

Abstract
Objectives: The white matter ischemic changes described in the literature are not specific and include white matter rarefaction, axonal damage, myelin degeneration, astrocytic fragmentation and beading of its processes (clasmatodendrosis), oligodendrocyte loss, and microglial activation. This morphological spectrum overlaps with morphological features of many other conditions. This retrospective study aims to describe the cellular changes (using immunohistochemical studies) in a spectrum of ischemic leukoencephalopathy. Methods: We studied 24 white matter ischemic injury cases with a well-documented interval from the ischemic event of interest. The autopsy reports were reviewed for the clinical information and pathological features to select the most representative areas for other stains and immunostains: luxol fast blue with hematoxylin and eosin (LFB-HE), anti-amyloid precursor protein (APP), anti-glial fibrillary acidic protein (GFAP), and anti-human leukocyte antigen (HLA-DR) antibodies. Results: The early changes detected in mild injury were axonal staining highlighted by APP immunostain and astrocytic and microglial reaction with no significant cellular loss. The most severe injury may lead to losing almost all cellular elements (complete infarct) and replacement by macrophages with time. Injuries in between resulted in a morphological spectrum of selective cellular injury (incomplete infarct), including apoptotic nuclei, axonal staining and swellings, clasmatodendrosis, and loss of ramified microglia. Conclusions: The pathological findings suggested that widespread axonal staining and swellings are early features followed or accompanied by loss of HLA-DR positive ramified microglia and then by astrocytes in ischemic leukoencephalopathy. The awareness of this morphological spectrum would prevent misdiagnosis, provide a better understanding of this condition and can guide future studies on this important and common subject.
Keywords: APP, HLA-DR, infarct, ischemic leukoencephalopathy, white matter
Introduction
Ischemia is commonly used to describe different levels of reduced (oligemia) to complete loss (ischemia) of blood supply. The gray matter ischemic changes are well documented in the literature [1-3]. However, the pathological features described in ischemic leukoencephalopathy are not specific and include white matter rarefaction, axonal damage, myelin degeneration, astrocytic fragmentation and beading of its processes (clasmatodendrosis), oligodendrocyte loss, and microglial activation [2,4]. In infarcts, the changes include complete loss of all white matter cellular components and replacement by macrophages with time.
These non-specific features overlap with many other white matter conditions such as toxic leukoencephalopathy [5], multifocal necrotizing leukoencephalopathy [6-8], acute demyelination [9,10], and leukoaraiosis [11]. This may reflect the limited studies of the white matter and the limited possible reactions/changes of the white matter to injuries. In animal studies, the most vulnerable cells in the white matter to hypoxic-ischemia are oligodendrocytes [12], and the white matter pathology precedes axonal damage [13,14]. This information is lacking in humans.
The pathology of ischemic leukoencephalopathy could be classified as acute changes as in ischemic injury with a predilection to the white matter [15] or as chronic hypoperfusion as in older individuals [16] or manifested as a part of small vessel disease [17]. The pathogenesis of ischemic leukoencephalopathy could be related to hypoperfusion secondary to the nature of the blood supply to the distal deep white matter [18], edematous changes accompanying hypoxia [19], metabolic acidosis, and other factors. In this study, we selected different forms that led to ischemic leukoencephalopathy including focal and global cerebral ischemia with and without cerebral edema.
A study of individual cellular changes in the white matter, using special stain and immunostains, could result in a better understanding of the effects of the ischemic injury on the white matter components. This study documents the cellular changes in the white matter in a spectrum of ischemic injuries and is expected to provide us with a better understanding of the effects of the ischemic injury on the white matter components.
Materials and methods
In this retrospective study, we searched the pathology archive for brain autopsy cases in a 10-year period with white matter involvement in ischemic insults. The autopsy reports were reviewed for the clinical information and pathology findings to select the most representative cases with a well-documented period of ischemia. Ethical approval for the study was obtained from the office of research ethics at the University of Western Ontario (Romeo number 105692).
Cases with a complicated medical history and no definite time frame that could be correlated to the pathological features were excluded from the analysis. The pathological slides were reviewed, and the areas of interest in each case were stained with: hematoxylin and eosin (HE) to assess morphological changes, luxol fast blue (LFB)-HE to assess myelin changes, anti-amyloid precursor protein (APP; Chemicon, Temecula, CA) to assess axonal reaction, anti-glial fibrillary acidic protein (GFAP; Dako, Carpinteria, CA) to assess astrocytic reaction, and anti-HLA-DR antibody (1:200; clone CR3/43; Dako, Carpinteria, CA) to assess microglial/macrophage reaction.
The mechanism of the ischemic events was divided into those representing global cerebral ischemia (GCI) and focal cerebral ischemia (FCI). Cases with FCI are divided into (1) macroscopic FCI where the lesions are larger than 5 mm and (2) microscopic FCI when the individual lesion is less than 5 mm in the largest dimension. The latter could be widespread and multifocal, but the size definition applies to every lesion and not to the whole region affected by the multifocal lesions. The cases ith GCI are divided into those with significant cerebral swelling and those without. The pathology of the latter group was published previously [20], and we re-assessed the pathology with particular attention to the cellular changes using immunostains (APP, GFAP, and HLA-DR).
