

Abstract
Adrenergic receptors are critical regulators of cardiac function with profound effects on cardiac output during sympathetic stimulation. Chronic stimulation of the adrenergic system of the heart under conditions of cardiac stress leads to cardiac dysfunction, hypertrophy, and ultimately failure. Emerging data have revealed that G protein coupled receptors in intracellular compartments are functionally active and regulate distinct cellular processes from those at the cell surface. β2 adrenergic receptors internalize onto endosomes in various cell types where they have recently been shown to continue to stimulate cAMP production to selectively regulate gene expression. Other studies have identified β1 adrenergic receptors at the nuclear envelope and the Golgi apparatus. Here we discuss data on signaling by β1 and β2 adrenergic receptors in the heart and the possible influence of their subcellular locations on their divergent physiological functions in cardiac myocytes, and in cardiac pathology. Understanding the relative roles of these receptors at these locations could have a significant impact on pharmacological targeting of these receptors for the treatment of heart failure and cardiac diseases.
Introduction
The sympathetic nervous system (SNS) has a fundamental role in maintaining cardiac output by regulating heart rate, cardiac contractility, and vascular resistance. The activity of SNS is mainly regulated by catecholamines including epinephrine (Epi) and norepinephrine (NE). These sympathetic neurotransmitters bind to β-adrenergic receptors (βARs) in the heart1. βARs are G protein coupled receptors (GPCRs) and have been extensively studied as paradigms for overall GPCR function, trafficking and structure. They are critical regulators of cardiac functions and are important therapeutic targets for treatment of cardiovascular diseases. The major focus of this review is on recent advances in our understanding of the different spatiotemporal features of β1 and β2 AR signaling, and how differential localization and trafficking may relate to distinct physiological and pathological roles in cardiac myocytes. We begin with an overview of the regulation of cardiac physiology by these receptors and then discuss potential location specific roles that may contribute to adrenergic physiology and pathology in the heart.
β-adrenergic receptor subtypes in the heart.
There are three subtypes of βARs present in the human heart, β1AR, β2AR, and β3AR. The healthy human heart comprises 80% β1ARs and 20% β2ARs, and lower expression of β3ARs 2,3. β1ARs and β2ARs share high homology (57% full-length identity)4, and both stimulate Gαs and cAMP production. Epi and NE bind to βARs in the heart with different affinities with NE targeting β1ARs with approximately 10-fold higher selectivity relative to β2ARs whereas Epi is non-selective for β1- and β2ARs5. Catecholamine-induced stimulation of βARs acutely improves cardiac performance in healthy human hearts by increasing heart rate (chronotropy) and contractility (inotropy). The simplistic view is that these receptors act at cell surface generating second messengers that diffuse throughout the cell to regulate cardiac cell physiology. This view has come under scrutiny in recent years. Cardiac myocytes have unique structural features that require a more detailed understanding of subcellular locations and functions of these GPCRs to establish a mechanistic picture of how they are involved in cardiac regulation and pathophysiology.
β-Adrenergic receptor subtypes in cardiac contraction and hypertrophy
Human Heart failure.
Heart failure is a complicated syndrome where the ability of the heart to pump blood declines over time due to chronic stress, resulting from hypertension or left ventricular dysfunction, leading to an inability to meet the metabolic demands of the body. It is well-recognized that hyperactivated adrenergic signaling, due to prolonged elevation of catecholamines and other neurohumoral factors, leads to the progression of maladaptive cardiac hypertrophy, myocyte apoptosis, fibrosis, and eventually heart failure likely accounting, at least in part, for the effectiveness of β-blockers in treatment of heart failure. In failing hearts, β1ARs are reduced from 80% of total βAR population to approximately 50% depending on disease severity, whereas the β2AR population remained unchanged6.
Mouse models of cardiac function and failure.
The first transgenic mice with cardiac-specific overexpression of β2ARs were created by Milano et al. in 19947. These mice exhibited enhanced atrial contractility and left ventricular function even in the absence of agonists possibly due to an increased level of receptors with basal constitutive activity. Later studies confirmed this observation8–10, however, high level over-expression of β2ARs, 300-fold over endogenous receptors, has detrimental effects on cardiac performance in the long term11. These long-term adverse consequences require β2ARs to be present at a high level while the positive inotropic effects from β2ARs possibly involve low to moderate level of β2ARs overexpression12. In contrast, studies on β1ARs consistently demonstrate that mice with increased (15-45 fold) cardiac-specific overexpression of β1ARs had enhanced cardiac contractility at young age but developed significant myocyte hypertrophy and reduced contractility as they aged13 14.
β1AR knockout mice, β2AR knockout mice or β1/β2AR double knockout mice have also been generated. The majority of mice lacking β1ARs die prenatally, but those that survive have blunted chronotropic and inotropic responses to isoproterenol (Iso), despite normal expression of β2ARs15. β2AR knockout mice, on the other hand, had normal resting heart rate and blood pressure, no prenatal death, and the chronotropic response to Iso was normal16. These studies indicated that β1ARs play a predominant role in regulating heart rate and contractility. Dual βAR knockout prevented increases in inflammatory cytokine production, fibrosis, and cardiac hypertrophic responses in a pressure overload transverse aortic constriction model (TAC)17. Zhao et al. confirmed that TAC-induced cardiac hypertrophy was abolished in the dual knockout mice18. Interestingly, they also found distinct regulation by these subtypes in pressure overload-induced hypertrophy18. Specifically, mice with β1AR deletion had similar hypertrophic responses compared to wild type, whereas, β2AR deletion led to development of exaggerated hypertrophy18. This indicated that coordination of signaling between these two subtypes is essential in mediating cardiac remodeling18. It is clear that β1ARs and β2ARs elicit markedly different outcomes in cardiac pathology, however, definitive answers as to the molecular mechanisms underlying these phenomena have not been fully elucidated.
Differential β1 and β2 adrenergic receptor signaling, internalization, and locations.
The marked differences in physiological and pathological outcomes downstream or either β1 or β2ARs occur despite the fact that they stimulate the same core signaling pathway (Gαs-AC-cAMP) and share overall sequence and structural homology. A large body of research has sought to decipher their different or even opposite effects on cardiac function. Possible mechanistic explanations include differences in β1AR and β2AR sarcolemmal microdomain distribution in the cardiac myocyte, subcellular trafficking and location, scaffolding interactions, and signal transduction. Some of these data are described below.
