

Abstract
Microenvironments tailored by multifunctional secondary coordination sphere groups can enhance catalytic performance at primary metal active sites in natural systems. Here, we capture this biological concept in synthetic systems by developing a family of iron porphyrins decorated with imidazolium (im) pendants for the electrochemical CO2 reduction reaction (CO2RR), which promotes multiple synergistic effects to enhance CO2RR and enables the disentangling of second-sphere contributions that stem from each type of interaction. Fe-ortho-im(H), which poises imidazolium units featuring both positive charge and hydrogen-bond capabilities proximal to the active iron center, increases CO2 binding affinity by 25-fold and CO2RR activity by 2,000-fold relative to the parent Fe tetraphenylporphyrin (Fe-TPP). Comparison with mono-functional analogs reveals that through-space charge effects have a greater impact on catalytic CO2RR performance compared to hydrogen bonding in this context.
Keywords: electrochemical carbon dioxide reduction, carbon dioxide capture, second-sphere effect, iron porphyrin, imidazolium
Graphical Abstract
Iron porphyrins decorated with imidazolium (im) pendants enable the disentangling of second-sphere contributions that stem from through-space charge and hydrogen-bond interactions for the electrochemical CO2 reduction reaction (CO2RR). Charge effects are the dominant contributor to observed improvements in CO2 conversion with hydrogen bonding effects augmenting CO2 capture affinity, resulting effective homogeneous electrocatalytic CO2RR in water.
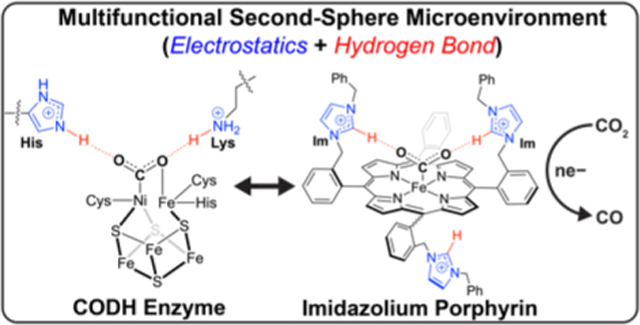
Rising global energy demands and fossil fuel use contribute to increasing concentrations of atmospheric carbon dioxide (CO2).[1] Capture and electrochemical reduction of CO2 offers a potentially powerful approach to converting this greenhouse gas into value-added chemical products.[2] The electrochemical CO2 reduction reaction (CO2RR) faces high kinetic and thermodynamic barriers, making it challenging to achieve requisite selectivity and activity, especially in aqueous media, where the abundance of protons favor the competing hydrogen evolution reaction (HER).[3] In this regard, enzyme biocatalysts such as Ni-Fe carbon monoxide dehydrogenases (CODH), enable the interconversion of CO2 and CO near the thermodynamic potential in neutral pH water[4] and provide inspiration for development of synthetic systems that can functionally mimic their chemistry.[2g, 3c] In particular, Ni-Fe CODH provides a microenvironment with a tailored secondary coordination sphere featuring two key histidine imidazolium (His93) and lysine (K563) ammonium pendants that are positioned proximally to the redox-active iron-sulfur cluster active site (Scheme 1A). These multifunctional second-sphere groups work in concert to help stabilize CO2-bound intermediates via both through-space electrostatic and hydrogen-bond interactions.[5] We now report that this concept can be captured in synthetic systems through the development of a family of iron porphyrins bearing multifunctional imidazolium pendants in the secondary coordination sphere (Scheme 1B). Combined electrostatic and hydrogen-bond interactions in Fe-ortho-im(H) result in enhanced CO2 binding and fast and selective electrochemical CO2 conversion in both organic and aqueous media, with a 2,000-fold increase in CO2RR activity relative to the parent Fe tetraphenylporphyrin (Fe-TPP) compound.[6] Comparison with the Fe-ortho-im(Me) analog that retains the charged functionality but lacks the C2-H hydrogen-bond moiety indicates that while hydrogen-bonding enhances CO2 capture, the proximal electrostatic component contributes the majority of the catalytic amplification observed in this system. This work establishes the use of multifunctional second-sphere pendants as an effective strategy for enhancing the performance of synthetic electrocatalysts and emphasizes the importance of disentangling relative contributions of second-sphere functionalities to inform catalyst design.