Results
The clinical summary of all cases is presented in Table 1. The first pattern of ischemic injury in the first two cases was macroscopic FCI. In Case 1, with nine days of survival, the gray matter in the grossly infarcted right middle frontal region showed scattered ischemic red neurons, infiltration of macrophages, and early microvascular reaction consistent with the survival period after the infarct. The adjacent affected white matter had geographic involvement with relatively preserved islands, ischemic core (Figure 1A, 1B), and transitional area (penumbra) (Figure 1A, 1C). The ischemic core showed diffuse myelin pallor with some stainable myelinated axons, coarse and fine vacuoles, and marked reduction in all cellular elements with only a few small round nuclei (Figure 1B). GFAP highlighted the complete loss of astrocytes with only background staining (Figure 1E, 1F). APP showed fragmented axonal staining and swellings (Figure 1I, 1J). HLA-DR showed complete loss of microglia with ramified processes but scattered macrophage phenotypes (Figure 1M, 1N). The white matter adjacent to the infarcted area (transitional/penumbra) showed selective cellular loss (i.e., incomplete infarct) with less myelin pallor with coarse and fine vacuoles and moderate reduction in all cellular elements (Figure 1C). GFAP in these areas highlighted astrocytes with large round cytoplasm and fragmented processes in the background (i.e., clasmatodendrosis) (Figure 1G) that changed to regular reactive astrocytes in the adjacent normal-appearing white matter (NAWM) (Figure 1H). Axonal staining and swelling are present in this transitional region (Figure 1K) but absent in the adjacent NAWM (Figure 1L). HLA-DR showed that most stainable cells are of macrophage phenotypes, only a few with the ramified phenotype (Figure 1O), while they were mainly of ramified phenotype in the adjacent NAWM (Figure 1P).
Table 1.
Clinical information
| Case | Age | Sex | Clinical and significant autopsy findings | Mechanism of ischemia | Survival after event |
|---|---|---|---|---|---|
| 1 | 69 | F | Stroke post-surgical cardiac valve repair and replacement. Wedge-shaped infarct at right frontoparietal area | Focal cerebral ischemia (FCI): macroscopic | 9 D |
| 2 | 65 | F | Complicated medical case. An infarct in the right parieto-occipital area 5 weeks before death | FCI: macroscopic | 35 D |
| 3 | 78 | M | Slurred speech and right-side weakness suggestive of stroke | FCI: microscopic (embolic) | 3 D |
| 4 | 60 | M | Post-operative aortic valve replacement is complicated with cardiac vegetations, sepsis, and septic emboli | FCI: microscopic (septic emboli) | 4 D |
| 5 | 24 | F | Poorly controlled type 1 diabetes mellitus and chronic diarrhea. Admitted for dehydration complicated by ARDS for 2 weeks | FCI: microscopic (embolic) | 2 W |
| 6 | 78 | F | Hypertensive right ganglionic hemorrhage and cardiac arrest 2 days before death | Cardiac arrest (CA)/global cerebral ischemia (GCI) and brain edema | 2 D |
| 7 | 45 | M | Hemorrhagic stroke secondary to right carotid dissection and cardiac arrest 3 days before death | CA/GCI and brain edema | 3 D |
| 8 | 56 | M | Cardiac transplant for hypertrophic cardiomyopathy developed coagulopathy, and brain swelling. Cardiac arrest 4 days before death | CA/GCI and brain edema | 4 D |
| 9 | 52 | M | Acute myocardial infarction (MI) | CA/GCI | 6 H |
| 10 | 65 | M | Gastrointestinal bleeding | CA/GCI | 7 H |
| 11 | 61 | M | Dyspnea, cough and unsteadiness for 7 days | CA/GCI | 9 H |
| 12 | 44 | M | History of melanoma. Hypokalemia secondary to tumor lysis syndrome | CA/GCI | 14 H |
| 13 | 39 | F | Acute chest infection | CA/GCI | 2 D |
| 14 | 65 | M | Acute MI | CA/GCI | 2 D |
| 15 | 67 | M | Previous history of MI, DM2, and COPD | CA/GCI | 3 D |
| 16 | 76 | F | Pneumonia. | CA/GCI | 3 D |
| 17 | 41 | F | Previous history of recurrent supraglottitis | CA/GCI | 3 D |
| 18 | 62 | M | Acute MI | CA/GCI | 4 D |
| 19 | 57 | F | Recent history of liver failure | CA/GCI | 4 D |
| 20 | 72 | F | CA at home | CA/GCI | 4 D |
| 21 | 52 | F | Shortness of breath and fever. Medical history indicated epilepsy and depression for 2 years | CA/GCI | 5 D |
| 22 | 59 | F | Acute MI. PMH: COPD, congestive heart failure, and epilepsy | CA/GCI | 6 D |
| 23 | 79 | M | Post right hip surgery | CA/GCI | 9 D |
| 24 | 68 | M | Post-cholecystectomy | CA/GCI | 14 D |
COPD: chronic obstructive pulmonary disease; D: days, DM: diabetes mellitus; H: hours; HF: heart failure; W: weeks.
Figure 1.
