

Abstract
The insulin-like growth factor receptor (IGF1R) pathway plays an important role in cancer progression. In breast cancer, the IGF1R pathway is linked to estrogen-dependent signaling. Regulation of IGF1R activity is complex and involves the actions of its ligands IGF1 and IGF2 and those of IGF-binding proteins (IGFBPs). Six IGFBPs are known that share the ability to form complexes with the IGFs, by which they control the bioavailability of these ligands. Besides, each of the IGFBPs have specific features. In this review, the focus lies on the biological effects and regulation of IGFBP5 in breast cancer. In breast cancer, estrogen is a critical regulator of IGFBP5 transcription. It exerts its effect through an intergenic enhancer loop that is part of the chromosomal breast cancer susceptibility region 2q35. The biological effects of IGFBP5 depend upon the cellular context. By inhibiting or promoting IGF1R signaling, IGFBP5 can either act as a tumor suppressor or promoter. Additionally, IGFBP5 possesses IGF-independent activities, which contribute to the complexity by which IGFBP5 interferes with cancer cell behavior.
Keywords: insulin-like growth factor binding protein 5, breast cancer, estrogen, transcriptional regulation, breast cancer susceptibility region
Introduction
Breast cancer (BC) is the most frequent cancer among women and leading cause of cancer-related death in women worldwide (1). As a systemic disease, it is characterized by early tumor cell dissemination, colonization of other organs and metastasis formation (2–4). It is generally accepted that BC is a cancer stem cell (CSC) disease, where CSCs drive cancer initiation, progression and metastasis (5, 6). Epithelial, quasi-mesenchymal and mesenchymal forms of CSCs have been identified and seem to have different functions in BC progression (7, 8).
BC is a heterogenous disease requiring subtype assessment to offer patients appropriate treatments. Classically, subtyping is performed by immunohistochemical examination of the expression of estrogen receptor α (ERα), progesterone receptor (PR) and the human epidermal growth factor receptor 2 (Her2). Approximately two-thirds of the BCs are positive for ERα. ERα expression is indicative of potential responsiveness of the cancer to ERα-targeting drugs (endocrine therapy), such as tamoxifen (TAM), fulvestrant (FULV), and aromatase inhibitors (9–11). Her2 overexpression, caused by amplification of its gene erbb2, defines a second major subtype (12). This subtype is potentially responsive to anti-Her2 antibodies or Her2 kinase inhibitors (13). A third subtype, triple-negative BC (TNBC), is devoid of ERα, PR- and Her2 and is routinely treated by chemotherapy (14). Molecular subtyping, based on mRNA profiling, revealed a heterogeneity of BCs beyond ERα, PR and Her2 expression, giving rise to the distinction of luminal A, luminal B, Her2-enriched and basal-like BCs (15). Clinically, the distinction between luminal A and the more aggressive luminal B, both predominantly ERα-positive BCs, is important (16).
In ERα-positive BCs, the insulin-like growth factor (IGF)/IGF1 receptor (IGF1R) pathway plays an important role in ERα-dependent regulation (17–19). IGF bioavailability and activity is controlled by IGF binding proteins (IGFBPs) (20–22). Hence, their abundances are crucial for the activity of the IGF/IGF1R pathway and, consequently, for ERα activity. Six IGFBPs are known (22). Of these, IGFBP5 may be of particular importance for IGF regulation in BC, as its gene is not only located in the vicinity of the BC susceptibility locus q35 on chromosome 2, but its activity also regulated by enhancer elements within this region (23–27). Moreover, ERα is a major driver of IGFBP5 transcription. Besides IGF-dependent effects, IGFBP5 shows also IGF-independent effects, which interferes with the behavior of BC cells (22).
This review intends to summarize the current knowledge on the biological effects of IGFBP5 in BC and on the regulation of its expression.