Scheme 1.

(A) Multifunctional histidine (His) and lysine (Lys) second-sphere protein residues proximal to the Fe-Ni cluster active site in the natural CODH biocatalyst inspire the design of synthetic Fe porphyrin catalysts decorated with multifunctional imidazolium pendants. (B) Fe porphyrins synthesized in this work enable disentangling of relative contributions of through-space electrostatic and C2-H hydrogen-bond interactions and positioning of second-sphere groups.
In contrast to what is observed for natural CO2RR biocatalysts, advances in synthetic CO2RR catalysts have largely focused on monofunctional second-sphere pendants,[7] including privileged Fe porphyrin scaffolds incorporating hydrogen-bonding groups such as phenols,[7a, 7c, 8] amides,[7e, 9] ureas,[7j, 7m, 7p, 10] guanidines, [11] and triazoles,[7i, 12] or through-space electrostatic functionalities such as trimethylanilinium[7d] [13] or imidazolium[7q, 14] cations. In this context, we reasoned that imidazoliums, when properly positioned, could serve as multifunctional second-sphere pendants that induce both through-space electrostatic and hydrogen-bonding effects, as well as offer the potential to disentangle the contributions that each type of functionality plays. Indeed, Nippe and colleagues reported that tethering of an imidazolium group improved activity in metal tricarbonyl-bipyridine CO2RR catalysts[15] and Aukauloo and colleagues observed through-space charge enhancement for CO2RR in iron porphyrins linked to distal imidazolium pendants through an aryl amide spacer.[7q, 14] Against this backdrop, we designed and synthesized a family of tetracationic imidazolium porphyrins that enabled systematic evaluation of positional tuning as well as hydrogen-bonding and through-space charge contributions. Specifically, we prepared C2-H and C2-methyl imidazolium (im) units at either the ortho [Fe-ortho-im(H), Fe-ortho-im(Me)] or para [Fe-para-im(H)] positions on the ancillary phenyl rings of a tetraphenylporphyrin scaffold (Scheme 1B). The target catalysts were synthesized in high yield in three steps, starting from appropriate ortho or para-(bromomethyl)benzaldehyde precursors (Scheme S1). Fe-ortho-im(H) places imidazolium units proximal to the Fe center to deliver both hydrogen-bond and charge stabilization functionalities to CO2-bound intermediates. In Fe-ortho-im(Me), the C2-H units are blocked with methyl groups to probe how through-space electrostatic effects alone influence CO2RR catalysis. Finally, the Fe-para-im(H) was synthesized to evaluate positional effects of the secondary imidazolium unit relative to the primary Fe center. Interestingly, incorporating imidazolium groups into the metalloporphyrin scaffold affords solubility in water, acetonitrile (MeCN), and dimethylformamide (DMF) (Figures S30–31, 33), in contrast to the vast majority of porphyrins whose solubility restricts their use as homogeneous electrocatalysts in DMF.[6–7, 7d, 7e, 7m, 7q, 14]
The redox behaviors of Fe-ortho-im(H), Fe-ortho-im(Me), and Fe-para-im(H) were characterized using cyclic voltammetry (CV) measurements in MeCN under an argon (Ar) atmosphere. The CVs show three distinct redox events corresponding to formal FeIII/II, FeII/I, and FeI/0 couples (Figure 1, Table S1). Scan rate-dependent data show a linear correlation between the peak current of the FeI/0 couples and the square root of the scan rate, indicating that all catalysts are freely diffusible under non-catalytic conditions (Figure S1). The observed formal FeI/0 potentials, , of the three porphyrins are similar (ca. −1.8 V vs. Fc+/Fc), indicating that the electronic properties of all members of this series are comparable. There is a 276 mV positive shift in the FeI/0 couple relative to the parent catalyst Fe-TPP[6] (Table S1), presaging that the cationic imidazolium units could activate CO2 at lower potentials, presumably by electrostatic stabilization of reduced iron species. Next, in order to decipher hydrogen-bond and through-space charge contributions in promoting CO2 capture prior to evaluation of CO2RR catalysis, we estimated the CO2 binding constants (KCO2) by measuring the shift in the FeI/0 wave under Ar and CO2 atmospheres in the absence of a proton source to prevent subsequent catalytic turnover (Table 1 and Figure S2). Fe-ortho-im(H) exhibits a KCO2 value of 65 M−1, representing a 5-fold increase in CO2 binding affinity over Fe-ortho-im(Me) and a 20-fold increase over Fe-para-im(H), suggesting that both through-space electrostatic and hydrogen-bonding interactions in the proximal ortho position stabilize the Fe-CO2 adduct.
