

Abstract
Lead (Pb) is a naturally occurring heavy metal, which can damage the brain and affect learning and memory. Sodium para-aminosalicylic acid (PAS-Na), a non-steroidal anti-inflammatory drug, can readily cross the blood-brain barrier. Our previous studies have found that PAS-Na alleviated Pb-induced hippocampal ultrastructural damage and neurodegeneration, but the mechanism has yet to be defined. Here, we investigated the molecular mechanisms that mediate Pb-induced apoptosis in hippocampal neurons, and the efficacy of PAS-Na in alleviating its effects. This work showed that juvenile developmental Pb exposure impaired rats cognitive ability by inducing apoptotic cell death in hippocampal neurons. Pb-induced neuronal apoptosis was accompanied by increased inositol 1,4,5-trisphosphate receptor (IP3R) expression and enhanced intracellular calcium [Ca2+]i levels, which resulted in increased phosphorylation of neuronal apoptosis signal-regulating kinase 1 (ASK1) and p38. Activation of ASK1 and p38 was blocked by IP3R inhibitor and a Ca2+ chelator. Importantly, PAS-Na treatment improved the Pb-induced effects on cognitive deficits in rats, concomitant with rescued neuronal apoptosis. In addition, PAS-Na reduced the expression of IP3R and the ensuing increase in intracellular Ca2+ and decreased the phosphorylation of ASK1 and p38 in Pb-exposed neurons. Taken together, this study demonstrates that the IP3R-Ca2+-ASK1-p38 signaling pathway mediates Pb-induced apoptosis in hippocampal neurons, and that PAS-Na, at a specific dose-range, ameliorates these changes. Collectively, this study sheds novel light on the cellular mechanisms that mediate PAS-Na efficacy, laying the groundwork for future research to examine the treatment potential of PAS-Na upon Pb poisoning.
Keywords: Lead, PAS-Na, IP3R, Ca2+, ASK1-p38 pathway, Neurotoxicity
1. Introduction
Lead (Pb) is a heavy metal which is used worldwide in several applications, especially in industry. Exposure to Pb may result in aberrant effects on the renal, hematopoietic, reproductive and central nervous system (CNS) (Flora et al., 2012). In the CNS, Pb has been shown to affect multiple regions, including the cerebellum, hippocampus and cerebral cortex (Liu et al., 2013). Once in the brain, Pb may cause various neurological deficits, including intellectual disability and behavioral deficits, and a possible link to Parkinson’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis has also been advanced (Jiang et al., 2008; Liu et al., 2013). In particular, the hippocampus, a brain region in the temporal lobe that is central to long-term memory formation, is a key target of Pb-induced effects (Bakulski et al., 2020). Once Pb crosses the blood–brain barrier (BBB) and the blood-cerebrospinal fluid (CSF) barrier into the brain and accumulates in the hippocampus (Ouyang et al., 2019), it may impair hippocampal structure and function. Although Pb affects the CNS of all ages of human and animals, the developing animals are particularly vulnerable to its toxicity, where it can disrupt cognitive and behavioral development, arresting children’s intellectual development, and culminating in cognitive and behavioral deficits (Shadbegian et al., 2019; Reuben et al., 2020). Unfortunately, the cognitive deficits caused by early-life Pb exposure persist into adulthood (Reuben et al., 2020). Thus, it is timely and meritorious to characterize the molecular mechanisms of Pb-induced neurotoxicity, and to develop novel diagnostic and therapeutic modalities.
The mechanisms by which Pb disrupts optimal brain function are complex and poorly understood. Nonetheless, cellular and molecular approaches have led to growing understanding of the effects of Pb on brain function. Several possible mechanisms of Pb neurotoxicity, such as disruption of normal neurotransmitter release, cytotoxicity, energy metabolism disorders, and epigenetic changes have been advanced, and reviewed elsewhere (Mitra and Sharma, 2019; Rocha and Trujillo, 2019). Moreover, considerable literature exists corroborating Ca2+ dysregulation and apoptosis as major mediators of Pb-induced neurotoxicity (Fan et al., 2013; Wang et al., 2015; Ouyang et al., 2019). Ca2+ plays a vital role in neurodegenerative processes, and its dysregulation can lead to a number of neurodegenerative diseases (Zündorf and Reiser, 2011). Regarding cell death, the release of Ca2+ from endoplasmic reticulum (ER) stores by inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs) has been implicated in multiple models of apoptosis (Fan et al., 2013). Wang et al. demonstrated that IP3R-mediated ER Ca2+ release mediates rat proximal tubular (rPT) cells apoptosis (Wang et al., 2015; Fang et al., 2021). It is noteworthy that apoptosis signal regulating kinase 1 (ASK1) is a key factor in regulating apoptosis, and is activated through a common Ca2+-dependent mechanism (Takeda et al., 2004). For example, Ca2+ influx activates calmodulin-dependent protein kinase type II (CaMKII) and CAMKII phosphorylates ASK1 (Takeda et al., 2004). Moreover, the increase in intracellular Ca2+ may induce calcium and integrin binding protein 1 (CIB1) dissociation from ASK1, leading to the association of ASK1 and tumor necrosis factor receptor-associated factor 2 (TRAF2) and subsequent activation of ASK1 (Shiizaki et al., 2013). Furthermore, overexpression of wild-type or the activated allele of ASK1 activates c-Jun N-terminal kinase (JNK) and p38 MAP kinase (p38), leading to the induction of apoptosis in multiple cell types secondary to the activation of mitochondria-dependent caspase (Tobiume et al., 2001). However, the roles of ASK1 signaling pathways in apoptosis have been controversial, as ASK1 also induces neuronal differentiation and survival in a model of PC12 cells (Takeda et al., 2000). Accordingly, whether and how Ca2+ signals regulate downstream signaling pathway in Pb-induced cells death in the hippocampus remains to be elucidated.
Sodium para-aminosalicylic acid (PAS-Na) is a non-steroidal anti-inflammatory drug. It has been shown to be effective in treatment of patients with manganism (Ky et al., 1992; Jiang et al., 2006). Additionally, para-aminosalicylic acid (PAS) as well as its salicylates PAS-Na acts as chelating agents for manganese (Mn), promoting the excretion of Mn in patients with manganism as well as Mn-exposed animals (Ky et al., 1992; Jiang et al., 2006; Zheng et al., 2009). In previous studies, we found that PAS-Na showed efficacy in reversing Mn-induced neurotoxicity, and its mode-of-action involved attenuation of oxidative stress (Peng et al., 2020), anti-inflammatory (Li et al., 2018; Peng et al., 2020) and anti-apoptotic (Yoon et al., 2009) effects, as well as restoration of amino acid neurotransmitter homeostasis (Li et al., 2019). Therefore, PAS-Na has been proven to be a promising drug for the treatment of Mn-induced neurotoxicity. Notably, our previous in vivo study has established by means of transmission electron microscope that PAS-Na therapy restored the pathological changes (including the myelin sheath structure delamination and fracture, neurofibril arrangement disorder and synaptic structure rupture) in hippocampal ultrastructure induced by Pb exposure (Deng et al., 2009). Furthermore, in vitro, PAS-Na was observed to lead to decrease Pb-induced PC12 cell apoptosis, accompanied by enhanced glutathione levels and a reduction in the ratio of Bax/Bcl-2 (He et al., 2017). Based on these observations, we speculated that PAS-Na may be an efficacious drug for the treatment of Pb-induced neurotoxicity. Accordingly, in the present study, we hypothesized that Pb induces hippocampal neuronal apoptosis via IP3R-Ca2+-ASK1-JNK/p38 signaling pathway. We aimed to verify this signaling pathway and investigated the interventional effects of PAS-Na treatment.
2. Materials and methods
2.1. Animals and experimental design
All animals were obtained from the Experimental Animal Center of Guangxi Medical University [SCXK (Gui) 2020–0004]. All animal experimental procedures were handled according to the international standards of animal care guidelines. The protocol was approved by the Animal Ethics Committee of Guangxi Medical University (approval number: 201806386). Animals were housed under standard laboratory conditions of light (a 12/12 h light/dark cycle), at temperature of 24 ± 2 °C and humidity range between 45% and 65%. Animals had ad lib access to food and water.
2.1.1. Experiment 1
To study the neurotoxicity of developmental Pb exposure in rats and the intervention effect of PAS-Na on it, we exposed Pb in Sprague-Dawley (SD) rats during the juvenile stage (from juvenile period to adult period). Sixty 24-day-old weaned rats were randomly divided into 6 groups (n = 10 per group): control, Pb exposure (Pb-treated), PAS-Na treatment (Pb + 80PAS, Pb + 160PAS and Pb + 240PAS groups) and C-PAS group. After 3 days of adaptive feeding, the rats in Pb-treated group, Pb + 80PAS, Pb + 160PAS and Pb + 240PAS groups were injected intraperitoneally (i.p.) with 10 mg/kg lead acetate (PbAc, Sigma-Aldrich, St. Louis, MO, USA) once a day for 4 weeks (28–55 days). At the same stage, the control and C-PAS group received i.p. injection sterile physiological saline. Subsequently, the Pb + 80PAS, Pb + 160PAS and Pb + 240PAS groups and C-PAS group were injected subcutaneously (s.c.) with 80, 160, 240 and 240 mg/kg PAS-Na (Sigma-Aldrich, St Louis, MO, USA), respectively, once a day, five days per week for another 2 consecutive weeks, while the control and Pb-treated group received s.c. injection of sterile physiological saline. The selected PAS-Na treatment dose (80, 160 and 240 mg/kg) for the in vivo experiments were based on our previous work (Li et al., 2018; Peng et al., 2020). These doses were shown to alleviated neuroinflammation and pyroptosis induced by manganese in rats’ brain.
2.1.2. Experiment 2
The purpose of this experiment was to investigate the neurotoxicity of subchronic (3 months) low-level Pb exposure in rats and the therapeutic effect of PAS-Na. After one week adaptation, four-week-old specific pathogen-free male SD rats were randomly selected into one of the 6 experimental groups (n = 10 per group): control, Pb exposure (Pb-treated), PAS-Na treatment (Pb + 80PAS, Pb + 160PAS and Pb + 240PAS groups) and C-PAS group. Same as the operation procedure of experiment 1, Pb-exposed rats were given 2 mg/kg lead acetate by intraperitoneal injection, once a day, 5 days a week for a total of 12 weeks. Next, the PAS-Na treatment was the same as in experiment 1, but the time was extended to 6 weeks. Both Pb and PAS-Na used in the in vivo experiments are soluble in sterile physiological saline. After animal behavioral testing, the rats were anesthetized with 10% chloral hydrate (i.p., 3.5 mL/kg) and sacrificed. Brain tissues (including hippocampus) were immediately dissected and collected in tube and stored at − 80 °C until experimental use.
2.2. Behavioral testing
2.2.1. Morris water maze test
The rats spatial learning and memory was investigated using the Morris water maze test. The test was designed in initial spatial training for 5 consecutive days and the probe trial was conducted at 6th day. At the beginning of each training trial, each animal was randomly placed with its face pointing to the pool wall at one of four different points (north, east, south and west). If the rat failed to locate the platform within 90 s, it was guided to the platform by the investigator and allowed to remain there for 15 s. Rats were underwent four training trials per day at 10-min intervals, each starting at the same time of day. A probe trial was administered at 6th day, the platform was removed and the rats were allowed to swim freely for 120 s. The escape latency and path length to reach the platform in each trial and the number of times a rat passed the platform were registered.
2.2.2. Y-maze test
The Y-maze forced alternation test was carried out to measure the performance of rats in a hippocampal-dependent spatial memory task as previously described (Dellu et al., 2000; Jung et al., 2008; Dinel et al., 2020). The Y-maze apparatus consisted of 3 identical arms (50 cm long, 18 cm wide and 35 cm high) with an angle of 120° between adjacent arms. Visual cues were located on the walls of the test chamber and remained constant throughout the test. The three arms were randomly assigned as the start arm, the novel arm and the other arm. The Y-maze test consists of 2 trials (acquisition trial and retrieval trial). During the acquisition trial, the novel arm was blocked by the baffle, and the rat was only allowed to explore 2 arms (the start arm and the other arms) undisturbed for 5 min. Retrieval trials were performed after a 30-min trial interval, the rat was again placed in the start arm and was allowed to access all three arms of the maze for 5 min. After each acquisition or retrieval trial, the corncob litter covered in the floor was mixed evenly, and all walls of the maze were cleaned to remove odor traces. The number of entries into each arm as well as the time spent in each arm during retrieval trials were record and the percentage of number and duration of visits in the novel arm/total arm were calculated. Based on the innate tendency of rodents to explore novel environments, the rat with intact spatial memory will enter and spend more time in the novel arm compared to the other arms of the maze (Kraeuter et al., 2019).