Estrogen receptor α
The transcription factor (TF) ERα is classically activated by estrogen (28). ERα binds directly to estrogen-responsive elements or indirectly to other DNA sites by tethering to TFs, such as activating protein-1 (AP-1) (29, 30). It regulates a plethora of genes through proximal promoters and/or distal enhancers (31, 32). The interaction with the transcriptional machinery is mediated by two domains, transactivation function (AF)-1 and the estrogen-regulated AF-2 (33). Typically, ERα cooperates with the pioneer TF Forkhead box protein A1 (FoxA1), which facilitates ERα chromatin binding (34). Phosphorylation has a critical impact on ERα activity (33). Phosphorylations at Ser-118 and Ser-167 within the AF-1 domain promote estrogen-dependent as well as estrogen-independent transcriptional activities of ERα. Such phosphorylations can be triggered by receptor tyrosine kinases (RTKs) through the PI3K/AKT/mTOR/p70S6K and the Ras/Raf/MEK1/ERK1/2 pathways. Endocrine resistance, the resistance to endocrine therapy, is often caused by permanent activation of RTKs (35–37). Of the RTKs, IGF1R is of particular importance for ERα-dependent transcription.
Involvement of the IGF/IGF1R pathway in the regulation of estrogen-dependent effects
Classically, insulin-like growth factor 1 receptor (IGF1R) either homodimerizes or forms heterodimers with the insulin receptor (IR) (38, 39). Under certain conditions, it can also heterodimerize with members of the Her RTK family (40). IGF1R activity is regulated by its ligands IGF1 and IGF2 and additionally modulated by six IGF binding proteins (IGFBPs) (20–22). By binding to IGF1 and IGF2, IGFBPs regulate their bioavailabilities. A tight control of IGF bioavailability is necessary, as, for instance, increased serum levels of liver-produced IGF1 correlates with higher risk of developing BC and other cancers (41). Like other RTKs, IGF1R activates the PI3K/AKT and Ras/Raf/MEK/ERK1/2 pathways, but involves the adapter proteins insulin receptor substrate 1/2 (IRS1/2) (17, 18, 42).
The IGF/IGF1R pathway modulates estrogen effects and ERα activity (17–19). IGF promotes ERα activity by stimulating ERα phosphorylation at Ser-167 (43). In turn, estrogen-activated ERα stimulates IGF1R gene transcription (17, 44, 45) and stabilizes IRS1 (46). ERα-independent effects of estrogen are executed by G-protein coupled receptor 30 (GPR30) (47). Activated by estrogen, GPR30 stimulates AKT and ERK1/2 phosphorylation (48–50). IGF1 and insulin stimulate the expression of GPR30 by involving a PKCδ/ERK1/c-Fos/AP-1 pathway (51, 52).
In primary BC, IGF1R is frequently expressed in luminal tumors (~50%) (53), likely reflecting its link to ERα signaling. In contrast, in the Her2-enriched or basal-like subtypes, only ~10% or ~20% of the tumors are positive for IGF1R, respectively. In luminal B tumors, higher IGF1R levels are associated with better prognosis, while, in luminal A tumors, IGF1R levels did not correlate with patients’ outcome (53). Its potential role in endocrine resistance has nevertheless stimulated research towards ways of targeting the IGF/IGFR signaling pathway (54).
