

Abstract
Incorporation of noncanonical amino acids (ncAAs) into proteins opens new opportunities in biotechnology and synthetic biology. Pyrrolysine (Pyl)-based ncAAs are some of the most predominantly used, but expression systems suffer from low yields. Here, we report a highly efficient cell-free protein synthesis (CFPS) platform for site-specific incorporation of Pyl-based ncAAs into proteins using amber suppression. This platform is based on cellular extracts derived from genomically recoded Escherichia coli lacking release factor 1 and enhanced through deletion of endonuclease A. To enable ncAA incorporation, orthogonal translation system (OTS) components (i.e., the orthogonal transfer RNA [tRNA] and orthogonal aminoacyl tRNA synthetase) were coexpressed in the source strain prior to lysis and the orthogonal tRNACUAPyl that decodes the amber codon was further enriched in the CFPS reaction via co-synthesis with the product. Using this platform, we demonstrate production of up to 442 ± 23 μg/mL modified superfolder green fluorescent protein (sfGFP) containing a single Pyl-based ncAA at high (>95%) suppression efficiency, as well as sfGFP variants harboring multiple, identical ncAAs. Our CFPS platform can be used for the synthesis of modified proteins containing multiple precisely positioned, genetically encoded Pyl-based ncAAs. We anticipate that it will facilitate more general use of CFPS in synthetic biology.
Keywords: cell-free protein synthesis, chemical biology, genetic code expansion, in vitro transcription and translation, noncanonical amino acids, synthetic biology
1 |. INTRODUCTION
Cell-free protein synthesis (CFPS) has emerged as a powerful and efficient technology platform for applied biotechnology.[1–4] In recent years, for example, CFPS systems have been applied to high-throughput protein production, enzyme screening, diagnostics, clinical scale production of therapeutics, genetic part and circuit characterization, glycoprotein synthesis, incorporation of noncanonical amino acids (ncAAs) into proteins for expanding the chemistry of life, and educational kits.[5–47] The driving forces behind the recent expansion of applications include advances in (i) extract optimization, (ii) source strain engineering, and (iii) the ability to activate cost-effective endogenous metabolism to fuel highly efficient CFPS.[3,21,26,48]
When compared to complementary in vivo protein production approaches, CFPS enjoys several key advantages. First, it provides an unprecedented ability to monitor, modify, and control reaction conditions by enabling easy substrate addition, product removal, and rapid sampling. For example, direct access to the reaction eliminates potential transport barriers interfering with the bioavailability of ncAAs. Second, CFPS systems are not affected by toxicity constraints that would be deleterious in living cells. Third, in vitro approaches offer rapid prototyping environments (i.e., hours to days) that are faster than in vivo standard testing settings that require time consuming cloning work (i.e., days to weeks).[3,6,49]
Given these advantages and recent technical improvements, CFPS is increasingly adapted for use in new application areas. In particular, the approach has been leveraged to advance efforts to expand the chemistry of life via ncAA incorporation into proteins.[24,46,50] Expansion of the amino acid repertoire with ncAAs unlocks otherwise inaccessible protein structures, functions, and sidechain chemistries, and has been used for production of antibody drug conjugates,[51,52] fluorescent probes for understanding biological systems,[53] more effective therapeutic proteins,[54] and phosphoproteins,[45] among others. The most established method to generate proteins containing ncAAs involves the use of engineered orthogonal transfer RNA (o-tRNA)/orthogonal aminoacyl-tRNA synthetase (o-aaRS) pairs sourced from phylogenetically distant organisms to repurpose the amber stop codon (TAG) as a coding channel for the incorporation of ncAAs.[55] These orthogonal translation system (OTS) components are evolved to be parallel to and independent from the host but operate in concert with the chassis organism’s native translation machinery to catalyze the cotranslational incorporation of ncAAs into nascent polypeptides in a sequence-defined manner. In Escherichia coli, amber suppression is limited by the activity of release factor 1 (RF1) which competes with loaded suppressor tRNAs at amber codons and catalyzes translational termination, often leading to the premature truncation of protein products at positions intended for ncAA incorporation using strains in which functional RF1 is intact.
A prominent, naturally occurring OTS is that for pyrrolysine (Pyl), commonly referred to as the 22nd amino acid. This amino acid is inserted into naturally occurring in-frame amber (TAG) stop codons in transcripts for methyltransferases of select bacteria,[56,57] and methanogenic archaea.[58] Notably, Pyl aminoacyl-tRNA synthetase (PylRS) and Pyl tRNA are orthogonal to the translation machinery in E. coli and other organisms,[59–61] enabling the incorporation of Pyl-based ncAAs into proteins in these hosts. Several studies in vivo have efficiently incorporated Pyl-based ncAAs into proteins and developed evolved Pyl OTS’.[62–65] CFPS systems for Pyl-based ncAAs have also been reported.[66]
Here, we expand upon existing work by developing and optimizing an efficient CFPS platform to produce proteins containing one or more Pyl-based ncAAs (Figure 1A). Specifically, we tested the incorporation of two ncAAs: (i) N6-(5-Norbornene-2-yloxycarbonyl)-l-lysine hydrochloride (hereafter referred to as pLysN) and (ii) N6-(propargyloxycarbonyl)-l-lysine hydrochloride (hereafter referred to as proCarb). Leveraging the open nature of the cell-free system, we first optimized the expression of modified proteins (i.e., those containing ncAAs) by adjusting the compositions of purified exogenous OTS components (e.g., tRNACUAPyl, PylRS, etc.). Next, we assessed the efficacy of enriching PylRS and tRNACUAPyl in the extracts via expression in the source strain. These optimizations were performed in CFPS systems derived from two different strains, one with and one without RF1. Compared to previous work using CFPS from RF1-deficient extracts,[66] our effort is unique in its use of a strain that lacks the nuclease gene endA, which stabilizes DNA concentration.[24] To our knowledge, the resulting CFPS platform synthesized the highest yields of modified proteins harboring single and multiple identical Pyl-based ncAAs yet reported, underscoring the importance of using modified extracts from genomically recoded organisms lacking RF1 for accurate and high-yielding ncAA incorporation.
FIGURE 1.
