

Abstract
The promiscuous nature of T-cell receptors (TCRs) allows T cells to recognize a large variety of pathogens, but makes it challenging to understand and control T-cell recognition1. Existing technologies provide limited information about the key requirements for T-cell recognition and the ability of TCRs to cross-recognize structurally related elements2,3. Here we present a ‘one-pot’ strategy for determining the interactions that govern TCR recognition of peptide–major histocompatibility complex (pMHC). We measured the relative affinities of TCRs to libraries of barcoded peptide–MHC variants and applied this knowledge to understand the recognition motif, here termed the TCR fingerprint. The TCR fingerprints of 16 different TCRs were identified and used to predict and validate cross-recognized peptides from the human proteome. The identified fingerprints differed among TCRs recognizing the same epitope, demonstrating the value of this strategy for understanding T-cell interactions and assessing potential cross-recognition before selection of TCRs for clinical development.
The antigen specificity of T cells is conferred by the TCR’s highly variable complementarity-determining regions (CDRs), which interact with the pMHC4. Cellular immunity requires a pool of naive T cells (the T-cell repertoire) that can recognize a multitude of potential pMHC antigens that may originate from infections or cellular transformation. If a given TCR could recognize only a single combination of peptide and MHC, an individual would need >1015 CD8+ T cells to provide efficient coverage of all potential foreign peptides, whereas it is estimated that an individual has only around 107–108 different T cells5–7. The promiscuity of TCRs allows the recognition of numerous different pMHCs by each T cell, which broadens the recognition space and ensures the effective recognition of most possible targets. A single TCR can interact with more than 1 million different peptide–MHC combinations1,8,9; however, owing to technical limitations, little is known about the extent of promiscuity for any particular TCR or the patterns that govern the exact hierarchical avidities to various pMHCs. Peptide–MHC display libraries in yeast10,11 have enabled interrogations of TCR cross-recognition and even identification of TCR targets without any preexisting knowledge of what that TCR recognizes12. Although such strategies theoretically offer an unbiased approach to determining the TCR recognition profile, not all possible positions are equally well represented and not all TCRs can be fully characterized12. Additionally, yeast display strategies have so far been developed only for the characterization of TCRs restricted to a few specific HLAs, and the technique is limited to a few specialized laboratories10,13. Another approach involves the functional interrogation of T cells exposed to peptides with only one fixed amino acid and a random composition for the rest of the sequence14,15; however such approaches require many TCR-expressing cells, and do not provide an in-depth hierarchy of binding interactions, as does the TCR fingerprinting presented here (Supplementary Fig. 1 and Supplementary Note 1).
Currently, the most widespread strategy for resolving potential cross-recognition of a TCR involves investigating the effect of single-position alanine substitutions on the reactivity of T cells2. This strategy is insufficient for describing the full TCR recognition profile (Supplementary Fig. 2). Here we present an approach that enables indepth characterization of the TCR recognition patterns that are decisive for pMHC interactions, which can be easily implemented in most immunology laboratories and is applicable across all foldable MHC molecules. We leverage the use of DNA barcode-labeled MHC multimers16, which allows a ‘one-pot’ strategy whereby the interaction of one clonal TCR with multiple related pMHC epitopes can be assessed simultaneously17. This is possible (i) because the sequencing-based readout of the DNA barcode-based MHC multimer analysis allows direct quantification of the relative interactions of a given TCR with multiple pMHC variants and (ii) because the high complexity of DNA barcodes enables the generation of large libraries of differently labeled pMHC multimers. On the basis of such analysis, a hierarchy of pMHC interactions can be determined. The feasibility of this approach for accurately determining the affinity-based hierarchy and the limitations related to alanine-only substitutions are presented in Supplementary Figure 2 and Supplementary Data 1 and 2, and the technical requirements for this strategy are described in Supplementary Notes 1 and 2.
We first investigated the recognition pattern of two different TCRs isolated from individual patients with Merkel cell carcinoma (MCC), each recognizing a different Merkel cell polyomavirus (MCPyV)-derived peptide: APNCYGNIPL (denoted as APN), restricted to HLA-B*0702, and EWWRSGGFSF (EWW), restricted to HLA-A*2402 (ref. 18). A collection of DNA barcode-labeled MHC multimers was produced for each TCR, containing all the peptides generated from sequentially substituting every single amino acid of the two original decamer peptides with all naturally occurring amino acids (n = 191 for each library) (Supplementary Data 3 and 4). For the HLA-B*0702APN-engaging TCR, these data showed that the original amino acids asparagine at position 3, tyrosine at position 5, and glycine at position 6 were essential for maintaining binding between the TCR and the MHC-bound peptide. In contrast, there was some flexibility at positions 2, 7, 8 and 10, and the amino acids present at positions 1, 4 and 9 seemed to be the least critical for the TCR to recognize the MHC-embedded peptide (Fig. 1a–c). For the HLA-A*2402EWW-engaging TCR, the glycine at position 7 and the phenylalanine at position 8 were critical for maintaining the interaction between the TCR and the MHC-bound peptide. Positions 2, 4, 5, 6, 9 and 10 were less restricted in terms of amino acid requirements, but did display some selectivity. Barely any effect was seen when amino acids at positions 1 and 3 were substituted (Fig. 1d–f). Moreover, when applying the same two MHC multimer libraries to screen peripheral blood mononuclear cells (PBMCs) from healthy donors, we found no noteworthy signal or weighted preference for any amino acids (Fig. 1a,d and Supplementary Figs. 3 and 4). Notably, results from screening of both TCRs showed that amino acid substitutions at the peptide–MHC anchor positions, which are predicted to impede peptide–MHC binding, can still allow the pMHC–TCR interaction (Fig. 1g,h and Supplementary Figs. 5 and 6). We experimentally validated this using an APNCYGNIPL-based alanine substitution library to assess pMHC binding to HLA-B*0702 by MHC ELISA19, which showed that peptides with 40% reduced MHC binding capacity (compared to the original sequence) retained a level of pMHC–TCR interaction similar to that of the original MHC-bound peptide (Supplementary Fig. 5). We also verified that all HLA-A*2402-EWWRSGGFSF peptide variants were stable during the course of the experiment by conformationally dependent enrichment of the pMHC library and by direct MHC tetramer staining using peptide–MHC complexes with various binding affinities (Supplementary Fig. 6). Consequently, the MHC anchor residues play a minor role in the TCR fingerprint (Fig. 1c,f), but are important for the natural presentation of peptides. This may be a consequence of the pMHC production strategy using ultraviolet-mediated peptide exchange technology20, which allows low-level stabilization of MHC complexes even when using peptide variants of very low affinity to a given MHC.
Figure 1.