IGFBP5 as a major regulator of the IGF/IGF1R pathway
IGFBPs consist of three distinct domains, the cysteine-rich N-terminal, the C-terminal and the linker domain (L-domain) (55). Of the six IGFBP genes, IGFBP5 is the most conserved one (55), whose cDNA was first isolated in 1991 (56, 57). The mature IGFBP5 protein shows an apparent molecular weight of 31-32kD (58, 59) and consists of 252 amino acids (60), of which aa 1-80 represent the N-terminal, 81-170 the L- and 171-252 the C-terminal domain (61) ( Figure 1 ). The L-domain serves as a substrate for various proteases, such as ADAM9, MMP-2, PSA, thrombin and PAPP-A, and can be O-glycosylated and phosphorylated (62–66). A fragment of IGFBP5 encompassing amino acids 40-92 within the N-terminal domain is sufficient for IGF1 binding (21). The interaction is predominantly based on hydrophobic contacts, whereby Val-49, Leu-70 and Leu-73 of the IGFBP5 protein play a crucial role (60). Hence, mutations between amino acids 68 and 74, including Leu-70 and Leu-73, abolishes IGF binding (59, 67). However, for efficient inhibitory action of IGFBP5 on IGF1-IGF1R binding, the C-terminus of IGFBP5 is also important (60). Consistent with this finding, a 23kD truncated version (aa 1-169), lacking the C-terminal domain, binds to IGF with lower affinity (68, 69). Mutational analyses confirmed the participation of the C-terminal domain in IGF binding (70, 71). Particularly, Gly-203 and Gln-209, which are part of the C-terminal heparin-binding domain (see below), are involved in IGF1/IGFBP5 interaction. In blood, IGF and IGFBP5 form a ternary complex with acid labile subunit (ALS), which binds to the C-terminal domain of IGFBP5 (72).
Figure 1.

Structure of the human mature IGFBP5 protein. Three major domains, the N-terminal (N), the linker (L) and the C-terminal (C) domain, can be distinguished, which fulfill different functions as indicated. For high affinity IGF binding, the C-terminal domain is required in addition to the primary IGF binding site (IB) within the N-terminal domain between aa 68 and 74. A nuclear localization signal (NLS) is located within the C-terminal domain between aa 201 and 218, which also mediates binding of the IGFBP5 protein to heparin.
Although IGFBP5 exerts an inhibitory effect on the IGF/IGFR pathway by binding to IGF, a supportive effect of IGFBP5 on this pathway has also been reported ( Figure 2 ). Recombinant IGFBP5 and IGF1 proteins together acted protective against ceramide-induced apoptosis of MCF-7 BC cells (73) and stimulated proliferation of human intestinal smooth muscle cells (74). In prostate cancer, where androgen deprivation causes prostate carcinoma and stromal cells to upregulate the IGFBP5 level (75, 76), overexpression of IGFBP5 enhances the anti-apoptotic and mitogenic activity of recombinant IGF1 protein, thereby helping to overcome castration-induced tumor regression (77, 78). Also, IGFBP5 enhances the anti-apoptotic effect of IGF on osteosarcoma cells (72). It has been proposed that, for the IGFBP5-promoting effect on IGF1, IGFBP5 presents and releases IGF1 to the receptor (73). Proteolysis of IGFBP5 may play a role in this process, in a way that IGFBP5 digestion by enzymes, like L-domain cutting PAPP-A, leads to a release of IGFBP5-bound IGF1 (72). Hence, not only is IGFBP5 an IGF inhibitor, but also seems to function as a reservoir for IGF, ready to release this growth factor, when required.
Figure 2.

IGFBP5 actions. (A, B) By forming an inhibitory complex with IGF, IGFBP5 can prevent IGF-induced activation of IGF1R at appropriate IGFBP5/IGF ratios. (C) IGF-bound IGFBP5 has also been suggested to function as an IGF storage complex. Sudden release of IGF due to IGFBP5 proteolysis can strongly increase the local IGF concentration, potentially enhancing the IGF effect on IGF1R. In this scenario, IGFBP5 acts as a promoter of IGF activity. (D, E) IGF-independent effects of IGFBP5 either caused by its interaction with a putative plasma membrane receptor (D) or caused by its ability to enter the cell and accumulate in the nucleus (E).
Based on data obtained with human intestinal smooth muscle cells, IGFBP5 may also indirectly promote IGF1 activity (79). IGFBP5 was shown to stimulate the secretion of IGF1 in an IGF-independent manner by activating ERK1/2 and stress-responsive p38 kinase. In turn, IGF1 promotes the expression of IGFBP5 to further fuel IGF1 production.