β1 and β2AR-dependent signaling pathways and contractility.
cAMP signaling downstream of β1AR stimulation increases Ca2+ transient amplitudes, at least in part, through PKA-dependent phosphorylation of a variety of intracellular proteins such as the type 2 ryanodine receptor (RyR2), phospohlamban, cardiac myosin binding protein-C (cMyBP-c) and Cav1.2 (L-type) Ca2+ channels. Our laboratory and others identified roles of EPAC proteins in adrenergic regulation of contractility, independent from PKA19,20. EPAC is an exchange factor for the small GTPase Rap, activated by directly binding cAMP21. Our studies indicated that EPAC-activated Rap directly binds to, and stimulates, a specific phospholipase (PLC) isoform, PLCε, leading to PKC activation, diacylglycerol (DAG) production and activation of Ca2+/calmodulin kinase II (CaMKII)22 ultimately resulting in phosphorylation of RyR219,20 and potentially other proteins to modulate Ca2+ transient amplitudes. Work by another group found that this pathway regulates SR Ca2+ leak19.
Compared to β1ARs, β2AR activation elicits a significantly smaller effect on cardiac inotropy23,24. Both receptors stimulate cAMP production although with different characteristics. β2AR-stimulated cAMP is highly compartmentalized whereas β1AR-cAMP signaling is more far-reaching and diffuse in cardiac myocytes25 (Fig. 1A). One hypothesis is that the cAMP produced downstream of β2AR cannot access PKA and EPAC to regulate the machinery involved in contraction. The relatively minor β2ARs-dependent increases in intracellular Ca2+ transient amplitudes are independent from cAMP-mediated protein phosphorylation23 or EPAC activation.
Figure 1.
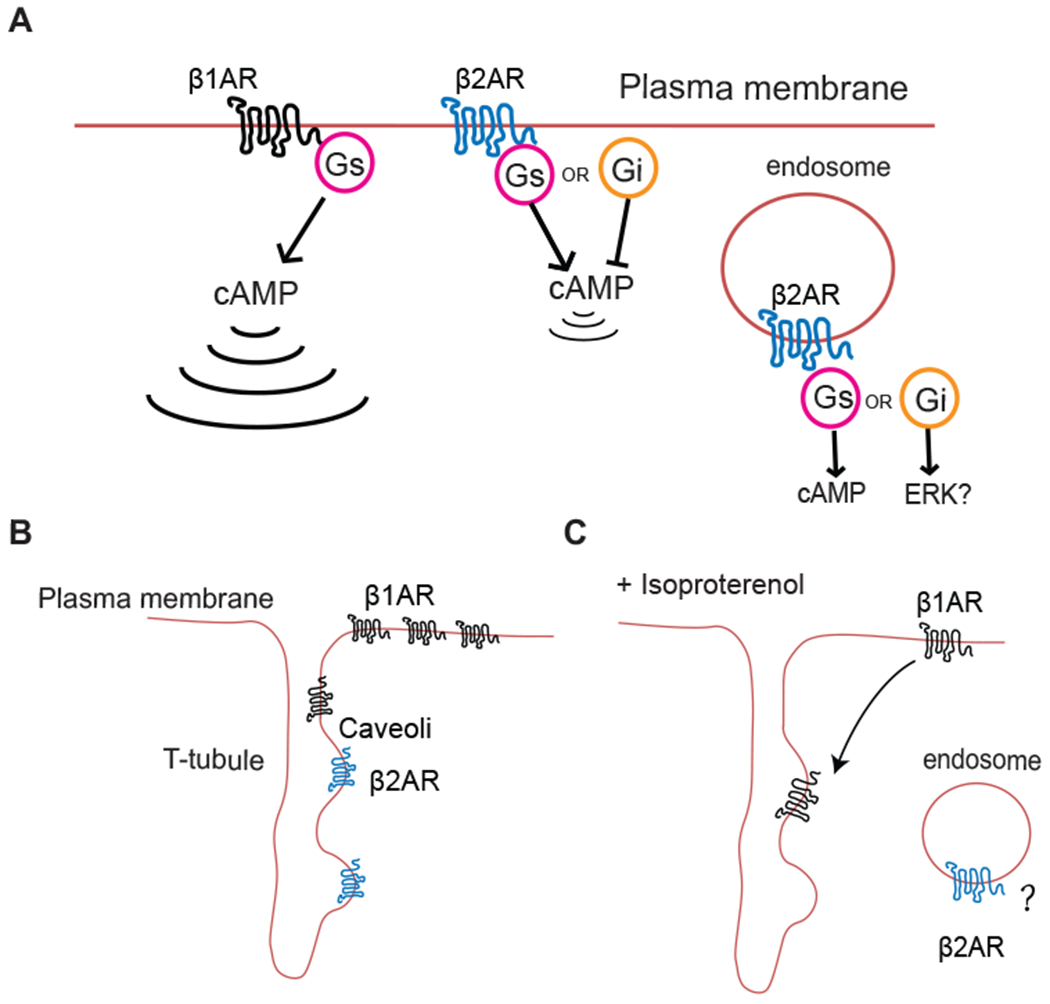
Discrete cAMP signals and microdomain distributions of βARs. A) β1AR-induced cAMP signaling is far-reaching, whereas β2AR-induced cAMP signaling is locally confined and Internalized β2ARs may continue to signal in myocytes. B) In healthy cardiomyocytes, β1ARs distribute widely across the cell surface33,34, while β2ARs are localized to T-tubules and caveoli. C) Stimulation with a βAR agonist induces a redistribution of β1ARs into T-tubules32 and possible internalization into endosomes, although this has not been directly confirmed in mature cardiac myocytes.
Another significant signaling difference between β2 and β1ARs is that β2ARs couple to both Gs and Gi proteins while β1ARs couple primarily to Gs. Functional studies in neonatal rat cardiac myocytes indicated that the initial transient contractile response to β2 activation is suppressed by a switch to Gi coupling26. Pertussis toxin treatment revealed a sustained contractile response in response to β2AR stimulation26,27. Activation β1ARs in the same system resulted in sustained signaling that was not affected by PTX treatment. Thus, differences in signaling properties (ie. coupling to different G proteins and pathways) of the receptors themselves could be responsible for some of the observed physiological differences.
Differential internalization of β1 vs. β2ARs.