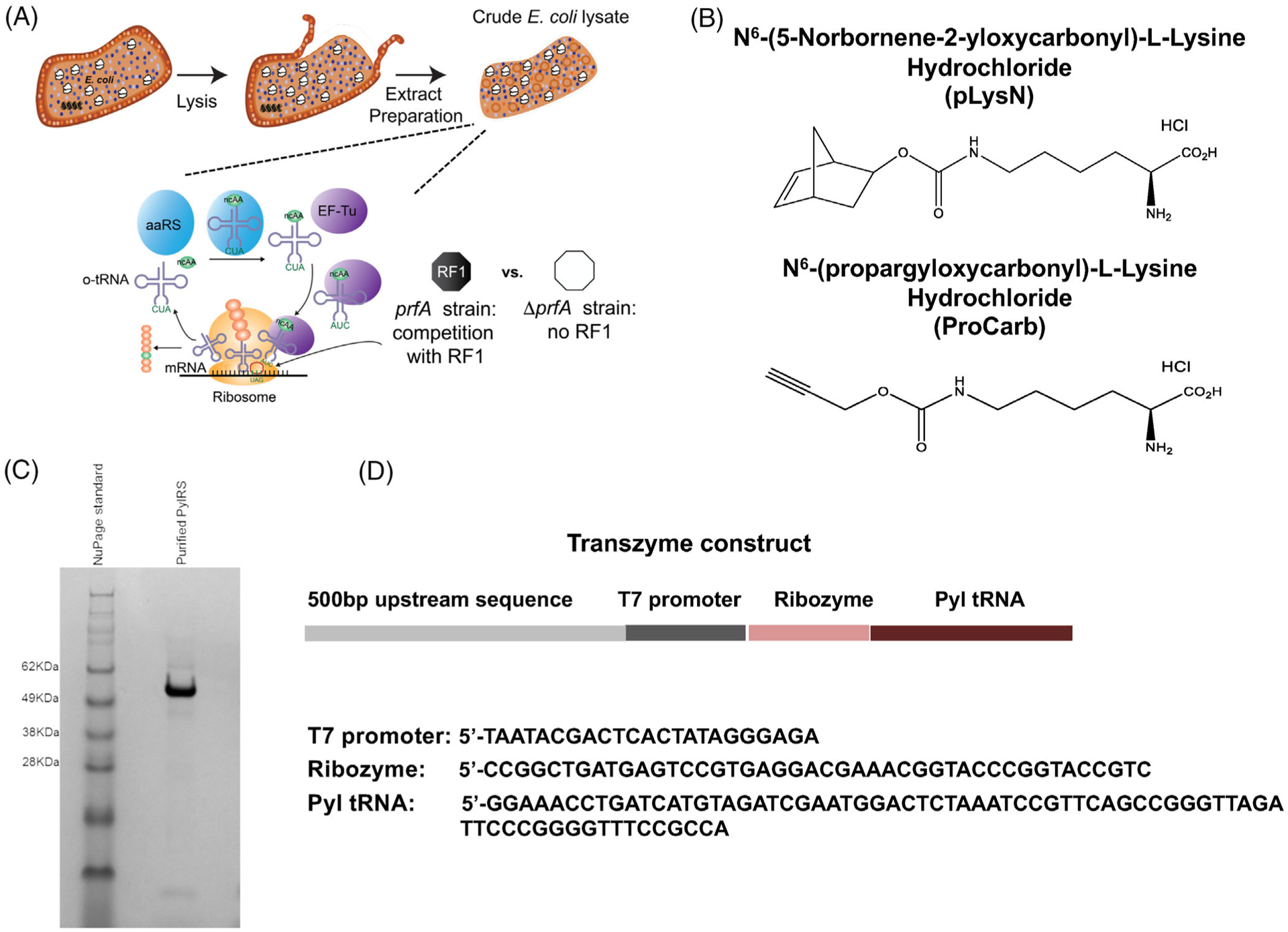
Development of an efficient cell-free platform for incorporation of pyrrolysine (Pyl)-based ncAAs into proteins. (A) Scheme showing the creation of a Pyl-based cell-free platform. Cell extracts containing cellular components required for transcription and translation of proteins are prepared from E. coli strains. DNA template encoding sfGFP containing single or multiple amber codon sites, Pyl tRNA, PylRS, Pyl-derived ncAA, T7 RNA polymerase, and other cofactors are added as necessary to activate the cell-free protein synthesis reaction. (B) Structures of the ncAAs used in this study are shown. (C) Purified PylRS was run on a 4%–12% NuPAGE SDS-polyacrylamide gel to assess for purity. The 51 kDa protein is >95% pure. Gel representative of n = 3 independent purifications. (D) The linear DNA template for the transzyme consisting of the T7 promoter, hammerhead Ribozyme, and Pyl tRNA is shown. Sequences of the individual components of the transzyme are indicated. ncAAs, noncanonical amino acids; sfGFP, superfolder green fluorescent protein
2 |. RESULTS
2.1 |. Expression of the Pyl aaRS and Pyl tRNA during extract preparation is important for protein production in the CFPS reaction
The goal of this work was to demonstrate high-level Pyl-based ncAA incorporation into proteins in a CFPS system. As a model, we tested the incorporation of pLysN and proCarb into superfolder green fluorescent protein (sfGFP) (Figure 1B). Creation of an efficient CFPS platform for incorporation of these ncAAs required supplementation of purified PylRS and a purified tRNACUAPyl construct into a suitable crude cell extract (Figure 1C, D). We first assessed the capacity for lysates derived from BL21 (DE3) supplemented with purified PylRS and tRNACUAPyl to incorporate pLysN into an in-frame amber stop codon in sfGFP at position 216 (sfGFPT216). Results indicated that this extract did not produce any measurable level of sfGFP as measured by fluorescence (Figure 2A). We hypothesized that this lack of full length sfGFP expression was due to the OTS being outcompeted by active RF1 in the lysates, and that this could be addressed by further enriching the OTS components in the reactions. To test this hypothesis, we expressed Pyl OTS components in the strain off of a plasmid during the exponential phase of cell growth.[67] The resulting extract, named BL21pEvol, was able to catalyze incorporation of pLysN into sfGFP in CFPS reactions as measured by fluorescence (Figure 2A).
FIGURE 2.
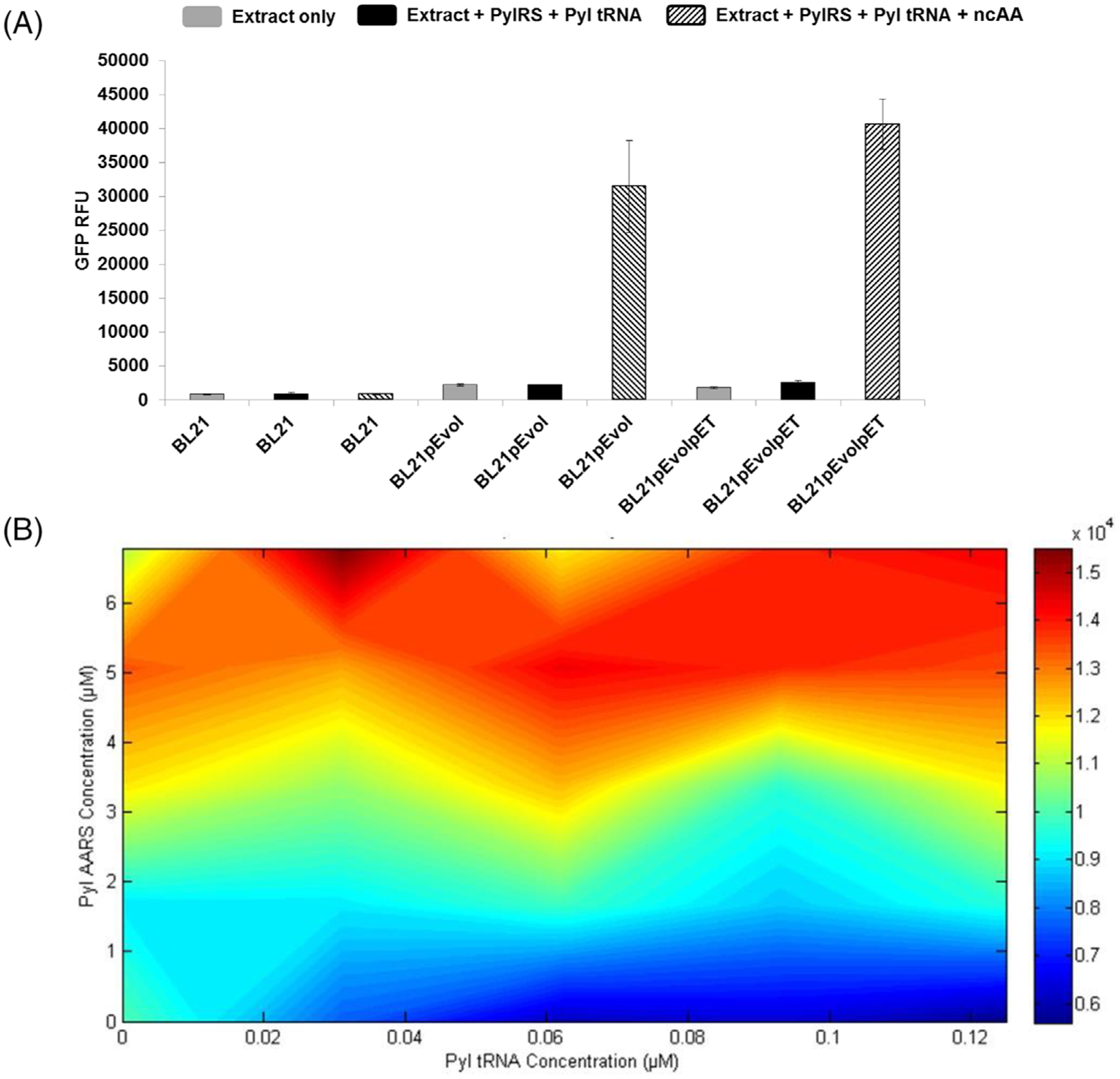
Expression of PylRS in cells before making the extract is important for amber suppression. All CFPS reactions were conducted with plasmid sfGFPT216 (amber position at 216) at 30°C for 20 h, and incorporated the Pyl-based ncAA pLysN. (A) sfGFP RFUs are graphed for CFPS reactions performed using lysates derived from the denoted strains in the presence and absence of ncAAs and supplemental OTS components as indicated. Three independent reactions (n = 3) were performed for each data point and one standard deviation is shown. (B) sfGFP yields (RFUs) of samples in BL21pEvol extract are plotted using MatLab software with increasing PylRS concentration on the y-axis and increasing tRNACUAPyl concentrations on the x-axis. Red color indicates the highest fluorescence values with blue indicating the lowest. Two independent reactions (n = 2) were performed for each data point. CFPS, cell-free protein synthesis; ncAAs, noncanonical amino acids; OTS, orthogonal translation system; RFU, relative fluorescence unit; sfGFP, superfolder green fluorescent protein
2.2 |. PylRS is limiting in CFPS reactions derived from BL21 pEvol extracts
We next set out to optimize the concentrations of supplemented PylRS and tRNACUAPyl in reactions performed using BL21pEvol extracts. Previous characterizations of PylRS have demonstrated up to a 10-fold reduction in binding affinity for non-Pyl substrates, and the enzyme’s low solubility is well-characterized.[68] Based on these observations, we hypothesized that a high concentration of PylRS would be required in CFPS reactions to overcome these limitations and increase ncAA incorporation. To test this, increasing concentrations of purified PylRS and tRNACUAPyl were added to CFPS reactions in various combinations, and the resulting yields of pLysN-containing protein (as measured via sfGFP fluorescence) were measured and plotted in MatLab in a two-point lattice (Figure 2B). As hypothesized, fluorescence values were observed to increase with increasing concentrations of PylRS but not with increasing tRNACUAPyl. These data supported our hypothesis that PylRS and not tRNACUAPyl was limiting in the reactions even at the highest concentration of PylRS evaluated. We were unable to concentrate the PylRS further due to solubility constraints, and therefore attempted to increase the concentration of PylRS in the extract by having the host strain further overexpress it during cell growth. To achieve this, BL21pEvol was transformed with a second plasmid encoding a single inducible copy of PylRS. As expected, when we attempted pLysN incorporation using the resulting extract (BL21pEvolpET) we observed a slight improvement over the BL21pEvol extract, suggesting that the additional PylRS facilitates incorporation of the ncAA (Figure 2A).