IGF-independent activities of IGFBP5
IGFBP5 can exert effects independently of its IGF-interfering activity. Such IGF-independent effects may be executed intracellularly (intrinsic effects) or may involve IGFBP5 binding to a putative plasma membrane receptor ( Figure 2 ). First indications for IGF-independent effects were provided by data showing that the IGFBP5 protein binds to the plasma membrane of osteoblasts and mesangial cells and is taken up by these cells (68, 69, 80, 81). The attachment to the plasma membrane can occur in the presence or the absence of the C-terminal domain, whereby the C-terminal domain contains a heparin-binding domain between aa 201-218, which allows membrane binding through heparin. The heparin-binding domain was also shown to be involved in IGF1-binding of the C-terminal domain, whereby it seems that IGF1 and heparin compete for binding to this site (71). The heparin-binding IGFBP5 (aa 201-218) fragment is able to stimulate migration of mesangial cells (82), while an IGFBP5 version lacking the C-terminus can increase proliferation of osteoblasts in an IGF-independent manner (69). Furthermore, the C-terminal domain shows biological activities that are distinct from that of the N-terminal domain (61).
Both IGF-dependent and IGF-independent effects were also detectable in transgenic mice. When overexpressed, wildtype IGFBP5, but not mutant IGFBP5 lacking a functional IGF-binding site was able to rescue embryonic development defects as induced by IGF2R loss (59) indicating, that for this effect, IGF binding was necessary. In contrast, overexpression of either IGFBP5 version caused a reduction in birth weight suggesting that IGFBP5-induced growth retardation did not rely on IGF-binding.
Part of the intrinsic effect of IGFBP5 seems to be based on its ability to enter the nucleus (83). The C-terminal domain contains a functional nuclear localization sequence (NLS, aa 201-218), which is identical to the heparin binding site (84–86). It interacts with importin β and the nucleolar protein nucleolin. Strikingly, an NLS-mutated version of IGFBP5 is able to fully support the IGF-induced anti-apoptotic effect on osteosarcoma cells (72), suggesting that the nuclear effects of IGFBP5 are independent of the IGF-depending ones.
One intracellular target of IGFBP5 is the Ras-Association Domain Family 1 C Protein (RASSF1C) (87). RASSF1C is a proto-oncogenic protein (88) that participates in driving invasion, metastasis and stem cell activity in BC (89). By physically interacting with RASSF1C, IGFBP5 may regulate ERK1/2 activity (87). Blocking the activity of RASSF1C resulted in loss of IGFBP5’s ability to stimulate ERK1/2 activity in osteoblasts. In BC cells, IGFBP5-induced ERK1/2 activation results in protection against apoptosis (90). IGFBP5 may also inhibit ERK1/2 activity. In lung cancer, IGFBP5 counteracted RASSF1C-induced ERK1/2-dependent stimulation of the stem cell gene Piwi like RNA-mediated gene silencing 1 (PIWIL1) resulting in re-sensitization of tumor cells to the anti-cancer drug betulinic acid (91). In osteoblasts, IGFBP5 has been found to interact with the heterodimer consisting of the vitamin D receptor and retinoid X receptor, thereby blocking vitamin D-dependent gene expression (92). IGFBP5 has also been shown to bind to Four and a Half LIM protein 2 (FHL2), though the significance of this interaction is not known yet (93).
IGF1-independent IGFBP5 effects based on extracellular interactions involve plasma membrane proteins, such as tumor necrosis factor receptor 1 (TNF1R), α2/β1 integrins and a 420 kD complex harboring a serine/threonine kinase (94–96). By binding of IGFBP5 to TNF1R, which requires its N- and L-domains (94), IGFBP5 interferes with TNFα activity. As shown with lymphoma cells, IGFBP5 blocks TNFα-dependent NFκB activation (94). Vice versa, TNFα prevents the L-domain-dependent interaction between IGFBP5 and TNF1R (94) and inhibits IGFBP5-dependent myoblast survival (97). On the other hand, IGFBP5 can also support TNFα activity and increases its NFκB-dependent pro-apoptotic action on MDA-MB-231 BC cells (98). In the latter case, however, it is not clear whether this effect of IGFBP5 involves its interaction with TNF1R.
