

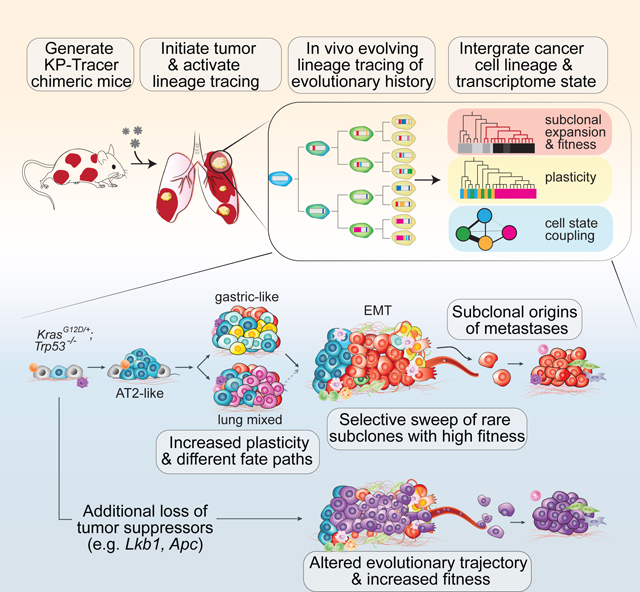
SUMMARY
Tumor evolution is driven by the progressive acquisition of genetic and epigenetic alterations that enable uncontrolled growth and expansion to neighboring and distal tissues. The study of phylogenetic relationships between cancer cells provides key insights into these processes. Here, we introduced an evolving lineage tracing system with a single-cell RNA-seq readout into a mouse model of Kras;Trp53(KP)-driven lung adenocarcinoma and tracked tumor evolution from single transformed cells to metastatic tumors at unprecedented resolution. We found that loss of the initial, stable alveolar-type2-like state was accompanied by transient increase in plasticity. This was followed by adoption of distinct transcriptional programs which enable rapid expansion and ultimately clonal sweep of stable subclones capable of metastasizing. Finally, tumors develop through stereotypical evolutionary trajectories, and perturbing additional tumor suppressors accelerates progression by creating novel trajectories. Our study elucidates the hierarchical nature of tumor evolution, and more broadly enables in-depth studies of tumor progression.
Keywords: Lineage Tracing, Tumor Evolution, Phylogenetics, Single Cell, Fitness, Plasticity, Transcriptome Heterogeneity, Genetically Engineered Mouse Model, Lung Cancer
eTOC Blurb
Yang et al. developed a genetically engineered mouse model of lung cancer capable of continuous lineage tracing with single-cell RNA-seq readout. They identified the subclonal dynamics of tumors, gene modules underlying expansion, transient increases in cellular plasticity, stereotypical evolutionary paths to aggressiveness across tumor genotypes, and the spatial and phylogenetic origins of metastases.
Graphical Abstract
INTRODUCTION
Cancer is an evolutionary process characterized by the dynamic interplay of cellular subpopulations, each driven by progressive genetic and epigenetic changes (Nowell 1976). Throughout this process, cancer cells can acquire phenotypic heterogeneity that increases fitness by enabling them to grow more aggressively, invade neighboring tissues, evade the immune system and therapeutic challenges, and metastasize to distant sites (Hanahan and Weinberg 2011; Vogelstein et al. 2013; McGranahan and Swanton 2017). Interrogating the molecular bases of subclonal selection and metastatic seeding, the origins of and transitions between transcriptional states, as well as the identities and genetic determinants of evolutionary paths that tumors undergo will not only illuminate fundamental principles governing tumor evolution, but also have immediate clinical implications (Black and McGranahan 2021). To fully understand these processes, it is essential to study the evolutionary dynamics giving rise to a tumor in its native setting, preferably in experimentally defined conditions (Amirouchene-Angelozzi et al. 2017).
Tumor phylogenetic analysis, the study of lineage relationships among the cells comprising the tumor population descended from a single transformed progenitor, can provide key insights into the dynamics of tumor progression. Classically, phylogenies have been constructed using naturally-occurring somatic genomic variations (mutations or copy-number variations [CNVs]) as natural lineage tracers. These efforts have illuminated several key evolutionary processes underpinning tumor development (Vogelstein et al. 1988; Sjöblom et al. 2006; Schwartz and Schäffer 2017; Ludwig et al. 2019; Gao et al. 2021; Gerstung et al. 2020; Sottoriva et al. 2015). including the acquisition of critical subclonal genetic or epigenetic changes (Gerlinger et al. 2014; Williams et al. 2018; Neftel et al. 2019), the timing and routes of metastatic dissemination (Turajlic and Swanton 2016; Hu and Curtis 2020), and the development of therapeutic resistance (Maynard et al. 2020; Powles et al. 2021; Abbosh et al. 2017; Kim et al. 2018; Salehi et al. 2021). While progress has been enabled by innovative computational methods (Potter et al. 2013; El-Kebir et al. 2016; Malikic et al. 2019; Satas et al. 2020), these studies are limited by the inherent variation in naturally-occurring somatic mutations, incomplete or low cell sampling, and other confounding variables (e.g. environmental exposures and genetic background), and are not amenable to further perturbations or functional studies.
Genetically engineered mouse models (GEMMs) of cancer provide a critical tool for modeling tumor progression as they allow one to study tumor evolution in its native microenvironment and experimentally defined conditions (Hann and Balmain 2001; Frese and Tuveson 2007). The KrasLSL-G12D/+; Trp53fl/fl (KP) model of lung adenocarcinoma allows tumor initiation via viral delivery of Cre recombinase to a small number of lung epithelial cells, leading to activation of oncogenic Kras, homozygous deletion of the p53 tumor suppressor gene, and clonal tumor outgrowth. It faithfully models the major steps of tumor evolution from nascent cell transformation to aggressive metastasis, recapitulating human lung adenocarcinoma progression both molecularly and histopathologically (Jackson et al. 2001; Jackson et al. 2005; Winslow et al. 2011). Moreover, recent work has revealed that substantial transcriptomic and epigenomic heterogeneity emerges during tumor evolution in this model (Marjanovic et al. 2020; LaFave et al. 2020), consistent with human tumors (Laughney et al. 2020). The tractability of this model provides an appealing opportunity to probe several unanswered, but crucial questions regarding how tumors evolve including: how a single transformed cell expands into an aggressive tumor, how various cell states relate to one another and contribute to tumor evolution, how different transcriptional states transition between each other, and how metastases and primary tumors are evolutionarily related.
Approaches that permit simultaneous measurements of cell lineage and cell state information have the potential to provide unique insights into these questions (Tammela and Sage 2020; Wagner and Klein 2020; Stadler et al. 2021). While previous studies have used synthetic “static” barcoding techniques to study clonal relationships (Bhang et al. 2015; Livet et al. 2007; Lan et al. 2017; Pei et al. 2017; Driessens et al. 2012; Schepers et al. 2012), studying the evolution of individual tumors at subclonal resolution remains challenging. This limitation is in large part due to the low mutational burden in GEMM tumors, thus offering little lineage resolution within individual tumors (Westcott et al. 2015; McFadden et al. 2016). The recent development of high resolution CRISPR/Cas9 evolving lineage tracing paired with single-cell RNA-seq (scRNA-seq) readouts overcomes these limitations. Generally, such continuous lineage-tracing approaches leverage Cas9-induced DNA cleavage and subsequent repair to progressively generate heritable insertions and deletions (“indels”) at synthetic DNA target sites engineered into the genomes of living cells (McKenna et al. 2016; Frieda et al. 2017; Kalhor et al. 2018; Chan et al. 2019; McKenna and Gagnon 2019). Importantly, these DNA target sites are transcribed into polyadenylated mRNAs, allowing them to be captured and profiled along with all other cellular mRNAs using scRNA-seq. In doing so, this approach makes it possible to directly link the current cell state (as measured by scRNA-seq) with its inferred or putative past lineage history (as captured by the lineage tracer), and to do so on a massive scale (Alemany et al. 2018; Spanjaard et al. 2018; Raj et al. 2018; Chan et al. 2019; Bowling et al. 2020). Recently, this technology has been introduced into cancer cell lines before transplanting them into mice to track metastatic behaviors in vivo (Simeonov et al. 2021; Quinn et al. 2021; Zhang et al. 2021).
Here, we have developed an autochthonous “KP-Tracer” mouse model which allows us to simultaneously initiate an engineered lineage tracing system and induce Kras and Trp53 oncogenic mutations in individual lung epithelial cells. This enabled continuous and comprehensive monitoring of the processes by which a single cell harboring oncogenic mutations evolves into an aggressive tumor. The resulting tumor phylogenies reveal that rare subclones drive tumor expansion by adopting distinct fitness-associated transcriptional programs. By integrating lineage and transcriptome data, we uncovered changes in cancer cell plasticity and parallel evolutionary paths of tumor evolution in this model, which could be profoundly altered by perturbing additional tumor suppressor genes commonly mutated in human tumors. We have also identified the subclonal origins, spatial locations and cellular states of metastatic progression. Collectively, this technology allowed us to reconstruct the lifespan of a tumor from a single transformed cell to a complex and aggressive tumor population at unprecedented scale and resolution.
RESULTS
KP-Tracer mouse enables continuous and high-resolution lineage tracing of tumor initiation and progression
To generate high-resolution tumor phylogenies, we developed a lineage-tracing competent mouse model of lung adenocarcinoma capable of months-long continuous cell lineage tracing (Fig 1A). Specifically, we engineered mouse embryonic stem cells (mESCs) harboring the conditional alleles KrasLSL-G12D/+ and Trp53fl/fl (KP) to additionally encode conditional SpCas9 and mNeonGreen fluorophore at the Rosa26 locus; Rosa26LSL-Cas9-P2A-mNeonGreen (KPCas9). We then engineered these mESCs with a refined version of our lineage tracing technology (Chan et al. 2019; Quinn et al. 2021). Specifically, we introduced a library of piggyBac transposon-based lineage tracing vector containing two essential components: first, target sites for lineage tracing, consisting of three cut sites positioned within the 3’ UTR of a mCherry fluorescent reporter and a 14-base-pair randomer integration barcode (“intBC”) to distinguish individual copies; and second, three constitutively expressed single-guide RNAs (sgRNAs) for directing Cas9 to each of the three individual cut-sites within the target sites, thereby generating indels for lineage tracing (Fig S1A). A key enabling feature is that the speed of tracing (i.e., indel generation kinetics) can be tuned to match the tumor developmental timescale by engineering mismatches between sgRNAs and target sites (Chan et al. 2019; Quinn et al. 2021). We isolated engineered mESC clones by fluorescence activated cell sorting (FACS) based on high mCherry expression (Fig S1B-C) and selected clones with 10–30 integrated target sites by quantitative PCR (qPCR) and DNA sequencing (Fig S1D-E). Finally, we generated chimeric mice (hereafter “KP-Tracer” mice) from five validated mESC clones to ensure evolutionary behavior was not idiosyncratic to a specific clone (Zhou et al. 2010; Premsrirut et al. 2011).
Figure 1. KP-Tracer mouse enables continuous and high-resolution lineage tracing of tumor initiation and progression.

(A) Generation of the KP-Tracer chimeric mouse and initiation of KP-Tracer tumors (STAR Methods). Five to six months after tumor initiation, individual tumors are dissociated into single cell suspension and single cell sequencing libraries are prepared. (B) Representative images of tumors from KP-Tracer mouse. Tumors are positive for mCherry and mNeonGreen. Scale bars = 5 mm. (C) Tumor lineage reconstruction data analysis pipeline. (D) Target site capture efficiency across tumors from mice generated from one representative mESC clone (2E1). Dots represent the average capture rate of a specific target site in a tumor. (E) Phylogeny with MULTI-seq, lenti-Cre-BC, and target site information for an example tumor. Each row represents a single cell and each column indicates barcode or target site information (ordered by the percentage of target sites detected across cells). Unique colors represent unique barcodes or indels, uncut sites are shown in light-gray, and missing data is indicated in white. (F) Comparison of phylogenetic distance (from the reconstructed tree) and allele edit distance (from target sites) for the example tumor in (E).
In KP-Tracer mice, intratracheal administration of lentivirus expressing Cre recombinase simultaneously initiates lung tumors by activating conditional oncogenic alleles and lineage tracing by inducing the expression of Cas9 which together with the expressed sgRNAs causes accumulation of indels in the target sites (DuPage et al. 2009). Previous static lineage tracing studies, using lentiviral barcoding or multi-color reporters, have shown that KP tumors induced with this strategy are clonal and homogenously contain oncogenic Kras;p53 mutations (Chuang et al. 2017; Caswell et al. 2014). To validate tumor clonality, we induced tumors with a barcoded lentiviral-Cre construct (lenti-Cre-BC) providing a unique clonal barcode for each tumor (Adamson et al. 2016).
Individual tumors with strong mCherry and mNeonGreen expression (indicating target site and Cre, respectively) and clear boundary separation from adjacent tumors were harvested 5–6 months after tumor initiation, microdissected, and dissociated completely to ensure unbiased cell sampling (Fig 1B; Table S1). After being labeled with Multiplexing Using Lipid-Tagged Indices for scRNA-seq (MULTI-seq) (McGinnis et al. 2019) and purified by FACS (STAR Methods), cancer cells were subjected to scRNA-seq analysis to measure cell state, lineage, sample identity, and tumor clonality. After integrating all four datasets for each cell (Fig 1C; STAR Methods), we proceeded with paired lineage and transcriptome measurements for 40,386 cells with a median of 9,680 UMIs and 2,877 genes detected across 35 tumors (29 primary tumors and 6 metastases; a median of 511 cells were detected per primary tumor). Importantly, target sites were consistently expressed across tumors (Fig 1D, S1F-G).
After preprocessing target site data based on lineage-tracing sequencing quality control and ensuring tumor clonality with lenti-Cre-BC information (Fig 1C; STAR Methods), we reconstructed phylogenies for each tumor with Cassiopeia (Jones et al. 2020). Figure 1E displays the inferred phylogeny and its corresponding indel status (summarized in an “allele heatmap”) of a single representative tumor, consisting of 772 cells. The resulting tree revealed a rich subclonal structure and deep lineage relationships, with a median depth of 12 and maximum depth of 15. As a validation of the integrity of our lineage reconstruction, we observed strong correlations between phylogenetic and allelic distances across our trees (Fig 1F; Table S1). With these high-resolution tumor phylogenies, we next turned to studying the relationship between subclonal dynamics and cellular state as determined by gene expression.
Rare subclones expand during tumor progression, marked by increased DNA copy number variation, cell cycle score, and fitness score
A key question in tumor evolution is how subclonal selection, based on the acquisition of growth-promoting genetic or epigenetic changes, and the resulting population dynamics lead to the expansion of aggressive subclones relative to other parts of the same tumor (Nowell 1976; McGranahan and Swanton 2017; Davis et al. 2017; Sottoriva et al. 2015). To examine the subclonal dynamics in KP tumors, we adapted a statistical test that compares the relative size of each subclone to what would be expected in a “neutral” model of evolution where no subclone is under selection (STAR Methods; (Griffiths and Tavaré 1998; Speidel et al. 2019)). Using this method on a high-quality subset (21/29) of primary tumors (Fig S1H; STAR Methods), we found examples of tumors that appeared to be neutrally evolving (i.e., with no evidence for positive selection) and tumors with subclones showing clear signs of positive selection (Fig 2A). Tumors predominantly had one or sometimes two subclones undergoing expansion, and across tumors there was a broad distribution in the proportion of cells within expansions (Fig 2B). The proportion of expanding cells in each tumor was poorly explained by individual technical covariates, including the age of the tumor (R2=0.25±0.14), the depth of the tumor phylogeny (R2=0.23±0.15), the number of cells in the tumor (R2=0.09±0.07), and the proportion of unique cell lineage states (R2=0.28±0.15, Fig S2A-D); though an additive linear model with all of these covariates was a stronger predictor (R2=0.52).
Figure 2. Rare subclones expand during tumor progression, marked by increased DNA copy number variation, cell cycle score, and fitness score.

(A) Example tumor phylogenies with expansions highlighted with red or purple branches. (B) The number of expansions and percentage of expanding cells across tumors. Tumors are ranked by the total percentage of cells in expanding subclones. (C) CNV numbers per cell (outer bar) in expanding (red) versus non-expanding (black) cells of an example tumor. (D) Comparison of CNV number per cell in expansions versus non-expansions (Permutation test, p<0.0001). (E) Comparison of cell cycle transcriptional scores of cells from the expanding and non-expanding subclones (two-sided Mann-Whitney U test, * p<0.05, ** p<0.01). Tumors without expansions are labeled as N/A. (F-H) Phylogenetic single-cell fitness scores in expansions. (F) A representative tumor phylogeny with single-cell fitness scores overlaid. (G) Single cell fitness scores in representative tumors. (H) Cancer cells from expansions have significantly higher single-cell fitness scores (two-sided Mann-Whitney U test, p < 0.0001).
See also Figure S2.
Several lines of evidence support the accuracy of the inferred phylogenies and subclonal dynamics. First, lineage trees inferred by an alternative phylogenetic reconstruction algorithm, Neighbor Joining, revealed consistent subclonal expansion proportions (Saitou and Nei 1987; Pearson’s ρ = 0.87, Fig S2E). Second, copy number variation (CNV) - a common feature for inferring subclonal structure in tumors (Tarabichi et al. 2021) - corroborated tumor subclonal structure. Specifically, despite the low-resolution lineages inferred from detected CNVs, in the majority of tumors (20/21) the relationships from subclonal CNVs were significantly similar to the relationships inferred from our Cas9 lineage-tracing trees (Fig S2G-I; Permutation Test; see STAR Methods). Furthermore, expanding subclones were significantly enriched for CNVs (Mann-Whitney U Test p < 0.0001, Fig 2C-D and Fig S2J) and independent subclonal expansions from the same tumor could harbor distinct CNV patterns (Fig S2K). Third, cancer cells in expansions had significantly higher expression of cell-cycle genes (Mann-Whitney U test; Fig 2E, S2F; STAR Methods). Together with our tumor spatial-lineage analysis (see below), these orthogonal data strongly support the fidelity of our tumor phylogeny and expansion calling and indicate the aggressive nature of subclonal expansions.
In population genetics, the relative “fitness” of a sample can be defined as the growth advantage of an individual compared to the rest of the population (Williams et al. 2018). The fine-scale structure of our lineages offers us the opportunity to predict fitness at single-cell resolution (Fig 2F; STAR Methods; Neher et al. 2014). This analysis revealed a spectrum of intratumoral fitness distributions across tumors (Fig 2G) with expanding cells consistently having higher single-cell fitness scores (Mann-Whitney U Test p < 0.0001, Fig 2F and 2H). Overall, these results argue that we can quantitatively infer the relative fitness of individual cells within a tumor and that cell fitness is consistent with the subclonal dynamics revealed by the tumor phylogeny.
Integration of phylodynamics and transcriptome uncovers fitness-associated gene programs for KP tumors
With quantitative measurements of single-cell fitness in each tumor, we next sought to identify the molecular features consistently associated with subclonal expansions. Consistent with KP tumor progression being driven largely by epigenetic rather than genetic changes (LaFave et al. 2020; Arnal-Estapé et al. 2020; Marjanovic et al. 2020), we observed that CNV profiles within expansions were largely inconsistent across tumors (Fig S2L). We therefore examined the transcriptomic differences underpinning expansion. By integrating the scRNA-seq data across tumors, we detected 15 distinct subpopulations characterized by marker genes consistent with previous work in the KP model: spanning from an early-stage Alveolar type 2 (AT2)-like population, characterized by expression of Lyz2 and Sftpc, to late-stage Epithelial-Mesenchymal transition (EMT)-related clusters characterized by expression of Vim, Twist1, and Zeb2 ((Marjanovic et al. 2020; LaFave et al. 2020); Fig 3A, S3A; Table S2). Notably, while normal AT2 cells appeared similar to the tumor AT2-like state, the transcriptome of cancer cells could be clearly distinguished from normal AT2 cells (Fig S3B; STAR Methods). Together, the agreement of transcriptomic states observed here and in previous studies implies that the continuous lineage tracing system did not strongly perturb tumor progression.
Figure 3. Integration of phylodynamics and transcriptome uncovers fitness-associated gene programs for KP tumors.

(A) Gene expression UMAP and clustering of cancer cells from KP-Tracer tumors. (B-C) Identification of a transcriptional FitnessSignature. (B) Differential expression analysis identifies genes positively (red) and negatively (blue) associated with single-cell fitness (C) Meta-analysis of fitness-associated genes across all KP tumors. (D) Gene expression UMAP annotated by individual cells’ single cell FitnessSignature scores (normalized to a 0–1 scale). (E) Average FitnessSignature scores of each Leiden cluster (normalized to 0–1). Colors reflect the Leiden clusters in (A). (F) Kaplan-Meier survival analysis of TCGA lung adenocarcinoma patients (n=495) stratified into high (red) and low (blue) groups based on gene expression of the derived transcriptional FitnessSignature. (Log-rank test, p=5e-4). (G) Gene expression UMAP annotated with transcriptional scores of the three fitness gene modules. (H) Heatmap of Z-normalized Pearson’s correlations between marker gene expression and fitness module scores for selected differentially expressed genes with manual annotations. Genes are colored by assigned fitness gene module; genes in black indicate helpful markers that did not appear in a fitness module. (I) Personality plots of three representative tumors displaying the fold change in fitness module scores of individual expansions compared to the non-expanding regions. Vertices indicate individual fitness modules. Axes are normalized to 0.4 – 2.2-fold change observed across tumors. Inner circle represents a fold change of 1 (no change) and values greater than 1 indicate the cells in expansions exhibiting enriched usage of the particular fitness gene module. Colors (see (H)) reflect the module a tumor expansion is characterized by.
Combining the aforementioned single-cell fitness scores with single-cell transcriptomes for each tumor, we next identified genes associated with changes in fitness for each tumor (Fig 3B; STAR Methods). We then utilized a majority-vote meta-analysis of differentially expressed genes across tumors to find genes consistently associated with fitness differences (Fig 3C; STAR Methods; Table S3). The resulting consensus genes associated with elevated fitness revealed broad transcriptomic changes and were enriched for gene sets associated with ribosome biogenesis, stem cell differentiation, and wound healing (Table S3).
The genes detected in our majority-vote meta-analysis represented a transcriptional program (hereafter referred to as the “FitnessSignature”) consistently associated with tumor expansions that could be used to describe state trajectories underlying tumor evolution. Indeed, the AT2-like cluster had the lowest FitnessSignature score while the Mesenchymal clusters scored highest (Fig 3D-E; STAR Methods). Interestingly, the ranking of Leiden clusters in between these extremes suggested that an increase in FitnessSignature was concomitant with dedifferentiation from the AT2-like state through various Gastric, Endoderm-like, or Lung Mixed states to an eventual Mesenchymal state (Fig 3D-E). Importantly, the FitnessSignature scores were significantly associated with poor prognosis in lung adenocarcinoma patients from The Cancer Genome Atlas (TCGA; The Cancer Genome Atlas Research Network 2014; Fig 3F; STAR Methods).