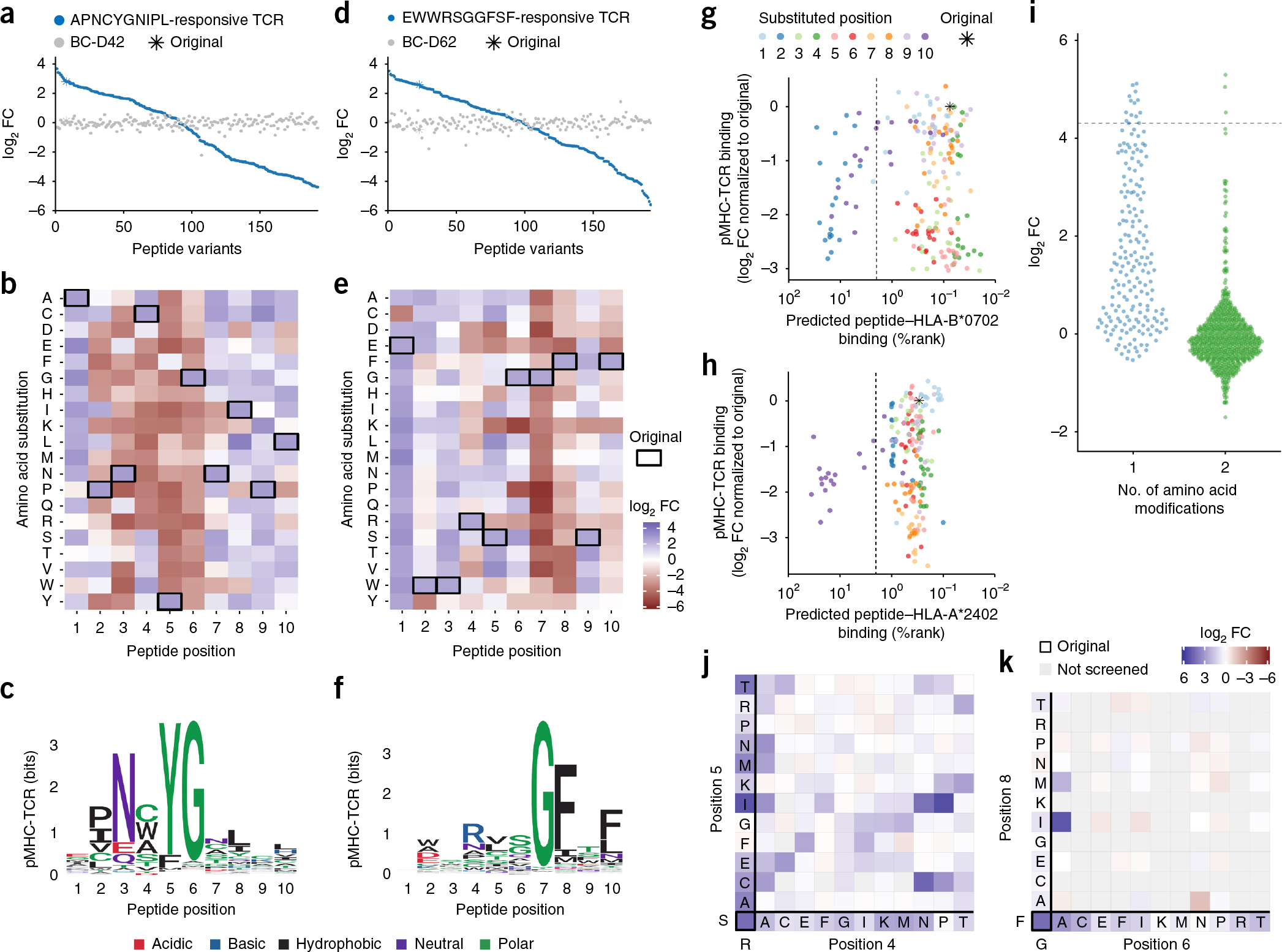
The fingerprints of two different TCRs that recognize MCC-derived peptides restricted to HLA-B*0702 or HLA-A*2402. (a–c) Results obtained from the DNA barcode-based analysis of T cells transduced with a TCR recognizing the HLA-B*0702 -restricted peptide APNCYGNIPL. The analysis was performed with all possible variations of peptides created by single-position amino acid substitutions. (a) The hierarchy of pMHC interactions expressed as log2FC of read counts relative to a triplicate baseline sample (see Supplementary Note 1). A healthy donor PBMC sample (BC-D42) was screened with the same MHC multimer panel in parallel. For both samples, the plotted order of log2FCs of each pMHC-associated DNA barcode is determined by the hierarchy obtained from screening the HLA-B*0702APN-responsive TCR. (b) Heat map of amino acid preferences of the HLA-B*0702APN-responsive TCR based on data from a. Each row represents a given amino acid and each column a position in the peptide sequence. The amino acids of the original peptide target are marked in black boxes. (c) Recognition pattern of the HLA-B*0702APN-interacting TCR, here visualized as a sequence logo based on the data from a and b. (d–f) Results obtained from the DNA barcode-based analysis of T cells transduced with a TCR recognizing the HLA-A*2402-restricted peptide EWWRSGGFSF. The analysis was performed with all possible variations of peptides created by single-position amino acid substitutions. Visualization of data corresponds to a–c. b and e are colored according to the same key. (g,h) Scatter plot of the predicted peptide binding, percentage rank (%rank, x axis) of all naturally occurring amino acid substitutions of APNCYGNIPL to HLA-B*0702 (g) or EWWRSGGFSF to HLA-A*2402 (h), in relation to the experimentally obtained TCR–pMHC interaction (y axis). The color indicates the position of the amino acid substitution. %rank < 2 (dotted line) marks the recommended cutoff of peptides that are considered binders to MHC. (i–k) Results from a parallel MHC multimer analysis of the TCR recognizing the HLA-A*2402 restricted peptide EWWRSGGFSF with a MHC multimer library composed of peptides with single amino acid substitutions corresponding to the one used in d–f, as well as double amino acid substitutions covering 12 naturally occurring amino acids, where positions 4–8 are substituted two amino acids at a time (n = 967; see full list in Supplementary Data 4 and 7). (i) The obtained log2FC values, grouped according to the number of substitutions, one (n = 191) or two (n = 776), within the peptide sequence. Dotted line at 4.30 indicates the original peptide. (j,k) Heat maps showing the log2FC obtained for peptides with amino acids substituted at (j) positions 4 and 5 simultaneously or (k) positions 6 and 8 simultaneously. Each row and column represents a given amino acid substitution (see heat map of all screened substitutions in Supplementary Fig. 9). The original amino acids are marked in bold and peptides in the same row or column are substituted at only one position, indicated with the one-letter code. j and k are colored according to the same key. All data are representative of duplicate analyses.
To gain a deeper understanding of the pMHC complexes, we next generated an in silico structure-based model of each of the original peptides: APNCYGNIPL bound to HLA-B*0702 and EWWRSGGFSF bound to HLA-A*2402 (Supplementary Figs. 7 and 8 and Supplementary Data 5 and 6). This served to visualize the ‘best fit’ of the respective peptides in the MHC pocket and could partially explain the amino acids essential for TCR recognition of pMHC, particularly the glycines at positions 6 and 7 of the HLA-B*0702- and A*2402-bound peptides, respectively. An unfavorable change in energy when substituting with any amino acid at these positions suggests that these glycines may be important to maintaining the peptide in a conformation that promotes TCR engagement, but that they are not necessarily direct interaction points for the TCR. Thus the TCR fingerprint illustrates (i) the amino acids essential for direct interaction between the TCR and the pMHC and (ii) the moieties crucial for maintaining a peptide conformation that favors such interactions.
