

Abstract
Aims
Frontotemporal dementias are neuropathologically characterised by frontotemporal lobar degeneration (FTLD). Intraneuronal inclusions of transactive response DNA‐binding protein 43 kDa (TDP‐43) are the defining pathological hallmark of approximately half of the FTLD cases, being referred to as FTLD‐TDP. The classification of FTLD‐TDP into five subtypes (Type A to Type E) is based on pathologic phenotypes; however, the molecular determinants underpinning the phenotypic heterogeneity of FTLD‐TDP are not well known. It is currently undetermined whether TDP‐43 post‐translational modifications (PTMs) may be related to the phenotypic diversity of the FTLDs. Thus, the investigation of FTLD‐TDP Type A and Type B, associated with GRN and C9orf72 mutations, becomes essential.
Methods
Immunohistochemistry was used to identify and map the intraneuronal inclusions. Sarkosyl‐insoluble TDP‐43 was extracted from brains of GRN and C9orf72 mutation carriers post‐mortem and studied by Western blot analysis, immuno‐electron microscopy and mass spectrometry.
Results
Filaments of TDP‐43 were present in all FTLD‐TDP preparations. PTM profiling identified multiple phosphorylated, N‐terminal acetylated or otherwise modified residues, several of which have been identified for the first time as related to sarkosyl‐insoluble TDP‐43. Several PTMs were specific for either Type A or Type B, while others were identified in both types.
Conclusions
The current results provide evidence that the intraneuronal inclusions in the two genetic diseases contain TDP‐43 filaments. The discovery of novel, potentially type‐specific TDP‐43 PTMs emphasises the need to determine the mechanisms leading to filament formation and PTMs, and the necessity of exploring the validity and occupancy of PTMs in a prognostic/diagnostic setting.
Keywords: mass spectrometry, neurodegeneration, phosphorylation, TDP‐43
Key Points.
In the brain of patients affected by hereditary FTD caused by GRN and C9orf72 mutations, novel post‐translational modifications (PTMs) of TDP‐43 have been identified.
In FTLD‐TDP Type A and Type B, the presence of neuronal intracytoplasmic TDP‐43 inclusions is associated with PTMs.
TDP‐43 intracytoplasmic inclusions contain TDP‐43 filaments as shown by immuno‐electron microscopy.
Post‐translational modifications are biologically relevant events that may have an important role in the phenotypic variability seen in FTLD‐TDP.
INTRODUCTION
Transactive response (TAR) DNA‐binding protein with molecular weight (MW) of 43 kDa (TDP‐43) is a highly conserved RNA/DNA‐binding protein. TDP‐43 is made of 414 amino acids, with an amino‐terminal (N‐term) domain containing a nuclear localisation signal (NLS), two RNA recognition motifs (RRM1 and RRM2) and glycine‐rich and glutamine/asparagine (Q/N)‐rich domains in the C‐terminal region (low‐complexity [LC] domain); these domains allow TDP‐43 to bind single‐stranded DNA, RNA and proteins [1, 2]. In normal brains, TDP‐43 is primarily found in the nucleus of neurons and glia; however, TDP‐43 is able to shuttle between the cytoplasm and the nucleus [3].
In many forms of frontotemporal dementia (FTD), amyotrophic lateral sclerosis (ALS) and in limbic‐predominant age‐related TDP‐43 encephalopathy (LATE), TDP‐43 is the main component of the intracellular inclusions found in the central nervous system [3, 4, 5, 6].
The identification and study of TDP‐43 post‐translational modifications (PTMs) are essential steps towards our understanding of diseases' pathogenetic mechanisms and for the development of potential diagnostic tools [7, 8, 9, 10, 11, 12, 13]. PTMs may regulate protein conformation, biophysical properties, protein interactions, function and subcellular localisation [14, 15]. Although PTMs occur physiologically, many have been detected in association with protein aggregates in diverse neurodegenerative diseases [14, 16, 17, 18, 19, 20, 21].
Previous studies have shown pathological TDP‐43 to be cleaved, ubiquitinated and hyperphosphorylated [3, 4]. Phosphorylation of two residues (serine [S] 409 and 410) has been observed in TDP‐43 associated with neurodegenerative disorders but not in control tissue [10, 11, 13, 22]. Using mass spectrometry (MS), PTMs of TDP‐43 have only been identified in ALS and ALS with frontotemporal lobar degeneration (FTLD); however, it remains to be determined which of the changes observed are disease specific [6, 23]. Data showing that in pathological conditions TDP‐43 may differ in susceptibility to proteolysis may suggest that diverse TDP‐43 conformations might characterise the numerous diseases associated with the presence of TDP‐43 aggregates, similarly to what has been found in disorders with tau pathology [24, 25, 26, 27, 28].
Among the clinical phenotypes of FTD, the most common are those associated with FTLD and tau or FTLD and TDP‐43 (FTLD‐Tau or FTLD‐TDP). Mutations in the granulin precursor (GRN) and C9orf72 genes have been found to be associated with various FTD phenotypes. In view of the neuropathologic heterogeneity of FTLD‐TDP, five distinct pathological subtypes (Type A to Type E) have been established [29, 30, 31]. FTLD‐TDP Type A is associated with mutations in both GRN and C9orf72, and Type B is associated with mutations in C9orf72 but not GRN; therefore, we asked the question whether, in dominantly inherited FTD associated with either GRN or C9orf72 mutations, a distinction of PTM patterns is possible through the analysis of the frontal cortex in two different subtypes of FTLD‐TDP. For the current experimental work, we carried out a proteomic analysis of frontal cortices, comparing those of GRN mutation carriers, FTLD‐TDP Type A, with those of C9orf72 mutation carriers with FTLD‐TDP Type B.
MATERIALS AND METHODS
See the supporting information for descriptions of Brain Tissue, Neuropathology, Genetics and Reagents.
PTM studies
Preparation of the sarkosyl‐insoluble fraction
Sarkosyl‐insoluble TDP‐43 was extracted from the grey matter of the frontal cortex (Figure S1). The tissue was homogenised in cold extraction buffer (10 mM Tris–HCl pH 7.4, 0.8 M NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM dithiothreitol and 10% sucrose, containing an inhibitor cocktail for proteases, phosphatases, deacetylases and deubiquitylating enzymes). Magnesium chloride, benzonase and sarkosyl were added to the sample to reach final concentrations of 3 mM, 1.25 U/μl and 1%, respectively. Sample was incubated at 37°C for 30 min and centrifuged at 3000 × g for 10 min at room temperature (RT). Supernatant (S1) was collected and centrifuged at 100,000 × g for 25 min at RT in a TLA‐110 rotor (Beckman) to generate a supernatant (S2; sarkosyl‐soluble fraction) and a pellet (P2). P2 was washed in extraction buffer containing protease and phosphatase inhibitors and centrifuged at 100,000 × g for 20 min, RT. The pellet generated (P3) was resuspended in 10 mM Tris–HCl pH 7.4, 150 mM NaCl or 5% sodium dodecyl sulphate (SDS)–Tris‐EDTA (TE) buffer and referred to as sarkosyl‐insoluble fraction (Figure S1). The supernatant (S3) was stored at −80°C.
Electrophoresis and immunoblot
TDP‐43 antibodies (Abs) and dilutions used are listed in Table S2. Phosphorylated sites targeted by these Abs are shown in Figure 1. Electrophoresis and immunoblot protocols are described in the supporting information.
FIGURE 1.
