

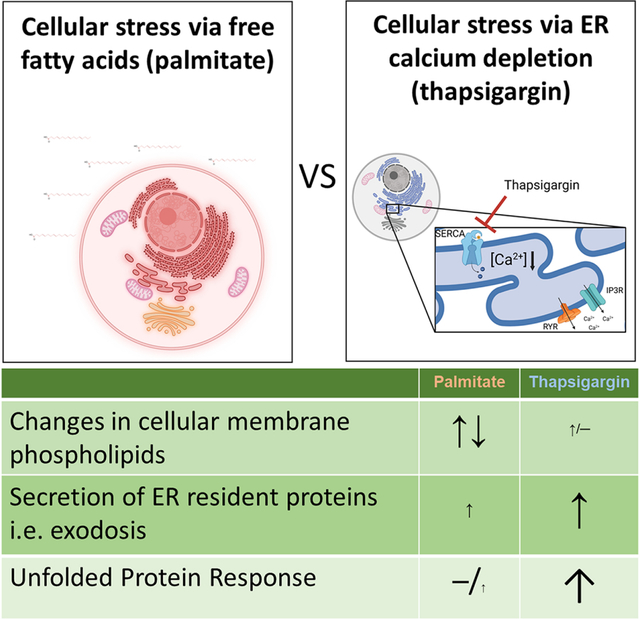
Abstract
The endoplasmic reticulum (ER) is an organelle that performs several key functions such as protein synthesis and folding, lipid metabolism and calcium homeostasis. When these functions are disrupted, such as upon protein misfolding, ER stress occurs. ER stress can trigger adaptive responses to restore proper functioning such as activation of the unfolded protein response (UPR). In certain cells, the free fatty acid palmitate has been shown to induce the UPR. Here, we examined the effects of palmitate on UPR gene expression in a human neuronal cell line and compared it with thapsigargin, a known depletor of ER calcium and trigger of the UPR. We used a Gaussia luciferase-based reporter to assess how palmitate treatment affects ER proteostasis and calcium homeostasis in the cells. We also investigated how ER calcium depletion by thapsigargin affects lipid membrane composition by performing mass spectrometry on subcellular fractions and compared this to palmitate. Surprisingly, palmitate treatment did not activate UPR despite prominent changes to membrane phospholipids. Conversely, thapsigargin induced a strong UPR, but did not significantly change the membrane lipid composition in subcellular fractions. In summary, our data demonstrate that changes in membrane lipid composition and disturbances in ER calcium homeostasis have a minimal influence on each other in neuronal cells. These data provide new insight into the adaptive interplay of lipid homeostasis and proteostasis in the cell.
Keywords: Endoplasmic reticulum, Unfolded Protein Response, UPR, exodosis, phospholipids, palmitate, ER calcium, lipidomics, cell signaling, lipids, brain lipids, thapsigargin
Graphical Abstract
1. Introduction
The endoplasmic reticulum (ER) is an intracellular organelle composed of a network of membranous tubules and sheets. The ER is the site of several critical cellular functions such as calcium homeostasis, protein synthesis and folding, and lipid metabolism. Disruption of these ER functions, for example protein misfolding, leads to ER stress. Following an accumulation of unfolded or misfolded proteins in the ER, the cell activates the unfolded protein response (UPR). The UPR is an adaptive cellular response to ER stress that is initiated by three ER-localized membrane proteins that act as protein-based sensors: IRE1 (Inositol-requiring enzyme 1), PERK (double-stranded RNA-activated protein kinase-like ER kinase) and ATF6 (activating transcription factor 6) (reviewed in (Walter and Ron, 2011; Hetz et al., 2020). Activation of the UPR pathways via these proteins cause attenuation of protein translation, increased expression of chaperone proteins, degradation of proteins (ER-associated degradation, ERAD) and, if the stress is prolonged, apoptosis (Walter and Ron, 2011; Hetz et al., 2020). The ER serves as the primary intracellular reservoir of calcium and depletion of ER calcium is a well-established activator of the UPR (reviewed in (Mekahli et al., 2011; Preissler et al., 2020). In addition, ER calcium depletion disrupts ER proteostasis by causing the secretion of resident ER proteins (Trychta et al., 2018) that are normally retained in the ER lumen via a C-terminal ER retention sequences (ERS) such as the prototypical “KDEL” sequence (Munro and Pelham, 1987). ERS-containing proteins that move from the ER to Golgi are recognized by the KDEL receptors (KDELRs) and returned to the ER lumen as part of the KDEL retrieval pathway (Munro and Pelham, 1987; Orci et al., 1997). Following ER calcium depletion, ERS-containing proteins leave the ER en masse and overwhelm the KDELRs resulting in the secretion of normally ER resident proteins in a phenomenon termed “exodosis” (Trychta et al., 2018). During exodosis there is both a loss-of-function inside the ER and a gain-of-function outside of the cell as intracellular proteins relocate to the extracellular space. Both consequences of exodosis have the potential to affect the metabolism of the secreting cell as well as impact the functioning of neighboring cells. Some ERS tails that undergo exodosis belong to proteins which are associated with lipid metabolism, such as carboxylesterase 1 and 2 (Ces1 and 2) (Ruby et al., 2017; Chalhoub et al., 2021; Lagrutta et al., 2021; Xu et al., 2021) and secreted phospholipase A2 XIIA and B (sPLA2XIIA and B) (Guan et al., 2011; Ee et al., 2014; Trychta et al., 2018). Given the ER is also the main site of lipid metabolism in the cell and subsequently regulates the lipid composition of other organelles, such as the Golgi, mitochondria, endosomes, plasma membrane, and lipid droplets (Phillips and Voeltz, 2016; Wu et al., 2018), it is unclear whether ER calcium depletion or the consequent loss of resident ER proteins, especially those proteins involved in lipid metabolism, alter membrane lipid composition in neuronal cells.
While the accumulation of misfolded proteins in the ER was initially characterized as the trigger of the UPR, more recent studies indicate that changes in the ER lipid composition also leads to activation of UPR pathways (Volmer et al., 2013; Hou et al., 2014; Halbleib et al., 2017). For example, treating cells with the saturated free fatty acid palmitate activates the UPR in macrophages (Kim et al., 2015), pancreatic cells (Cunha et al., 2008), liver cells (Achard and Laybutt, 2012; Trentzsch et al., 2020), leukemia cells (Piccolis et al., 2019) and cardiomyocytes (Yamamoto et al., 2020). Gwiazda et al. (2009) demonstrated that palmitate caused UPR associated with ER calcium depletion in pancreatic β-cells. However, not much is known about how palmitate affects the UPR or ER calcium in neuronal cells.
Here we examine cellular lipid composition, UPR and exodosis in a human neuronal cell line following treatment with two known inducers of ER stress associated with ER calcium depletion and changes in lipid composition, thapsigargin and palmitate, respectively. We have previously described a Gaussia luciferase (GLuc)-based reporter termed GLuc-SERCaMP (secreted ER calcium modulated protein) that can be used to monitor exodosis triggered in vitro and in vivo (Henderson et al., 2014). The exodosis phenomenon is triggered by ER calcium depletion, so the GLuc-SERCaMP can be viewed as a de facto indicator of ER calcium depletion (Henderson et al., 2014; Wires et al., 2017). In the current study, we used palmitate to modify the membrane lipid composition of neuroblastoma SH-SY5Y cells and used our GLuc-SERCaMP reporter to study the effects on ER resident protein secretion and indirectly ER calcium. We also measured mRNA levels for a set of UPR markers following palmitate treatment to further gauge the effects of lipid modifications on proteostasis. In addition, using a cell fractioning method (Hayashi and Su, 2007) to enrich the cellular membranes into different fractions and mass spectrometry to detect glycerophospholipids we studied whether calcium depletion and subsequent exodosis influence membrane phospholipid composition. Overall, we examined changes in ER lipid composition and ER proteostasis caused by two different triggers of ER stress in a human neuronal cell to study the interplay between primary ER functions.
2. Materials and Methods
2.1. Cell culture
SH-SY5Y human neuroblastoma cells (ATCC, CRL-2266) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM + GlutaMAX, 4.5 g/L D-glucose, 110 mg/L Sodium pyruvate, Thermo Fisher Scientific, Waltham, MA) supplemented with 10 % (v/v) Bovine Growth Serum (BGS, GE Life Sciences), penicillin (10 Units/mL) and streptomycin (10 μg/ml) (Thermo Fisher Scientific). Cells were kept in a humified incubator with 5.5 % CO2 at 37°C. SH-SY5Y-GLuc-SERCaMP cells, stably expressing GLuc-ASARTDL were maintained in similar conditions. For the electron microscopy experiments SH-SY5Y cells were grown in DMEM (Sigma-Aldrich, St Louis, MO) supplemented with 10% fetal bovine serum (Gibco, Invitrogen, Thermo Fisher Scientific, Waltham, MA) and 1% Pen-Strep (Gibco, Waltham, MA).
2.2. Chemicals and Reagents
Palmitic acid (#P0500, dissolved in ethanol) and thapsigargin (#T9033, dissolved in DMSO) were from Sigma-Aldrich. Prior to treatment palmitic acid was bound to fatty acid free bovine serum albumin (A7030, Sigma-Aldrich) by first dissolving BSA in DMEM (without supplements) at 20 % (w/v) concentration and then adding 10x palmitic acid to the solution. The solution was then placed on a shaker at 42°C for 30 minutes to allow for binding of palmitic acid to BSA. The BSA/palmitic acid solutions were then diluted 1:10 with DMEM and final concentration of 5 % BGS, 1 % Penicillin/streptomycin and 2 % BSA. For the dose range of palmitic acid from 200–600 μM the molar ratio range of palmitic acid to BSA was 0.83:1 – 2:1, with the ratio for the 400 μM palmitic acid being 1.33:1. Thapsigargin and vehicle treatments were prepared in similar medium (DMEM with 5 % BGS, 1 % penicillin/streptomycin and 2 % BSA).
2.3. Cell morphology
For the brightfield and confocal microscope imaging the cells were plated in 96-well plates with glass like polymer bottom (cat# P96-1.5P, Cellvis, Mountain View, CA) 1.2 × 104 cells/well. For the DsRed2-ER images the cells were reverse transfected with the pDsRed2-ER construct (100 ng DNA/well, Clontech, cat# 632409) simultaneously to their plating. Transfection was done with Lipofectamine 3000 (cat# L300015, Invitrogen), according to the manufacturer’s instructions. The following day (non-transfected cells) or 48 hours later (pDsRed-ER transfected cells) the cells were treated with vehicle, 100 nM thapsigargin or 400 μM palmitate for 24 hours. After the treatment, the live cells were imaged with Evos FL Auto 2 imaging system (Thermo Fisher Scientific Cat#AMAFD2000). After the brightfield images were taken, the cells were rinsed with PBS and fixed with 4 % paraformaldehyde in PBS for 15 min at RT. After fixing, the cells were rinsed three times with PBS followed by blocking and permeabilizing with PBS containing 0.1 % Triton-X and 10 % Goat serum (Fisher cat#16210-064) for 1 h at RT. The cells were then incubated overnight at +4°C with primary antibodies diluted in the blocking buffer. The following day the cells were first washed three times with PBS, incubated with secondary antibodies diluted in blocking buffer for 2 h at RT and finally washed again three times with PBS. DAPI nucleus stain was added to the cells (1 μg/ml) in the second last wash. After the staining the cells were imaged with Nikon Eclipse Ti2 A1 Confocal microscope using Nikon 20x Plan Apo 0.75 DIC N2 air objective or 60x Plan Apo 1.4 NA oil objective (Nikon Instruments, Melville, NY). Z-stack images (1024×1024) were taken with 1 μm step size and 0.5 pixel size (20x images) or 0.2 μm step size and 0.07 μm pixel size (60x images). To analyze the ER volume we used Imaris v9.7.2 (Oxford Instruments) software surface module and measured MANF immunoreactivity as a volumetric reconstruction. We used 5 regions of interest (ROIs) per cell and each ROI was 50 pix * 50 pix * 12 steps or 3.5 μm * 3.5 μm * 2.4 μm and max ROI volume is 29.4 μm^3.