2.3. Inductively coupled plasma-mass spectrometry (ICP-MS) of Pb and Ca2+ levels
Hippocampus tissues (50 mg) were digested with 4 mL of 65% nitric acid (Merck ppb, USA) at 120–180 °C for 1 h. After cooling to room temperature, the samples were transferred to a 120–180 °C graphite heating plates to evaporate the acid until the liquid reached about 0.5 mL. Each sample was adjusted to 8 mL with ultrapure double distilled water. The Pb and Ca2+ concentrations in hippocampus tissues were determined by ICP-MS the same as the previous study (Li et al., 2020).
2.4. Primary hippocampal neuron cultures and treatments
Neonatal SD rats (postnatal 0–24 h) were decapitated and transferred to anatomical fluid (50% Hanks Balanced Salts, 4.2 mM NaHCO3, 1% HEPES, 33 mM glucose, 0.01% gentamysin, 45 mM BSA, 12 mM MgSO4, NaOH adjusts pH to 7.3). Next, the brains were dissected out and the hippocampi were isolated and dissociated (the hippocampus can be distinguished given its crescent-shaped structure in the medial surface of the cortical hemisphere) into a 60 mm Petri dish containing 2 mL anatomical fluid on ice. The collected hippocampi were mechanically fragmented and placed in papain (Worthington, USA) for 30 min at 37 °C. Then hippocampal neurons were added to plating medium containing 84% MEM (Gibco, USA), 10% horse serum (Hyclone, Logan, Utah, USA), 5% glucose (Sigma-Aldrich, St. Louis, MO, USA), 1% Glutamax (Gibco, USA) to terminate the enzymatic activity of papain. Hippocampal neurons were separated using a 45 μm cell filter and plated at a density of 40,000–80,000 cells/well (depending upon the intended experiment) on poly-L-lysine-coated plates in a humidified incubator in an atmosphere of 5% CO2 at 37 °C. Four hours later, the plating medium was changed to Neurobasal-A medium (Gibco, USA) supplemented with 2% B-27 supplement (Gibco, USA), 1% Glutamax, and 0.5% (v/v) penicillin–streptomycin (Solarbio, Beijing, China). Half of the old medium was replaced by the same volume of fresh medium every 3 days. After culturing for 7 days, the hippocampal neurons were used for further experiments. Hippocampal neurons were incubated with 50 μM PbAc for 24 h, then the neurons were treated with 100, 200 or 400 μM PAS-Na for 24 h to obtain PAS-Na treatment models. The selected Pb exposure concentration was determined with a cell viability assay (Fig. 2). The PAS-Na treatment dose in vitro experiment was based on our previous research that 100–400 μM PAS-Na significantly attenuated manganese-induced BV2 cells pyroptosis (Peng et al., 2020). Sterile double distilled water was used as the solvent for Pb and PAS-Na in vitro experiments. All the inhibitors used in the present study were added into the wells 30 min before Pb treatment.
Fig. 2.
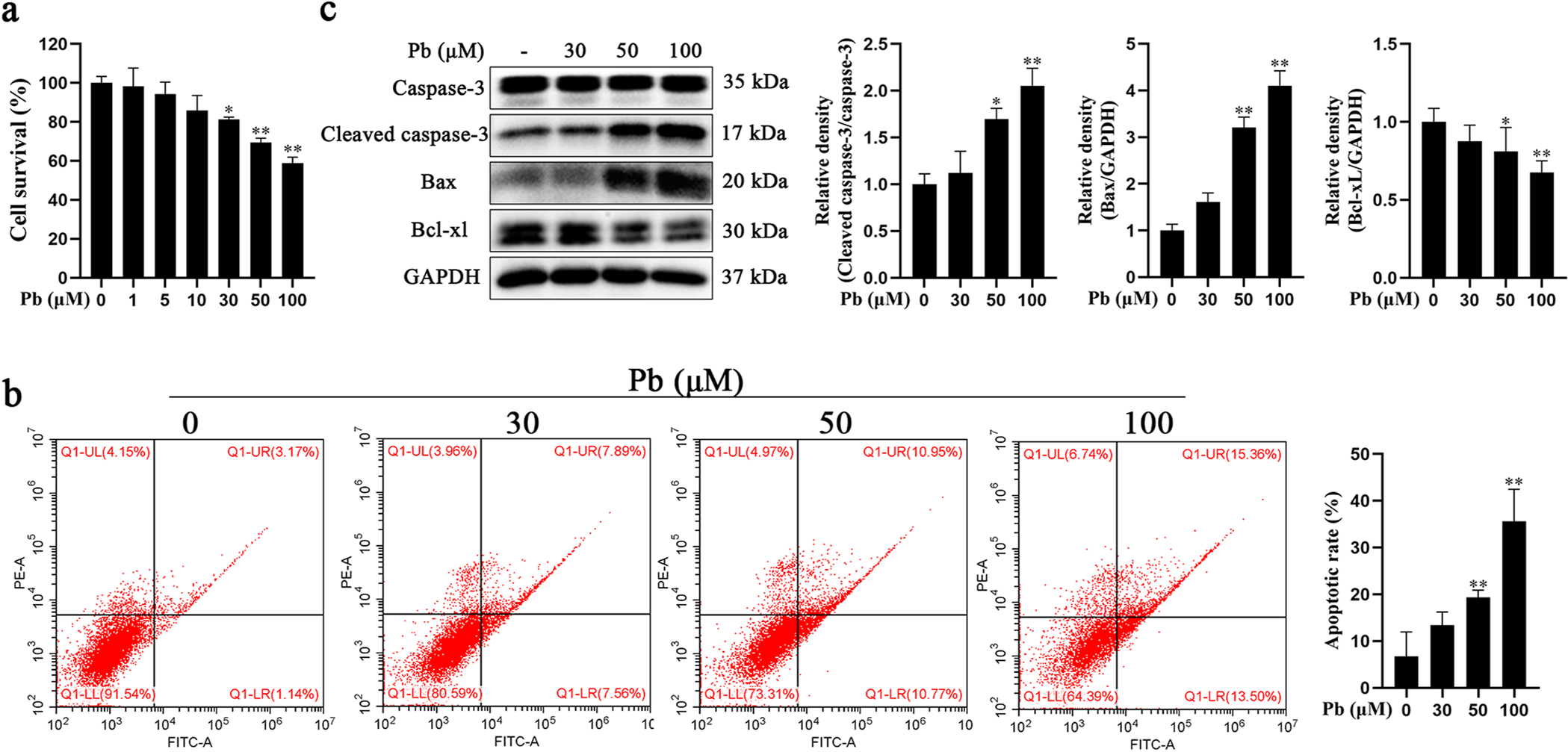
Pb exposure induced apoptosis in primary hippocampal neurons. a-c Primary hippocampal neurons were treated with increased concentrations of PbAc for 24 h. a Cell viability was determined with CCK8. b Neuronal apoptotic rate was determined by flow cytometry analysis. Early apoptotic cells (AV+/PI ) are shown in the lower right quadrant and late apoptotic cells (AV+/PI+) in upper right quadrant. c Protein levels of cleaved caspase-3, Bax and Bcl-xL were determined by western blotting. The protein expression was normalized by GAPDH or corresponding total protein content. * P < 0.05 and * * P < 0.01, compared to the control group.
2.5. CCK8 assay
Primary hippocampal neurons in 96-well plates were treated with Pb (0, 1, 5, 10, 30, 50 and 100 μM) for 24 h. Subsequently, 100 μL of 0.5% CCK-8 (Solarbio, Beijing, China) solution was added to each well. Cell viability was determined after one hour by measuring the absorbance with a microplate reader at 450 nm. Each experiment was performed independently three times in triplicate wells.
2.6. Annexin V-FITC/propidium iodide (PI) staining
After Pb and PAS-Na treatment, hippocampal neurons were digested with 0.25% EDTA-free trypsin and collected. To quantify cellular death, FITC Annexin V staining was carried out (BD Pharmingen, San Diego, USA) in the dark for 10 min, then stained with Propidium Iodide (BD Pharmingen, San Diego, USA) in the dark for 5 min. Fluorescence labeling was analyzed by two-color flow cytometry (CytoFLEX FCM, Beckman, USA) according to the manufacturer’s protocol. Ten thousand cells were analyzed for each sample. At least three independent experiments with different cell batches of cells were carried out.
2.7. Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling (TUNEL) staining
Rat hippocampi were coronally sliced and fixed in 4% paraformaldehyde for over 24 h, followed with paraffin embedding. The hippocampal sections were processed using the TUNEL assay to label apoptotic cells in accordance with the manufacturer’s instructions for the TUNEL kit (Servicebio, Wuhan, China). After deparaffinization and rehydrate, sections were digested with proteinase K for 20 min at 37 °C for DNA exposure, and treated with 3% H2O2 to block endogenous peroxidation activity. The resulted DNA strand breaks were labelled with a terminal deoxynucleotidyl transferase (TdT) enzyme, with dUTP molecules conjugated to horseradish peroxidase. The sections were then rinsed and visualized with 3,3′-diaminobenzidine (DAB) and counterstained with hematoxylin. Brown-red-stained cell nuclei were considered positive apoptotic cells, while blue-stained nuclei were considered as negative cells. Images of hippocampus including the CA1, CA3 and DG regions were captured using a ZEISS Axio Imager M2p Microscope Imaging System. Positive brown-red cells and total cells from each hippocampal section of three animals per group were counted and the percentage of positive cells for each group was calculated.
2.8. Ca2+ imaging experiments
At the end of the designated treatments, hippocampal neurons in 24-well plates were loaded with 5 μM Fluo4, AM (Solarbio, Beijing, China) for 30 min in dark at 37 °C. Then cells were gently washed with PBS for 3 times. Images were captured using an EVOS Cell Imaging System and were quantified with the ImageJ software under identical threshold conditions. All Fluo-4 fluorescence imaging assays were independently replicated at least three times.
2.9. Real-time quantitative polymerase chain reaction (RT-qPCR)
Total RNA of hippocampal neuronal was isolated using TRIzol reagent (TaKaRa, Tokyo, Japan). The RNA samples were transcribed into cDNA and quantified by RT-qPCR as previously described (Li et al., 2019). Each experiment was carried out independently three times in triplicate wells. The primers were obtained from Invitrogen Inc. (Shanghai, China), and the sequences were as follows: IP3R2, forward 5′-ACGGCATCGAGAGGACCTGTG-3′ and reverse 5′-GTCACCAACTCCGCCACCATTC-3′; IP3R3, forward 5′-TCACCACCACGGAGCAGGAC-3′ and reverse 5′-TGCGGCGGGAGAACCAGTAG-3′; β-actin, forward 5′-GTGCTATGTTGCTCTAGACTTCG-3′ and reverse 5′-ATGCCACAGGATTCCAT ACC-3′.
2.10. Western blotting and antibodies
At the end of the designated treatments, hippocampal neurons were lysed in cell lysis buffer with 1 mM proteinase inhibitor cocktail (APExBIO, Houston, USA) and 1 mM phosphatase Inhibitor cocktail (APExBIO, Houston, USA) for 30 min. After 15 min centrifugation at 12,000 rpm at 4 °C, the supernatant was collected. Protein concentrations were determined with the BCA assay kit (Beyotime, Beijing, China). Thirty μg protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and the separated proteins were transferred to 0.22 μm polyvinylidene fluoride membranes (Millipore, Darmstadt, Germany). Membranes were blocked in 5% non-fat dried milk in TBST (Solarbio, Beijing, China) for 1 h at room temperature, then incubated overnight at 4 °C with the following primary antibodies. Membranes were rinsed 3-times with TBST for 5 min each, and subsequently incubated at room temperature for 1 h with the secondary antibodies. Membranes were treated with Super Signal West Femto substrate (Bridgen, Beijing, China) and detected using the intelligent gel imaging system iBright FL1000 (Thermo Fisher, USA). The band density was quantified with ImageJ software. Each experiment was performed independently at least three times. The antibodies used were as follows : antibodies against IP3R (07–1210, 1:500) were purchased from Merck Millipore (New Jersey, USA), phospho-ASK1 (Ser966, PA5–105027, 1:1000) were purchased from Thermo Fisher Scientific (Waltham, Massachusetts, USA), total ASK1 (#2532, 1:1000), phospho-JNK (Thr183/Tyr185, #4668, 1:1000), JNK (#9252, 1:1000), phospho-p38 (Thr180/Tyr182, #4511, 1:1000), p38 (#8690, 1:1000), cleaved caspase-3 (#9664, 1:1000), caspase-3 (#14220, 1:1000), Bax (#14796, 1:1000), Bcl-xL (#2764, 1:1000) and GAPDH (#2118, 1:1000) from Cell Signaling Technology (Danvers, Massachusetts, USA).