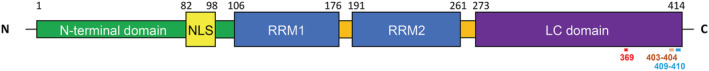
Schematic representation of TDP‐43 protein domains. Numbers on the top of the diagram represent the residues delimiting each domain; numbers on the bottom of the diagram represent potentially phosphorylated sites targeted by antibodies used in this study. LC, low‐complexity; NLS, nuclear localisation signal; RRM, RNA recognition motif
Immuno‐gold electron microscopy
A 1.5 μl droplet of the 1:20 sarkosyl‐insoluble TDP‐43 fraction was pipetted onto carbon nickel transmission electron microscopy (TEM) grids (300 mesh) and blotted dry. The grid was blocked in 0.1% gelatin in phosphate‐buffered saline (PBS) for 20 min; then, excess solution was blotted off with filter paper. The primary Ab (Table S2) diluted 1:50 in 0.1% gelatin in PBS was added for 1 h at RT. The grid was then washed three times in 0.1% gelatin in PBS for 5 min each wash. Grids were incubated in secondary Ab (Table S2) diluted 1:40 in 0.1% gelatin in PBS for 1 h at RT. Secondary Ab was washed off three times in 0.1% gelatin in PBS and blotted dry. Negative staining was performed with NanoVan for 5 s at RT. TEM images were taken on a Tecnai G2 Spirit Twin scope equipped with an AMT CCD Camera. Negative controls were prepared to omit primary Abs to assess the possible cross‐reaction of the secondary Ab with the sample.
Enzymatic digestion
Sarkosyl‐insoluble TDP‐43 preparations in 5% SDS–TE buffer were reduced with 5 mM tris(2‐carboxyethyl)phosphine (TCEP) at RT for 30 min and alkylated using a final concentration of 10 mM chloroacetamide (CAA) for 30 min in the dark at RT. Samples were then diluted to 1% SDS in 8 M urea and 100 mM Tris pH 8.5 for a modified filter‐aided sample preparation (FASP) digestion. Each sample was split loaded onto four FASP spin filters and centrifuged at 14,000 × g for 15 min at RT. The 30 kDa filters were washed with 2 × 200 μl 8 M urea followed by 2 × 200 μl 50 mM Tris–HCl, pH 8.5. Samples were then digested overnight at 25° C with either Trypsin/Lys‐C (two filters each with 3 μg in 50 mM Tris–HCl, pH 8.5) or chymotrypsin (two filters each with 12.5 μg in 100 mM Tris–HCl, 10 mM CaCl2, pH 7.8). Filters were moved to clean collection tubes and centrifuged at 15,000 × g for 10 min, and the membranes were rinsed with 40 μl of 50 mM Tris–HCl followed by 50 μl of 0.5 M sodium chloride solution. The eluted peptides were acidified with trifluoroacetic acid (TFA, 0.5% final v/v). Trypsin and chymotrypsin digestions were cleaned up using Waters SepPak 50 mg vacuum manifold cartridges (with a 1 ml wash of 0.1% TFA and elution in 70% acetonitrile 0.1% formic acid [FA]).
Phosphopeptide enrichment
Peptides were dried in a speed vacuum, and phosphopeptides were enriched using a High‐Select TiO2 phosphopeptide enrichment kit. The elution from the TiO2 tips was dried and resuspended in 28 μl 0.1% FA for phosphopeptide liquid chromatography with tandem MS (LC–MS/MS). The flow‐through and wash from the tips were retained, desalted on a Waters SepPak column as previously described, resuspended in 50 μl 0.1% FA and run as ‘non‐phospho’ samples.
Liquid chromatography with tandem MS
Each sample was run in technical duplicate on an Easy nano LC1200 coupled to a Fusion Lumos Orbitrap Eclipse using an EasySpray 25 cm column. Phosphopeptides (6 μl) were analysed in a 90 min EtHCD/HCD decision tree method and 120 min CID with MSA method, whereas non‐phosphopeptides (3 μl) were analysed on 180 min gradient EtHCD/HCD and CID methods. Full gradient and LC–MS/MS settings are described below:
EtHCD/HCD method: 6–35% B (80% acetonitrile and 0.1% FA) over 85 min, 35–80% B over 3 min and 80–6% B over 2 min all at 400 nl/min. Orbitrap was operated at 120,000 resolution, APD and IC on, with standard AGC, auto max IT, minimum intensity of 2e4 and 60 s dynamic exclusion excluding isotopes. MS2 settings (3 s cycle time) were operated with a decision tree of EtHC‐HCD options both with Orbitrap resolution 50,000, 1.7 m/z isolation window, standard AGC and dynamic IT. EtHCD prioritised precursors with highest charge state/lowest m/z for calibrated ETD plus 20% HCD. HCD‐only scans were operated with 30% CE.
CID with multistage activation phosphopeptide method: LC: 4–28% B (80% acetonitrile and 0.1% FA) over 110 min, 28–80% B over 7 min, 80% B hold for 1 min and 80–6% B over 2 min all at 400 nl/min. Orbitrap was operated at 120,000 resolution, 400–1600 m/z, APD and IC on, with standard AGC, auto max IT, minimum intensity of 2e4 and 60 s dynamic exclusion excluding isotopes. MS2 settings (3 s cycle time) were operated at CID of 35%, Orbitrap resolution of 15,000, 1 m/z isolation window, standard AGC, dynamic IT and multistage activation with a neutral mass loss of 97.9673 on.
CID for non‐phosphopeptides: 6–35% B (80% acetonitrile and 0.1% FA) over 160 min, 35–80% B over 10 min, 80% hold for 4 min and 80–6% B over 4 min all at 400 nl/min. Orbitrap was operated at 120,000 resolution, 400–1600 m/z, APD and IC on, with standard AGC, auto max IT, minimum intensity of 5e3 and 60 s dynamic exclusion excluding isotopes. MS2 settings (3 s cycle time) were operated at CID of 35%, ion trap rapid resolution, 1 m/z isolation window, standard AGC and dynamic IT.
Data analysis
Data were analysed using PEAKS Studio Xpro (Bioinformatics Solutions Inc.). Each sample resulted in eight separate LC–MS/MS raw files loaded as samples with corresponding protease, fragmentation and detection parameters into a single PEAKS project. PEAKS database search was performed using the reviewed human UniProt database and common contaminants with 10 ppm precursor error and 0.5 Da fragment tolerances. Semi‐specific digest mode was utilized, with protease and fragmentation type specified at the sample level; a maximum of three missed cleavages were allowed with up to three variable modifications of N‐terminal (N‐term) acetylation, (M, H, W) oxidation, (S, T, Y) phosphorylation, C carbamidomethylation and K ubiquitination. PEAKS PTM and SPIDER were performed, and data were exported with a 1% false discovery rate cut‐off at the peptide level, AScore ≥10, mutation ion intensity ≥5 and de novo scores ≥80%. AScore is a localisation score assigned to modifications on peptide as the −10 log of a p value corresponding to the probability that the modification occurs at the reported position compared with other possible positions. Ion intensity is the minimum relative fragment ion intensity required for accurate localisation of a mutation or PTM on a specific amino acid. The de novo score applies a threshold cut‐off for de novo sequence tags not identified in database search algorithms.
RESULTS
Neuropathology
The tissue studied neuropathologically was obtained from the cerebral hemisphere contralateral to that used for biochemical studies (Table S1). The frontal and temporal cortices of the five GRN and the two C9orf72 mutation carriers showed numerous intracellular TDP‐43 aggregates. Neuronal loss ranged from moderate to severe and was most prominent in the brain areas that contained the TDP‐43 aggregates. TDP‐43‐immunoreactive neuronal cytoplasmic inclusions (NCIs) and neurites ranged from sparse to frequent and were primarily seen in the second layer of the cerebral cortex, where spongiosis of the neuropil was also consistently present. In the GRN cases, TDP‐43 NCIs were consistently found in the frontal cortex (Figure 2), temporal cortex and hippocampus, including the dentate gyrus. Neuronal nuclear inclusions were infrequently detected in the cerebral cortex. All GRN cases but Case 2 had TDP‐43 NCIs in the striatum; sparse to frequent NCIs were identified in the amygdala and inferior olivary nucleus, thalamus (Cases 1 and 4) and midbrain (Case 2).
FIGURE 2.