Consistent with previous studies showing increased transcriptional heterogeneity during KP tumor evolution (Marjanovic et al. 2020), we observed that tumors occupied qualitatively different transcriptional states (Fig S3E). This progression could be categorized into three non-overlapping gene modules decomposed from the FitnessSignature (Fig S3F-G; STAR Methods): Module 1 contained genes enriched for gastric and endoderm signatures (Tff1, Hnf4a, Gkn2), Module 2 contained a subset of EMT marker genes and some neuronal genes (Hmga2, Inhba, Gap43) and Module 3 contained classical mesenchymal and pro-metastasis genes (Vim, Twist1, Cdh2, Cd109, Runx2) (Fig 3G-H; Table S3). Additionally, tumor subclonal expansions could preferentially employ a particular module, though some expansions exhibited co-expression of multiple modules (Fig 3I, S3I-J; STAR Methods). Importantly, the expression of each of these modules was predictive of worse patient survival in the TCGA lung adenocarcinoma cohort (Fig S3H; STAR Methods). Collectively, these results argue that increased cell fitness in lung adenocarcinoma can be achieved via at least three distinct transcriptional modules.
Intratumoral transcriptional heterogeneity is driven by transient increases in plasticity of cell states
We next investigated the dynamics of intratumoral transcriptional diversity, as such behavior is can be a driver of tumor aggressiveness and therapeutic resistance (Patel et al. 2014; Rathert et al. 2015; Shaffer et al. 2017; Kim et al. 2018; Marjanovic et al. 2020; Maynard et al. 2020). In our model, tumors varied widely in the transcriptional states they occupied, rarely being dominated by a single state. While tumors with low FitnessSignature scores were enriched for the AT2-like state, increases in the Fitness score were associated with Gastric-like, Lung Mixed, and Mesenchymal states (Fig S4A). Moreover, tumors had generally similar levels of transcriptional state heterogeneity, as measured by Shannon’s Entropy Index ((Marjanovic et al. 2020; LaFave et al. 2020); Fig S4B).
How is this intratumoral diversity established and maintained? In principle, this diversity reflected by the entropy index can be achieved either by rare transitions and stable commitment to distinct states or by frequent transitions between these states. Lineage tracing is uniquely positioned to distinguish these two models as it directly reports how intermixed transcriptomic states are in subclonal lineages, thus providing a measure of effective plasticity. Interestingly, tumor subclones exhibited varying amounts of plasticity: some tumor subclones were dominated by a single transcriptomic state, suggesting strong stability (Fig 4A), while others were characterized by strong mixing between transcriptomic states (Fig 4B). Using tumor phylogenies, we estimated the frequency of cellular state changes for each tumor to create an empirical measurement of the tree plasticity (hereafter referred to as the “EffectivePlasticity” score) and extended this measure to a single-cell statistic (“scEffectivePlasticity”) by averaging together the EffectivePlasticity scores for all the subclades that contained a particular cell (Quinn et al. 2021; STAR Methods). Importantly, this scEffectivePlasticity statistic was consistent with alternative approaches that quantified the effective plasticity by comparing transcriptional states between cells with similar indel states (without relying on trees; Fig S4C-E) or by computing dissimilarity in gene expression profiles between nearest neighbors on the phylogeny (Fig S4F-H; STAR Methods).
Figure 4. Intratumoral transcriptional heterogeneity is driven by transient increases in plasticity of cell states.

(A-B) Representative tumors with (A) low EffectivePlasticity and (B) high EffectivePlasticity. Outer bar indicates the Leiden cluster of single cells (as in 3A). Selected clades are highlighted on the gene expression UMAP to the right of phylogenies. (C-D) Quantification of scEffectivePlasticity for each transcriptional state (Leiden cluster) for tumors in (A) and (B). Each dot represents a single cell’s EffectivePlasticity. (E) Distribution of mean EffectivePlasticity scores for each Leiden cluster across KP tumors. Each dot represents a Leiden cluster’s mean EffectivePlasticity within a tumor. Leiden clusters are ranked by the mean of the distribution across tumors. (F) scEffectivePlasticity score overlaid onto the gene expression UMAP. Cells marked in grey are from metastases and not included. (G) Relationship between tumor average FitnessSignature and EffectivePlasticity. Three representative phylogenies are displayed with Leiden cluster annotations (outer circle). (H) A model describing changes of transcriptome heterogeneity and EffectivePlasticity following tumor progression.
See also Figure S4.
In two representative tumors, we observed that cells from the AT2-like state exhibited consistently low scEffectivePlasticity, whereas other states like the Gastric- and AT1-like state had elevated scEffectivePlasticity scores (Fig 4C-D). To systematically quantify the relative effective plasticity of different cell states, we averaged scEffectivePlasticity scores for each Leiden cluster on a tumor-by-tumor basis (Fig 4E). Mesenchymal (Leiden clusters 1 & 2) and AT2-like clusters (Leiden cluster 4) represented the most stable states, while the previously reported “High Plasticity Cell State” (Marjanovic et al. 2020; Leiden cluster 5) , Gastric-Like (Leiden clusters 3, 8, 12) and Endoderm-like states (Leiden cluster 0) exhibited high EffectivePlasticity (Fig 4F).
We next investigated the relationship of tumor plasticity, as measured by EffectivePlasticity, and aggressiveness, as measured by the FitnessSignature. While previous studies have indicated that transcriptional heterogeneity is a hallmark of tumor progression (Marjanovic et al. 2020), we found that the average EffectivePlasticity score was maximized when the FitnessSignature score was in the intermediate regime and minimized when the FitnessSignature was on the low or high extremes (Fig 4G and S4I-J). Taken together, these findings support a model of tumor progression whereby loss of AT2-like state unlocked high plasticity enabling rapid, parallel transitions to generate high transcriptomic heterogeneity, which permitted selection of increasingly stable states with higher-fitness and ultimately resulted in subclonal expansion and tumor progression (Fig 4H).
Mapping the phylogenetic relationships between cell states reveals common paths of tumor evolution
In principle, the observed cellular plasticity and subsequent transcriptional heterogeneity in the KP model could arise from either random or structured evolutionary paths through transcriptional states. To investigate the consistency of evolutionary paths across tumors, we developed a statistic termed “Evolutionary Coupling”, which extends a clonal coupling statistic (Weinreb et al. 2020; Wagner et al. 2018) to quantify the phylogenetic distance between pairs of cell states (STAR Methods).
Applying this approach to individual tumors uncovered distinct coupling patterns between transcriptomic states. In one example tumor, the Lung Mixed state was more closely related to the High Plasticity state than to the AT2-like state (Fig 5A-B). In another tumor, the Gastric-like and High Plasticity states clustered together, while the AT1-like and Early Gastric states clustered together (Fig 5C-D). Relationships for these two tumors were consistent with alternative definitions for inter-state coupling, inferred directly from the indel information (without relying on trees; Fig S5A-B; STAR Methods) or based on local neighborhoods on the tree (Fig S5C-D; STAR Methods); these statistics were generally consistent across trees (Fig S5E).
Figure 5. Mapping the phylogenetic relationships between cell states reveals common paths of tumor evolution.

(A-D) Transcriptional state relationships of representative tumors are quantified with Evolutionary Couplings. (A, C) Phylogenies of tumors 3435_NT_T1 and 3513_NT_T3 with overlaid Leiden cluster annotations (colors from Fig 3A). (B, D) Corresponding normalized Evolutionary Couplings between Leiden clusters in each tumor. (E) UMAP projection of KP tumor Evolutionary Couplings annotated by identified “Fate Clusters” (see Fig S5F). Dots correspond to tumors. (F) Aggregated Evolutionary Couplings between transcriptional states of tumors from each Fate Cluster visualized on the gene expression UMAP. Thickness of bars reflect the average magnitude of couplings across tumors in a Fate Cluster. (G) Gene expression UMAP annotated by Phylotime of single cells from tumors in Fate Cluster 1 (top) and 2 (bottom) (normalized to 0–1). Cells from tumors that do not appear in the Fate Cluster of interest are shown in gray. (H) Significant gene expression changes along Phylotime for Fate Cluster 1 and 2 across Phylotime quantiles. Genes are annotated by their assigned Fate Cluster. Colors in heatmap are library-normalized gene expression, Z-normalized across quantiles of both Fate Clusters. (I) Summary of major paths of KP tumor progression. Solid lines indicate direct evidence of Evolution Couplings; dotted lines indicate couplings likely involving unobserved intermediate states; gray lines indicate couplings that are supported by rare examples.
A data-driven hierarchical clustering of the full set of tumors based on their transcriptional state occupancy and Evolutionary Couplings revealed that tumors could be classified into three distinct groups (“Fate Clusters”; Fig 5E and S5F; STAR Methods; Table S4). While some transcriptional states were shared between Fate Cluster 1 and 2 (including the AT2-like, AT1-like, and High-Plasticity states), Fate Cluster 1 was predominantly distinguished by couplings that include the Gastric-like (Leiden clusters 3, 8, and 12) and Endoderm-like states (Leiden cluster 0; Fig 5F, left, Fig S5G) and Fate Cluster 2 by evolution towards the Lung Mixed state (Leiden cluster 10; Fig 5F, middle, Fig S5G). Fate Cluster 3 was more difficult to interpret as it lacked couplings with the AT2-like state and instead was dominated by high-fitness states, such as early EMT (Leiden clusters 7 and 13) and Mesenchymal states (Leiden cluster 1 and 2; Fig 5F, right, Fig S5G).
We thus hypothesized the majority of differences between tumors was driven by tendencies towards Fate Cluster 1 or 2. Indeed, Principal Component Analysis (PCA) on Evolutionary Couplings and state composition revealed that the first two principal components explained a substantial amount of the observed variance (~32%; Fig S5H) and couplings involving the Gastric & Endoderm states (Fate Cluster 1; Leiden clusters 3, 8, 0) or the Mixed Lung state (Fate Cluster 2; Leiden cluster 10) were among the strongest features distinguishing tumors (Fig S5I). Taken together, these distinct coupling patterns argue that tumor progression from the initial AT2 state preferentially follows one of two non-overlapping evolutionary paths, characterized by Fate Clusters 1 and 2, to aggressive states like those found in Fate Cluster 3.
To characterize the transcriptional changes that underlie these two alternative fates (Fate Cluster 1 & 2), we developed “Phylotime”: a single-cell statistic that quantifies the evolutionary distance between an individual cell and cells in the progenitor, AT2-like state (STAR Methods). Importantly, estimates of Phylotime were consistent with different metrics for approximating distances on the tree: either by the absolute number of mutations or the number of mutation-bearing edges (Fig S5J-K). Integrating Phylotimes from tumors within Fate Clusters 1 and 2 confirmed two separate evolutionary routes (Fig 5G) and highlighted distinct transcriptional changes associated with Phylotime along each route (Fig 5H; STAR Methods; Table S5). Specifically, while expression of early markers like Lyz2 and Sftpc were shared in early Phylotime of both Fate Clusters, late Phylotime in Fate Cluster 1 was enriched for gastric and endoderm markers like Gkn2, whereas late Phylotime in Fate Cluster 2 was characterized by markers of airway progenitors, such as Sox2 and Scgb1a1, and markers of tumor propagating cells, like Cd24a and Itgb4. Although Fate Cluster 3 tumors generally had poor couplings with earlier states, our data suggest that tumors can evolve from either the Fate Cluster 1 or Fate Cluster 2 into an EMT state and progress to late-stage Mesenchymal states (Fig S5L). Overall, our analysis provides evidence that KP tumors could evolve predominantly through one of two major paths with one towards Gastric-like and Endoderm-like state, and the other through the Mixed-Lung state, with distinct transcriptional changes associated with each evolutionary trajectory (summarized in Fig 5I).
Loss of tumor suppressors alters tumor transcriptome, plasticity and evolutionary trajectory
Tumor suppressor genes regulate diverse cellular activities and their loss is associated with increased tumor aggressiveness (Weinberg 1991; Sherr 2004); however, it remains unclear how these genes affect tumor evolutionary dynamics in vivo. Here, we combined genetic perturbations with our quantitative phylodynamic approaches to interrogate how additional oncogenic mutations altered KP tumor evolutionary trajectories.
We focused on two frequently mutated tumor suppressors in human lung adenocarcinoma, LKB1 and APC (Ding et al. 2008; The Cancer Genome Atlas Research Network 2014; Skoulidis et al. 2015). Both genes have been studied extensively in both human and mouse models and appear to regulate progression through distinct mechanisms (Ji et al. 2007; Carretero et al. 2010; Nguyen et al. 2009; Hollstein et al. 2019; Tammela et al. 2017; Murray et al. 2019; Kerk et al. 2021; Parsons et al. 2021). We engineered our lenti-Cre-BC vector to carry an additional sgRNA targeting Lkb1 or Apc, such that delivery of this vector simultaneously initiated tumor induction, lineage tracing, and disruption of the targeted tumor suppressor gene. With this system, we collected data from 18,321 cells across 57 KP tumors with Lkb1 knockout (24 primary and 33 metastatic tumors; referred to as KPL tumors), and 13,825 cells across 35 KP tumors with Apc knockout (23 primary and 12 metastatic tumors; referred to as KPA tumors). Targeting of either Lkb1 or Apc increased tumor burden (Rogers et al. 2018), but did not appear to alter the number and relative size of subclonal expansions (Fig S6A-B). Yet, genes associated with tumor fitness were largely distinct across genetic backgrounds (Fig S6C; Table S3).
To examine whether perturbations alter the transcriptional landscape of KP tumors, we integrated transcriptional states of KPL and KPA tumors with the prior KP dataset. While many cells could be classified into existing Leiden clusters identified in the KP analysis, the additional perturbations also created four new transcriptional states (Fig 6A; STAR Methods). As expected from Apc’s role as a negative regulator of Wnt signaling (Barker et al. 2009), Axin2 expression was high in the three KPA-specific clusters, indicative of elevated Wnt signaling (Fig S6D), as was the expression of Wnt antagonists such as Notum and Nkd1 which were recently reported to increase the ability of cancer cells to compete with the neighboring niche in human APC mutant colon tumors ((Flanagan et al. 2021; van Neerven et al. 2021); Fig S6D; Table S3). Moreover, targeting of Lkb1 or Apc resulted in changes to the relative occupancies of transcriptomic states: KPL tumors were primarily enriched in the Pre-EMT state (Leiden cluster 9), while KPA tumors were enriched in Apc-specific early, mesenchymal, and metastatic states (Leiden clusters 15, 16, and 17; Fig 6B-C and S6E).
Figure 6. Loss of tumor suppressors alters tumor transcriptome, plasticity and evolutionary trajectory.

(A) Batch corrected and integrated gene expression UMAP of all cancer cells from KP, KPL and KPA tumors annotated by 19 Leiden clusters (STAR Methods). (B) Density plots of cancer cells from KP, KPL and KPA tumors on the UMAP. (C) Enrichment of genotypes in each Leiden cluster. Enrichments below 1 are colored blue; enrichments above 1 are colored red. (D) Median EffectivePlasticity scores in selected Leiden clusters across genotypes (one-sided Mann-Whitney U Test, *p≤0.05, n.s. = not significant). (E) Genes up-regulated (red) and down-regulated (blue) in the Pre-EMT state of KPL tumors compared to KP and KPA tumors combined. (F) PCA of Evolutionary Coupling and transcriptional state proportion vectors for all tumors analyzed across genotypes. Each dot represents a tumor.(G) Biplot of top 10 features per principal component from PCA analysis shown in (F). Evolutionary Couplings are shown as tuples (x, y); transcriptional state proportions are shown as a single number x indicating Leiden cluster ID. (H) Summary of major evolutionary paths in KPL and KPA tumors. Solid lines indicate direct evidence of Evolution Couplings between transcriptome states, dotted lines indicate couplings that likely involve unobserved intermediate cell states.
Interestingly, although most cell states had comparable EffectivePlasticity across tumor genotypes (Fig S6F), the Pre-EMT state (Leiden cluster 9) in KPL tumors had significantly less EffectivePlasticity, indicating stabilization of this cell state (p < 0.05, Mann-Whitney U Test; Fig 6D). We next identified genes differentially expressed in cells from KPL tumors in the Pre-EMT cluster (Fig 6E; Table S2; STAR Methods), which included gene programs that can promote pro-metastatic chromatin remodeling (Sox17; Pierce et al. 2021), tumor progression (Ifitm1 and loss of Gata6; Yan et al. 2019; Cheung et al. 2013), metastatic ability (Mmp7; He et al. 2018), and tumor fitness by modulating cancer-immune cell interaction (Cd24a, Il33, and loss of Apoe; Sinjab et al. 2021; Li et al. 2019; Tavazoie et al. 2018). These together potentially explain why the Pre-EMT state was uniquely stabilized in KPL tumors.
To examine how loss of tumor suppressors altered evolutionary trajectories, we performed PCA on the transcriptional state occupancy and Evolutionary Couplings of individual tumors and found that tumors broadly segregated according to their genotypes (Fig 6F; STAR Methods; Table S4). Specifically, KPA tumors created a unique trajectory including a coupling between the AT2-like and the Apc-early states (Leiden clusters 4 and 16), while KPL tumors were characterized by couplings between the Pre-EMT state and nearby states (Fig 6G).
In summary, although the targeting of the tumor suppressors Lkb1 or Apc both increased tumor growth, their effects on cell states, plasticity and paths of evolution varied substantially. Specifically, KPL tumors quickly progressed to and became stabilized in the Pre-EMT state, while KPA tumors largely exploited a distinct path through new Apc-specific states (Fig S6G and summarized in Fig 6H and Table S4). Together, our analyses highlight how lineage tracing offers rich information for dissecting the multifaceted role of tumor suppressors in tumor evolution.
Metastases originate from spatially localized, expanding subclones of primary tumors
Metastases account for 90% of cancer mortality yet remain difficult to study because of their spatially and temporally sporadic nature (Ganesh and Massagué 2021). An outstanding question is how metastases originate from the primary tumor. Here we integrated lineage tracing with spatial and transcriptomic information to investigate the subclonal origins and evolution of metastases.
We first focused on a single primary tumor, which consisted of two independent subclonal expansions (3724_NT_T1; Fig 2B), and its four related metastases (three in liver and one in soft tissue; Fig 7A, S7A). We performed multi-regional analysis of the primary tumor (Fig 7A, inset) and inferred a combined phylogeny relating all cells in the primary tumor and metastases. Integrating lineage-spatial information revealed that individual metastases originated from distinct spatial locations (Fig 7A-C; STAR Methods), and phylogenetically originated from specific subclonal expansions in the primary tumor (Fig 7C-D).
Figure 7. Metastases originate from spatially localized, expanding subclones of primary tumors.

(A) Multi-region analysis of tumor-metastasis family 3724_NT_T1. Top left inset showed the relative spatial location of tumor pieces. The phylogeny of the primary tumor and metastases is annotated via peripheral radial tracks for each color-coded region of the tumor (matching the inset) and four metastases. (B) Heatmap of Evolutionary Couplings of primary tumor pieces (black) and 4 related metastases (matching colors in (A)) from the 3724_NT_T1 tumor-metastasis family.
(C) Summary of the spatial-phylogenetic relationship of the tumor-metastasis family 3724_NT_T1. (D) Single-cell phylogenetic distance of each metastasis to the non-expanding and expanding subclones in its related primary tumor. Each box represents the distribution of phylogenetic distances from a metastasis to a defined region of its related primary tumor (one-sided Mann-Whitney U test are indicated: ***p<0.0001, n.s. = not significant). (E-F) Gene expression UMAP annotated by metastases and their original subclones in 3724_NT_T1. Cells that are not relevant to the comparison in each panel are shown in gray. (G) Transcriptional distances between expanding regions of 3724_NT_T1 and its four metastases (one-sided Mann-Whitney U test are indicated: **p < 0.001, ***p<0.0001).
See also Figure S7.
To investigate the consistency of these results, we extended this phylogenetic analysis to five other tumor-metastasis families, across KP, KPL, and KPA backgrounds. Importantly, metastases were consistently more closely related phylogenetically to specific subclonal expansions regardless of tumor genotype (Fig 7D and Fig S7D). Collectively, our results argue that metastases generally originated from subclonal expansions within primary tumors. Independent metastases from the same primary tumor could arise from spatially and phylogenetically distinct subclones.
We next evaluated to what degree metastases preserved the transcriptional state of their origins in the primary tumor. Analysis of metastases arising from an example primary tumor (3724_NT_T1) revealed that liver metastases were more similar to the subclone from which they originated, whereas the soft tissue metastasis evolved to a new transcriptional state (Fig 7E-F). This was further quantified by measurements of total transcriptional distance between each metastasis and the subclonal expansions in the metastatic primary tumor (Fig 7G). Liver metastases were significantly more similar to its originating subclonal expansion (p < 0.0001, one-sided Mann-Whitney U Test), while the soft tissue metastasis did not clearly resemble its subclonal origin (Fig 7G; STAR Methods). Consistently, metastases from KP, KPL, and KPA mice were significantly more similar, as measured by transcriptional state, to their respective expanding subclades in the primary tumor as compared to non-expanding regions, further suggesting that progression at the primary site is a prerequisite for metastasis (LaFave et al. 2020; Fig S7E).
In addition, our high-resolution lineage tracing offered evidence of complex metastatic behaviors, including multi-subclonal seeding from a primary tumor to the lymph node, and cross-seeding from one metastatic primary tumor to another primary tumor, or from one metastasis to another (Fig S7A-C). Collectively, these results highlight the ability of phylogenetic analysis to trace the origins and evolution of metastases.
DISCUSSION
In this study, we have developed a genetically engineered mouse model of lung adenocarcinoma that allows Cre-inducible initiation of oncogenic mutations and simultaneous continuous in vivo lineage tracing of tumor development over many months, paired with a single-cell transcriptomic readout. This model system enabled us to track at an unprecedented resolution the recurring patterns of tumor evolution from activation of oncogenic mutations in single cells as they grow into large, aggressive, and ultimately metastatic tumors. Three principles emerged from our study, linking together tumor phylodynamics, fitness, plasticity, parallel evolutionary trajectories, origins of metastasis, and genetic determinants of tumor evolution.