A potential limitation of the current technology is the lack of assessment for TCR interactions arising from mutual amino acid variations at several defined positions. To determine the potential impact of such mutual variations, we designed a library consisting of peptides with two independent alterations at every position from 4 to 8. To minimize the library size, only high-affinity HLA-A*2402 binding peptides were included (n = 776, Supplementary Data 7). The majority of the mutually substituted peptides showed decreased TCR interaction properties compared to single-position amino acid variations when screened in parallel (Fig. 1i). Several mutually beneficial amino acid combinations could be identified at positions 4 and 5 (Fig. 1j and Supplementary Fig. 9), reflecting the preferences and flexibility determined by the original fingerprint at these positions (Fig. 1f). Notably, alanine at position 6 specifically allowed isoleucine or methionine at position 8 (Fig. 1k), which are the only alternative residues tolerated at this position as determined by the original fingerprint. For the restricted positions 7 and 8, no alternative amino acid combinations were tolerated despite the mutual substitutions (Supplementary Fig. 9). TCR recognition of selected peptides with multiple substituted amino acids was confirmed by direct MHC tetramer staining (Supplementary Fig. 9). Thus, although the mutual substitutions did reveal favorable interactions and limit the combinatorial space for possible peptide interaction partners, they did not show unique interaction requirements that were not accounted for in the original fingerprint.
We next set out to resolve the recognition pattern of different TCRs recognizing the same target. We first investigated two murine transgenic TCR cell lines, OT-1 and OT-3, which have been reported to have high and low functional avidity, respectively, to the H-2Kb-restricted peptide SIINFEKL21. These T cells were screened with a library of 153 DNA barcode-labeled MHC multimers holding single amino acid substitutions of the SIINFEKL peptide (Supplementary Data 8). The hierarchy of pMHCs (Fig. 2a,b and Supplementary Fig. 10) was again used to generate individual TCR fingerprints to visualize the amino acids critical for TCR recognition (Fig. 2c,d). Both TCRs were highly dependent on the original amino acids at positions 6 (glutamic acid) and 7 (lysine). However, while the OT-1 TCR was more flexible at these positions, tolerating other amino acids with the same properties, it had a higher dependence on the original amino acids at positions 1 (serine) and 4 (asparagine) than the OT-3 TCR. T cell staining using fluorescently labeled MHC multimers carrying one of seven SIINFEKL variants supported the different binding properties of the two TCRs (Supplementary Fig. 11).
Figure 2.
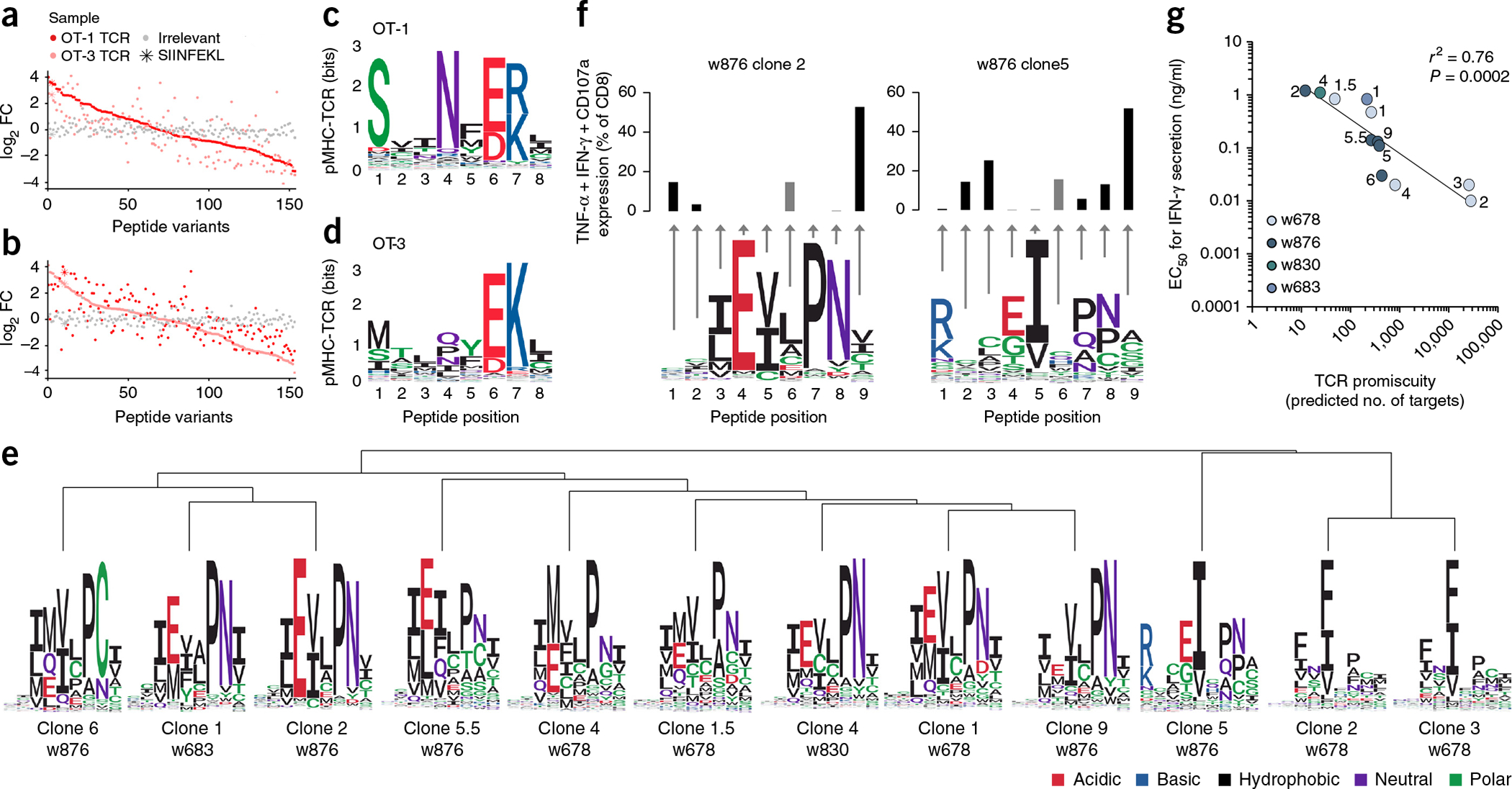
Diverse recognition patterns of TCRs recognizing the same pMHC epitope. (a,b) The TCR-pMHC interaction hierarchy obtained from DNA barcode-based analysis of mouse OT-1 (dark red) and OT-3 (light red) T cells, both recognizing the H-2Kb-restricted peptide SIINFEKL. The analyses were performed with all possible variations of peptides created by single-position amino acid substitutions. log2FC of read counts relative to a triplicate baseline (see Supplementary Note 1) is plotted according to the hierarchy obtained from the OT-1 T cells (a) or the OT-3 T cells (b), compared to the signal obtained using T cells from wild-type C57BL/6 mice (irrelevant), all screened with the same MHC multimer panel. (c,d) The different TCR fingerprints obtained from the screening of (c) OT-1 and (d) OT-3 derived T cells. The OT-1 and OT-3 T cells were screened once. See Supplementary Figure 10 for the read counts of the corresponding data and Supplementary Figure 11 for single fluorescence-based MHC multimer stainings of a range of SIINFEKL variants. (e) TCR fingerprints of 12 MCC clones all originally identified for their recognition of the HLA-A*0201-restricted peptide KLLEIAPNC. The fingerprints are clustered according to the similarity of their recognition pattern. Data are representative of duplicate analyses. (f) Bar plots showing cytokine secretion after stimulating the clonal T cells with peptides containing alanine substitutions at the indicated positions compared to the obtained TCR fingerprints (from e) of clone 2 and clone 5, w876. The gray bars indicate the original peptide, which has an alanine at position 6. Cytokine secretion was determined once (individual frequencies are shown in Supplementary Fig. 12). TNF, tumor necrosis factor; IFN, interferon. (g) Correlation between the number of targets estimated for each TCR, based on data from e (x axis), and the obtained half-maximal effective concentration (EC50) values of each clone (y axis). Each dot represents one T-cell clone. Dots of the same color indicate clones derived from one patient. R2 is based on Pearson’s r on the log-transformed values (n = 12 individual T cell clones).