For the electron microscopy, after 24h of treatment with vehicle, palmitate or thapsigargin, cells grown on glass coverslips were fixed with 2% glutaraldehyde (Sigma-Aldrich) in 0.1 M sodium cacodylate (Electron Microscopy sciences (EMS), Hatfield, PA) buffer, pH 7.4, for 20 min at RT, washed twice with cacodylate buffer, and postfixed with 1% reduced osmium tetroxide (EMS) in cacodylate buffer for 1 h on ice. Samples were further washed with cacodylate, buffer and dehydrated with a series of ethanols (70, 96, and finally 100 % EtOH) and infiltrated with Epon (TAAB 812) for 2 h at RT prior to 14 h polymerization at 60 °C (as described in (Seemann et al., 2000). 60 nm or 100 nm thin sections were picked on Pioloform-coated single-slot copper grids (2mm × 1mm Slot Copper Grid, from EMS) and post-stained with uranyl acetate (SPI Supplies, West Chester, PA) and lead citrate (3 % solution, Leica). Transmission electron microscopy images were acquired using a Hitachi HT780 (Hitachi High-Technologies Corporation, Tokyo, Japan) operating at 100 kV, equipped with Gatan Rio 9 bottom-mounted CMOS camera (Gatan, Pleasanton, CA) with 6000X magnification and processed with ImageJ (version 1.53f, imagej.nih.gov).
2.4. Gaussia luciferase secretion assay
SH-SY5Y-GLuc-SERCaMP cells (Henderson et al., 2014) were plated in 96-well plate at 1.2 × 104 cells/well and after 24 h 5 μL samples of the cell culture medium were taken to determine the pre-treatment GLuc SERCAMP levels. After taking the samples the cells were treated with either thapsigargin or palmitate by full medium exchange. Twenty-four hours after treatment, 5 μL samples of cell culture medium was collected, and an ATP assay was run. The medium samples were collected into white walled 96-well plates (Fisher, cat# 3912) and the luciferase activity was measured with a plate reader (BioTek Synergy 2, Winooski, VT) following injection of 100 μL of 10 μM coelenterazine (Regis Technologies, Morten grove, IL) in PBS. Measurements were done at 25°C, with a sensitivity of 100 and 0.5 s integration time, as described previously (Henderson et al 2014).
2.5. Cell viability (ATP and blue/green) assays
Cells were assayed using the CellTiter-Glo luminescent cell viability assay (G7570, Promega, WI), that measures intracellular ATP levels. The assay was done according to the manufacturer’s instructions and the luminescence was measured with a plate reader (BioTek Synergy 2, Winooski, VT). For Blue/Green cell viability assay SH-SY5Y cells were plated on 96-well plates and treated similarly to Gaussia Luciferase secretion assay. After 24-hour treatment the cells were stained with ReadyProbes Cell Viability Imaging kit, Blue/Green (cat# R37609, Invitrogen) according to the manufacturer’s instructions and imaged with EVOS FL Auto 2 Imaging System (Invitrogen). The number of blue (total) and green (dead/damaged) cells were counted from the images with Nikon NISElements (NIS-Elements AR Analysis 4.30.02 64-bit, Nikon Instruments, Melville, NY).
2.6. Gene expression analysis
Cells were grown in 96-well plates (1.2 × 104 cells/well) and treated with thapsigargin (60 nM) or palmitate (400 μM) for either 8 or 24 h. Cells were lysed and the total RNA was extracted with an RNAdvance Total RNA isolation kit (Beckman Coulter Inc, Brea, CA), according to the manufacturer’s instructions. The integrity and concentration of the isolated RNA was determined by an Agilent Bioanalyzer using an Agilent RNA 6000 Nanokit (Agilent, Santa Clara, CA). 0.5–0.7 μg of RNA in a 40 μL reaction mix was transcribed into cDNA using an iScript cDNA Synthesis kit (Bio-Rad, Hercules, CA). After the reaction 20 μL of DNAse free water was added to the samples and 5 μL of this diluted cDNA was added in duplicates to 20 μL of reaction mix consisting of ddPCR Supermix for probes (no dUTP, Bio-Rad), 450 nM of each primer, and 125 nM of probe (with FAM, 6-carboxyfluorescein; HEX, Hexachlorofluorescein; or VIC, Aequorea Victoria Green Fluorescent Protein fluorophore). The primer and probe sequences are listed in Table 1, all the primers and probes were from Integrated DNA Technologies (IDT, Coralville, IA). MANF, CHOP and GADD34 were quantified using Taqman gene expression assays (ThermoFisher Scientific, cat# Hs00180640_m1, Hs01090850_m1, Hs00169585_m1). Samples were run on automated droplet generator (QX100, Bio-Rad), followed by PCR (T100 Thermal cycler, Bio-Rad) of 39 amplification repeats (amplification sequence 95°C 10 min, 94°C 30 s, 60°C 1 min). The signal in the droplets was measured by droplet reader (QX200, Bio-Rad). No reverse transcriptase (NRT) and no template (NTC) were used as negative controls and to control for contamination. The signal from UPR markers was normalized to the geometric mean of the housekeeping genes ubiquitin-conjugating enzyme 2i (Ube2i) and RNA polymerase II (RNA pol II).
Table 1.
Primers and probes used for measuring UPR activation.
| Target | Forward | Reverse | probe |
|---|---|---|---|
| Ube2i | gtgtgcctgtccatcttagag | gctgggtcttggatatttggttc | caaggactggaggccagccatcac |
| RNApol II | gcaccacgtccaatgacattg | ggagccatcaaaggagatgac | acggcttcaatgcccagcaccg |
| ASNS | ggattggctgccttttatcagg | ggcttctttcagctgcttcaac | tggactccagcttggttgctgcc |
| BiP | gttgtggccactaatggagatac | ggagtttctgcacagctctattg | acgctggtcaaagtcttctccaccca |
| Erdj4 | gccatgaagtaccaccctg | ccactagtaaaagcactgtgtc | ctgcaatctctctgaattttgcttcagc |
2.7. Cell fractioning
The cell fractioning was done as previously described (Vance, 1990; Hayashi and Su, 2007). Briefly, SH-SY5Y cells (2.5 × 106) were plated in 15 cm dishes (5 dishes per one sample), 24 h after plating the cells were treated with vehicle (2× 10−3 % EtOH and 5.6 × 10−5 % DMSO), thapsigargin (60 nM) or palmitate (400 μM). Both treatments had the same amount of DMSO and ethanol. Treatments were done by full medium exchange. After 24 h of treatment cells were collected by scraping them in ice-cold PBS on ice. The cells were spun down (500 × g, 5 min, 4°C), the pellet was weighed and resuspended in 5 × volume of homogenizing buffer (H-B, 10 mM HEPES (4-(2-hydproxyethyl)-1-piperazineethanesulfonic acid) pH 7.4, 0.25 M sucrose) and homogenized with a Dounce glass homogenizer. Samples were centrifugated (600 × g, 5 min, 4°C) and the supernatant was collected. The pellet was resuspended in 600 μl of H-B and centrifugated as before. The supernatant was collected and combined with the first supernatant and the pellet was collected as the P1 fraction. The supernatant was then centrifugated (10 300 × g, 20 min, 4°C) and the supernatant and pellet were collected. The pellet was re-suspended in isolation medium (I-M, 5 mM HEPES, pH 7.4, 225 mM mannitol, 1 mM EGTA) pipetted on top of Percoll (Sigma) medium (25 mM HEPES, pH 7.4, 225 mM mannitol, 1 mM EGTA, 30 % (v/v) Percoll) and centrifugated (Swinging bucket rotor, 95 000 × g with no braking, 30 min, 4°C). The MAM and mitochondria fractions were collected. The mitochondria fraction was washed 3 times with I-M (centrifugated between the washes 10 500 × g, 10 min, 4°C) and the MAM fraction was washed once with isolation medium 2 (I-M2, 25 mM HEPES, pH 7.4, 225 mM mannitol, 1 mM EGTA) and the pellet was collected as the crude MAM fraction. The supernatant containing MAM and the supernatant from the first ultracentrifugation step were centrifugated (100 000 × g, 1 h, 4°C) and the pellets were collected as P3 (microsome fraction) and purified MAM. The supernatant on top of the P3 fraction was collected as the cytosolic fraction. A schematic presentation of the fractioning workflow and a table with the protein yields for each fraction can be found in the supplement (Supplemental S1 and S2).
2.8. Western blot analysis
The protein concentrations of the cellular fractions were measured using colorimetric RC DC assay kit (#5000122, Bio-Rad). 15 μg of protein per sample (except for mitochondria fractions, that were too dilute to take 15 μg, from which maximum volume of sample was run) was run on a Bis-Tris 4–12% gel (NP0321, NuPage, Invitrogen, MA) with MOPS Buffer (200V, 45 min) and transferred to a 0.2 micron PVDF membrane (IB24001, Invitrogen) with iBlot2 (Invitrogen). Immediately after the transfer the blot was stained for total proteins (Total Stain Q, AC2225, Azure Biosystems), dried, and imaged with a Sapphire Biomolecular Imager (488 nm laser, Azure Biosystems). After imaging the total protein stain the membranes were re-hydrated with methanol, washed with distilled water followed by PBS, and blocked with blocking buffer (cat# MB070, Rockland, PA) for 1 h at room temperature (RT) and then probed with primary antibodies at 4°C overnight in blocking buffer. The following day the blots were washed and probed with secondary antibodies. The blots were scanned with Odyssey scanner (LI-COR Biosciences, Lincoln, NE). The optical density of each protein band was normalized to the optical density of the total proteins of the corresponding sample lane. Optical densities were measure using ImageJ (version 1.53f, imagej.nih.gov). All the used antibodies are listed in Table 2.
Table 2.
Antibodies and the dilutions they were used in.
| Antibody | Source | Identifier | dilution | Method |
|---|---|---|---|---|
| Rabbit anti-DGAT1 | Abcam | Ab181180 | 1:8000 | WB |
| Mouse anti-Sigma 1 Receptor | Santa Cruz | sc-137075 | 1:500 | WB |
| Mouse anti-cytochrome C | BDPharmingen | #556433 | 1:1000 | WB |
| Rabbit anti-SERCA | Cell signaling | 4388S | 1:2000 | WB |
| Mouse anti-Na/K ATPase | NovusBio | NB300-146SS | 1:8000 | WB |
| Mouse anti-TGN38 | Santa Cruz | sc-166594 | 1:1000 | WB |
| Goat anti-Rabbit IRDye 800CW | LiCor | 926-32211 | 1:8000 | WB |
| Goat anti-Mouse IRDye 680 RD | LiCor | 926-68070 | 1:8000 | WB |
| Rabbit anti-MANF | YenZym | 56AP#2 | 1:500 | ICC |
| Mouse anti-GM130 | BD | 610822 | 1:500 | ICC |
| Goat anti-Rabbit AlexaFluor 568 | Invitrogen | A11004 | 1:2000 | ICC |
| Goat anti-Mouse AlexaFluor 488 | Invitrogen | A11027 | 1:2000 | ICC |
ICC, Immunocytochemistry; WB, Western blotting.