First, tumors were driven by rare subclonal expansions that utilized distinct fitness-associated transcriptional programs and enabled both tumor progression at the primary site and metastasis to distant tissues. The expansions identified by tree topology argue for subclonal selection, distinct from evolutionary models lacking selective sweeps observed in other cancer types (Sottoriva et al. 2015). The identification of gene expression states associated with tumor fitness revealed a set of transcriptional fitness modules underlying KP-Tracer tumor development. Importantly, these signatures of aggressive tumors found in our mouse model were predictive of the outcome of human disease. Despite the higher somatic mutation burden and longer developing timescales of human tumors (Campbell et al. 2017; Jamal-Hanjani et al. 2017; Gerstung et al. 2020; Hill et al. 2021), our data uncovered critical fitness gene programs that are conserved in both mouse and human lung adenocarcinomas. Notably, we found that metastases consistently originated from expanding subclones, regardless of additional loss of Lkb1 or Apc. They often retained the same transcriptional state as their original subclones but could further adopt distinct transcriptional states. This underscored the importance of tumor progression at the primary site in enabling metastasis (Caswell et al. 2014; Turajlic and Swanton 2016; Hu et al. 2020; LaFave et al. 2020), and argues against alternative models in which metastases arise early during tumor evolution (Hüsemann et al. 2008; Podsypanina et al. 2008; Klein 2009; Rhim et al. 2012; Sottoriva et al. 2015).
Second, our analysis revealed that tumor progression is accompanied by transient increases in lineage plasticity. This period of high plasticity is followed by clonal sweeps of subclones with aggressive cell states that can remain stable even following metastasis to new environments. Our ability to monitor how often cells are transitioning between transcriptomic states also allowed us to untangle the relationship between intratumoral heterogeneity and lineage plasticity, and shed light on the dynamics of the transcriptomic heterogeneity observed in the KP mouse model and human NSCLC (Marjanovic et al. 2020; Laughney et al. 2020). The finding that KP tumors progress via parallel, rapid transitions between cell states is consistent with previous work suggesting that epigenetic instability is a major driver of tumor progression in this model (LaFave et al. 2020; Marjanovic et al. 2020). Given the essential role of cellular plasticity in tumor progression and therapeutic resistance (Chaffer et al. 2013; Easwaran et al. 2014; Ge et al. 2017; Flavahan et al. 2017; Yuan et al. 2019; Quintanal-Villalonga et al. 2020), the ability of our lineage tracing system to quantitatively explore plasticity provides a critical tool for understanding the role that cell state plasticity plays in various aspects of tumor evolution.
Third, tumors evolved through stereotypical trajectories and introduction of additional oncogenic mutations increased the speed of tumor evolution by creating new evolutionary trajectories. Traditionally, while cellular trajectories inferred by pseudotemporal approaches have proved to be a versatile tool for scRNA-seq datasets (Trapnell et al. 2014; La Manno et al. 2018), they make the inviolable assumption that transcriptional similarity indicates developmental relationship (Tritschler et al. 2019). Overcoming this, our measurement of cell state coupling directly from phylogenies enabled the discovery of two distinct evolutionary paths that are substantiated by transcriptional differences. Moreover, CRISPR targeting of tumor suppressors Lkb1 and Apc altered the cellular plasticity and observed evolutionary paths in a genotype-specific way, which can be explained by alterations in transcriptional landscape. Collectively, our approach offers an orthogonal and more quantitative evaluation of the multifaceted role genes play in tumor evolution as compared to traditional growth-based fitness analysis. Future studies combining the KP-Tracer model and high-throughput in vivo functional genomics will be foundational in assessing the evolutionary consequences of any genes of interest in lung adenocarcinoma progression (Winters et al. 2018).
In summary, our results represent the first report of tracing the evolutionary history of a tumor from a single transformed cell to an aggressive tumor using a CRISPR-based lineage tracer in an autochthonous mouse model. The continuous and high-resolution tumor lineage tracing in this setting offers a major advance in tumor evolution modeling by enabling quantitative inference of fitness landscapes, cellular plasticity, evolutionary paths, origins of metastases, and the role of tumor suppressors in altering all these facets of tumor development. With the expanding lineage tracing toolkit and integration of other emerging data modalities, we expect that the experimental and computational framework presented here will greatly improve future efforts at building high-dimensional, quantitative, and predictive models of tumor evolution, thus shedding light on new therapeutic strategies.
Limitations of the study
Our findings highlight several opportunities for future efforts. First, we were limited in our ability to describe the directionality of transitions or to rule out the possibility of unobserved intermediates. This issue could be resolved experimentally by harvesting samples from multiple time points of tumor development, or expanding our lineage-tracing technology to develop multichannel molecular recorders for simultaneous recording of marker gene expression of intermediate states (Frieda et al. 2017; Tang and Liu 2018). Alternatively, enhancing the interpretability of branch lengths by engineering a “molecular clock” or probabilistic models of Cas9 editing (Park et al. 2021) could aid in the reconstruction of unobserved intermediate states (Ouardini et al. 2021). Second, our fitness-inference approach assumes that evolution occurs via small effect size mutations, which may overlook the impact of mutations with large impact such as CNVs in other tumor models (Neher et al. 2014). Third, future integration of emerging data modalities with lineage tracing, such as combined genomic, multiomic and spatial analysis (Mimitou et al. 2021; Ma et al. 2020; Lee et al. 2014; Stickels et al. 2021; Chow et al. 2021), will illuminate how genetic and epigenetic changes and the tumor microenvironment influence tumor evolution.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Jonathan Weissman (weissman@wi.mit.edu).
Materials Availability
Plasmids generated in this study are being submitted to Addgene. All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Raw single-cell RNA-sequencing data has been deposited at the NCBI Sequence Read Archive database and are publicly available as of the date of the publication. Accession numbers are listed in the key resources table. Processed single-cell data, reconstructed phylogenies, derived statistics, interactive VISION (DeTomaso et al., 2019) and PhyloVision (Jones et al., 2022) reports have been deposited at Zenodo and are publicly available as of the date of the publication. DOIs are listed in the key resources table.
All original code is available on Github (https://github.com/mattjones315/KPTracer-release) and has been deposited at Zenodo and is publicly available as of the date of the publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Chimeric Lineage Tracing Mouse Model
All mouse experiments described in this study were approved by the Massachusetts Institute of Technology Institutional Animal Care and Use Committee (IACUC) (institutional animal welfare assurance no. A-3125–01). A male mouse embryonic stem cell (mESC) line harboring the conditional alleles KrasLSL-G12D/+ and Trp53fl/fl (KP) was engineered with the lineage tracer cassettes. The engineered and selected mESC clones were injected into blastocysts from albino B6 or CD1 background for chimera making as previously described (Zhou et al. 2010). We chose to use the chimeric mice strategy because the multiple, random integration of lineage tracing target sites in the genome makes it challenging for breeding stable strains. Both male and female mice with more than 10% chimerism based on coat color were used in this study. Tumors were initiated by intratracheal infection of mice with lentiviral vectors expressing Cre recombinase (DuPage, Dooley, and Jacks 2009). Five total mESC clones were used in this study to avoid idiosyncrasy in clonal behavior and analyses were performed on all tumors combined. Lenti-Cre-BC vector was co-transfected with packaging vectors (delta8.2 and VSV-G) into HEK-293T cells using polyethylenimine (Polysciences). The supernatant was collected at 48h post-transfection, ultracentrifuged at 25,000 r.p.m. for 90 min at 4C, and resuspended in phosphate-buffered saline (PBS). 8–12-week-old chimeras were infected intratracheally with lentiviral vectors, including lenti-Cre-BC-sgNT (2×107 PFU) or lenti-Cre-BC-sgLkb1 (4×106 PFU) or lenti-Cre-BC-sgApc (1×107 PFU) to achieve similar aging time after tumor initiation.
METHOD DETAILS
Lenti-sgRNA-Cre-Barcode vector
The lenti_sgRNA_Cre_barcode vector was derived from a previously described Perturb-seq lentiviral vector (Adamson et al., 2016), pBA439, with the following changes: the two loxP sites were removed by site-directed mutagenesis (SDM) using oDYT001 and oDYT002 followed by oDYT009 and oDYT010; the Puro-BFP was removed using restriction sites NheI and PacI and was replaced by Cre that was PCR amplified using oDYT003 and oDYT004 via Gibson assembly; a ubiquitous chromatin opening element (UCOE) that was PCR amplified using oDYT005 and oDYT006 was introduced using restriction sites NsiI and NotI via Gibson assembly. oDYT007 and oDYT008 (containing EcoRI and SbfI sites for subsequent barcode cloning) were then annealed and ligated using restriction sites BclI and PacI. Three different sgRNAs of interest were then cloned into the resulting vector using pairs of top and bottom strand sgRNA oligos: sgNT (non-targeting) (oDYT011 and oDYT012), sgLkb1 (oDYT013 and oDYT014), and sgApc (oDYT015 and oDYT016) were each annealed and ligated using restriction sites BlpI and BstXI to form pDYT003, pDYT004, and pDYT005 respectively. These sgRNAs have been used and validated previously (Rogers et al. 2017, 2018). Finally, a whitelist barcode oligo pool consisting of 249,959 unique 16-nucleotide barcodes where every barcode has a Levenshtein distance of >3 from every other barcode was designed. The whitelist barcode library was PCR amplified then introduced at the 3’UTR region of Cre in each of the three constructs using restriction sites EcoRI and SbfI.
Lineage tracer vector (Target site & triple sgRNAs)
The lineage tracer vectors pDYT001 and pDYT002 were derived from previously described target site plasmids, PCT 60–62 (Chan et al. 2019; Quinn et al. 2021; Jones et al. 2020). A loxP site was first removed from both PCT61 and PCT62 using oDYT017 and oDYT018 via site-directed mutagenesis. The triple sgRNA cassettes driven by distinct U6 promoters in PCT61 and PCT62 were then PCR amplified using oDYT019 and oDYT020 and introduced into the PCT60 backbone using restriction sites XbaI and NotI via Gibson assembly. Finally, the target site barcode library was PCR amplified from a previously described gene fragment from PCT48 (Jones et al. 2020), using oDYT021 and oDYT022 and introduced into the two resulting vectors above using restriction sites PacI and HpaI to form pDYT001 and pDYT002, which contain the triple guide cassette from PCT61 and PCT62 respectively. The target site library consists of a 14-bp random integration barcode and three target sites (ade2, bri1, whtB), which are complementary to the three sgRNAs.
Lineage tracing embryonic stem cell engineering
KP*17 is an embryonic stem (ES) cell line derived from C57BL/6–129/Sv F1 background engineered with conditional alleles KrasLSL_G12D/+; p53fl/fl. ES cells were maintained with JM8 media (500mL: 82.9% Knockout DMEM (Gibco Cat#10829–018), 15% FBS (Hyclone Cat#SV30014), 1% GlutaMax (Gibco Cat#35050–061), 1% Non-essential amino acids (Thermo Fisher Scientific Cat#11140050), 0.1% 2-mercaptoethanol (Sigma Cat#M-7522), 500,000U Recombinant Mouse LIF Protein (Millipore Cat#ESG1107)) with feeders. KP*17 was first targeted using CRISPR-assisted HDR to generate Rosa26LSL-Cas9-P2A-mNeonGreen which was validated for correct targeting by PCR and southern blot and validated for Cas9 activity. The lineage tracing transposon vectors were then introduced together with transposase vector (SBI) by transfection. Three passages after transfection, mESCs were purified by FACS based on mCherry expression and expanded as individual clones.
Target site integration number was quantified as the following: We first used fluorescence-based readout to examine mCherry expression of each ES cell clone in 96 well format, which allowed us to narrow down the ES clone candidates with relatively high expression of mCherry (the reporter of lineage tracer library). Then we used quantitative genomic PCR to count the number of lineage tracer genome integration in each ES cell clone by amplifying the target site regions (oDYT062 and oDYT063) and normalized to a 2N locus, β-actin, in the genome (oDYT060 and oDYT061). Samples were run in triplicates and the reactions were performed on a QuantStudio 6 Flex Real-Time PCR System. In this study, we used the following ES clones in the tumor analysis due to a combination of high chimeric rate and good target site capture: 1D5, 2E1, 1C4, 2F4 and 2H9. Clones 1D5, 1C4 were engineered with pDYT001 and clones 2E1, 2F4 and 2H9 were engineered with pDYT002. All five clones were used independently for generating chimeric mice in this study and no major difference in their lineage tracing performance was observed.
Sample preparation and purification of cancer cells
Tumors were harvested and single cell suspension was prepared as described in (Chuang et al. 2017) and (Denny et al. 2016). Primary tumors and metastases were dissociated using a digestion buffer (DMEM/F12, 5mM HEPES, DNase, Collagenase IV, Dispase, Trypsin-EDTA) and incubated at 37 °C for 30 min. After dissociation, the samples were quenched with twice the volume of cold quench solution (L-15 medium, FBS, DNase). The cells were then filtered through a 40um cell strainer, spun down at 1000rpm for 5 min, resuspended in 2mL ACK Lysing Buffer, and incubated at room temperature for 1–2 min. Lysis was then stopped with the addition of 10mL DMEM/F12 followed by the spinning down and resuspending of the samples in 1mL FACS buffer. Cells within the pleural fluid were collected immediately after euthanasia by making a small incision in the ventral aspect of the diaphragm followed by introduction of 1 ml of PBS. Cells were stained with antibodies to CD45 (30-F11, Biolegend Cat#103112), CD31 (390, Biolegend Cat#102410), F4/80 (BM8, Biolegend Cat#123116), CD11b (Biolegend Cat#101212) and Ter119 (Biolegend Cat#116212) to exclude cells from the hematopoietic and endothelial lineages. DAPI was used to stain dead cells.
Cells were then labeled by MULTI-seq (McGinnis et al. 2019) in 100ul PBS buffer containing 5ul lipid anchor (50uM) and 2.5ul of barcode oligos (100uM) for 10 min on ice and then 6ul co-anchor (50uM) 10 min on ice. Cells were washed and resuspended with ice-cold FACS buffer to prevent aggregation. DAPI was used to exclude dead cells. FACS Aria sorters (BD Biosciences) were used for cell sorting. Live cancer cells were sorted based on positive expression of mCherry and mNeonGreen as well as negative expression of hematopoietic and endothelial lineage markers (mCherry+, mNeonGreen+, CD45-, CD31-, Ter119-, F4/80-, DAPI-). High purity of the resulting cancer cells has been confirmed in previous studies using similar fluorescent reporter systems (Caswell et al. 2014; Chuang et al. 2017; LaFave et al. 2020). Live normal lung cells were sorted based on negative expression of mNeonGreen, and hematopoietic and endothelial lineage markers. Datasets were further filtered for normal cells analytically via gene expression analyses (see section below “Single-cell transcriptome processing for KP-Tracer NT data”) and by removing cells with low editing efficiencies (see section below “Single-cell lineage tracing preprocessing pipeline and quality control filtering”).
Single-cell RNAseq library preparation
Single-cell RNA-seq libraries were prepared using 10x_3’_V2 kit according to the 10x user guide, except for the following modification. After cDNA amplification, the cDNA pool is split into two fractions. Half of the cDNA pool are used for scRNA-seq library construction and proceed as directed in the 10x user guide.
Target site library preparation
To prepare the Target Site libraries, the amplified cDNA libraries were further amplified with Target Site-specific primers containing Illumina-compatible adapters and sample indices (oDYT023-oDYT038, forward:5′CAAGCAGAAGACGGCATACGAGATNNNNNNNNGTCTCGTGGGCTCGGAG ATGTGTATAAGAGACAGAATCCAGCTAGCTGTGCAGC; reverse:5′-AATGATACGGCGACCACCGAGATCTACACNNNNNNNNTCTTTCCCTACACGACGCT CTTCCGATCT; “N” denotes sample indices) using Kapa HiFi ReadyMix (Roche), as described in (Jones et al. 2020). Approximately 30 fmol of template cDNA was used per sample, divided between four identical reactions to avoid possible PCR induced library biases. PCR products were purified and size-selected using SPRI magnetic beads (Beckman) and quantified by BioAnalyzer (Agilent).
MULTI-seq library preparation
The MULTI-seq libraries were prepared as described in (McGinnis et al.), using a custom protocol based on the 10x Genomics Single Cell V2 and CITE-seq workflows. Briefly, the 10x workflow was followed up until complementary DNA amplification, where 1μl of 2.5μM MULTI-seq additive primer (oDYT039) was added to the cDNA amplification master mix. After amplification, MULTIseq barcode and endogenous cDNA fractions were separated using a 0.6X solid phase reversible immobilization (SPRI) size selection. To further purify the MULTI-seq barcode, we increased the final SPRI ratio in the barcode fraction to 3.2X reaction volumes and added 1.8X reaction volumes of 100% isopropanol (Sigma-Aldrich). Eluted barcode cDNA was then quantified using QuBit before library preparation PCR using primers oDYT040 and oDYT041-oDYT048 (95 °C, 5′; 98 °C, 15′; 60 °C, 30′; 72 °C, 30′; eight cycles; 72 °C, 1′; 4 °C hold). TruSeq RPIX: 5′-CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCCTTGGCACCCGAG AATTCCA-3′ TruSeq P5 adaptor: 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATC T-3′ Following library preparation PCR, the library was size-selected by a 1.6X SPRI clean-up prior to sequencing.
Lenti_Cre_BC library preparation
The Lenti_Cre_BC library amplification protocol was adapted from the Perturb-seq protocol (Adamson et al., 2016). 4 parallel PCR reactions were constructed containing 30ng of final scRNA-seq library as template, oDYT049, and indexed oDYT050-oDYT059, and amplified using KapaHiFi ReadyMix according to the following PCR protocol: (1) 95C for 3 min, (2) 98C for 15 s, then 70C for 10 s (16–24 cycles, depending on final product amount). Reactions were repooled during 0.8X SPRI selection, and then fragments of length ~390bp were quantified by bioanalyzer. Lenti_Cre_BC libraries were sequenced as spike-ins alongside the parent RNA-seq libraries.
Sequencing
Sequencing libraries from each sample were pooled to yield approximately equal coverage per cell per sample; scRNA gene expression libraries, Target Site amplicon libraries, MULTI-seq amplicon libraries and Lenti-Cre-BC amplicon libraries were pooled in an approximately 10:3:1:1 molar ratio for sequencing, aiming for at least 70,000 total reads per cell. The libraries were sequenced using a custom sequencing strategy on the NovaSeq platform (Illumina) in order to read the full-length Target Site amplicons. Sample identities were read as indices (I1: 8 cycles, R1: 26 cycles, R2: 290 cycles). Only the first 98 bases per read were used for analysis in the RNA expression libraries to mask the longer reads required to sequence the Target Sites.
QUANTIFICATION AND STATISTICAL ANALYSIS
Single-cell lineage tracing preprocessing pipeline and quality control filtering
Each cell was sequenced in four sequencing libraries: a MULTI-seq library (for identifying sample identity), a target site library (for reconstructing phylogenies), an RNA-seq library (for measuring transcriptional states), and a Lenti-Cre-BC library (for verifying clonal identity). First, the scRNA-seq was processed using the 10X CellRanger pipeline (version 2.1.1) with the mm10 genome build. Then, each cell barcode identified from the 10X pipeline was assigned to a sample using the MULTI-seq library, which was processed with the deMULTIplex R package (version 1.0.2; (McGinnis et al. 2019)). Cells identified as doublets or without a discernible MULTI-seq label were filtered out from downstream analysis.
Next, we processed the Target Site library using the previously described Cassiopeia preprocessing pipeline (Jones et al. 2020; Quinn et al. 2021). Briefly, reads with identical cellBC and UMI were collapsed into a single, error-corrected consensus sequence representing a single expressed transcript. Consensus sequences were identified within a cell based on a maximum of 10 high-quality mismatches (PHRED score greater than 30) and an edit distance less than 2 (default pipeline parameters). UMIs within a cell reporting more than one consensus sequence were resolved by selecting the consensus sequence with more reads. Each consensus sequence was aligned to the wild-type reference Target Site sequence using a local alignment strategy, and the intBC and indel alleles were called from the alignment. Cells with fewer than 2 reads per UMI on average or fewer than 10 UMIs overall were filtered out. These data are summarized in a molecule table which records the cellBC, UMI, intBC, indel allele, read depth, and other relevant information. Cells that were assigned to Normal lung tissue via a MULTI-seq barcode or had more than 80% of their TargetSites uncut were assigned as “Normal” and not used for downstream lineage reconstruction tasks.
Lenti-Cre-BC libraries were processed using a custom pipeline combining Cassiopeia transcript collapsing, filtering, and quantification and a probabilistic assignment strategy based on the Perturb-seq gRNA calling pipeline (Adamson et al. 2016). First, sequencing reads were collapsed based on a maximum sequence edit distance of 2 and 3 high-quality sequences mismatches and then cells with fewer than 2 average reads per UMI or 2 UMIs overall were filtered out. Then, Lenti-Cre-BC sequencing reads were compared to the reference sequence and barcode identities were extracted and error-corrected by comparing each extracted barcode to a whitelist of Lenti-Cre-BC sequences, allowing for an edit distance of 3. Then, the count distributions for each unique Lenti-Cre-BC were inspected to remove barcodes that represented background noise. Next, a Lenti-Cre-BC coverage matrix was formed, summarizing the ratio between reads and number of UMIs for each barcode in each cell. Cell coverages were normalized to sum to the median number of coverages across the matrix and log2-normalized. Finally, with this matrix we adapted the Perturb-seq gRNA calling pipeline to assign barcode identity to cells (Adamson et al. 2016). To do so, we fit a Guassian kernel density function to the coverage distribution for each barcode and then determined a threshold separating “foreground” from “background” based on the relative extrema of the distribution (after removing the 99th percentile of the coverage distribution). Cells whose coverage values fell above the threshold were assigned that particular Lenti-Cre-BC. Cells that received more than one assignment or no assignment at all were marked as ambiguous.
After pre-processing each of these libraries, we called clonal populations, created character matrices, and reconstructed phylogenies for each clonal population (see sections below “Tree Reconstruction with Cassiopeia” and “Calling clonal populations and creating character matrices”). In this, we removed cells that contained few edited sites as this could indicate normal cell contamination (i.e. inactivity of Cas9) and identified consensus sets of intBCs per mES Clone (see section below “Creating a consensus intBC set for mESC clones”) that were used for tree reconstruction. After tree reconstruction, we used the Lenti-Cre-BC data to remove cells within each tumor that contained strong evidence of different clonal origin (see section below “Cell Filtering with Lenti-Cre-BC”). Finally, we computed important clone-level quality-control statistics used for identifying clones with sufficient information for phylodynamic analysis (see section below “Tree Quality Control for Fitness Inference”).
Across all three datasets (KP, KPL and KPA), this pipeline left us with 72,328 cells with high-quality Target Site information.