We then examined 12 different TCRs derived from four patients with MCC22 (Supplementary Data 9). These TCRs all recognized the same HLA-A*0201-restricted nonamer peptide KLLEIAPNC, derived from the common region of the oncogenic proteins, large and small T antigen, of MCPyV. The individual TCR recognition patterns were determined by screening each clone with the same library of 192 DNA barcode-labeled MHC multimers holding the KLLEIAPNC peptide variants (Supplementary Data 10). Notably, we observed a substantial variance in the TCR fingerprints of these TCRs, even among those derived from the same patient. The most notable recurring pattern was a preference for the hydrophobic amino acids isoleucine, phenylalanine or valine at position 5 (Fig. 2e), although the influence of these amino acids varied between the TCRs. The pattern of similarity was clearly evident when clustering the 12 TCR fingerprints in a hierarchical manner (Fig. 2e and Supplementary Fig. 12). A weaker restriction at position 5 appeared to be associated with a strong requirement for proline at position 7 and, in some cases, asparagine at position 8. Furthermore, several TCRs required glutamic acid or methionine at position 4; these positions were sometimes more important for TCR recognition than the hydrophobic amino acid at position 5. Clone 5 from patient w876 had an additional preference for amino acids with a positive side chain, lysine or arginine, at position 1. We validated two of the TCR fingerprints with distinct characteristics (clones 2 and 5 from patient w876) by assessing the functional capacity of the corresponding T-cell clones for recognition of alanine substitution variants of KLLEIAPNC (Fig. 2f and Supplementary Fig. 12). Although the TCR recognition pattern is ultimately determined by the TCR sequence, the specific involvement of α and β variable regions did not explain all of the differences observed between the TCR fingerprints (Supplementary Data 9). The distinct TCR recognition patterns of the individual clones also imply a quantifiable difference in the number of potential peptide sequences that may be recognized by each TCR. On the basis of the fingerprint profile, we calculated for each clone any possible substitution that resulted in a similar or enhanced pMHC–TCR interaction compared to the original peptide sequence. This simplistic approach does not consider potential mutual substitution biases (for example, unfavorable amino acid combinations) and defines a fixed number of MHC anchor residues. The 12 TCRs analyzed here may recognize a range of 12 to 28,080 different peptide sequences with similar or increased affinity compared to the original KLLEIAPNC peptide (Supplementary Data 11). Of note, we observed that the number of potential targets for a given TCR inversely correlated to the functional avidity of the interrogated T cells (Fig. 2g). This indicates that the TCR recognition pattern may not only be useful for further characterizing TCRs with respect to their range of potential pMHC targets, but may also hint at their functional capacity.
To investigate the range of potential pMHC targets, we next used the TCR fingerprints of the 12 HLA-A*0201 KLLEIAPNC-specific T-cell clones (Fig. 2e) to predict peptides from the entire human proteome that may be potentially cross-recognized by each TCR. We used the Find Individual Motif Occurrences (FIMO) software package23 to create a priority list based on the likelihood of cross-recognition between the TCR and the human proteome. On the basis of the top 1,000 sequences from this priority list, for each TCR, we generated a correlation matrix illustrating the overlap of potential cross-recognition between the different clones (Supplementary Fig. 12). This overlap follows the patterns of the TCR fingerprint, with clones 2 and 3 (from w678) sharing very similar profiles, which are quite distinct from the other clones, and clone 5 (from w876) having a unique profile. From the priority list, we applied the top ten sequences of each TCR (Supplementary Data 12), along with the original peptide, to experimentally evaluate the cross-recognition through one combined DNA barcode-labeled MHC multimer library (n = 75) used to stain all 12 T-cell clones. We were able to confirm some level of recognition against 25 of the 75 peptides (Fig. 3a and Supplementary Data 12). We again observed distinct cross-recognition properties of clones 2 and 3 from patient w678 and clone 5 from patient w876. For the most prominent hit (related to clone 5), we confirmed the cross-recognition of peptide (KTVGIYPNA) by conventional fluorescence-based tetramer staining (Fig. 3b) and by intracellular cytokine staining after stimulation with peptide-pulsed HLA-A*0201-expressing cells (Fig. 3c). This peptide had a 44% sequence overlap with the original peptide. From the pool of cross-reactive peptides, the vast majority were predicted not to bind HLA-A*0201 (Fig. 3a and Supplementary Data 12), minimizing the actual risk of clinically relevant cross-recognition. However, low-affinity peptides have been identified as targets for T-cell recognition24. The exemplified cross-recognized peptide KTVGIYPNA is predicted as a low-affinity ligand (netMHCpan percentage rank score of 3.2), but with a confirmed functional recognition (Fig. 3c). It is derived from ST6 N-acetylgalactosaminide (Supplementary Data 13) and expressed at medium to high levels in myocytes. Consequently, we predict that muscle tissue would be at direct risk of attack if one were to apply such TCR in a clinical setting.
Figure 3.