2.9. Lipid extraction and mass spectrometry
Total lipids were extracted from cellular fractions (80 μg of protein/sample) using a modified Folch extraction method (Folch et al 1957). Five μL of 2:1 v/v of chloroform (CHL)/methanol (MeOH) per μg of protein was added to the cell fractions. A lipid internal standard mixture in 2:1 v/v of CHL/MeOH (glycerolphosphocholine (PC, cat# 8503258P) 10:0/10:0 at 50μg/mL; glycerolphosphatidylethanolamine (PE, cat# 850700P) 10:0/10:0 at 50μg/mL; phosphatidylglycerol (PG, cat# 840434P) 10:0/10:0 at 60μg/mL, Avanti Polar Lipids (Alabaster, Al)) was added at 12 μL per 80 μg of protein and was included in the total volume of CHL/MeOH added to the samples. The cellular fractions were sonicated and vortexed. Next, 3 μL of water per μg of protein was added to the cell fractions. The sample mixture was again vortexed and centrifuged. The lower organic phase was collected and evaporated to dryness using nitrogen. The samples were resuspended in 160 μL of 95:5 v/v MeOH/CHL and stored at −80 °C until mass analysis. Samples were analyzed in an Q Exactive mass spectrometer (Thermo Fisher Scientific, San Jose, CA) with an electrospray ionization source coupled with an auto sampler (Ultimate 3000 HPLC, Thermo Fisher). Data was recorded in positive and negative ion mode with a m/z range of 400–1000 and a mass resolution setting of 100,000. In positive ion mode, samples were diluted 1:10 (v/v) in 10 mM ammonium acetate in methanol and 2:3 (v/v) in methanol for negative ion mode. Ten μL of this diluted sample mixture was injected into the instrument. PC, PE, PG, PI, PS, and CL species number equals the total length and number of the acyl chains, with “a” representing 1,2 diacyl species and “p” representing a 1-O-(10-alkenyl)-2-acyl (plasmalogen) species. Lipids were assigned based on a database search with a mass error less than 3 ppm in positive ion mode and less than 5 ppm in negative ion mode. We used our lipid database for peaks annotation and intensities of all annotated lipids in each sample were automatically recorded in an Excel sheet using R software (www.r-project.org). To obtain the relative quantities of lipids, the data were normalized with the intensity of the internal standards as follows: PC and SM species normalized by PC standard in positive ion mode, PE species normalized by PE standard in positive ion mode, and PG, PI, PS, CL species normalized by PG standard in negative ion mode.
2.10. Statistical analyses
All the statistical analyses were performed using Graphpad Prism version 8.4.3 for Windows (Graphpad Software, San Diego, CA, US, www.graphpad.com). Data is presented as average +SEM, unless stated otherwise in the figure legend. The N for each experiment and the statistical tests used are described in the figure legends. The formulas used to calculate protein yields of the fractions, enrichment of markers and distribution of marker proteins in the fractions are shown in supplement (Supplemental S3).
3. Results
3.1. Both palmitate and thapsigargin treatments cause distinct ultrastructural changes in cell morphology
It was previously reported that in some cell types, especially secretory cells, ER stress and UPR activation lead to expansion of ER i.e. increased surface area and volume of rough ER (Sriburi et al., 2007). Here we use the human neuroblastoma cell line SH-SY5Y cells which are extensively used as a neuronal surrogate in research and our lab has used SH-SY5Y cells in the identification of exodosis (Trychta et al., 2018) to first examine the effects of a 24-hour exposure to thapsigargin (100 nM) or palmitate (400 μM) on cellular morphology. We examined the SH-SY5Y morphology using different microscopy techniques, starting with brightfield microscopy (Fig. 1 A–C). Neither palmitate nor thapsigargin caused robust changes in the overall morphology of the cells. To further assess the effects of these drugs on ER and Golgi, we immunostained the cells for MANF and GM130, respectively, and imaged using confocal microscope (Fig. 1 D–F 20x, G-I 60x). We did not observe any overt changes to the morphology of the ER or Golgi. Similar results were observed with a fluorescent reporter localized to the ER (Supplemental S4). Using an endogenous ER lumenal marker, we measured the ER volume and no significant differences were observed among the treatments (Supplemental S5). Finally, we used transmission electron microscopy to examine ultrastructural changes in the cells in response to drug treatments. We observed signs of cellular stress for both palmitate and thapsigargin based on greater heterogeneity in cell morphology compared to control cells (Fig. 1 J–L). Palmitate treatment correlated with robust changes in the mitochondria, mainly swelling and disruption of cristae. Thapsigargin treatment caused less robust mitochondrial changes, although some cristae disruption was visible. However, thapsigargin caused changes in the ER structure i.e. convoluted or spiraling membranes (Fig. 1K), indicative of ER-phagy (Bernales et al., 2006). These ER-phagy structures were present in 50 % of the thapsigargin treated cells (on average 1 ER-phagy structure per cell) but they were not observed in the palmitate treated cells or control cells (Fig. 1J and 1L).
Figure 1. Representative images of the SH-SY5Y morphology following thapsigargin or palmitate treatment.
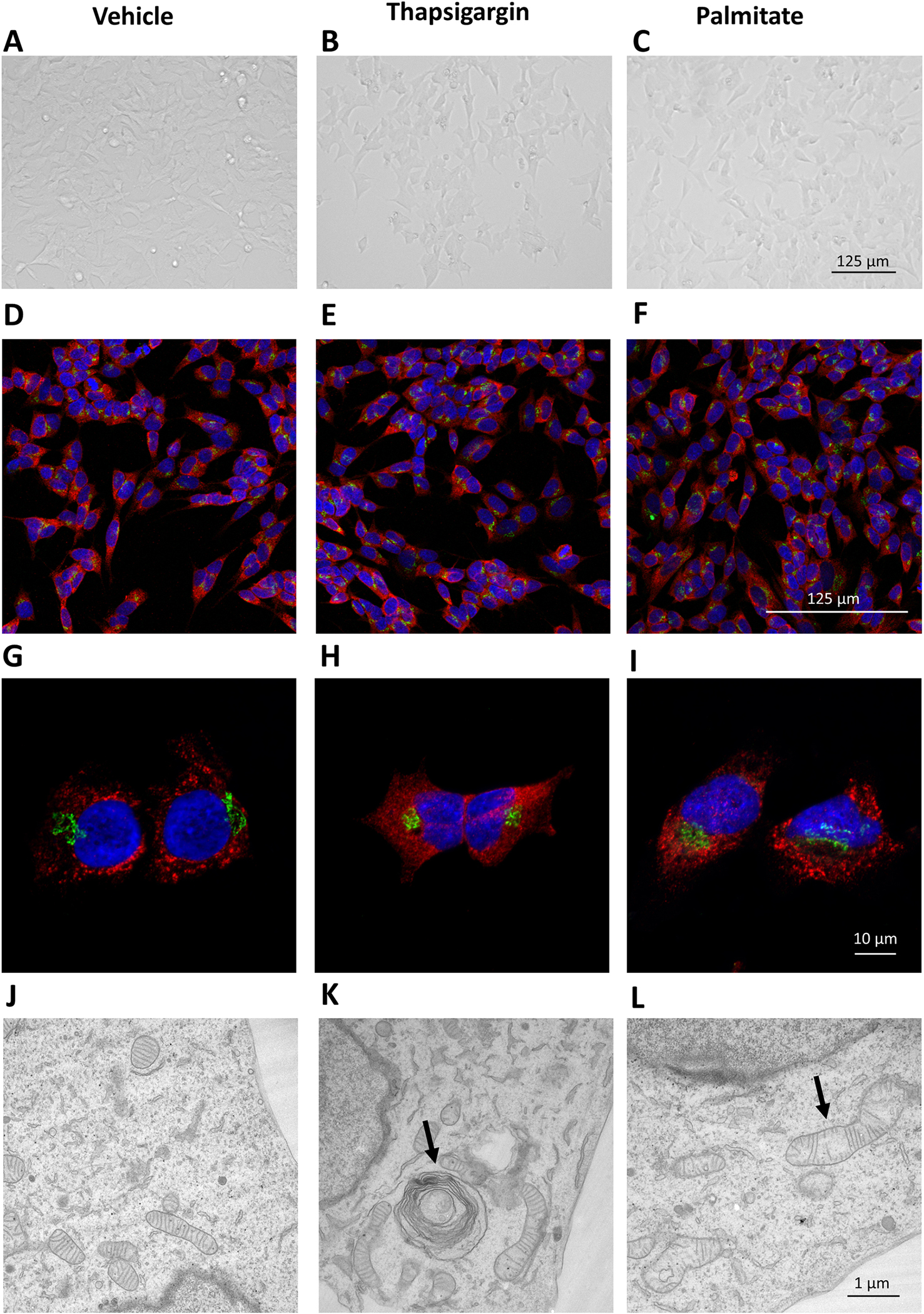
Brightfield images of SH-SY5Y cells treated with vehicle (A), 100 nM thapsigargin (B) and 400 μM palmitate (C) for 24 hours. 20X Confocal images of SH-SY5Y cells treated with vehicle (D), 100 nM thapsigargin (E) and 400 μM palmitate (F) and 60X Confocal images of SH-SY5Y cells treated with vehicle (G), 100 nM thapsigargin (H) and 400 μM palmitate (I) stained with anti-MANF (red) and anti-GM130 (green) antibodies (markers of ER and Golgi, respectively). EM images of SH-SY5Y cells after 24-hour treatment with vehicle (J), 100 nM thapsigargin (K) and 400 μM palmitate (L). The most prevalent changes i.e., convoluted spiraling membranes (K) and disrupted mitochondria cristae (L) in cellular structures are indicated with arrows.
3.2. Thapsigargin causes robust UPR activation and exodosis compared to palmitate treatment
Thapsigargin causes a dose dependent increase in ER exodosis as monitored using GLuc-SERCaMP secretion as a readout of ER resident protein secretion ((Henderson et al., 2014); Fig. 2A). At the highest concentration of thapsigargin used (100 nM), GLuc-SERCaMP secretion was approximately 12-fold higher than in the vehicle treated cells. Palmitate also increased GLuc-SERCaMP secretion, but to a much lesser extent (2-fold) relative to vehicle (Fig. 2B). At lower concentrations neither of the treatments significantly reduced cell viability as measured using an ATP assay, only 100 nM thapsigargin slightly decreased the viability (Fig. 2 A and B). At the lowest exodosis inducing concentrations of palmitate (400 uM) and thapsigargin (60 nM), a small increase in dead cells was observed (5.5+/−1.5% total for palmitate, 4+/−0.9% total thapsigargin; Supplemental S6).
Figure 2. Effects of thapsigargin and palmitate on exodosis and UPR in SH-SY5Y cells.