Calling clonal populations and creating character matrices
In this study, each clonal population corresponded to a primary tumor or metastatic family. Tumors were identified with two approaches: first, by deconvolution with MULTI-seq (and filtering with Lenti-Cre-BC information; see below in section “Cell Filtering with Lenti-Cre-BC”); and second, by separating cells based on differing intBC sets. In the second approach, we used Cassiopeia to identify non-overlapping intBC sets and classify cells using the “call-lineages” command-line tool. Once clonal populations were identified, consensus intBC sets were identified (see “Creating a consensus intBC set for mESC clones” below). All summarized molecular information for a given cell (cellBC, number of UMI, intBC, indel allele, read depth, etc) along with the assigned clonal identity were summarized in an allele table. Then, character matrices were formed for each clonal population, summarizing mutation information across the N cells in a population and their M cut-sites. Characters (i.e., cut-sites) with more than 80% missing information or containing a mutation that was reported in greater than 98% of cells were filtered out for downstream tree reconstruction.
Creating a consensus intBC set for mESC clones
Given that each mouse is generated from a specific mESC clone, we expected tumors from each mouse would maintain the same set of intBCs as the parental mESC clone. To identify this consensus set of intBCs, we stratified tumors based on which mESC clone they originated from, and within these groups computed the proportion of tumors that reported a given intBC in at least 10% of cells. We determined cutoffs separating reproducible intBCs from irreproducible intBCs for each mES clone separately. These consensus intBC sets were used for downstream reconstruction of phylogenies.
Tree Reconstruction with Cassiopeia
Trees for each clonal population (see “Calling clonal populations and creating character matrices” above) were reconstructed with Cassiopeia-Hybrid (Jones et al. 2020). Briefly, Cassiopeia-Hybrid infers phylogenies by first splitting cells into clusters using a “greedy” criterion (Cassiopeia-Greedy) until a user-defined criteria is met at which point each cluster of cells is reconstructed using a near-optimal Steiner-Tree maximum-parsimony algorithm (Cassiopeia-ILP). We compared the parsimony of trees generated using two different greedy criterions - both criterions employed work by first identifying a mutation and subsequently splitting cells based on whether or not this mutation was observed in a cell. First, we used the original Cassiopeia-Greedy criterion, which identifies mutations to split cells on by using the frequency and probability of mutations. Second we applied a compatibility-based criterion which prioritizes mutations based on character-compatibility (see section “Compatibility-based greedy heuristic for tree reconstruction” below). We proceeded with the more parsimonious tree. In one specific case, (3515_Lkb1_T1), we observed that the lineage tracing alleles were not adequately captured with phylogenetic inference of the primary tumor alone. To handle this, we rebuilt the tree of the tumor-metastasis family and then subset the phylogeny to consist of only the cells from the primary tumor - resulting in a clonal phylogeny that appeared to be better supported by allelic information.
In most inferences, we used indel priors computed with Cassiopeia to select mutations with a Cassiopeia-Greedy algorithm as well as weight edges during the Steiner-Tree search with Cassiopeia-ILP. Generally, we used an LCA-based cutoff to transition between Cassiopeia-Greedy and Cassiopeia-ILP as previously described (Quinn et al. 2021). Clone-specific parameters are reported in Table S1.
Compatibility-based greedy heuristic for tree reconstruction
A rare, but simple case for phylogenetic inference is that of perfect phylogeny in which every character (or cut-site) is binary (that is, can be cut or uncut) and mutates at most one time. In this regime, every pair of characters is “compatible” -- that is, given two binary characters i and j, the sets of cells that report a character i as mutated are non-overlapping with the set of cells that report character j as mutated, or one set of cells is completely contained within the other.
In this approach, we used a heuristic, called the compatibility index, to measure how far a pair of characters is from compatibility. To do so, we first “binarized” our character matrices by treating each unique (cut-site, mutation) pair as a binary character. (To note this binarization procedure is possible because of the irreversibility of Cas9 mutations and discussed in our previous work (Jones et al. 2020).) Then, we found the character that had deviated the least from perfect phylogeny, that is violated compatibility the least. To find this character, we first built a directed “compatibility-graph”, where individual nodes represented characters and edges between nodes represented deviations from compatibility. Each edge from character to was weighted as follows:
where and are two incompatible characters, is the number of cells reporting character , and is the prior probability of character mutating. For the purposes of building this compatibility matrix, missing data was ignored (this is, no node in the graph corresponded to a missing state). A character to split cells with was identified by minimizing the sum of weights emitted from the node:
where Out(c) denotes the set of edges with c as a source. This process was repeated until the tree was resolved completely, or a criterion was reached as in Cassiopeia-Hybrid.
Cell Filtering with Lenti-Cre-BC
After performing tree reconstruction for each clonal population, leaves were annotated with Lenti-Cre-BC information and evaluated manually for filtering. Specifically, in tumors with adequate Lenti-Cre-BC information, we identified subclades (defined here as clades that joined directly to the root) that clearly had divergent Lenti-Cre-BC information. This combined Lenti- Cre-BC and lineage analysis helped minimize the influence of lenti-Cre-BC dropout in single-cell experiments. These subclades were subsequently removed and cells were filtered out from the phylogenetic analysis. In one case (3513_NT_T4 and 3513_NT_T5), two tumor populations were split from a parental tumor (3513_NT_N2), reconstructed, and used in downstream analyses.
CNV analysis
Chromosomal copy number variations (CNV) were inferred with the InferCNV R package (version 1.2.1), which predicts CNVs based on single-cell gene expression data. InferCNV was run in ‘subclusters’ analysis mode using ‘random_trees’ as the subclustering method. Genes with less than one cell were filtered with the ‘min_cells_per_gene’ option, and no clipping was performed on centered values (‘max_centered_threshold’ set to ‘NA’). The cutoff for the minimum average read count per gene among reference cells was set to 0.1, per software recommendation for 10x data. CNV prediction was performed with the ‘i6’ Hidden Markov Model, whose output CNV states were filtered with the included Bayesian mixture model with a threshold of 0.2 to find the most confident CNVs. All other options were set to their default values.
Each tumor sample was processed independently with normal lung cells (identified solely from the MULTI-seq deconvolution pipeline) as the reference cells. The number of CNVs for each cell was computed by counting the number of CNV regions predicted. We filtered cells with CNV counts greater than three standard deviations away from the mean of each tumor, in addition to cells with greater than or equal to 20 predicted CNVs. When comparing CNV counts of cells in expansions against those of cells in non-expansions, statistical significance was computed with a one-sided permutation test and the Mann-Whitney U-test, both of which yielded the same results.
We applied hierarchical clustering with a euclidean distance metric and the “ward” linkage to identify CNV clusters of cells within each tumor. For each clustering induced by cutting the hierarchical clustering dendrogram at different heights, we computed the probability that a cell and its nearest neighbor on the Cassiopeia tree were in the same hierarchical cluster (“nearest neighbor probability”). These clusters ranged from most coarse-grained (low cutoff height) to the most fine-grained (high cutoff height). When there were multiple nearest neighbors, pseudocounts were used by taking the fraction of nearest neighbors that were in the same cluster. We performed nonparametric Permutation Tests for each unique clustering by shuffling the cluster assignments of the cells and computing the nearest neighbor probability using these assignments.
Tree Quality Control for Fitness Inference
Trees were subjected to quality control before identifying subclones under positive selection and single-cell fitness inference. We employed two quality control metrics: first, a measure of subclonal diversity known as “percent unique indel states”, defined as the proportion of cells that reported a unique set of character states (i.e., mutations). Second, we also filter lineage trees based on the level of “unexhausted target sites” defined as the proportion of characters (i.e., specific cut sites) that were not dominated by a single mutation (i.e, more than 98% of cells contained the same mutation). These metrics describe the diversity and depth of the lineage trees, and enable filtering out tumors with poor lineage tracing quality (i.e., lineage tracing capacity became saturated too early during tumor development). Using these two metrics, we filtered out tumors that had less than 10% unique indel states or less than 20% unexhausted target sites. Additionally, tumors with too few cells recovered (fewer than 100 cells) were ignored for this analysis because of a lack of power to confidently quantify subclonal behavior.
Identifying subclonal selection (i.e., expansions)
Subclones undergoing positive selection were identified by comparing the number of cells contained in the subclone to its direct “sisters” (i.e. branches emanating directly from the parent of a subclone of interest) and computing a probability of this observation with a coalescent model. Specifically, consider a node in a particular tree with children stored in the set . Let denote the number of leaves below a particular node (and observe that N = nv=c Cnc). Under the coalescent model, we can compute a probability indicating how likely a subclone under would have exactly leaves given had total leaves as follows:
Finally, we computed the probability that a subclone under would have at least leaves given had total leaves is:
Nodes with probabilities , at least a depth of 1 from the root, and containing subclades with at least 15% of the total tree population were annotated as undergoing an “expansion”. In the analysis presented in this study, we additionally filtered out nodes annotated as “expanding” if they contained another node in their subtree that was also expanding. Expansion proportions were calculated as the fraction of the tree consisting of cells residing in any subclade called as “expanding”.
Inferring single-cell fitness
To compute single-cell fitness, we used the “infer_fitness” function from the jungle package (publicly available at https://github.com/felixhorns/jungle) which implements a previously described probabilistic method for inferring relative fitness coefficients between samples in a clonal population (Neher et al. 2014). Because some trees contained exhausted lineages (i.e., those in which all target sites were saturated with edits), after filtering out trees that did not pass quality control (see section “Tree quality control for fitness inference” above), we pre-processed branch lengths on each phylogeny such that branches had a length of 0 if no mutations separated nodes and 1 if not. In essence, this collapses uninformative edges in the fitness inference and helps control for lineage exhaustion. After this procedure, we were left with fitness estimates for each leaf in a phylogeny, normalized to other cells within the phylogeny.
Tumor fitness differential expression
Genes differentially expressed along the fitness continuum within each tumor were identified with a linear regression approach. Specifically, given a cell , we can model the expression of some gene according to the cell’s fitness score as follows:
Where is the count-normalized expression of gene in cell (we used the median number of UMI counts across the dataset to normalize expression level) and is the number of genes detected in the cell. Only genes appearing in more than 10 cells were retained for differential expression analysis. Linear models were fit using Julia’s GLM package (v1.3.7). Significances were computed using a Likelihood Ratio Test, comparing the model above to a model only using the as a predictor. P-values were FDR corrected using the Benjamini-Hochberg procedure (Benjamini and Hochberg 1995). Log2fold-changes were computed by comparing the average expression of a gene in the top vs bottom 10th percentile of fitness scores.
Meta-analysis and derivation of the FitnessSignature
The transcriptional FitnessSignature was derived from the results of individual tumor fitness differential expressions with a majority-vote meta-analysis. This approach ranks genes based on the number of times that a gene is differentially expressed (FDR < 0.05 and ) and the consistency of its direction. We used the MetaVolanoR R package (version 1.0.1) to perform this majority-vote analysis, which computed both of these values. We identified consistently differentially expressed genes for our transcriptional FitnessSignature if a gene appeared to show up at least 2 times in the same direction, and if the ratio between frequency and consistency was greater than 0.5.
Fitness module identification
We determined transcriptional fitness gene modules using the Hotspot package (version 0.9.0; (DeTomaso and Yosef 2021)). To do so, we first subset our processed single-cell expression matrix (see section below “Single-cell transcriptome analysis for KP-Tracer data”) to contain only the 1,183 genes in the FitnessSignature that were positively associated with fitness. Then, using Hotspot we identified fitness-related genes that were significantly autocorrelated with the scVI latent space using the “danb” observation model and 211 neighbors (the square-root of the number of cells in the expression matrix). After this procedure, genes with an FDR of less than 0.05 were retained for downstream clustering. We then computed pairwise local autocorrelations with Hotspot and clustered genes using these pairwise statistics with the “create_modules” function in Hotspot (minimum gene threshold of 100, FDR threshold of 0.05, core_only=False). This procedure identified three modules that were used for downstream analysis.
Single-cell transcriptome processing for KP-Tracer NT data
The scRNA-seq was processed using the 10X CellRanger pipeline (version 2.1.1) with the mm10 genome build. Cells were assigned to a sample using the MULTI-seq pipeline described above (see section “Single-cell preprocessing pipeline”). After quantification, informative genes were identified using the Fano filtering process implemented in VISION (DeTomaso et al. 2019), and raw counts were batch-corrected (using the batch-harvest data, indicating when a batch of mice were sacrificed as the batch variable) and projected into a shared latent space of 10 dimensions with scVI (Lopez et al. 2018). Cells were initially clustered with the Leiden algorithm as implemented in Scanpy (Wolf et al. 2018; Traag et al. 2019), and two clusters dominated by cells annotated as normal and cells that could not be confidently mapped to a tumor via MULTI-seq or Lenti-Cre-BC analysis (see section “Single-cell preprocessing pipeline” and “Cell Filtering with Lenti-Cre-BC” above) were removed from downstream analysis. Clusters were then manually re-clustered to obtain segmentations that aligned with gene expression patterns. After this process, we were left with a total of high-quality 58,022 cells with single-cell transcriptomic profiles from KP mouse tumors. Single-cell counts were normalized by the median UMI count across cells and logged to obtain log-normalized data. Gene markers for each Leiden cluster were identified using the Wilcox rank-sums test on the log-normalized gene counts with the Scanpy package (Wolf et al. 2018).
Integration of normal lung epithelium transcriptomes
scRNA-seq data of cells obtained from various tissues in sample L46 were quantified using the 10X CellRanger pipeline (version 2.1.1) with the mm10 genome build. Cells were assigned to a sample (one of 4 tissues) using the CellRanger multi procedure. After quantification and sample assignment, cells with fewer than 200 UMIs and genes appearing in fewer than 1% of cells were filtered out. This left us with 14,424 high-quality cells. A low-dimensional embedding was inferred using scVI on the dataset with the 4000 most highly-variable genes (using the “seurat_v3” flavor of Scanpy’s highly_variable_genes function). Transcriptional clusters were identified using the Leiden community detection algorithm. One cluster of 329 cells consisted of normal lung cells and expressed gene markers Nkx2–1, Sftpc, and Scgb1a1; we isolated and annotated this cluster as normal lung epithelial cells (primarily AT2 and club cells).
This dataset of 329 normal lung epithelial cells (isolated from the L46 sample, as described above) was integrated into the scRNA-seq dataset of KP tumors (see section “Single-cell transcriptome processing for KP-Tracer NT data”) using scVI. Specifically, we used scVI to batch-correct these two datasets and project all cells into a common coordinate system. Then, we visualized this scVI batch-corrected embedding with UMAP.
Differential expression analysis of Chuang et al
TPM-normalized RNA-seq data were downloaded from GEO accession GSE84447. Samples were split into early and late-stage tumor groups based on the author annotations: tumors annotated with “KPT-E” were assigned to the early stage group and tumors with “TnonMet” or “TMet” annotations were assigned to the late group. Then, we log-normalized the TPM counts and used the limma R package (version 3.36.3) to infer differentially expressed genes with the “eBayes” function. Genes passing an FDR threshold of 0.05 and log2-fold-change threshold of 1 (in either direction) were called differentially expressed and used for comparison with the FitnessSignature described in this study.
FitnessSignature analysis of Marjanovic et al
Raw expression count matrices were downloaded directly from GEO, accession number GSE152607. Gene counts were normalized to transcript length, to account for read depth artifacts in the Smart-Seq2 protocol. VISION (DeTomaso et al. 2019) was used to compute FitnessSignature scores (using the FitnessSignature gene set described in our study) for each cell in the dataset and scores were averaged within time points of KP mice.
Survival analysis with TCGA lung adenocarcinoma tumors
The fitness signature genes including 1183 up-regulated genes and 1027 down-regulated genes from mice experiments were converted to corresponding genes from the H. sapiens genome (build hg19), resulting in 1126 up- and 970 down-regulated human genes, respectively. FitnessSignature with only up-related genes was denoted as FSU, FitnessSignature with only down-related genes was denoted as FSD. TCGA Lung adenocarcinoma cohort with RNAseq data (n=495) were stratified into FSU-High, FSU-Low, FSD-High, and FSD-Low according to median expression of sum of FitnessSignature genes, then, patients harboring genes with FSU-High and FSD-Low formed a group, patients containing FSU-Low and FSD-High gene expression formed another group. Subsequently, these two groups were used for survival analysis using the survival package in R (version 3.2.11). The survival analysis was invoked with the call “survfit(Surv(Time, Event) ~ Group)” where “Group” is the FitnessSignature-based stratification. Kaplan–Meier curve is shown with a log-rank statistical test. For fitness gene module 1, 2, and 3 analyses, patients were divided into module gene expression of High and Low based on the median of the sum of gene expression, followed by survival analysis.
Fitness Module Enrichment
Each of the three fitness gene module scores (computed with VISION) were normalized to the range [0, 1] across all NT cells. All NT cells in non-expansions were defined as the background cells, and the background module scores were calculated by averaging the normalized module scores of these cells. Additionally, the module scores of cells in each expansion were averaged to obtain the psuedo-bulk module score for each expansion. These module scores were divided by the background module scores, yielding the module enrichment score (i.e. fold-change versus background) per fitness module. These scores were plotted on a personality plot for visualization. Every expansion was assigned (non-exclusively) to the three fitness modules using a permutation test to test whether the cells in the expansion exhibited a significant increase in fitness module score compared to non-expanding background cells (p < 0.05).
Calculation of single-cell and Leiden cluster EffectivePlasticity
EffectivePlasticity for each tumor was computed by first calculating a normalized parsimony score for the tumor tree, with respect to the Leiden cluster identities at the leaves, using the Fitch-Hartigan algorithm (Fitch 1971; Hartigan 1973). Briefly, this procedure begins by assigning cluster identities to the leaves of the tree, and then calculates the minimum number of times a transition between cluster identities must have happened ancestrally in order to account for the pattern observed at the leaves. To compare scores across trees, we normalize these parsimony scores by the number of edges in the tree, thus giving the EffectivePlasticity score. In all analyses, we filtered out cells that were part of Leiden clusters that were represented in less than 2.5% of the total size of the tree.
In order to generate single-cell EffectivePlasticity (“scEffectivePlasticity”), we computed the EffectivePlasticity for each subtree rooted at a node on the path from the root to a leaf and averaged these scores together. This score thus represents the average EffectivePlasticity of every subtree that contains a single cell.
To generate average EffectivePlasticity for each Leiden cluster, we first stratified cells in each tumor according to the Leiden cluster. Then, we averaged together scores within each tumor for each Leiden cluster, thus providing a distribution of EffectivePlasticity for each Leiden cluster.
Calculation of the Allelic EffectivePlasticity score
The Allelic EffectivePlasticity score provided a “tree-agnostic” measurement of a cell’s effective plasticity. Qualitatively, the score measures the proportion of cells that are found in a different Leiden cluster than their closest relative (as determined by the modified edit distance between two cells’ character states; see section “Allelic Coupling” for the definition of this distance metric). Importantly, if a cell has more than one closest relative, each of their votes are normalized by the number of equally close relatives this cell has. More formally, the single-cell Allelic EffectivePlasticity was defined as:
Where indicates the set of a cell’s closest relatives, as measured by modified edit distance, indicates the Leiden cluster that cell resides in, and is an indicator function that is 1 if the two Leiden clusters are the same and 0 otherwise. The Allelic EffectivePlasticity of a tumor is the average of these scores:
Calculation of the L2 EffectivePlasticity score
The L2 EffectivePlasticity score served as an alternative tree-based score that accounted for random noise at the boundary between two Leiden clusters, as opposed to treating each Leiden Cluster as a point. As with the EffectivePlasticity score, we first found nearest-neighbors of each cell using the phylogenies and considered neighbors found in a different Leiden cluster than . Yet, in contrast to the EffectivePlasticity score, we distinctly used an L2-distance in the 10 dimensional scVI latent space to obtain a measure of how distinct the neighbor was. Mathematically, the single-cell L2 EffectivePlasticity score was defined as:
Where indicates the set of a cell’s closest relatives, as found with the phylogeny, and xi indicates the 10-dimensional embedding of cell ’s single-cell expression profile in scVI space. The L2 EffectivePlasticity of a tumor was defined as the average across all leaves in the tumor.
Evolutionary Coupling
Evolutionary Coupling is the normalized phylogenetic distance between any pair of variables on a tree. Mathematically, given two states and that can be used to label a subset of the leaves of the tree, we compute the average distance between these states:
where is the number of leaves with state denotes the set of cells in set , and denotes the phylogenetic distance between leaves. There are multiple ways to score , and here we used the number of mutated edges for our analysis (i.e., the number of edges separating two leaves and that carried at least one mutation). To normalize these distances, we compare to a random background generated by shuffling the leaf assignments 2000 times. Then, to obtain background-normalized scores, we Z-normalize to the random distribution :
This score is obtained for all pairs of states in a tumor that pass a 2.5% proportion threshold (i.e., we filter out cells in states that fall below this threshold). Then, from the matrix of all background-normalized phylogenetic distances, (such that is equal to ), we compute the Evolutionary Couplings between two states and by Z-normalizing :
Evolutionary Couplings presented in Fig 5B and 5D are normalized as:
Where denotes all the Evolutionary Couplings between states in a given tumor.
Allelic Coupling
We used modified edit distances between cells to compute an Allelic Coupling score that could be used to assess consistency of the Evolutionary Coupling results. Here, we used a modified edit distance, , that scored the distance between sample and at the character:
The allelic distance between two samples and is . We used these distances instead of phylogenetic distances to compute the coupling statistic described in the section above entitled “Evolutionary Coupling” and called this new coupling statistic “Allelic Coupling”.
K-nearest-neighbor (KNN) Coupling
K-nearest-neighbor (KNN) coupling was computed by using as the distance to the neighbor in the Evolutionary Coupling statistic. We used the same phylogenetic distance described in the section entitled “Evolutionary Coupling” to compute the neighbor and used for the analysis.
Fate clustering
To identify separate fates in the KP-Tracer dataset, we first computed Evolutionary Couplings in each tumor for all pairs of states. To remove noise intrinsic to the clustering, we filtered out clusters that accounted for less than 2.5% of the tumor. As a phylogenetic distance metric, we used the number of mutated edges (i.e., any edge that contained at least one mutation was given a weight of 1 and otherwise the edge was weighted as 0). Before computing Evolutionary Couplings, we preprocessed the lineages such that each leaves with the same Leiden cluster were grouped together (see section entitled “Preprocessing lineages with respect to states”).
After calculating the Evolutionary Coupling for all pairs of states within each tumor, we concatenated all vectors of Evolutionary Coupling together into a matrix. We additionally converted Evolutionary Couplings to similarities by exponentiating these values (i.e, ). As additional features for this clustering, we also added Leiden cluster proportions to each tumor’s vector of couplings. Then we Z-normalized across features to compare tumors and clustered this transformed matrix using a hierarchical clustering approach in the python scipy package (version 1.6.1). We used a Euclidean metric and the “ward” linkage method. We identified three clusters from this hierarchical clustering, corresponding to our three Fate Clusters. These three Fate Clusters were visualized using Uniform Manifold Approximation and Projection (UMAP) on the Evolutionary Coupling and Leiden cluster proportion concatenated matrix. Important couplings were identified using Principal Component Analysis on the same Evolutionary Coupling concatenated matrix.