We set out to determine whether expressing PylRS during chassis strain growth is equivalent to or more or less effective than adding exogenous purified PylRS to reactions. To assess this, extracts were prepared from cultures with varying amounts of PylRS overexpression and utilized in CFPS both with and without supplementation with purified PylRS to incorporate pLysN into sfGFPT216. Results indicate that initially it is important to express the Pyl OTS components in the chassis strain, as adding exogenous PylRS alone is not sufficient to facilitate pLysN incorporation (Figures 2A, 3A). However, once the extract has a certain concentration of PylRS and tRNACUAPyl (as in the BL21pEvol extract), further addition of exogenous PylRS is approximately equivalent to expressing a second PylRS-encoding plasmid during chassis strain growth (as in the BL21pEvolpET extract). The system was further improved by supplementing BL21pEvolpET extract with additional purified exogenous PylRS to yield an optimized system (Figure 3A).
FIGURE 3.
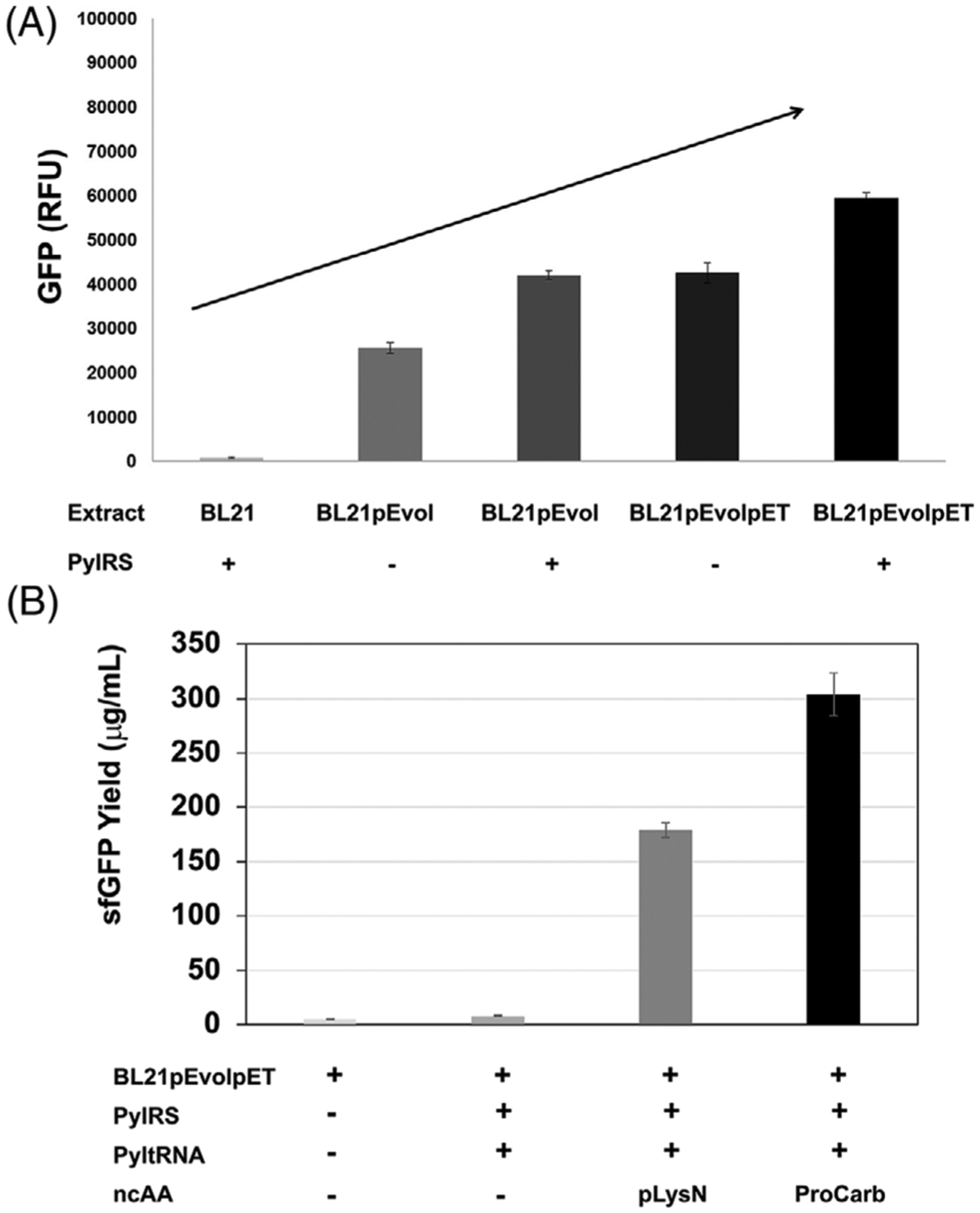
Additional PylRS expression in-strain further increases production of proteins with Pyl-ncAAs. (A) sfGFP produced from samples in the presence (+) and absence (−) of supplemental purified PylRS are compared. Strains used to prepare extracts are indicated and all samples contain pLysN. (B) sfGFP yields (in μg/mL) are compared for samples in the presence (+) and absence (−) of ncAAs pLysN and ProCarb in BL21pEvolpET extract. PylRS and tRNACUAPyl presence (+) and absence (−) in each sample are indicated. These data show the sfGFP produced from samples in which a single amber codon is suppressed (position T216). Three independent reactions (n = 3) were performed for each data point and one standard deviation is shown. ncAAs, noncanonical amino acids; sfGFP, superfolder green fluorescent protein
2.3 |. CFPS enables distinct incorporation of Pyl-based ncAAs at high levels
We next applied BL21pEVOLpET-derived lysates supplemented with tRNACUAPyl transzyme for the incorporation of the ncAA proCarb and compared its incorporation to that of pLysN. For each of these ncAAs, reactions were assembled directed to synthesize sfGFPT216 both in the presence and absence of the ncAA of interest (Figure 3B). Synthesis of 304 ± 17 μg/mL and 180 ± 9 μg/mL of modified sfGFP was observed when proCarb and pLysN were added to reactions, respectively. Next, we assembled reactions using all three BL21-derived extracts that were supplemented with radioactive 14C-Leucine and used autoradiography to visualize radiolabeled sfGFP produced with and without the addition of proCarb and pLysN. This analysis revealed only truncated sfGFP in the BL21 extract both with and without the ncAAs (Supplementary Figure S1A). This is expected as in the absence of Pyl OTS components this extract is unable to incorporate the ncAAs into the amber position at amino acid 216 and therefore translation stalls at this position yielding no full-length product. The other two extracts (BL21pEVOL and BL21pEVOLpET) do generate full length sfGFP upon addition of the ncAAs; however, most of the product is still truncated which is to be expected in an extract that still has active RF1 competing with ncAA incorporation at amber codons (Supplementary Figure S1A).