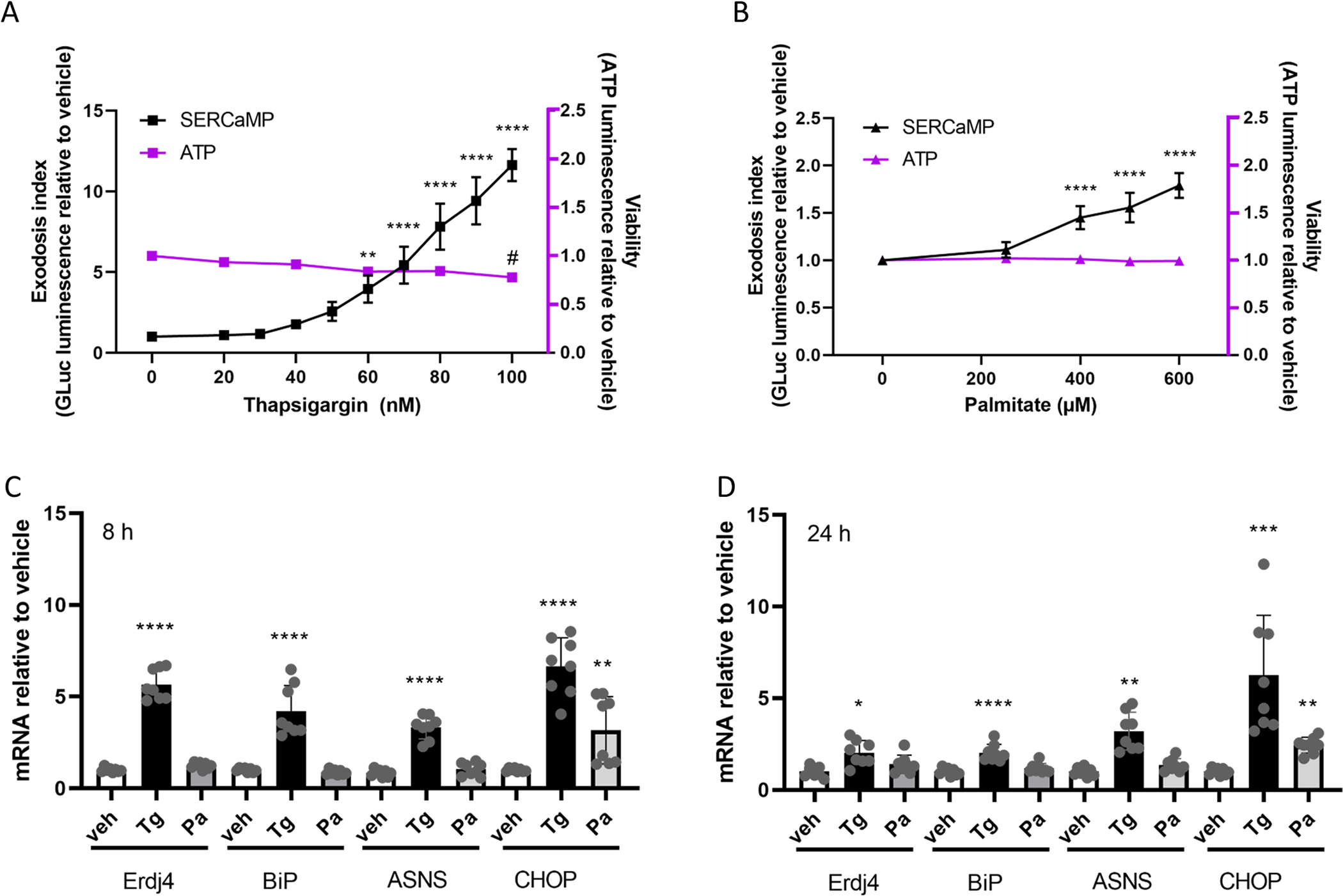
Thapsigargin (A) and palmitate (B) increase exodosis in SH-SY5Y GLuc-SERCaMP cells dose-dependently (measured as the amount of Luminescence signal in the cell culture medium normalized to vehicle, referred to as exodosis index), without affecting the viability of the cells as measured by cellular ATP levels. In SH-SY5Y cells thapsigargin (60 nM) increased the mRNA levels of all studied UPR markers after an 8-hour exposure (C) whereas palmitate (400 μM) increased only CHOP mRNA expression. After 24-hour exposure (D) to thapsigargin the mRNA levels of all UPR markers were still increased compared to vehicle, but to a lesser extent than at 8 hours. After 24-hour treatment with palmitate, only CHOP mRNA levels were increased. One-way ANOVAs with Bonferroni multiple comparisons test were run for both thapsigargin and palmitate treatments separately for exodosis and viability data (A and B) and for each UPR marker (C and D) separately, comparing each treatment to vehicle control (n=8–12). Significances are as follows; */# p<0.05, ** p<0.01, ***p<0.005, ****p<0.001. Pa, palmitate; Tg, thapsigargin; veh, vehicle.
Both palmitate and thapsigargin have been shown to induce UPR. To avoid triggering high levels of cell death or decrease in viability, we chose to use the lowest concentrations of palmitate (400 μM) and thapsigargin (60 nM) that caused significant increase in exodosis in SH-SY5Y cells, to compare their effects on the UPR pathways by measuring the mRNA levels of Erdj4 (IRE1 pathway), BIP (ATF6 pathway), ASNS, CHOP (PERK pathways), MANF (ATF6 pathway) and GADD34 (PERK pathway). Palmitate caused about a 4-fold increase in CHOP mRNA levels, whereas thapsigargin treatment led to about an 8-fold increase. Both treatments increased the CHOP mRNA at 8 h post-treatment, and the effect was maintained at 24 h post-treatment (Fig. 2 C and D). Palmitate did not increase the mRNA levels of any of the other UPR genes measured at any timepoint, whereas thapsigargin increased Erdj4, BiP, and ASNS (4–6 fold after 8 hours, and 2–5 fold after 24 hours, Fig. 2 C and D). In addition, thapsigargin treatment increased the mRNA levels of MANF and GADD34, whereas palmitate slightly increased GADD34 (1.5 fold) but not MANF (Supplemental S6). We tested higher concentrations of palmitate to see if they would increase the mRNA levels of the UPR markers. We also did similar palmitate treatments to human hepatocellular carcinoma cell line Hep3B to see if there was a difference between cell types. An 8 hour treatment with 100 nM thapsirgargin increased the mRNA levels of all the markers in both cell lines, whereas 800 μM palmitate did not (Supplemental S7A). After 24-hour treatment even with 1 mM palmitate we did not see increase in BiP, MANF or ASNS in SH-SY5Y and a slight, less than 2-fold increase in Erdj4 with 600 μM and 1 mM concentrations of palmitate. GADD34 mRNA expression was increased 2.3-fold with 1 mM palmitate in SH-SY5Y cells. However, we saw a more robust, statistically significant increase in the mRNA levels of all the markers in Hep3B cells, indicating that the response is indeed cell line dependent and the lower concentrations of palmitate are capable of eliciting a UPR (Supplemental S7B). In addition, we did a longer (48 h) palmitate treatment (400 and 600 μM) to SH-SY5Y cells, but this did not increase the mRNA levels of any of the markers, except CHOP (Supplemental S7C).
3.3. The distribution of membrane proteins following subcellular fractionation are not affected by thapsigargin or palmitate
To examine effects of thapsigargin and palmitate on the lipid composition of different membrane compartments of the cells, we utilized a cell fractionation method developed to yield an enriched mitochondria associated membrane (MAM) fraction (Hayashi and Su, 2007). The MAM is a raft-like structure within the ER membrane known to be enriched with several enzymes central to lipid metabolism (Rusinol et al., 1994; Hayashi and Su, 2007; Hayashi et al., 2009; Raturi and Simmen, 2013). UPR sensor PERK is also enriched in MAM (Verfaillie et al., 2012). To validate the fractionation procedure, we used a set of subcellular membrane protein markers for each of the different fractions. Like previous reports (Hayashi and Su, 2007), many of the proteins used as markers were present in more than one fraction, but they are enriched in particular fractions. We used Sigma1 Receptor as a marker for MAM, SERCA and DGAT1 as markers for ER fractions (crude MAM or “cMAM”, MAM and P3 a.k.a. microsome fraction), cytochrome c for mitochondria, Na/K ATPase for plasma membrane and TGN38 as a marker for trans-Golgi membranes. P1 fraction consists of nuclei primarily, but also has some cellular debris and possibly unhomogenized cells in it. The used method was developed to enrich MAM fraction and therefore the crude MAM fraction contains part of the MAM fraction, but also some markers of Golgi (TGN38) and mitochondria (Cytochrome C). The distribution of marker proteins between the fractions remained relatively constant between treatments as we did not observe shifts in the protein marker from one membrane compartment to another after 24 hours treatment with thapsigargin or palmitate (Fig. 3, Supplemental S8). Images of representative whole blots are available in the Supplemental data (Supplemental S9).
Figure 3. Marker proteins vary in abundance between different membrane fractions but are not altered by treatment of palmitate or thapsigargin.
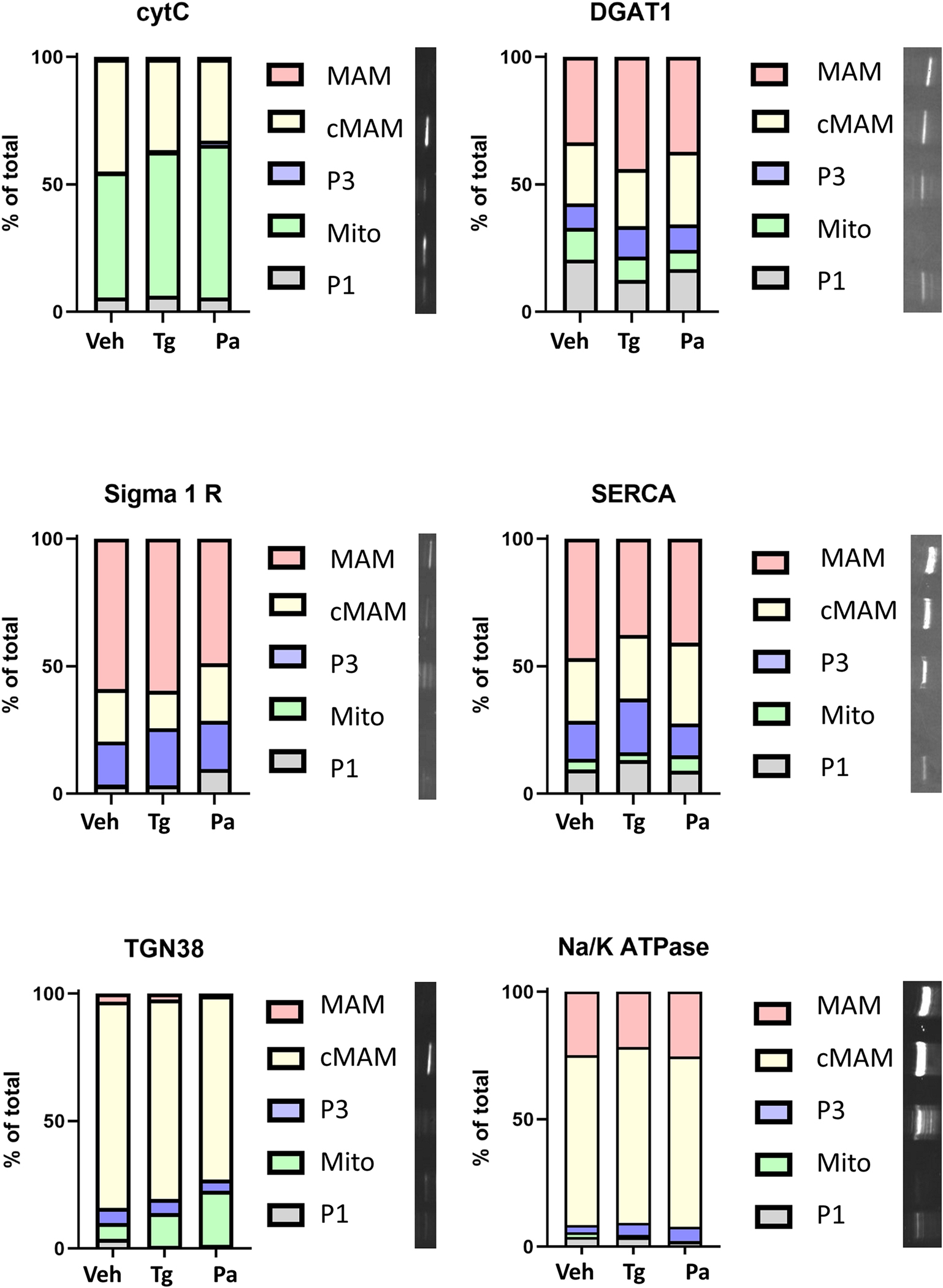
There was no statistically significant shift in the marker proteins between the different fractions following 24-hour treatment with thapsigargin (60 nM) or palmitate (400 μM). Different proteins were detected by Western blot and their relative levels in the fractions are presented in the bar graphs. Representative blots of the proteins from vehicle treated cells are presented to the right of the graph key (blots are cropped and rotated 90° for presentation). The fractions (P1, nuclei, some cell debris; mito, mitochondria; P3, microsome (ER); MAM, mitochondria associated membrane; cMAM, crude MAM that includes ER, Golgi and some mitochondria) in the blot are in the same order as the graph key. CytC, cytochrome C; DGAT1, Diacylglycerol O-acyltransferase 1; Pa, palmitate; SERCA, Sarco(endo)plasmic reticulum calcium ATPase; Tg, thapsigargin; TGN38, Trans-Golgi network integral membrane protein 38.