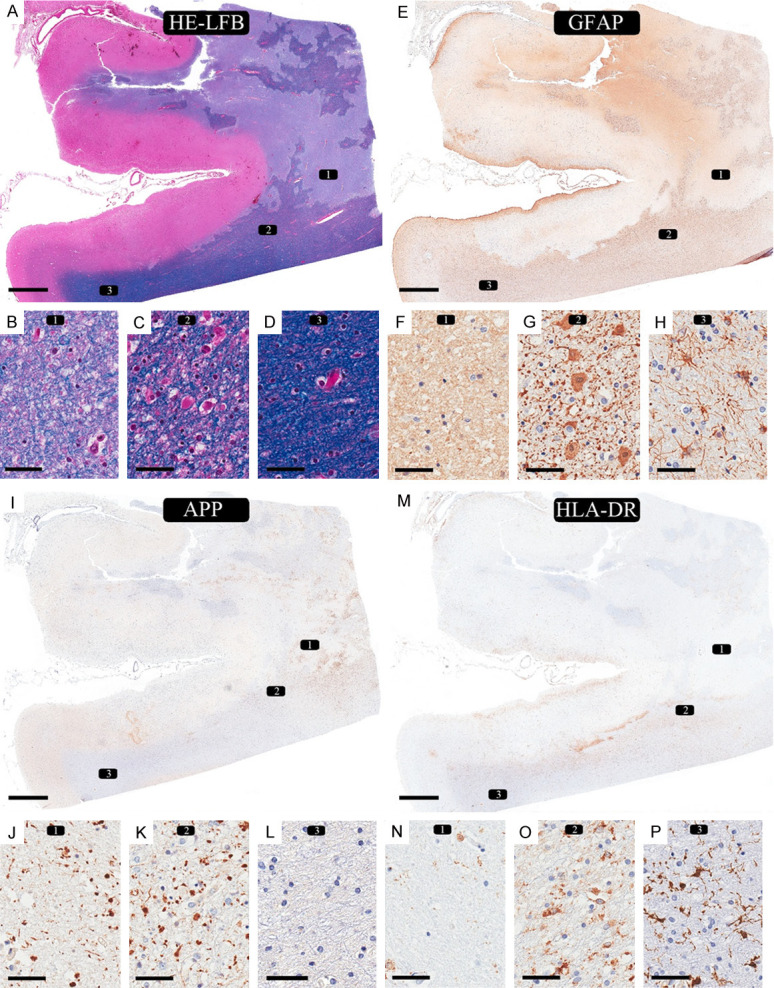
Ischemic changes in macroscopic focal cerebral ischemia: 9 days of survival (case 1). (A, E, I, M) Infarcted region in the right middle frontal area. An overview of the involved cortex and underlying white matter shows a geographic pattern of ischemic changes in white matter with some relatively preserved islands. (A: LFB-HE, E: GFAP, I: APP, M: HLA-DR). The white matter shows three different regions: (1) necrotic zone, (2) transitional area (penumbra), and (3) normal-appearing white matter (NAWM). Rectangle 1 represents the necrotic zone. (B) LFB-H&E shows myelin pallor, ghost of oligodendrocytes with few scattered viable nuclei and fine vacuoles. (F) GFAP immunostain shows mainly background staining but no viable cells. (J) APP immunostain shows axonal staining, and small spheroids. (N) HLA-DR immunostain shows loss of all ramified microglia, fragmented, broken processes in the background, and only scattered macrophage phenotype. Rectangle 2 represents the area adjacent to the infarct (i.e., penumbra). (C) There is reduced myelin staining compared to the normal-appearing white matter (NAWM) in (D). (G) GFAP highlights the astrocytes with swollen cytoplasmic staining and fragmented processes/“dot-like” staining in the background (clasmatodendrosis). (K) APP immunostain shows axonal staining, and small spheroids. (O) HLA-DR shows a mixture of microglia with predominant round/macrophage phenotype and few ramified phenotypes. Rectangle 3 represents the area with NAWM. There are no observable pathological changes by myelin stain (D) and APP immunostain (L). (H) GFAP highlights reactive astrocytes with multiple thin intact processes. (P) HLA-DR immunostain shows many ramified microglia in the background. (LFB-HE: A-D; GFAP: E-H; APP: I-L; HLA-DR: M-P. Scale bars: 4 mm (A, E, I, M), 50 µm (B-D, F-H, J-L, N-P).
In case 2, with 35 days of survival, the affected gray matter showed morphological features consistent with the survival period with complete loss of all cellular components and replacement with macrophages (Figure 2A, 2B and 2F). The infarcted white matter showed similar changes (Figure 2B), representing the ischemic core changes with time. The white matter adjacent to the infarcted region (penumbra) showed vacuolated white matter, loss of myelinated axons, myelin ovoids, reduced number of oligodendrocytes (Figure 2C), and gemistocytic astrocytes (Figure 2D) compared to reactive fibrillary astrocytosis with thin processes in the NAWM (Figure 2E). APP immunostain showed no stainable axons, and HLA-DR highlighted macrophage phenotype in the infarcted area, predominant round/macrophage type in adjacent white matter (Figure 2G), and ramified morphology in adjacent NAWM (Figure 2H).
Figure 2.
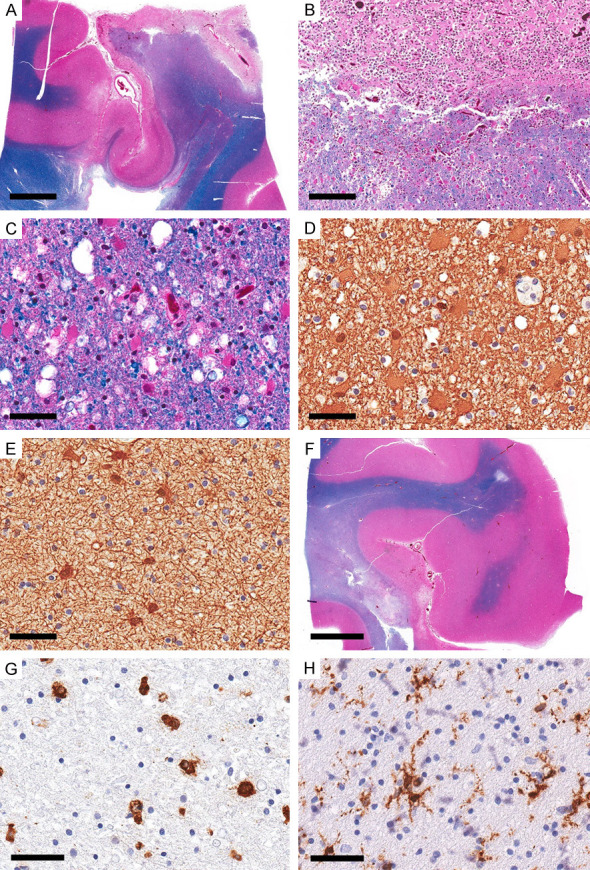
Ischemic changes in macroscopic focal cerebral ischemia: 5 weeks survival case 2). Representative sections from the infarcted area in the right parieto-occipital region. (A) Overview section showing cortical and subcortical necrosis. (LFB-HE). (B) There is a good demarcation of the necrotic zone formed mainly by macrophages consistent with 5-week survival. (LFB-HE). (C) The adjacent white matter shows coarse vacuoles and reactive astrocytes (LFB-HE). (D) GFAP highlights the gemistocytes with dense cytoplasmic staining (GFAP). (E) GFAP in the adjacent NAWM show reactive astrocytic reaction with thin processes (GFAP). (F) The overview section from the adjacent region shows a small focus of cortical necrosis, white matter necrosis and the adjacent area of myelin pallor (LFB-HE). (G, H) HLA-DR immunostain in the area of myelin pallor shows mainly round/macrophage phenotype with rare ramified phenotype compared to (H) prominent ramified phenotype in NAWM (HLA-DR). Scale bars: 4 mm (A, F), 200 µm (B), 50 µm (C-E, G, H).