Figure 1.

Cyclic voltammograms (CVs) of imidazolium (im)-functionalized porphyrins under Ar. Conditions: 0.3 mM catalyst, 0.1 M TBAPF6 in MeCN; scan rate is 100 mV s−1.
Table 1.
Electrochemical properties of iron porphyrin catalysts
| Catalyst | [a] | [b] | LogTOFmax[c] | KIE[d] | FEco[e] |
|---|---|---|---|---|---|
|
| |||||
| Fe–ortho-im(H) | −1.78 | 65 | 9.1 | 8.7 | 100 |
| Fe–ortho-im(Me) | −1.79 | 12 | 7.5 | 9.2 | 100 |
| Fe–para-im(H) | −1.79 | 3 | 4.9 | 6.5 | 28 |
| FeTPP | −2.06 | 2.58 | 5.8 | — | 98 |
(V vs. Fc+/Fc) obtained from CVs under argon and catalyst concentration (0.3 mM).
KCO2 (M−1) data was obtained from measuring CVs under a CO2 atmosphere without 2,2,2-trifluoroethanol (TFE) at a scan rate of 1.0 V s−1.
Maximum turnover frequency (TOFmax) values were estimated using foot-of-the-wave (FOWA) under CO2 with 3.0 M TFE.
Kinetic isotope effect (KIE) values represent the ratio of kH/kD measured using water as the proton source.
FE (%) data for CO was obtained as an average of three controlled potential electrolyses (CPEs) under CO2 with 1.5 M TFE and catalyst concentration (10 μM). All experiments in this table were performed in 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6)/MeCN, conditions used for FeTPP are provided in the reference.
Upon addition of 2,2,2-trifluoroethanol (TFE) as a proton source, all three Fe porphyrins show large catalytic current enhancement under a CO2 atmosphere, with Fe-ortho-im(H) showing the highest catalytic current and most positive onset potential for catalysis, followed by Fe-ortho-im(Me), with Fe-para-im(H) being the least active of the three congeners (Figure 2A, S3). To estimate the turnover frequency (TOF) values free from secondary phenomena (e.g. substrate consumption, product inhibition, catalyst degradation), we applied the foot-of-the-wave analysis (FOWA) introduced by Savéant and colleagues (Tables 1, S2, S3, Figure S4).[7d, 16] We observed a linear relationship between LogTOFmax values and the concentration of added TFE (Figure 2B), demonstrating that the rates display a first-order dependence on the acid concentration. The observed TOF values with 3.0 M TFE for the Fe-ortho-im(H) catalyst (1.3 × 109 s−1) represent a 14,000-fold increase over the positional Fe-para-im(H) congener (8.7 × 104 s−1) and 2,000-fold increase over Fe-TPP-ClO4 in MeCN (6.5 × 105 s−1),[6] but only a 40-fold increase over the Fe-ortho-im(Me) derivative (3.2 × 107 s−1) that retains the imidazolium positive charge but lacks the C2-H hydrogen-bond capability. The data indicate that electrostatic effects dominate catalytic amplification over hydrogen-bond interactions in this context. We applied current-limiting plateau as a complementary method to confirm the observed trend in the TOFmax values from FOWA. The trend in activity derived from the current-limiting plateau method [Fe-ortho-im(H) > Fe-ortho-im(Me) > Fe-para-im(H)] matches that observed using FOWA, yet with lower absolute values that can be attributed to lower-limit estimations using this workflow. (Table S4 and Figures S16–18). We note that the turnover frequencies in all cases represent the initial catalytic nature of the system and do not reflect the eventual loss of catalytic activity.