3.4. Palmitate, not thapsigargin, significantly changes the phospholipid composition in cellular membrane fractions
Lipids were isolated from the subcellular fractions that had been characterized by the presence of protein markers and mass spectrometry was used to identify lipid species present in each of these fractions and their relative amounts (normalized to the internal standards) (Fig. 4). The fractions showed different changes in lipid composition in response to thapsigargin and palmitate treatments (Fig. 4 and 5). When compared to vehicle treated cells, palmitate treatment caused prominent differences in relative levels of lipids in the P1, P3, cMAM and MAM fractions, but did not similarly change the lipids in the mitochondria fraction (Fig. 5). Thapsigargin had little or no effect on lipids in all the fractions. For palmitate, the greatest changes in lipids were observed in the MAM fraction, which is the part of ER where most of the lipid metabolizing enzymes are enriched (Rusinol et al., 1994; Raturi and Simmen, 2013).
Figure 4. Palmitate treatment of SH-SY5Y cells causes changes in the phospholipid composition of different cellular fractions but thapsigargin does not.
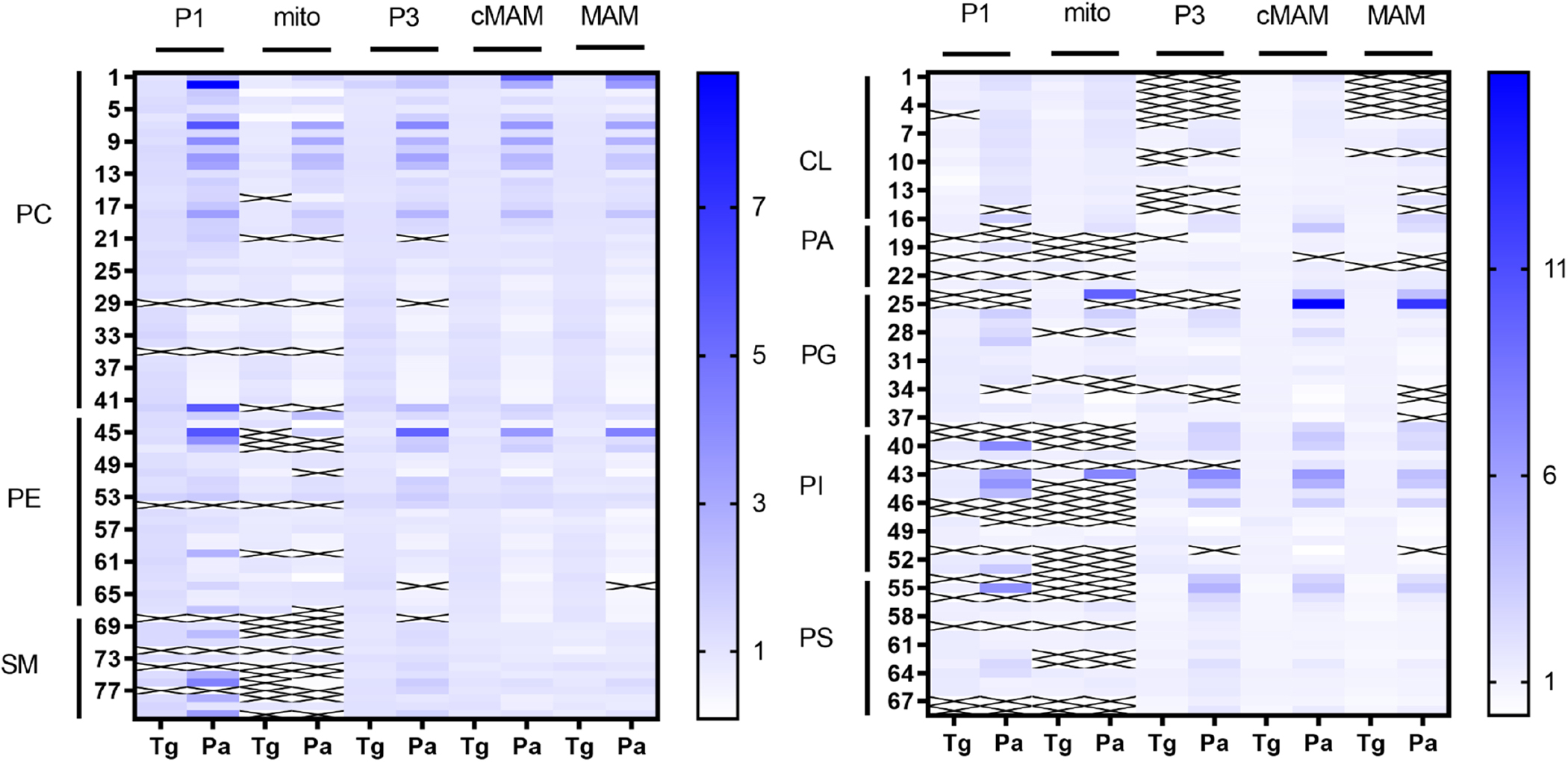
Heatmaps showing the increase or decrease of different lipid species in the studied fractions following treatments, an “X” in the heatmap indicates that the lipid species was not detected. Numbers indicate different lipid species and a list of the corresponding lipids can be found in supplement (Supplemental S9). CL, cardiolipins; cMAM, crude MAM fraction; MAM, mitochondria associated membrane; PA, phosphatidic acid; Pa, palmitate; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; PI, phosphatidylinositol; PS, phosphatidylserine; SM, sphingomyelin; Tg, thapsigargin; veh, vehicle.
Figure 5. In SH-SY5Y cells palmitate causes robust changes in the phospholipids of different fractions, whereas thapsigargin does not.
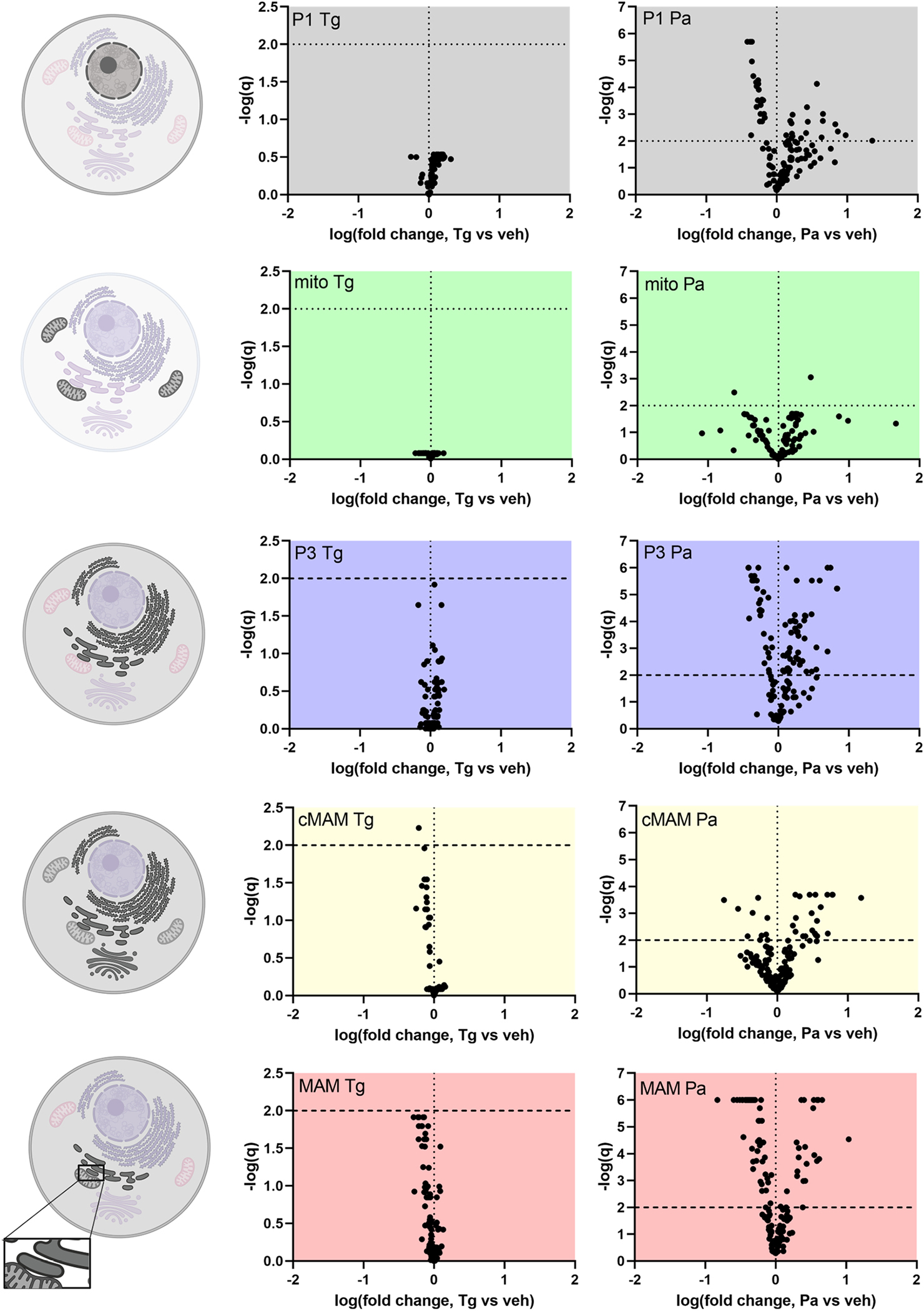
Volcano plots showing the changes in lipid species in different fractions and graphical keys (left) to the cellular membranes that the fractions represent. The darker the membrane indicates it contributes to the corresponding fraction based on protein markers. The different fractions represent following membranes: P1, nuclei, some cell debris; mito, mitochondria; P3, microsome (ER); cMAM, ER, Golgi, some mitochondria; MAM, mitochondria associated membrane. There is also some plasma membrane present in P1, P3, cMAM and MAM fractions. Every dot in the volcano plot represents one phospholipid species detected by mass spectrometry. The phospholipids are blotted according to the logarithm of the fold change compared to vehicle (X-axis) and their q-value obtained from multiple t-tests (false discovery rate approach, Two-stage step-up method of Benjamini, Krieger and Yekutieli, FDR(q) set to 1%). Lipid species above the horizontal line (negative logarithmic q-value 2) are statistically significantly different from vehicle. CL, cardiolipins; cMAM, crude MAM fraction; MAM, mitochondria associated membrane; PA, phosphatidic acid; Pa, palmitate; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; PI, phosphatidylinositol; PS, phosphatidylserine; SM, sphingomyelin; Tg, thapsigargin; veh, vehicle. Cell images were created with BioRender.