2.4 |. Genomically recoded strain lacking RF1 and endonuclease A circumvents problem of protein truncation
We next pursued a strategy to circumvent product truncation due to premature RF1-mediated translational termination. We reasoned that this could be achieved by preparing lysates from a chassis organism lacking the gene encoding RF1, as has been reported before.[66] One such organism is E. coli strain C321.ΔA, which has been genomically recoded to replace all 321 TAG amber codons with TAA codons enabling the complete knockout of the RF1-encoding gene prfA from the strain.[24,46,69,70] Previous works have demonstrated that lysates derived from this strain and its derivatives show a significantly increased ability to incorporate ncAAs in CFPS.[24,46] We therefore selected a derivative of C321.ΔA deficient in endonuclease A (C321.ΔA.ΔendA, henceforth known as rEcoli) and compared extracts derived from this strain to the BL21-derived extracts. Of note, extracts from C321.ΔA.ΔendA (i.e., rEcoli) produce more protein than those from C321.ΔA.[24]
We produced crude S30 extract from rEcoli cells carrying a plasmid encoding Pyl OTS components (strain rEcolipEvol). Experiments to determine if this extract was limited by PylRS or tRNACUAPyl revealed that addition of extra PylRS or Pyl tRNACUAPyl did not significantly improve the incorporation of ncAAs (Supplementary Figure S2). As with the BL21-derived extracts, we tested the amount of sfGFPT216X produced in the rEcoli extracts both with and without the addition of pLysN or proCarb. We observed synthesis of 349 ± 79 and 442 ± 22.8 μg/mL of sfGFPT216 with pLysN and proCarb, respectively (Figure 4A). As expected, these yields were higher than what was produced by the best performing BL21 extract (Figure 3). Autoradiography experiments to visualize protein showed a reduction in the amount of truncated protein and a corresponding increase in the amount of full-length sfGFP (Supplementary Figure S1B).
FIGURE 4.

rEcoli extract allows for high yielding biosynthesis of proteins containing Pyl-based ncAAs. (A) sfGFP produced from cell-free reactions in which a single amber codon is suppressed (position T216). The presence/absence of the listed OTS components is indicated for each condition. (B) sfGFP produced from cell-free reactions in which two (2TAG), three (3TAG), and five (5TAG) amber codons are suppressed. The presence/absence of the listed OTS components is indicated for each condition. All reactions were performed using lysates derived from strain rEcolipEvol. Three independent reactions (n = 3) were performed for each data point and one standard deviation is shown. ncAAs, noncanonical amino acids; sfGFP, superfolder green fluorescent protein
2.5 |. Cell-free rEcolipEvol extracts can incorporate multiple ncAAs
We next assessed if cell-free extracts from C321.ΔA.ΔendA had the ability to incorporate multiple, identical ncAAs into sfGFP. To test this, we used our rEcolipEvol lysate to synthesize sfGFP constructs featuring two, three, or five TAG codons in the presence of either pLysN or proCarb (Figure 4B). The system was able to synthesize ~82 and 111 μg/mL of sfGFP2TAG and ~46 and 99 μg/mL of sfGFP3TAG featuring pLysN or proCarb, respectively. sfGFP5TAG synthesis was not detected above background using either ncAA. These results were confirmed and quantified using top-down mass spectrometry (i.e., MS analysis of whole, intact proteins), which showed >95% ncAA incorporation at amber codons in the sfGFP variants containing 1, 2, and 3 TAGs (Figure 5, Supplementary Table S1).
FIGURE 5.

Top-down mass spectrometry demonstrates efficient incorporation of ncAAs into sfGFP. pLysN and ProCarb were incorporated into sfGFP using the indicated extracts. sfGFP variants containing (A) 1TAG (sfGFPT216), (B) 2TAG (sfGFP S2T216) or 3TAG (sfGFPS2N212T216) were synthesized by the various extracts as indicated. In each example, colored peaks correspond to the expected mass. The second peak under the red arrow in panel (A) corresponds to the mass of the modified protein retaining the initiator methionine residue. All other samples have this methionine cleaved off. Exact mass and mass shifts are indicated in Supplementary Table S1. Data representative of three independent reactions. ncAAs, noncanonical amino acids; sfGFP, superfolder green fluorescent protein
3 |. DISCUSSION
Here, we present the construction of a cell-free platform for site-specific genetically encoded incorporation of single or multiple Pyl-based ncAAs into proteins based on RF1 deleted extracts. Expression of the orthogonal PylRS and tRNACUAPyl in the cells prior to preparation of the extract was instrumental in creating a robust and efficient S30 extract that was then optimized by supplementation with exogenous purified components. Furthermore, use of extracts derived from the genomically recoded RF1-deficient E. coli strain C321.ΔA.ΔendA resulted in the ability to generate significantly larger quantities of full-length protein, as well as the ability to incorporate more than one ncAA into proteins, as compared to BL21-based extracts. Top-down mass spectrometry confirmed the high degree of ncAA incorporation and purity of the full-length samples.
Our final optimized CFPS system can produce up to ~440 or ~349 μg/mL of sfGFP featuring a single incorporation of proCarb or pLysN, respectively, as determined by active fluorescence (i.e., active protein). The described CFPS system may provide several advantages over in vivo approaches, namely, improved bioavailability of the Pyl-derived ncAAs and increased yields. Reports in the literature for incorporation of ncAAs using the Pyl OTS in vivo suggest yields between ~1 and 40 μg/mL.[63,71] As a result, we anticipate that the cell-free gene expression platform developed here will be useful for synthetic and chemical biology.
4 |. EXPERIMENTAL SECTION
4.1 |. PylRS purification
An overnight culture of BL21(DE3) transformed with pET21aMmPyl was inoculated into 1 L of LB (1:100 dilution) and grown at 250 rpm and 37°C until OD 0.1 (600 nm). At this point the cells were moved to 42°C for heat shock treatment and grown at 250 rpm until OD 0.5 (600 nm). Protein production was induced by adding 0.5 mm isopropyl-β-d-thiogalactopyranoside (Sigma, St. Louis, MO) and cells were moved to a 33°C incubator and grown at 250 rpm for 2.5 h. Cells were harvested at 6000× g for 15 min at 4°C, washed with 1X PBS buffer, and stored at −80°C. The frozen cell pellet was thawed in lysis buffer (100 mm HEPES pH 7.2, 500 mm NaCl, 5 mm BME) and lysed using a homogenizer at 20,000–25,000 psi. After clarification by centrifuging at 12,000 × g at 4°C for 15 min, imidazole was added to the supernatant at a final concentration of 10 mm and it was loaded onto 2 mL of Ni-NTA agarose slurry (Qiagen) that had been washed twice with 1X PBS. The beads were rotated for 1 h at room temp and spun down at 5000 × g at 4°C for 4 min. The beads were washed twice with 10 mL wash buffer (100 mm Hepes pH 7.2, 500 mm NaCl, 50 mm imidazole) by slow rotation at room temp for 25 min followed by pelleting the beads as before. The His-tagged PylRS was eluted in 1 mL of elution buffer (100 mm Hepes pH 7.2, 500 mm NaCl, 750 mm imidazole) with rotation for 20 min. The beads were spun down and the eluate was collected and dialyzed against an excess of dialysis buffer (100 mm Hepes pH 7.2, 10 mm MgCl2, 10 mm KCl) overnight at 4°C to remove imidazole. Buffer was exchanged once in the middle of this dialysis. Protein purity was confirmed by 4%–12% NuPAGE SDS-PAGE (Life Technologies, Grand Island, NY). Concentrations were determined by Quick-Start Bradford protein assay kit (Bio-Rad, Hercules, CA) and the protein was stored at −80°C.