The second pattern of ischemic injury (microscopic FCI) was present in three cases. In case 3, there were at least five small foci of well-circumscribed white matter lesions with changes consistent with the survival period of 3 days distributed in the left and right frontal regions, left parietooccipital, and the midbrain. Other white matter lesions with chronic morphological features were excluded from the assessment. All lesions showed similar features with prominent vacuolization, axonal swellings, crossing normal-appearing myelinated fibers, reduced oligodendrocytes, no visible astrocytes, and no macrophages (Figure 3A, 3B, 3G and 3H). APP immunostaining highlighted the prominent axonal spheroids (Figure 3C, 3I), and GFAP highlighted a marked reduction in astrocytes, background staining of thin processes but only a few astrocytes with visible cell bodies with no swelling (Figure 3D). HLA-DR showed near-complete loss of all phenotypes in the lesions but preserved ramified phenotype in the surrounding areas (Figure 3E, 3F). Case 4 showed four lesions (left and right frontal white matter and the pons) with similar characteristics to those described in case 3. It also showed multiple foci of septic emboli in both gray and white matter with micro-abscesses and microglial nodules. Case 5, with 14 days of survival, showed numerous small well-circumcised white matter lesions, some with associated microhemorrhages, distributed in all brain regions (Figure 4A), with similar morphological features consistent with the duration of 2 weeks. The lesions showed few myelinated axons but a prominent loss of oligodendrocytes (Figure 4B). APP immunostaining highlighted axonal staining in these foci but was less prominent than those in cases 3 and 4 (Figure 4C, 4D). GFAP staining was completely lost in the center of the lesions, while the surrounding area showed fibrillary astrocytosis (Figure 4E). HLA-DR highlighted round/macrophage phenotype in the lesions, prominent at the periphery of larger foci, and ramified morphology in surrounding areas (Figure 4F).
Figure 3.
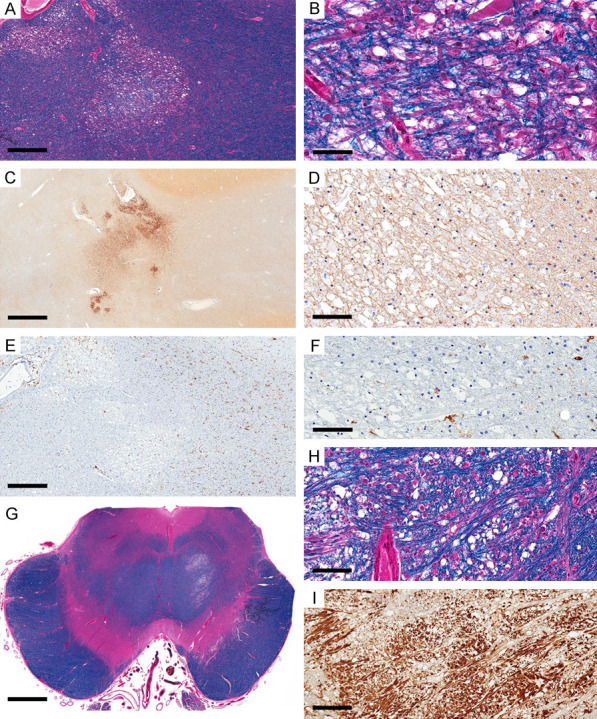
Ischemic lesions in microscopic focal cerebral ischemia, with 3 days of survival (case 3). Focal area with selective cellular loss in the left frontal region’s white matter (WM) with prominent vacuoles (LFB). (B) Higher magnification (A) shows prominent axonal swelling in the vacuolated area, myelinated fibers, and reduced oligodendrocytes. (C) APP immunostain highlights prominent axonal swelling and staining in the vacuolated WM and adjacent areas (APP). (D) GFAP shows background staining but no astrocytic swelling (GFAP). (E, F) HLA-DR is almost completely lost in the affected area that matches the area of APP axonal staining (HLA-DR). (G-I) Similar morphological changes in affected focus in the pons (LFB-HLA: G, H; APP: I). Scale bars: 500 µm (A, E), 50 µm (B), 2 mm (C), 100 µm (D, F, H), 3 mm (G), 200 µm (I).
Figure 4.