In transfected cells β2 adrenergic receptors robustly internalize while β1ARs also do but to a lesser extent28. Initial studies in NRVMs isolated from β1/β2 AR double knockout mice transduced with either flag epitope tagged-β1 or β2 ARs indicated that β2ARs robustly internalize into endosomes but β1ARs do not29,30. This difference was demonstrated to result from differential scaffolding via PDZ interacting proteins at the C-termini of βARs. Subsequent studies presented conflicting results in NRVMs where flag-β1ARs were observed to internalize31. Both studies explored the role of scaffolding via a PDZ ligands at the C-terminus.
Neonatal myocytes are a very useful model system for studying cardiomyocyte function but do not have the complex subcellular architecture of a mature adult myocyte. In particular, mature myocytes have a highly organized network of T-tubules that are extended invaginations of the sarcolemma. This network of tubules contains L-type Ca2+ channels directly opposed to Ryr2 in the SR where excitation-contraction coupling occurs. A recent study examined the location of β1ARs in adult ventricular myocytes (AVMs) with adenoviral mediated expression of flag-β1ARs32. Here, under non stimulated conditions β1 ARs were broadly expressed at the cell surface but not in T-tubules (Fig. 1B). Upon stimulation with a high concentration of Iso (10 μM), flag-β1 ARs redistributed to T-tubules without any visible receptor internalization into endosomes (Fig. 1C). β2ARs were not examined in this study. More recently, studies were done with a novel fluorescent derivative of the βAR antagonist carazolol to label βARs expressed at native levels in mature cardiac myocytes33. Current antibodies against β1ARs are not reliable in the detection of endogenous receptors. Using sophisticated image analysis, it was shown that endogenous β1ARs are present at the cell surface and in T-tubules. On the other hand, β2ARs are present only at the T-tubules consistent with a restricted pool of cAMP generated at this location. Due to the nature of ligand used for these studies, agonist-dependent changes in receptor location could not be monitored. Interestingly the results with native receptors had important differences from those with expressed receptors where expressed β2ARs were observed at both the cell surface and in T-tubules emphasizing the importance of examining endogenous receptors where possible.
βARs in caveolae
Caveolae are microdomains in the plasma membrane enriched in sphingolipids and cholesterol and caveolin. These microdomains promote interactions between signaling proteins that sort into these domains based in part on their lipid modification. Cardiac myocytes are enriched in caveolae and adrenergic receptor subtypes differentially sort into these domains with β2ARs selectively partitioning into caveolae, while the majority of β1ARs excluded from these domains34,35. Experimental manipulations that disrupted caveolae led to stronger coupling of β2ARs to cAMP production suggesting that residence in caveolae limits cAMP signaling from β2ARs35. Caveolar compartmentation of βARs has been reviewed in detail elsewhere34.
Signaling by internalized βARs from endosomes.
While internalization of GPCRs has been observed for many years, it had been assumed that the purpose of internalization was to desensitize the receptor. Development of genetically encodable nanobody-based sensors36 or engineered Gα subunits (fluorescent protein tagged miniG proteins) 37 has enabled direct monitoring of the activation state of βARs in living cells at a subcellular level. Using Nanobody 80 fused to GFP (NB80-GFP), which detects activated βARs, and Nb37-GFP, which detects active Gαs, Irannejad et al. found activated β2ARs coupled to Gαs activation at early endosomes. Using inhibitors of receptor internalization it was shown internalized receptors contributed to the overall cAMP response to an agonist in some cell types36. Following up on this work, it was found that signaling from internal receptors was selectively associated with expression of a subset of genes38. Ferrandon et al. also identified roles for intracellular GPCRs in regulation of sustained cAMP generation downstream of parathyroid hormone receptors39. The existence or role of signaling by internalized activated β2ARs has not been investigated in cardiac myocytes, or in the heart.
Resident Intracellular β1ARs
Nuclear Envelope-
Initial evidence for intracellular localization of βARs in cardiomyocytes was reported by Boivin et al. in 200640. Using immunocytochemistry in adult cardiomyocytes they showed that in addition to cell surface and T-tubules localizations, β1ARs but not β2ARs were localized on the periphery of nuclear envelope40. These intracellular β1ARs activated AC and stimulated RNA synthesis in isolated nuclear fractions possibly through Gαs40 or other signaling pathways including ERK1/2 and p3841. Interestingly these investigators also identified β3ARs on the nuclear envelope where β3 but not β1ARs regulated NO production42. Other GPCRs have been found at the nuclear envelope in CMs including endothelin and α1-adrenergic receptors and have been demonstrated to stimulate phospholipase C activation and subsequent nuclear Ca2+ increases43,44.
Golgi apparatus.
In 2017, Irannejad et al. found a functional pool of β1ARs at Golgi apparatus that is not delivered via receptor endocytosis in HeLa cells28. However, β2ARs were not detected at this location. This suggests an additional possible mechanism underlying the divergence in βAR subtype functions in the heart. Activation and signaling from Golgi-β1AR was independent from receptors at the cell surface28. In cardiac myocytes, our laboratory demonstrated activation of a signaling pathway downstream of Golgi resident β1ARs45 (Fig. 2A). These studies arose from the observation that the relatively cell impermeant β-AR agonist, Iso (partition coefficient (LogP) = −0.24) was unable to stimulate activation of a muscle specific AKAP (mAKAP)/EPAC/PLCε complex at the nuclear envelope/Golgi interface46, while cAMP analogs and forskolin robustly activated this pathway. It was previously shown that this complex regulates cardiac hypertrophy and hypertrophic gene expression47. This suggested that there may be a local site of cAMP generation deeper within the myocyte, and indeed, stimulation with a cell permeant βAR agonist, dobutamine (LogP = 3.0), revealed activation of the endogenous mAKAP/EPAC/PLCε pathway that results in hydrolysis of phosphatidylinositol 4-phosphate (PI4P) at the Golgi to produce local diacylglycerol. This involved β1ARs and not β2ARs since a selective β2AR antagonist had no effect in these experiments. Finally, specific blockade of βAR signaling at the Golgi with a Golgi-targeted βAR inhibitor, Nanobody 80 (NB80), demonstrated that endogenous Golgi β1ARs were required for stimulation of cardiomyocyte hypertrophy by NE or dobutamine45. These studies specifically examined the intracellular mAKAP/EPAC/PLCε pathway, but it is possible or even likely, that other pathways are regulated downstream of Golgi resident β1ARs (Fig. 2A). This pathway remains to be investigated in animal models of hypertrophy and heart failure.
Figure 2. Intracellular β1AR activation and signaling.