Figure 2.

Cyclic voltammetry (CV) experiments. Forward CV traces of imidazolium (im)-functionalized porphyrins under (A) CO2 in the presence of 3 M TFE. (B) Calculated logTOFmax as a function of TFE concentration. (C) Catalytic Tafel plots. Conditions: 0.3 mM catalyst, 0.1 M TBAPF6 in MeCN; scan rate is 100 mV s−1. i0p represents the cathodic peak height of the formal FeII/I couple under Ar. TOFmax values were determined by FOWA (see SI for details).
We next moved on to construct catalytic Tafel plots in which the LogTOF values vary as a function of applied overpotential, where the most efficient electrocatalysts function with high TOFs at low overpotentials. The three catalysts reach a plateau (LogTOFmax) at relatively low overpotentials (ca. 0.15 V, Figure 2C), with Fe-ortho-im(H) exhibiting higher TOFs over all applied potentials relative to its congeners. Benchmarking[17] Fe-ortho-im(H), Fe-ortho-im(Me), and Fe-para-im(H) indicates that Fe-ortho-im(H) is among the fastest homogeneous molecular catalysts for electrochemical CO2RR reported to date (Table S3 and Figures S5, S6). In addition, to determine the contributions of proton transfer in CO2RR catalysis, we measured kinetic isotope effects (KIE) for all three catalysts using varying concentrations of H2O or D2O as the proton source (Table 1, Figures S7–15). Large normal primary KIEs were observed for all three catalysts (6.5 to 9.2), suggesting that proton transfer is involved in the rate-determining step regardless of imidazolium positioning.
With data in hand showing that these second-sphere imidazolium pendants promote enhanced CO2RR activity, we then performed controlled potential electrolysis (CPE) experiments to measure catalyst stability and product distribution (Figure 3). CPE measurements were conducted in a CO2-saturated NBu4PF6/MeCN electrolyte using a glassy carbon electrode with as low as 10 μM catalyst loading (Figures 3, S16). Fe-ortho-im(H) is highly selective for CO2RR over HER, generating CO as a product over a wide potential range with Faradaic efficiencies (FE) that exceed 95% at overpotentials[6, 18] above 0.2 V (Figures 3A, S17). CPEs at −1.87 V vs. Fc+/Fc (η = 0.3 V) showed that both Fe-ortho-im(H) and Fe-ortho-im(Me) were selective towards CO production (FE ≈ 100%) with more charge passed using Fe-ortho-im(H), whereas Fe-para-im(H) shows a significant loss in selectivity for CO production (FE ≈ 28%). (Tables 1, S4, Figures 3B, S18–19). Although Fe-para-im(H) is stable in bulk solution, electrode deposition happens gradually during electrolysis, passivating the working electrode and decreasing the current, causing the low observed FE (Figure S25). Additionally, FT-IR recorded before and after electrolysis under CO2 using Fe-ortho-im(H) and Fe-ortho-im(Me) were similar (Figures S23–S24), showing no emergence of the N-heterocyclic carbene-CO2 band (υ C=O at 1625–1675 cm−1) under the experimental detection limits (Figures S23).
Figure 3.

Controlled-potential electrolysis (CPE) experiments. (A) Faradaic efficiencies as a function of overpotential in CPE experiment using Fe-ortho-im(H) in 0.1 M TBAPF6 in MeCN; overpotential values are reported as the difference between the and −1.54 vs. Fc+/Fc for MeCN. CPEs of imidazolium-functionalized porphyrins under CO2 (B) at −1.87 V (vs. Fc+/Fc) in 0.1 M TBAPF6 in MeCN and (C) in 0.1 M aqueous PBS (pH of 6.8) at −0.95 V (vs. NHE). Catalyst concentration in all CPEs (10 μM). (See SI for details).