The ratio between phosphatidylcholine and phosphatidylethanolamine (PC:PE) is important for maintaining SERCA function and consequently the ER calcium (Fu et al., 2011; Funai et al., 2013; Paran et al., 2015; reviewed in: van der Veen et al 2017). In our experiments, palmitate induced a statistically significant increase in the PC:PE ratio in all fractions except for mitochondria (Fig. 6A). Our method only allows for the quantification of lipids relative to internal standards and therefore the PC:PE ratio is not an absolute molar ratio but a ratio of the total normalized PC species over the total normalized PE species. When looking at all the PC and PE species in the MAM fraction, regardless of their abundance, the PC species both increase and decrease in relative levels (Fig. 6B) whereas the PE species are mainly decreasing (Fig. 5B). Closer inspection of the 10 most abundant PC species (Fig. 7A and B) and 10 most abundant PE species (Fig. 7C and D) in the MAM confirms that the most abundant PE species (except for PE38p:4 and PE38p:5) are decreasing upon palmitate treatment, whereas the increase in the total PC species is due to increases in PC32a:1 and PC34a:1 (Fig. 7D), which is to be expected as they reflect the incorporation of palmitate. The list of all lipids detected by mass spectrometry is provided in the Supplement (S6).
Figure 6. Palmitate but not thapsigargin changes the relative abundances of two phospholipid groups phosphatidylcholines (PC) and phosphatidylethanolamines (PE) in subcellular fractions.
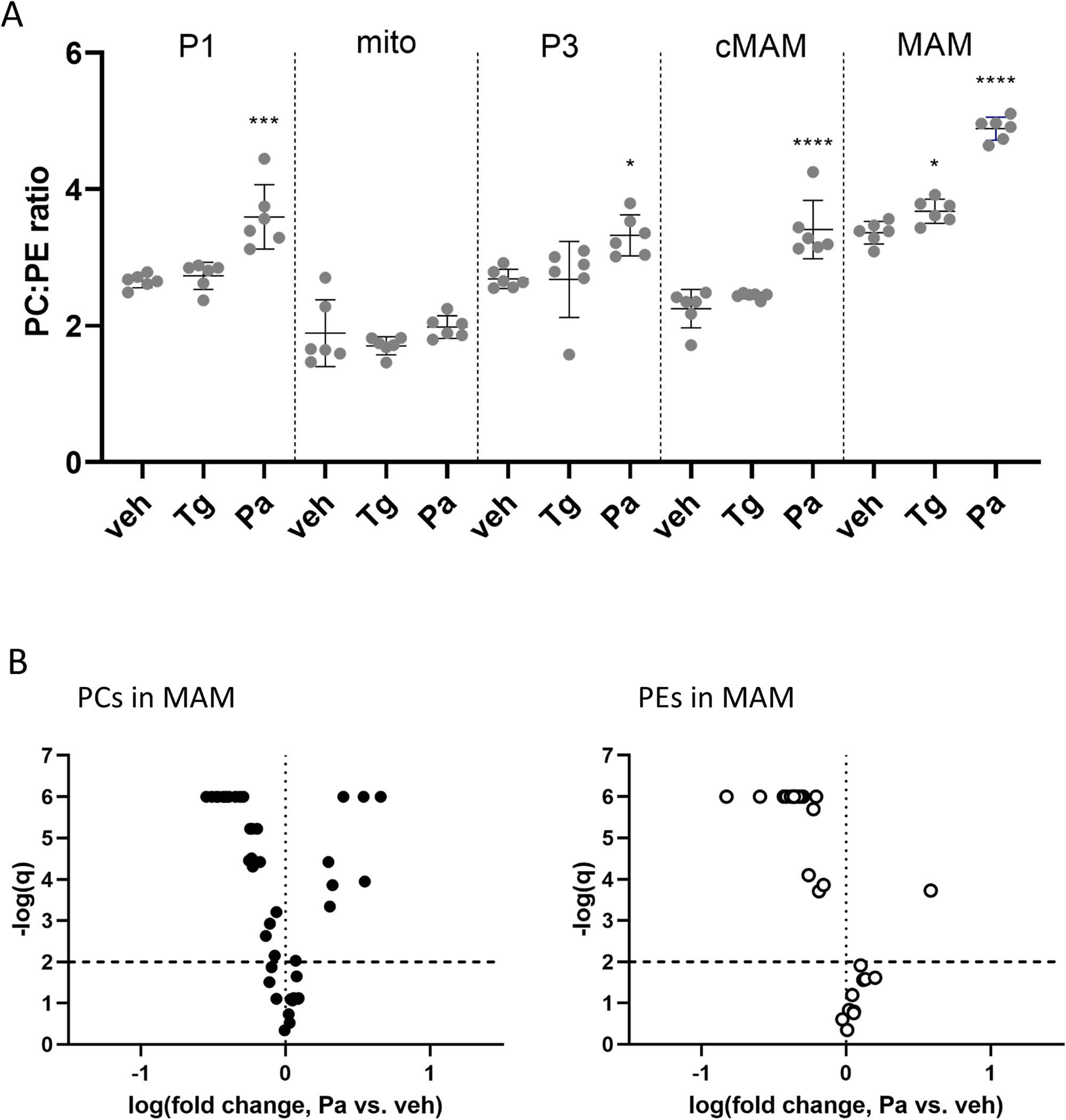
(A) The PC:PE ratio changes in different fractions following the 24-hour thapsigargin (Tg, 60 nM) or palmitate (Pa, 400 μM) treatments. The PC:PE ratios are not molar ratios but were calculated from the summed signals of all the PC species and all the PE species detected by mass spectrometry and normalized to an internal standard. One-way ANOVAs with Dunnett’s test run within every fraction (n=6), significances are as follows, * p<0.05, ** p<0.01, ***p<0.005, ****p<0.001. Volcano plots (B) showing the change in PC and PE species in the MAM fraction following 24-hour exposure to palmitate (400 μM). The phospholipids are plotted according to the logarithm of the fold change compared to vehicle (X-axis) and the logarithm of FDR, the false discovery rate (q; Y-axis), where q-value is obtained from multiple t-tests (Two-stage step-up method of Benjamini, Krieger and Yekutieli, FDR(q) set to 1%). Lipid species above the horizontal line (negative logarithmic q-value 2) are statistically significantly different from vehicle. PC, phosphatidylcholine; PE, phosphatidylethanolamine.
Figure 7. Palmitate changes the relative abundance of several most prevalent PC and PE species in the MAM fraction, whereas thapsigargin only changes three.
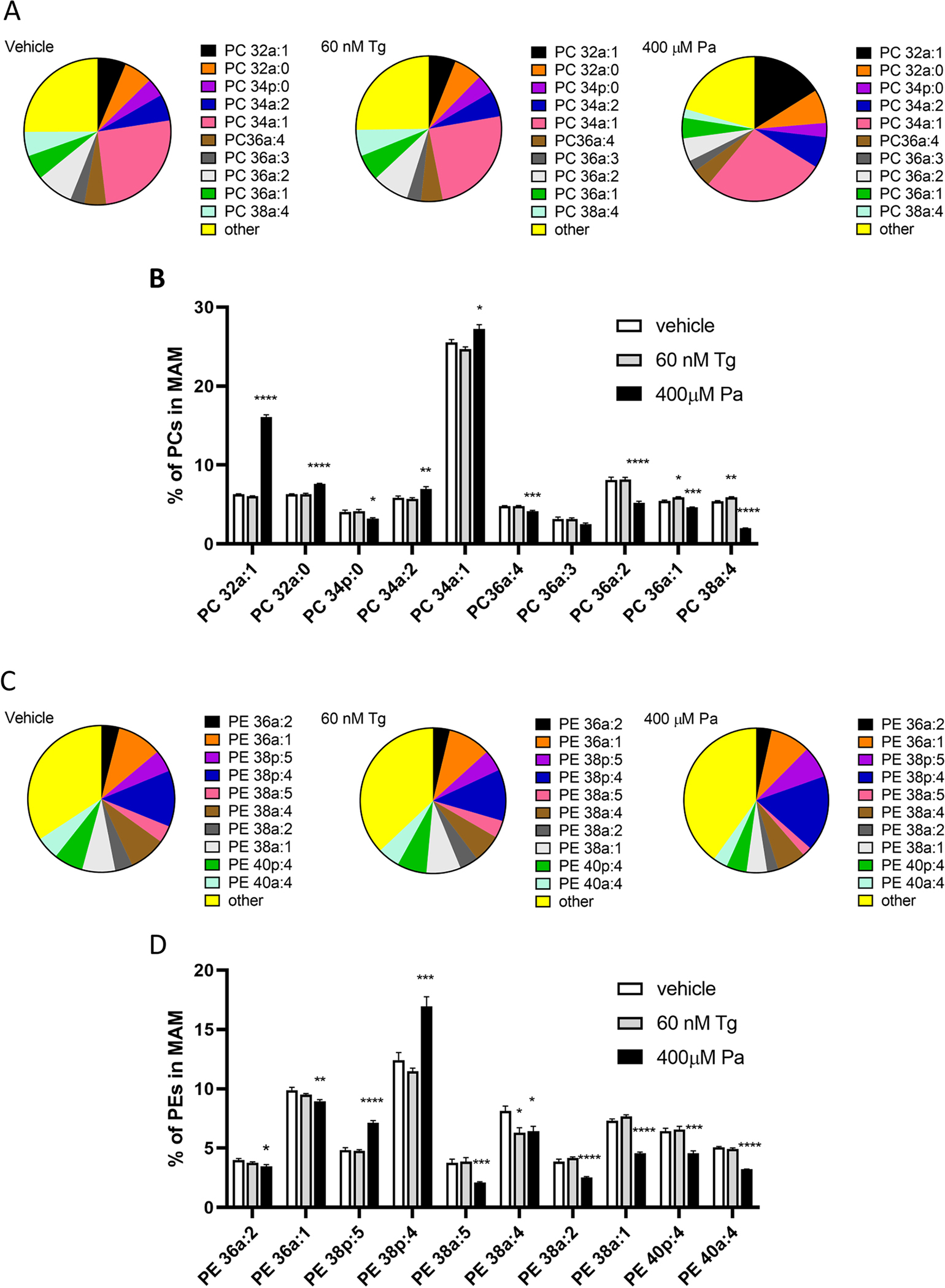
Changes in the 10 most abundant Phosphatidylcholine (PC) (A, B) and Phosphatidylethanolamine (PE) (C, D) species, measured with mass spectrometry, in the MAM fraction after 24-hour exposure to either thapsigargin or palmitate. All the lipid species were first normalized to internal standard of each sample and then the normalized signals were summed together (within a treatment) for PC and PE species separately, the percentages were calculated for each lipid species using the sum of all the species as 100%. One-way ANOVAS with Bonferroni multiple comparisons were run within every lipid species (n=6), significances are as follows * p<0.05, ** p<0.01, ***p<0.005, ****p<0.001.