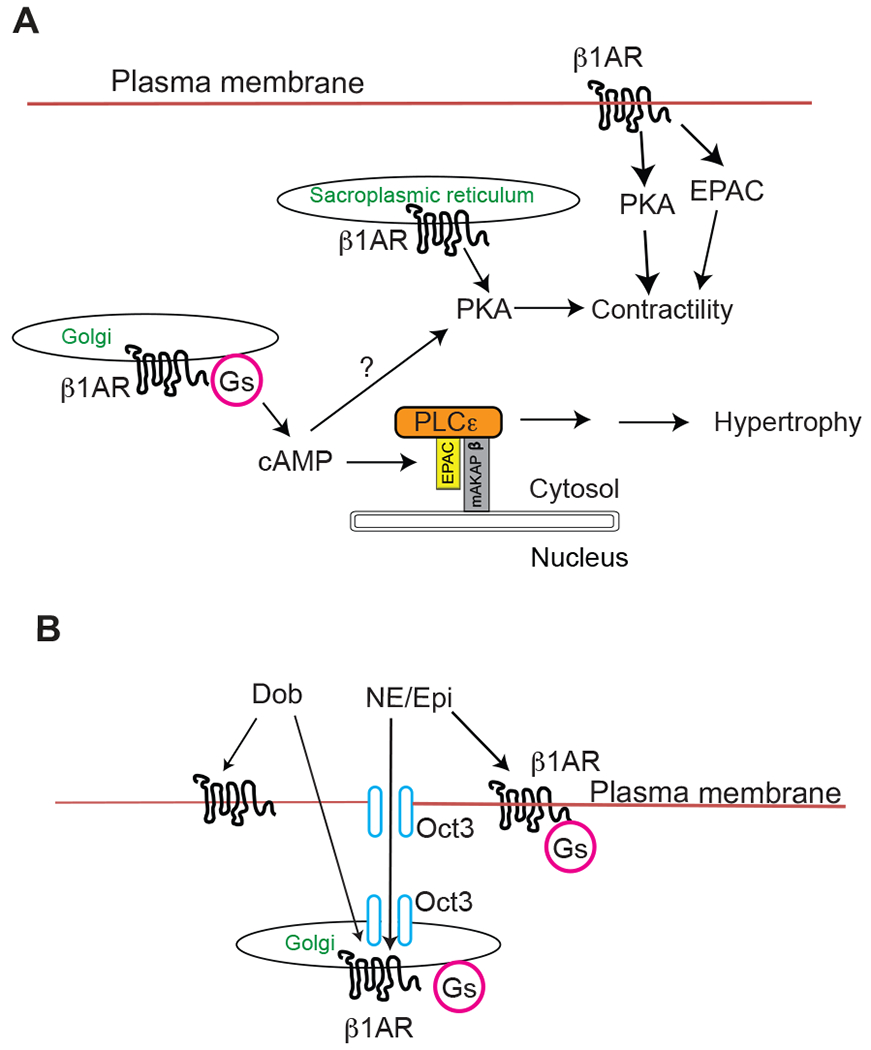
A) β1ARs are resident in the plasma membrane, Golgi apparatus and sarcoplasmic reticulum. Activation of Golgi-β1ARs generates a local cAMP pool that induces cardiomyocyte hypertrophy through mAKAP/EPAC/PLCε pathway45. Golgi b1 ARs likely also regulate PKA and may contribute to contractile or other processes. β1ARs at sarcoplasmic reticulum modulate contractility through PKA-dependent phospholamban phosphorylation54. Plasma membrane resident β1ARs can aksi regulate contractility via PKA, or CamKII downstream of EPAC20. B) The organic cation transporter, OCT3 facilitate the uptake of the membrane impermeant endogenous catecholamines across the sarcolemma and Golgi membrane as well as the nuclear envelope membrane (not shown). Dobutamine, a membrane permeant agonist, does not rely on OCT3 to across the membrane.
Organic Cation Transporters provide access of catecholamines to intracellular receptors.
For intracellular adrenergic systems to operate physiologically they must be accessed by endogenous neurotransmitters. The endogenous catecholamines, Epi and NE, are ligands of βARs but are membrane impermeant. How these physiological relevant ligands access their intracellular targets in the Golgi and elsewhere must be explained. A particular organic cation transporter subtype, OCT3, has been shown to facilitate the uptake of catecholamines across the plasma membrane/sarcolemma, and is also found in the Golgi, and nuclear membranes of cultured cardiac cells44. An OCT3-dependent mechanism has been previously reported to be involved in uptake of NE into cardiac myocytes for regulation of α-adrenergic receptors located at the nuclear envelope 44. In our studies of Golgi β1ARs, activation of the EPAC/PLCε pathway by NE required OCT3, both in NRVMs and AVMs. We have also shown that blockade of OCT3 prevents catecholamine-induced cardiomyocyte hypertrophy45 in NRVMs and, as discussed below, blunts the contractile response in cardiac myocytes and in animals48.
These data support a physiologically relevant role of intracellular βARs and provide a mechanism by which catecholamines can cross the membranes to act on the intracellular receptors. Interestingly, corticosterone released by the adrenal gland blocks OCT3 activity in vitro. It is possible that corticosterone might exert direct effects in cardiomyocytes in vivo by inhibiting OCT3 activity49 thereby regulating intracellular β-AR-related cardiac functions. Supporting this view, clinical evidence suggests that adrenal insufficiency is associated with cardiac dysfunction50,51.
In our in vitro studies Iso was poorly membrane permeant52. However, it is possible Iso could be transported into cells through OCT3 and potentially engage internal β1ARs at a high concentration. This, at least in part, could account for its ability to induce cardiac hypertrophy in vivo. In vitro, a non-permissive temperature for the transporter, a lack of sufficient transporter expression, or low concentrations of Iso could explain the failure to observe internal βAR activation.
Physiological roles for β1AR compartmentation.