Observing that the cationic nature of the imidazolium pendants enabled solubility of these catalysts in water (Figures S29–30, 32), we next proceeded to evaluate their performance for homogeneous electrochemical CO2RR in aqueous media (Figure 3C, Figure S29–30). We were pleased to find that both Fe-ortho-im(H) and Fe-ortho-im(Me) exhibit exceptionally high rates for CO2RR in aqueous KCl solution, with TOFmax values of 4.6 × 106 and 2.3 × 106 s−1, respectively (Figures S21–22, Table S5). The Fe-para-im(H) complex was not sufficiently soluble under these conditions for evaluation. The observed TOF value of Fe-ortho-im(H) is only 2-fold higher than that of Fe-ortho-im(Me), confirming that through-space charge effects dominate over hydrogen-bonding ones these imidazolium systems. Moreover, CPE experiments under CO2 in 0.1 M KCl electrolyte (initial pH of 4.0) showed that both Fe-ortho-im(H) and Fe-ortho-im(Me) catalysts retained their high selectivity for CO2RR over HER, even in aqueous media with an abundance of protons, with FE > 90% for CO product (Figure S23, Table S6). Additionally, we performed CPE experiments in aqueous phosphate solutions buffered to near neutral pH (6.8), where all three catalysts were soluble. The Fe-ortho-im(H) and Fe-ortho-im(Me) complexes were again more selective for CO formation (FE 70%) compared to Fe-para-im(H) counterpart (FE 50%), with <5% H2 evolution observed for all three catalysts, even in aqueous conditions with an abundance of available protons (Figures 3C, S24, Table S7). Interestingly, these data indicate that imidazolium groups are superior at suppressing HER compared to related cationic anilinium Fe-porphyrin catalysts under similar conditions, where the latter systems show ca. 50% FE for H2 production in water.[8d] To probe the effect of hydrophobicity stemming from the N-benzyl imidazolium groups on catalytic activity, we prepared an analog with N-methyl groups: Fe-ortho-im(H)-N(Me) (see SI for details). This derivative remained selective for CO over HER (Table S8 and Figure S52), corroborating the beneficial role provided by imidazolium groups.
To close, we have presented a family of iron porphyrins functionalized with multifunctional imidazolium pendants in the secondary coordination sphere. These pendants promote synergistic through-space electrostatic and hydrogen-bonding interactions to improve activity and selectivity for homogeneous electrochemical CO2RR, as well as enable us to disentangle contributions for each of these individual second-sphere effects. Specifically, comparison of Fe-ortho-im(H) and Fe-ortho-im(Me) versus a para-substituted counterpart reveals that while electrostatic effects contribute a majority of the observed catalytic enhancements, synergistic hydrogen-bonding effects further augment CO2 capture affiniity and CO2RR conversion rates. Indeed, the calculated geometry optimization of the Fe-CO2 adduct of Fe-ortho-im(H) using the density-functional tight-binding (DFTB) method (Figure 4) suggests that the imidazolium pendants orient themselves towards the CO2 ligand and form short-hydrogen bonds between the C2-H groups and bound CO2 (1.97 and 2.33 Å), reminiscent to what is found in the active site of Ni-Fe CODH enzymes (2.63 and 2.88 Å) (Figures 4, S26).[8d] This compound, which possesses both electrostatic and hydrogen-bonding capabilities, shows the highest activity and selectivity for CO2RR in the series, with rate enhancements of 2,000-fold over the parent Fe-TPP compound, enabling efficient, homogeneous CO2RR in both organic and aqueous media. This work provides a starting point for designing second-sphere pendants that enable synergistic, multifunctional effects to enhance catalytic performance for a broader array of chemical transformations, in particular avoiding competing hydrogen evolution pathways even in proton-rich aqueous media.