Preprocessing lineages with respect to states
In some lineages, we observed that polytomies (or non-bifurcating) subclades were created at the very bottom of the tree due to the saturation of target site edits. Because this could artificially appear to make cellular states more closely related than they actually were, we took a conservative approach to making conclusions about cellular relationships between leaves in such polytomies. Specifically, we first assigned states from a state space to each leaf in a tree according to some function for all leaves in the tree. Then, for all polytomies that contained at least unique states or more, we created extra splits in the tree for each unique state. More formally:
PREPROCESS-LINEAGE (Tree):
for v in Tree:
states = []
If len(children(v)) < 3:
continue
for c in children(v):
if is_leaf(c):
states.append(s(c))
If len(unique(states)) > 2:
for state in unique(states):
Tree.add_edge(v, ‘new-node-{state}’)
for c in children(v):
If s(c) == state:
Tree.add_edge(‘new-node-{state}’, c)
Tree.remove_edge(v, c)
return Tree
Aggregating Evolutionary Coupling across Fate Cluster
To create a consensus Evolutionary Coupling map across the tumors in a Fate Cluster, we first computed the average Evolutionary Coupling between all pairs of states in a tumor as described previously. Then, we computed an average Evolutionary Coupling for each pair of states, normalizing by the number of tumors that this pair appeared in above the requisite 2.5% threshold. Critically, we removed patterns that were driven by a small proportion of cells, we only considered states that appeared in at least 2.5% of the total number of cells across all tumors in a Fate Cluster.
Phylotime
Phylotime was defined as the distance to the first ancestor that could have been a particular state. To approximate the Phylotime in this study, we defined the initial AT2-like state (Leiden cluster 4) as the ground state, and inferred the sets of states for each ancestor with the Fitch-Hartigan bottom-up algorithm (Fitch 1971; Hartigan 1973). Then, in each tumor, we computed the phylogenetic distance separating each cell from its closest ancestor that could have been an AT2-like cell, as determined with the Fitch-Hartigan bottom-up algorithm. Phylogenetic distances were defined as the number of non-zero-length branches (though we compare the consistency of Phylotime to a distance metric that uses the number of mutations along each edge in Fig S5J,K). In this way, Phylotime is proportional to the number of generations elapsed since the more recent ancestral node that, under a maximum-parsimony approach, could have been an AT2-like cell. Here, the tree structure is advantageous in modeling divergence times from the AT2-like state because it can account for homoplasy (i.e., the same mutation occurring independently) and convergent phenotypic evolution events (i.e., the same transcriptomic state being reached separately, as opposed to pseudotime statistics estimated from single-cell transcriptomes (Trapnell et al., 2014)). events. Thus, it is preferable, in principle, to comparing the mutation states directly between a leaf and all AT2-like cells. Phylotime within each tumor was normalized to a 0–1 scale. Once every tumor was analyzed this way, Phylotime across tumors was merged by performing an average-based smoothing across the transcriptional space: specifically, for each cell, we found the 5 closest neighbors in transcriptional space (in the low-dimensional scVI latent space) and averaged Phylotimes within this neighborhood. After integrating together Phylotime in this manner, the final distribution across tumors was normalized once again to a 0–1 scale.
Phylotime differential expression
Genes associated with Phylotime in each Fate Cluster were identified using the Tradeseq package (Van den Berge et al. 2020). Specifically, for each Fate Cluster, lowly-expressed genes were filtered if they were detected in fewer than 10% of cells and high-variance genes were identified with the Fano filtering procedure implemented in VISION (DeTomaso et al. 2019). Then, in each cluster, expression models were fitted with the “fitGAM” function and genes associated with a specific segment of Phylotime were identified with the “associationTest” function. P-values were FDR corrected using the Benjamini-Hochberg procedure (Benjamini and Hochberg 1995), and significant genes were retained if they had an FDR below 0.05 and a mean log2-fold-change above 0.5. Smoothed expression profiles were predicted with the Tradeseq package using the models fit from the fitGAM procedure and genes were subsequently clustered into those expressed early and late. Gene set enrichment analysis was performed using the enrichR R package (version 3.0) after converting gene names from mm10 to GRCh38. We used the Biological Process gene ontology, ChEA, and MsigDB Hallmark gene sets. Informative genes were manually selected from the set of genes passing the significance and effect-size thresholds, and manually clustered for display in Figure 5.
Integrating transcriptomes of KP-Lkb1 and KP-Apc data
The scRNA-seq data was processed using the 10X CellRanger pipeline (version 2.1.1) with the mm10 genome build. Cells were assigned to a sample using the MULTI-seq pipeline as described above (see section “Single-cell preprocessing pipeline”) to form a raw count matrix consisting of cells from KP, KPL, or KPA mice. Cells with fewer than 200 genes detected, greater than 15% of mitochondrial reads, or greater than 7000 genes detected were filtered out. Cells were batch-corrected and projected into 20 latent dimensions using scVI (Lopez et al. 2018) with 2 hidden layers and the library batch as a batch covariate on the top 4000 most variable genes, as detected with Scanpy’s “highly_variable_genes” function with the “seruat_v3” flavor (Wolf, Angerer, and Theis 2018). Clusters were identified with the Leiden algorithm (Traag, Waltman, and van Eck 2019) with manual parameter selection to obtain an acceptable resolution. All normal cells and seven additional clusters with high proportions of normally-annotated cells (as with MULTI-seq or via the lineage-tracing data) were filtered out for downstream analysis (a total of 2,209 cells in the entire dataset).
To perform label transfer from the KP-Tracer dataset, we first labeled all KP cells in the integrated dataset with previous annotations and labeled all new cells with “Unknown”. Then, we used scANVI (Xu et al. 2021) to predict labels of cells from KPL and KPA mice using 40 latent dimensions, 2 hidden layers, and a dropout rate of 0.2. Upon inspecting predictions, we elected to keep predictions made by scANVI for the majority of cells, with the exception of 5 new Leiden clusters identified by clustering the scVI latent space. Additionally, we elected to merge one new Leiden cluster with the Pre-EMT state because key gene expression markers across these two states were consistent. After this process, we were left with a total of 104,197 high-quality cell transcriptomes.
Differential expression analysis of Pre-EMT state
The single-cell RNA count matrix was first count-normalized to the median number of UMI counts across cells and log-transformed. Then, cells assigned to the Pre-EMT state were separated into three non-overlapping sets according to their genotype (KP, KPL, or KPA). Differentially expressed genes in the KPL subset of cells in the Pre-EMT cluster were identified by comparing these cells to all other cells with Scanpy using a t-test on log-normalized count matrix with the top 5000 most variable genes. Highlighted genes were selected from the set genes passing an FDR cutoff of 0.05 and a log2FC cutoff of 1.
Evolutionary Trajectory Analysis of KPL and KPA Tumors
The evolutionary trajectories from KPL and KPA mice were analyzed identically to the KP tumors as described in the previous section entitled “Fate Clustering”. Briefly, each tumor was described as a vector of Leiden cluster proportions and exponentiated Evolutionary Couplings (i.e, ). Vectors were concatenated together and Z-normalized across features. The resulting matrix was decomposed and analyzed using Principal Component Analysis (PCA) and informative features were identified by evaluating the features with highest principal component loadings.
Evolutionary Coupling of 3724_NT_T1 Tumor-Metastasis Family
Using the tumor-metastasis family tree for 3724_NT_T1 and associated metastases, we computed the Evolutionary Couplings between each microdissected piece of the primary tumor (T1–15) and each metastasis (the statistic is described in the section entitled “Evolutionary Coupling”). Normalized Evolutionary Couplings (E) were computed as described previously.
Phylogenetic distances on Tumor-Metastasis Family trees
In each of the tumor-metastasis families (defined as a tumor containing both a primary tumor and a large enough metastatic population) analyzed in Fig 7 and S7, we first reconstructed trees encompassing all cells in the primary and metastatic tumors (referred to as a “tumor-metastasis family” tree). Then, we stratified cells in the primary tumor by the expansions called with our expansion-calling statistic (see above, “Identifying subclonal selection”). If a cell was not part of an expansion, it was labeled as “non-expansion”. Then, for each cell in a metastatic tumor, we computed the average modified phylogenetic distance to all primary tumor cells in the tumor-metastasis family tree. The modified phylogenetic distance was computed as the sum of branch lengths, where each branch length was defined as the number of mutations separating each node from one another (as inferred using Camin-Sokal parsimony - i.e., irreversibility of mutations)..
Transcriptional distances on Tumor-Metastasis Family trees
Tumor-metastasis family trees were inferred and stratified as described above (see “Phylogenetic distances on Tumor-Metastasis Family trees”) and Euclidean distance was used to measure transcriptomic differences between metastatic cells and primary tumor subpopulations.
Distribution comparisons and statistical significance
All statistical tests comparing the distribution of continuous values are indicated in the appropriate figure legend. Mann-Whitney U tests were performed using the ranksums function in the scipy.stats python package with sidedness specified in the figure legend. All boxplots present the quartiles of the distribution and whiskers show the rest of the distribution. Outliers of boxplots are determined using as being 1.5x the inter-quantile range.
Supplementary Material
(A) The piggyBac transposon-based lineage tracing vector libraries used to engineer the KP-Tracer mice contained (1) a triple-guideRNA cassette and (2) a target site library cassette with a 14bp integration barcode (“intBC”) and three CRISPR/Cas9 cut sites on the 3’ UTR of an mCherry reporter gene. (B) Enrichment of mESC population with high lineage-tracer expression based on high mCherry expression (a reporter indicating lineage tracer expression). These cells are then single-cell cloned before generating chimeric KP-Tracer mice. (C) Representative images of specific mCherry positive mESC clones that express the lineage tracing vectors. (D) Copy number of lineage tracing vectors across 5 mouse embryonic stem cell (mESC) clones used in this study measured by genomic qPCR are shown. (E-F) Detection of unique lineage tracing target site intBCs for a representative mESC clone (1D5) using (E) DNA-sequencing and (F) scRNA-seq. A consensus set of target sites intBCs for each mESC clone was determined by selecting intBCs detected in at least 40% of all tumors derived from that mESC clone. (G) The consensus intBC pivot table across all five mESC clones used in this study to generate KP-Tracer mice. Each row is a single cell and is annotated with which mESC clone it came from. Each column is a unique intBC. Colors in the heatmap indicate whether or not an intBC was detected in a given cell. (H) Quality-control filtering of tumor phylogenies for subclonal expansion analyses. Quality of lineage-tracing data was assessed with two metrics: first, the percentage of cells that contained a unique set of mutations (“% unique indel state”; STAR Methods); and second, the percentage of target sites that had to be filtered because of low-diversity (“target site saturation”; STAR Methods). Tumors with less than 5% overall unique indel state, greater than 80% target site saturation, or fewer than 100 cells were filtered out.
(A-D) Phylogenetic features of tumor lineages and their predictiveness (as measured with R2) on the expansion proportion of a tumor. Features evaluated were (A) age, (B) median tree depth, (C) size measured in the number of cells, and (D) proportion of unique cells. (E) Expansion proportion of tumors measured from Neighbor-Joining trees versus Cassiopeia trees. The percentage of cells in expansions were highly consistent between these two tree reconstruction strategies (Pearson’s correlation = 0.87). (F) Comparison of cell-cycle scores inferred from transcriptomic profiles in expanding versus non-expanding tumor subclones, identified from Neighbor-Joining trees (** p < 0.01). (G-H) Representative example of comparison between hierarchical clustering of CNVs and Cassiopeia-reconstructed phylogeny. (G) The inferred CNVs are shown for the representative tumor, with the largest two clusters, identified via hierarchical clustering, indicated by the colorbar. (H) These two clusters are also indicated with unique colors on the Cassiopeia-reconstructed tumor phylogeny. The good correlation between CNV status and tumor phylogeny indicates the accuracy of tree reconstruction. (I) Heatmap displaying the probabilities that a cell and its nearest neighbor on the Cassiopeia-reconstructed phylogeny are in the same CNV cluster (size of circles). These probabilities were calculated for each tumor at various depths of the CNV hierarchical clustering dendrogram. The depth that yielded the most coarse-grained clusters were set to have a cutoff height of 1, with higher cutoff heights indicating finer clusters. The majority of Cassiopeia-reconstructed phylogenies were significantly consistent with CNV clusters (color of circles; Permutation Test) at all clustering resolutions. (J) A comparison of CNV counts in expanding versus non-expanding portions of tumors (* p < 0.05, ** p < 0.01, *** p < 0.001). (K) An example of distinct CNV regions of cells from a single tumor. This tumor underwent two independent clonal expansions (red branches; left), each of which exhibited distinct CNV patterns (red bars; right). (L) An aggregated view of the CNV “hotspots” across subclonal expansions from all tumors. Each horizontal bar represents a chromosome, and the intensity of color indicates the number of subclonal expansions exhibiting a CNV in a region (STAR Methods). Regions that more often exhibited copy number gains are indicated in red (left); genomic regions that more often exhibited copy number losses are indicated in blue (right).
(A) Gene markers for each Leiden cluster identified in the processed scRNA-seq latent space. Dot size indicates the percent of cells expressing the marker. Color indicates mean expression level. (B) Integration of normal lung epithelial cells with KP-Tracer dataset. Normal lung epithelial cells were isolated from an independent dataset and integrated with KP-Tracer tumors using scVI (STAR Methods). Leiden cluster annotations from analysis of KP-Tracer tumors are shown (top) and normal cells are highlighted against tumor cells (bottom). (C) Gene set comparison between the FitnessSignature described in this study and KP tumor progression-associated genes described in (Chuang et al. 2017). Overlap significance assessed with a hypergeometric test (*** = p < 1e-5). (D) Average transcriptional FitnessSignature score in KP tumors harvested at 12-week, 20-week, and 30-week timepoints from (Marjanovic et al. 2020). (E) Representative examples of tumors occupying distinct regions of the transcriptional space. Cells from the tumor of interest are shown in red, and all other cells are shown in gray. (F) Hotspot autocorrelation heatmap and clustering of genes that appear in the FitnessSignature and are positively associated with fitness. Gene modules are identified by distinct color strips on the left. Values in the heatmap are Z-normalized pairwise autocorrelation scores between genes. The dendrogram linking genes is shown for the columns. (G) Z-normalized mean fitness gene module signature scores of each Leiden cluster. (H) Kaplan-Meier plots for TCGA human lung adenocarcinoma patients with respect to genes in each fitness module. Curves are shown comparing overall survival of patient groups whose tumors have high (red) versus low (blue) expression of individual fitness gene modules, as determined by the median fitness module score. P-values from a log-rank test are indicated. (I) Fitness module enrichment personality plots. Each corner of the triangle represents the fold enrichment of an expansion’s fitness module expression over expectation (non-expanding background). Independent expansions in each tumor are shown in unique colors (blue or orange). (J) Venn diagram illustrating the classification of expansions to gene modules based on a p-value threshold of 0.05 using a permutation test against non-expanding background.
(A) Leiden cluster proportions for each KP-Tracer tumor. The fraction of cells in each Leiden cluster is shown for each tumor in a stacked bar plot, where each Leiden cluster is indicated by the unique color introduced in Fig 3A. Tumors are ordered by mean FitnessSignature score. (B) Shannon’s Entropy statistic for each tumor, computed with the Leiden cluster proportions; tumors are ordered by mean FitnessSignature score. (C) Allelic EffectivePlasticity score overlaid onto two-dimensional gene expression UMAP is shown. Allelic EffectivePlasticity is an alternative way to quantify EffectivePlasticity by comparing transcriptional states between cells with similar lineage tracing indel states without using lineage trees. (D) Comparison of Allelic EffectivePlasticity to scEffectivePlasticity (Pearson’s correlation = 0.73). Each point represents a single cell. (E) Comparison of mean tumor Allelic EffectivePlasticity to tumor EffectivePlasticity (Pearson’s correlation = 0.96). Each point represents a tumor. (F) L2 EffectivePlasticity score overlaid onto two-dimensional gene expression UMAP is shown. L2 EffectivePlasticity is another alternative way to quantify EffectivePlasticity by computing dissimilarity in gene expression profiles between nearest neighbors on the phylogeny. (G) Comparison of single-cell L2 EffectivePlasticity to scEffectivePlasticity (Pearson’s correlation = 0.69). Each point represents a single cell. (H) Comparison of mean tumor L2 EffectivePlasticity to mean tumor EffectivePlasticity (Pearson’s correlation = 0.95). Each point represents a tumor. (I) Comparison of scEffectivePlasticity to single-cell FitnessSignature scores. Each point represents a single cell. (J) Weighted mean EffectivePlasticity vs mean FitnessSignature for each transcriptional state (Leiden cluster). The weighted Mean EffectivePlasticity for each Leiden cluster was determined by first computing the mean scEffectivePlasticity for each Leiden cluster in a tumor, and then averaging these values together. Each point represents a tumor.
(A-D) Two alternative statistics measuring couplings between states from lineage tracing data are used to corroborate the Evolutionary Coupling results for the representative tumors 3435_NT_T1 and 3513_NT_T3 shown in Figure 5A-D. The comparisons between Allelic Coupling and Evolutionary Coupling for (A) 3435_NT_T1 and (B) 3513_NT_T3 are consistent (Pearson’s correlation = 0.94 and 0.99, respectively). The comparisons between KNN Coupling and Evolutionary Coupling for (C) 3435_NT_T1 and (D) 3513_NT_T3 are consistent (Pearson’s correlation = 0.97 and 0.86, respectively). Red line indicates the symmetrical y=x relationship. (E) Cumulative density function for Pearson’s correlation of Allelic Coupling and KNN Coupling statistics with Evolutionary Couplings for all KP-Tracer tumors. Median correlations are indicated with vertical bars and annotated with the median correlation value. (F) Clustering of tumors based on Evolutionary Coupling and Leiden cluster proportion statistics reveals features that distinguish different Fate Clusters. Three clusters are identified by unbiased clustering, corresponding to Fate Clusters 1, 2, and 3. Fate Cluster is annotated on top of each unique color in the first row of the heatmap. Values/colors in the heatmap are normalized across tumors, and each row corresponds to a feature (either an Evolutionary Coupling or Leiden cluster proportion). Evolutionary couplings are indicated by a tuple of the form (x, y) and Leiden cluster proportions are indicated by a single number of the form x. We focus on showing features that distinguish different clusters, and uninformative features, identified as non-significant by a Mann-Whitney U test (p > 0.1), are not shown. (G) Heatmap of state proportions for each Fate Cluster across Leiden clusters. The value of the ith row and jth column indicate the fraction of cells found in the jth Leiden Cluster across all tumors in the ith Fate Cluster. (H) Principal Component Analysis (PCA) of tumor Evolutionary Coupling and Leiden cluster proportion vectors. Each dot is a tumor. Tumors are colored by their Fate Cluster, as identified with the hierarchical clustering shown in Fig S5E. The percent of variance explained is indicated on each axis. (I) Biplot of PCA of Evolutionary Coupling and Leiden cluster composition vectors, where each arrow indicates the loading of the feature with respect to the first two principal components. The top 10 features for the first two principal components are shown; arrows are annotated with the feature label. The percent of variance explained is indicated on each axis. Features of the form (x, y) represent Evolutionary Couplings between state x and state y; features of the form x represent the proportion of cells found in Leiden cluster x. (J-K) Comparison of Phylotime statistics computed using weighted and binary tree branch lengths for (J) Fate Cluster 1 and (K) Fate Cluster 2 (STAR Methods). Correlations are strong for both Fate Clusters (Pearson’s correlation = 0.94 and correlation = 0.98, respectively). (L) Selected Evolutionary Couplings of individual tumors displayed on gene expression UMAP illustrating connections between transcriptional states (Leiden clusters) of interest. From left: the first plot shows the Evolutionary Couplings within a representative tumor in Fate Cluster 1. The second plot shows the Evolutionary Couplings within a representative tumor in Fate Cluster 2. The third plot shows couplings between Fate Cluster 1 (Leiden clusters 3 and 5) and Late stage transcriptome states (Leiden cluster 9). The fourth plot shows couplings between Fate Cluster 1 (Leiden clusters 3 and 5) and high fitness transcriptome states (Leiden cluster 7 and 9). The last plot shows couplings between Fate Cluster 1 (Leiden clusters 3, 5 and 14) and high fitness transcriptome states (Leiden cluster 9 and 13). These results offer evidence of potential transition from early, low fitness to late, high fitness transcriptome states during tumor evolution.
(A-B) Subclonal expansion dynamics of (A) KPL and (B) KPA tumors. Independent expansions are colored with black, orange or blue and measured with the percentage of cells in the expanding subclone. (C) Overlap of genes associated with high and low fitness for KP, KPL and KPA tumors. (D) Gene markers for newly identified Leiden clusters in the KP, KPL and KPA integrated analysis. Dots are sized by the fraction of cells expressing a marker and colored by the mean expression of the gene marker in a Leiden cluster. (E) Leiden cluster proportions for each KPL (left) and KPA (right) tumor. (F) Distribution of the mean EffectivePlasticity for each Leiden cluster, averaged within each tumor, compared across genotypes. Leiden clusters 6, 11, 17, 18 are not shown because they lacked enough tumors across genotypes to make comparisons. (G) Evolutionary Couplings of different transcriptional states in three representative tumors reveals evolutionary paths in KPL and KPA tumors. Transcriptional states that are represented by at least 2.5% of cells in each tumor are used. 3515_Lkb1_T1 is a representative KPL tumor. The left plot shows the lineage relationship of transcriptional states in this KPL tumor and the right plot summarizes Evolutionary Couplings on the gene expression UMAP illustrating connections between Leiden clusters 4, 0 and 9. 3777_Apc_T1 is a representative KPA tumor. The left plot shows the lineage relationship of transcriptional states in this KPL tumor and the right plot summarizes Evolutionary Couplings on the gene expression UMAP illustrating connections between Leiden clusters 4 and 16. 3765_Apc_T1 is another representative KPA tumor. The left plot shows the lineage relationship of transcriptional states in this KPL tumor and the right plot summarizes Evolutionary Couplings on the gene expression UMAP illustrating connections between Leiden clusters 4, 16, 13, 7 and 1.
