Radiology 2001;221:251–55 10.1148/radiol.2211001712 [DOI] [PubMed] [Google Scholar]
- 48.Bartynski WS. Posterior reversible encephalopathy syndrome, Part 1: fundamental imaging and clinical features. AJNR Am J Neuroradiol 2008;29:1036–42 10.3174/ajnr.A0928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stuckey SL, Goh TD, Heffernan T, et al. Hyperintensity in the subarachnoid space on FLAIR MRI. AJR Am J Roentgenol 2007;189:913–21 10.2214/AJR.07.2424 [DOI] [PubMed] [Google Scholar]
- 50.Moraal B, Wattjes MP, Geurts JJG, et al. Improved detection of active multiple sclerosis lesions: 3D subtraction imaging. Radiology 2010;255:154–63 10.1148/radiol.09090814 [DOI] [PubMed] [Google Scholar]
- 51.VandeVrede L, Gibbs DM, Koestler M, et al. Symptomatic amyloid-related imaging abnormalities in an APOE ε4/ε4 patient treated with aducanumab. Alzheimers Dement (Amst) 2020;12:e12101 10.1002/dad2.12101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haller S, Haacke EM, Thurnher MM, et al. Susceptibility-weighted imaging: technical essentials and clinical neurologic applications. Radiology 2021;299:3–26 10.1148/radiol.2021203071 [DOI] [PubMed] [Google Scholar]
- 53.Nandigam RK, Viswanathan A, Delgado P, et al. MR imaging detection of cerebral microbleeds: effect of susceptibility-weighted imaging, section thickness, and field strength. AJNR Am J Neuroradiol 2009;30:338–43 10.3174/ajnr.A1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haller S, Vernooij MW, Kuijer JP, et al. Cerebral microbleeds: imaging and clinical significance. Radiology 2018;287:11–28 10.1148/radiol.2018170803 [DOI] [PubMed] [Google Scholar]
- 55.Viswanathan A, Chabriat H. Cerebral microhemorrhage. Stroke 2006;37:550–55 10.1161/01.STR.0000199847.96188.12 [DOI] [PubMed] [Google Scholar]
- 56.Koenig LN, McCue LM, Grant E, et al. Lack of association between acute stroke, post-stroke dementia, race, and β-amyloid status. Neuroimage Clin 2021;29:102553 10.1016/j.nicl.2020.102553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cordonnier C, van der Flier WM, Sluimer JD, et al. Prevalence and severity of microbleeds in a memory clinic setting. Neurology 2006;66:1356–60 10.1212/01.wnl.0000210535.20297.ae [DOI] [PubMed] [Google Scholar]
- 58.Siemers ER, Sundell KL, Carlson C, et al. Phase 3 solanezumab trials: secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement 2016;12:110–20 10.1016/j.jalz.2015.06.1893 [DOI] [PubMed] [Google Scholar]
- 59.Zonneveld HI, Goos JD, Wattjes MP, et al. Prevalence of cortical superficial siderosis in a memory clinic population. Neurology 2014;82:698–704 10.1212/WNL.0000000000000150 [DOI] [PubMed] [Google Scholar]
- 60.Shoamanesh A, Akoudad S, Himali JJ, et al. Cortical superficial siderosis in the general population: the Framingham Heart and Rotterdam studies. Int J Stroke 2021;16:798–808 10.1177/1747493020984559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barkhof F, Daams M, Scheltens P, et al. An MRI rating scale for amyloid-related imaging abnormalities with edema or effusion. AJNR Am J Neuroradiol 2013;34:1550–55 10.3174/ajnr.A3475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cummings J, Aisen P, Apostolova LG, et al. Aducanumab: appropriate use recommendations. J Prev Alzheimers Dis 2021;8:398–410 10.14283/jpad.2021.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cummings J, Rabinovici GD, Atri A, et al. Aducanumab: appropriate use recommendations update. J Prev Alzheimers Dis 2022;9:221–30 10.14283/jpad.2022.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aisen PS, Cummings J, Doody R, et al. The future of anti-amyloid trials. J Prev Alzheimers Dis 2020;7:146–51 10.14283/jpad.2020.24 [DOI] [PubMed] [Google Scholar]
- 65.Wattjes MP, Lutterbey GG, Harzheim M, et al. Higher sensitivity in the detection of inflammatory brain lesions in patients with clinically isolated syndromes suggestive of multiple sclerosis using high field MRI: an intraindividual comparison of 1.5 T with 3.0 T. Eur Radiol 2006;16:2067–73 10.1007/s00330-006-0195-4 [DOI] [PubMed] [Google Scholar]