Figure 4.

Density-functional tight-binding (DFTB)-optimized structure of the [Fe−CO2]2− adduct on Fe-ortho-im(H). Hydrogen atoms and bromides were omitted for clarity except for the C1 imidazolium hydrogens. H white, C grey, N blue, O red; Fe orange.
Supplementary Material
Acknowledgments
This work was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, via the Division of Chemical Sciences, Geosciences, and Bioscience of the U.S. Department of Energy at Lawrence Berkeley National Laboratory (Grant No. DE-AC02–05CH11231 to C.J.C.) M.R.N. acknowledges support from an NSERC (Canada) postdoctoral fellowship and P.D.T. acknowledges NSF for a graduate fellowship. We acknowledge Drs. Kathleen A. Durkin and Dave Small at the Molecular Graphics and Computation Facility (MGCF) (NIH S10OD023532) for their advice regarding DFTB calculations. We acknowledge the use of CoC-NMR instruments supported in part by NIH S10OD024998. C.J.C. is a CIFAR Fellow.
References
- [1].Kätelhön A, Meys R, Deutz S, Suh S, Bardow A, Proceedings of the National Academy of Sciences 2019, 116, 11187–11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Hori Y, in Modern Aspects of Electrochemistry (Eds.: Vayenas CG, White RE, Gamboa-Aldeco ME), Springer New York, New York, NY, 2008, pp. 89–189; [Google Scholar]; b) Benson EE, Kubiak CP, Sathrum AJ, Smieja JM, Chemical Society Reviews 2009, 38, 89–99; [DOI] [PubMed] [Google Scholar]; c) Appel AM, Bercaw JE, Bocarsly AB, Dobbek H, DuBois DL, Dupuis M, Ferry JG, Fujita E, Hille R, Kenis PJA, Kerfeld CA, Morris RH, Peden CHF, Portis AR, Ragsdale SW, Rauchfuss TB, Reek JNH, Seefeldt LC, Thauer RK, Waldrop GL, Chemical Reviews 2013, 113, 6621–6658; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Francke R, Schille B, Roemelt M, Chemical Reviews 2018, 118, 4631–4701; [DOI] [PubMed] [Google Scholar]; e) Zhao S, Jin R, Jin R, ACS Energy Letters 2018, 3, 452–462; [Google Scholar]; f) Luna PD, Hahn C, Higgins D, Jaffer SA, Jaramillo TF, Sargent EH, Science 2019, 364, eaav3506; [DOI] [PubMed] [Google Scholar]; g) Proppe AH, Li YC, Aspuru-Guzik A, Berlinguette CP, Chang CJ, Cogdell R, Doyle AG, Flick J, Gabor NM, van Grondelle R, Hammes-Schiffer S, Jaffer SA, Kelley SO, Leclerc M, Leo K, Mallouk TE, Narang P, Schlau-Cohen GS, Scholes GD, Vojvodic A, Yam VW-W, Yang JY, Sargent EH, Nature Reviews Materials 2020, 5, 828–846. [Google Scholar]
- [3].a) Nitopi S, Bertheussen E, Scott SB, Liu X, Engstfeld AK, Horch S, Seger B, Stephens IEL, Chan K, Hahn C, Nørskov JK, Jaramillo TF, Chorkendorff I, Chemical Reviews 2019, 119, 7610–7672; [DOI] [PubMed] [Google Scholar]; b) Boutin E, Merakeb L, Ma B, Boudy B, Wang M, Bonin J, Anxolabéhère-Mallart E, Robert M, Chemical Society Reviews 2020, 49, 5772–5809; [DOI] [PubMed] [Google Scholar]; c) Smith PT, Nichols EM, Cao Z, Chang CJ, Accounts of Chemical Research 2020, 53, 575–587; [DOI] [PubMed] [Google Scholar]; d) Saha P, Amanullah S, Dey A, Accounts of Chemical Research 2022, 55, 134–144. [DOI] [PubMed] [Google Scholar]