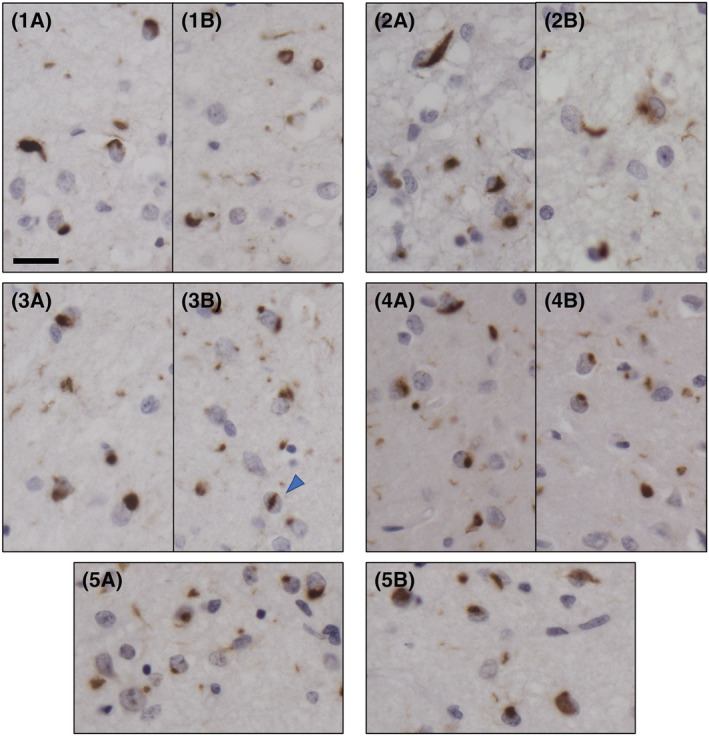
TDP‐43 immunohistochemistry in the frontal cortex of five GRN mutation carriers (Cases 1–5). TDP‐43 immunoreactive intracellular inclusions and neuropil deposits in two fields of the second layer of the frontal cortex (A, B). Neuronal cytoplasmic inclusions (NCIs), a neuronal intranuclear inclusion (NII, arrowhead) and granular deposits are seen in the neuropil. The pattern of TDP‐43 inclusions is consistent with Mackenzie Type A classification. Scale = 20 μm; antibody: pS409/410
In the two samples obtained from the C9orf72 mutation carriers (Cases 6 and 7), TDP‐43 aggregates were present in frontal (Figure 3), temporal and parietal cortices as well as in the hippocampus, including the dentate gyrus. Case 6 had NCIs also in the insular cortex, caudate and putamen, thalamus and medulla. Case 7 had NCIs in the amygdala and striatum. Sparse NCIs were seen in the occipital cortex in one brain. TDP‐43 aggregates were not identified in the cerebellum. Variable numbers of neurites and NCIs were present in the white matter.
FIGURE 3.
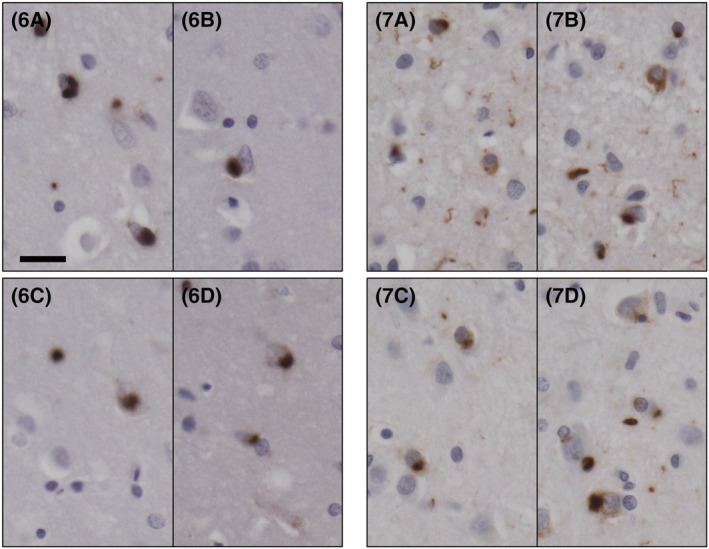
TDP‐43 immunohistochemistry in the frontal cortex of two C9orf72 mutation carriers (Cases 6 and 7). TDP‐43 immunoreactive intracellular inclusions and neuropil deposits in two fields of the second layer of the frontal cortex (A, B) and of the fifth layer of the frontal cortex (C, D). Neuronal cytoplasmic inclusions (NCIs) and diffuse granular NCIs (dNCIs) are present. The pattern of TDP‐43 inclusions is consistent with Mackenzie Type B classification. Scale = 20 μm; antibody: pS409/410
Characterisation of sarkosyl‐insoluble TDP‐43 by immunolabelling techniques
The TDP‐43 extraction protocol was designed to maximise the solubility of TDP‐43 with benzonase while preserving the nature of all TDP‐43 species using inhibitors for proteases, phosphatases, deacetylases and deubiquitylating enzymes [26] (Figure S1). Western blots (WBs) of sarkosyl‐soluble and sarkosyl‐insoluble fractions from FTLD‐TDP and control (CTR) cases with phosphoserine (pS) 409/410 Ab consistently showed the presence of phosphorylated TDP‐43 only in the sarkosyl‐insoluble fraction of FTLD‐TDP cases (Figures 4A,C,D and S2). Full‐length phosphorylated TDP‐43 and several C‐terminal fragments (CTFs) were also present (Figures 4A,C,D and S2). A small amount of full‐length non‐phosphorylated at S409/410 TDP‐43 was also detected in the sarkosyl‐insoluble pellet of the FTLD‐TDP Type A and CTR samples after double‐immunolabelling WB with pS409/410 and C‐terminal pan TDP‐43 Abs (Figure 4B), as previously described [6, 11]. A comparison between Type A and Type B cases is shown in Figure 4C,D. Immunoblotting of FTLD‐TDP Type A and Type B cases with pS409/410 Ab revealed the presence of full‐length phosphorylated TDP‐43 migrating around 45 kDa, enriched CTFs with MWs in the range of 17–25 kDa, and a multitude of bands with MW between 25–44 kDa and above 50 kDa (Figure 4C,D). The electrophoretic pattern of the CTFs in the two FTLD‐TDP types was similar (Figure 4D). All FTLD‐TDP Type A and Type B cases positively immunoreacted with pS403/404 Ab, but, interestingly, prominent bands were observed at MW ≥ 37 kDa, whereas CTFs were less immunoreactive (Figure 4C,D). Immunoblotting with pS369 Ab generated biochemical profiles reminiscent of those observed with the pS409/410 Ab, in Type A and Type B cases; additionally, significant immunoreactivity was displayed by species with MW ≥ 70 kDa and by a ~38 kDa band in three FTLD‐TDP Type A and one Type B specimen (Figure 4C,D). Ubiquitination was detected with Ubi‐1 Ab and, as expected, affected mostly high MW species in all samples (Figure 4C,D). Although the presence of numerous proteins in the sarkosyl‐insoluble fraction makes it impossible to determine the number and identity all polypeptides recognised by the Ubi‐1 Ab, ubiquitination of TDP‐43 has been already established [3].
FIGURE 4.