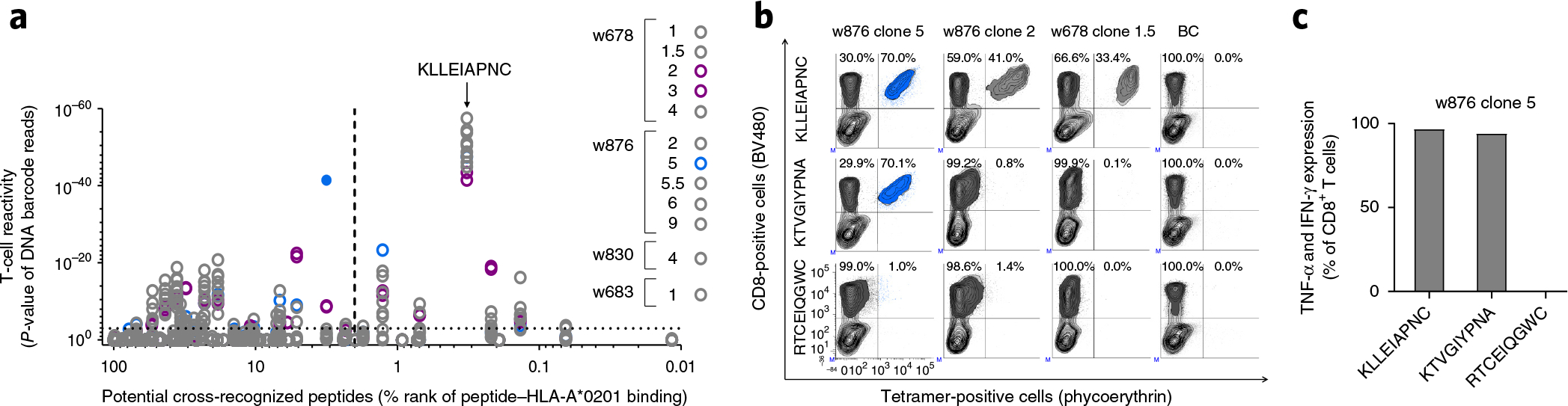
Cross-reactivity of HLA-A*A0201KLL-responsive TCRs. (a) Screening for T-cell recognition of 75 peptides that are potentially cross-recognized by one or more of the 12 clonal T cells that have the HLA-A*A0201-restricted KLLEIAPNC peptide as original target. For each clone the top ten potential cross-reactive peptides were synthetized and used to screen for TCR cross-recognition using DNA barcode-labeled MHC multimers. Total library size was 75 peptides (Supplementary Data 12). The P-values resulting from the DNA barcode-based screen of all 75 pMHC multimers and all 12 clones are plotted (y axis) according to percentage rank score (%rank, x axis). Dotted line at y = 3 represent the selected threshold of false-discovery rate < 0.1%. Dotted line at x = 2 marks the recommended cutoff of peptides that are considered binders to MHC. The closed symbol indicates a response that was also confirmed by staining with fluorescently labeled MHC tetramers. The T-cell clones were screened twice. See Online Methods for statistical processing. (b) Contour plots from the fluorescently based tetramer screening of three clones that all recognize the original HLA-A*0201-restricted KLLEIAPNC peptide. Tetramers are generated from either the original peptide target (KLLEIAPNC), a peptide (KTVGIYPNA) that was cross-recognized by the TCR of clone 5, w876, in a, or a peptide (RTCEIQGWC) that was not recognized by any of the clones in a. The clones were spiked into a healthy donor PBMC sample (BC) in equal amounts. The percentage of total CD8+ T cells is indicated within the contour plots. (c) The frequency of cytokine-producing cells of CD8+ T cells after stimulating clone 5, w876, with HLA-A*0201-expressing cells pulsed with the indicated nonamer peptide. TNF, tumor necrosis factor; IFN, interferon. Tetramer staining and cytokine secretion were determined once.
In summary, our data demonstrate the feasibility of a one-pot approach for generation of TCR fingerprints and the utility of this method for characterizing potential TCR cross-recognition. This is valuable because the promiscuity of TCRs provides an intrinsic challenge to the use of TCRs in clinical applications and increases the risk of autoimmune reactions25. Although TCRs from patients have undergone thymic selection, the strategies applied for TCR gene therapy may override additional peripheral tolerance mechanisms that normally work to avoid cross-recognition of healthy tissues. Consequently, critical adverse events may arise from the use of natural TCRs26. Additionally, many TCR transgenic strategies have involved the optimization of TCR affinity, for example through deep mutational scanning27,28, without understanding the impact of such modifications in terms of pMHC recognition profiles.
Direct evidence for pathological effects from TCR cross-recognition stem from a clinical trial of adoptive cell therapy using such an affinity-optimized TCR for transduction of T cells3,29. Severe adverse events were observed in this trial due to cross-recognition of healthy tissue, including two cases of fatal toxicity linked to T-cell crossreactivity between the melanoma-associated antigen (MAGE-A3) and a titin-derived peptide expressed in healthy cardiac cells3,29. With only 55% sequence overlap between those two peptides, this case highlights the great challenge facing preclinical evaluations of TCRs3,29,30. The fingerprinting strategy presented here provides a screening tool for understanding the TCR recognition profile in greater detail before selection of TCRs intended for clinical development.
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
ONLINE METHODS
Ethical approval.
All healthy donor material was collected under approval by the Scientific Ethics Committee of the Capital Region, Denmark, and written informed consent was obtained according to the Declaration of Helsinki. Collection of MCC patient material was approved by the Fred Hutchinson Cancer Research Center Institutional Review Board and conducted according to the Declaration of Helsinki principles. Informed consent was received from all participants. Splenocytes from OT1 and OT3 transgenic mice were collected from collaborators, under regular approval from the national committee of animal health (approval no. M165–15, University of Lund, Sweden).
Cell samples.
Peripheral blood mononuclear cells (PBMCs) from healthy donors were isolated from whole blood by density centrifugation on Lymphoprep (Axis-Shield PoC) and cryopreserved at −150 °C in FCS (Gibco) + 10% DMSO. Mouse spleen suspensions were obtained by mashing the full spleen through a 70-μm cell strainer (Fischer Scientific). Red blood cells were lysed with RBC Lysis buffer (BioLegend) and used directly or cryopreserved at −150 °C in FCS (Gibco) + 10% DMSO.
Generation of DNA barcodes and dextran conjugation.
Attachment of 5′ biotinylated AxBy DNA barcodes to PE- and streptavidin-conjugated dextran was performed as described in ref. 16. Oligonucleotides containing distinct 25mer nucleotide sequences31 were purchased from LGC Biosearch Technologies (Denmark), and PE- and streptavidin-conjugated dextran was provided by Immudex (Denmark) and FINA Biosolutions LCC (USA). All oligonucleotides carry a 6-nt unique molecular identifier32.
Peptide libraries.
Most of the collections of peptide variants applied in this study were designed by sequentially substituting every single position of the full original peptide sequence with all naturally occurring amino acids. Some libraries also included a number of length and position variants, extending the peptide to either the C- or N-terminal direction of the full protein sequence (Supplementary Data 1, 2 and 10). For the double substitution library applied in Figure 1i–k and Supplementary Figure 9, the variants comprise peptides with two independent amino acid substitutions at all positions from 4 to 8. These positions were substituted with all combinations of 12 different amino acids (T, R, P, N, M, K, I, G, F, E, C and A) to minimize the total library size. For the same purpose only high-affinity peptides (%rank < 0.5) were synthesized and included in the MHC multimer analysis, accumulating to 782 different peptides that were substituted at two positions simultaneously (Supplementary Data 7). Peptides were purchased from Pepscan (Pepscan Presto) and dissolved to 10 mM in DMSO.
MHC monomer production.
UV-sensitive ligands were synthesized as previously described19,20,33. In brief, recombinant HLA-A*0201, HLA-A*2402, HLA-B*0702 and H-2Kb heavy chains and human or mouse β2 microglobulin light chain were produced in Escherichia coli. HLA and H-2 heavy and light chains were refolded with UV-sensitive ligands and purified as described in ref. 34. Specific peptide–MHC complexes were generated by UV-mediated peptide exchange19,20,33,35.
The stability of various peptide–MHC complexes generated through UV-exchange was investigated further as described in Supplementary Note 3.
Generation of DNA barcode-labeled peptide–MHC multimer libraries.
DNA barcode-labeled peptide–MHC multimers, all carrying a common fluorescent PE label, were generated as previously described16. Immediately before staining, barcode-labeled MHC multimers were centrifuged for 5 min at 3,300g and pooled (0.0036–2.3 pmol of each pMHC per sample) to enable parallel staining. An aliquot of ~5 μL of the MHC multimer reagent pool was stored at −20 °C for baseline analysis.
Staining with DNA barcode-labeled multimers.