4.2 |. Cell extract preparation
The engineering of E. coli strain C321.ΔA.ΔendA was described previously.[24] All E. coli cells were grown in 2xYTPG media (Tryptone 16 g/L, Yeast extract 10 g/L, NaCl 5 g/L, K2HPO4 7 g/L KH2PO4 3 g/L, glucose 18 g/L) at 34°C. To enable in-cell expression of Pyl OTS components, strains were transformed with plasmid pEVOL-Pyl.[67] To drive higher overexpression levels of PylRS, some strains were additionally transformed with plasmid pET21aMmPyl. Cells harboring pEVOL-Pyl were induced at OD 0.4 (600 nm) with 0.1% arabinose. Cells harboring pET21aMmPyl were additionally induced with 0.5 mm isopropyl-β-d-thiogalactopyranoside. After induction, cells were grown further until OD 3.0 (600 nm). Cells were pelleted by centrifuging for 15 min at 6000 × g at 4°C, washed twice with cold S30 buffer (10 mm trisacetate pH 8.2, 14 mm magnesium acetate, 60 mm potassium acetate, 1 mm dithiothreitol),[72] and stored at −80°C. Thawed cells were suspended in 1 mL of S30 buffer per gram cells and lysed using a sonicator (Q-Sonica Model CL-18) using 50% amplitude and three pulses (45 s on and 59 s off) on ice. Three microliters of DTT (1 m) was added per mL of sample and the lysate was clarified by spinning at 12,000 × g for 10 min at 4°C. Clarified supernatant was transferred to a fresh tube and incubated for 80 min at 120 rpm at 37°C to optimize the extract activity, after which it was again centrifuged for 15 min at 15,000 × g at 4°C. The final clarified supernatant was flash-frozen using liquid nitrogen and stored at −80°C until use. Total protein concentration of the extracts was approximately 55 mg/mL, as measured by Quick-Start Bradford protein assay kits (Bio-Rad, Hercules, CA).
4.3 |. CFPS
CFPS reactions were performed as described previously[73] using a modified PANOx-SP system.[21] Briefly, 15 μL of CFPS reaction in a 1.5 mL microcentrifuge tube was prepared by mixing the following components: 1.2 mm ATP; 0.85 mm each of GTP, UTP, and CTP; 34.0 μg/mL folinic acid; 170.0 μg/mL of E. coli tRNA mixture; 100 μg/mL T7 RNA polymerase; 2 mm each of 20 standard amino acids; 0.33 mm nicotinamide adenine dinucleotide (NAD); 0.27 mm coenzyme-A (CoA); 1.5 mm spermidine; 1 mm putrescine; 4 mm sodium oxalate; 130 mm potassium glutamate; 10 mm ammonium glutamate; 12 mm magnesium glutamate; 33 mm phosphoenolpyruvate (PEP); 200 ng of plasmid DNA, 6.8 μm PylRS(where indicated); 0.045 μm tRNACUAPyl (where indicated); 2 mm pLysN or proCarb (where indicated); and 27% v/v of cell extract. Assembled reactions were incubated for 20 h at 30°C.
4.4 |. Quantification of the synthesized sfGFP
Total protein yields were quantified by determining radioactive 14C-Leucine incorporation using trichloroacetic acid (TCA) precipitation onto paper strips.[74] Radioactivity of TCA-precipitated samples was measured using liquid scintillation counting (MicroBeta2, PerkinElmer, Waltham, MA). Active sfGFP protein yields were quantified by measuring fluorescence of the product and converting it to concentration (μg/mL) according to a standard curve.[73] For quantification via fluorescence, 2 μL of CFPS reaction was added in the middle of the flat bottom of 96 well half area black plates along with 48 μL nuclease-free water (Costar 3694; Corning Incorporated, Corning, NY). sfGFP was excited at 485 nm while measuring emission at 528 nm with a 510 nm cut-off filter. The fluorescence of sfGFP was converted to concentration (μg/mL) according to a standard curve described previously.[73]
4.5 |. Autoradiography analysis
Radioactive 14C-Leucine was added in CFPS reactions. Five microliters of each reaction was heated at 90°C with 10 mm DTT and LDS sample loading buffer from Novex (Life Technologies) and loaded onto a 4%–12% NuPAGE SDS-PAGE gel. The gel was stained using Simply Blue Safe Stain (Invitrogen), destained in water, and soaked in Gel Drying Solution (Bio-Rad, Hercules, CA) for 30 min, fixed with cello-phane films, dried without applying heat overnight in GelAir Dryer (Bio-Rad, Hercules, CA), and exposed for 48 h on a Storage Phosphor Screen (GE Healthcare Biosciences, Pittsburgh, PA). The autoradiogram was scanned using a Storm Imager (GE Healthcare Biosciences, Pittsburgh, PA) and analyzed using Quantity One software (Bio-Rad, Hercules, CA).
4.6 |. Preparation of sfGFP proteins in vitro for mass spectrometry
Multiple CFPS reactions were set up (up to 20 reactions per sample) as described above. After CFPS samples were combined and the product was purified using 0.2 mL gravity-flow Strep–Tactin Sepharose minicolumns (IBA GmbH, Gottingen, Germany). Eluted protein samples were concentrated using Microcon centrifugal filter columns YM-10 (Millipore, Billerica, MA). Concentrations were determined by Quick-Start Bradford protein assay kit (Bio-Rad, Hercules, CA). The samples were analyzed by top-down mass spectrometry as detailed in the following section.
4.7 |. Mass spectrometry
The purified protein was analyzed by nano-capillary LC-MS using a 100 mm × 75 μm ID PLRP-S column in-line with an Orbitrap Elite (ThermoFisher, Waltham, MA). All MS methods included the following events: (i) FT scan, m/z 400–2000, 120,000 resolving power and (ii) data-dependent MS/MS on the top 2 peaks in each spectrum from scan event 1 using higher-energy collisional dissociation (HCD) with normalized collision energy of 25, isolation width 50 m/z, and detection of ions with resolving power of 60,000. All data were analyzed using QualBrowser, part of the Xcalibur software packaged with the ThermoFisher Orbitrap Elite.
4.8 |. Preparation of ncAAs
N6-(5-Norbornene-2-yloxycarbonyl)-l-lysine hydrochloride (pLysN) and N6-(propargyloxycarbonyl)-l-lysine hydrochloride (proCarb) were synthesized by the Center for Molecular Innovation & Drug Discovery. The methods are detailed in the Supplementary Information section. Both ncAAs were soluble in nuclease-free water. Stock solutions of 500 mm were made for use in experiments.
4.9 |. Plasmids and cloning
Plasmid pEvolPylRSWT was a kind gift from Dr. E. Lemke.[75] Plasmids sfGFPWT and sfGFPT216 were a kind gift from Dr. B. Bundy.[76] Plasmid sfGFP-5TAG (sfGFP D36xK101xE132xD190x E213x) was described previously.[73] Plasmid sfGFP-2TAG (sfGFPS2xT216x) was created by introducing an amber codon at the S2 site in sfGFPT216x and sfGFP-3TAG (sfGFPS2xT216x) was created by introducing an amber codon at the S2 site in sfGFPN212xT216x that has been previously described.[73] The amber codon was introduced at S2 by performing PCR using primers S2-f forward primer and S2-r reverse primer (Supplementary Table S2) with Phusion High-Fidelity DNA polymerase (New England Biolabs, Ipswich, MA) at 98°C for 30 s, with 30 cycles of 98°C for 10 s, 49°C for 30 s, and 72°C for 3 min, and a final extension of 72°C for 5 min followed by gel extraction of the band and ligation. Plasmid pET21aMmPyl was created as follows. The wildtype PylRS DNA sequence was amplified using primers AR108 and AR109 (Supplementary Table S2) from pEvolPylRSWT and the PCR product was digested with Not1 and Nde1. This digested product was ligated with vector pET21a (EMD Millipore) that had also been digested with Not1 and Nde1 to get the PylRS gene upstream of a fused C-terminal His tag. Sequences for the plasmids described here can be found in the Supplementary Information (Supplementary Sequences).
4.10 |. Construction of linear DNA templates for expressing Pyl tRNA
A plasmid pY71-GB1f was created to contain the transzyme sequence[70] composed of the DNA sequence of the T7 promoter followed by the hammerhead Ribozyme and the Pyl tRNA. Briefly, gBlock GB1 (Supplementary Table S2) was obtained (Integrated DNA technologies, Coralville, IA) and digested with BglII and Sal1 and ligated into pY71 plasmid using BglII and SalI restriction sites. The final sequence for plasmid pY71-GB1f can be found in the Supplementary Information (Supplementary Sequences). The linear DNA template was created by amplifying the transzyme sequence as well as 500 basepairs of upstream sequence in the plasmid using primers T7500up forward primer (Supplementary Table S2) and AR045 reverse primer. The PCR was performed using Phusion High-Fidelity DNA polymerase (New England Biolabs, Ipswich, MA) at 98°C for 30 s, with 30 cycles of 98°C for 40 s, 58°C for 40 s, and 72°C for 1 min, and a final extension of 72°C for 5 min. The PCR was purified using the E.Z.N.A. Cycle Pure Kit (Omega Biotech) and quantified using a nanodrop 2000c (Thermo Scientific).
Supplementary Material
NOVELTY STATEMENT.