2.11. Statistical analysis
Experimental differences were determined by SPSS 21.0 software (IBM SPSS Statistics for Windows, Chicago, IL, USA). If the data variances were homogeneity, comparisons among different groups were analyzed with one-way ANOVA followed by the Tukey’s post hoc test. Otherwise, the rank sum test was required. All data are expressed as mean ± SD for a minimum of three independent experiments. Significance was assigned at P < 0.05.
3. Results
3.1. Effect of PAS-Na on cognitive ability in Pb-exposed rats
As shown in Table 1, the concentrations of Pb and Ca2+ were significantly increased in the hippocampus after 4 weeks of Pb exposure. Treatment with 80, 160 and 240 mg/kg PAS-Na for 2 weeks significantly reduced Pb levels and 240 mg/kg PAS-Na significantly decreased Ca2+ levels in the hippocampus (Table 1). The Morris water maze is a well-established behavioural test to monitor learning and memory deficits in rodents (Salman et al., 2019). To evaluate the effects of Pb exposure in rats on spatial learning and memory, and the ability of PAS-Na to ameliorate these effects, rats were trained for 5 days and a probe trial was performed on the sixth day. After 4 weeks of developmental Pb exposure during the juvenile stage and 2 weeks of cessation of exposure, as compared with the control group, the escape latency and swimming distance on the 4th and 5th training day were significantly increased in Pb-exposed rats (Fig. 1a, b). In addition, Pb-exposed rats treated with 240 mg/kg PAS-Na for 2 weeks showed lesser escape latency on the 4th and 5th training day and lesser swimming distance on the 5th training day compared to the Pb-treated rats (Fig. 1a, b). There was no significant difference in the probe time between Pb-treated and the control groups during the juvenile Pb exposure (Fig. 1c).
Table 1.
Effect of PAS-Na treatment on Pb and Ca2+ concentrations in hippocampus (μg/g) of the Pb-exposed rats.
| Group | Control | Pb | Pb + 80PAS | Pb + 160PAS | Pb + 240PAS | 240PAS |
|---|---|---|---|---|---|---|
|
| ||||||
| Pb | 0.08 ± 0.02 | 2.27 ± 0.23** | 1.36 ± 0.12## | 1.28 ± 0.26## | 1.17 ± 0.09## | 0.17 ± 0.01 |
| Ca2+ | 558.6 ± 74.97 | 994.40 ± 11.49** | 1082.0 ± 167.70 | 756.30 ± 65.33 | 686.60 ± 59.10# | 590.40 ± 39.88 |
Rats were treated with 10 mg/kg PbAc for 4 weeks during juvenile perior, followed by 80, 160 or 240 mg/kg PAS-Na treatment for 2 weeks.
Data represent mean ± SD, N = 3.
P < 0.05 or
P < 0.01 is presented as there was significance as vs. the Controls.
P < 0.05 or
P < 0.01 is presented as there was significance as vs. Pb-exposed group.
Fig. 1.
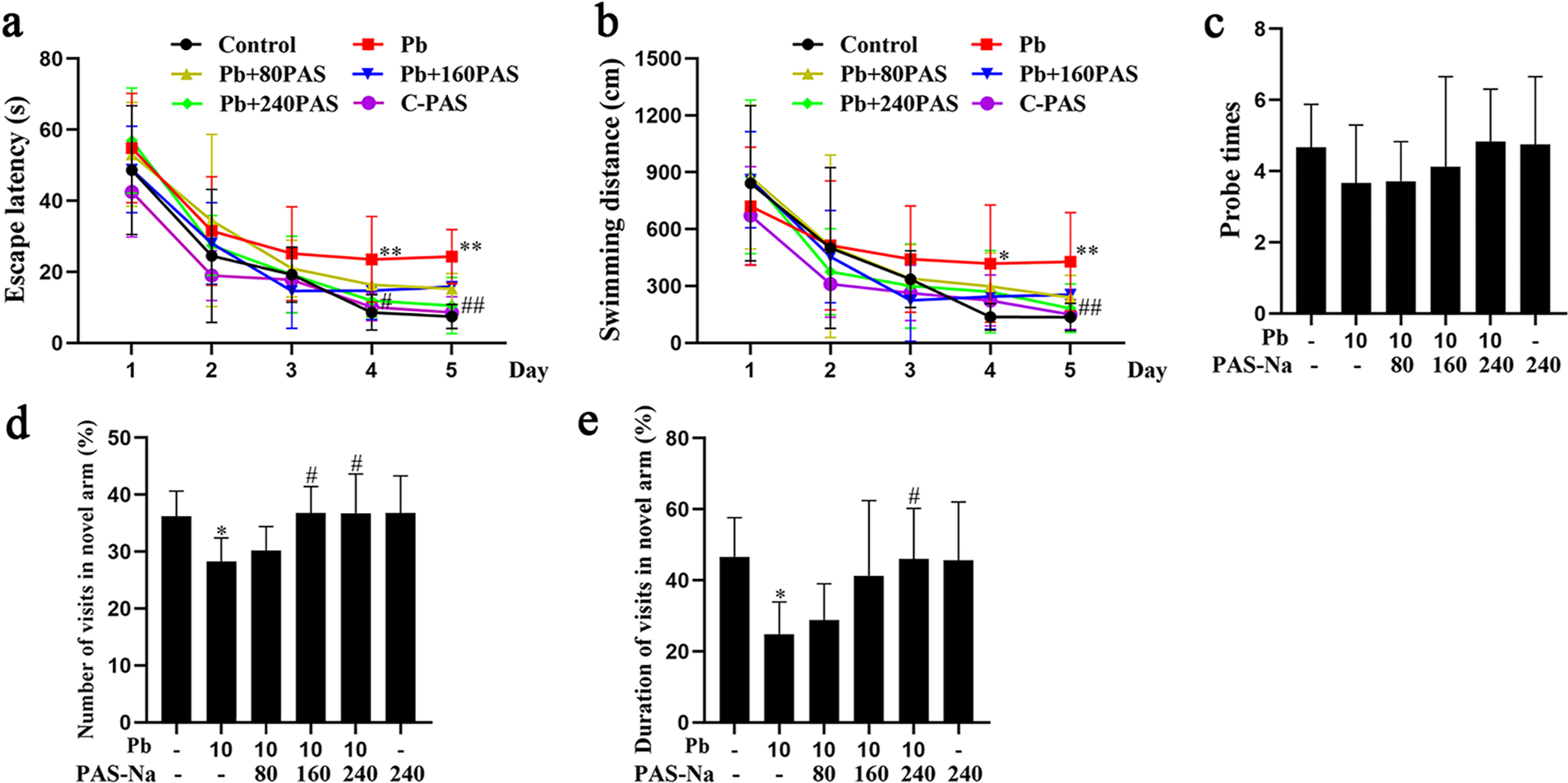
Effect of PAS-Na on cognitive ability in juvenile developmental Pb-exposed rats. a-e Rats were treated with 10 mg/kg PbAc for 4 weeks during the juvenile period, followed by 80, 160 or 240 mg/kg PAS-Na treatment for 2 weeks. Spatial learning and memory performance was evaluated in the Morris water maze. a Escape latency. b Swimming distance. c Number of times the rats crossed over the platform in the probe trial (Probe times) (n = 8 per group). Next, a Y-maze test was carried out to measure the rats spatial memory performance. Percentages of visit numbers (d) and duration (e) into the novel arm during the 5 min of the retrieval trial (n = 8 per group). * P < 0.05 and **P < 0.01, compared to the control group. #p < 0.01 and ##P < 0.01, compared to the Pb-treated group.
The Y-maze alternation test measures a rodent’s disposition to explore a new environment and is generally considered a measure of spatial memory (Conrad et al., 2003; Jung et al., 2008). Thus, the Y-maze test was used next to evaluate the effect of juvenile developmental Pb exposure on spatial memory. As shown in Fig. 1d and e, after 10 mg/kg Pb exposure for 4 weeks in juvenile, Pb-treated rats exhibited a less level of novelty exploration as compared to the control groups, showing less entries into the novel arm and spending less time in this arm during the 5 min of the retrieval trial. As compared with the Pb exposure group, Pb-exposed rats also treatment with 160 and 240 mg/kg PAS-Na for 2 weeks made more visited to the novel arm (Fig. 1d). Likewise, rats in the 240 mg/kg PAS-Na treatment groups spent significantly more time in the novel arm (Fig. 1e).
3.2. Pb exposure induced primary hippocampal neurons apoptosis
To determine optimal concentrations in vitro, we first assessed the effect of Pb on hippocampal neuron viability. Hippocampal neurons were treated with increasing concentrations of Pb (0–100 μM) for 24 h, and cell viability was measured with the CCK-8 assay. As shown in Fig. 2a, Pb-induced concentration-dependent hippocampal neuronal death. Moreover, the results of the annexin V-FITC/PI double staining showed that the percentage of apoptotic neurons (6.71%, 13.43%, 19.31% and 35.65%, respectively) observed increased in a concentration-dependent manner (Fig. 2b). Next, we used western blots to determine apoptosis-associated proteins (caspase-3, Bax and Bcl-xl) expression in Pb-exposed hippocampal neurons. As illustrated in Fig. 2c, cleaved caspase-3 and Bax expression levels increased and Bcl-xl expression levels decreased upon Pb exposure in a concentration-dependent manner. Cleaved-caspase-3, Bax and Bcl-xl expression levels in the 50 μM and 100 μM Pb exposure groups showed a significantly difference compared with the control group (P < 0.05).
3.3. PAS-Na decreased the Pb-induced apoptosis in hippocampal neurons
Next, we explored whether PAS-Na can inhibit the Pb-induced hippocampal neuron apoptosis. Hippocampal neurons were treated with Pb (50 μM) for 24 h, followed by PAS-Na (100, 200 or 400 μM) for additional 24 h. Flow cytometry results showed that apoptosis in Pb-treated neurons was significantly increased compared with the controls (P < 0.01, Fig. 3a). PAS-Na (200 and 400 μM) significantly decreased apoptosis compared to the Pb-treated group. However, 100 μM PAS-Na had no effect on Pb-induced increases in neuronal apoptosis. PAS-Na treatment alone did not have notable toxic effects on apoptosis (Fig. 3). Consistently, western blotting results showed that 200 and 400 μM PAS-Na significantly reduced the Pb-induced increase in the expression levels of cleaved caspase-3 and Bax in Pb-treated hippocampal neurons (P < 0.05, Fig. 3b).
Fig. 3.