4. Discussion
In the current study we explored the interplay of ER functions, namely lipid metabolism, calcium homeostasis, and proteostasis in a human neuronal cell line. By altering the membrane phospholipids (palmitate) and disrupting the ER calcium homeostasis (thapsigargin), we found that the change in membrane lipid composition did not significantly disrupt proteostasis and that disrupting the calcium homeostasis did not change the membrane lipid composition in a human neuronal cell line. Data from previous studies suggest that palmitate triggers ER stress in various cell types (Cunha et al., 2008; Gwiazda et al., 2009; Achard and Laybutt, 2012; Volmer et al., 2013; Hou et al., 2014; Kim et al., 2015; Halbleib et al., 2017; Macchioni et al., 2018; Piccolis et al., 2019; Yamamoto et al., 2020) however, we observed a minimal change to ER proteostasis (i.e. UPR and exodosis, also an indicator of ER calcium depletion (Henderson et al., 2014)) despite dramatic changes in subcellular membrane lipid composition. In contrast, causing depletion of ER calcium by thapsigargin resulted in a robust activation of UPR and change in ER proteostasis, but there were only minor changes in the composition of subcellular membrane lipids. These data imply that in neuronal cells, the key ER functions are not tightly coupled, but appear to work independently when the other is challenged.
Recent studies found that palmitate is present in high levels not only in plasma, but also in cerebrospinal fluid (CSF) in obese individuals (Melo et al., 2020) suggesting that neurons get exposed to relatively high levels of palmitate. Even though most of the fatty acid metabolism in the brain takes place in astrocytes (Escartin et al., 2007; Magnan et al., 2015), and neurons can transfer toxic fatty acid species to astrocytes for metabolism or storage (Liu et al., 2015; Ioannou et al., 2019), neuronal cells can also incorporate the excess fatty acids into the cellular membranes which can affect the membrane properties (Rapoport, 1999; Bruce et al., 2017). Indeed, high fat diet has been shown to change the brain phospholipid composition in mice (Pakiet et al., 2019; Sighinolfi et al., 2021) and high fat diet, especially diet rich in saturated fatty acids, such as palmitate, is deleterious for memory and learning (Freeman et al., 2014; Melo et al., 2020). Previous studies reporting the effects of palmitate on UPR activation were focused on pancreatic and liver cells and the results have been contradictory or inconclusive with different read outs for UPR activation making comparisons between studies challenging (Cunha et al., 2008; Achard and Laybutt, 2012; Volmer et al., 2013; Hou et al., 2014; Halbleib et al., 2017; Trentzsch et al., 2020; Yamamoto et al., 2020). Collectively, the data imply that changes in lipid composition can activate the UPR in non-neuronal cells and our data from Hep3B cells supports these observations. However, in this study we focused on neuronal cells and used SH-SY5Y cells, a human cell line of neuronal origin widely used in models of neurodegeneration and previously used by our lab in the identification of exodosis (Trychta et al., 2018), to examine palmitate effects on membrane composition and ER proteostasis in neurons. Although we saw prominent changes in the phospholipids composition of all the ER fractions (P3, crude MAM and MAM) following palmitate treatment and slight increases in exodosis, no changes were observed in the UPR target genes used to reflect activation of the three UPR pathways (ATF6, IRE1 and PERK). These findings are similar to a previous mouse study showing 24 hour post-palmitate did not alter UPR gene expression in the three brain regions analyzed (Moon et al., 2014). High fat diet and, specifically, diets high in palmitate (palm oil) can be harmful to some types of neurons and we found palmitate increased CHOP mRNA levels compared to vehicle at the 8 hours and 24 hours after treatment. CHOP upregulation is associated with ER stress and the UPR but also indicates apoptosis (Li et al., 2014). We observed a slight increase in number of dead cells upon palmitate treatment which might indicate increased apoptosis in these cells. Our results suggest that neuronal cells lack the ability to respond to high levels of saturated fatty acids in a manner that would alleviate ER stress and may explain a previous study that high fat diet induces apoptosis in neurons (Nyamugenda et al., 2019). A recent study by Rizzo et al (2021) highlighted the differences between cell lines in handling of exogenous fatty acids. They compared two breast cancer cell lines treated with palmitate or docosahexaenoic acid (DHA) and reported differences between these two cell lines in the incorporation of fatty acids into cellular membranes and the activity of fatty acid metabolizing enzymes (Rizzo et al., 2021). The lack of UPR activation also suggests that protein folding in the ER is maintained despite substantial changes in its membrane lipid composition, or that the UPR sensors are incapable of signaling in the modified lipid environment. Additional studies are needed to understand the effects of excess free fatty acids on neuronal proteostasis.
Although we did not observe UPR activation by palmitate, there was an increase in secretion of GLuc-SERCaMP, a reporter of exodosis or the mass departure of ER resident proteins. The exodosis phenomenon has been shown to be dependent on ER calcium depletion and may be considered a surrogate readout of ER calcium depletion (Henderson et al., 2014). We observed a significant increase in secretion of GLuc-SERCaMP following palmitate although it was not as striking as thapsigargin, an inhibitor of SERCA which pumps calcium from the cytosol into the ER lumen. Palmitate has been shown to cause ER calcium depletion in mouse podocytes via activation of IP3 receptors (Xu et al., 2015). Palmitate treatment also inhibits the SERCA pump by modulating the ER membrane properties (or the activity of SERCA pump directly) and decreasing SERCA pump expression (Gustavo Vazquez-Jimenez et al., 2016). SERCA pump activity has also been shown to depend on the thickness of the membrane, i.e. the length of the fatty acyl chains of the phospholipids, it is embedded in (Caffrey and Feigenson, 1981). SERCA pump activity was highest in membranes composed of phosphatidylcholine with intermediate length fatty acyl chains, whereas changes towards either shorter or longer fatty acid tails decreased the activity (Caffrey and Feigenson, 1981). Another way palmitate could affect the SERCA function is by changing the ratio between the two most common phospholipids of the cellular membranes, phosphatidylcholines and phosphatidylethanolamines (PC:PE). Changes of this ratio in either direction can be detrimental for the cell, for example, an increase in PC:PE ratio was linked to disrupted SERCA function in mouse liver microsomes (Fu et al., 2011), whereas decreased ratio led to disrupted membrane integrity in the mouse liver cells (Li et al., 2006) and activation of UPR in mouse liver (Gao et al., 2015). In these studies the decrease in PC:PE ratio was observed in phosphatidylethanolamine N-methyltransferase knock-out (Pemt−/−) mice, that are incapable of converting PE to PC. The decrease of PC:PE ratio was exacerbated in these mice upon both choline deficient (Li et al., 2006) and high fat diet (Li et al., 2006) and both of these diets made Pemt−/− mice more susceptible to steatohepatitis. Although most of the studies regarding modified PC:PE ratios have been performed in hepatocytes, some studies highlight the importance of the correct ratio in other cell types such as intestinal and skeletal muscle cells (Funai et al., 2013; Paran et al., 2015; van der Veen et al., 2017). In our study, palmitate treatment led to modest increase in exodosis along with an increase in the relative PC:PE ratio, especially in the MAM fraction where IP3Rs are concentrated (Hayashi and Su, 2007). Our data show that palmitate changes the ER membrane composition with modest effects to ER calcium (as measured via exodosis reporter). However, further studies are needed to examine effects of palmitate on ER calcium changes in neurons.
In summary, the saturated free fatty acid palmitate causes significant changes to lipid composition of subcellular membranes without an associated activation of the UPR indicating that ER proteostasis is maintained despite overt changes in lipid composition of the ER membrane in neuronal cells. Furthermore, disruption of ER calcium homeostasis by thapsigargin leads to robust exodosis and UPR responses reflecting changes to ER proteostasis but with little effect on the membrane phospholipid composition after 24-hour exposure. These studies provide new insight into the complex dynamics of lipid homeostasis and ER proteostasis within neuronal cells, and their response to disturbances in critical ER functions.
Supplementary Material
Highlights.
Free fatty acids (FFA) alter subcellular membrane composition in neuronal cells
FFA changes membrane lipids but does not trigger unfolded protein response
Triggering ER stress in neuronal cells doesn’t affect the membrane lipid composition
ER lipid composition and proteostasis minimally impact each other in neuronal cells
Acknowledgements
We thank Dr. Tsung-Ping Su, Dr. Yuko Yasui and Dr. Hsiang-en Wu for access to equipment and technical assistance with the cell fractioning assay and the Electron Microscopy Unit of the Institute of Biotechnology, University of Helsinki, for support in the EM study. We thank Dr Helena Vihinen and Dr. Mikko Airavaara (U Helsinki) for their input on the EM studies and Dr. Kathleen Trychta for critical reading of the manuscript. This work was supported by the Intramural Research Programs at the National Institute on Drug Abuse (NIDA) and the National Institute of Environmental Health Sciences (NIEHS). Funding was also provided by the Sigrid Jusélius Foundation (E. Jss.).
Footnotes
Declaration of Interest statement
All the authors of the submitted manuscript “Palmitate and thapsigargin have contrasting effects on ER membrane lipid composition and ER proteostasis in neuronal cells” declare no conflicts of interest.
CrediT Author statement
MHJ, Conceptualization, Data curation, Formal analysis, Visualization, Writing - original draft, review & editing
SJ, Data curation, Formal analysis, Writing - review & editing
JK, Data curation, Formal analysis, Writing - review & editing
IP, Data curation, Writing - review & editing
SS, Data curation, Writing - review & editing
JH, Data curation, Writing - review & editing
EJ, Supervision, Writing - review & editing, Funding acquisition, Resources
LD, Supervision, Writing - review & editing, Funding acquisition, Resources
BKH, Supervision, Funding acquisition, Resources, Conceptualization, Writing - review & editing
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability statement
All data is available upon request from the corresponding authors.