(A) Lineage indel heatmap of the 3724_NT_T1 tumor-metastasis family, summarizing the allelic information (indels) from the target sites confirming the separate origin of the soft tissue and liver metastatic tumors. In the Lineage indel heatmap, each row represents a single cell and each column represents a cut site of the lineage tracer. Unique indels are shown in unique colors, uncut target sites are indicated in gray, and missing data is indicated in white. The reconstructed lineage based on the accumulated indel patterns using Cassiopeia are shown on the left. The corresponding sample ID for each cell is labeled on the right. (B-C) Subclonal origin and the metastatic routes for 3515_Lkb1_T1 tumor-metastasis family. (B) Lineage indel heatmap of 3515_Lkb1_T1 tumor-metastasis family, indicating indel alleles supporting the subclonal origins, the relative order and the routes of metastases and (C) a model summarizing these metastatic behaviors. (D) More supporting examples of expanding subclones giving rise to metastases across genotypes for 3513_NT_T1 (left), 3508_Apc_T2 (center), and 3519_Lkb1_T1 (right). (E) Comparison of transcriptional distance between metastatic tumors and cells in non-expanding and expanding regions of the primary tumor phylogeny for 3513_NT_T1, 3508_Apc_T2, 3519_Lkb1_T1, 3457_Apc_T1, and 3515_Lkb1_T1 metastasis families. All significances are indicated from a one-sided Mann-Whitney U test: *** indicates p < 0.001, ** indicates p < 0.01, and * indicates p < 0.05.
Tumor Sample information, tree reconstruction parameters & quality-control statistics. This table contains information about all tumor samples regarding their timing, ES clones, mice and genetic perturbations, the Cassiopeia parameters used to reconstruct trees, the parsimony scores, depths, and indel-phylogeny distance correlations.
Differential expression gene lists of individual Leiden clusters. Reports the effect sizes and significances genes across various differential expression analyses performed throughout the study, including the differentially expressed genes of Leiden clusters in KP tumors, in tumors of all three genotypes combined (KP, KPL and KPA) and differentially expressed genes of the PreEMT transcriptional states in KPL versus the other tumors.
Phylogenetic fitness majority-vote gene association signatures and modules. This table reports the low- and high-fitness-associated-genes from the majority-vote meta-analysis of KP, KPL and KPA tumors individually. The gene set enrichment analysis for the fitness-associated genes in KP mice is included. We also include the gene lists for the three gene modules from the Hotspot analysis of high fitness-associated genes of the KP tumors.
Evolutionary coupling and leiden cluster proportions for individual tumors (including all KP, KPL and KPA tumors) analyzed for Figures 5 and 6.
Phylotime differential gene expression analysis. This table contains the differentially expressed genes following early to late Phylotime in tumors from Fate Cluster 1 and 2.
Primers and plasmids used in this manuscript.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, enzymes, and antibodies | ||
| Collagenase Type IV | Thermo Fisher Scientific | Cat#: 17104019 |
| Dispase | Thermo Fisher Scientific | Cat#: 17105041 |
| Trypsin | Thermo Fisher Scientific | Cat#: 25200056 |
| ACK | Thermo Fisher Scientific | Cat#: A1049201 |
| DNase I | Millipore Sigma | SKU 11284932001 |
| UltraPure BSA | Thermo Fisher Scientific | Cat#: AM2618 |
| Anti-mouse CD45 Monoclonal Antibody, APC | BioLegend | Cat#: 103111 |
| Anti-mouse CD31 Monoclonal Antibody, APC | BioLegend | Cat#102410 |
| Anti-mouse CD11b Monoclonal Antibody, APC | BioLegend | Cat#: 101212 |
| Anti-mouse F4/80 Monoclonal Antibody, APC | BioLegend | Cat#: 123116 |
| Anti-mouse Ter119 Monoclonal Antibody, APC | BioLegend | Cat#: 116212 |
| MULTI-seq lipid anchor and co-anchor | McGinnis et al. 2019 | Generated by the Gartner lab |
| Knockout DMEM | Gibco | Cat#10829-018 |
| Fetal Bovine Serum | Hyclone | Cat#SV30014 |
| GlutaMax | Gibco | Cat#35050-061 |
| Non-essential amino acids | Thermo Fisher Scientific | Cat#11140050 |
| 2-mercaptoethanol | Sigma | Cat#M-7522 |
| Recombinant Mouse LIF Protein | Millipore | Cat#ESG1107 |
| Critical commercial assays | ||
| SPRI Bead | Beckman Coulter | A63881 |
| KAPA HiFi HotStart ReadyMix | KAPA Biosystems | KK2601 |
| Chromium Single Cell 3’ Library & Gel Bead Kit v2 | 10x Genomics | PN-120237 |
| Chromium Single Cell A Chip Kit | 10x Genomics | PN-1000009 |
| Chromium i7 Multiplex Kit | 10x Genomics | PN-120262 |
| Qiagen Plasmid Giga kit | Qiagen | cat. no. 12191 |
| Site-directed mutagenesis kit | New England Biolabs | E0554S |
| Agilent Technologies High Sensitivity DNA Kit | Fisher Scientific | NC1738319 |
| Super PiggyBac transposase | System Biosciences | PB210PA-1 |
| Deposited data | ||
| Raw data from KP-Tracer mice (scRNA-seq, MULTI-seq, target site, and Lenti-Cre-BC) | This manuscript | PRJNA803321 |
| Processed data for KP-Tracer tumors | This manuscript | https://doi.org/10.5281/zenodo.5847461 |
| Oligonucleotides | ||
| oDYT011 sgNT oligo top tTAGCTCTtAAACCGCGGAGCCGAATACCTCGCCAACAag | This manuscript | N/A |
| oDYT012 sgNT oligo bottom TTGGCGAGGTATTCGGCTCCGCGGTTTaAGAGC | This manuscript | N/A |
| oDYT013 sgLkb1 oligo top tTAGCTCTtAAACTTGTGACTGCGGCCCACCACCAACAag | This manuscript | N/A |
| oDYT014 sgLkb1 oligo bottom TTGGTGGTGGGCCGCAGTCACAAGTTTaAGAGC | This manuscript | N/A |
| oDYT015 sgApc oligo top tTAGCTCTtAAACCGGAGTGAAACTACGCTCAAcCAACAag | This manuscript | N/A |
| oDYT016 sgApc oligo bottom TTGgTTGAGCGTAGTTTCACTCCGGTTTaAGAGC | This manuscript | N/A |
| oDYT019 gibson_3xsg_piggy FWD GACTGGATTCCTTTTTTAGGGCCCATTGGTctagaCGTGACCGAGCTTGTC | This manuscript | N/A |
| oDYT020 gibson_3xsg_piggy REV CGGGGAAAAAGCCATGTTTAAACGcggccgcctaatggatcctagtactcgaG | This manuscript | N/A |
| oDYT021 gibson_TS1.1gB_ FWD catggacgagctgtacaagtaaTGAATTAATtaaGTCACGAATCCAGCTAGCTG | This manuscript | N/A |
| oDYT022 gibson_TS1.1gB_ REV CCATTATAAGCTGCAATAAACAAGTTTCCTTAGCCGCTAATAGGTGAGCAGTTAACACCTGCAGGAGCGATGG | This manuscript | N/A |
| oDYT023-030 10x_target site amplification_primer_FAATGATACGGCGACCACCGAGATCTACACNNNNNNNNTCTTTCCCTACACGACGCTCTTCCGATCT | This manuscript | N/A |
| oDYT031-038 10x_target site amplification_primer_RCAAGCAGAAGACGGCATACGAGANNNNNNNNTGTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGAATCCAGCTAGCTGTGCAGC | This manuscript | N/A |
| oDYT039 MULTIseq spike-in CCTTGGCACCCGAGAATTCC | This manuscript | N/A |
| oDYT040 MULTIseq P5 AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT | This manuscript | N/A |
| oDYT041-48 MULTIseq P7 CAAGCAGAAGACGGCATACGAGATNNNNNNNNGTGACTGGAGTTCCTTGGCACCCGAGAATTCC | This manuscript | N/A |
| oDYT049 P5 universal for Lenti-BC AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT | This manuscript | N/A |
| oDYT050-059 P7 for Lenti-BC CAAGCAGAAGACGGCATACGAGATNNNNNNNNAGTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACCTCCCTAGCAAACTGGGGCACAAG | This manuscript | N/A |
| Software and Code | ||
| 10X cellranger | https://support.10xgenomics.com/single-cell-geneexpression/software/pipelines/latest/installation | v2.1.1 |
| deMULTIplex | https://github.com/chris-mcginnisucsf/MULTI-seq | v1.0.2 |
| inferCNV | https://github.com/broadinstitute/infercnv | v1.11.1 |
| scanpy | https://github.com/theislab/scanpy | 1.7.0rc1 |
| jungle | https://github.com/felixhorns/jungle | N/A |
| Hotspot | DeTomaso & Yosef, 2021 | v0.9.1 |
| Evolutionary Coupling | This study | https://doi.org/10.5281/zenodo.6354596 |
| Phylotime | This study | https://doi.org/10.5281/zenodo.6354596 |
| EffectivePlasticity | This study | https://doi.org/10.5281/zenodo.6354596 |
| Subclonal expansion detection | This study | https://doi.org/10.5281/zenodo.6354596 |
| Cassiopeia tree reconstruction algorithms | Jones et al, 2020 and this sutdy | https://doi.org/10.5281/zenodo.6354596 |
HIGHLIGHTS.
KP-tracer mice enable continuous, high-resolution in vivo cancer lineage tracing.
Rare subclones with distinct expression programs expand during tumor evolution.
Lineage tracing reveals cellular plasticity and evolutionary paths.
Metastases derive from spatially localized, expanding subclones of the tumor.
ACKNOWLEDGMENTS
We thank Marco Jost, Jeffrey Hussmann, Luke Koblan, Yocef Ouadah, Lindsay LaFave, Luke Gilbert, Julien Sage, Xin Ye, Brittany Adamson, Sebastian Prillo, and all members of the Weissman, Jacks and Yosef labs for helpful discussions. We thank Liming Tao, Demi Sandel, Caterina Colon, Laura Liao, Kieren Marini, Alejandro Sweet-Cordero, Danielle Dionne, Toni Delorey, Jenna Pfiffner-Borges, Orit Rozenblatt-Rosen and Aviv Regev for technical help. We thank Joan Kanter, Cristen Muresan, Karen Yee, Judy Teixeira for administrative support. We thank the UCSF Center for Advanced Technology and the Chan Zuckerberg Biohub for assistance with high-throughput sequencing. We thank UCSF Flow Cytometry Facility, UCSF Cell and Genome Engineering Core, MIT Koch Institute Animal Facility, MIT Swanson Biotechnology Center Flow Cytometry Facility.
Research reported in this publication was supported in part by the NCI Cancer Target Discovery And Development (CTD^2) and the NIH Centers of Excellence in Genomic Science (CEGS), the NCI Cancer Center Support (core) grant P30-CA14051, the Howard Hughes Medical Institute, and the Ludwig Center at MIT. D.Y. is supported by a Damon Runyon Cancer Research Foundation Postdoctoral Fellowship (DRG-2238-18). M.G.J. is supported by a UCSF Discovery Fellowship. S.N. is supported by a pre-doctoral Training Grant T32GM007287 and a Howard Hughes Medical Institute Gilliam Award. J.M.R. is supported by the NIH F31NS115380. J.J.Q. is supported by a NIH NIGMS F32GM125247. F.H. is supported by a Helen Hay Whitney Foundation Fellowship. C.S.M. is supported by the NIH-NCI F31CA257349. D.M.P. is supported by the NIH-NIGMS F32GM128366. M.M.C. is a Gordon and Betty Moore fellow of the Life Sciences Research Foundation. J.S.W. and T.J. were supported by the Howard Hughes Medical Institute and the Ludwig Center at MIT. T.J. is supported by the Break Through Cancer Foundation, Johnson & Johnson Lung Cancer Initiative, and The Lustgarten Foundation. T.G.B received funding support from the National Institutes of Health (R01CA231300, U54CA224081, R01CA204302, R01CA211052 and R01CA169338).
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
Footnotes
DECLARATION OF INTERESTS
J.S.W. declares outside interest in 5 AM Venture, Amgen, Chroma Medicine, KSQ Therapeutics, Maze Therapeutics, Tenaya Therapeutics, and Tessera Therapeutics. T.J. is a member of the Board of Directors of Amgen and Thermo Fisher Scientific, co-Founders of Dragonfly Therapeutics and T2 Biosystems, and the President of Break Through Cancer. T.J. serves on the Scientific Advisory Board of Dragonfly Therapeutics, SQZ Biotech, and Skyhawk Therapeutics. None of these affiliations represent a conflict of interest with respect to this study. T.G.B. is an advisor to Array Biopharma, Revolution Medicines, Novartis, AstraZeneca, Takeda, Springworks, Jazz Pharmaceuticals, Relay Therapeutics, Rain Therapeutics, Engine Biosciences, and receives research funding from Novartis, Strategia, Kinnate, and Revolution Medicines. J.M.R. consults for Maze Therapeutics and Waypoint Bio. Z.J.G. is an equity holder in Scribe biosciences and Provenance bio, and a member of the SAB of Serotiny Bio.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abbosh Christopher, Birkbak Nicolai J., Wilson Gareth A., Jamal-Hanjani Mariam, Constantin Tudor, Salari Raheleh, Le Quesne John, et al. 2017. “Phylogenetic ctDNA Analysis Depicts Early-Stage Lung Cancer Evolution.” Nature 545 (7655): 446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson Britt, Norman Thomas M., Jost Marco, Cho Min Y., Nuñez James K., Chen Yuwen, Villalta Jacqueline E., et al. 2016. “A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response.” Cell 167 (7): 1867–82.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alemany Anna, Florescu Maria, Baron Chloé S., Peterson-Maduro Josi, and van Oudenaarden Alexander. 2018. “Whole-Organism Clone Tracing Using Single-Cell Sequencing.” Nature 556 (7699): 108–12. [DOI] [PubMed] [Google Scholar]
- Amirouchene-Angelozzi Nabil, Swanton Charles, and Bardelli Alberto. 2017. “Tumor Evolution as a Therapeutic Target.” Cancer Discovery 7(8), pp.805–817. [DOI] [PubMed] [Google Scholar]
- Arnal-Estapé Anna, Cai Wesley L., Albert Alexandra E., Zhao Minghui, Stevens Laura E., López-Giráldez Francesc, Patel Kiran D., et al. 2020. “Tumor Progression and Chromatin Landscape of Lung Cancer Are Regulated by the Lineage Factor GATA6.” Oncogene 39 (18): 3726–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker Nick, Ridgway Rachel A., van Es Johan H., van de Wetering Marc, Begthel Harry, van den Born Maaike, Danenberg Esther, Clarke Alan R., Sansom Owen J., and Clevers Hans. 2009. “Crypt Stem Cells as the Cells-of-Origin of Intestinal Cancer.” Nature 457 (7229): 608–11. [DOI] [PubMed] [Google Scholar]
- Benjamini Yoav, and Hochberg Yosef. 1995. “Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing.” Journal of the Royal Statistical Society 57 (1): 289–300. [Google Scholar]
- Bhang Hyo-Eun C., Ruddy David A., Krishnamurthy Radhakrishna Viveksagar, Caushi Justina X., Zhao Rui, Hims Matthew M., Singh Angad P., et al. 2015. “Studying Clonal Dynamics in Response to Cancer Therapy Using High-Complexity Barcoding.” Nature Medicine 21 (5): 440–48. [DOI] [PubMed] [Google Scholar]
- Black James R. M., and McGranahan Nicholas. 2021. “Genetic and Non-Genetic Clonal Diversity in Cancer Evolution.” Nature Reviews. Cancer 21 (6): 379–92. [DOI] [PubMed] [Google Scholar]
- Bowling Sarah, Sritharan Duluxan, Osorio Fernando G., Nguyen Maximilian, Cheung Priscilla, Rodriguez-Fraticelli Alejo, Patel Sachin, et al. 2020. “An Engineered CRISPR-Cas9 Mouse Line for Simultaneous Readout of Lineage Histories and Gene Expression Profiles in Single Cells.” Cell 181 (6): 1410–22.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell Brittany B., Light Nicholas, Fabrizio David, Zatzman Matthew, Fuligni Fabio, de Borja Richard, Davidson Scott, et al. 2017. “Comprehensive Analysis of Hypermutation in Human Cancer.” Cell 171 (5): 1042–56.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carretero Julian, Shimamura Takeshi, Rikova Klarisa, Jackson Autumn L., Wilkerson Matthew D., Borgman Christa L., Buttarazzi Matthew S., et al. 2010. “Integrative Genomic and Proteomic Analyses Identify Targets for Lkb1-Deficient Metastatic Lung Tumors.” Cancer Cell 17 (6): 547–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caswell Deborah R., Chuang Chen-Hua, Yang Dian, Chiou Shin-Heng, Cheemalavagu Shashank, Kim-Kiselak Caroline, Connolly Andrew, and Winslow Monte M. 2014. “Obligate Progression Precedes Lung Adenocarcinoma Dissemination.” Cancer Discovery 4 (7): 781–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer Christine L., Marjanovic Nemanja D., Lee Tony, Bell George, Kleer Celina G., Reinhardt Ferenc, D’Alessio Ana C., Young Richard A., and Weinberg Robert A. 2013. “Poised Chromatin at the ZEB1 Promoter Enables Breast Cancer Cell Plasticity and Enhances Tumorigenicity.” Cell, 154(1): 61–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan Michelle M., Smith Zachary D., Grosswendt Stefanie, Kretzmer Helene, Norman Thomas M., Adamson Britt, Jost Marco, et al. 2019. “Molecular Recording of Mammalian Embryogenesis.” Nature 570 (7759): 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung William K. C., Zhao Minghui, Liu Zongzhi, Stevens Laura E., Cao Paul D., Fang Justin E., Westbrook Thomas F., and Nguyen Don X. 2013. “Control of Alveolar Differentiation by the Lineage Transcription Factors GATA6 and HOPX Inhibits Lung Adenocarcinoma Metastasis.” Cancer Cell 23 (6): 725–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow Ke-Huan K., Budde Mark W., Granados Alejandro A., Cabrera Maria, Yoon Shinae, Cho Soomin, Huang Ting-Hao, et al. 2021. “Imaging Cell Lineage with a Synthetic Digital Recording System.” Science 372(6538), p.eabb3099. [DOI] [PubMed] [Google Scholar]
- Chuang Chen-Hua, Greenside Peyton G., Rogers Zoë N., Brady Jennifer J., Yang Dian, Ma Rosanna K., Caswell Deborah R., et al. 2017. “Molecular Definition of a Metastatic Lung Cancer State Reveals a Targetable CD109–Janus kinase–Stat Axis.” Nature Medicine 23 (3): 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis Alexander, Gao Ruli, and Navin Nicholas. 2017. “Tumor Evolution: Linear, Branching, Neutral or Punctuated?” Biochimica et Biophysica Acta, Reviews on Cancer 1867 (2): 151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny Sarah K., Yang Dian, Chuang Chen-Hua, Brady Jennifer J., Lim Jing Shan, Grüner Barbara M., Chiou Shin-Heng, et al. 2016. “Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility.” Cell 166 (2): 328–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeTomaso David, Jones Matthew, Subramaniam Meena, Ashuach Tal, Ye Chun J., and Yosef Nir. 2019. “Functional Interpretation of Single-Cell Similarity Maps.” Nature Communications, 10(1), pp.1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeTomaso David, and Yosef Nir. 2021. “Hotspot Identifies Informative Gene Modules across Modalities of Single-Cell Genomics.” Cell Systems 12 (5): 446–56.e9. [DOI] [PubMed] [Google Scholar]
- Ding Li, Getz Gad, Wheeler David A., Mardis Elaine R., McLellan Michael D., Cibulskis Kristian, Sougnez Carrie, et al. 2008. “Somatic Mutations Affect Key Pathways in Lung Adenocarcinoma.” Nature 455 (7216): 1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driessens Gregory, Beck Benjamin, Caauwe Amélie, Simons Benjamin D., and Blanpain Cédric. 2012. “Defining the Mode of Tumour Growth by Clonal Analysis.” Nature 488 (7412): 527–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage Michel, Dooley Alison L., and Jacks Tyler. 2009. “Conditional Mouse Lung Cancer Models Using Adenoviral or Lentiviral Delivery of Cre Recombinase.” Nature Protocols 4 (7): 1064–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easwaran Hariharan, Tsai Hsing-Chen, and Baylin Stephen B. 2014. “Cancer Epigenetics: Tumor Heterogeneity, Plasticity of Stem-like States, and Drug Resistance.” Molecular Cell 54 (5): 716–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Kebir Mohammed, Satas Gryte, Oesper Layla, and Raphael Benjamin J. 2016. “Inferring the Mutational History of a Tumor Using Multi-State Perfect Phylogeny Mixtures.” Cell Systems 3 (1): 43–53. [DOI] [PubMed] [Google Scholar]
- Fitch Walter M. 1971. “Toward Defining the Course of Evolution: Minimum Change for a Specific Tree Topology.” Systematic Biology 20 (4): 406–16. [Google Scholar]
- Flanagan Dustin J., Pentinmikko Nalle, Luopajärvi Kalle, Willis Nicky J., Gilroy Kathryn, Raven Alexander P., Mcgarry Lynn, et al. 2021. “NOTUM from Apc-Mutant Cells Biases Clonal Competition to Initiate Cancer.” Nature 594 (7863): 430–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan William A., Gaskell Elizabeth, and Bernstein Bradley E. 2017. “Epigenetic Plasticity and the Hallmarks of Cancer.” Science 357 (6348), p.eaal2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese Kristopher K., and Tuveson David A. 2007. “Maximizing Mouse Cancer Models.” Nature Reviews. Cancer 7 (9): 645–58. [DOI] [PubMed] [Google Scholar]
- Frieda Kirsten L., Linton James M., Hormoz Sahand, Choi Joonhyuk, Chow Ke-Huan K., Singer Zakary S., Budde Mark W., Elowitz Michael B., and Cai Long. 2017. “Synthetic Recording and in Situ Readout of Lineage Information in Single Cells.” Nature 541 (7635): 107–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh Karuna, and Massagué Joan. 2021. “Targeting Metastatic Cancer.” Nature Medicine 27 (1): 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Ruli, Bai Shanshan, Henderson Ying C., Lin Yiyun, Schalck Aislyn, Yan Yun, Kumar Tapsi, et al. 2021. “Delineating Copy Number and Clonal Substructure in Human Tumors from Single-Cell Transcriptomes.” Nature Biotechnology 39 (5): 599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayoso Adam, Lopez Romain, Xing Galen, Boyeau Pierre, Wu Katherine, Jayasuriya Michael, Melhman Edouard, et al. 2022. “A Python library for probabilistic analysis of single-cell omics data.” Nature Biotechnology, 40(2), pp.163–166. [DOI] [PubMed] [Google Scholar]
- Gerlinger Marco, McGranahan Nicholas, Dewhurst Sally M., Burrell Rebecca A., Tomlinson Ian, and Swanton Charles. 2014. “Cancer: Evolution within a Lifetime.” Annual Review of Genetics 4, 215–36. [DOI] [PubMed] [Google Scholar]
- Gerstung Moritz, Jolly Clemency, Leshchiner Ignaty, Dentro Stefan C., Gonzalez Santiago, Rosebrock Daniel, Mitchell Thomas J., et al. 2020. “The Evolutionary History of 2,658 Cancers.” Nature 578 (7793): 122–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Yejing, Gomez Nicholas C., Adam Rene C., Nikolova Maria, Yang Hanseul, Verma Akanksha, Pei-Ju Lu Catherine, et al. 2017. “Stem Cell Lineage Infidelity Drives Wound Repair and Cancer.” Cell 169 (4): 636–50.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths RC, and Tavaré Simon. 1998. “The Age of a Mutation in a General Coalescent Tree.” Communications in Statistics. Stochastic Models 14 (1–2): 273–95. [Google Scholar]
- Hanahan Douglas, and Weinberg Robert A. 2011. “Hallmarks of Cancer: The next Generation.” Cell 144 (5): 646–74. [DOI] [PubMed] [Google Scholar]
- Hann Byron, and Balmain Allan. 2001. “Building ‘validated’ Mouse Models of Human Cancer.” Current Opinion in Cell Biology, 13(6), pp.778–784 [DOI] [PubMed] [Google Scholar]
- Hartigan JA 1973. “Minimum Mutation Fits to a Given Tree.” Biometrics 29 (1): 53–65. [Google Scholar]
- He Weiling, Zhang Hui, Wang Yuefeng, Zhou Yanbin, Luo Yifeng, Cui Yongmei, Jiang Neng, et al. 2018. “CTHRC1 Induces Non-Small Cell Lung Cancer (NSCLC) Invasion through Upregulating MMP-7/MMP-9.” BMC Cancer 18 (1): 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill William, Caswell Deborah R., and Swanton Charles. 2021. “Capturing Cancer Evolution Using Genetically Engineered Mouse Models (GEMMs).” Trends in Cell Biology, 31(12), pp.1007–1018 [DOI] [PubMed] [Google Scholar]