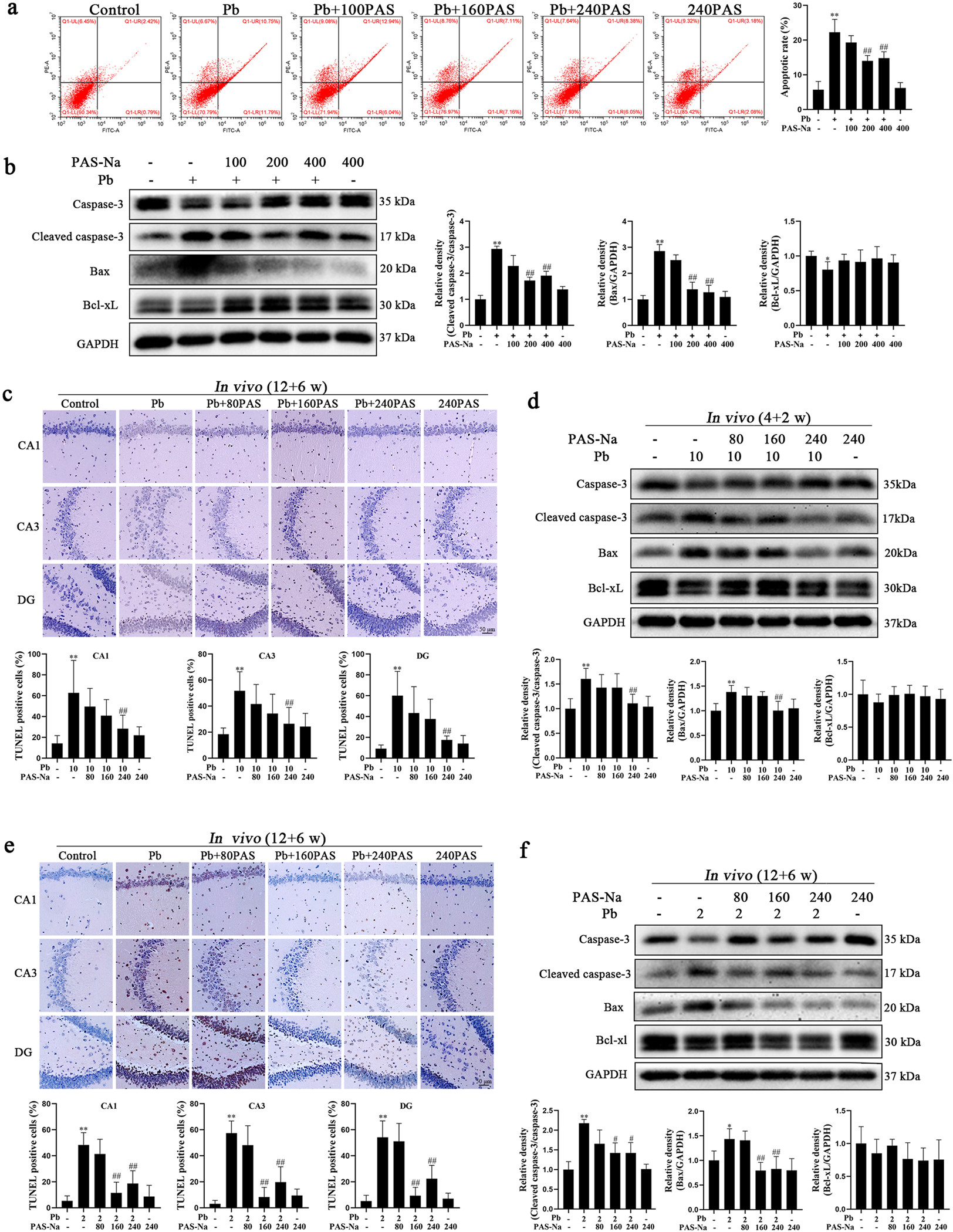
PAS-Na treatment decreased the Pb-induced hippocampal neuron apoptosis. a, b Primary hippocampal neurons were treated with 50 μM PbAc for 24 h, followed by 100, 200 or 400 μM PAS-Na treatment for 24 h. a Apoptosis in hippocampal neurons was determined using flow cytometry analysis. b Protein levels of cleaved caspase-3, Bax and Bcl-xL were determined by western blotting. c, d Rats were treated with 10 mg/kg PbAc for 4 weeks during juvenile, followed by 80, 160 or 240 mg/kg PAS-Na treatment for 2 weeks. e, f Rats were treated with 2 mg/kg PbAc for 12 weeks, followed by 80, 160 or 240 mg/kg PAS-Na treatment for 6 weeks. c, e Apoptotic nuclei in hippocampal tissues of rats were determined using TUNEL assay. Brown-red-stained cell nuclei were considered positive apoptosis cells, while blue-stained nuclei were identified as negative cells. Scale bar = 50 μm. d, f Protein level of cleaved caspase-3, Bax and Bcl-xL were determined by western blotting. The protein expression was normalized by GAPDH or corresponding total protein content. * P < 0.05 and * * P < 0.01, compared to the control group. #P < 0.05 and ##P < 0.01, compared to the Pb-treated group.
To characterize the apoptotic activity in hippocampal tissues in vivo after Pb exposure and the intervention effect of PAS-Na, hippocampal sections were labelled using a TUNEL assay. After juvenile developmental Pb exposure for 4 weeks, a significant number of apoptotic neurons was localized to the hippocampus in Pb-treated rats (Fig. 3c). We observed that in the juvenile Pb-exposure groups, TUNEL-positive cells were evenly distributed in the CA1, CA3 and DG regions of hippocampus, and the TUNEL-positive product were strongly labeled. Interestingly, 240 mg/kg PAS-Na treatment for 2 weeks showed the efficacy of reducing development Pb exposure induced apoptosis in hippocampal neurons (Fig. 3c). Consistently, western blotting results showed that the apoptotic protein levels of cleaved caspase-3 and Bax in the hippocampus were significantly enhanced after 4 weeks of juvenile Pb exposure, while 240 mg/kg PAS-Na treatment for 2 weeks significantly decreased cleaved caspase-3 and Bax levels (Fig. 3d). Interestingly, following subchronic Pb exposure for 12 weeks, the increased number of apoptotic neurons in the hippocampal CA1, CA3 and DG areas were found in Pb-treated rats, which were reduced upon treatment with 160 and 240 mg/kg PAS-Na (Fig. 3e). Furthermore, both 160 and 240 mg/kg PAS-Na treatment for 6 weeks significantly reduced the Pb-induced increase in the expression of cleaved caspase-3 and Bax in the hippocampus (Fig. 3 f). Taken together, these results demonstrated that at the certain dose range, PAS-Na efficiently ameliorated the Pb-induced apoptotic effect in rat hippocampus.
3.4. PAS-Na mitigated Pb-induced increase of intracellular Ca2+ in primary hippocampal neurons
It is well established that increased intracellular Ca2+ concentrations may result in cell death (Orrenius et al., 2003). To determine whether the Pb-induced apoptosis was associated with increased intracellular Ca2+, we tested intracellular free calcium concentrations ([Ca2+]i) by Fluo4, AM. Once Fluo-4, AM enters the cells, it is cleaved by the intracellular esterase to form Fluo-4, which combines with Ca2+ and produces strong green fluorescence. Otherwise, Fluo-4 exists as a free ligand, and is non-fluorescent. Fluorescent images showed that Pb treatment increased [Ca2+]i in hippocampal neurons in a concentration-dependent manner compared with the controls (Fig. 4a). Furthermore, PAS-Na exposure at three concentrations (100, 200 and 400 μM) significantly mitigated the Pb-induced [Ca2+]i increase in hippocampal neurons (Fig. 4b). Treatment with PAS-Na alone minimally affected [Ca2+]i in hippocampal neurons. These results suggest that Pb-induced increased [Ca2+]i in hippocampal neurons, and PAS-Na blocked this increase.
Fig. 4.
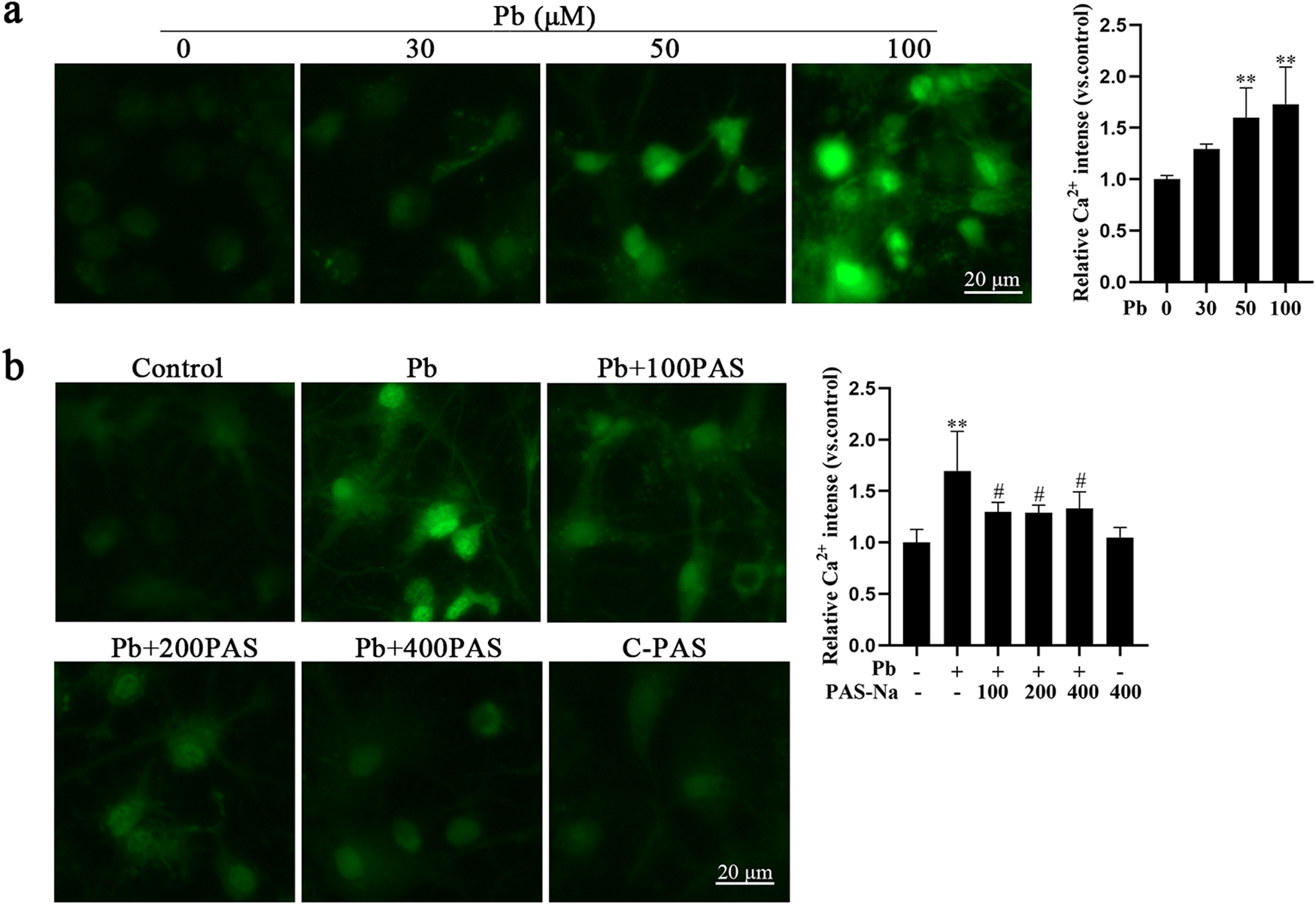
PAS-Na treatment mitigated the Pb-induced increase in intracellular Ca2+ in primary hippocampal neurons. a Primary hippocampal neurons were treated with increased concentrations of PbAc for 24 h. b Primary hippocampal neurons were treated with 50 μM PbAc for 24 h, followed by 100, 200 or 400 μM PAS-Na treatment for 24 h. a, b Representative fluorescent images of intracellular Ca2+ and quantifications of corresponding fluorescence intensity. Scale bar = 20 μm. * P < 0.05 and * * P < 0.01, compared to the control group. ##P < 0.05 and ##P < 0.01, compared to the Pb-treated group.
3.5. Pb-induced intracellular Ca2+ augment was associated with IP3R activation, which was blocked by PAS-Na
IP3R is an important ion channel that releases Ca2+ from the ER (Chan, 2013). To assess whether Pb-induced increase in [Ca2+]i is mediated by IP3R activation, primary hippocampal neurons were pre-treated with 50 μM 2-APB (an IP3R inhibitor) for 30 min. As detected by EVOS cell imaging system, [Ca2+]i increased following Pb treatment in hippocampal neurons were significantly repressed by 2-APB (Fig. 5a), suggesting that Pb mediated Ca2+ transient by activating IP3R. Moreover, the Pb-induced IP3R activation was also documented by western blotting and RT-qPCR analysis, corroborating increased IP3R protein and gene expression, respectively (Fig. 5b, c).
Fig. 5.