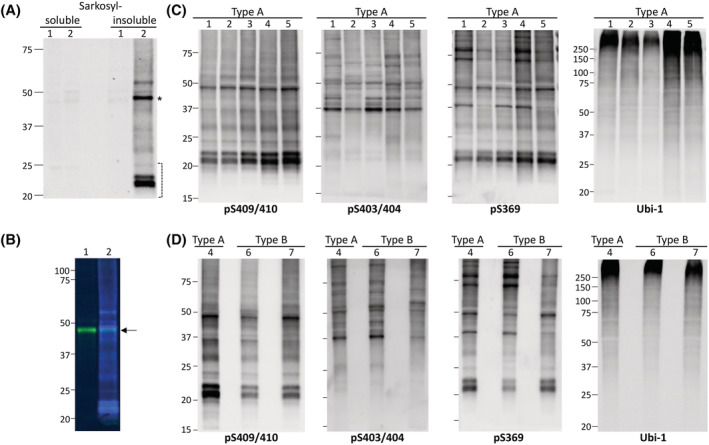
Isolation and biochemical characterisation of sarkosyl‐insoluble TDP‐43 in FTLD‐TDP Type A and Type B cases. (A) Representative Western blot (WB) of sarkosyl‐soluble and ‐insoluble fractions of a control (CTR) (1) and FTLD‐TDP Type A (2) case with pS409/410 antibody (Ab); phosphorylated TDP‐43 is only detected in the insoluble fraction of the Type A case. (B) Sarkosyl‐insoluble fractions from CTR (1) and FTLD‐TDP Type A (2) simultaneously immunoblotted with pS409/410 (blue) and C‐terminal pan TDP‐43 (green) Abs. A band of ~43 kDa (arrow) is observed in both preparations, corresponding to insoluble, non‐phosphorylated full‐length TDP‐43. TDP‐43 isolated from the frontal cortex of Type A (1–5) (C) and Type B (6 and 7) (D) cases immunoreacted with Abs targeting phosphorylated serines 409/410, 403/404 and 369, generating similar electrophoretic signatures within and between sets of samples. Ubiquitination affected mostly high molecular weight (MW) bands in all specimens; Ab: Ubi‐1. Asterisk indicates insoluble phosphorylated full‐length TDP‐43, and bracket indicates C‐terminal fragments (CTFs). Position and size (kDa) of MW markers are indicated on the left side of each WB
Sarkosyl‐insoluble preparations were also assessed for the presence of TDP‐43 filaments using immuno‐gold labelling and TEM (Figure 5). TDP‐43 filaments from both Type A and Type B cases were immunopositive for pS409/410 and pS403/404 Abs, with Ab labelling decorating the length of the fibrils in all cases (Figure 5). Although the pS369 Ab was previously reported to rarely recognise TDP‐43 Type A by immunohistochemistry, pS369 reactivity was observed in isolated TDP‐43 filaments from all our cases, including FTLD‐TDP Type A, suggesting this epitope to be exposed and accessible using this specific technique (Figure 5). No TDP‐43 filaments were detected in CTR samples.
FIGURE 5.
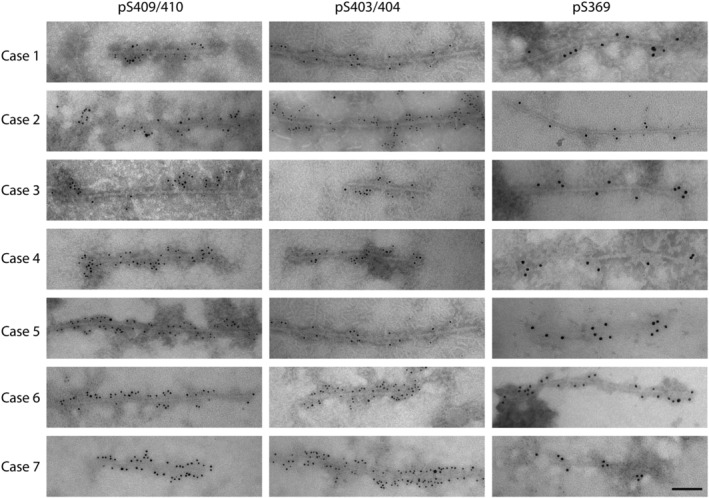
Characterisation of sarkosyl‐insoluble TDP‐43 filaments extracted from the frontal cortex of FTLD‐TDP Type A and Type B cases. TDP‐43 filaments were detected in all sarkosyl‐insoluble preparations. Immuno‐electron microscopy (EM) experiments utilised the same phospho‐antibodies (Abs) used for the biochemical study (Figure 4), targeting pS409/410, pS403/404 and pS369. Cases 1–5: FTLD‐TDP Type A; Cases 6 and 7: FTLD‐TDP Type B. Scale = 100 nm
Identification of PTMs by MS
Sarkosyl‐insoluble preparations from all FTLD‐TDP and CTR cases were subjected to LC–MS/MS to detect PTMs. A modified filter‐aided digestion (FASP) technique was used to compensate for the insoluble nature of the samples. Both Trypsin/Lys‐C and chymotrypsin digestions were performed to maximise TDP‐43 sequence coverage. Potential de novo peptides, which would not be created from specific digestion conditions, were excluded from the coverage count shown in the final table. Although only PTMs with high localisation to a particular site (AScore ≥10) were considered in the analysis of all samples, potentially modified sites for each modification are also listed and indicated in yellow in Tables 1, 2 and S3–S6. In the CTR samples, few TDP‐43 peptides were identified with an average sequence coverage of 32% (Table S7); no potentially biologically relevant PTM (e.g., phosphorylation) changes were detected. MS analyses of FTLD‐TDP sarkosyl‐insoluble samples identified significantly more numerous TDP‐43 peptides (average sequence coverage was 64% and 61%, in Type A and Type B, respectively) (Table S7) than in controls as well as peptides containing multiple modifications, which were potentially biologically relevant. This difference in results between pathological samples, which had abundant phosphorylated TDP‐43 and a minimal amount of insoluble, full‐length non‐phosphorylated TDP‐43, and CTR specimens, which lacked phosphorylated TDP‐43, indicated that the contribution of insoluble, full‐length and non‐phosphorylated TDP‐43 to PTM profiles was negligible.
TABLE 1.
Phosphorylated sites identified by mass spectrometry
| TDP‐43 residue | FTLD‐TDP Type A | FTLD‐TDP Type B | Controls | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 | Case 9 | Case 10 | |
| S20 | ‐ | ‐ | ‐ | ‐ | ||||||
| T25 | ‐ | ‐ | ‐ | ‐ | ||||||
| T88 | ||||||||||
| S91 | ||||||||||
| S92 | ||||||||||
| S183 | ‐ | |||||||||
| S212 | ‐ | ‐ | ||||||||
| Y214 | ‐ | ‐ | ||||||||
| S242 | ‐ | ‐ | ‐ | |||||||
| S254 | ‐ | ‐ | ‐ | |||||||
| S258 | ‐ | ‐ | ‐ | |||||||
| S266 | ‐ | ‐ | ‐ | |||||||
| S273 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ||||
| S305 | ‐ | ‐ | ||||||||
| S342 | ||||||||||
| S347 | ||||||||||
| S350 | ||||||||||
| S369 | ‐ | ‐ | ||||||||
| S373 | ‐ | ‐ | ||||||||
| Y374 | ‐ | ‐ | ||||||||
| S375 | ‐ | |||||||||
| S377 | ‐ | |||||||||
| S379 | ‐ | |||||||||
| S387 | ||||||||||
| S389 | ||||||||||
| S393 | ||||||||||
| S395 | ||||||||||
| S403 | ‐ | ‐ | ||||||||
| S404 | ‐ | ‐ | ||||||||
| S410 | ‐ | ‐ | ||||||||
Note: Green cell indicates a confidently localised phosphosite (AScore ≥10). Yellow cell indicates a potentially phosphorylated site–phosphorylated peptide with ambiguous localisation. Grey cell indicates that only an unmodified residue was identified. ‘‐’ indicates a residue not identified by LC–MS/MS. AScore is a localisation score assigned to modifications on peptide as the −10 log of a p value corresponding to the probability that the modification occurs at the reported position compared with other possible positions.
TABLE 2.
N‐term acetylated sites identified by mass spectrometry
| TDP‐43 residue | FTLD‐TDP Type A | FTLD‐TDP Type B | Controls | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 | Case 9 | Case 10 | |
| S2 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| M85 | ||||||||||
| C198 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |||
| T199 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ||||
| M202 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ||||
| G215 | ‐ | ‐ | ||||||||
| A235 | ‐ | ‐ | ‐ | |||||||
Note: Green cell indicates a confidently localised N‐term acetylated site (AScore ≥10). Grey cell indicates that only an unmodified residue was identified. ‘‐’ indicates residue not identified by LC–MS/MS.
Multiple PTMs, which may be potentially considered biologically relevant, were recovered in all FTLD‐TDP cases, including phosphorylation, N‐term acetylation, cysteine oxidation to cysteic acid and deamidation (Figure 6 and Tables 1, 2, S3 and S4). These modifications were mapped on the human TDP‐43 sequence (Figure 6). Additional modifications were also detected, such as oxidation, ammonia loss, sulfation, pyro‐Glu from glutamine, dihydroxy, dihydroxy methylglyoxal adduct, dehydration, formylation, sulphone, sodium adduct, tryptophan oxidation to kynurenine, carboxyethyl, the addition of carbon to cysteine, carbamidomethylation, carbamylation, cyano and 2‐amino‐3‐oxo‐butanoic acid (Tables S5 and S6). Several of these are modifications known to occur during sample processing (e.g., oxidation) or electrospray ionisation MS (e.g., water loss, ammonia loss and metal adducts) [32]. However, although oxidation is sometimes associated with sampling processing, it is impossible to rule out that some sites that have been reported previously may have a biological significance [6, 23, 33]. For this reason, this modification has been included in our analysis.