The interaction of IGFBP5 to α2/β1 integrins causes MCF-7 cells to stronger adhere to the extracellular matrix (95). This effect involves activation of the GTPase Cdc42 and AKT as well as inactivation of the stress-response kinase p38. Interestingly, Cdc42 was also shown to be activated in the migration-stimulatory effect of the heparin-binding IGFBP5 fragment (aa 201-218) on mesangial cells (82).
A 420 kD plasma membrane complex had been identified to interact with IGFBP5 (96). Full length IGFBP5, IGFBP5 (aa 1-169) lacking the C-terminal domain and the IGFBP5 (201-218) harboring the heparin-binding domain were all found to be able to bind to this complex and to activate a serine/threonine kinase within this complex.
Cellular source of IGFBP5
Cancer cells, including BC cells, express and secret IGFBP5 (99–101). In low-grade invasive BC, ~50% of the cancer cells show well detectable protein levels of IGFBP5 in the cytoplasm (102). IGFBP5 is also found in the nucleus of BC cells (103). The expression level of IGFBP5 is lower in normal breast, in normal tissue adjacent to the cancer lesion and in benign breast lesions (102, 104–106).
The microenvironment may also contribute to IGFBP5 production in cancer. Particularly fibroblasts, which in cancers are found in a modified form, called cancer-associated fibroblasts (CAFs) (107, 108), are a major source of secreted IGFBP5 protein in normal tissues (109–111). Attached to extracellular matrix (ECM) proteins, such as collagen, fibronectin and laminin, IGFBP5 is protected against proteolytic cleavage and can exert its effects on other cells. IGFBP5 can also feed back on fibroblasts by stimulating these cells to produce more ECM proteins, such as collagen 1A1 (109). Interestingly, in BC specimens, collagen 1A1 mRNA levels correlate with those of IGFBP5 (112). Excessive secretion of IGFBP5 by fibroblasts can block epithelial cell proliferation after injury, induce fibrosis and lead to premature senescence of fibroblasts (86, 109, 113–115). Senescent fibroblasts can acquire a senescence-associated secretory phenotype which can promote tumor progression (116) indicating that IGFBP5 may be involved in the generation of CAFs. In prostate cancer, stromal cells, particularly fibroblasts, were found to be the major source of IGFBP5 (117, 118) suggesting that fibroblasts contribute to IGFBP5 production also in cancer.
IGFBP5 as a potential prognostic/predictive biomarker in breast cancer
A Japanese study on a total of 167 BC patients revealed a correlation of higher IGFBP5 expression with poorer disease-free survival for patients with ERα-positive tumors, whereas no such correlation was found, when patients with ERα-negative tumors were included in the analysis (119). In contrast, a Chinese study on 108 patients showed that higher IGFBP5 mRNA levels were significantly associated with poorer disease-free survival for patients with ERα-negative tumors, but not for those with ERα-positive tumors (106). A third, US-American study on 76 patients showed a correlation of higher IGFBP5 protein expression with worse metastasis-, relapse-free and overall survival irrespective of the ERα status (104). An in silico study revealed that higher IGFBP5 RNA was associated with poorer overall survival in luminal A, but not in luminal B cancer (90). In sum, all these studies show that higher IGFBP5 expression indicates poorer prognosis, though the studies remain inconclusive in regard to the role of the ERα status. It is unclear if the correlation between IGFBP5 and prognosis is coincidential or causitive in nature. IGFBP5 shows diverse effects on apoptosis, cell-ECM attachment and cellular migration (see below).