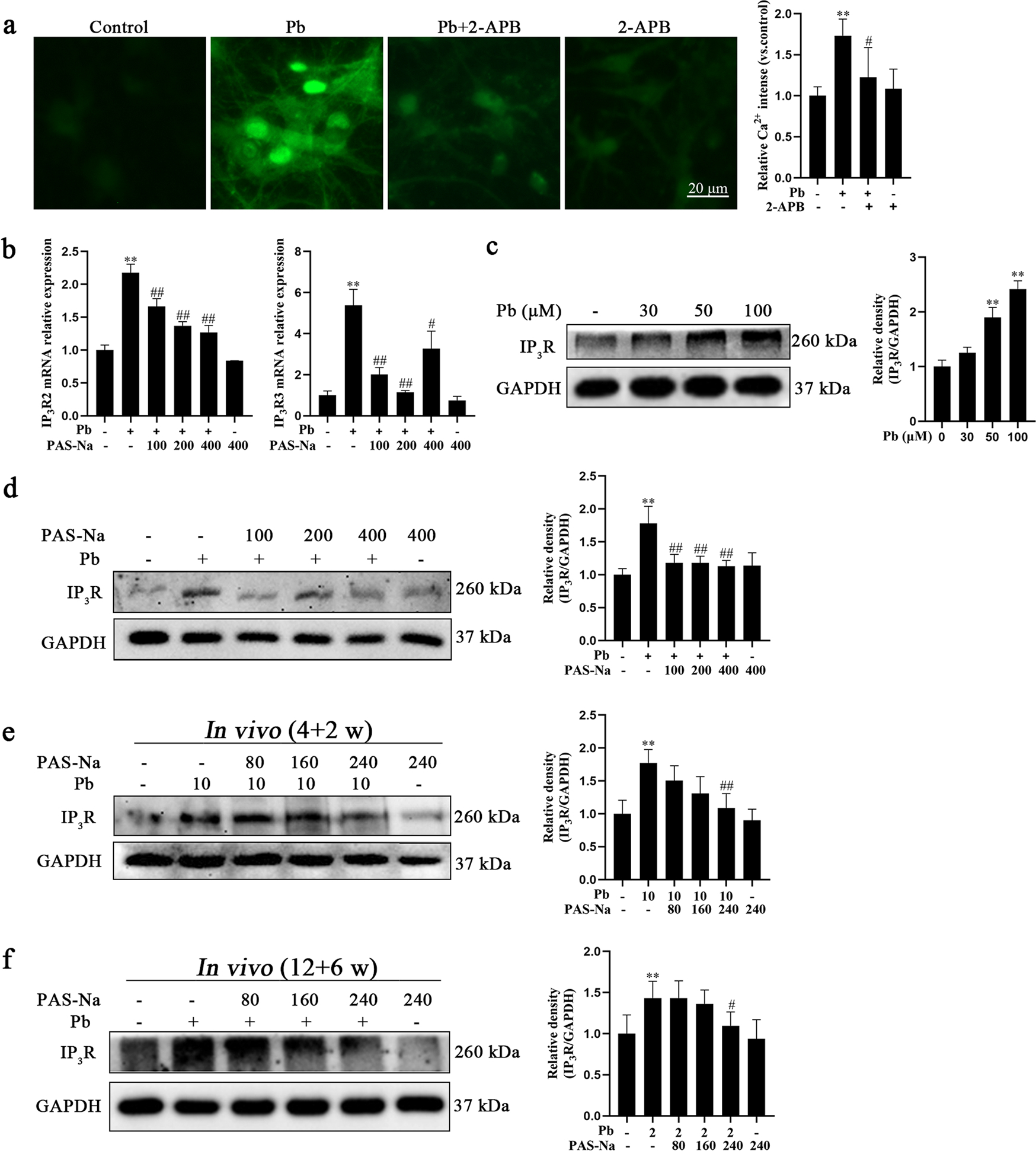
Pb-induced intracellular Ca2+ augment was associated with IP3R activation, which was blocked by PAS-Na. a Primary hippocampal neurons were treated with 50 μM PbAc in the presence or absence of pretreatment with 2-APB for 24 h. Representative fluorescent images of intracellular Ca2+ and quantifications of fluorescence intensity. Scale bar = 20 μm. b, d Primary hippocampal neurons were treated with 50 μM PbAc for 24 h, followed by 100, 200 or 400 μM PAS-Na treatment for 24 h. b The mRNA expressions of IP3R2 and IP3R3 were determined by RT-qPCR. c Primary hippocampal neurons were treated with increased concentrations of PbAc for 24 h. e Rats were treated with 10 mg/kg PbAc for 4 weeks during the juvenile period, followed by 80, 160 or 240 mg/kg PAS-Na treatment for 2 weeks. f Rats were treated with 2 mg/kg PbAc for 12 weeks, followed by 80, 160 or 240 mg/kg PAS-Na treatment for 6 weeks. c-f Protein level of IP3R was determined by western blotting. The protein expression was normalized by GAPDH. *P < and **P < 0.01, compared to the control group. #P < 0.05 and ##P < 0.01, compared to the Pb-treated group.
Next, we determined whether PAS-Na inhibited the Pb-induced IP3R activation in hippocampal neurons. There are three IP3R isoforms: IP3R1, IP3R2 and IP3R3. The mRNA expression of IP3R2 and IP3R3 in Pb-treated group were significantly higher compared to the control group (P < 0.01, Fig. 5b), while no significant changes were found for IP3R1 mRNA (data not shown). Notably, compared with the Pb-treated group, IP3R2 mRNA expression was reduced by PAS-Na in a concentration-dependent manner (Fig. 5b). Consistently, IP3R3 mRNA expression was restored by PAS-Na at the three tested concentrations (100, 200 and 400 μM) to levels statistically indistinguishable from the controls, as compared to the Pb-treated group. The expression of IP3R protein in Pb-treated hippocampal neurons was increased in a concentration-dependent manner (Fig. 5c). Notably, 100, 200 and 400 μM PAS-Na treatment significantly repressed IP3R protein expression in vitro (Fig. 5d). Consistently, after juvenile Pb exposure for 4 weeks following 2 weeks of PAS-Na treatment, we observed increased expression of IP3R protein in Pb-exposed rats was restored by treatment with 240 mg/kg PAS-Na (Fig. 5e). Likewise, Pb exposure for 12 weeks significantly increased IP3R protein expression, which was decreased by 240 mg/kg PAS-Na treatment for 6 weeks (Fig. 5 f). Treatment with PAS-Na alone had no effect on IP3R2, IP3R3 mRNA and IP3R protein expressions. Taken together, the above results indicate that PAS-Na might inhibit the Pb-induced IP3R activation in hippocampal neurons by decreasing IP3R gene and protein expression.
3.6. Pb activated ASK1/p38 by increasing intracellular Ca2+, and PAS-Na blocked its activation
ASK1 is a member of the mitogen-activated protein kinase kinase kinase (MAPKKK) family. ASK1 has been reported to activate JNK and p38 resulting in apoptosis (Tobiume et al., 2001). Our results showed that the phosphorylation levels of ASK1 at Ser966 (the activation site) and p38 at Thr180/Tyr182 (the activation site) in hippocampal neurons were increased in concentration-dependent manners after Pb exposure 24 h, while no significant changes were found for JNK compared to control (Fig. 6a). To investigate whether there is a link between ASK1-p38 apoptotic signaling pathway activation and Pb-induced elevation in [Ca2+]i, we tested the phosphorylation levels of ASK1 and p38 in hippocampal neurons co-treated with Pb and 2-APB or BAPTA-AM (a Ca2+ chelator). As shown in Fig. 6b, Pb significantly increased the phosphorylation of ASK1 and p38, which were blocked by 2-APB to levels statistically indistinguishable from the controls, as compared to the Pb-treated group. It should be noted that Pb-induced activation of ASK1 and p38 also decreased in pre-treated BAPTA-AM hippocampal neurons (Fig. 6c), suggesting that Pb-induced ASK1 and p38 activation were correlated with increased intracellular [Ca2+]i. Next, we used a p38 specific inhibitor, SB203580, pretreating primary hippocampal neurons for 30 min prior to Pb. As shown in Fig. 6d, SB203580 significantly reduced apoptotic neuronal death induced by Pb treatment. Consistently, SB203580 was able to repress caspase-3 cleavage and decrease the expression of Bax in Pb-treated hippocampal neurons (Fig. 6e). These results confirmed the essential roles of IP3R-Ca2+-ASK1-p38 in Pb-induced cell death.
Fig. 6.
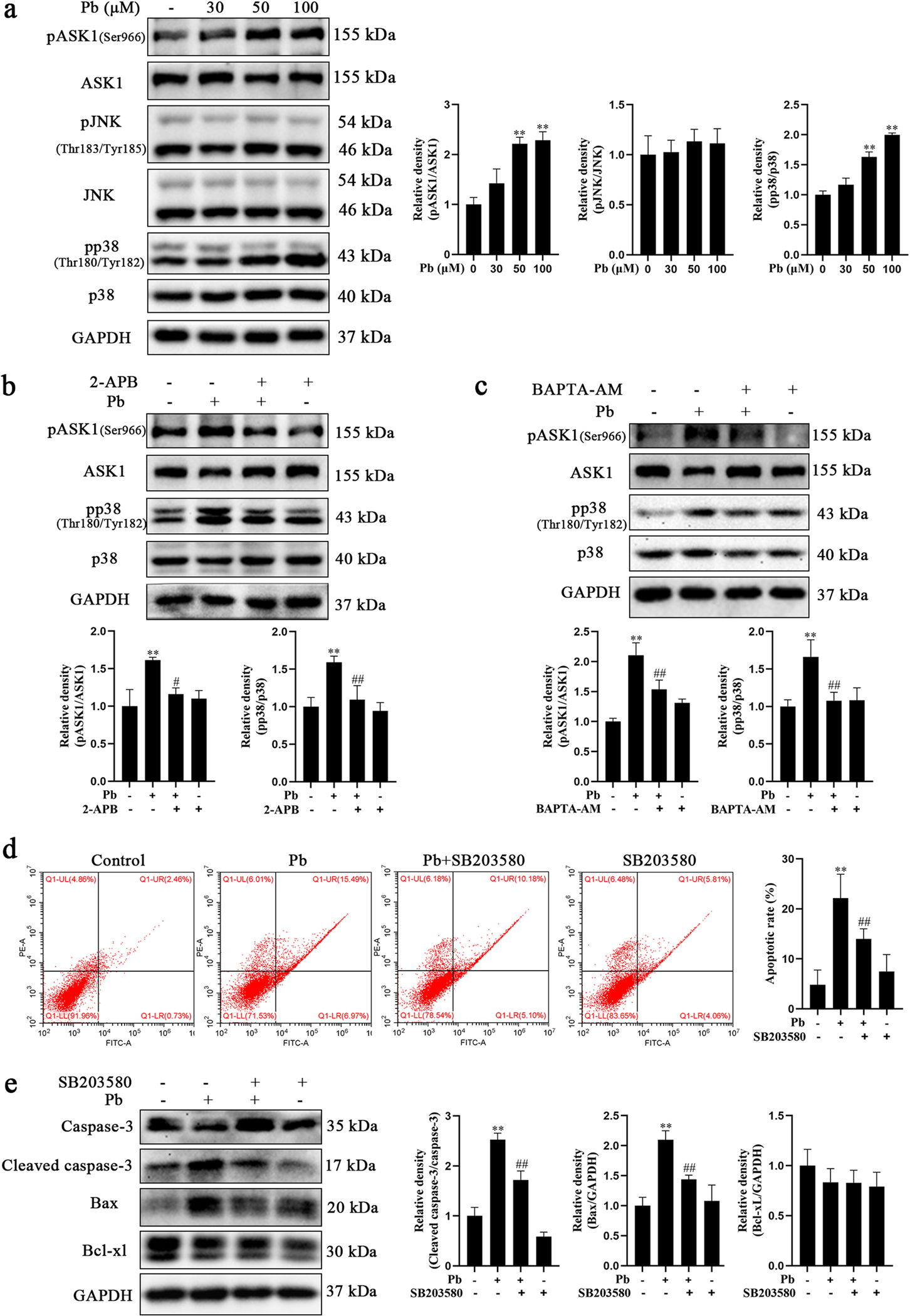
Pb-induced hippocampal neurons apoptosis though IP3R-Ca2+-ASK1-p38 signaling pathway. a Primary hippocampal neurons were treated with increased concentrations of PbAc for 24 h. The activations of ASK1, JNK and p38 were determined by western blotting. b, c Primary hippocampal neurons were treated with 50 μM PbAc in the presence or absence of pretreatment with 2-APB or BAPTA-AM for 24 h. The activations of ASK1 and p38 were determined by western blotting. d, e Primary hippocampal neurons were treated with 50 μM PbAc in the presence or absence of pretreatment with SB203580 for 24 h. d Hippocampal neuron apoptosis was determined by flow cytometry analysis. e Protein level of cleaved caspase-3, Bax and Bcl-xL were determined using western blotting. The protein expression was normalized by GAPDH or corresponding total protein content. * P < 0.05 and * * P < 0.01, compared to the control group. #P < 0.05 and ##P < 0.01, compared to the Pb-treated group.
Given that PAS-Na suppressed the Pb-induced increased levels of IP3R expression and Ca2+ production, next, we investigated whether it could repress the Pb-induced ASK1/p38 activation. As shown in Fig. 7a, 200 and 400 μM PAS-Na significantly mitigated the Pb-induced ASK1 phosphorylation in primary hippocampal neurons. While the phosphorylation of p38 in PAS-Na treatment group was decreased compared to the Pb-treated group, it did not attain statistical significance. More importantly, in vivo, we further corroborated that the level of ASK1 and p38 phosphorylation in the hippocampal of juvenile Pb-exposed group were significantly decreased by 240 mg/kg PAS-Na treatment for 2 weeks (Fig. 7b). Treatment with 160 and 240 mg/kg PAS-Na for 6 weeks significantly reduced ASK1 and p38 phosphorylation levels in the hippocampal of subchronic Pb exposed group (Fig. 7c). Taken together, these results demonstrate that PAS-Na ameliorates the Pb-induced hippocampal neuronal apoptosis via the IP3R-Ca2+-ASK1-p38 signaling pathway.