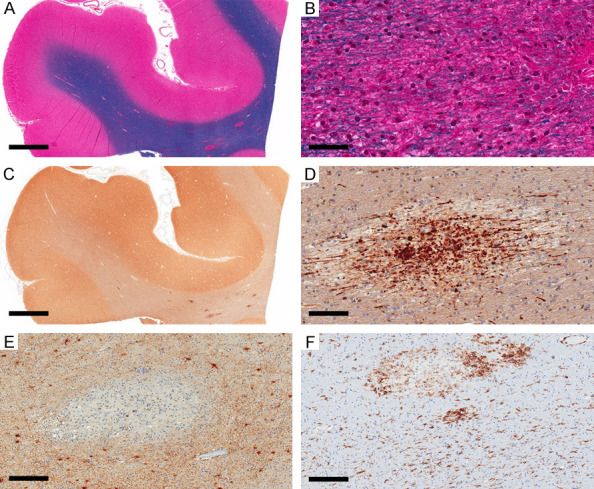
Ischemic lesions in microscopic focal cerebral ischemia, with 14 days of survival (case 5). (A) Multifocal well-circumscribed regions in the right frontal area, some with hemorrhage (LFB-HE). (B) Higher magnification of one of these foci showing prominent axonal swellings (LFB-HE). (C, D) APP immunostain highlights the axonal swelling in these foci (APP). (E) GFAP is completely lost in the affected foci (GFAP). (F) HLA-DR shows round/macrophage phenotype in the affected foci and ramified phenotype in the surrounding white matter (HLA-DR). Scale bars: 3 mm (A, C), 50 µm (B), 100 µm (C), 200 µm (E, F).
The third pattern of ischemic injury (GCI) cases were divided into those with no associated significant brain swelling and cases with gross cerebral swelling/edema associated with intracerebral hemorrhage (cases 6-8 and survival periods of 2-3 days). Cases from the first pattern (cases 9-24) were described in detail in our previous study [20]. In this study, we concentrated on HLA-DR, GFAP, and APP immunostaining patterns in these cases. These cases showed, consistent with previous observations [20], slight myelin pallor with fine vacuoles (Figure 5A), fibrillary astrocytes (Figure 5B), widespread HLA-DR staining ramified microglia (Figure 5C, 5D) in all cases. There were focal areas of astrocytic swelling (Figure 5E) in 8/16 cases and focal areas of mild APP staining (Figure 5F) in 5/16 cases. Apart from the small foci of possible embolic infarcts, there were no areas of complete loss of HLA-DR ramified microglia. Some foci showed mild fragmentation of microglial processes, possibly indicating the milder effect of the global hypoperfusion, but this finding was difficult to study and confirm owing to the spectrum of the microglial morphological changes upon activation.
Figure 5.
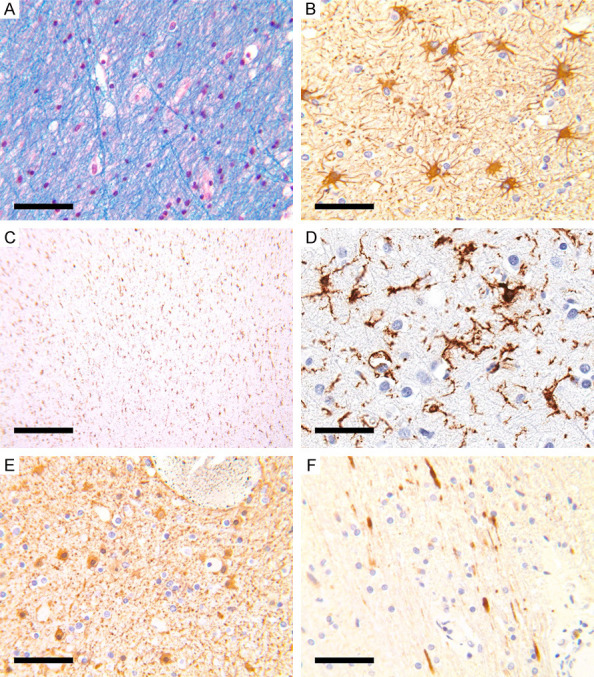
Hypoperfusion with minimal edema. (A) Slight myelin pallor with fine vacuoles. (B) GFAP showed reactive astrocytosis. (C, D) HLA-DR staining showed widespread ramified microglia. (E) Focal area of astrocytic swelling (GFAP). (F) Focal area of mild APP staining (APP). Scale bars: 50 µm (A, B, D, E), 500 µm (C).
For the cases with GCI and brain swellings, the affected gray matter in these cases showed early ischemic red neurons consistent with the survival period. The involved white matter showed myelin pallor and fine and course vacuoles (Figure 6A). The white matter adjacent to the hemorrhage showed astrocytic cytoplasmic swellings with fragmented processes (clasmatodendrosis) with GFAP immunostain (Figure 6B); prominent, widespread axonal staining with APP immunostain (Figure 6C); and fragmented microglial processes with HLA-DR immunostain (Figure 6D). The white matter away from the hemorrhagic foci showed similar features on LFB-HE (Figure 6E). GFAP in most examined areas showed reactive astrocytosis with thin processes and no cytoplasmic swelling (Figure 6F), while APP immunostain showed widespread axonal staining (Figure 6G). HLA-DR showed predominant microglia with ramified phenotype and at most mild fragmentation in the processes (Figure 6H).
Figure 6.