Incorporation of ncAAs into proteins opens new opportunities in chemical and synthetic biology, as well as biotechnology. Pyrrolysine-based ncAAs are some of the most predominantly used, but expression systems suffer from low yields. Here, we report a robust and efficient cell-free protein synthesis platform for site-specific incorporation of pyrrolysine-based ncAAs into proteins using amber suppression. This platform is based on cellular extracts derived from genomically recoded Escherichia coli lacking release factor 1 (RF1) and deficient in endonuclease A. We anticipate that the platform will facilitate more general use of cell-free protein synthesis in systems and synthetic biology.
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation (NSF) (MCB-1716766), the Army Research Office (W911NF-20-1-0195, W911NF-18-1-0200), Army Contracting Command (W52P1J-21-9-3023), and the David and Lucile Packard Foundation (2011-37152). This research was also carried out in collaboration with the National Resource for Translational and Developmental Proteomics under Grant P41 GM108569 from the National Institute of General Medical Sciences, National Institutes of Health and supported by the Sherman Fairchild Foundation. The ncAA synthesis was completed by Christopher Holmquist at ChemCore in the Center for Molecular Innovation and Drug Discovery which is supported in part by the Chicago Biomedical Consortium with support from The Searle Funds at The Chicago Community Trust. The authors would like to thank Dr. Farren Isaacs for the kind gift of the genomically recoded E. coli strain; Dr. Javin Oza for cloning of the sfGFP2TAG plasmid; Dr. Seok Hoon Hong for cloning the sfGFP5TAG plasmid; Alaksh Choudhury for help with Matlab coding; Mark Anderson for assistance with Chemdraw; and Dr. E. Lemke for plasmid pEvolPylRSWT.
Footnotes
CONFLICT OF INTEREST
Michael C Jewett has a financial interest in Pearl Bio and his interests are reviewed and managed by Northwestern University in accordance with their conflict-of-interest policies. All other authors declare no competing interests.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
Data available upon request from authors.
REFERENCES
- 1.Hammerling MJ, Krüger A, & Jewett MC (2020). Strategies for in vitro engineering of the translation machinery. Nucleic Acids Research, 48, 1068–1083. 10.1093/nar/gkz1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bogart JW, Cabezas MD, Vögeli B, Wong DA, Karim AS, & Jewett MC (2021). Cell-free exploration of the natural product chemical space. ChemBioChem, 22, 84–91. 10.1002/cbic.202000452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carlson ED, Gan R, Hodgman CE, & Jewett MC (2012). Cell-free protein synthesis: Applications come of age. Biotechnology Advances, 30, 1185–1194. 10.1016/j.biotechadv.2011.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silverman AD, Karim AS, & Jewett MC (2020). Cell-free gene expression: an expanded repertoire of applications. Nature Reviews Genetics, 21, 151–170. 10.1038/s41576-019-0186-3 [DOI] [PubMed] [Google Scholar]
- 5.Sun ZZ, Hayes CA, Shin J, Caschera F, Murray RM, & Noireaux V (2013). Protocols for Implementing an Escherichia coli based TX-TL cell-free expression system for synthetic biology. Journal of Visualized Experiments, e50762. 10.3791/50762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hodgman CE, & Jewett MC (2012). Cell-free synthetic biology: thinking outside the cell. Metabolic Engineering, 14, 261–269. 10.1016/j.ymben.2011.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karim AS, & Jewett MC (2016). A cell-free framework for rapid biosynthetic pathway prototyping and enzyme discovery. Metabolic Engineering, 36, 116–126. 10.1016/j.ymben.2016.03.002 [DOI] [PubMed] [Google Scholar]
- 8.Marshall R, Maxwell CS, Collins SP, Jacobsen T, Luo ML, Begemann MB, Gray BN, January E, Singer A, He Y, Beisel CL, & Noireaux V (2018). Rapid and scalable characterization of CRISPR technologies using an E. coli cell-free transcription-translation system. Molecular Cell, 69, 146–157.e3. 10.1016/j.molcel.2017.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang A, Nguyen PQ, Stark JC, Takahashi MK, Donghia N, Ferrante T, Dy AJ, Hsu KJ, Dubner RS, Pardee K, Jewett MC, & Collins JJ (2018). BioBits™ explorer: A modular synthetic biology education kit. Science Advances, 4, eaat5105. 10.1126/sciadv.aat5105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rasor BJ, Vögeli B, Landwehr GM, Bogart JW, Karim AS, & Jewett MC (2021). Toward sustainable, cell-free biomanufacturing. Current Opinion in Biotechnology, 69, 136–144. 10.1016/j.copbio.2020.12.012 [DOI] [PubMed] [Google Scholar]
- 11.Lee J, Schwieter KE, Watkins AM, Kim DS, Yu H, Schwarz KJ, Lim J, Coronado J, Byrom M, Anslyn EV, Ellington AD, Moore JS, & Jewett MC (2019). Expanding the limits of the second genetic code with ribozymes. Nature Communications, 10, 5097. 10.1038/s41467-019-12916-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hammerling MJ, Fritz BR, Yoesep DJ, & Kim DS, Carlson ED, & Jewett MC (2020). In vitro ribosome synthesis and evolution through ribosome display. Nature Communications, 11, 1108. 10.1038/s41467-020-14705-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silverman AD, Akova U, Alam KK, & Jewett MC, Lucks JB (2020). Design and optimization of a cell-free atrazine biosensor. ACS Synthetic Biology, 9, 671–677. 10.1021/acssynbio.9b00388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee J, Schwarz KJ, Kim Do S., Moore JS, & Jewett MC (2020). Ribosome-mediated polymerization of long chain carbon and cyclic amino acids into peptides in vitro. Nature Communications, 11, 4304. 10.1038/s41467-020-18001-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karim AS, Dudley QM, Juminaga A, Yuan Y, Crowe SA, Heggestad JT, Garg S, Abdalla T, Grubbe WS, Rasor BJ, Coar DN, Torculas M, Krein M, Liew F, Quattlebaum A, Jensen RO, Stuart JA, Simpson SD, Köpke M, & Jewett MC (2020). In vitro prototyping and rapid optimization of biosynthetic enzymes for cell design. Nature Chemical Biology, 16, 912–919. 10.1038/s41589-020-0559-0 [DOI] [PubMed] [Google Scholar]
- 16.Dudley QM, Karim AS, Nash CJ, & Jewett MC (2020). In vitro prototyping of limonene biosynthesis using cell-free protein synthesis. Metabolic Engineering, 61, 251–260. 10.1016/j.ymben.2020.05.006 [DOI] [PubMed] [Google Scholar]
- 17.Hershewe J, Kightlinger W, & Jewett MC (2020). Cell-free systems for accelerating glycoprotein expression and biomanufacturing. Journal of Industrial Microbiology and Biotechnology, 47, 977–991. 10.1007/s10295-020-02321-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stark JC, Jaroentomeechai T, Moeller TD, Hershewe JM, Warfel KF, Moricz BS, Martini AM, Dubner RS, Hsu KJ, Stevenson TC, Jones BD, Delisa MP, & Jewett MC (2021). On-demand biomanufacturing of protective conjugate vaccines. Science Advances, 7, eabe9444. 10.1126/sciadv.abe9444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin RW, Majewska NI, Chen CX, Albanetti TE, Jimenez RBC, Schmelzer AE, Jewett MC, & Roy V (2017). Development of a CHO-based cell-free platform for synthesis of active monoclonal anti-bodies. ACS Synthetic Biology, 6, 1370–1379. 10.1021/acssynbio.7b00001 [DOI] [PubMed] [Google Scholar]
- 20.Silverman AD, Kelley-Loughnane N, Lucks JB, & Jewett MC (2019). Deconstructing cell-free extract preparation for in vitro activation of transcriptional genetic circuitry. ACS Synthetic Biology, 8, 403–414. 10.1021/acssynbio.8b00430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jewett MC, & Swartz JR (2004). Mimicking the Escherichia coli cytoplasmic environment activates long-lived and efficient cell-free protein synthesis. Biotechnology and Bioengineering, 86, 19–26. 10.1002/bit.20026 [DOI] [PubMed] [Google Scholar]