References
- Achard CS, Laybutt DR (2012) Lipid-induced endoplasmic reticulum stress in liver cells results in two distinct outcomes: adaptation with enhanced insulin signaling or insulin resistance. Endocrinology 153:2164–2177. [DOI] [PubMed] [Google Scholar]
- Bernales S, McDonald KL, Walter P (2006) Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol 4:e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce KD, Zsombok A, Eckel RH (2017) Lipid Processing in the Brain: A Key Regulator of Systemic Metabolism. Front Endocrinol (Lausanne) 8:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey M, Feigenson GW (1981) Fluorescence quenching in model membranes. 3. Relationship between calcium adenosinetriphosphatase enzyme activity and the affinity of the protein for phosphatidylcholines with different acyl chain characteristics. Biochemistry 20:1949–1961. [DOI] [PubMed] [Google Scholar]
- Chalhoub G, Kolleritsch S, Maresch LK, Taschler U, Pajed L, Tilp A, Eisner H, Rosina P, Kien B, Radner FPW, Schicho R, Oberer M, Schoiswohl G, Haemmerle G (2021) Carboxylesterase 2 proteins are efficient diglyceride and monoglyceride lipases possibly implicated in metabolic disease. J Lipid Res 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha DA, Hekerman P, Ladriere L, Bazarra-Castro A, Ortis F, Wakeham MC, Moore F, Rasschaert J, Cardozo AK, Bellomo E, Overbergh L, Mathieu C, Lupi R, Hai T, Herchuelz A, Marchetti P, Rutter GA, Eizirik DL, Cnop M (2008) Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J Cell Sci 121:2308–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ee SM, Lo YL, Shui GH, Wenk MR, Shin EJ, Kim HC, Ong WY (2014) Distribution of Secretory Phospholipase A2 XIIA in the Brain and its Role in Lipid Metabolism and Cognition. Mol Neurobiol 50:60–75. [DOI] [PubMed] [Google Scholar]
- Escartin C, Pierre K, Colin A, Brouillet E, Delzescaux T, Guillermier M, Dhenain M, Deglon N, Hantraye P, Pellerin L, Bonvento G (2007) Activation of astrocytes by CNTF induces metabolic plasticity and increases resistance to metabolic insults. J Neurosci 27:7094–7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman LR, Haley-Zitlin V, Rosenberger DS, Granholm AC (2014) Damaging effects of a high-fat diet to the brain and cognition: a review of proposed mechanisms. Nutr Neurosci 17:241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, Hotamisligil GS (2011) Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473:528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funai K, Song H, Yin L, Lodhi IJ, Wei X, Yoshino J, Coleman T, Semenkovich CF (2013) Muscle lipogenesis balances insulin sensitivity and strength through calcium signaling. J Clin Invest 123:1229–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, van der Veen JN, Vance JE, Thiesen A, Vance DE, Jacobs RL (2015) Lack of phosphatidylethanolamine N-methyltransferase alters hepatic phospholipid composition and induces endoplasmic reticulum stress. Biochim Biophys Acta 1852:2689–2699. [DOI] [PubMed] [Google Scholar]
- Guan M, Qu L, Tan W, Chen L, Wong CW (2011) Hepatocyte nuclear factor-4 alpha regulates liver triglyceride metabolism in part through secreted phospholipase A(2) GXIIB. Hepatology 53:458–466. [DOI] [PubMed] [Google Scholar]
- Gustavo Vazquez-Jimenez J, Chavez-Reyes J, Romero-Garcia T, Zarain-Herzberg A, Valdes-Flores J, Manuel Galindo-Rosales J, Rueda A, Guerrero-Hernandez A, Olivares-Reyes JA (2016) Palmitic acid but not palmitoleic acid induces insulin resistance in a human endothelial cell line by decreasing SERCA pump expression. Cell Signal 28:53–59. [DOI] [PubMed] [Google Scholar]
- Gwiazda KS, Yang TL, Lin Y, Johnson JD (2009) Effects of palmitate on ER and cytosolic Ca2+ homeostasis in beta-cells. Am J Physiol Endocrinol Metab 296:E690–701. [DOI] [PubMed] [Google Scholar]
- Halbleib K, Pesek K, Covino R, Hofbauer HF, Wunnicke D, Hanelt I, Hummer G, Ernst R (2017) Activation of the Unfolded Protein Response by Lipid Bilayer Stress. Mol Cell 67:673–684 e678. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Su TP (2007) Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131:596–610. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Rizzuto R, Hajnoczky G, Su TP (2009) MAM: more than just a housekeeper. Trends Cell Biol 19:81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson MJ, Wires ES, Trychta KA, Richie CT, Harvey BK (2014) SERCaMP: a carboxy-terminal protein modification that enables monitoring of ER calcium homeostasis. Mol Biol Cell 25:2828–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Zhang K, Kaufman RJ (2020) Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol 21:421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou NS, Gutschmidt A, Choi DY, Pather K, Shi X, Watts JL, Hoppe T, Taubert S (2014) Activation of the endoplasmic reticulum unfolded protein response by lipid disequilibrium without disturbed proteostasis in vivo. Proc Natl Acad Sci U S A 111:E2271–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannou MS, Jackson J, Sheu SH, Chang CL, Weigel AV, Liu H, Pasolli HA, Xu CS, Pang S, Matthies D, Hess HF, Lippincott-Schwartz J, Liu Z (2019) Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 177:1522–1535 e1514. [DOI] [PubMed] [Google Scholar]
- Kim SK, Oh E, Yun M, Lee SB, Chae GT (2015) Palmitate induces cisternal ER expansion via the activation of XBP-1/CCTalpha-mediated phospholipid accumulation in RAW 264.7 cells. Lipids Health Dis 14:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagrutta LC, Layerenza JP, Bronsoms S, Trejo SA, Ves-Losada A (2021) Nuclear-lipid-droplet proteome: carboxylesterase as a nuclear lipase involved in lipid-droplet homeostasis. Heliyon 7:e06539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Guo Y, Tang J, Jiang J, Chen Z (2014) New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim Biophys Sin (Shanghai) 46:629–640. [DOI] [PubMed] [Google Scholar]
- Li Z, Agellon LB, Allen TM, Umeda M, Jewell L, Mason A, Vance DE (2006) The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab 3:321–331. [DOI] [PubMed] [Google Scholar]
- Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, Li Z, Hui J, Graham BH, Quintana A, Bellen HJ (2015) Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160:177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macchioni L, Petricciuolo M, Davidescu M, Fettucciari K, Scarpelli P, Vitale R, Gatticchi L, Orvietani PL, Marchegiani A, Marconi P, Bassotti G, Corcelli A, Corazzi L (2018) Palmitate lipotoxicity in enteric glial cells: Lipid remodeling and mitochondrial ROS are responsible for cyt c release outside mitochondria. Biochim Biophys Acta Mol Cell Biol Lipids 1863:895–908. [DOI] [PubMed] [Google Scholar]
- Magnan C, Levin BE, Luquet S (2015) Brain lipid sensing and the neural control of energy balance. Mol Cell Endocrinol 418 Pt 1:3–8. [DOI] [PubMed] [Google Scholar]
- Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L (2011) Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo HM et al. (2020) Palmitate Is Increased in the Cerebrospinal Fluid of Humans with Obesity and Induces Memory Impairment in Mice via Pro-inflammatory TNF-alpha. Cell Rep 30:2180–2194 e2188. [DOI] [PubMed] [Google Scholar]
- Moon ML, Joesting JJ, Lawson MA, Chiu GS, Blevins NA, Kwakwa KA, Freund GG (2014) The saturated fatty acid, palmitic acid, induces anxiety-like behavior in mice. Metabolism 63:1131–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S, Pelham HR (1987) A C-terminal signal prevents secretion of luminal ER proteins. Cell 48:899–907. [DOI] [PubMed] [Google Scholar]
- Nyamugenda E, Trentzsch M, Russell S, Miles T, Boysen G, Phelan KD, Baldini G (2019) Injury to hypothalamic Sim1 neurons is a common feature of obesity by exposure to high-fat diet in male and female mice. J Neurochem 149:73–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci L, Stamnes M, Ravazzola M, Amherdt M, Perrelet A, Sollner TH, Rothman JE (1997) Bidirectional transport by distinct populations of COPI-coated vesicles. Cell 90:335–349. [DOI] [PubMed] [Google Scholar]
- Pakiet A, Jakubiak A, Czumaj A, Sledzinski T, Mika A (2019) The effect of western diet on mice brain lipid composition. Nutr Metab (Lond) 16:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paran CW, Zou K, Ferrara PJ, Song H, Turk J, Funai K (2015) Lipogenesis mitigates dysregulated sarcoplasmic reticulum calcium uptake in muscular dystrophy. Biochim Biophys Acta 1851:1530–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips MJ, Voeltz GK (2016) Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol 17:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolis M, Bond LM, Kampmann M, Pulimeno P, Chitraju C, Jayson CBK, Vaites LP, Boland S, Lai ZW, Gabriel KR, Elliott SD, Paulo JA, Harper JW, Weissman JS, Walther TC, Farese RV, Jr. (2019) Probing the Global Cellular Responses to Lipotoxicity Caused by Saturated Fatty Acids. Mol Cell 74:32–44 e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissler S, Rato C, Yan Y, Perera LA, Czako A, Ron D (2020) Calcium depletion challenges endoplasmic reticulum proteostasis by destabilising BiP-substrate complexes. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport SI (1999) In vivo fatty acid incorporation into brain phospholipids in relation to signal transduction and membrane remodeling. Neurochem Res 24:1403–1415. [DOI] [PubMed] [Google Scholar]
- Raturi A, Simmen T (2013) Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM). Biochim Biophys Acta 1833:213–224. [DOI] [PubMed] [Google Scholar]
- Rizzo AM, Colombo I, Montorfano G, Zava S, Corsetto PA (2021) Exogenous Fatty Acids Modulate ER Lipid Composition and Metabolism in Breast Cancer Cells. Cells 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby MA, Massart J, Hunerdosse DM, Schonke M, Correia JC, Louie SM, Ruas JL, Naslund E, Nomura DK, Zierath JR (2017) Human Carboxylesterase 2 Reverses Obesity-Induced Diacylglycerol Accumulation and Glucose Intolerance. Cell Reports 18:636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusinol AE, Cui Z, Chen MH, Vance JE (1994) A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J Biol Chem 269:27494–27502. [PubMed] [Google Scholar]
- Seemann J, Jokitalo EJ, Warren G (2000) The role of the tethering proteins p115 and GM130 in transport through the Golgi apparatus in vivo. Mol Biol Cell 11:635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sighinolfi G, Clark S, Blanc L, Cota D, Rhourri-Frih B (2021) Mass spectrometry imaging of mice brain lipid profile changes over time under high fat diet. Sci Rep 11:19664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriburi R, Bommiasamy H, Buldak GL, Robbins GR, Frank M, Jackowski S, Brewer JW (2007) Coordinate regulation of phospholipid biosynthesis and secretory pathway gene expression in XBP-1(S)-induced endoplasmic reticulum biogenesis. J Biol Chem 282:7024–7034. [DOI] [PubMed] [Google Scholar]
- Trentzsch M, Nyamugenda E, Miles TK, Griffin H, Russell S, Koss B, Cooney KA, Phelan KD, Tackett AJ, Iyer S, Boysen G, Baldini G (2020) Delivery of phosphatidylethanolamine blunts stress in hepatoma cells exposed to elevated palmitate by targeting the endoplasmic reticulum. Cell Death Discov 6:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trychta KA, Back S, Henderson MJ, Harvey BK (2018) KDEL Receptors Are Differentially Regulated to Maintain the ER Proteome under Calcium Deficiency. Cell Rep 25:1829–1840 e1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veen JN, Kennelly JP, Wan S, Vance JE, Vance DE, Jacobs RL (2017) The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim Biophys Acta Biomembr 1859:1558–1572. [DOI] [PubMed] [Google Scholar]
- Vance JE (1990) Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem 265:7248–7256. [PubMed] [Google Scholar]
- Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A, Agostinis P (2012) PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 19:1880–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volmer R, van der Ploeg K, Ron D (2013) Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci U S A 110:4628–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334:1081–1086. [DOI] [PubMed] [Google Scholar]
- Wires ES, Trychta KA, Back S, Sulima A, Rice KC, Harvey BK (2017) High fat diet disrupts endoplasmic reticulum calcium homeostasis in the rat liver. J Hepatol 67:1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Carvalho P, Voeltz GK (2018) Here, there, and everywhere: The importance of ER membrane contact sites. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Nam SM, Kim JH, Das R, Choi SK, Nguyen TT, Quan X, Choi SJ, Chung CH, Lee EY, Lee IK, Wiederkehr A, Wollheim CB, Cha SK, Park KS (2015) Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis 6:e1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YY, Pan XL, Hu SW, Zhu YD, Bawa FC, Li YY, Yin LY, Zhang YQ (2021) Hepatocyte-specific expression of human carboxylesterase 2 attenuates nonalcoholic steatohepatitis in mice. Am J Physiol-Gastr L 320:G166–G174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Endo J, Kataoka M, Matsuhashi T, Katsumata Y, Shirakawa K, Isobe S, Moriyama H, Goto S, Shimanaka Y, Kono N, Arai H, Shinmura K, Fukuda K, Sano M (2020) Palmitate induces cardiomyocyte death via inositol requiring enzyme-1 (IRE1)-mediated signaling independent of X-box binding protein 1 (XBP1). Biochem Biophys Res Commun 526:122–127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available upon request from the corresponding authors.