Interestingly, under certain treatment conditions, higher IGFBP5 expression was associated with better outcome. Of the 153 TAM-treated BC patients with mostly lobular cancer, those with higher tumoral protein expression of IGFBP5 had a higher probability to survive (120). Also, a study on 371 patients revealed that patients who received a chemotherapy less likely relapsed, if the nuclear IGFBP5 protein level was higher (103). This was not the case for patients who were never chemotreated. Hence, higher IGFBP5 protein expression may predict better response to endocrine and chemo therapy. In vitro studies suggest a causative role of IGFBP5 in anti-estrogen response (see below).
IGFBP5’s effects on apoptosis, adhesion and migration
The Wnt pathway is critically involved in mammary development (121). Its deregulation by the mouse mammary tumor virus (MMTV) causes BC in mice. Hence, inhibition of this pathway by frizzled 8 receptor-immunoglobulin G Fc fusion protein (Fzd8CRD) leads to tumor regression (122). Part of this regression is caused by IGFBP5. Upregulated by Fzd8CRD, IGFBP5 inhibits the pro-survival effects of the IGF/IGF1R pathway. An inverse relationship between Wnt pathway activity and IGFBP5 expression is also found during mammary gland involution (122). Here again IGFBP5 acts pro-apoptotically and prevents IGF-depending survival of epithelial cells (122–126). Consistent with these results, IGFBP5 knock-out mice showed delayed involution after weaning (127). Since the proliferative activity of the breast epithelium is linked to the risk of developing BC in mice (128), the anti-mitogenic activity of IGFBP5 on breast epithelium fits well with the finding that enhancer activities that potentially increase IGFBP5 transcription are associated with lower BC risk (25–27, 129).
The Wnt pathway is also important for survival of basal-like BCs/TNBCs (6), which have much in common with the MMTV-induced BCs (121). The Wnt pathway might be responsible for the low IGFBP5 expression in basal-like BCs/TNBCs compared to that in other BCs (122). When TNBC cell lines MDA-MB-231 and Hs578T were exposed to IGFBP5, cells underwent apoptosis (98, 130, 131). This pro-apoptotic activity of IGFBP5 is likely to be IGF-independent, as both cell lines do not have a functional IGF survival pathway (130).
However, under certain conditions, IGFBP5 can switch from a pro-apoptotic factor to an anti-apoptotic factor. In the presence of pro-apoptotic ceramide, IGFBP5 inhibits apoptosis of Hs578T cells by promoting protein kinase C-dependent conversion of ceramide to anti-apoptotic sphingosine-1-phosphate (73, 132).
Also ERα-positive MCF-7 cells show a link between IGFBP5 and apoptosis, though the data are not conclusive. One study shows that PI3K inhibition, which causes apoptosis in this cell line (133), leads to higher transcription of the IGFBP5 gene causing ERK1/2 activity to rise and the expression of pro-apoptotic Bim to decline to counteract apoptosis (90). In contrast, in our study, MCF-7 cells responded to IGFBP5 knock-down by upregulation of AKT activity and IGF1R expression suggesting a pro-apoptotic activity of IGFBP5 in these cells (134). Yet another study demonstrated that recombinant IGFBP5 protein had little effect on MCF-7 cell survival, unless cells were treated with ceramide, in which case it counteracted ceramide-induced apoptosis (73). The dosage of IGFBP5 (overexpression vs. loss) and potential differences in IGF involvement in these effects may explain the conflicting results. In fact, increasing the dose of recombinant IGFBP5 was shown to switch IGFBP5 from being an anti-apoptotic to a pro-apoptotic agent in the presence of ceramide (73). And, MCF-7 cells have a functional IGF/IGF1R pathway allowing IGF-dependent IGFBP5 effects to come into play in addition to its IGF-independent effects.
Recombinant IGFBP5 protein can increase cell-ECM interactions. This was found with MCF-7 cells and required the C-terminal domain and α2/β1 integrins (95). The effect coincided with reduced cell-cell contacts and lower migratory activity. In non-transformed mouse mammary gland (NMuMG) cells, IGFBP5-induced changes in cell-ECM attachment activity depended upon the cellular phenotype (103). In cells with epithelial phenotype, IGFBP5 enhanced both cell-ECM interaction and migratory activity, while, in mesenchymal cells, it decreased attachment to ECM and had no effect on migration. These results emphasize again the cell context-dependent behavior of IGFBP5.