Fig. 7.
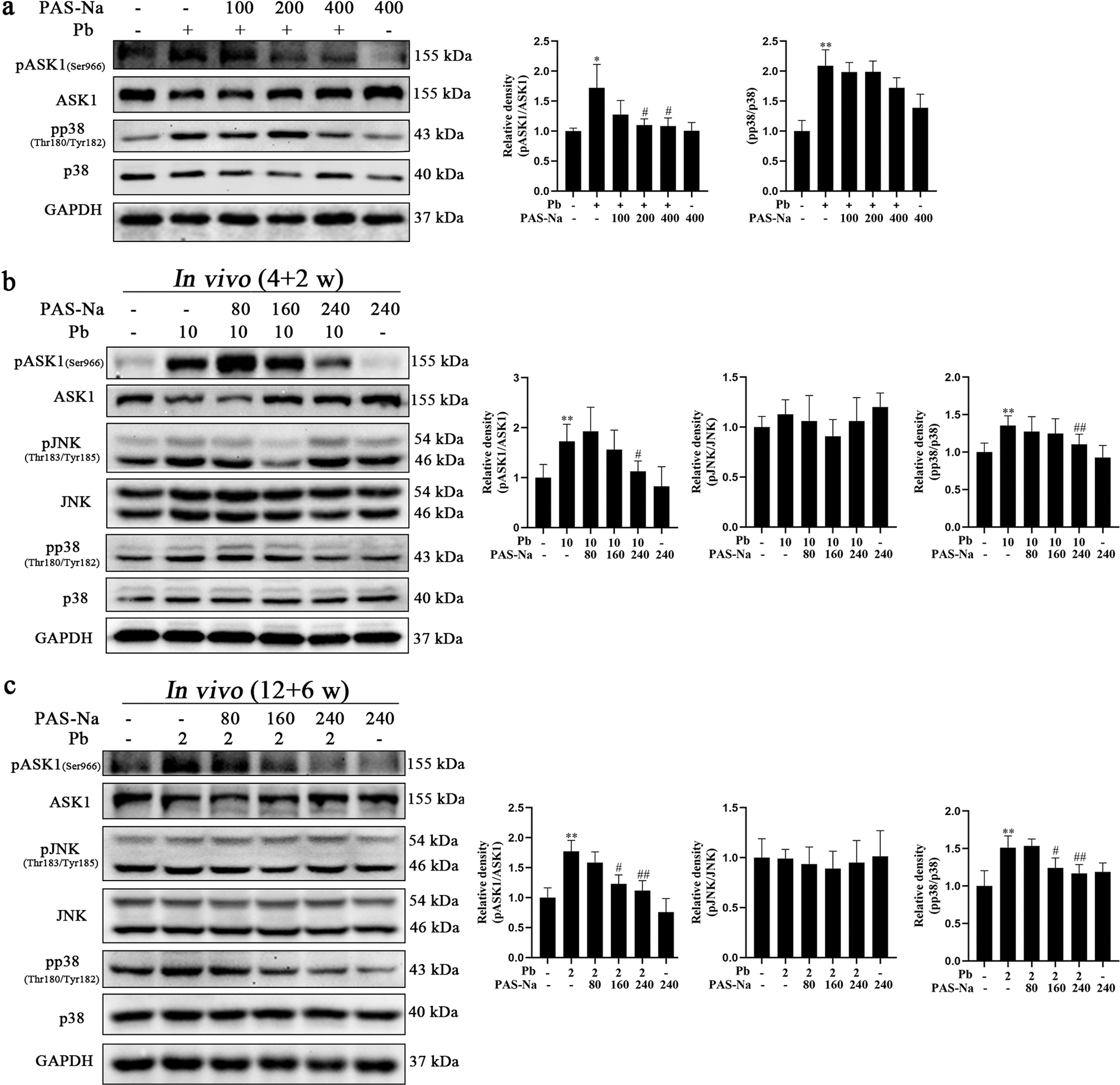
PAS-Na treatment reduced the activation of ASK1 and p38 induced by Pb in hippocampal neurons. a Primary hippocampal neurons were treated with 50 μM PbAc for 24 h, followed by 100, 200 or 400 μM PAS-Na treatment for 24 h. The activations of ASK1 and p38 were determined by western blotting. b Rats were treated with 10 mg/kg PbAc for 4 weeks during the juvenile period, followed by 80, 160 or 240 mg/kg PAS-Na treatment for 2 weeks. c Rats were treated with 2 mg/kg PbAc for 12 weeks, followed by 80, 160 or 240 mg/kg PAS-Na treatment for 6 weeks. The activations of ASK1, JNK and p38 were determined by western blotting. The phosphorylated protein expression was normalized by total protein content. * P < 0.05 and * * P < 0.01, compared to the control group. #P < 0.05 and ##P < 0.01, compared to the Pb-treated group.
4. Discussion
The hippocampus plays a critical role in cognitive function. Our earlier study has reported that occupational Pb exposure resulted in hippocampal ultrastructural and functional changes in Pb-smelting and Pb-battery manufacturing workers (Jiang et al., 2008). Notably, currently no treatments are clinically available for Pb-induced hippocampal damage. Herein, we report that PAS-Na treatment rescued Pb-induce hippocampal cells death both in vitro and in vivo. Moreover, PAS-Na significantly mitigated the Pb-induced increase of intracellular Ca2+ and inhibited Pb-induced IP3R activation, which is associated with ER Ca2+ release (Fan et al., 2013). In addition, PAS-Na significantly reduced the Pb-induced activation of the ASK1-p38 apoptotic signaling pathway. PAS-Na ameliorated the Pb-induced hippocampal neuronal apoptosis may secondary to IP3R-Ca2+-ASK1-p38 signaling pathway activation. Collectively, these findings establish the therapeutic potential of PAS-Na for Pb-induced neurotoxicity.
In this novel study, we found that juvenile developmental Pb exposure in rats resulted in cognitive dysfunction, indicated that the developing brain is sensitive to Pb exposure. It is noteworthy that the dosing, initial time of administration and duration of Pb exposure are all key factors in the outcome of Pb exposure-induced behaviors (Schneider et al., 2012). In particular, the timing of developmental Pb exposure can influence a variety of outcomes, as children are particularly vulnerable to Pb toxicity. Importantly, we found that high-dose PAS-Na (240 mg/kg) treatment ameliorated juvenile developmental Pb exposure-induced cognitive dysfunctions, possibly due to the rescue of Pb-induce hippocampal cells death by PAS-Na treatment.
Ca2+ signaling is critically important for both the initiation and effectuation of cell death. High levels of intracellular free Ca2+ concentration have been shown to contribute to hippocampal functional impairment after early-life and lifetime Pb exposure (Ouyang et al., 2019). In the present study, quantitative Fluo-4 fluorescence imaging (Fig. 4a) confirmed that upon Pb exposure hippocampal neuronal apoptosis was associated with increased intracellular Ca2+ levels. It has been previously reported that Pb exposure caused apoptosis in rat rPT cells by promoting elevation in intracellular and mitochondrial Ca2+, and that the Pb-induced apoptosis was markedly inhibited by IP3R and Ca2+ inhibitors (Wang et al., 2015). Abnormal Ca2+ release from IP3R has been posited to act as an important apoptotic signal. Herein, we demonstrated that IP3R protein level was increased after Pb exposure both in vitro and in vivo (Fig. 5c–f), while 2-APB effectively decreased [Ca2+]i in Pb-treated hippocampal neurons (Fig. 5a), indicating that the Pb-induced intracellular Ca2+ increase was associated with increased levels of IP3R. Importantly, this result is further corroborated by other studies (Fan et al., 2013). Interestingly, among the three IP3R isoforms, only IP3R2 and IP3R3 mRNA expression were significantly elevated in Pb-treated hippocampal neurons (Fig. 5b). Notably, it has been previously shown that in Pb-treated rPT cells, mRNA expression of both IP3R1 and IP3R2 isoforms was significantly up-regulated (Wang et al., 2015). These findings differ from ours, possibly due to the Pb exposure paradigm, and given that expression patterns and subcellular distributions of the three IP3R isoforms are heterogeneous in different tissues/cell types (Ivanova et al., 2014; Bartok et al., 2019). Nonetheless, it was noted that IP3R2 and IP3R3 are more effective than IP3R1 to release Ca2+ from the ER to mitochondria (Mendes et al., 2005; Bartok et al., 2019). These findings suggest that Pb increases transcriptionally the levels of select IP3R subtypes to trigger the opening of the IP3R Ca2+ channels. However, an interaction of Pb with the IP3R was also posited to be associated with this effect, given the structural similarity of Pb and Ca2+, as both are divalent metals with analogous transport mechanisms (Fang et al., 2021). Pb can readily bind Ca2+ domains (Ca2+ binding sites) and replacing Ca2+, activating or inhibiting protein kinase C (depending on Pb concentration) (Kasten-Jolly and Lawrence, 2018), which has been closely associated with synaptic plasticity, learning and memory processes (Nihei et al., 2001). Furthermore, Pb may mimic and simulate Ca2+ signaling to activate the phospholipase C, which leads to the production of a second messenger IP3, which, in turn, binds to the IP3R, triggering the opening of the IP3R Ca2+ channels and release of Ca2+ from internal stores to the cytosol (Marchi et al., 2012; Lee et al., 2021). Overall, these observations emphasize that IP3R-modulation of ER-associated Ca2+ release might play a critical role in Pb-induced neuronal death.
ASK1 is a well-known mediator of apoptosis. ASK1 is activated in response to stress, including ER, oxidative stress as well as death receptor ligands (Shiizaki et al., 2013). The results from the present study showed that ASK1 activation was essential for hippocampal neuron apoptosis in response to Pb exposure, as evident by the Pb-induced increase in the phosphorylation of ASK1 at Ser966 linearly and in a concentration-dependent. Moreover, the activation of ASK1 by Pb was mediated secondary to increased intracellular Ca2+ transient, as the phosphorylation of ASK1 was repressed by both 2-APB and BAPTA-AM (Fig. 6b, c). Nonetheless, Pb failed to activate JNK, although it has been reported to act as a downstream regulator of ASK1 activation in response to environmental stresses (Tobiume et al., 2001; Tesch et al., 2016). In contrast, Pb induced the activation of p38 in hippocampal neurons, which was blocked by an IP3R inhibitor and a Ca2+ inhibitor (Fig. 6b, c). It is likely that the elevated Ca2+ levels are hence responsible for p38 activation in Pb-treated hippocampal neurons. Sun et al. reported that aconitine-induced rat ventricular myocytes apoptosis secondary to Ca2+ overload and p38 phosphorylation, while the p38 specific inhibitor SB203580 effectively mitigated cells apoptosis (Sun et al., 2014). In agreement, the present study corroborated the pro-apoptotic role of p38 activation in Pb-exposed neurons, as the up-regulation of apoptotic rate of Pb-exposed neurons as well as the high expression of the apoptosis-associated protein (cleaved caspase-3 and Bax) was decreased by the p38 inhibitor (Fig. 6d, e). These findings support the notion that Ca2+ and p38 play a critical role in apoptosis. Collectively, Pb-induced rat hippocampal neurons apoptosis appears to be mediated via the IP3R-Ca2+-ASK1-p38 signaling pathway.
PAS-Na has shown efficacy in the treatment of Mn-induced neurotoxicity (Ky et al., 1992; Jiang et al., 2006; Li et al., 2018; Peng et al., 2020). Herein, PAS-Na significantly reduced Pb-induced hippocampal neurons apoptosis in vitro and in vivo (Fig. 3), raising the possibility of PAS-Na therapy for Pb-induced neurotoxicity. PAS-Na was originally discovered in our previous study to possess anti-lipid peroxidation properties, protecting PC12 cells from Pb-induced apoptosis secondary to increased intracellular levels of glutathione (He et al., 2017). Hence, we posited that PAS-Na antagonized Pb-induced apoptotic cells death, at least in part, given its anti-oxidation properties. However, the present study suggested that the mechanism by which PAS-Na alleviated Pb-induced apoptosis may also be mediated by the lowering of intracellular Ca2+ levels. On one hand, PAS-Na inhibited Pb-induced IP3R activation by reducing IP3R gene and protein expressions (Fig. 5b, d, e). Interestingly, PAS-Na reduced the Pb-induced increase in IP3R2 mRNA expression in a dose-dependent manner, while there was a spike in IP3R3 mRNA expression at 400 μM PAS-Na treatment for Pb-treated neurons. The reason may be associated with the different sensitivity of IP3R2 and IP3R3 to PAS-Na. IP3R3 was preferentially expressed in cultured cells and was sensitive to extracellular stimulations (Mendes et al., 2005; Ivanova et al., 2014). IP3R3 may have high affinity or high sensitivity to PAS-Na in cultured hippocampal neurons, contributing to the effectiveness of lower dose of PAS-Na (100 and 200 μM) to reduce the high mRNA expression of IP3R3 in Pb-exposed neurons. On the other hand, PAS-Na may act as metal chelator to combine with Ca2+ and decrease intracellular Ca2+ levels. Moreover, the molecular mechanisms involved in the efficacy of PAS-Na in decreasing Pb-induced cell apoptosis may also be associated with reduction in ASK1 and p38 expression, since PAS-Na effectively decreased the phosphorylation of ASK1 and p38 both in vitro and in vivo. Therefore, the molecular mechanism of PAS-Na in protecting cells needs further study.