FIGURE 6.
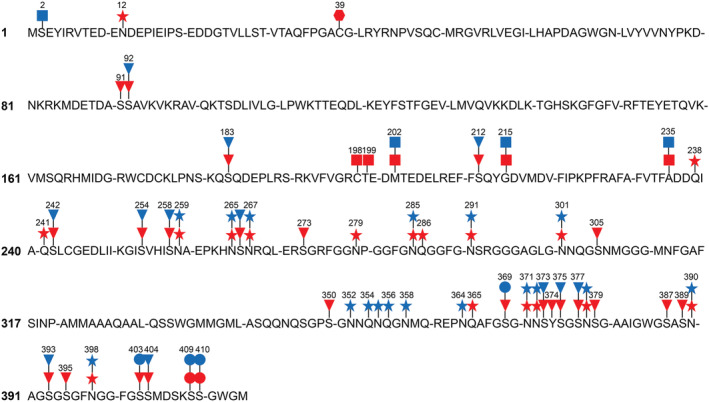
Potentially biologically relevant TDP‐43 post‐translational modifications (PTMs) in FTLD‐TDP Type A and Type B. Phosphorylation (triangles), N‐terminal acetylation (squares), deamidation (stars) and cysteine oxidation to cysteic acid (hexagon) modifications identified by mass spectrometry were mapped on FTLD‐TDP Type A (red) and Type B (blue) TDP‐43 amino acid sequence. Circles identify phosphorylated residues detected by immunolabelling techniques in the same preparations. Only mass spectrometry PTMs with high localisation at a specific site (AScore ≥10) are indicated
MS analyses overall detected 23 confidently localised phosphosites in FTLD‐TDP Type A and Type B samples (Figure 6 and Table 1). Twelve were common to both groups, with three phosphosites (S183, S242 and S266) being detected in all Type A and Type B cases (Figure 6 and Table 1). Eleven sites were exclusively identified in Type A specimens, all but one located in the LC domain (Figures 1 and 6 and Table 1). No Type B‐specific phosphorylation changes were revealed in our analyses (Figure 6 and Table 1).
Phosphorylated S369 and S403 residues were detected by MS only in Type A samples, although by WB and immuno‐electron microscopy (EM), these residues appeared phosphorylated in both groups of pathological cases (Figures 4 and 5 and Table 1). Phosphorylated S409 and S410 were not identified by MS, although the appropriate Ab detected them in all cases (Figures 4 and 5 and Table 1). Not all the cases within each group displayed the same sets of phosphorylated residues (Table 1).
In the C‐terminal region of TDP‐43 (peptides S375–W385, G386–F397, F398–W412 and G402–W412), almost every tryptic or chymotryptic peptide had multiple potentially phosphorylated sites (Table 1). We identified peptides containing up to three phosphorylation sites in this region. In our analysis, the region S375–S410 showed the highest levels of multiply phosphorylated peptides (Table 1).
Peptide N‐term acetylation was detected at six different sites on TDP‐43 from Type A and Type B cases (Figure 6 and Table 2). M202, G215 and A235 sites were shared by Type A and Type B cases (Figure 6 and Table 2). C198 and T199 were exclusively identified in Type A cases, whereas a peptide containing S2 was only recovered in one Type B sample (Figure 6 and Table 2). A235 was N‐term acetylated in both Type B cases, whereas N‐term acetylated S2, M202 and G215 were only observed in one Type B case (Table 2). Two Type A cases showed N‐term acetylation at T199, M202, G215 and A235, and one only at C198 (Table 2). N‐term acetylation is suggestive of biologically relevant proteolysis, and the sites we identified would represent N‐terminal sites of generated CTFs.
Cysteine oxidation to cysteic acid was also detected in one Type A case, at residue 39, which is part of the N‐terminal region of the protein (Figures 1 and 6 and Table S3).
Confidently localised deamidated sites were detected at 23 TDP‐43 residues, although two sites (N301 and N398) were also observed in CTR samples, and for this reason, they were excluded from further analyses (Table S4). Six changes were recovered exclusively in Type A samples, six in Type B specimens alone, whereas nine were uncovered in both groups (Table S4). Most of the deamidation changes were detected in the LC domain, although one was found in the N‐terminal domain, three were found in the RRM2 and two in regions outside TDP‐43 domains (Figure 1 and Table S4). Deamidation at residue N378 was detected in all Type A cases; all Type B cases displayed deamidation at N259 and N291. Other changes were uncovered in fewer Type A and Type B cases (Table S4).
Oxidation of methionine, histidine or tryptophan was uncovered at 10 sites of the TDP‐43 sequence (Table S5). Of the eight sites not observed in CTR, one site was in RRM1, one was in RRM2 and six were part of the LC domain (Figure 1 and Table S5). M162 and W385 were only oxidised in Type A cases, whereas all others were modified in both types (Table S5).
DISCUSSION
Inclusions of TDP‐43 in neurons are a defining pathological hallmark of multiple forms of sporadic and inherited FTD and ALS. The development of a classification of FTLD‐TDP into five subtypes has proven to be useful towards the effort directed to the identification of the molecular determinants of the phenotypic heterogeneity associated with TDP‐43 proteinopathies [30, 31, 34]. Thus, the investigation of well‐characterised cases of FTLD‐TDP associated with GRN and C9orf72 mutations became a necessary step. The aim of the current study was to identify TDP‐43 PTMs in five cases of FTLD‐TDP Type A with GRN mutations, two cases of FTLD‐TDP Type B with C9orf72 repeat expansions and three controls free of TDP‐43 pathology. This type of study has not been previously done in dominantly inherited FTD, and it is currently unknown whether comorbidities influence PTMs or if different brain regions are associated with specific TDP‐43 PTMs. In the FTLD‐TDP and CTR cases studied here, the frontal cortex was selected in view of the florid TDP‐43 pathology and the almost complete absence of co‐pathologies. Nevertheless, in some cases, mild age‐related vascular pathology was present in areas other than the frontal cortex, and two cases had mild amyloid‐β deposits.
Among the numerous post‐translationally modified TDP‐43 residues in TDP‐43 proteinopathies of Type A and Type B, caused by GRN mutations and C9orf72 repeat expansions, respectively, the phosphorylated and N‐term acetylated residues emerge as being of particular interest, also in view of the fact that some of them were identified for the first time.
A comparison between the current results on genetically determined FTD and those previously published on ALS and ALS with FTLD showed important differences related to the site and number of the phosphorylated amino acids, in particular within the C‐terminal region, which is known to be hyperphosphorylated in pathological conditions [6, 23]. Specifically, the amino acid sequence between residues 267 and 368 appears to be the most heterogeneous with respect to phosphorylation patterns [6, 23]. In contrast, phosphorylation events related to the sequences between amino acids 92 and 266 are comparable for FTLD‐TDP Type A and Type B but differ from those in ALS and ALS with FTLD [6, 23]. Of the 23 confidently localised phosphosites reported in this study, 11 novel sites were identified with two being in the NLS region, three in RRM2, four in the prion‐like LC domain, but two were related to regions outside the three aforementioned domains [6, 23, 35]. Three of the four novel phosphosites identified in the LC domain (S273, S350, Y374 and S377) were previously reported as being phosphorylated after in vitro phosphorylation using casein kinase 1 [36]. The results reported here are the first to describe these modifications as being present in human brain. Previously, phosphorylation at S273 was only identified in studies exploring the phosphoproteomics of genomically annotated breast cancers and cancerous cells [37, 38]. Not all the phosphorylated sites detected in this study were reported in the in vitro phosphorylation assay previously mentioned [36]; however, it is conceivable that other kinases such as tau‐tubulin kinase 1 or cell division cycle 7 may be involved in TDP‐43 phosphorylation [39, 40]. Peptides that were phosphorylated at multiple residues were also frequently identified, a finding suggestive of sequential kinase activity at adjacent sites and consistent with the recent evidence of sequential TDP‐43 phosphorylation in vitro with type‐specific timelines [41].