Cryopreserved cells were thawed and washed in RPMI + 10% FCS, then washed in barcode-cytometry buffer (PBS + 0.5% BSA + 100 μg/mL herring DNA + 2 mM EDTA) and incubated for 30 min at 37 °C in the presence of 50 nM dasatinib. Cells (0.5 × 106–2 × 106) were incubated for 15 min at 37 °C with the DNA-barcoded MHC multimer pool in a total volume of 80 μL (final concentration of each distinct pMHC, 0.036–23 nM). Next, a 5× antibody mix composed of CD8-PerCP (Invitrogen MHCD0831) (final dilution 1:50) or BV510 (BD 563256, clone RPA-T8) (final dilution 1:25) or BV480 (BD 566121, clone RPA-T8) (final dilution 1:50), dump channel antibodies (CD4-FITC (BD 345768) (final dilution 1:80), CD14-FITC (BD 345784) (final dilution 1:32), CD19-FITC (BD 345776) (final dilution 1:16), CD40-FITC (Serotech MCA1590F) (final dilution 1:40), and CD16-FITC (BD 335035) (final dilution 1:64)) and a dead cell marker (LIVE/DEAD Fixable Near-IR; Invitrogen L10119) (final dilution 1:1,000) was added and incubated for 30 min at 4 °C. Cells were washed three times in barcode-cytometry buffer and fixed in 1% paraformaldehyde (PFA). If the cells were not analyzed within 24 h, they were washed twice and resuspended in barcode-cytometry buffer. Cells were analyzed within a week after multimer staining. For staining of mouse splenocytes, OT1 and OT3 T cells, the following antibodies were used: CD8a-BV480 (BD 566096, clone 53–6.7) and CD3-FITC (BioLegend 100206, clone 145–2C11).
Cell sorting.
Cells were sorted on a FACSAriaFusion (BD) into tubes containing 100 μL of barcode-cytometry buffer (tubes were saturated with PBS + 2% BSA in advance). Using FACSDiva software, we gated on single, live, CD8-positive and ‘dump’ (CD4, 14, 16, 19 and 40)-negative lymphocytes and sorted all multimer-positive (PE) cells within this population. The sorted cells were centrifuged for 10 min at 5,000g and the buffer was removed. The cell pellet was stored at −80 °C in a minimal amount of residual buffer (<20 μL). The gating strategy is shown in Supplementary Figure 13.
DNA barcode amplification.
DNA barcode amplification was performed as previously described16. PCR amplification was conducted on isolated cells (in <20 μL of buffer) or on a stored aliquot of the MHC multimer reagent pool (diluted 50,000× in the final PCR), which was used as the baseline to determine the number of DNA barcode reads within an unprocessed MHC multimer reagent library. PCR products were purified with a QIAquick PCR Purification kit (Qiagen, 28104). The amplified DNA barcodes were sequenced at Sequetech (USA) using an Ion Torrent PGM 316 or 318 chip (Life Technologies).
Processing of sequencing data derived from multimer-associated DNA barcodes.
Sequencing data were processed by the software package Barracoda, available online at http://www.cbs.dtu.dk/services/barracoda. This tool identifies the barcodes used in a given experiment, assigns sample ID and pMHC specificity to each barcode, and counts the total number of reads and clonally reduced reads for each pMHC-associated DNA barcode. Furthermore, it accounts for barcode enrichment, expressed as log2FC, based on methods designed for the analysis of RNA-seq data. See details in “Statistical analyses.”
Normalization of log2FC values relative to the original peptide–MHC.
To compare the log2FC values of the original peptide with those obtained from the peptides with an amino acid substitution, all log2FCs were normalized using the formula , where z is the normalized log2FC, x is the log2FC of the peptide variation, σ is the s.d. of ll log2FCs, and ω is the log2FC of the original peptide. Applied in Figure 1g,h and Supplementary Figures 2 and 5.
Generation of fluorescently labeled MHC tetramers.
MHC tetramers were assembled on PE-conjugated streptavidin (BioLegend, Nordic Biosite, Denmark) as previously described36,37 and acquired on a BD LSR Fortessa. Gating strategy exemplified in Supplementary Figure 13.
T-cell functional assays.
T-cell functionality was evaluated from EC50 values (interferon-γ secretion) or through intracellular cytokine stainings as described in Supplementary Note 4.
TCR gene capture.
For HLA-B*0702APN- and HLA-A*2402EWW-responsive CD8+ T cells, TCR gene capturing was performed as previously described38. Briefly, DNA isolated from MCC CD8+ T cells was sheared to fragments of 500–600 bp with a Covaris system (S-series, D10%, I5, C/b 200, 30 s), and the resulting DNA fragments were purified with SPRI beads (Agencourt). Sequence library preparation was performed using the TruSeq DNA Sample Preparation kit (Illumina) with the adaptation of only seven cycles for the final library amplification. Illumina TruSeq 6-bp indexes (as designed by the manufacturer) were used for multiplexing. Multiplexed TCR captures were performed using a custom-designed Agilent SureSelect bait library with the following adaptations: pools of six to eight DNA libraries were captured with 1:10 of a bait reaction and block 3 in the hybridization mixture was replaced with a custom NKI block 3. The NKI block 3 consisted of equal amounts of two DNA oligonucleotides (IDT-DNA, Iowa, USA) at 16.6 μg μl−1:NKI 3.1 5′-AGATCGGAAGAGCACACGTCTGAACTCCAGTCACNNNNNNATCTCGTATGCCGTCTTCTGCTTG/3′ddC/−3′ and NKI 3.2 5′-CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGAC GTGTGCTCTTCCGATCT/3′ddC/−3′. Captured library fragments were split into two fractions and PCR enrichment (15 cycles) was performed using the Illumina P5 and P7 oligonucleotides (IDT-DNA, Iowa, USA): P5 primer 5′-AATGATACGGCGACCACCGAGATCT-3′ and P7 primer 5′-CAAGCAGAAGACGGCATACGAG-3′.Quantification of PCR reactions was validated on a BioAnalyzer DNA Chip (Agilent) and reactions were combined in equal amounts and diluted (10 nM) afterwards. Paired-end sequencing was performed using the Illumina Hiseq2000 platform with a read length of 75–100 bp. CDR3 TCR sequences were identified from the sequencing data as previously reported39.
KLL-specific CD8+ T cell clones.
Simultaneous sequencing of TCRα and TCRβ repertoires was performed as described in ref. 40.
TCR transduction.
Retroviral transduction was performed as previously described41. Briefly, Phoenix-A cells were used as packaging cells and were transfected with 10 μg of retroviral plasmid DNA. Virus supernatant was harvested 2 d after transfection and was either used immediately or was snap-frozen and stored at −80 °C. PBMCs were activated by CD3/CD28 beads (human T cell expander; Invitrogen/Dynal) in a 1:2 ratio (cell/bead). After 30 min of incubation at room temperature, non-CD3+ cells were removed by magnetic separation. Cells were incubated in RPMI media + 10% human serum containing IL-15 (Peprotech; 5 ng/mL) and rh-IL-2 (Novartis; 100 IU/mL). For bead-based transduction, beads were incubated with retronectin overnight at 4 °C. Beads were washed with PBS and blocked with 2% BSA following a 2-h incubation with virus supernatant (1 × 107–2 × 107 beads per milliliter virus supernatant). PMBCs were incubated with virus-coated beads at a 1:10 ratio (cell/bead) for 24 h at 37 °C. The transduction efficiency was determined 5 d after transfection and was always over 60%.