Role of IGFBP5 in anti-cancer drug response
Evidence has been accumulated that IGFBP5 plays a role in the responsiveness to anti-cancer drugs in BC ( Figure 3 ). Depending on the anti-cancer drug, IGFBP5 either sensitizes or de-sensitizes BC to such drugs.
Figure 3.
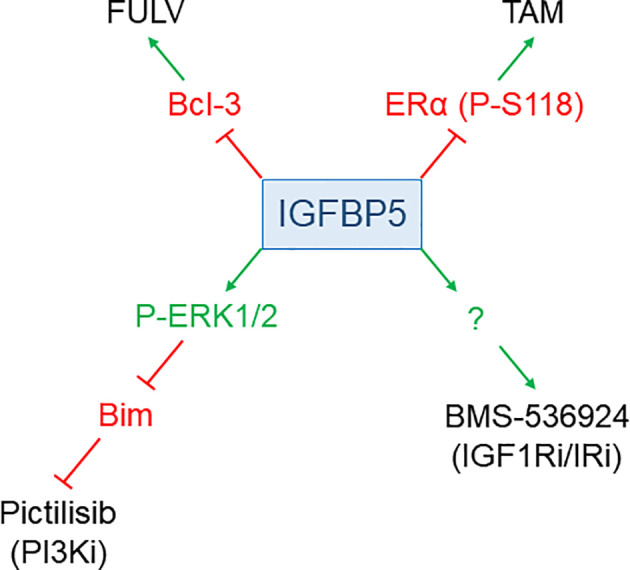
IGFBP5 modulates drug responses of BC cells. IGFBP5 counteracts resistance to the anti-estrogens FULV and TAM by either inhibiting the expression of Bcl-3 or by preventing phosphorylation of ERα at S118, respectively. On the other hand, IGFBP5 de-sensitizes cells to the PI3K inhibitor (i) GDC-0941, involving ERK1/2 activation and downregulation of pro-apoptotic Bim, and to the dual IGF1R/IR inhibitor BMS-536924 through a not yet defined mechanism.
Anti-estrogens
One frequent event leading to endocrine resistance is permanent activation of the PI3K/AKT signaling pathway by highly active RTKs (37). RTKs involved in endocrine resistance include Her family members, FGFR and IGF1R. The role of IGF1R in endocrine resistance is complicated as the IGF1R pathway is linked to ERα activity (see above). As estrogen stimulates IGF1R expression, FULV by inhibiting ERα has the opposite effect (45) and lower levels of IGF1R are found in FULV- and TAM-resistant MCF-7 sublines (135, 136). Nevertheless, the remaining IGF1R protein may still be important for growth of FULV-treated MCF-7 cells (136), as it could be hyperactivated by a FULV-induced rise in IGF1 expression (45) or by upregulation of the IGF2 level and simultaneous downregulation of the IGFBP5 level (134). However, a clinical study on the potential association of IGF1R expression and prognosis did not confirm a critical role of IGF1R in endocrine resistance (137). Nor did an IGF1R-targeting drug (ganitumab) improve patients’ outcome in an endocrine resistance setting (138).
IGFBP5 may play a role in anti-estrogen resistance independent of the IGF/IGF1R pathway. Overexpressed in MCF-7 cells, IGFBP5 was found to inhibit estrogen-dependent phosphorylation of the ERα protein at Ser-118 leading to a reduction in ERα-dependent growth (101). Though the IGF/IGF1R pathway is critically linked to ERα-activity, the inhibitory effects of IGFBP5 on ERα-activity did not rely on the IGF/IGF1R pathway. Importantly, Ser-118 phosphorylation was found to be indicative for TAM resistance in primary BC (139) and can be induced by TAM and FULV treatment (33). Hence, by blocking ERα phosphorylation at Ser-118, IGFBP5 may counteract anti-estrogen resistance.