The phosphorylation of S369, S403, S404, S409 and S410 was consistently observed in all FTLD‐TDP cases by WB and immuno‐EM, but not by MS. Deposits immunoreactive to pS409/410 Ab were observed in all FTLD‐TDP preparations also by immunohistochemistry. Thus, discrepancies between different technical approaches emerged, an observation that requires the reconsideration of multiple technical and biological aspects inherent to the current study. First, the confident identification and localisation of phosphorylation sites on TDP‐43 by MS are challenging due to the high number of serine residues in close proximity to each other, most of which could potentially be modified. Second, the database search parameters used for this study allowed for up to three variable modifications per peptide, a typical setting in proteomic studies; however, increasing the number of potential variable modifications greatly increases computing time and can potentially cause false discovery rate issues. Third, the chymotryptic peptides created at the C‐terminal end of TDP‐43 are likely highly hydrophilic and may not be retained in the current sample preparation workflow (solid‐phase extraction workup requires binding to reverse‐phase material). Fourth, increasing phosphorylation (negatively charged PTM) is expected to decrease the ability of the peptides to be positively charged and thus detectable by positive ion MS. Therefore, the triple or quadruple phosphorylated peptides encompassing S303, S304, S409 and S410 may not be identified within the current LC–MS/MS workflow. The development of targeted MS methods for TDP‐43 and other neurodegeneration proteins will be essential for consistent PTM profiling.
Does the formation of TDP‐43 filaments occur in all TDP‐43 proteinopathies? Presence of TDP‐43 filaments in sarkosyl‐insoluble preparations has been a matter of discussion; however, Arseni et al. have shown the presence and structure of TDP‐43 filaments in ALS with FTLD [6, 42]. Phosphorylation may be relevant to the conformation of TDP‐43 in pathological conditions. In this study, the presence of TDP‐43 filaments in the FTLD‐TDP sarkosyl‐insoluble preparations was unequivocally demonstrated using immuno‐gold labelling and TEM. Research on the potential relationship between PTMs and filament formation represents an important direction in present and future research. As already stated, phosphorylation in FTLD‐TDP Type A and Type B differs from that in ALS with FTLD, suggesting that different PTM patterns may only be compatible with specific structures and supporting the hypothesis that different TDP‐43 structures exist [6, 23]. It is worth noting that the two phosphorylation sites S305 and S350 are not compatible with the TDP‐43 structure reported in ALS with FTLD [6]. In this study, the evidence of an inconsistent presence of phosphorylation of residues S305 and S350 might represent important phosphorylation changes occurring at different stages of filament formation and also might suggest the possibility of a difference in the structure of the filament core [6].
In the LC–MS/MS data analysis, a semi‐tryptic search strategy to identify non proteolytic N‐terminal fragments was implemented. N‐terminal peptide acetylation was observed, a result that would indicate the occurrence of a proteolytic processing of TDP‐43 at several sites in FTLD‐TDP Type A and Type B, with only one location having previously been identified [23, 43, 44]. It should be noted that since no size‐based separation was used prior to TDP‐43 digestion, validation of proteolytic cleavage at specific sites would be required. All but one N‐term acetylated site identified was located within the RRM2 and in proximity of previously identified N‐terminal sites, a finding supporting the hypothesis that N‐term acetylated residues may represent the N‐terminal amino acid of CTFs [23, 43, 44].
In addition to phosphorylation and N‐term acetylation, other potentially biologically relevant PTMs were identified, including cysteine oxidation to cysteic acid, deamidation and oxidation. Evidence of cysteine oxidation to cysteic acid was found at C39 in a single case of FTLD‐TDP Type A. This residue has been implicated in the oligomerisation process of TDP‐43 [17, 45, 46]. Seven novel deamidated residues and one novel oxidation site (W385) were also observed [6, 23].
In the FTLD‐TDP samples analysed, there was no MS evidence of lysine acetylation, ubiquitination and SUMOylation [17]. In fact, in the frontal cortex of FTLD‐TDP Type A and Type B, acetylated lysine residues were not detected by LC–MS/MS at TDP‐43 K82 and K145 residues, a result that differs from the finding of K82 acetylation in ALS brain tissue [23]. It is noteworthy that using an Ab to acetylated K145, immunohistochemical studies of the spinal cord in ALS and of the brain in FTD have shown immunoreactivity in ALS but not in FTD cases [47]. Furthermore, TDP‐43 ubiquitinated and SUMOylated sites were not detected; however, it has been suggested that the identification of such sites is challenging in view of the size of the modified products, the low stoichiometry of ubiquitinated proteins and the diversity of the resulting ubiquitin chains [48]. In addition, chymotryptic peptides would not have the well‐characterised ubiquitin remnant Gly–Gly, which is detectable after trypsin digestion [49]. The identification by MS of products following ubiquitination and SUMOylation requires most likely improved methods of TDP‐43 purification as well as modified peptide enrichment (i.e., ubiquitin enrichment by PTMScan, Cell Signaling).
The value of the present study resides in the analysis and comparison of TDP‐43 PTMs occurring in the human frontal cortex in FTLD‐TDP Type A and Type B associated to dominantly inherited FTDs caused by GRN and C9orf72 mutations, respectively. In sarkosyl‐insoluble preparations from the frontal cortex of patients affected by FTD caused by mutations in GRN and C9orf72 and rich in intracellular inclusions of TDP‐43 in neurons, TDP‐43 filaments were demonstrated by EM in both negative‐stained and immuno‐gold EM preparations with three different Abs targeting phosphorylated epitopes in TDP‐43. Furthermore, among the multiple PTMs of TDP‐43 found in both Type A and Type B, some appear to be specific for one type and others are present in both types. Remarkably, multiple phosphorylation sites were unique to Type A FTLD‐TDP. Some TDP‐43 PTMs were novel in that they were not previously seen in other TDP‐43 proteinopathies.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
ETHICS STATEMENT
All procedures were performed under protocols approved by the Institutional Review Board at Indiana University School of Medicine. Written informed consent for research was obtained from all patients or legal guardians. All patients' data and samples were coded and handled in accordance with NIH guidelines to protect patients' identities.
AUTHOR CONTRIBUTIONS
L. C., B. G. and K. L. N. conceived and coordinated the study; L. C. carried out the protein purifications and the biochemical studies; L. C. and E. H. D. carried out the mass spectrometry; G. I. H. carried out the transmission electron microscopy; H. J. G. and R. V. carried out the genetic studies; R. R. carried out the histological and immunohistochemical preparations; B. G. and K. L. N. carried out the neuropathological studies; L. C., E. H. D., G. I. H., H. J. G., E. B., R. V., B. G. and K. L. N. analysed the data; L. C., E. H. D., G. I. H., M. H. J., B. G. and K. L. N. prepared the iconography for publication; L. C., E. H. D., G. I. H., H. J. G., B. G. and K. L. N. contributed to the manuscript; B. G. edited the final version. All authors read and approved the final manuscript.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/nan.12836.
Supporting information
Figure S1. Isolation of sarkosyl‐insoluble TDP‐43 from brain tissue. Schematic representation of the protocol used to generate sarkosyl‐ soluble and ‐insoluble fractions.
{kind=link}
Figure S2. Absence of phosphorylated TDP‐43 in sarkosyl‐soluble fractions from all cases and in sarkosyl‐insoluble fractions from controls. Phosphorylated TDP‐43 is not detected in the sarkosyl‐soluble fractions of FTLD‐TDP Type A (Cases 1–5), FTLD‐TDP Type B (Cases 6–7), and CTR (Cases 8–10) cases (a), nor in sarkosyl‐insoluble fractions of the three CRT (cases 8–10) (b). Sarkosyl‐insoluble fraction of a FTLD‐TDP Type A (Case 2) is shown in both a and b as a positive control. Ab: pS409/410. Position and size (kDa) of molecular weight (MW) markers are indicated on the left side of each WB.