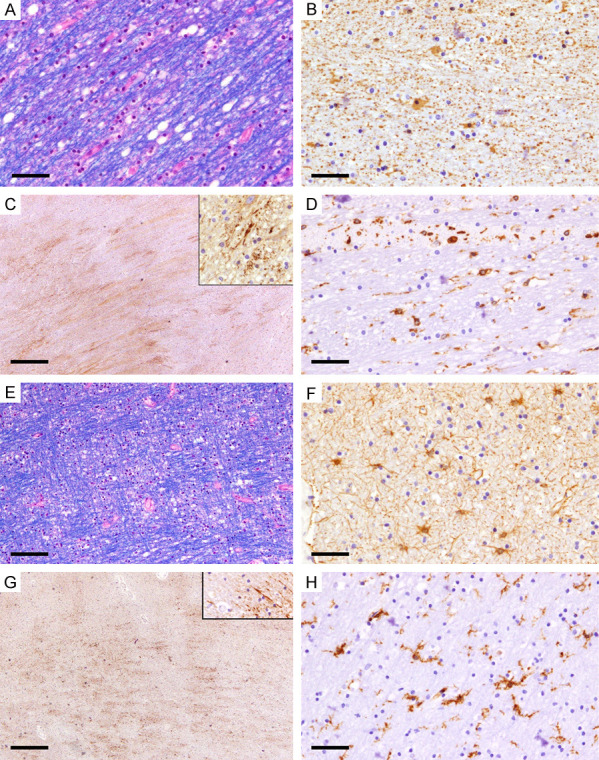
Hypoperfusion with significant edema. (A-D) A representative from case 6. (A) Fibers of the internal capsule on the same side of the frontal intracerebral hemorrhage show large vacuoles with smooth borders and separation of the myelinated fibers (LFB-HE). (B) GFAP highlights astrocytes with cytoplasmic swelling and fragmented processes in the background (GFAP). (C) APP immunostain show widespread axonal staining along with small spheroids (APP) (D) HLA-DR shows microglia with round/macrophage phenotype and a few with ramified phenotype but with fragmented processes (HLA-DR). (E-H) A representative from case 7. (E) White matter from the right frontal regions opposite to the site of the ganglionic hemorrhage shows fine and large vacuoles and separation of the myelinated fibers similar to (A) (LFB). (F) Reactive astrocytes with thin processes, no swollen cytoplasm, and no fragmented processes in the background (GFAP). (G) APP immunostain show widespread axonal staining along with small spheroids (APP). (H) HLA-DR mainly highlights microglia with ramified phenotype and no significant fragmentation of the processes (HLA-DR). Scale bars: 50 µm (A, B, D, F, H), 100 μm (C, E), 500 μm (G).
Discussion
The known pathological changes in ischemic leukoencephalopathy are not specific, and they include white matter rarefaction, axonal damage, myelin degeneration, fragmentation and beading of astrocytic processes (clasmatodendrosis), oligodendrocyte loss, and microglial activation [4]. These changes overlap with many other white matter pathologies. Ischemic changes in the gray matter are well documented, and a general estimate of the time and severity of the injury can be correlated with the changes [1-3]. In this study, we documented the cellular changes in the white matter, highlighted by different special stains and immunostains, which revealed a morphological spectrum of white matter changes.
The first pattern of injury, “complete infarct/pan necrosis”, represents the end stage of the ischemic injury. This pattern is present in the macroscopic FCI necrotic cores and some areas of the microscopic FCI. The early lesions are characterized by near-complete loss of all cells (i.e., subcortical neurons, oligodendrocytes, astrocytes, and microglia), axonal fragmentation and swellings highlighted by APP immunostains, and myelin pallor but not complete loss of myelin stain with LFB-HE special stain. GFAP and HLA-DR immunostains confirmed the complete loss of astrocytes and microglia. The progression of this lesion is characterized by the replacement of the necrotic core by macrophages which are stained by HLA-DR immunostain.
The second injury pattern represents the spectrum of “incomplete” infarcts/selective cellular loss. This pattern was observed in the transitional zone adjacent to the ischemic core (penumbra) in macroscopic FCI, most microscopic FCI lesions, and a few GCI lesions (mildest form). The defining feature of this pattern is cellular loss of any cell type in the affected area and hence represents a large spectrum of pathology. The histological features in the ischemic penumbra confirm their proposed existence in the white matter [21].
In macroscopic FCI, the reduced blood flow in the transitional zone (penumbra) between the necrotic center and NAWM causes a selective cellular loss/“incomplete infarct” [4]. It could also result in activation proliferation of microglia, oligodendrocytes, and astrocytes [3]. We found the following changes in this area: axonal injury/swellings with myelin pallor, astrocytic swelling and fragmentation of its processes, fragmentation of microglial processes and transformation to macrophage phenotype, and reduced number of oligodendrocytes. With time, astrocytes become gemistocytic, proportion of myelinated axons are lost, and axonal swelling becomes smaller and eventually disappears, leaving variably sized vacuoles.
In microscopic FCI, the spectrum of morphological changes of the “incomplete infarct” resembles the changes in the “penumbra” described above. These include apoptotic bodies in the lesions, axonal staining (denote reversible disruption of axonal flow [22]) and swelling highlighted by APP immunostain, partial loss of myelinated axons and myelin pallor, microglia reaction ranging from complete loss, and fragmentation of processes to transformation to macrophage phenotype.
This selectivity should reflect the different cellular vulnerabilities to the hypoperfusion. In gray matter, neurons are considered the most vulnerable to hypoperfusion and in selective necrosis in cortical areas of macaque monkeys [23,24]. In rats, the oligodendrocytes were considered the most vulnerable cells in the white matter [12], while we do not know which cells are the most vulnerable in human white matter. Combining the pathological results of “incomplete infarcts”, we found that axonal staining and swelling are the earliest recognizable pathology in these areas accompanied by apoptotic bodies. We could not identify the cells of origin of these apoptotic bodies, and they can be oligodendrocytes, astrocytes, or microglia. We found complete loss of ramified microglia in a few lesions with some stainable astrocytic processes. This finding may indicate that ramified microglia are more vulnerable than astrocytes, but further investigation with many cases are required to prove this observation. The association between the formation of axonal spheroids and the loss of ramified microglia in early ischemic lesions is consistent with our observation of adult-onset leukoencephalopathy with axonal spheroids (ALAS) [25]. However, the microglial loss precedes the axonal spheroid formation in ALAS, which is expected in a disease that primarily affects the microglia [25].