- 22.Bujara M, Schümperli M, Pellaux R, Heinemann M, & Panke S (2011). Optimization of a blueprint for in vitro glycolysis by metabolic real-time analysis. Nature Chemical Biology, 7, 271–277. 10.1038/nchembio.541 [DOI] [PubMed] [Google Scholar]
- 23.Jaroentomeechai T, Stark JC, Natarajan A, Glasscock CJ, Yates LE, Hsu KJ, Mrksich M, Jewett MC, & Delisa MP (2018). Single-pot glycoprotein biosynthesis using a cell-free transcription-translation system enriched with glycosylation machinery. Nature Communications, 9, 2686. 10.1038/s41467-018-05110-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin RW, Des Soye BJ, Kwon Y-C, Kay J, Davis RG, Thomas PM, Majewska NI, Chen CX, Marcum RD, Weiss MG, Stoddart AE, Amiram M, Ranji Charna AK, Patel JR, Isaacs FJ, Kelleher NL, Hong SH, & Jewett MC (2018). Cell-free protein synthesis from genomically recoded bacteria enables multisite incorporation of noncanonical amino acids. Nature Communications, 9, 1203. 10.1038/s41467-018-03469-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pardee K, Slomovic S, Nguyen PQ, Lee JW, Donghia N, Burrill D, Ferrante T, Mcsorley FR, Furuta Y, Vernet A, Lewandowski M, Boddy CN, Joshi NS, & Collins JJ (2016). Portable, on-demand biomolecular manufacturing. Cell, 167, 248–259.e12. 10.1016/j.cell.2016.09.013 [DOI] [PubMed] [Google Scholar]
- 26.Zawada JF, Yin G, Steiner AR, Yang J, Naresh A, Roy SM, Gold DS, Heinsohn HG, & Murray CJ (2011). Microscale to manufacturing scale-up of cell-free cytokine production – a new approach for shortening protein production development timelines. Biotechnology and Bioengineering, 108, 1570–1578. 10.1002/bit.23103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mcnerney MP, Zhang Y, Steppe P, Silverman AD, Jewett MC, & Styczynski MP (2019). Point-of-care biomarker quantification enabled by sample-specific calibration. Science Advances, 5, eaax4473. 10.1126/sciadv.aax4473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takahashi MK, Tan X, Dy AJ, Braff D, Akana RT, Furuta Y, Donghia N, Ananthakrishnan A, & Collins JJ (2018). A low-cost paper-based synthetic biology platform for analyzing gut microbiota and host biomarkers. Nature Communications, 9, 3347. 10.1038/s41467-018-05864-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pardee K, Green AA, Takahashi MK, Braff D, Lambert G, Lee JW, Ferrante T, Ma D, Donghia N, Fan M, Daringer NM, Bosch I, Dudley DM, O’connor DH, Gehrke L, & Collins JJ (2016). Rapid, rapid, low-cost detection of Zika virus using programmable biomolecular components. Cell, 165, 1255–1266. 10.1016/j.cell.2016.04.059 [DOI] [PubMed] [Google Scholar]
- 30.Verosloff M, Chappell J, Perry KL, Thompson JR, & Lucks JB (2019). PLANT-Dx: A molecular diagnostic for point-of-use detection of plant pathogens. ACS Synthetic Biology, 8, 902–905. 10.1021/acssynbio.8b00526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pardee K, Green AA, Ferrante T, Cameron DE, Daleykeyser A, Yin P, & Collins JJ (2014). Paper-based synthetic gene networks. Cell, 159, 940–954. 10.1016/j.cell.2014.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stark JC, Huang A, Hsu KJ, Dubner RS, Forbrook J, Marshalla S, Rodriguez F, Washington M, Rybnicky GA, Nguyen PQ, Hasselbacher B, Jabri R, Kamran R, Koralewski V, Wightkin W, Martinez T, & Jewett MC (2019). BioBits Health: Classroom activities exploring engineering, biology, and human health with fluorescent readouts. ACS Synthetic Biology, 8, 1001–1009. 10.1021/acssynbio.8b00381 [DOI] [PubMed] [Google Scholar]
- 33.Calhoun KA, & Swartz JR (2005). An economical method for cell-free protein synthesis using glucose and nucleoside monophosphates. Biotechnology Progress, 21, 1146–1153. 10.1021/bp050052y [DOI] [PubMed] [Google Scholar]
- 34.Jewett MC, Calhoun KA, Voloshin A, Wuu JJ, & Swartz JR (2008). An integrated cell-free metabolic platform for protein production and synthetic biology. Molecular Systems Biology, 4, 220. 10.1038/msb.2008.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bundy BC, Franciszkowicz MJ, & Swartz JR (2008). Escherichia coli-based cell-free synthesis of virus-like particles. Biotechnology and Bioengineering, 100, 28–37. 10.1002/bit.21716 [DOI] [PubMed] [Google Scholar]
- 36.Thoring L, Dondapati SK, Stech M, Wüstenhagen DA, & Kubick S (2017). High-yield production of -œdifficult-to-express-proteins in a continuous exchange cell-free system based on CHO cell lysates. Scientific Reports, 7, 11710. 10.1038/s41598-017-12188-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelwick R, Webb AJ, Macdonald JT, & Freemont PS (2016). Development of a Bacillus subtilis cell-free transcription-translation system for prototyping regulatory elements. Metabolic Engineering, 38, 370–381. 10.1016/j.ymben.2016.09.008 [DOI] [PubMed] [Google Scholar]
- 38.Kwon YC, & Jewett MC (2015). High-throughput preparation methods of crude extract for robust cell-free protein synthesis. Scientific Reports, 5, 8663. 10.1038/srep08663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moore SJ, et al. (2018). Rapid acquisition and model-based analysis of cell-free transcription – translation reactions from nonmodel bacteria. Proceedings of the National Academy of Sciences of the United States of America, 115, E4340–E4349. 10.1073/pnas.1715806115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chappell J, Jensen K, & Freemont PS (2013). Validation of an entirely in vitro approach for rapid prototyping of DNA regulatory elements for synthetic biology. Nucleic Acids Research, 41, 3471–3481. 10.1093/nar/gkt052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun ZZ, Yeung E, Hayes CA, Noireaux V, & Murray RM (2014). Linear DNA for rapid prototyping of synthetic biological circuits in an Escherichia coli based TX-TL cell-free system. ACS Synthetic Biology, 3, 387–397. 10.1021/sb400131a [DOI] [PubMed] [Google Scholar]
- 42.Garamella J, Marshall R, Rustad M, & Noireaux V (2016). The all E. coli TX-TL Toolbox 2.0: A platform for cell-free synthetic biology. ACS Synthetic Biology, 5, 344–355. 10.1021/acssynbio.5b00296 [DOI] [PubMed] [Google Scholar]
- 43.Takahashi MK, Chappell J, Hayes CA, Sun ZZ, Kim J, Singhal V, Spring KJ, Al-Khabouri S, Fall CP, Noireaux V, Murray RM, & Lucks JB (2015). Rapidly characterizing the fast dynamics of RNA genetic circuitry with cell-free transcription – translation (TX-TL) systems. ACS Synthetic Biology, 4, 503–515. 10.1021/sb400206c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kightlinger W, Lin L, Rosztoczy M, Li W, Delisa MP, Mrksich M, & Jewett MC (2018). Design of glycosylation sites by rapid synthesis and analysis of glycosyltransferases. Nature Chemical Biology, 14, 627–635. 10.1038/s41589-018-0051-2 [DOI] [PubMed] [Google Scholar]
- 45.Oza JP, Aerni HR, Pirman NL, Barber KW, Ter Haar CM, Rogulina S, Amrofell MB, Isaacs FJ, Rinehart J, & Jewett MC (2015). Robust production of recombinant phosphoproteins using cell-free protein synthesis. Nature Communications, 6, 8168. 10.1038/ncomms9168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Des Soye BJ, Gerbasi VR, Thomas PM, Kelleher NL, & Jewett MC (2019). A highly productive, one-pot cell-free protein synthesis platform based on genomically recoded Escherichia coli. Cell Chemical Biology, 26, 1743–1754.e9. 10.1016/j.chembiol.2019.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Des Soye BJ, Davidson SR, Weinstock MT, Gibson DG, & Jewett MC (2018). Establishing a high-yielding cell-free protein synthesis platform derived from Vibrio natriegens. ACS Synthetic Biology, 7, 2245–2255. 10.1021/acssynbio.8b00252 [DOI] [PubMed] [Google Scholar]
- 48.Jewett MC, & Swartz JR (2004). Substrate replenishment extends protein synthesis with an in vitro translation system designed to mimic the cytoplasm. Biotechnology and Bioengineering, 87, 465–471. 10.1002/bit.20139 [DOI] [PubMed] [Google Scholar]
- 49.Ranji A, Wu JC, Bundy BC, & Jewett MC (2013). Transforming synthetic biology with cell-free systems. Synthetic Biology: Tools and Applications, 277–301. doi: 10.1016/B978-0-12-394430-6.00015-7 [DOI] [Google Scholar]