Table S1. Case Data
Table S2. Antibodies used in the study
Table S3. Cysteine oxidation to cysteic acid site identified by mass spectrometry
Table S4. Deamidated sites identified by mass spectrometry
Table S5. Oxidized (M, W, H) sites identified by mass spectrometry
Table S6. Additional PTMs identified in the samples by mass spectrometry
Table S7. Overview of TDP‐43 mass spectrometry coverage
ACKNOWLEDGEMENTS
We thank the families of the patients for donating brain tissue. Dr Benjamin Ryskeldi‐Falcon shared the protocol for the extraction of TDP‐43 filaments and provided valuable discussions. This work was supported in part by grants from the US National Institutes of Health (P30 AG010133 to B. G. and U01 NS110437 to B. G. and R. V.), by the Indiana University (IU) Health Strategic Research Initiative (to K. L. N.) and by the Department of Pathology and Laboratory Medicine, Indiana University School of Medicine (IUSM). The mass spectrometry studies were carried out by the IUSM Center for Proteome Analysis. Acquisition of the IUSM Proteomics core instrumentation used for this project was provided by the IU Precision Health Initiative. The proteomics work was supported, in part, by the Indiana Clinical and Translational Sciences Institute, by Award Number UL1TR002529 from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award and the Cancer Center Support Grant for the IU Simon Comprehensive Cancer Center (Award Number P30CA082709) from the National Cancer Institute. We obtained one sample from the National Centralized Repository for Alzheimer's Disease and Related Dementias (NCRAD), which receives government support under a cooperative agreement grant (U24 AG021886) awarded by the National Institute on Aging (NIA). We thank contributors who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible. The content of this study is the sole responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Cracco L, Doud EH, Hallinan GI, et al. Distinguishing post‐translational modifications in dominantly inherited frontotemporal dementias: FTLD‐TDP Type A (GRN) vs Type B (C9orf72). Neuropathol Appl Neurobiol. 2022;48(6):e12836. doi: 10.1111/nan.12836
Funding information National Institutes of Health, Grant/Award Numbers: P30AG010133, U01NS110437, UL1TR002529, P30CA082709, U24AG021886; Indiana University Health Strategic Research Initiative; Indiana University Precision Health Initiative; Department of Pathology and Laboratory Medicine, Indiana University
Contributor Information
Laura Cracco, Email: lcracco@iu.edu.
Bernardino Ghetti, Email: bghetti@iu.edu.
Kathy L. Newell, Email: klnewell@iu.edu.
DATA AVAILABILITY STATEMENT
Mass spectrometry raw data and tabulated results for TDP‐43 peptides are uploaded to the MassIVE repository, under Accession Number MSV000089242: https://massive.ucsd.edu/ProteoSAFe/private-dataset.jsp?task=49d0affc18ef4f46a892e3dfb2d75ae5.
REFERENCES
- 1. Buratti E, Dörk T, Zuccato E, Pagani F, Romano M, Baralle FE. Nuclear factor TDP‐43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001;20(7):1774‐1784. doi: 10.1093/emboj/20.7.1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang HY, Wang IF, Bose J, Shen CK. Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics. 2004;83(1):130‐139. doi: 10.1016/S0888-7543(03)00214-3 [DOI] [PubMed] [Google Scholar]
- 3. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130‐133. doi: 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- 4. Arai T, Hasegawa M, Akiyama H, et al. TDP‐43 is a component of ubiquitin‐positive tau‐negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602‐611. doi: 10.1016/j.bbrc.2006.10.093 [DOI] [PubMed] [Google Scholar]
- 5. Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): consensus working group report. Brain. 2019;142(6):1503‐1527. doi: 10.1093/brain/awz099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arseni D, Hasegawa M, Murzin AG, et al. Structure of pathological TDP‐43 filaments from ALS with FTLD. Nature. 2022;601(7891):139‐143. doi: 10.1038/s41586-021-04199-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brady OA, Meng P, Zheng Y, Mao Y, Hu F. Regulation of TDP‐43 aggregation by phosphorylation and p62/SQSTM1. J Neurochem. 2011;116(2):248‐259. doi: 10.1111/j.1471-4159.2010.07098.x [DOI] [PubMed] [Google Scholar]
- 8. Gruijs da Silva LA, Simonetti F, Hutten S, et al. Disease‐linked TDP‐43 hyperphosphorylation suppresses TDP‐43 condensation and aggregation. EMBO J. 2022;e108443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eck RJ, Kraemer BC, Liachko NF. Regulation of TDP‐43 phosphorylation in aging and disease. Geroscience. 2021;43(4):1605‐1614. doi: 10.1007/s11357-021-00383-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hasegawa M, Arai T, Nonaka T, et al. Phosphorylated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64(1):60‐70. doi: 10.1002/ana.21425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Inukai Y, Nonaka T, Arai T, et al. Abnormal phosphorylation of Ser409/410 of TDP‐43 in FTLD‐U and ALS. FEBS Lett. 2008;582(19):2899‐2904. doi: 10.1016/j.febslet.2008.07.027 [DOI] [PubMed] [Google Scholar]
- 12. Li HY, Yeh PA, Chiu HC, Tang CY, Tu BP. Hyperphosphorylation as a defense mechanism to reduce TDP‐43 aggregation. PLoS ONE. 2011;6(8):e23075. doi: 10.1371/journal.pone.0023075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Neumann M, Kwong LK, Lee EB, et al. Phosphorylation of S409/410 of TDP‐43 is a consistent feature in all sporadic and familial forms of TDP‐43 proteinopathies. Acta Neuropathol. 2009;117(2):137‐149. doi: 10.1007/s00401-008-0477-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sternburg EL, Gruijs da Silva LA, Dormann D. Post‐translational modifications on RNA‐binding proteins: accelerators, brakes, or passengers in neurodegeneration? Trends Biochem Sci. 2022;47(1):6‐22. doi: 10.1016/j.tibs.2021.07.004 [DOI] [PubMed] [Google Scholar]
- 15. François‐Moutal L, Perez‐Miller S, Scott DD, Miranda VG, Mollasalehi N, Khanna M. Structural insights into TDP‐43 and effects of post‐translational modifications. Front Mol Neurosci. 2019;12:301. doi: 10.3389/fnmol.2019.00301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aebersold R, Agar JN, Amster IJ, et al. How many human proteoforms are there? Nat Chem Biol. 2018;14(3):206‐214. doi: 10.1038/nchembio.2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Buratti E. TDP‐43 post‐translational modifications in health and disease. Expert Opin Ther Targets. 2018;22(3):279‐293. doi: 10.1080/14728222.2018.1439923 [DOI] [PubMed] [Google Scholar]
- 18. Farina S, Esposito F, Battistoni M, Biamonti G, Francia S. Post‐translational modifications modulate proteinopathies of TDP‐43, FUS and hnRNP‐A/B in amyotrophic lateral sclerosis. Front Mol Biosci. 2021;8:693325. doi: 10.3389/fmolb.2021.693325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wesseling H, Mair W, Kumar M, et al. Tau PTM profiles identify patient heterogeneity and stages of Alzheimer's disease. Cell. 2020;183(6):1699‐713.e13. doi: 10.1016/j.cell.2020.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mair W, Muntel J, Tepper K, et al. FLEXITau: quantifying post‐translational modifications of tau protein in vitro and in human disease. Anal Chem. 2016;88(7):3704‐3714. doi: 10.1021/acs.analchem.5b04509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hubbard EE, Heil LR, Merrihew GE, et al. Does data‐independent acquisition data contain hidden gems? A case study related to Alzheimer's disease. J Proteome Res. 2022;21(1):118‐131. doi: 10.1021/acs.jproteome.1c00558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Riva N, Gentile F, Cerri F, et al. Phosphorylated TDP‐43 aggregates in peripheral motor nerves of patients with amyotrophic lateral sclerosis. Brain. 2022;145(1):276‐284. doi: 10.1093/brain/awab285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kametani F, Obi T, Shishido T, et al. Mass spectrometric analysis of accumulated TDP‐43 in amyotrophic lateral sclerosis brains. Sci Rep. 2016;6(1):23281. doi: 10.1038/srep23281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tsuji H, Nonaka T, Yamashita M, et al. Epitope mapping of antibodies against TDP‐43 and detection of protease‐resistant fragments of pathological TDP‐43 in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Biochem Biophys Res Commun. 2012;417(1):116‐121. doi: 10.1016/j.bbrc.2011.11.066 [DOI] [PubMed] [Google Scholar]