At present, edetate calcium disodium (CaNa2EDTA) remains the drug of choice for the treatment of Pb poisoning (Sakthithasan et al., 2018). In addition, dimercaptosuccinic acid, penicillamine, and succimer are also recognized as antidotes for Pb poisoning (Gracia and Snodgrass, 2007; Sakthithasan et al., 2018). In fact, the aforementioned Pb poisoning antidotes have the same detoxification mechanism: acting as Pb chelator. While undoubtedly chelation therapy has greatly contributed to lowering the morbidity and mortality associated with occupational Pb exposure, it has failed to rescue Pb-induced cognitive defects associated neurotoxicity. The presence of contains amino, carboxyl and hydroxyl functional groups in PAS-Na (Nelson et al., 2010), may render this molecule effective as a Pb chelating therapeutic agent. In the present in vitro study, to avoid the direct complexation of PAS-Na with Pb, we added PAS-Na after the exposures to Pb to mimic a physiological real-life-scenario treatment paradigm. The results showed that at a certain concentration, PAS-Na mitigated Pb-induced apoptosis in hippocampal neurons, likely reflecting the inhibition of IP3R-Ca2+-ASK1-p38 signaling pathway. It is noteworthy, the regulation of Ca2+ release via IP3R-Ca2+ channels has been reported as a potential treatment for several neurological diseases (Chan, 2013). Therefore, IP3R is likely a critical target of PAS-Na for the treatment of Pb-induced neurotoxicity. Moreover, unlike CaNa2EDTA, PAS-Na can readily cross the blood-brain barrier (Hong et al., 2011), which is essential for an efficacious therapeutic treatment of Pb-induced neurotoxicity. Notably, in manganism patients, intravenous PAS-Na (receiving 6 g dissolved in 500 mL of 10% glucose) per day, 4 days a week, over 3.5 months, improved the symptoms of manganism (Ky et al., 1992; Jiang et al., 2006). However, there remains a lack of epidemiological studies to support the efficacy of PAS-Na in the treatment of Pb poisoning, and the optimal dose of PAS-Na for Pb poisoning has yet to be determined. Overall, whether PAS-Na can be used to treat Pb-induced neurotoxicity requires additional in vitro, in vivo and ultimately clinical studies.
Here, we found that Pb-induced neuronal apoptosis was secondary to intracellular Ca2+ disturbance followed by ASK1-p38 apoptotic pathway activation. PAS-Na showed efficacy in reversing these Pb-induced effects (Fig. 8). However, this study has several limitations. While our findings suggest that 160 and 240 mg/kg PAS-Na have effects on reducing hippocampal apoptosis, the optimal treatment dose and duration have yet to be determined. In addition, although PAS-Na was shown to reduce the Pb-induced increases in hippocampal Pb and Ca2+, the pharmacokinetics of its transport and accumulation in the hippocampus, and its mode-of-action in chelating divalent metals have yet to be characterized. Moreover, due to the side effects of PAS-Na on the gastrointestinal system, its clinical application should be explored. Future researches should focus on developing PAS-Na analogs to achieve more efficient chelation effect and increase its clinical utility.
Fig. 8.
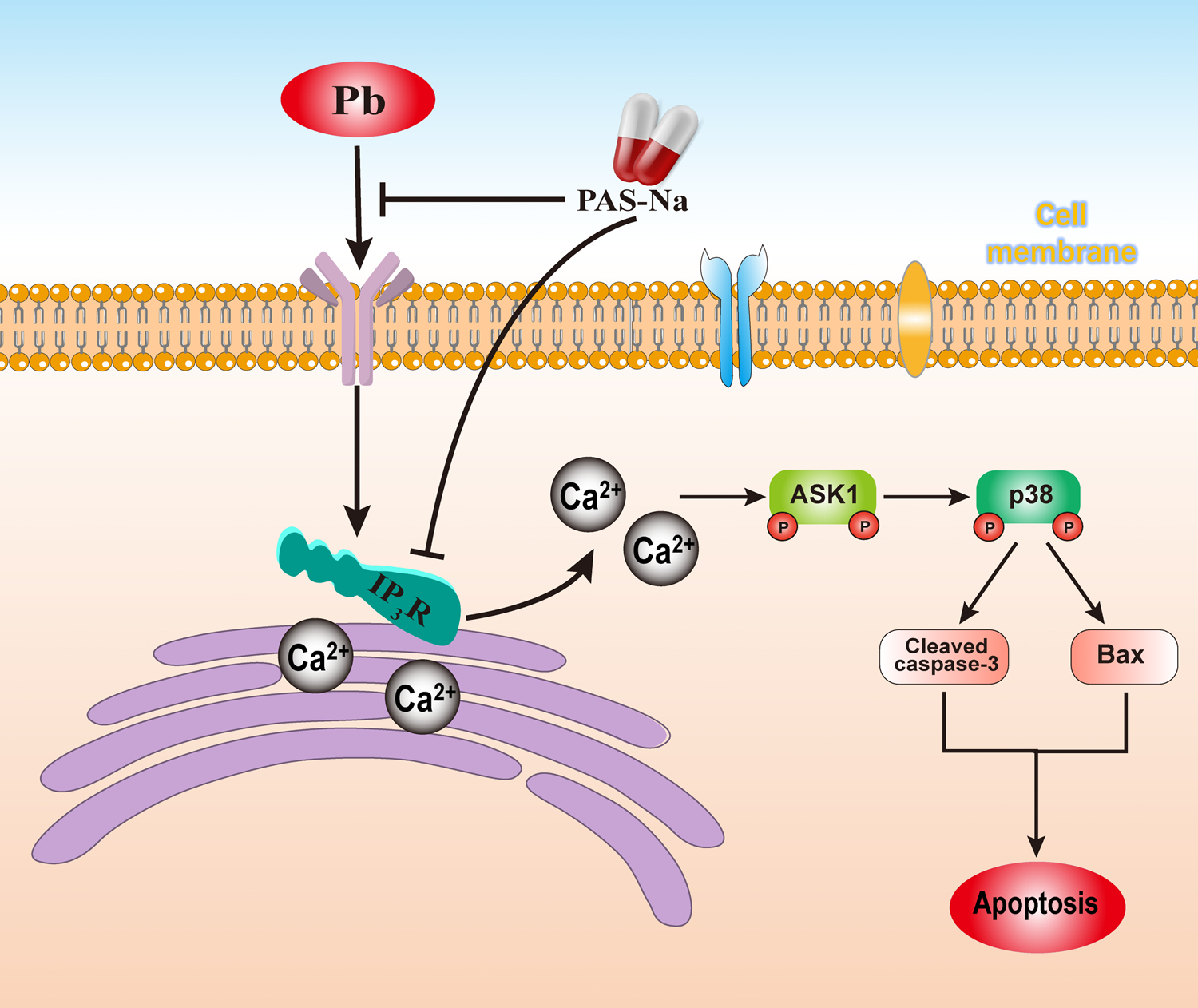
Schematic diagram summarizing the role of PAS-Na in Pb-induced neuronal apoptosis.
In conclusion, the present study demonstrated that Pb-induced apoptosis in hippocampal neurons is probably mediated by IP3R-Ca2+-ASK1-p38 signaling pathway activation. PAS-Na at select concentrations (160 and 240 mg/kg in vivo and 200 and 400 μM in vitro), afforded efficacy in reducing the Pb-induced apoptosis in hippocampal neurons, likely associated with inhibition of IP3R-modulation of intracellular Ca2+ signaling, and the ensuing decrease in the phosphorylation of ASK1 and p38. These findings provide novel insight to cellular and molecular mechanisms of PAS-Na-induced therapy, and merit future studies to characterize its therapeutic efficacy for Pb-induced neurotoxicity.
Acknowledgements
Funding support was provided by grants from the National Natural Science Foundation of China (NSFC 81773476 and 82160626). MA was supported in part by grants from the National Institute of Environmental Health Sciences (NIEHS) R01ES07331 and R01ES10563. The authors thank Prof. Hui-Dong Zhang from Sichuan University and Prof. Guo-Dong Lu from Guangxi Medical University for their useful suggestions. The authors are grateful to Prof. Yan-Ning Li from Guangxi Medical University for her guidance in statistical analysis methods.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Zhao-cong Li, Lei-lei Wang and Yue-song Zhao implemented the research process and performed the statistical analysis. Dong-jie Peng, Jing Chen and Si-yang Jiang assisted in establishing animal models and implementing behavioral experiments. Lin Zhao participated in part of the in vitro experiments. Zhao-cong Li wrote the paper. Michael Aschner polished the grammar and gave many suggestions. Shao-jun Li and Yue-ming Jiang designed and led the study. All authors read and approved the final manuscript.