What is the physiological significance of having separate pools of β1AR at the Golgi apparatus or nuclear envelope, and the sarcolemma? A possible function might be to separate short term sympathetic stimulation from changes in gene expression. In this scenario, during fight or flight responses, acute catecholamine release would access receptors at the cell surface but would be insufficient in duration and magnitude to access intracellular pathways through OCT3. The kinetics of Golgi β1AR activation in cardiac myocytes and Hela cells are significantly slower than activation of these receptors at the cell surface28,45 perhaps due to the kinetic properties of the transporter. The efficiency and rate of uptake of catecholamines depend on the Km, kcat and expression level of OCT3. The Km’s of OCT3 for Epi and NE are in the high μM range (~500-1000 μM) 53 while affinities for the βARs are in the low μM range (1-15 μM)5. One might expect that during acute stimulation, surface β1ARs would be rapidly stimulated but NE or EPI may not reach sufficient concentrations long enough for significant uptake by OCT3. Under chronic cardiac stress and sustained catecholamine elevation, NE and Epi could achieve sufficient concentrations long enough to accumulate intracellularly and access the Golgi pool of β1ARs to regulate gene expression through the mAKAP/EPAC/PLCε pathway. This mechanism would prevent inappropriate activation of PLCε-dependent cardiac hypertrophic responses to acute catecholamine exposure. Also, with sustained sympathetic stimulation and development of heart failure β1AR signaling from plasma membrane is significantly blunted. Under these conditions Golgi-β1AR mediated signaling may become more prevalent. Having separate internal β1AR pools also likely to allows cells to generate precise signals that regulate specific cellular responses instead of activating uncontrolled global signal events. These speculations remain to be experimentally verified.
Sarcoplasmic reticulum.
A recent study showed that a population of β1ARs was localized to the SR, and regulated contractility through local PKA activation (discussed below) and phospholamban phosphorylation54 (Fig. 2A). It was also shown that β1ARs coimmunoprecipitated with SERCA2 supporting an SR location, although it is possible these interactions could be in other compartments or between compartments (Fig. 2A). Here, OCT3 knockout animals had blunted contractile responses to Iso and NE supporting the idea that OCT3 transporters have physiological significance beyond neurotransmitter clearance.
Compartmentalized cAMP signaling in cardiac myocytes.
cAMP microdomains.
The accumulating evidence discussed above support the emerging concept of functional pools of βARs at various subcellular compartments that contribute to cAMP compartmentalization to optimally exert physiological cardiac outcomes. There is ample evidence that cAMP is tightly regulated at a nanodomain level, rather than simply diffusing from the site of generation, to optimally regulate different downstream effectors. Early in 1980s, the working hypothesis of cAMP compartmentalization and subcellular pools of cAMP-dependent protein kinases downstream of β-AR activation in cardiomyocytes was proposed55. This concept became generally accepted when optical probes (fluorescence resonance energy transfer, FRET-based reporters) were exploited to visualize distinct subcellular cAMP pools56,57. By targeting the sensors to specific subcellular locations in living cells or even in living animals, intracellular cAMP levels could be measured in a real-time, quantitative, and spatiotemporal manner. This tool is extremely useful especially in the architecturally complicated cells like ventricular cardiac myocytes. In one series of experiments a modified FRET sensor “CUTie” was targeted to the different sites known to regulate cardiac excitation-contraction coupling (ECC) including the sarcolemma, SR, and myofilaments in isolated cardiac myocytes58. They found distinct regulation of cAMP pools at these sites with significantly smaller and delayed response at myofilaments compared to sarcolemma and SR58. Importantly, they also showed that when cAMP compartmentation was abolished using phosphodiesterase (PDE) inhibitors the inotropic response was blunted in cardiomyocytes, indicating the importance of the precise control of local cAMP level in the contractility58.
These approaches were also employed to study cAMP generation by βARs located at the SR54. In these experiments blockade or knockout of OCT3 inhibited NE-dependent cAMP generation detected by an SR-targeted PKA regulated FRET reporter (SR-AKAR). This implied, but did not directly show, that βARs localized to the SR are responsible for local cAMP production at the SR. Targeting the βAR inhibitor, NB80, to the SR would more directly address a role for βARs at the SR. Similar studies have not yet been used to monitor cAMP production downstream of Golgi localized βARs.
cAMP PDEs control cAMP microdomains.
Where cAMP signaling is terminated is a major contributor to restricting microdomains of cAMP. The enzymes responsible for cAMP degradation, cAMP-phosphodiesterases (PDEs), consist of 11 members (PDE1-11). Different PDE isoforms have unique subcellular locations, substrate specificity and regulatory mechanisms, therefore, localization of specific PDE isoforms to specific myocyte subdomains shape cAMP signaling locally leading to specific biological outcomes without broadly altering cAMP concentration within cells. PDEs 1-5 and 8-10 are reported to be expressed in the heart59. Dysregulation of PDE isoform expression level, subcellular sites and activation have been implicated in cardiac diseases.
PDEs are selectively anchored to signalosomes via anchoring proteins including A kinase anchoring proteins (AKAPs), which also bring together PKA, adenylyl cyclase (AC), and other cAMP-effectors in many cell types including the heart. For example, mAKAPβ, scaffolds PDE4D3, AC5, PKA, EPAC, PLCε, protein kinase (PKD), and other proteins, at the nuclear membrane, to regulate cardiac hypertrophy60–62. AKAP12 scaffolds PDE4D and β2ARs63,64. β2AR-dependent cAMP production is limited by PDE4, since blockade of PDE4D results in sustained cAMP production65. PDE5 also binds to β2ARs under conditions of diabetic cardiomyopathy66. β1AR-dependent cAMP signaling has also been shown to regulated through scaffolding to PDE4D67. Thus, PDE scaffolding to βARs modulates cAMP production downstream of both receptor subtypes, but likely does so to different extents.
PDEs limit access of cAMP generated at the cell surface to the mAKAP/EPAC/PLCε signaling pathway at the Golgi-nuclear envelope interface.
As discussed above, treatment of cells with the cell impermeant agonist Iso is unable to stimulate activation of mAKAP/EPAC/PLCε at the Golgi/nuclear envelope interface. This indicates that something must be restricting access of cAMP to this complex. We identified PDE3 as a key PDE preventing access of cAMP to the mAKAP/EPAC/PLCε complex in cardiac myocytes52 (Fig. 3). Inhibition of PDE3 revealed activation of PLCε at the NE/Golgi interface in the absence of ligand stimulation and led to development of cardiomyocyte hypertrophy in NRVMs. Treatment with Iso in combination with PDE3 inhibitor led to an enhanced response. A second pool of cAMP controlled by PDEs 2 and 9A, opposed activation of PLCε at the Golgi through a PKA-dependent mechanism which would be predicted to oppose hypertrophy (Fig. 3). This evidence again emphasizes the importance of balanced cAMP compartmentalized signaling in cardiac functions and shows that cAMP signaling can be either detrimental or protective depending on where or how it is regulated in the heart.
Figure 3. Balancing pools of cAMP signaling to PLCε by PDE isoforms.