- 25. Tsuji H, Arai T, Kametani F, et al. Molecular analysis and biochemical classification of TDP‐43 proteinopathy. Brain. 2012;135(11):3380‐3391. doi: 10.1093/brain/aws230 [DOI] [PubMed] [Google Scholar]
- 26. Laferrière F, Maniecka Z, Pérez‐Berlanga M, et al. TDP‐43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat Neurosci. 2019;22(1):65‐77. doi: 10.1038/s41593-018-0294-y [DOI] [PubMed] [Google Scholar]
- 27. Porta S, Xu Y, Restrepo CR, et al. Patient‐derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP‐43 pathology in vivo. Nat Commun. 2018;9(1):4220. doi: 10.1038/s41467-018-06548-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shi Y, Zhang W, Yang Y, et al. Structure‐based classification of tauopathies. Nature. 2021;598(7880):359‐363. doi: 10.1038/s41586-021-03911-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Neumann M, Lee EB, Mackenzie IR. Frontotemporal lobar degeneration TDP‐43‐immunoreactive pathological subtypes: clinical and mechanistic significance. In: Ghetti B, Buratti E, Boeve B, Rademakers R, eds. Frontotemporal Dementias: Emerging Milestones of the 21st Century. Cham: Springer International Publishing; 2021:201‐217. doi: 10.1007/978-3-030-51140-1_13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mackenzie IR, Neumann M. Reappraisal of TDP‐43 pathology in FTLD‐U subtypes. Acta Neuropathol. 2017;134(1):79‐96. doi: 10.1007/s00401-017-1716-8 [DOI] [PubMed] [Google Scholar]
- 31. Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD‐TDP pathology. Acta Neuropathol. 2011;122(1):111‐113. doi: 10.1007/s00401-011-0845-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li Q, Shortreed MR, Wenger CD, et al. Global post‐translational modification discovery. J Proteome Res. 2017;16(4):1383‐1390. doi: 10.1021/acs.jproteome.6b00034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Silva CJ, Erickson‐Beltran M. Detecting differences in prion protein conformation by quantifying methionine oxidation. ACS Omega. 2022;7(3):2649‐2660. doi: 10.1021/acsomega.1c04989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 2009;117(1):15‐18. doi: 10.1007/s00401-008-0460-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neumann M, Frick P, Paron F, Kosten J, Buratti E, Mackenzie IR. Antibody against TDP‐43 phosphorylated at serine 375 suggests conformational differences of TDP‐43 aggregates among FTLD–TDP subtypes. Acta Neuropathol. 2020;140(5):645‐658. doi: 10.1007/s00401-020-02207-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kametani F, Nonaka T, Suzuki T, et al. Identification of casein kinase‐1 phosphorylation sites on TDP‐43. Biochem Biophys Res Commun. 2009;382(2):405‐409. doi: 10.1016/j.bbrc.2009.03.038 [DOI] [PubMed] [Google Scholar]
- 37. Mertins P, Mani DR, Ruggles KV, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534(7605):55‐62. doi: 10.1038/nature18003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou H, Di Palma S, Preisinger C, et al. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J Proteome Res. 2013;12(1):260‐271. doi: 10.1021/pr300630k [DOI] [PubMed] [Google Scholar]
- 39. Liachko NF, McMillan PJ, Strovas TJ, et al. The tau tubulin kinases TTBK1/2 promote accumulation of pathological TDP‐43. PLoS Genet. 2014;10(12):e1004803. doi: 10.1371/journal.pgen.1004803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liachko NF, McMillan PJ, Guthrie CR, Bird TD, Leverenz JB, Kraemer BC. CDC7 inhibition blocks pathological TDP‐43 phosphorylation and neurodegeneration. Ann Neurol. 2013;74(1):39‐52. doi: 10.1002/ana.23870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. De Rossi P, Lewis AJ, Furrer J, et al. FTLD‐TDP assemblies seed neoaggregates with subtype‐specific features via a prion‐like cascade. EMBO Rep. 2021;22(12):e53877. doi: 10.15252/embr.202153877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jiang YX, Cao Q, Sawaya MR, et al. Amyloid fibrils in FTLD‐TDP are composed of TMEM106B and not TDP‐43. Nature. 2022;605(7909):304‐309. doi: 10.1038/s41586-022-04670-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nonaka T, Kametani F, Arai T, Akiyama H, Hasegawa M. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP‐43. Hum Mol Genet. 2009;18(18):3353‐3364. doi: 10.1093/hmg/ddp275 [DOI] [PubMed] [Google Scholar]
- 44. Igaz LM, Kwong LK, Chen‐Plotkin A, et al. Expression of TDP‐43 C‐terminal fragments in vitro recapitulates pathological features of TDP‐43 proteinopathies. J Biol Chem. 2009;284(13):8516‐8524. doi: 10.1074/jbc.M809462200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bozzo F, Salvatori I, Iacovelli F, et al. Structural insights into the multi‐determinant aggregation of TDP‐43 in motor neuron‐like cells. Neurobiol Dis. 2016;94:63‐72. doi: 10.1016/j.nbd.2016.06.006 [DOI] [PubMed] [Google Scholar]
- 46. Jiang LL, Xue W, Hong JY, et al. The N‐terminal dimerization is required for TDP‐43 splicing activity. Sci Rep. 2017;7(1):6196. doi: 10.1038/s41598-017-06263-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cohen TJ, Hwang AW, Restrepo CR, Yuan CX, Trojanowski JQ, Lee VM. An acetylation switch controls TDP‐43 function and aggregation propensity. Nat Commun. 2015;6(1):5845. doi: 10.1038/ncomms6845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Udeshi ND, Mertins P, Svinkina T, Carr SA. Large‐scale identification of ubiquitination sites by mass spectrometry. Nat Protoc. 2013;8(10):1950‐1960. doi: 10.1038/nprot.2013.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sheng Z, Wang X, Ma Y, et al. MS‐based strategies for identification of protein SUMOylation modification. Electrophoresis. 2019;40(21):2877‐2887. doi: 10.1002/elps.201900100 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Isolation of sarkosyl‐insoluble TDP‐43 from brain tissue. Schematic representation of the protocol used to generate sarkosyl‐ soluble and ‐insoluble fractions.
Figure S2. Absence of phosphorylated TDP‐43 in sarkosyl‐soluble fractions from all cases and in sarkosyl‐insoluble fractions from controls. Phosphorylated TDP‐43 is not detected in the sarkosyl‐soluble fractions of FTLD‐TDP Type A (Cases 1–5), FTLD‐TDP Type B (Cases 6–7), and CTR (Cases 8–10) cases (a), nor in sarkosyl‐insoluble fractions of the three CRT (cases 8–10) (b). Sarkosyl‐insoluble fraction of a FTLD‐TDP Type A (Case 2) is shown in both a and b as a positive control. Ab: pS409/410. Position and size (kDa) of molecular weight (MW) markers are indicated on the left side of each WB.
Table S1. Case Data
Table S2. Antibodies used in the study
Table S3. Cysteine oxidation to cysteic acid site identified by mass spectrometry
Table S4. Deamidated sites identified by mass spectrometry
Table S5. Oxidized (M, W, H) sites identified by mass spectrometry
Table S6. Additional PTMs identified in the samples by mass spectrometry
Table S7. Overview of TDP‐43 mass spectrometry coverage
Data Availability Statement
Mass spectrometry raw data and tabulated results for TDP‐43 peptides are uploaded to the MassIVE repository, under Accession Number MSV000089242: https://massive.ucsd.edu/ProteoSAFe/private-dataset.jsp?task=49d0affc18ef4f46a892e3dfb2d75ae5.