- Hollstein Pablo E., Eichner Lillian J., Brun Sonja N., Kamireddy Anwesh, Svensson Robert U., Vera Liliana I., Ross Debbie S., et al. 2019. “The AMPK-Related Kinases SIK1 and SIK3 Mediate Key Tumor-Suppressive Effects of LKB1 in NSCLC.” Cancer Discovery 9 (11): 1606–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüsemann Yves, Geigl Jochen B., Schubert Falk, Musiani Piero, Meyer Manfred, Burghart Elke, Forni Guido, et al. 2008. “Systemic Spread Is an Early Step in Breast Cancer.” Cancer Cell 13 (1): 58–68. [DOI] [PubMed] [Google Scholar]
- Hu Zheng, and Curtis Christina. 2020. “Looking Backward in Time to Define the Chronology of Metastasis.” Nature Communications 11 (1): 3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Zheng, Li Zan, Ma Zhicheng, and Curtis Christina. 2020. “Multi-Cancer Analysis of Clonality and the Timing of Systemic Spread in Paired Primary Tumors and Metastases.” Nature Genetics 52 (7): 701–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, and Tuveson DA 2001. “Analysis of Lung Tumor Initiation and Progression Using Conditional Expression of Oncogenic K-Ras.” Genes & Development 15 (24): 3243–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson Erica L., Olive Kenneth P., Tuveson David A., Bronson Roderick, Crowley Denise, Brown Michael, and Jacks Tyler. 2005. “The Differential Effects of Mutant p53 Alleles on Advanced Murine Lung Cancer.” Cancer Research 65 (22): 10280–88. [DOI] [PubMed] [Google Scholar]
- Jamal-Hanjani Mariam, Wilson Gareth A., McGranahan Nicholas, Birkbak Nicolai J., Watkins Thomas B. K., Veeriah Selvaraju, Shafi Seema, et al. 2017. “Tracking the Evolution of Non-Small-Cell Lung Cancer.” The New England Journal of Medicine 376 (22): 2109–21. [DOI] [PubMed] [Google Scholar]
- Ji Hongbin, Ramsey Matthew R., Hayes D. Neil, Fan Cheng, McNamara Kate, Kozlowski Piotr, Torrice Chad, et al. 2007. “LKB1 Modulates Lung Cancer Differentiation and Metastasis.” Nature 448 (7155): 807–10. [DOI] [PubMed] [Google Scholar]
- Jones Matthew G., Khodaverdian Alex, Quinn Jeffrey J., Chan Michelle M., Hussmann Jeffrey A., Wang Robert, Xu Chenling, Weissman Jonathan S., and Yosef Nir. 2020. “Inference of Single-Cell Phylogenies from Lineage Tracing Data Using Cassiopeia.” Genome Biology 21 (1): 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones Matthew G., Rosen Yanay, and Yosef Nir. 2021. “PhyloVision: Interactive Software for Integrated Analysis of Single-Cell Transcriptomic and Phylogenetic Data.” bioRxiv. 10.1101/2021.09.13.460142 [DOI] [PMC free article] [PubMed]
- Kalhor Reza, Kalhor Kian, Mejia Leo, Leeper Kathleen, Graveline Amanda, Mali Prashant, and Church George M. 2018. “Developmental Barcoding of Whole Mouse via Homing CRISPR.” Science 361 (6405), p.eaat9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerk Samuel A., Papagiannakopoulos Thales, Shah Yatrik M., and Lyssiotis Costas A. 2021. “Metabolic Networks in Mutant KRAS-Driven Tumours: Tissue Specificities and the Microenvironment.” Nature Reviews. Cancer 21 (8): 510–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Charissa, Gao Ruli, Sei Emi, Brandt Rachel, Hartman Johan, Hatschek Thomas, Crosetto Nicola, Foukakis Theodoros, and Navin Nicholas E. 2018. “Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing.” Cell 173 (4): 879–93.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Christoph A. 2009. “Parallel Progression of Primary Tumours and Metastases.” Nature Reviews. Cancer 9 (4): 302–12. [DOI] [PubMed] [Google Scholar]
- LaFave Lindsay M., Kartha Vinay K., Ma Sai, Meli Kevin, Isabella Del Priore Caleb Lareau, Naranjo Santiago, et al. 2020. “Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma.” Cancer Cell 38 (2): 212–28.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manno La, Gioele Ruslan Soldatov, Zeisel Amit, Braun Emelie, Hochgerner Hannah, Petukhov Viktor, Lidschreiber Katja, et al. 2018. “RNA Velocity of Single Cells.” Nature 560 (7719): 494–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Xiaoyang, Jörg David J., Cavalli Florence M. G., Richards Laura M., Nguyen Long V., Vanner Robert J., Guilhamon Paul, et al. 2017. “Fate Mapping of Human Glioblastoma Reveals an Invariant Stem Cell Hierarchy.” Nature 549 (7671): 227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughney Ashley M., Hu Jing, Campbell Nathaniel R., Bakhoum Samuel F., Setty Manu, Lavallée Vincent-Philippe, Xie Yubin, et al. 2020. “Regenerative Lineages and Immune-Mediated Pruning in Lung Cancer Metastasis.” Nature Medicine, 26(2), pp.259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Je Hyuk, Daugharthy Evan R., Scheiman Jonathan, Kalhor Reza, Yang Joyce L., Ferrante Thomas C., Terry Richard, et al. 2014. “Highly Multiplexed Subcellular RNA Sequencing in Situ.” Science 343 (6177): 1360–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Amy, Herbst Rebecca H., Canner David, Schenkel Jason M., Smith Olivia C., Kim Jonathan Y., Hillman Michelle, et al. 2019. “IL-33 Signaling Alters Regulatory T Cell Diversity in Support of Tumor Development.” Cell Reports 29 (10): 2998–3008.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livet Jean, Weissman Tamily A., Kang Hyuno, Draft Ryan W., Lu Ju, Bennis Robyn A., Sanes Joshua R., and Lichtman Jeff W. 2007. “Transgenic Strategies for Combinatorial Expression of Fluorescent Proteins in the Nervous System.” Nature 450 (7166): 56–62. [DOI] [PubMed] [Google Scholar]
- Lopez Romain, Regier Jeffrey, Cole Michael B., Jordan Michael I., and Yosef Nir. 2018. “Deep Generative Modeling for Single-Cell Transcriptomics.” Nature Methods 15 (12): 1053–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig Leif S., Lareau Caleb A., Ulirsch Jacob C., Christian Elena, Muus Christoph, Li Lauren H., Pelka Karin, et al. 2019. “Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics.” Cell 176 (6): 1325–39.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malikic Salem, Jahn Katharina, Kuipers Jack, Sahinalp S. Cenk, and Beerenwinkel Niko. 2019. “Integrative Inference of Subclonal Tumour Evolution from Single-Cell and Bulk Sequencing Data.” Nature Communications 10 (1): 2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjanovic Nemanja Despot, Hofree Matan, Chan Jason E., Canner David, Wu Katherine, Trakala Marianna, Hartmann Griffin G., et al. 2020. “Emergence of a High-Plasticity Cell State during Lung Cancer Evolution.” Cancer Cell 38 (2): 229–46.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Sai, Zhang Bing, LaFave Lindsay M., Earl Andrew S., Chiang Zachary, Hu Yan, Ding Jiarui, et al. 2020. “Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin.” Cell 183 (4): 1103–16.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Ashley, McCoach Caroline E., Rotow Julia K., Harris Lincoln, Haderk Franziska, Kerr D. Lucas, Yu Elizabeth A., et al. 2020. “Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing.” Cell 182 (5): 1232–51.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden David G., Politi Katerina, Bhutkar Arjun, Chen Frances K., Song Xiaoling, Pirun Mono, Santiago Philip M., et al. 2016. “Mutational Landscape of EGFR-, MYC-, and Kras-Driven Genetically Engineered Mouse Models of Lung Adenocarcinoma.” Proceedings of the National Academy of Sciences of the United States of America 113 (42): E6409–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis Christopher S., Patterson David M., Winkler Juliane, Conrad Daniel N., Hein Marco Y., Srivastava Vasudha, Hu Jennifer L., et al. 2019. “MULTI-Seq: Sample Multiplexing for Single-Cell RNA Sequencing Using Lipid-Tagged Indices.” Nature Methods 16 (7): 619–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan Nicholas, and Swanton Charles. 2017. “Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future.” Cell 168 (4): 613–28. [DOI] [PubMed] [Google Scholar]
- McInnes Leland, Healy John, and Melville James. 2018. “UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction.” arXiv [stat.ML] preprint at arXiv:1802.03426.
- McKenna Aaron, Findlay Gregory M., Gagnon James A., Horwitz Marshall S., Schier Alexander F., and Shendure Jay. 2016. “Whole-Organism Lineage Tracing by Combinatorial and Cumulative Genome Editing.” Science 353 (6298), p.aaf7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna Aaron, and Gagnon James A. 2019. “Recording Development with Single Cell Dynamic Lineage Tracing.” Development 146 (12). 10.1242/dev.169730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou Eleni P., Lareau Caleb A., Chen Kelvin Y., Zorzetto-Fernandes Andre L., Hao Yuhan, Takeshima Yusuke, Luo Wendy, et al. 2021. “Scalable, Multimodal Profiling of Chromatin Accessibility, Gene Expression and Protein Levels in Single Cells.” Nature Biotechnology, 39(10), pp.1246–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray Christopher W., Brady Jennifer J., Tsai Min K., Li Chuan, Winters Ian P., Tang Rui, Andrejka Laura, et al. 2019. “An LKB1–SIK Axis Suppresses Lung Tumor Growth and Controls Differentiation.” Cancer Discovery, 9(11), pp.1590–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Neerven Sanne M., de Groot Nina E., Nijman Lisanne E., Scicluna Brendon P., van Driel Milou S., Lecca Maria C., Warmerdam Daniël O., et al. 2021. “Apc-Mutant Cells Act as Supercompetitors in Intestinal Tumour Initiation.” Nature 594 (7863): 436–41. [DOI] [PubMed] [Google Scholar]
- Neftel Cyril, Laffy Julie, Filbin Mariella G., Hara Toshiro, Shore Marni E., Rahme Gilbert J., Richman Alyssa R., et al. 2019. “An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma.” Cell 178 (4): 835–49.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher Richard A., Russell Colin A., and Shraiman Boris I. 2014. “Predicting Evolution from the Shape of Genealogical Trees.” eLife 3, p.e03568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network, The Cancer Genome Atlas Research. 2014. “Comprehensive Molecular Profiling of Lung Adenocarcinoma.” Nature, 511, pp.543–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Don X., Chiang Anne C., Zhang Xiang H-F, Kim Juliet Y., Kris Mark G., Ladanyi Marc, Gerald William L., and Massagué Joan. 2009. “WNT/TCF Signaling through LEF1 and HOXB9 Mediates Lung Adenocarcinoma Metastasis.” Cell 138 (1): 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell PC 1976. “The Clonal Evolution of Tumor Cell Populations.” Science 194 (4260): 23–28. [DOI] [PubMed] [Google Scholar]
- Ouardini Khalil, Lopez Romain, Jones Matthew G., Prillo Sebastian, Zhang Richard, Jordan Michael I., and Yosef Nir. 2021. “Reconstructing Unobserved Cellular States from Paired Single-Cell Lineage Tracing and Transcriptomics Data.” bioRxiv preprint available at 10.1101/2021.05.28.446021. [DOI]
- Park Jihye, Lim Jung Min, Jung Inkyung, Heo Seok-Jae, Park Jinman, Chang Yoojin, Kim Hui Kwon, et al. 2021. “Recording of Elapsed Time and Temporal Information about Biological Events Using Cas9.” Cell 184 (4): 1047–63.e23. [DOI] [PubMed] [Google Scholar]
- Parsons Marie J., Tammela Tuomas, and Dow Lukas E. 2021. “WNT as a Driver and Dependency in Cancer.” Cancer Discovery, 11(10), pp.2413–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel Anoop P., Tirosh Itay, Trombetta John J., Shalek Alex K., Gillespie Shawn M., Wakimoto Hiroaki, Cahill Daniel P., et al. 2014. “Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma.” Science 344 (6190): 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Weike, Feyerabend Thorsten B., Rössler Jens, Wang Xi, Postrach Daniel, Busch Katrin, Rode Immanuel, et al. 2017. “Polylox Barcoding Reveals Haematopoietic Stem Cell Fates Realized in Vivo.” Nature 548 (7668): 456–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce Sarah E., Granja Jeffrey M., Corces M. Ryan, Brady Jennifer J., Tsai Min K., Pierce Aubrey B., Tang Rui, et al. 2021. “LKB1 Inactivation Modulates Chromatin Accessibility to Drive Metastatic Progression.” Nature Cell Biology 23, 915–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podsypanina Katrina, Du Yi-Chieh Nancy, Jechlinger Martin, Beverly Levi J., Hambardzumyan Dolores, and Varmus Harold. 2008. “Seeding and Propagation of Untransformed Mouse Mammary Cells in the Lung.” Science 321 (5897): 1841–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter Nicola E., Ermini Luca, Papaemmanuil Elli, Cazzaniga Giovanni, Vijayaraghavan Gowri, Titley Ian, Ford Anthony, Campbell Peter, Kearney Lyndal, and Greaves Mel. 2013. “Single-Cell Mutational Profiling and Clonal Phylogeny in Cancer.” Genome Research 23 (12): 2115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powles Thomas, Assaf Zoe June, Davarpanah Nicole, Banchereau Romain, Szabados Bernadett E., Yuen Kobe C., Grivas Petros, et al. 2021. “ctDNA Guiding Adjuvant Immunotherapy in Urothelial Carcinoma.” Nature 595(7867), pp.432–437. [DOI] [PubMed] [Google Scholar]
- Premsrirut Prem K., Dow Lukas E., Kim Sang Yong, Camiolo Matthew, Malone Colin D., Miething Cornelius, Scuoppo Claudio, et al. 2011. “A Rapid and Scalable System for Studying Gene Function in Mice Using Conditional RNA Interference.” Cell 145 (1): 145–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn Jeffrey J., Jones Matthew G., Okimoto Ross A., Nanjo Shigeki, Chan Michelle M., Yosef Nir, Bivona Trever G., and Weissman Jonathan S. 2021. “Single-Cell Lineages Reveal the Rates, Routes, and Drivers of Metastasis in Cancer Xenografts.” Science, 371(6532), p.eabc1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanal-Villalonga Álvaro, Chan Joseph M., Yu Helena A., Pe’er Dana, Sawyers Charles L., Sen Triparna, and Rudin Charles M. 2020. “Lineage Plasticity in Cancer: A Shared Pathway of Therapeutic Resistance.” Nature Reviews. Clinical Oncology 17 (6): 360–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj Bushra, Wagner Daniel E., McKenna Aaron, Pandey Shristi, Klein Allon M., Shendure Jay, Gagnon James A., and Schier Alexander F. 2018. “Simultaneous Single-Cell Profiling of Lineages and Cell Types in the Vertebrate Brain.” Nature Biotechnology 36 (5): 442–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert Philipp, Roth Mareike, Neumann Tobias, Muerdter Felix, Roe Jae-Seok, Muhar Matthias, Deswal Sumit, et al. 2015. “Transcriptional Plasticity Promotes Primary and Acquired Resistance to BET Inhibition.” Nature 525 (7570): 543–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim Andrew D., Mirek Emily T., Aiello Nicole M., Maitra Anirban, Bailey Jennifer M., McAllister Florencia, Reichert Maximilian, et al. 2012. “EMT and Dissemination Precede Pancreatic Tumor Formation.” Cell, 148(1–2), pp.349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers Zoë N., McFarland Christopher D., Winters Ian P., Naranjo Santiago, Chuang Chen-Hua, Petrov Dmitri, and Winslow Monte M. 2017. “A Quantitative and Multiplexed Approach to Uncover the Fitness Landscape of Tumor Suppression in Vivo.” Nature Methods 14 (7): 737–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers Zoë N., McFarland Christopher D., Winters Ian P., Seoane Jose A., Brady Jennifer J., Yoon Stephanie, Curtis Christina, Petrov Dmitri A., and Winslow Monte M. 2018. “Mapping the in Vivo Fitness Landscape of Lung Adenocarcinoma Tumor Suppression in Mice.” Nature Genetics 50 (4): 483–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, and Nei M. 1987. “The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees.” Molecular Biology and Evolution 4 (4): 406–25. [DOI] [PubMed] [Google Scholar]
- Salehi Sohrab, Kabeer Farhia, Ceglia Nicholas, Andronescu Mirela, Williams Marc J., Campbell Kieran R., Masud Tehmina, et al. 2021. “Clonal Fitness Inferred from Time-Series Modelling of Single-Cell Cancer Genomes.” Nature 595 (7868): 585–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satas Gryte, Zaccaria Simone, Mon Geoffrey, and Raphael Benjamin J. 2020. “SCARLET: Single-Cell Tumor Phylogeny Inference with Copy-Number Constrained Mutation Losses.” Cell Systems 10 (4): 323–32.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepers Arnout G., Snippert Hugo J., Stange Daniel E., van den Born Maaike, van Es Johan H., van de Wetering Marc, and Clevers Hans. 2012. “Lineage Tracing Reveals Lgr5+ Stem Cell Activity in Mouse Intestinal Adenomas.” Science 337 (6095): 730–35. [DOI] [PubMed] [Google Scholar]
- Schwartz Russell, and Schäffer Alejandro A. 2017. “The Evolution of Tumour Phylogenetics: Principles and Practice.” Nature Reviews. Genetics 18 (4): 213–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer Sydney M., Dunagin Margaret C., Torborg Stefan R., Torre Eduardo A., Emert Benjamin, Krepler Clemens, Beqiri Marilda, et al. 2017. “Rare Cell Variability and Drug-Induced Reprogramming as a Mode of Cancer Drug Resistance.” Nature 546 (7658): 431–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr Charles J. 2004. “Principles of Tumor Suppression.” Cell, 116(2), pp.235–246. [DOI] [PubMed] [Google Scholar]
- Simeonov Kamen P., Byrns China N., Clark Megan L., Norgard Robert J., Martin Beth, Ben Stanger Z, Shendure Jay, McKenna Aaron, and Lengner Christopher J. 2021. “Single-Cell Lineage Tracing of Metastatic Cancer Reveals Selection of Hybrid EMT States.” Cancer Cell, 39(8), pp.1150–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinjab Ansam, Han Guangchun, Treekitkarnmongkol Warapen, Hara Kieko, Brennan Patrick M., Dang Minghao, Hao Dapeng, et al. 2021. “Resolving the Spatial and Cellular Architecture of Lung Adenocarcinoma by Multiregion Single-Cell Sequencing.” Cancer Discovery, 11(10), pp.2506–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöblom Tobias, Jones Siân, Wood Laura D., Parsons D. Williams, Lin Jimmy, Barber Thomas D., Mandelker Diana, et al. 2006. “The Consensus Coding Sequences of Human Breast and Colorectal Cancers.” Science 314 (5797): 268–74. [DOI] [PubMed] [Google Scholar]
- Skoulidis Ferdinandos, Byers Lauren A., Diao Lixia, Papadimitrakopoulou Vassiliki A., Tong Pan, Izzo Julie, Behrens Carmen, et al. 2015. “Co-Occurring Genomic Alterations Define Major Subsets of KRAS-Mutant Lung Adenocarcinoma with Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities.” Cancer Discovery 5 (8): 860–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottoriva Andrea, Kang Haeyoun, Ma Zhicheng, Graham Trevor A., Salomon Matthew P., Zhao Junsong, Marjoram Paul, et al. 2015. “A Big Bang Model of Human Colorectal Tumor Growth.” Nature Genetics 47 (3): 209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanjaard Bastiaan, Hu Bo, Mitic Nina, Pedro Olivares-Chauvet Sharan Janjuha, Ninov Nikolay, and Junker Jan Philipp. 2018. “Simultaneous Lineage Tracing and Cell-Type Identification Using CRISPR--Cas9-Induced Genetic Scars.” Nature Biotechnology 36 (5): 469–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speidel Leo, Forest Marie, Shi Sinan, and Myers Simon R. 2019. “A Method for Genome-Wide Genealogy Estimation for Thousands of Samples.” Nature Genetics 51 (9): 1321–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler T, Pybus OG, and Stumpf MPH 2021. “Phylodynamics for Cell Biologists.” Science 371 (6526), p.eaah6266. [DOI] [PubMed] [Google Scholar]
- Stickels Robert R., Murray Evan, Kumar Pawan, Li Jilong, Marshall Jamie L., Di Bella Daniela J., Arlotta Paola, Macosko Evan Z., and Chen Fei. 2021. “Highly Sensitive Spatial Transcriptomics at near-Cellular Resolution with Slide-seqV2.” Nature Biotechnology 39 (3): 313–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammela Tuomas, and Sage Julien. 2020. “Investigating Tumor Heterogeneity in Mouse Models.” Annual Review of Cancer Biology 4 (1): 99–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammela Tuomas, Sanchez-Rivera Francisco J., Cetinbas Naniye Malli, Wu Katherine, Joshi Nikhil S., Helenius Katja, Park Yoona, et al. 2017. “A Wnt-Producing Niche Drives Proliferative Potential and Progression in Lung Adenocarcinoma.” Nature 545 (7654): 355–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Weixin, and Liu David R. 2018. “Rewritable Multi-Event Analog Recording in Bacterial and Mammalian Cells.” Science, 360(6285), p.eaap8992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarabichi Maxime, Salcedo Adriana, Deshwar Amit G., Máire Ni Leathlobhair Jeff Wintersinger, Wedge David C., Peter Van Loo Quaid D. Morris, and Boutros Paul C. 2021. “A Practical Guide to Cancer Subclonal Reconstruction from DNA Sequencing.” Nature Methods 18 (2): 144–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie Masoud F., Pollack Ilana, Tanqueco Raissa, Ostendorf Benjamin N., Reis Bernardo S., Gonsalves Foster C., Kurth Isabel, et al. 2018. “LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer.” Cell 172 (4): 825–40.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traag VA, Waltman L, and van Eck NJ 2019. “From Louvain to Leiden: Guaranteeing Well-Connected Communities.” Scientific Reports 9 (1): 5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell Cole, Cacchiarelli Davide, Grimsby Jonna, Pokharel Prapti, Li Shuqiang, Morse Michael, Lennon Niall J., Livak Kenneth J., Mikkelsen Tarjei S., and Rinn John L. 2014. “The Dynamics and Regulators of Cell Fate Decisions Are Revealed by Pseudotemporal Ordering of Single Cells.” Nature Biotechnology 32 (4): 381–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritschler Sophie, Maren Büttner David S. Fischer, Lange Marius, Bergen Volker, Lickert Heiko, and Theis Fabian J. 2019. “Concepts and Limitations for Learning Developmental Trajectories from Single Cell Genomics.” Development 146 (12), p.dev170506. [DOI] [PubMed] [Google Scholar]
- Turajlic Samra, and Swanton Charles. 2016. “Metastasis as an Evolutionary Process.” Science 352 (6282): 169–75. [DOI] [PubMed] [Google Scholar]