Affinity predictions.
The binding affinity resulting from each of the amino acid substitutions of the original peptide sequences was predicted using NetMHCpan 4.0 (ref. 42). The %rank is the rank of the predicted binding affinity compared to a set of random natural peptides. This measure is not affected by inherent bias of certain MHC molecules toward higher or lower mean predicted affinities. Strong binders are defined as having %rank < 0.5, and weak binders as %rank < 2.
Modeling of MHC-bound peptides.
The in silico structure-based pMHC model of HLA-B*0702APN and HLA-A*2402EWW with the original peptide embedded in the MHC binding pocket was made using MODELLER43, and the conformation of the original peptide was then optimized using the robotics-based kinematic closure (KIC)44 protocol from Rosetta45.
The program FoldX46 was used to model all single amino acid substitutions of the original peptide and to predict their effect on the interaction as the difference between the predicted binding energy of the MHC to the mutated and original peptide, respectively: ΔΔ = Δamino acid substitution − Δoriginal. A ΔΔ > 0 indicates that a given substitution has destabilizing properties, and a ΔΔ < 0 indicates that a given substitution has stabilizing properties.
Generation of sequence logos.
TCR interaction with pMHC.
The TCR fingerprints were created on the basis of the log2FC values calculated by Barracoda. The amino acid substitution setup that we applied allowed us to assign a single log2FC value to each amino acid residue at each position in the peptide sequence. The log2FC of the original peptide was assigned to each of the respective amino acid residues at each position. In this way a position-specific scoring matrix (PSSM) for each clone was calculated, with the number of rows corresponding to the number of positions in the peptide and the number of columns corresponding to the number of naturally occurring amino acids. We then used softmax to normalize each row of the PSSMs to sum to 1, and the resulting position-specific frequency matrices were then converted into Shannon logos47.
Energy-based sequence logos of peptide–MHC.
The sequence logo of the structurally predicted peptide–MHC binding preference for HLA-B*0702APN and HLA-A*2402EWW was made using the energies from the FoldX analysis of each amino acid substitution to generate a PSSM in which each row represents positions in the peptide and each column represent an amino acid substitution. We then normalized each row in the PSSM. All FoldX energies were normalized using the following formula:
where Nij is the normalized FoldX energy for each position in the normalized PSSM and Aij is the FoldX energies in the ith row and jth column of the PSSM. The PSSMs were normalized so that each position summed to 1, and the sequence logos were then generated with the Shannon method in Seq2Logo48. We calculated the information content I for each position in the peptide using the following formula:
where pa is the normalized FoldX energy for each amino acid substitution. This information content is shown in this structural pMHC model of HLA-B*0702APN and HLA-A*2402EWW in Supplementary Figures 7 and 8 and Supplementary Data 5 and 6.
Predicted peptide binding affinities.
The predicted peptide binding affinity of each amino acid substitution of the original peptide sequence (shown in Fig. 1g,h) was found using NetMHCpan-4.0 (ref. 49).
Principal component analysis.
To visualize the inter-Shannon-logo distances, we flattened each PSSM to a vector with elements corresponding to the number of naturally occurring amino acids (n = 20) times the number of peptide positions (n = 9). We then stacked the flattened PSSMs to form a matrix with the number of rows corresponding to the number of clones and number of columns as before. On this combined matrix, we could then perform a PCA and visualize by standard methods. Hierarchical clustering was performed using PC1–PC12, and we visualized the distances using a dendrogram. Applied in Figure 2e and Supplementary Figure 12.
Estimating the TCR promiscuity.
For estimating the TCR promiscuity—i.e., the estimated number of cross-recognized peptides by a given TCR—the log-ratios were calculated by transforming the log2FCs given by Barracoda for each TCR to fold change using base 2 and then dividing each fold change with the fold change of the original peptide. The calculated fold-change ratios were then applied to estimate the number of possible cross-recognized peptides calculated per TCR using standard combinatorics on the positional set of log ratios larger than zero. In this calculation the anchor positions (2 and ∞) were included with the numbers 2 and 3 respectively, reflecting the selectivity of HLA-A*0201 (determined from the ‘naturally presented ligands’ given by NetMHCpan-4.0)49. The calculations for H-2Kb followed the same rationale, with the anchor positions (3, 5 and 8) included with the numbers 4, 4 and 3, respectively. The obtained values for TCR promiscuity were plotted against the experimentally obtained EC50 values22 (Fig. 2g) and are listed in Supplementary Data 11.
Prediction of cross-reactive peptides related to MCC clones.
From each Shannon logo, cross-reactive peptides were predicted from the corresponding PSSMs using the Find Individual Motif Occurrences (FIMO) software package23, which searches the human proteome for sequences that match each logo. For each MCC clone, the ten peptides with the highest likelihood for cross-recognition to the given TCR (lowest P-value) were synthesized.
Signature of potential cross-recognition.
The correlation matrix (Supplementary Fig. 12) was constructed on the basis of the output from the FIMO database. For each clone, the corresponding top 1,000 peptides (based on P-values) were retrieved. The total pool of top peptides from all clones was then, per peptide, scored against the Shannon PSSMs using the sum of positional scores. The set of scores per clone was then correlated all-against-all using Pearson’s correlation coefficient. Lastly, the set of correlations per clone was clustered using hierarchical clustering.
Statistical analyses.
The statistical processing of DNA barcode reads was developed in ref. 16 and is based on methods designed for the analysis of RNA-seq data, implemented in the R package edgeR49. Fold changes in read counts mapped to a given sample relative to mean read counts mapped to triplicate baseline samples are estimated using normalization factors determined by the trimmed mean of M-values method50. P values (applied in Fig. 3a) were calculated by comparing the read counts from each experiment (n = 2 individual samples and 75 individual DNA barcodes) with the mean baseline sample reads (n = 3) using a negative binomial distribution with a fixed dispersion parameter set to 0.1. False-discovery rates (FDRs) were estimated using the Benjamini–Hochberg method. Specific barcodes with an FDR < 0.1% were defined as significant. At least 1/1,000 reads associated with a given DNA barcode relative to the total number of DNA barcode reads in that given sample was set as the threshold to avoid false-positive detection of T-cell populations due to low number of reads in the baseline samples.
The statistical analyses in Figure 2g were conducted using GraphPad Prism 7. Normal distribution was assessed using a Shapiro–Wilk normality test, suggesting log transformation of the data. The statistical test (two-tailed) and Pearson correlation is therefore performed on log-transformed data.
Code availability.
All relevant code is available from the authors. For DNA barcode analysis, the tool Barracoda is available online at http://www.cbs.dtu.dk/services/barracoda.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability.
All relevant data are available from the authors. TCR sequences and expression vectors must be obtained through a material transfer agreement.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank all Merkel cell carcinoma patients and healthy donors contributing material to this study; T. Joeris, W. Agace and K. Lahl (Lund University, Lund, Sweden) for sharing of splenocytes from OT-1 and OT-3 transgenic mice; B. Rotbøl and T. Tamhane for excellent technical assistance; and S.R.H.’s group for scientific discussions. This research was funded in part through the European Research Council, StG 677268 NextDART, Lundbeck Foundation Fellowship R190–2014-4178 (to S.R.H.), the Independent Research Fund Denmark (DFF 4004–00422 and DFF 7014–00055, to S.R.H. and M.N.), NIH NCI Cancer Center Support Grant P30 CA015704, K24-CA-139052 and R01 CA176841, Kelsey Dickson Team Science Courage Research Team Award, Prostate Cancer Foundation Award 15CHAS04, and the UW MCC Patient Gift Fund (to P.N.).