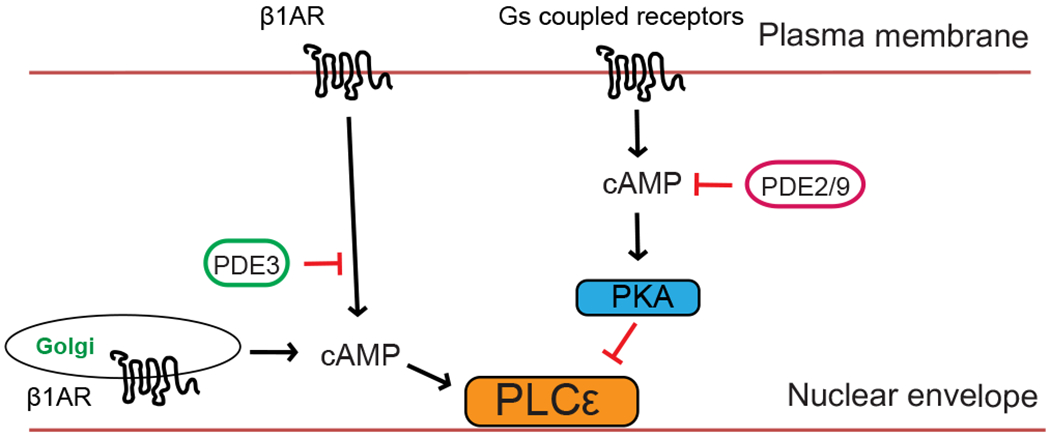
Distinct cAMP pools are differentially controlled by PDEs 2/9 and PDE3. PDE3 prevents cAMP diffusion to the nuclear envelope/Golgi limiting activation of PLCε at that location. A distinct pool of cAMP regulated by PDEs 2/9 at plasma membrane opposes the activity of PLCε through a PKA dependent mechanism46.
Conclusions and future directions.
It is well established that under both normal and pathologic conditions, βARs play an important role in initiating and regulating signaling pathways involved in cardiac function. Due to the inhibitory effects of β-blockers on myocardial contractility, they were initially considered as contraindicated in the treatment of heart failure. In mid 1970s, small-scale trials were conducted to show an improved outcome of β-blockers in patients with cardiac dysfunction68,69. Today, β-blockers such as carvedilol, bisoprolol, and metoprolol along with other medications are amongst first-line treatment used in patients with stable, mild, moderate, and severe heart failure, even though the exact mechanism of action of β-blockers remain incompletely understood. Studies have shown several potential mechanisms of β-blockers that contribute to their beneficial effects on heart failure. The mechanisms include but are not limited to antagonizing neurohormonal stimulatory effects of hyper-βAR mediated hypertrophic and proapoptotic effects on cardiomyocytes, slowing heart rate, lowering blood pressure, reducing myocardial oxygen consumption, and restoring the ratio of β1ARs and β2ARs70,71. A large body of evidence shows favorable effects of β-blockers in heart failure and reversal of cardiac remodeling. However, not all β-blockers showed the same beneficial effects. Currently available β-blockers are divided based on their different interactions with β1 and β2 subtypes. Bisoprolol, metoprolol, and nebivolol are β1-selective blockers while carvedilol is a non-selective β-blocker. In addition to subtype selectivity, a new principle for functional selectivity could be based on the accessibility to the subcellular locations. Bisoprolol, carvedilol and metoprolol which show beneficial effects in heart failure tend to be hydrophobic while sotalol, a membrane impermeant β-blocker, is not used to treat heart failure. Hydrophobicity might be an important factor for a clinically effective β-blocker based on the discovery of functional roles of intracellular βAR signaling in cardiac contractility, hypertrophy, and subsequent heart failure. Furthermore, elucidation of the distinct signaling properties of βAR signaling at the cell surface vs. the interior of the cell, and the balance between cardiac protective versus toxic effects could provide critical insights into the development of new strategies for heart failure.
A genetically encoded, and nanobody based β-AR blocker, NB80, has been recently utilized to specifically target βARs intracellularly28,45 by fusing different targeting sequences to its N terminus. This approach combined with adeno-associated virus (AAV) based gene delivery system opens the possibility of targeting select subcellular pools of receptors with a high specificity in specific cell types. Investigation of βARs at various compartments and their common or unique physiological roles will provide substantial and comprehensive information of the roles of βARs in cardiac functions.
References
- 1.Brodde OE β1- and β2-adrenoceptors in the human heart: Properties, function, and alterations in chronic heart failure. Pharmacol. Rev 43, 203–242 (1991). [PubMed] [Google Scholar]
- 2.Ihl-Vahl R, Eschenhagen T, Kübler W, Marquetant R, et al. Differential regulation of mRNA specific for β1- and β2-adrenergic receptors in human failing hearts. Evaluation of the absolute cardiac mRNA levels by two independent methods. J. Mol. Cell. Cardiol 28, 1–10 (1996). [DOI] [PubMed] [Google Scholar]
- 3.Schobesberger S, Wright PT, Poulet C, Sanchez-Alonso JL, et al. β-Adrenoceptor redistribution impairs NO/cGMP/PDE2 signalling in failing cardiomyocytes. Elife 9, 1–15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frielle T, Collins S, Daniel KW, Caron MG, et al. Cloning of the cDNA for the human β1-adrenergic receptor. Proc. Natl. Acad. Sci. U. S. A 84, 7920–7924 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frielle T, Daniel KW, Caron MG & Lefkowitz RJ Structural basis of β-adrenergic receptor subtype specificity studied with chimeric β1/β2-adrenergic receptors. Proc. Natl. Acad. Sci. U. S. A 85, 9494–9498 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, et al. Decreased catecholamine sensitivity and β-adrenergic-receptor density in failing human hearts. New Engl. J. Med 307, 205–211 (1982). [DOI] [PubMed] [Google Scholar]
- 7.Milano CA, Allen LF, Rockman HA, Dolber PC, et al. Enhanced myocardial function in transgenic mice overexpressing the β2-adrenergic receptor. Science 264, 582–586 (1994). [DOI] [PubMed] [Google Scholar]
- 8.Dorn GW, Tepe NM, Lorenz JN, Koch WJ, et al. Low- and high-level transgenic expression of β2-adrenergic receptors differentially affect cardiac hypertrophy and function in Gαq-overexpressing mice. Proc. Natl. Acad. Sci. U. S. A 96, 6400–6405 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tevaearai HT, Eckhart AD, Walton GB, Keys JR, et al. Myocardial gene transfer and overexpression of β2-adrenergic receptors potentiates the functional recovery of unloaded failing hearts. Circulation 106, 124–129 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Rengo G, Zincarelli C, Femminella GD, Liccardo D, et al. Myocardial β2-adrenoceptor gene delivery promotes coordinated cardiac adaptive remodelling and angiogenesis in heart failure. Br. J. Pharmacol 166, 2348–2361 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Boer OJ, van der Wal AC, Houtkamp MA, Ossewaarde JM, et al. Age-dependent cardiomyopathy and heart failure phenotype in mice overexpressing β2-adrenergic receptors in the heart. Cardiovasc. Res 48, 448–454 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Liggett SB, Tepe NM, Lorenz JN, Canning AM, et al. Early and delayed consequences of β2-adrenergic receptor overexpression in mouse hearts: Critical role for expression level. Circulation 101, 1707–1714 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Engelhardt S, Hein L, Wiesmann F & Lohse MJ Progressive hypertrophy and heart failure in