- 50.Des Soye BJ, Patel JR, Isaacs FJ, & Jewett MC (2015). Repurposing the translation apparatus for synthetic biology. Current Opinion in Chemical Biology, 28, 83–90. 10.1016/j.cbpa.2015.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Axup JY, Bajjuri KM, Ritland M, Hutchins BM, Kim CH, Kazane SA, Halder R, Forsyth JS, Santidrian AF, Stafin K, Lu Y, Tran H, Seller AJ, Biroc SL, Szydlik A, Pinkstaff JK, Tian F, Sinha SC, Felding-Habermann B, … Schultz PG (2012). Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proceedings of the National Academy of Sciences of the United States of America, 109, 16101–16106. 10.1073/pnas.1211023109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim CH, Axup JY, Lawson BR, Yun H, Tardif V, Choi SH, Zhou Q, Dubrovska A, Biroc SL, Marsden R, Pinstaff J, Smider VV, & Schultz PG (2013). Bispecific small molecule-“antibody conjugate targeting prostate cancer. Proceedings of the National Academy of Sciences of the United States of America, 110, 17796–17801. 10.1073/pnas.1316026110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chatterjee A, Guo J, Lee HS, & Schultz PG (2013). A genetically encoded fluorescent probe in mammalian cells. Journal of the American Chemical Society, 135, 12540–12543. 10.1021/ja4059553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cho H, Daniel T, Buechler YJ, Litzinger DC, Maio Z, Putnam A-MH, Kraynov VS, Sim BC, Bussell S, Javahishvili T, Kaphle S, Viramontes G, Ong M, Chu S, Gc B, Lieu R, Knudsen N, Castiglioni P, Norman TC, … Kimmel BE (2011). Optimized clinical performance of growth hormone with an expanded genetic code. Proceedings of the National Academy of Sciences of the United States of America, 108, 9060–9065. 10.1073/pnas.1100387108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang L, Brock A, Herberich B, & Schultz PG (2001). Expanding the genetic code of Escherichia coli. Science, 292, 498–500. 10.1126/science.1060077 [DOI] [PubMed] [Google Scholar]
- 56.Herring S, Ambrogelly A, Polycarpo CR, & Soll D (2007). Recognition of pyrrolysine tRNA by the Desulfitobacterium hafniense pyrrolysyl-tRNA synthetase. Nucleic Acids Research, 35, 1270–1278. 10.1093/nar/gkl1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y, & Gladyshev VN (2007). High content of proteins containing 21st and 22nd amino acids, selenocysteine and pyrrolysine, in a symbiotic deltaproteobacterium of gutless worm Olavius algarvensis. Nucleic Acids Research, 35, 4952–4963. 10.1093/nar/gkm514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.James CM, Ferguson TK, Leykam JF, & Krzycki JA (2001). The amber codon in the gene encoding the monomethylamine methyltransferase isolated from Methanosarcina barkeri is translated as a sense codon. The Journal of Biological Chemistry, 276, 34252–34258. 10.1074/jbc.M102929200 [DOI] [PubMed] [Google Scholar]
- 59.Blight SK, Larue RC, Mahapatra A, Longstaff DG, Chang E, Zhao G, Kang PT, Green-Church KB, Chan MK, & Krzycki JA (2004). Direct charging of tRNACUA with pyrrolysine in vitro and in vivo. Nature, 431, 333–335. 10.1038/nature02895 [DOI] [PubMed] [Google Scholar]
- 60.Fekner T, & Chan MK (2011). The pyrrolysine translational machinery as a genetic-code expansion tool. Current Opinion in Chemical Biology, 15, 387–391. 10.1016/j.cbpa.2011.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Polycarpo C, Ambrogelly A, Bérubé A, Winbush SM, Mccloskey JA, Crain PF, Wood JL, & Söll D (2013). An aminoacyl-tRNA synthetase that specifically activates pyrrolysine. Proceedings of the National Academy of Sciences of the United States of America, 101, 12450–12454. 10.1073/pnas.0405362101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaya E, Vrabel M, Deiml C, Prill S, Fluxa VS, & Carell T (2012). A genetically encoded norbornene amino acid for the mild and selective modification of proteins in a copper-free click reaction. Angewandte Chemie International Edition in English, 51, 4466–4469. 10.1002/anie.201109252 [DOI] [PubMed] [Google Scholar]
- 63.Lang K, Davis L, Torres-Kolbus J, Chou C, Deiters A, & Chin JW (2012). Genetically encoded norbornene directs site-specific cellular protein labelling via a rapid bioorthogonal reaction. Nature Chemistry, 4, 298–304. 10.1038/nchem.1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Neumann H, Wang K, Davis L, Garcia-Alai M, & Chin JW (2010). Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature, 464, 441–444. 10.1038/nature08817 [DOI] [PubMed] [Google Scholar]
- 65.Wang YS, Fang X, Wallace AL, Wu B, & Liu WR (2012). A rationally designed pyrrolysyl-tRNA synthetase mutant with a broad substrate spectrum. Journal of the American Chemical Society, 134, 2950–2953. 10.1021/ja211972x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chemla Y, Ozer E, Schlesinger O, & Noireaux V, Alfonta L (2015). Genetically expanded cell-free protein synthesis using endogenous pyrrolysyl orthogonal translation system. Biotechnology and Bioengineering, 112, 1663–1672. 10.1002/bit.25587 [DOI] [PubMed] [Google Scholar]
- 67.Young TS, Ahmad I, Yin JA, & Schultz PG (2010). An enhanced system for unnatural amino acid mutagenesis in E. coli. Journal of Molecular Biology, 395, 361–374. 10.1016/j.jmb.2009.10.030 [DOI] [PubMed] [Google Scholar]
- 68.Wan W, Tharp JM, & Liu WR (2014). Pyrrolysyl-tRNA synthetase: An ordinary enzyme but an outstanding genetic code expansion tool. Biochimica et Biophysica Acta, 1844, 1059–1070. 10.1016/j.bbapap.2014.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lajoie MJ, Rovner AJ, Goodman DB, Aerni H-R, Haimovich AD, Kuznetsov G, Mercer JA, Wang HH, Carr PA, Mosberg JA, Rohland N, Schultz PG, Jacobson JM, Rinehart J, Church GM, & Isaacs FJ (2013). Genomically recoded organisms expand biological functions. Science, 342, 357–360. 10.1126/science.1241459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Albayrak C, & Swartz JR (2013). Cell-free co-production of an orthogonal transfer RNA activates efficient site-specific non-natural amino acid incorporation. Nucleic Acids Research, 41, 5949–5963. 10.1093/nar/gkt226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang YS, Fang X, Chen HY, Wu B, Wang ZU, Hilty C, & Liu WR (2013). Genetic incorporation of twelve meta-substituted phenylalanine derivatives using a single pyrrolysyl-tRNA synthetase mutant. ACS Chemical Biology, 8, 405–415. 10.1021/cb300512r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Swartz JR, Jewett MC, & Woodrow KA (2004). In eds Balbás P. &.Lorence A (Eds.), Methods in molecular biology, vol. 267: Recombinant gene expression, (pp. 169–182). Humana Press. [DOI] [PubMed] [Google Scholar]
- 73.Hong SH, Ntai I, Haimovich AD, Kelleher NL, Isaacs FJ, & Jewett MC (2014). Cell-free protein synthesis from a release factor 1 deficient Escherichia coli activates efficient and multiple site-specific nonstandard amino acid incorporation. ACS Synthetic Biology, 3, 398–409. 10.1021/sb400140t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Michel-Reydellet N, Woodrow K, & Swartz J (2005). Increasing PCR fragment stability and protein yields in a cell-free system with genetically modified Escherichia coli extracts. Journal of Molecular Microbiology and Biotechnology, 9, 26–34. 10.1159/000088143 [DOI] [PubMed] [Google Scholar]
- 75.Plass T, Milles S, Koehler C, Szymański J, Mueller R, Wießler M, Schultz C, & Lemke EA (2012). Amino acids for Diels-Alder reactions in living cells. Angewandte Chemie International Edition in English, 51, 4166–4170. 10.1002/anie.201108231 [DOI] [PubMed] [Google Scholar]
- 76.Bundy BC, & Swartz JR (2010). Site-specific incorporation of p-propargyloxyphenylalanine in a cell-free environment for direct protein-protein click conjugation. Bioconjugate Chemistry, 21, 255–263. 10.1021/bc9002844 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data available upon request from authors.