Footnotes
COMPETING INTERESTS
A.K.B. and S.R.H. are co-inventors on a patent covering the use of DNA barcode-labeled MHC multimers (WO2015185067 and WO2015188839). N.J.M., C.D.C., D.M.K. and P.N. are co-inventors on a patent application filed by their employer, University of Washington, concerning the HLA-A*0201-restricted TCR sequences.
Any Supplementary Information and Source Data files are available in the online version of the paper.
References
- 1.Sewell AK Why must T cells be cross-reactive? Nat. Rev. Immunol. 12, 669–677 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Obenaus M et al. Identification of human T-cell receptors with optimal affinity to cancer antigens using antigen-negative humanized mice. Nat. Biotechnol. 33, 402–407 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Cameron BJ et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 5, 197ra103 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis MM & Bjorkman PJ T-cell antigen receptor genes and T-cell recognition. Nature 334, 395–402 (1988). [DOI] [PubMed] [Google Scholar]
- 5.Mason D A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol. Today 19, 395–404 (1998). [DOI] [PubMed] [Google Scholar]
- 6.Arstila TP et al. A direct estimate of the human αβ T cell receptor diversity. Science 286, 958–961 (1999). [DOI] [PubMed] [Google Scholar]
- 7.Robins HS et al. Comprehensive assessment of T-cell receptor β-chain diversity in αβ T cells. Blood 114, 4099–4107 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cornberg M & Wedemeyer H Hepatitis C virus infection from the perspective of heterologous immunity. Curr. Opin. Virol. 16, 41–48 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Wooldridge L et al. A single autoimmune T cell receptor recognizes more than a million different peptides. J. Biol. Chem. 287, 1168–1177 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Birnbaum ME et al. Deconstructing the peptide-MHC specificity of T cell recognition. Cell 157, 1073–1087 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adams JJ et al. Structural interplay between germline interactions and adaptive recognition determines the bandwidth of TCR-peptide-MHC cross-reactivity. Nat. Immunol. 17, 87–94 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gee MH et al. Antigen identification for orphan T cell receptors expressed on tumor-infiltrating lymphocytes. Cell 172, 549–563.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sibener LV et al. Isolation of a structural mechanism for uncoupling T cell receptor signaling from peptide-MHC binding. Cell 174, 672–687.e27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wooldridge L et al. CD8 controls T cell cross-reactivity. J. Immunol. 185, 4625–4632 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schaubert KL et al. Generation of robust CD8+ T-cell responses against subdominant epitopes in conserved regions of HIV-1 by repertoire mining with mimotopes. Eur. J. Immunol. 40, 1950–1962 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bentzen AK et al. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat. Biotechnol. 34, 1037–1045 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Bentzen AK & Hadrup SR Evolution of MHC-based technologies used for detection of antigen-responsive T cells. Cancer Immunol. Immunother. 66, 657–666 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lyngaa R et al. T-cell responses to oncogenic Merkel cell polyomavirus proteins distinguish patients with Merkel cell carcinoma from healthy donors. Clin. Cancer Res. 20, 1768–1778 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodenko B et al. Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat. Protoc. 1, 1120–1132 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Toebes M et al. Design and use of conditional MHC class I ligands. Nat. Med. 12, 246–251 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Enouz S, Carrié L, Merkler D, Bevan MJ & Zehn D Autoreactive T cells bypass negative selection and respond to self-antigen stimulation during infection. J. Exp. Med. 209, 1769–1779 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller NJ et al. Tumor-infiltrating Merkel cell polyomavirus-specific t cells are diverse and associated with improved patient survival. Cancer Immunol. Res. 5, 137–147 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grant CE, Bailey TL & Noble WS FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee PP et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med. 5, 677–685 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Albert LJ & Inman RD Molecular mimicry and autoimmunity. N. Engl. J. Med. 341, 2068–2074 (1999). [DOI] [PubMed] [Google Scholar]
- 26.van den Berg JH et al. Case report of a fatal serious adverse event upon administration of T cells transduced with a MART-1-specific T-cell receptor. Mol. Ther. 23, 1541–1550 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fowler DM & Fields S Deep mutational scanning: a new style of protein science. Nat. Methods 11, 801–807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harris DT et al. Deep mutational scans as a guide to engineering high affinity t cell receptor interactions with peptide-bound major histocompatibility complex. J. Biol. Chem. 291, 24566–24578 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linette GP et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122, 863–871 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morgan RA et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 36, 133–151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu Q, Schlabach MR, Hannon GJ & Elledge SJ Design of 240,000 orthogonal 25mer DNA barcode probes. Proc. Natl. Acad. Sci. USA 106, 2289–2294 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kivioja T et al. Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods 9, 72–74 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Chang CXL et al. Conditional ligands for Asian HLA variants facilitate the definition of CD8+ T-cell responses in acute and chronic viral diseases. Eur. J. Immunol. 43, 1109–1120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hadrup SR et al. High-throughput T-cell epitope discovery through MHC peptide exchange. Methods Mol. Biol. 524, 383–405 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Frøsig TM et al. Design and validation of conditional ligands for HLA-B*08:01, HLAB*15:01, HLA-B*35:01, and HLA-B*44:05. Cytometry A 87, 967–975 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Hadrup SR et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat. Methods 6, 520–526 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Andersen RS et al. Parallel detection of antigen-specific T cell responses by combinatorial encoding of MHC multimers. Nat. Protoc. 7, 891–902 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Linnemann C et al. High-throughput identification of antigen-specific TCRs by TCR gene capture. Nat. Med. 19, 1534–1541 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Bolotin DA et al. Next generation sequencing for TCR repertoire profiling: platform-specific features and correction algorithms. Eur. J. Immunol. 42, 3073–3083 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Han A, Glanville J, Hansmann L & Davis MM Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat. Biotechnol. 32, 684–692 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kühlcke K et al. Retroviral transduction of T lymphocytes for suicide gene therapy in allogeneic stem cell transplantation. Bone Marrow Transplant. 25(Suppl. 2), S96–S98 (2000). [DOI] [PubMed] [Google Scholar]
- 42.Nielsen M & Andreatta M NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 8, 33 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fiser A & Sali A ModLoop: automated modeling of loops in protein structures. Bioinformatics 19, 2500–2501 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Stein A & Kortemme T Improvements to robotics-inspired conformational sampling in Rosetta. PLoS One 8, e63090 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaufmann KW, Lemmon GH, Deluca SL, Sheehan JH & Meiler J Practically useful: what the Rosetta protein modeling suite can do for you. Biochemistry 49, 2987–2998 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schymkowitz J et al. The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–W388 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wagih O ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics 33, 3645–3647 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Thomsen MCF & Nielsen M Seq2Logo: a method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res. 40, W281–W287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jurtz V et al. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 199, 3360–3368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robinson MD & Oshlack A A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are available from the authors. TCR sequences and expression vectors must be obtained through a material transfer agreement.