β1-adrenergic receptor transgenic mice. Proc. Natl. Acad. Sci. U. S. A 96, 7059–7064 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bisognano JD, Weinberger HD, Bohlmeyer TJ, Pende A, et al. Myocardial-directed overexpression of the human β1-adrenergic receptor in transgenic mice. J. Mol. Cell. Cardiol 32, 817–830 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Rohrer DK, Desai KH, Jasper JR, Stevens ME, et al. Targeted disruption of the mouse β1-adrenergic receptor gene: Developmental and cardiovascular effects. proc. natl. acad. sci. usa 93, 7375–7380 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chruscinski AJ, Rohrer DK, Schauble E, Desai KH, et al. Targeted disruption of the β2 adrenergic receptor gene. J. Biol. Chem 274, 16694–16700 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Kiriazis H, Wang K, Xu Q, Gao XM, et al. Knockout of β1- and β2-adrenoceptors attenuates pressure overload-induced cardiac hypertrophy and fibrosis. Br. J. Pharmacol 153, 684–692 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao M, Fajardo G, Urashima T, Spin JM, et al. Cardiac pressure overload hypertrophy is differentially regulated by β-adrenergic receptor subtypes. Am J Physiol Heart Circ Physiol 301, 1461–1470 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pereira L, Métrich M, Fernández-velasco M, Lucas A, et al. The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J. Physiol 583, 685–694 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oestreich EA, Malik S, Goonasekera SA, Blaxall BC, et al. Epac and phospholipase Cepsilon regulate Ca2+ release in the heart by activation of protein kinase Cepsilon and calcium-calmodulin kinase II. J. Biol. Chem 284, 1514–1522 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bos JL Epac proteins: multi-purpose cAMP targets. Trends Biochem. Sci 31, 680–686 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Hothi SS, Gurung IS, Heathcote JC, Zhang Y, et al. Epac activation, altered calcium homeostasis and ventricular arrhythmogenesis in the murine heart. Pflugers Arch. Eur. J. Physiol 457, 253–270 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao RP, Hohl C, Altschuld R, Jones L, et al. β2-Adrenergic receptor-stimulated increase in cAMP in rat heart cells is not coupled to changes in Ca2+ dynamics, contractility, or phospholamban phosphorylation. J. Biol. Chem 269, 19151–19156 (1994). [PubMed] [Google Scholar]
- 24.Kaumann AJ, Hall JA, Murray KJ, Wells FC, et al. A comparison of the effects of adrenaline and noradrenaline on human heart: The role of β1- and β2-adrenoceptors in the stimulation of adenylate cyclase and contractile force. Eur. Heart J 10, 29–37 (1989). [DOI] [PubMed] [Google Scholar]
- 25.Nikolaev VO, Bünemann M, Schmitteckert E, Lohse MJ, et al. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching β1-adrenergic but locally confined β2-adrenergic receptor-mediated signaling. Circ. Res 99, 1084–1091 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Devic E, Xiang Y, Gould D & Kobilka B β-Adrenergic receptor subtype-specific signaling in cardiac myocytes from β1 and β2 adrenoceptor knockout mice. Mol. Pharmacol 60, 577–583 (2001). [PubMed] [Google Scholar]
- 27.Xiao R, Avdonin P, Zhou Y, Cheng H, et al. Coupling of β2 -adrenoceptor to Gi proteins and its physiological relevance in murine cardiac myocytes. (1999). [DOI] [PubMed] [Google Scholar]
- 28.Irannejad R, Pessino V, Mika D, Huang B, et al. Functional selectivity of GPCR-directed drug action through location bias. Nat. Chem. Biol 13, 799–806 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiang Y, Devic E & Kobilka B The PDZ binding motif of the β1 adrenergic receptor modulates receptor trafficking and signaling in cardiac myocytes. J. Biol. Chem 277, 33783–33790 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Xiang Y & Kobilka B The PDZ-binding motif of the β2-adrenoceptor is essential for physiologic signaling and trafficking in cardiac myocytes. Proc. Natl. Acad. Sci. U. S. A 100, 10776–10781 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Nooh MM & Bahouth SW Role of AKAP79/150 protein in β1-adrenergic receptor trafficking and signaling in mammalian cells. J. Biol. Chem 288, 33797–33812 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nooh MM, Mancarella S & Bahouth SW Novel paradigms governing β1-adrenergic receptor trafficking in primary adult rat cardiac myocytes. Mol. Pharmacol 94, 862–875 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bathe-Peters M, Gmach P, Boltz HH, Einsiedel J, et al. Visualization of β-adrenergic receptor dynamics and differential localization in cardiomyocytes. Proc. Natl. Acad. Sci. U. S. A 118, 1–10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steinberg SF β2-Adrenergic receptor signaling complexes in cardiomyocyte caveolae/lipid rafts. J. Mol. Cell. Cardiol 37, 407–415 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Rybin VO, Xu X, Lisanti MP & Steinberg SF Differential targeting of β-adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae: A mechanism to functionally regulate the cAMP signaling pathway. J. Biol. Chem 275, 41447–41457 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, et al. Conformational biosensors reveal GPCR signalling from endosomes. Nature 495, 534–538 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wan Q, Okashah N, Inoue A, Nehme R, et al. Mini G protein probes for active G protein– coupled receptors (GPCRs) in live cells. J. Biol. Chem 293, 7466–7473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsvetanova NG & von Zastrow M Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat. Chem. Biol 10, 1061–1065 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrandon S, Feinstein TN, Castro M, Wang B, et al. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat. Chem. Biol 5, 734–742 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boivin B, Lavoie C, Vaniotis G, Baragli A, et al. Functional β-adrenergic receptor signalling on nuclear membranes in adult rat and mouse ventricular cardiomyocytes. Cardiovasc. Res 71, 69–78 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Vaniotis G, Del Duca D, Trieu P, Rohlicek CV, et al. Nuclear β-adrenergic receptors modulate gene expression in adult rat heart. Cell. Signal 23, 89–98 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vaniotis G, Glazkova I, Merlen C, Smith C, et al. Regulation of cardiac nitric oxide signaling by nuclear β-adrenergic and endothelin receptors. J. Mol. Cell. Cardiol 62, 58–68 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Branco Ana F, and Allen BG. G Protein-coupled receptor signaling in cardiac nuclear membranes. J Cardiovasc Pharmacol. 