The pathological spectrum of this pattern of injury overlaps with many other pathological processes such as (1) some form of acute demyelination [26] (where hypoxic ischemia was considered the most likely pathogenesis in this pattern of acute demyelination) [10]; (2) spongiform leukoencephalopathy characterized by numerous white matter vacuoles and usually considered secondary to “toxic” effects of the drugs that could also be secondary to hypoxic/ischemic etiology [27]; (3) multifocal necrotizing leukoencephalopathy (MNL), especially in cases with pontine involvement. This pathology is described as a complication of acute leukemias and lymphoma treated with chemotherapy and radiation [28], acquired immunodeficiency syndrome [6], postcardiac transplant [7], and septic shock [29]. The mechanism of necrosis in MNL is unknown but is thought to be related to inflammatory cytokine production [6]. As such, we recommend that when encountering the pathology of one of these conditions, the possibility of “ischemic” injury should first be excluded before entertaining other possibilities.
The third pattern of injury is GCI, and we separated this pattern into cases with significant brain edema from those without the symptom. We previously studied cardiac arrest cases with no significant brain edema and found that widespread ramified microglial staining by HLA-DR and astrocytosis were consistent with the changes in the white matter [20]. While these could be considered the earliest observable pathological findings, their non-specificity limited their clinical interpretation. In chronic lesions, the white matter pathology includes rarefaction with degeneration of myelin, oligodendroglial apoptosis, astrocytosis, and microglial proliferation [16], while cases with GCI accompanied by significant brain edema showed more severe injuries in the form of prominent, widespread axonal staining by APP immunostain, consistent astrocytic swelling, and areas of fragmented microglial processes with HLA-DR immunostain.
The primary limitations in this study are the limited number of cases and the fact that essential variables such as the severity and the duration cannot be controlled in human studies, and the results are best interpreted as “most likely”.
In conclusion, we found that: (1) Axonal staining seems to be the earliest recognizable pathology with the aid of APP immunostain. (2) Mild ischemic injuries resulted in microglial and astrocytic proliferation, which most likely limited the effects of the ischemia. (3) Complete loss of microglia and astrocytes occurs in severe injuries. (4) The selective cellular injury is the most difficult to interpret and can be misinterpreted as other injuries.
Disclosure of conflict of interest
None.
References
- 1.Chuaqui R, Tapia J. Histologic assessment of the age of recent brain infarcts in man. J Neuropathol Exp Neurol. 1993;52:481–489. doi: 10.1097/00005072-199309000-00006. [DOI] [PubMed] [Google Scholar]
- 2.Mena H, Cadavid D, Rushing EJ. Human cerebral infarct: a proposed histopathologic classification based on 137 cases. Acta Neuropathol. 2004;108:524–530. doi: 10.1007/s00401-004-0918-z. [DOI] [PubMed] [Google Scholar]
- 3.Margaritescu O, Mogoanta L, Pirici I, Pirici D, Cernea D, Margaritescu C. Histopathological changes in acute ischemic stroke. Rom J Morphol Embryol. 2009;50:327–339. [PubMed] [Google Scholar]
- 4.Kalaria R, Ferrer I, Love S. Vascular disease, hypoxia and related conditions. In: Love S, Perry A, Ironside J, Budka H, editors. Greenfield’s neuropathology. 9th ed. Florida: CRS Press; 2015. pp. 59–209. [Google Scholar]
- 5.Filley CM, Kleinschmidt-DeMasters BK. Toxic leukoencephalopathy. N Engl J Med. 2001;345:425–432. doi: 10.1056/NEJM200108093450606. [DOI] [PubMed] [Google Scholar]
- 6.Anders KH, Becker PS, Holden JK, Sharer LR, Cornford ME, Hansen LA, Hamilton R, Vinters HV. Multifocal necrotizing leukoencephalopathy with pontine predilection in immunosuppressed patients: a clinicopathologic review of 16 cases. Hum Pathol. 1993;24:897–904. doi: 10.1016/0046-8177(93)90140-c. [DOI] [PubMed] [Google Scholar]
- 7.Premji S, Kang L, Rojiani MV, Kresak J, Rojiani AM. Multifocal Necrotizing Leukoencephalopathy: expanding the clinicopathologic spectrum. J Neuropathol Exp Neurol. 2019;78:340–347. doi: 10.1093/jnen/nlz003. [DOI] [PubMed] [Google Scholar]
- 8.Marker DF, Kofler JK, Mettenburg JA, Agha ME, Wiley CA. Multifocal Necrotizing Leukoencephalopathy with preferential microglia toxicity in a patient treated with chimeric antigen receptor T-cells and review of the literature. J Neuropathol Exp Neurol. 2020;79:1115–1121. doi: 10.1093/jnen/nlaa099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 10.Aboul-Enein F, Rauschka H, Kornek B, Stadelmann C, Stefferl A, Bruck W, Lucchinetti C, Schmidbauer M, Jellinger K, Lassmann H. Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol. 2003;62:25–33. doi: 10.1093/jnen/62.1.25. [DOI] [PubMed] [Google Scholar]