- Van den Berge Koen, de Bézieux Hector Roux, Street Kelly, Saelens Wouter, Cannoodt Robrecht, Saeys Yvan, Dudoit Sandrine, and Clement Lieven. 2020. “Trajectory-Based Differential Expression Analysis for Single-Cell Sequencing Data.” Nature Communications 11 (1): 1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein Bert, Fearon Eric R., Hamilton Stanley R., Kern Scott E., Preisinger Ann C., Leppert Mark, Smits Alida M. M., and Bos Johannes L. 1988. “Genetic Alterations during Colorectal-Tumor Development.” New England Journal of Medicine 319 (9): 525–32. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, and Kinzler KW 2013. “Cancer Genome Landscapes.” Science, 339(6127), pp.1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner Daniel E., and Klein Allon M. 2020. “Lineage Tracing Meets Single-Cell Omics: Opportunities and Challenges.” Nature Reviews. Genetics 21 (7): 410–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner Daniel E., Weinreb Caleb, Collins Zach M., Briggs James A., Megason Sean G., and Klein Allon M. 2018. “Single-Cell Mapping of Gene Expression Landscapes and Lineage in the Zebrafish Embryo.” Science 360 (6392): 981–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA 1991. “Tumor Suppressor Genes.” Science 254 (5035): 1138–46. [DOI] [PubMed] [Google Scholar]
- Weinreb Caleb, Rodriguez-Fraticelli Alejo, Camargo Fernando D., and Klein Allon M. 2020. “Lineage Tracing on Transcriptional Landscapes Links State to Fate during Differentiation.” Science 367 (6479), p.eaaw3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westcott Peter M. K., Halliwill Kyle D., To Minh D., Rashid Mamunur, Rust Alistair G., Keane Thomas M., Delrosario Reyno, et al. 2015. “The Mutational Landscapes of Genetic and Chemical Models of Kras-Driven Lung Cancer.” Nature 517 (7535): 489–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams Marc J., Werner Benjamin, Heide Timon, Curtis Christina, Barnes Chris P., Sottoriva Andrea, and Graham Trevor A. 2018. “Quantification of Subclonal Selection in Cancer from Bulk Sequencing Data.” Nature Genetics 50 (6): 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow Monte M., Dayton Talya L., Verhaak Roel G. W., Kim-Kiselak Caroline, Snyder Eric L., Feldser David M., Hubbard Diana D., et al. 2011. “Suppression of Lung Adenocarcinoma Progression by Nkx2–1.” Nature 473 (7345): 101–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters Ian P., Murray Christopher W., and Winslow Monte M. 2018. “Towards Quantitative and Multiplexed in Vivo Functional Cancer Genomics.” Nature Reviews. Genetics 19 (12): 741–55. [DOI] [PubMed] [Google Scholar]
- Wolf F. Alexander, Angerer Philipp, and Theis Fabian J. 2018. “SCANPY: Large-Scale Single-Cell Gene Expression Data Analysis.” Genome Biology 19 (1): 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Chenling, Lopez Romain, Mehlman Edouard, Regier Jeffrey, Jordan Michael I., and Yosef Nir. 2021. “Probabilistic Harmonization and Annotation of Single-Cell Transcriptomics Data with Deep Generative Models.” Molecular Systems Biology 17 (1): e9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Jun, Jiang Ying, Lu Jianfeng, Wu Jianhui, and Zhang Mingfang. 2019. “Inhibiting of Proliferation, Migration, and Invasion in Lung Cancer Induced by Silencing Interferon-Induced Transmembrane Protein 1 (IFITM1).” BioMed Research International 2019 (May). 10.1155/2019/9085435. [DOI] [PMC free article] [PubMed] [Retracted]
- Yuan Salina, Norgard Robert J., and Ben Stanger Z 2019. “Cellular Plasticity in Cancer.” Cancer Discovery. 9(7), pp.837–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Weijie, Bado Igor L., Hu Jingyuan, Wan Ying-Wooi, Wu Ling, Wang Hai, Gao Yang, et al. 2021. “The Bone Microenvironment Invigorates Metastatic Seeds for Further Dissemination.” Cell 184 (9): 2471–86.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Yinghui, Rideout William M. 3rd, Zi Tong, Bressel Angela, Reddypalli Shailaja, Rancourt Rebecca, Woo Jin-Kyeung, et al. 2010. “Chimeric Mouse Tumor Models Reveal Differences in Pathway Activation between ERBB Family- and KRAS-Dependent Lung Adenocarcinomas.” Nature Biotechnology 28 (1): 71–78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) The piggyBac transposon-based lineage tracing vector libraries used to engineer the KP-Tracer mice contained (1) a triple-guideRNA cassette and (2) a target site library cassette with a 14bp integration barcode (“intBC”) and three CRISPR/Cas9 cut sites on the 3’ UTR of an mCherry reporter gene. (B) Enrichment of mESC population with high lineage-tracer expression based on high mCherry expression (a reporter indicating lineage tracer expression). These cells are then single-cell cloned before generating chimeric KP-Tracer mice. (C) Representative images of specific mCherry positive mESC clones that express the lineage tracing vectors. (D) Copy number of lineage tracing vectors across 5 mouse embryonic stem cell (mESC) clones used in this study measured by genomic qPCR are shown. (E-F) Detection of unique lineage tracing target site intBCs for a representative mESC clone (1D5) using (E) DNA-sequencing and (F) scRNA-seq. A consensus set of target sites intBCs for each mESC clone was determined by selecting intBCs detected in at least 40% of all tumors derived from that mESC clone. (G) The consensus intBC pivot table across all five mESC clones used in this study to generate KP-Tracer mice. Each row is a single cell and is annotated with which mESC clone it came from. Each column is a unique intBC. Colors in the heatmap indicate whether or not an intBC was detected in a given cell. (H) Quality-control filtering of tumor phylogenies for subclonal expansion analyses. Quality of lineage-tracing data was assessed with two metrics: first, the percentage of cells that contained a unique set of mutations (“% unique indel state”; STAR Methods); and second, the percentage of target sites that had to be filtered because of low-diversity (“target site saturation”; STAR Methods). Tumors with less than 5% overall unique indel state, greater than 80% target site saturation, or fewer than 100 cells were filtered out.
(A-D) Phylogenetic features of tumor lineages and their predictiveness (as measured with R2) on the expansion proportion of a tumor. Features evaluated were (A) age, (B) median tree depth, (C) size measured in the number of cells, and (D) proportion of unique cells. (E) Expansion proportion of tumors measured from Neighbor-Joining trees versus Cassiopeia trees. The percentage of cells in expansions were highly consistent between these two tree reconstruction strategies (Pearson’s correlation = 0.87). (F) Comparison of cell-cycle scores inferred from transcriptomic profiles in expanding versus non-expanding tumor subclones, identified from Neighbor-Joining trees (** p < 0.01). (G-H) Representative example of comparison between hierarchical clustering of CNVs and Cassiopeia-reconstructed phylogeny. (G) The inferred CNVs are shown for the representative tumor, with the largest two clusters, identified via hierarchical clustering, indicated by the colorbar. (H) These two clusters are also indicated with unique colors on the Cassiopeia-reconstructed tumor phylogeny. The good correlation between CNV status and tumor phylogeny indicates the accuracy of tree reconstruction. (I) Heatmap displaying the probabilities that a cell and its nearest neighbor on the Cassiopeia-reconstructed phylogeny are in the same CNV cluster (size of circles). These probabilities were calculated for each tumor at various depths of the CNV hierarchical clustering dendrogram. The depth that yielded the most coarse-grained clusters were set to have a cutoff height of 1, with higher cutoff heights indicating finer clusters. The majority of Cassiopeia-reconstructed phylogenies were significantly consistent with CNV clusters (color of circles; Permutation Test) at all clustering resolutions. (J) A comparison of CNV counts in expanding versus non-expanding portions of tumors (* p < 0.05, ** p < 0.01, *** p < 0.001). (K) An example of distinct CNV regions of cells from a single tumor. This tumor underwent two independent clonal expansions (red branches; left), each of which exhibited distinct CNV patterns (red bars; right). (L) An aggregated view of the CNV “hotspots” across subclonal expansions from all tumors. Each horizontal bar represents a chromosome, and the intensity of color indicates the number of subclonal expansions exhibiting a CNV in a region (STAR Methods). Regions that more often exhibited copy number gains are indicated in red (left); genomic regions that more often exhibited copy number losses are indicated in blue (right).
(A) Gene markers for each Leiden cluster identified in the processed scRNA-seq latent space. Dot size indicates the percent of cells expressing the marker. Color indicates mean expression level. (B) Integration of normal lung epithelial cells with KP-Tracer dataset. Normal lung epithelial cells were isolated from an independent dataset and integrated with KP-Tracer tumors using scVI (STAR Methods). Leiden cluster annotations from analysis of KP-Tracer tumors are shown (top) and normal cells are highlighted against tumor cells (bottom). (C) Gene set comparison between the FitnessSignature described in this study and KP tumor progression-associated genes described in (Chuang et al. 2017). Overlap significance assessed with a hypergeometric test (*** = p < 1e-5). (D) Average transcriptional FitnessSignature score in KP tumors harvested at 12-week, 20-week, and 30-week timepoints from (Marjanovic et al. 2020). (E) Representative examples of tumors occupying distinct regions of the transcriptional space. Cells from the tumor of interest are shown in red, and all other cells are shown in gray. (F) Hotspot autocorrelation heatmap and clustering of genes that appear in the FitnessSignature and are positively associated with fitness. Gene modules are identified by distinct color strips on the left. Values in the heatmap are Z-normalized pairwise autocorrelation scores between genes. The dendrogram linking genes is shown for the columns. (G) Z-normalized mean fitness gene module signature scores of each Leiden cluster. (H) Kaplan-Meier plots for TCGA human lung adenocarcinoma patients with respect to genes in each fitness module. Curves are shown comparing overall survival of patient groups whose tumors have high (red) versus low (blue) expression of individual fitness gene modules, as determined by the median fitness module score. P-values from a log-rank test are indicated. (I) Fitness module enrichment personality plots. Each corner of the triangle represents the fold enrichment of an expansion’s fitness module expression over expectation (non-expanding background). Independent expansions in each tumor are shown in unique colors (blue or orange). (J) Venn diagram illustrating the classification of expansions to gene modules based on a p-value threshold of 0.05 using a permutation test against non-expanding background.
(A) Leiden cluster proportions for each KP-Tracer tumor. The fraction of cells in each Leiden cluster is shown for each tumor in a stacked bar plot, where each Leiden cluster is indicated by the unique color introduced in Fig 3A. Tumors are ordered by mean FitnessSignature score. (B) Shannon’s Entropy statistic for each tumor, computed with the Leiden cluster proportions; tumors are ordered by mean FitnessSignature score. (C) Allelic EffectivePlasticity score overlaid onto two-dimensional gene expression UMAP is shown. Allelic EffectivePlasticity is an alternative way to quantify EffectivePlasticity by comparing transcriptional states between cells with similar lineage tracing indel states without using lineage trees. (D) Comparison of Allelic EffectivePlasticity to scEffectivePlasticity (Pearson’s correlation = 0.73). Each point represents a single cell. (E) Comparison of mean tumor Allelic EffectivePlasticity to tumor EffectivePlasticity (Pearson’s correlation = 0.96). Each point represents a tumor. (F) L2 EffectivePlasticity score overlaid onto two-dimensional gene expression UMAP is shown. L2 EffectivePlasticity is another alternative way to quantify EffectivePlasticity by computing dissimilarity in gene expression profiles between nearest neighbors on the phylogeny. (G) Comparison of single-cell L2 EffectivePlasticity to scEffectivePlasticity (Pearson’s correlation = 0.69). Each point represents a single cell. (H) Comparison of mean tumor L2 EffectivePlasticity to mean tumor EffectivePlasticity (Pearson’s correlation = 0.95). Each point represents a tumor. (I) Comparison of scEffectivePlasticity to single-cell FitnessSignature scores. Each point represents a single cell. (J) Weighted mean EffectivePlasticity vs mean FitnessSignature for each transcriptional state (Leiden cluster). The weighted Mean EffectivePlasticity for each Leiden cluster was determined by first computing the mean scEffectivePlasticity for each Leiden cluster in a tumor, and then averaging these values together. Each point represents a tumor.
(A-D) Two alternative statistics measuring couplings between states from lineage tracing data are used to corroborate the Evolutionary Coupling results for the representative tumors 3435_NT_T1 and 3513_NT_T3 shown in Figure 5A-D. The comparisons between Allelic Coupling and Evolutionary Coupling for (A) 3435_NT_T1 and (B) 3513_NT_T3 are consistent (Pearson’s correlation = 0.94 and 0.99, respectively). The comparisons between KNN Coupling and Evolutionary Coupling for (C) 3435_NT_T1 and (D) 3513_NT_T3 are consistent (Pearson’s correlation = 0.97 and 0.86, respectively). Red line indicates the symmetrical y=x relationship. (E) Cumulative density function for Pearson’s correlation of Allelic Coupling and KNN Coupling statistics with Evolutionary Couplings for all KP-Tracer tumors. Median correlations are indicated with vertical bars and annotated with the median correlation value. (F) Clustering of tumors based on Evolutionary Coupling and Leiden cluster proportion statistics reveals features that distinguish different Fate Clusters. Three clusters are identified by unbiased clustering, corresponding to Fate Clusters 1, 2, and 3. Fate Cluster is annotated on top of each unique color in the first row of the heatmap. Values/colors in the heatmap are normalized across tumors, and each row corresponds to a feature (either an Evolutionary Coupling or Leiden cluster proportion). Evolutionary couplings are indicated by a tuple of the form (x, y) and Leiden cluster proportions are indicated by a single number of the form x. We focus on showing features that distinguish different clusters, and uninformative features, identified as non-significant by a Mann-Whitney U test (p > 0.1), are not shown. (G) Heatmap of state proportions for each Fate Cluster across Leiden clusters. The value of the ith row and jth column indicate the fraction of cells found in the jth Leiden Cluster across all tumors in the ith Fate Cluster. (H) Principal Component Analysis (PCA) of tumor Evolutionary Coupling and Leiden cluster proportion vectors. Each dot is a tumor. Tumors are colored by their Fate Cluster, as identified with the hierarchical clustering shown in Fig S5E. The percent of variance explained is indicated on each axis. (I) Biplot of PCA of Evolutionary Coupling and Leiden cluster composition vectors, where each arrow indicates the loading of the feature with respect to the first two principal components. The top 10 features for the first two principal components are shown; arrows are annotated with the feature label. The percent of variance explained is indicated on each axis. Features of the form (x, y) represent Evolutionary Couplings between state x and state y; features of the form x represent the proportion of cells found in Leiden cluster x. (J-K) Comparison of Phylotime statistics computed using weighted and binary tree branch lengths for (J) Fate Cluster 1 and (K) Fate Cluster 2 (STAR Methods). Correlations are strong for both Fate Clusters (Pearson’s correlation = 0.94 and correlation = 0.98, respectively). (L) Selected Evolutionary Couplings of individual tumors displayed on gene expression UMAP illustrating connections between transcriptional states (Leiden clusters) of interest. From left: the first plot shows the Evolutionary Couplings within a representative tumor in Fate Cluster 1. The second plot shows the Evolutionary Couplings within a representative tumor in Fate Cluster 2. The third plot shows couplings between Fate Cluster 1 (Leiden clusters 3 and 5) and Late stage transcriptome states (Leiden cluster 9). The fourth plot shows couplings between Fate Cluster 1 (Leiden clusters 3 and 5) and high fitness transcriptome states (Leiden cluster 7 and 9). The last plot shows couplings between Fate Cluster 1 (Leiden clusters 3, 5 and 14) and high fitness transcriptome states (Leiden cluster 9 and 13). These results offer evidence of potential transition from early, low fitness to late, high fitness transcriptome states during tumor evolution.
(A-B) Subclonal expansion dynamics of (A) KPL and (B) KPA tumors. Independent expansions are colored with black, orange or blue and measured with the percentage of cells in the expanding subclone. (C) Overlap of genes associated with high and low fitness for KP, KPL and KPA tumors. (D) Gene markers for newly identified Leiden clusters in the KP, KPL and KPA integrated analysis. Dots are sized by the fraction of cells expressing a marker and colored by the mean expression of the gene marker in a Leiden cluster. (E) Leiden cluster proportions for each KPL (left) and KPA (right) tumor. (F) Distribution of the mean EffectivePlasticity for each Leiden cluster, averaged within each tumor, compared across genotypes. Leiden clusters 6, 11, 17, 18 are not shown because they lacked enough tumors across genotypes to make comparisons. (G) Evolutionary Couplings of different transcriptional states in three representative tumors reveals evolutionary paths in KPL and KPA tumors. Transcriptional states that are represented by at least 2.5% of cells in each tumor are used. 3515_Lkb1_T1 is a representative KPL tumor. The left plot shows the lineage relationship of transcriptional states in this KPL tumor and the right plot summarizes Evolutionary Couplings on the gene expression UMAP illustrating connections between Leiden clusters 4, 0 and 9. 3777_Apc_T1 is a representative KPA tumor. The left plot shows the lineage relationship of transcriptional states in this KPL tumor and the right plot summarizes Evolutionary Couplings on the gene expression UMAP illustrating connections between Leiden clusters 4 and 16. 3765_Apc_T1 is another representative KPA tumor. The left plot shows the lineage relationship of transcriptional states in this KPL tumor and the right plot summarizes Evolutionary Couplings on the gene expression UMAP illustrating connections between Leiden clusters 4, 16, 13, 7 and 1.
(A) Lineage indel heatmap of the 3724_NT_T1 tumor-metastasis family, summarizing the allelic information (indels) from the target sites confirming the separate origin of the soft tissue and liver metastatic tumors. In the Lineage indel heatmap, each row represents a single cell and each column represents a cut site of the lineage tracer. Unique indels are shown in unique colors, uncut target sites are indicated in gray, and missing data is indicated in white. The reconstructed lineage based on the accumulated indel patterns using Cassiopeia are shown on the left. The corresponding sample ID for each cell is labeled on the right. (B-C) Subclonal origin and the metastatic routes for 3515_Lkb1_T1 tumor-metastasis family. (B) Lineage indel heatmap of 3515_Lkb1_T1 tumor-metastasis family, indicating indel alleles supporting the subclonal origins, the relative order and the routes of metastases and (C) a model summarizing these metastatic behaviors. (D) More supporting examples of expanding subclones giving rise to metastases across genotypes for 3513_NT_T1 (left), 3508_Apc_T2 (center), and 3519_Lkb1_T1 (right). (E) Comparison of transcriptional distance between metastatic tumors and cells in non-expanding and expanding regions of the primary tumor phylogeny for 3513_NT_T1, 3508_Apc_T2, 3519_Lkb1_T1, 3457_Apc_T1, and 3515_Lkb1_T1 metastasis families. All significances are indicated from a one-sided Mann-Whitney U test: *** indicates p < 0.001, ** indicates p < 0.01, and * indicates p < 0.05.
Tumor Sample information, tree reconstruction parameters & quality-control statistics. This table contains information about all tumor samples regarding their timing, ES clones, mice and genetic perturbations, the Cassiopeia parameters used to reconstruct trees, the parsimony scores, depths, and indel-phylogeny distance correlations.
Differential expression gene lists of individual Leiden clusters. Reports the effect sizes and significances genes across various differential expression analyses performed throughout the study, including the differentially expressed genes of Leiden clusters in KP tumors, in tumors of all three genotypes combined (KP, KPL and KPA) and differentially expressed genes of the PreEMT transcriptional states in KPL versus the other tumors.
Phylogenetic fitness majority-vote gene association signatures and modules. This table reports the low- and high-fitness-associated-genes from the majority-vote meta-analysis of KP, KPL and KPA tumors individually. The gene set enrichment analysis for the fitness-associated genes in KP mice is included. We also include the gene lists for the three gene modules from the Hotspot analysis of high fitness-associated genes of the KP tumors.
Evolutionary coupling and leiden cluster proportions for individual tumors (including all KP, KPL and KPA tumors) analyzed for Figures 5 and 6.
Phylotime differential gene expression analysis. This table contains the differentially expressed genes following early to late Phylotime in tumors from Fate Cluster 1 and 2.
Primers and plasmids used in this manuscript.
Data Availability Statement
Raw single-cell RNA-sequencing data has been deposited at the NCBI Sequence Read Archive database and are publicly available as of the date of the publication. Accession numbers are listed in the key resources table. Processed single-cell data, reconstructed phylogenies, derived statistics, interactive VISION (DeTomaso et al., 2019) and PhyloVision (Jones et al., 2022) reports have been deposited at Zenodo and are publicly available as of the date of the publication. DOIs are listed in the key resources table.
All original code is available on Github (https://github.com/mattjones315/KPTracer-release) and has been deposited at Zenodo and is publicly available as of the date of the publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.