References
- Bakulski KM, Dou JF, Thompson RC, Lee C, Middleton LY, Perera BPU, Ferris SP, Jones TR, Neier K, Zhou X, Sartor MA, Hammoud SS, Dolinoy DC, Colacino JA, 2020. Single-cell analysis of the gene expression effects of developmental lead (Pb) exposure on the mouse hippocampus. J. Toxicol. Sci 176 (2), 396–409. 10.1093/toxsci/kfaa069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartok A, Weaver D, Golenár T, Nichtova Z, Katona M, Bánsághi S, Alzayady KJ, Thomas VK, Ando H, Mikoshiba K, Joseph SK, Yule DI, Csordás G, Hajnóczky G, 2019. IP(3) receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. J. Nat. Commun. 10 (1), 3726. 10.1038/s41467-019-11646-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CC, 2013. Modulating Ca2+ release by the IP3R/Ca2+ channel as a potential therapeutic treatment for neurological diseases. J. Pharm. Pat. Anal. 2 (5), 629–636. 10.4155/ppa.13.42. [DOI] [PubMed] [Google Scholar]
- Conrad CD, Grote KA, Hobbs RJ, Ferayorni A, 2003. Sex differences in spatial and non-spatial Y-maze performance after chronic stress. J. Neurobiol. Learn Mem. 79 (1), 32–40. 10.1016/s1074-7427(02)00018-7. [DOI] [PubMed] [Google Scholar]
- Dellu F, Contarino A, Simon H, Koob GF, Gold LH, 2000. Genetic differences in response to novelty and spatial memory using a two-trial recognition task in mice. J. Neurobiol. Learn Mem. 73 (1), 31–48. 10.1006/nlme.1999.3919. [DOI] [PubMed] [Google Scholar]
- Deng XF, Ou SY, Jiang YM, Chen HB, Deng X, Lu S, Wang K, Jiang YH, Li G, Lu JP, 2009. Effects of sodium para-aminosalicylic acid on hippocampal ultramicro-structure of subchronic lead-exposed rats. J. J. Toxicol. 23 (3), 213–216. 10.16421/j.cnki.1002-3127.2009.03.011. [DOI] [Google Scholar]
- Dinel AL, Lucas C, Guillemet D, Layé S, Pallet V, Joffre C, 2020. Chronic supplementation with a mix of salvia officinalis and salvia lavandulaefolia improves morris water maze learning in normal adult C57Bl/6J mice. J. Nutr 12 (6) 10.3390/nu12061777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G, Zhou F, Feng C, Wu F, Ye W, Wang C, Lin F, Yan J, Li Y, Chen Y, Bi Y, 2013. Lead-induced ER calcium release and inhibitory effects of methionine choline in cultured rat hippocampal neurons. J. Toxicol. Vitr. 27 (1), 387–395. 10.1016/j.tiv.2012.06.019. [DOI] [PubMed] [Google Scholar]
- Fang Y, Lu L, Liang Y, Peng D, Aschner M, Jiang Y, 2021. Signal transduction associated with lead-induced neurological disorders: a review. J. Food Chem. Toxicol. 150, 112063 10.1016/j.fct.2021.112063. [DOI] [PubMed] [Google Scholar]
- Flora G, Gupta D, Tiwari A, 2012. Toxicity of lead: a review with recent updates. J. Inter. Toxicol. 5 (2), 47–58. 10.2478/v10102-012-0009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracia RC, Snodgrass WR, 2007. Lead toxicity and chelation therapy. J. Am. J. Health Syst. Pharm. 64 (1), 45–53. 10.2146/ajhp060175. [DOI] [PubMed] [Google Scholar]
- He SN, Qin WX, Lu YH, Li K, Luo YN, Yuan ZX, Jiang XL, Mo YH, Li WJ, Jiang YM, 2017. Effect of sodium para-aminosalicylic acid on apoptosis of PC12 cells induced by lead-exposure. J. Chin. J. Pharmacol. Toxicol 31 (02), 159–164. 10.3867/j.issn.1000-3002.2017.02.06. [DOI] [Google Scholar]
- Hong L, Jiang W, Pan H, Jiang Y, Zeng S, Zheng W, 2011. Brain regional pharmacokinetics of p-aminosalicylic acid and its N-acetylated metabolite: effectiveness in chelating brain manganese. J. Drug Metab. Dispos. 39 (10), 1904–1909. 10.1124/dmd.111.040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova H, Vervliet T, Missiaen L, Parys JB, De Smedt H, Bultynck G, 2014. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. J. Biochim Biophys. Acta 1843 (10), 2164–2183. 10.1016/j.bbamcr.2014.03.007. [DOI] [PubMed] [Google Scholar]
- Jiang YM, Long LL, Zhu XY, Zheng H, Fu X, Ou SY, Wei DL, Zhou HL, Zheng W, 2008. Evidence for altered hippocampal volume and brain metabolites in workers occupationally exposed to lead: a study by magnetic resonance imaging and (1)H magnetic resonance spectroscopy. J. Toxicol. Lett. 181 (2), 118–125. 10.1016/j.toxlet.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YM, Mo XA, Du FQ, Fu X, Zhu XY, Gao HY, Xie JL, Liao FL, Pira E, Zheng W, 2006. Effective treatment of manganese-induced occupational Parkinsonism with p-aminosalicylic acid: a case of 17-year follow-up study. J. J. Occup. Environ. Med 48 (6), 644–649. 10.1097/01.jom.0000204114.01893.3e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung WR, Kim HG, Kim KL, 2008. Ganglioside GQ1b improves spatial learning and memory of rats as measured by the Y-maze and the Morris water maze tests. J. Neurosci. Lett 439 (2), 220–225. 10.1016/j.neulet.2008.05.020. [DOI] [PubMed] [Google Scholar]
- Kasten-Jolly J, Lawrence DA, 2018. The cationic (calcium and lead) and enzyme conundrum. J. J. Toxicol. Environ. Health B Crit. Rev. 21 (6–8), 400–413. 10.1080/10937404.2019.1592728. [DOI] [PubMed] [Google Scholar]
- Kraeuter AK, Guest PC, Sarnyai Z, 2019. The Y-maze for assessment of spatial working and reference memory in mice. J. Methods Mol. Biol. 1916, 105–111. 10.1007/978-1-4939-8994-2_10. [DOI] [PubMed] [Google Scholar]
- Ky SQ, Deng HS, Xie PY, Hu W, 1992. A report of two cases of chronic serious manganese poisoning treated with sodium para-aminosalicylic acid. J. Br. J. Ind. Med 49 (1), 66–69. 10.1136/oem.49.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Rosales JL, Byun HG, Lee KY, 2021. D,L-Methadone causes leukemic cell apoptosis via an OPRM1-triggered increase in IP3R-mediated ER Ca(2+) release and decrease in Ca(2+) efflux, elevating [Ca(2+)](i). J. Sci. Rep 11 (1), 1009. 10.1038/s41598-020-80520-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SJ, Qin WX, Peng DJ, Yuan ZX, He SN, Luo YN, Aschner M, Jiang YM, Liang DY, Xie BY, Xu F, 2018. Sodium P-aminosalicylic acid inhibits subchronic manganese-induced neuroinflammation in rats by modulating MAPK and COX-2. J. Neurotoxicology. 64, 219–229. 10.1016/j.neuro.2017.06.012. [DOI] [PubMed] [Google Scholar]
- Li ZC, Wang F, Li SJ, Zhao L, Li JY, Deng Y, Zhu XJ, Zhang YW, Peng DJ, Jiang YM, 2019. Sodium para-aminosalicylic acid reverses changes of glutamate turnover in manganese-exposed rats. J. Biol. Trace Elem. Res. 10.1007/s12011-019-02001-0. [DOI] [PubMed] [Google Scholar]
- Li ZC, Wang F, Li SJ, Zhao L, Li JY, Deng Y, Zhu XJ, Zhang YW, Peng DJ, Jiang YM, 2020. Sodium para-aminosalicylic acid reverses changes of glutamate turnover in manganese-exposed rats. J. Biol. Trace Elem. Res 197 (2), 544–554. 10.1007/s12011-019-02001-0. [DOI] [PubMed] [Google Scholar]
- Liu KS, Hao JH, Zeng Y, Dai FC, Gu PQ, 2013. Neurotoxicity and biomarkers of lead exposure: a review. J. Chin. Med Sci. J. 28 (3), 178–188. 10.1016/s1001-9294(13)60045-0. [DOI] [PubMed] [Google Scholar]
- Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, Pinton P, 2012. Selective modulation of subtype III IP3R by Akt regulates ER Ca2+ release and apoptosis. J. Cell Death Dis. 3 (5), e304 10.1038/cddis.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, Rodrigues MA, Gomez MV, Nathanson MH, Leite MF, 2005. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J. J. Biol. Chem. 280 (49), 40892–40900. 10.1074/jbc.M506623200. [DOI] [PubMed] [Google Scholar]
- Mitra P, Sharma P, 2019. Novel direction in mechanisms underlying lead toxicity: evidence and prospective. J. Indian J. Clin. Biochem 34 (2), 121–122. 10.1007/s12291-019-00829-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M, Huggins T, Licorish R, Carroll MA, Catapane EJ, 2010. Effects of p-Aminosalicylic acid on the neurotoxicity of manganese on the dopaminergic innervation of the cilia of the lateral cells of the gill of the bivalve mollusc, Crassostrea virginica. J. Comp. Biochem Physiol. C. Toxicol. Pharm 151 (2), 264–270. 10.1016/j.cbpc.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nihei MK, McGlothan JL, Toscano CD, Guilarte TR, 2001. Low level Pb(2+) exposure affects hippocampal protein kinase C gamma gene and protein expression in rats. J. Neurosci. Lett. 298 (3), 212–216. 10.1016/s0304-3940(00)01741-9. [DOI] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P, 2003. Regulation of cell death: the calcium-apoptosis link. J. Nat. Rev. Mol. Cell Biol 4 (7), 552–565. 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Ouyang L, Zhang W, Du G, Liu H, Xie J, Gu J, Zhang S, Zhou F, Shao L, Feng C, Fan G, 2019. Lead exposure-induced cognitive impairment through RyR-modulating intracellular calcium signaling in aged rats. J. Toxicol. 419, 55–64. 10.1016/j.tox.2019.03.005. [DOI] [PubMed] [Google Scholar]
- Peng D, Li J, Deng Y, Zhu X, Zhao L, Zhang Y, Li Z, Ou S, Li S, Jiang Y, 2020. Sodium para-aminosalicylic acid inhibits manganese-induced NLRP3 inflammasome-dependent pyroptosis by inhibiting NF-κB pathway activation and oxidative stress. J. J. Neuroinflamm. 17 (1), 343. 10.1186/s12974-020-02018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuben A, Elliott M, Caspi A, 2020. Implications of legacy lead for children’s brain development. J. Nat. Med. 26 (1), 23–25. 10.1038/s41591-019-0731-9. [DOI] [PubMed] [Google Scholar]
- Reuben A, Elliott ML, Abraham WC, Broadbent J, Houts RM, Ireland D, Knodt AR, Poulton R, Ramrakha S, Hariri AR, Caspi A, Moffitt TE, 2020. Association of childhood lead exposure with MRI measurements of structural brain integrity in midlife. J. Jama. 324 (19), 1970–1979. 10.1001/jama.2020.19998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha A, Trujillo KA, 2019. Neurotoxicity of low-level lead exposure: History, mechanisms of action, and behavioral effects in humans and preclinical models. J. Neurotoxicology 73, 58–80. 10.1016/j.neuro.2019.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakthithasan K, Lévy P, Poupon J, Garnier R, 2018. A comparative study of edetate calcium disodium and dimercaptosuccinic acid in the treatment of lead poisoning in adults. J. Clin. Toxicol. (Philos. ) 56 (11), 1143–1149. 10.1080/15563650.2018.1478424. [DOI] [PubMed] [Google Scholar]
- Salman T, Nawaz S, Ikram H, Haleem DJ, 2019. Enhancement and impairment of cognitive behaviour in Morris water maze test by methylphenidate to rats. J. Pak. J. Pharm. Sci. 32 (3), 899–903. [PubMed] [Google Scholar]
- Schneider JS, Anderson DW, Talsania K, Mettil W, Vadigepalli R, 2012. Effects of developmental lead exposure on the hippocampal transcriptome: influences of sex, developmental period, and lead exposure level. J. Toxicol. Sci. 129 (1), 108–125. 10.1093/toxsci/kfs189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadbegian R, Guignet D, Klemick H, Bui L, 2019. Early childhood lead exposure and the persistence of educational consequences into adolescence. J. Environ. Res. 178, 108643 10.1016/j.envres.2019.108643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiizaki S, Naguro I, Ichijo H, 2013. Activation mechanisms of ASK1 in response to various stresses and its significance in intracellular signaling. J. Adv. Biol. Regul. 53 (1), 135–144. 10.1016/j.jbior.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Sun GB, Sun H, Meng XB, Hu J, Zhang Q, Liu B, Wang M, Xu HB, Sun XB, 2014. Aconitine-induced Ca2+ overload causes arrhythmia and triggers apoptosis through p38 MAPK signaling pathway in rats. J. Toxicol. Appl. Pharm 279 (1), 8–22. 10.1016/j.taap.2014.05.005. [DOI] [PubMed] [Google Scholar]
- Takeda K, Hatai T, Hamazaki TS, Nishitoh H, Saitoh M, Ichijo H, 2000. Apoptosis signal-regulating kinase 1 (ASK1) induces neuronal differentiation and survival of PC12 cells. J. J. Biol. Chem. 275 (13), 9805–9813. 10.1074/jbc.275.13.9805. [DOI] [PubMed] [Google Scholar]
- Takeda K, Matsuzawa A, Nishitoh H, Tobiume K, Kishida S, Ninomiya-Tsuji J, Matsumoto K, Ichijo H, 2004. Involvement of ASK1 in Ca2+-induced p38 MAP kinase activation. J. EMBO Rep. 5 (2), 161–166. 10.1038/sj.embor.7400072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesch GH, Ma FY, Nikolic-Paterson DJ, 2016. ASK1: a new therapeutic target for kidney disease. J. Am. J. Physiol. Ren. Physiol 311 (2), F373–F381. 10.1152/ajprenal.00208.2016. [DOI] [PubMed] [Google Scholar]
- Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T, Ichijo H, 2001. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. J. EMBO Rep. 2 (3), 222–228. 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang ZK, Jiao P, Zhou XP, Yang DB, Wang ZY, Wang L, 2015. Redistribution of subcellular calcium and its effect on apoptosis in primary cultures of rat proximal tubular cells exposed to lead. J. Toxicol. 333, 137–146. 10.1016/j.tox.2015.04.015. [DOI] [PubMed] [Google Scholar]
- Yoon H, Kim DS, Lee GH, Kim JY, Kim DH, Kim KW, Chae SW, You WH, Lee YC, Park SJ, Kim HR, Chae HJ, 2009. Protective effects of sodium para-amino salicylate on manganese-induced neuronal death: the involvement of reactive oxygen species. J. J. Pharm. Pharm. 61 (11), 1563–1569. 10.1211/jpp/61.11.0017. [DOI] [PubMed] [Google Scholar]
- Zheng W, Jiang YM, Zhang Y, Jiang W, Wang X, Cowan DM, 2009. Chelation therapy of manganese intoxication with para-aminosalicylic acid (PAS) in Sprague-Dawley rats. J. Neurotoxicology. 30 (2), 240–248. 10.1016/j.neuro.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zündorf G, Reiser G, 2011. Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. J. Antioxid. Redox Signal 14 (7), 1275–1288. 10.1089/ars.2010.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]