65, 89–90 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Wright CD, Chen Q, Baye NL, Huang Y, et al. Nuclear α1-adrenergic receptors signal activated ERK localization to caveolae in adult cardiac myocytes. Circ. Res 103, 992–1000 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nash CA, Wei W, Irannejad R & Smrcka AV Golgi localized β1-adrenergic receptors stimulate Golgi PI4P hydrolysis by PLCε to regulate cardiac hypertrophy. Elife 8, 1–22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nash CA, Brown LM, Malik S, Cheng X, et al. Compartmentalized cyclic nucleotides have opposing effects on regulation of hypertrophic phospholipase C ε signaling in cardiac myocytes. J. Mol. Cell. Cardiol 121, 51–59 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang L, Malik S, Pang J, Wang H, et al. Phospholipase Cε hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell 153, 216–227 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiang Y, Rybin VO, Steinberg SF & Kobilka B Caveolar localization dictates physiologic signaling of β2-adrenoceptors in neonatal cardiac myocytes. J. Biol. Chem 277, 34280–34286 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Gründemann D, Koschker AC, Haag C, Honold C, et al. Activation of the extraneuronal monoamine transporter (EMT) from rat expressed in 293 cells. Br. J. Pharmacol 137, 910–918 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krug JJ Cardiac arrest secondary to Addison’s disease. Ann. Emerg. Med 15, 735–737 (1986). [DOI] [PubMed] [Google Scholar]
- 51.Krishnamoorthy A, Mentz RJ, Hyland KA, McMillan EB, et al. A crisis of the heart: An acute reversible cardiomyopathy bridged to recovery in a patient with addison’s disease. ASAIO J. 59, 668–670 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Nash CA, Brown LM, Malik S, Cheng X, et al. Compartmentalized cyclic nucleotides have opposing effects on regulation of hypertrophic phospholipase Cε signaling in cardiac myocytes. J. Mol. Cell. Cardiol 121, 51–59 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Duan H & Wang J Selective transport of monoamine neurotransmitters by human plasma membrane monoamine transporter and organic cation transporter 3. J. Pharmacol. Exp. Ther 335, 743–753 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Y, Shi Q, Li M, Zhao M, et al. Intracellular β1-adrenergic receptors and organic cation transporter 3 mediate phospholamban phosphorylation to enhance cardiac contractility. Circ. Res 246–261 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buxton ILO & Brunton LL Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J. Biol. Chem 258, 10233–10239 (1983). [PubMed] [Google Scholar]
- 56.Zaccolo M & Pozzan T Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 295, 1711–1715 (2002). [DOI] [PubMed] [Google Scholar]
- 57.DiPilato LM, Cheng X & Zhang J Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc. Natl. Acad. Sci. U. S. A 101, 16513–16518 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Surdo NC, Berrera M, Koschinski A, Brescia M, et al. FRET biosensor uncovers cAMP nano-domains at β-adrenergic targets that dictate precise tuning of cardiac contractility. Nat. Commun 8, 1–14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen S & Yan C An update of cyclic nucleotide phosphodiesterase as a target for cardiac diseases. Expert Opin. Drug Discov 16, 183–196 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dodge KL, Khouangsathiene S, Kapiloff MS, Mouton R, et al. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 20, 1921–1930 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, et al. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 437, 574–578 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kapiloff MS, Piggott LA, Sadana R, Li J, et al. An adenylyl cyclase-mAKAPβ signaling complex regulates cAMP levels in cardiac myocytes. J. Biol. Chem 284, 23540–23546 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qasim H & McConnell BK Akap12 signaling complex: Impacts of compartmentalizing camp-dependent signaling pathways in the heart and various signaling systems. J. Am. Heart Assoc 9, 1–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fan GF, Shumay E, Wang HY & Malbon CC The scaffold protein gravin (cAMP-dependent protein kinase-anchoring protein 250) binds the β2-adrenergic receptor via the receptor cytoplasmic Arg-329 to Leu-413 domain and provides a mobile scaffold during desensitization. J. Biol. Chem 276, 24005–24014 (2001). [DOI] [PubMed] [Google Scholar]
- 65.Xiang Y, Naro F, Zoudilova M, Jin SLC, et al. Phosphodiesterase 4D is required for β2 adrenoceptor subtype-specific signaling in cardiac myocytes. Proc. Natl. Acad. Sci. U. S. A 102, 909–914 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.West TM, Wang Q, Deng B, Zhang Y, et al. Phosphodiesterase 5 Associates with β2 adrenergic receptor to modulate cardiac gunction in Type 2 diabetic hearts. J. Am. Heart Assoc 8, 1–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fu Q, Kim S, Soto D, De Arcangelis V, et al. A long lasting β1 adrenergic receptor stimulation of cAMP/protein kinase A (PKA) signal in cardiac myocytes. J. Biol. Chem 289, 14771–14781 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Waagstein F, Hjalmarson A & Varnauskas E Effect of chronic β-adrenergic receptor blockade in congestive cardiomyopathy. Br. Heart J 37, 1022 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Swedberg K, Hjalmarson A, Waagstein F, W. I. Prolongation of survival in congestive cardiomyopathy by β-receptor blockade. Lancet 1374–1376 (1979). [DOI] [PubMed] [Google Scholar]
- 70.Gilbert EM, Olsen SL, Renlund DG & Bristow MR β-adrenergic receptor regulation and left ventricular function in idiopathic dilated cardiomyopathy. Am. J. Cardiol 71, (1993). [DOI] [PubMed] [Google Scholar]
- 71.Heilbrunn SM, Shah P, Bristow MR, Valantine HA, et al. Increased β-receptor density and improved hemodynamic response to catecholamine stimulation during long-term metoprolol therapy in heart failure from dilated cardiomyopathy. Circulation 79, 483–490 (1989). [DOI] [PubMed] [Google Scholar]