- [4].a) Armstrong FA, Hirst J, Proceedings of the National Academy of Sciences 2011, 108, 14049–14054; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang VC-C, Ragsdale SW, Armstrong FA, ChemBioChem 2013, 14, 1845–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Drennan CL, Heo J, Sintchak MD, Schreiter E, Ludden PW, Proceedings of the National Academy of Sciences 2001, 98, 11973–11978; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jeoung J-H, Dobbek H, Science 2007, 318, 1461–1464. [DOI] [PubMed] [Google Scholar]
- [6].Kosugi K, Kondo M, Masaoka S, Angewandte Chemie International Edition 2021, 60, 22070–22074. [DOI] [PubMed] [Google Scholar]
- [7].a) Costentin C, Drouet S, Robert M, Savéant J-M, Science 2012, 338, 90–94; [DOI] [PubMed] [Google Scholar]; b) Yang JY, Smith SE, Liu T, Dougherty WG, Hoffert WA, Kassel WS, DuBois MR, DuBois DL, Bullock RM, Journal of the American Chemical Society 2013, 135, 9700–9712; [DOI] [PubMed] [Google Scholar]; c) Costentin C, Passard G, Robert M, Savéant J-M, Proceedings of the National Academy of Sciences 2014, 111, 14990–14994; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Azcarate I, Costentin C, Robert M, Savéant J-M, Journal of the American Chemical Society 2016, 138, 16639–16644; [DOI] [PubMed] [Google Scholar]; e) Nichols Eva M., Derrick JS, Nistanaki SK, Smith PT, Chang CJ, Chemical Science 2018, 9, 2952–2960; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Haviv E, Azaiza-Dabbah D, Carmieli R, Avram L, Martin JML, Neumann R, Journal of the American Chemical Society 2018, 140, 12451–12456; [DOI] [PubMed] [Google Scholar]; g) Chapovetsky A, Welborn M, Luna JM, Haiges R, Miller TF, Marinescu SC, ACS Central Science 2018, 4, 397–404; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Nichols EM, Chang CJ, Organometallics 2019, 38, 1213–1218; [Google Scholar]; i) Sen P, Mondal B, Saha D, Rana A, Dey A, Dalton Transactions 2019, 48, 5965–5977; [DOI] [PubMed] [Google Scholar]; j) Gotico P, Boitrel B, Guillot R, Sircoglou M, Quaranta A, Halime Z, Leibl W, Aukauloo A, Angewandte Chemie International Edition 2019, 58, 4504–4509; [DOI] [PubMed] [Google Scholar]; k) Chapovetsky A, Liu JJ, Welborn M, Luna JM, Do T, Haiges R, Miller Iii TF, Marinescu SC, Inorganic Chemistry 2020, 59, 13709–13718; [DOI] [PubMed] [Google Scholar]; l) Margarit CG, Asimow NG, Gonzalez MI, Nocera DG, The Journal of Physical Chemistry Letters 2020, 11, 1890–1895; [DOI] [PubMed] [Google Scholar]; m) Gotico P, Roupnel L, Guillot R, Sircoglou M, Leibl W, Halime Z, Aukauloo A, Angewandte Chemie International Edition 2020, 59, 22451–22455; [DOI] [PubMed] [Google Scholar]; n) Kinzel NW, Werlé C, Leitner W, Angewandte Chemie International Edition 2021, 60, 11628–11686; [DOI] [PMC free article] [PubMed] [Google Scholar]; o) Amanullah S, Saha P, Dey A, Journal of the American Chemical Society 2021, 143, 13579–13592; [DOI] [PubMed] [Google Scholar]; p) Zhang C, Dragoe D, Brisset F, Boitrel B, Lassalle-Kaiser B, Leibl W, Halime Z, Aukauloo A, Green Chemistry 2021, 23, 8979–8987; [Google Scholar]; q) Khadhraoui A, Gotico P, Leibl W, Halime Z, Aukauloo A, ChemSusChem 2021, 14, 1308–1315; [DOI] [PubMed] [Google Scholar]; r) Drover MW, Chemical Society Reviews 2022, 51, 1861–1880; [DOI] [PubMed] [Google Scholar]; s) Johnson EM, Liu JJ, Samuel AD, Haiges R, Marinescu SC, Inorganic Chemistry 2022, 61, 1316–1326. [DOI] [PubMed] [Google Scholar]