- 11.Pantoni L, Garcia JH. Pathogenesis of leukoaraiosis: a review. Stroke. 1997;28:652–659. doi: 10.1161/01.str.28.3.652. [DOI] [PubMed] [Google Scholar]
- 12.Pantoni L, Garcia JH, Gutierrez JA. Cerebral white matter is highly vulnerable to ischemia. Stroke. 1996;27:1641–1646. doi: 10.1161/01.str.27.9.1641. [DOI] [PubMed] [Google Scholar]
- 13.Kurumatani T, Kudo T, Ikura Y, Takeda M. White matter changes in the gerbil brain under chronic cerebral hypoperfusion. Stroke. 1998;29:1058–1062. doi: 10.1161/01.str.29.5.1058. [DOI] [PubMed] [Google Scholar]
- 14.Sozmen EG, Kolekar A, Havton LA, Carmichael ST. A white matter stroke model in the mouse: axonal damage, progenitor responses and MRI correlates. J Neurosci Methods. 2009;180:261–272. doi: 10.1016/j.jneumeth.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ginsberg MD, Hedley-Whyte ET, Richardson EP. Hypoxic-ischemic leukoencephalopathy in man. Trans Am Neurol Assoc. 1974;99:213–216. [PubMed] [Google Scholar]
- 16.Simpson JE, Fernando MS, Clark L, Ince PG, Matthews F, Forster G, O’Brien JT, Barber R, Kalaria RN, Brayne C, Shaw PJ, Lewis CE, Wharton SB MRC Cognitive Function and Ageing Neuropathology Study Group. White matter lesions in an unselected cohort of the elderly: astrocytic, microglial and oligodendrocyte precursor cell responses. Neuropathol Appl Neurobiol. 2007;33:410–419. doi: 10.1111/j.1365-2990.2007.00828.x. [DOI] [PubMed] [Google Scholar]
- 17.Gouw AA, Seewann A, van der Flier WM, Barkhof F, Rozemuller AM, Scheltens P, Geurts JJ. Heterogeneity of small vessel disease: a systematic review of MRI and histopathology correlations. J Neurol Neurosurg Psychiatry. 2011;82:126–135. doi: 10.1136/jnnp.2009.204685. [DOI] [PubMed] [Google Scholar]
- 18.Nonaka H, Akima M, Hatori T, Nagayama T, Zhang Z, Ihara F. Microvasculature of the human cerebral white matter: arteries of the deep white matter. Neuropathology. 2003;23:111–118. doi: 10.1046/j.1440-1789.2003.00486.x. [DOI] [PubMed] [Google Scholar]
- 19.Feigin I, Budzilovich G, Weinberg S, Ogata J. Degeneration of white matter in hypoxia, acidosis and edema. J Neuropathol Exp Neurol. 1973;32:125–143. doi: 10.1097/00005072-197301000-00008. [DOI] [PubMed] [Google Scholar]
- 20.Alturkustani M, Ang LC. Acute hypoxic-ischemia in cardiac arrest encephalopathy causes only minimal demyelination. Neuropathology. 2016;36:413–420. doi: 10.1111/neup.12287. [DOI] [PubMed] [Google Scholar]
- 21.Falcao AL, Reutens DC, Markus R, Koga M, Read SJ, Tochon-Danguy H, Sachinidis J, Howells DW, Donnan GA. The resistance to ischemia of white and gray matter after stroke. Ann Neurol. 2004;56:695–701. doi: 10.1002/ana.20265. [DOI] [PubMed] [Google Scholar]
- 22.Petty MA, Wettstein JG. White matter ischaemia. Brain Res Brain Res Rev. 1999;31:58–64. doi: 10.1016/s0165-0173(99)00025-9. [DOI] [PubMed] [Google Scholar]
- 23.Marcoux FW, Morawetz RB, Crowell RM, DeGirolami U, Halsey JH Jr. Differential regional vulnerability in transient focal cerebral ischemia. Stroke. 1982;13:339–346. doi: 10.1161/01.str.13.3.339. [DOI] [PubMed] [Google Scholar]
- 24.DeGirolami U, Crowell RM, Marcoux FW. Selective necrosis and total necrosis in focal cerebral ischemia. Neuropathologic observations on experimental middle cerebral artery occlusion in the macaque monkey. J Neuropathol Exp Neurol. 1984;43:57–71. doi: 10.1097/00005072-198401000-00005. [DOI] [PubMed] [Google Scholar]
- 25.Alturkustani M, Zhang Q, Alyamany B, Ang LC. Loss of ramified microglia precedes axonal spheroid formation in adult-onset leukoencephalopathy with axonal spheroids. Free Neuropathol. 2020;1:27. doi: 10.17879/freeneuropathology-2020-2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alturkustani M, Bahakeem B, Zhang Q, Ang LC. Different pathological processes for acute white matter lesions in multiple sclerosis. Malays J Pathol. 2020;42:187–194. [PubMed] [Google Scholar]
- 27.Alturkustani M, Ang LC, Ramsay D. Pathology of toxic leucoencephalopathy in drug abuse supports hypoxic-ischemic pathophysiology/etiology. Neuropathology. 2017;37:321–328. doi: 10.1111/neup.12377. [DOI] [PubMed] [Google Scholar]
- 28.Rubinstein LJ, Herman MM, Long TF, Wilbur JR. Disseminated necrotizing leukoencephalopathy: a complication of treated central nervous system leukemia and lymphoma. Cancer. 1975;35:291–305. doi: 10.1002/1097-0142(197502)35:2<291::aid-cncr2820350202>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 29.Sharshar T, Gray F, Poron F, Raphael JC, Gajdos P, Annane D. Multifocal necrotizing leukoencephalopathy in septic shock. Crit Care Med. 2002;30:2371–2375. doi: 10.1097/00003246-200210000-00031. [DOI] [PubMed] [Google Scholar]