- [8].a) Costentin C, Passard G, Robert M, Savéant J-M, Journal of the American Chemical Society 2014, 136, 11821–11829; [DOI] [PubMed] [Google Scholar]; b) Bonin J, Chaussemier M, Robert M, Routier M, ChemCatChem 2014, 6, 3200–3207; [Google Scholar]; c) Bonin J, Robert M, Routier M, Journal of the American Chemical Society 2014, 136, 16768–16771; [DOI] [PubMed] [Google Scholar]; d) Costentin C, Robert M, Savéant J-M, Tatin A, Proceedings of the National Academy of Sciences 2015, 112, 6882–6886; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Guo K, Li X, Lei H, Zhang W, Cao R, ChemCatChem 2020, 12, 1591–1595. [Google Scholar]
- [9].Manoranjan N, Won DH, Kim J, Woo SI, Journal of CO2 Utilization 2016, 16, 486–491. [Google Scholar]
- [10].a) Gotico P, Halime Z, Aukauloo A, Dalton Transactions 2020, 49, 2381–2396; [DOI] [PubMed] [Google Scholar]; b) Pugliese E, Gotico P, Wehrung I, Boitrel B, Quaranta A, Ha-Thi M-H, Pino T, Sircoglou M, Leibl W, Halime Z, Aukauloo A, Angewandte Chemie International Edition 2022, 61, e202117530; [DOI] [PubMed] [Google Scholar]; c) Derrick J, Loipersberger M, Nistanaki S, Rothweiler A, Head-Gordon M, Nichols E, Chang C, J. Am. Chem. Soc. 2022, 144, 26, 11656–11663 [DOI] [PubMed] [Google Scholar]
- [11].Margarit CG, Schnedermann C, Asimow NG, Nocera DG, Organometallics 2019, 38, 1219–1223. [Google Scholar]
- [12].Williams CK, Lashgari A, Tomb JA, Chai J, Jiang JJ, ChemCatChem 2020, 12, 4886–4892. [Google Scholar]
- [13].a) Rao H, Bonin J, Robert M, ChemSusChem 2017, 10, 4447–4450; [DOI] [PubMed] [Google Scholar]; b) Rao H, Schmidt LC, Bonin J, Robert M, Nature 2017, 548, 74–77; [DOI] [PubMed] [Google Scholar]; c) Rao H, Bonin J, Robert M, Chemical Communications 2017, 53, 2830–2833. [DOI] [PubMed] [Google Scholar]
- [14].Khadhraoui A, Gotico P, Boitrel B, Leibl W, Halime Z, Aukauloo A, Chemical Communications 2018, 54, 11630–11633. [DOI] [PubMed] [Google Scholar]
- [15].a) Sung S, Kumar D, Gil-Sepulcre M, Nippe M, Journal of the American Chemical Society 2017, 139, 13993–13996; [DOI] [PubMed] [Google Scholar]; b) Sung S, Li X, Wolf LM, Meeder JR, Bhuvanesh NS, Grice KA, Panetier JA, Nippe M, Journal of the American Chemical Society 2019, 141, 6569–6582. [DOI] [PubMed] [Google Scholar]
- [16].Costentin C, Savéant J-M, Journal of the American Chemical Society 2018, 140, 16669–16675. [DOI] [PubMed] [Google Scholar]
- [17].a) Stratakes BM, Dempsey JL, Miller AJM, ChemElectroChem 2021, 8, 4161–4180; [Google Scholar]; b) Costentin C, Drouet S, Robert M, Savéant J-M, Journal of the American Chemical Society 2012, 134, 11235–11242. [DOI] [PubMed] [Google Scholar]
- [18].Sinha S, Warren JJ, Inorganic Chemistry 2018, 57, 12650–12656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.