Another mechanism by which IGFBP5 could inhibit the acquisition of anti-estrogen resistance is by keeping the expression of B-cell leukemia/lymphoma 3 (Bcl-3) at bay (134). This IκB-like protein, able to activate the NFκB survival pathway (140), shows higher expression in TAM-resistant BCs (134, 141). By upregulating Bcl-3 expression, CAFs or mesenchymal stem cells (MSCs), stromal cells known to be involved in drug resistance (108), de-sensitize MCF-7 and BT474 cells to FULV (134). The CAF/MSC-induced increase in the Bcl-3 level, which subsequently leads to FULV tolerance, could be mimicked by IGFBP5 knock-down and did not depend upon the PI3K/AKT pathway suggesting that IGFBP5 controls Bcl-3 expression in an IGF-independent manner.
Knock-down of IGFBP5 in MCF-7 cells was also shown to induce TAM resistance (120). Furthermore, TAM-resistant MCF-7 cells were found to express IGFBP5 at a lower level than parental cells and could be re-sensitized to TAM by exposure to recombinant IGFBP5 protein. Though another study confirmed the lower IGFBP5 level in TAM-resistant MCF-7 cells, it failed to show a causal link between IGFBP5 expression and TAM resistance (142). In sum, there is evidence that IGFBP5 can counteract the acquisition of anti-estrogen resistance in an IGF-independent manner, though more research is needed to confirm this hypothesis.
PI3K and IGF1R inhibitors
Given the important role of the PI3K/AKT pathway in endocrine resistance, PI3K inhibitors are used to prolong survival of breast cancer patients who developed resistant tumors (143). Resistance of MCF-7 cells to the PI3K inhibitor GDC-0941 (Pictilisib) has been shown to be accompanied by increased expression of IGFBP5 (90). The GDC-0941-dependent upregulation of the IGFBP5 level is caused by the conversion of methylated to acetylated Lys-27 of the histone 3 protein resulting in increased IGFBP5 promoter activity, whereby lysine methylase KDM6B is a key enzyme in demethylating Lys-27. Therefore, inhibition of KDM6B or knock-down of IGFBP5 re-sensitized cells to GDC-0941. The protective effect of IGFBP5 was shown to involve the activation of ERK1/2, which, in turn, suppressed the expression of the pro-apoptotic protein Bim. Hence, though IGFBP5 sensitizes BC cells to anti-estrogens, it promotes resistance to PI3K inhibitors applied to fight anti-estrogen resistance. This is another example pointing to the dual nature of IGFBP5 in cancer cell biology.
Also resistance of MCF-7 cells to BMS-536924, a dual inhibitor of IGF1R and insulin receptor, coincides with higher levels of IGFBP5 (104). There is evidence for a causative role of IGFBP5 in the acquisition of this resistance, as its knock-down re-sensitized resistant cells to BMS-536924, while its overexpression in parental cells had the opposite effect. Also, in a panel of 93 cell lines, established from breast, colon and lung cancers, lower IGFBP5 levels were indicative for a strong response to the IGF1R-specific human monoclonal antibody figitumumab (144). Furthermore, in bladder cancer, higher resistance to the selective IGF1R kinase inhibitor AEW541 coincided with higher IGFBP5 levels and lower IGF1R activity (145).
In sum, these data not only suggest that tumorally expressed IGFBP5 may serve as a useful marker to assess responsiveness of cancer cells to IGF1R inhibitors, but also that IGFBP5 is causatively involved in the acquisition of resistance to these drugs.
Regulation of IGFBP5 expression
A number of TFs, non-TFs, non-coding RNAs as well as steroids have been identified to affect IGFBP5 expression ( Figure 4 ). In BC, its regulation by estrogen is of particular importance, as it links IGFBP5 to BC risk.
Figure 4.
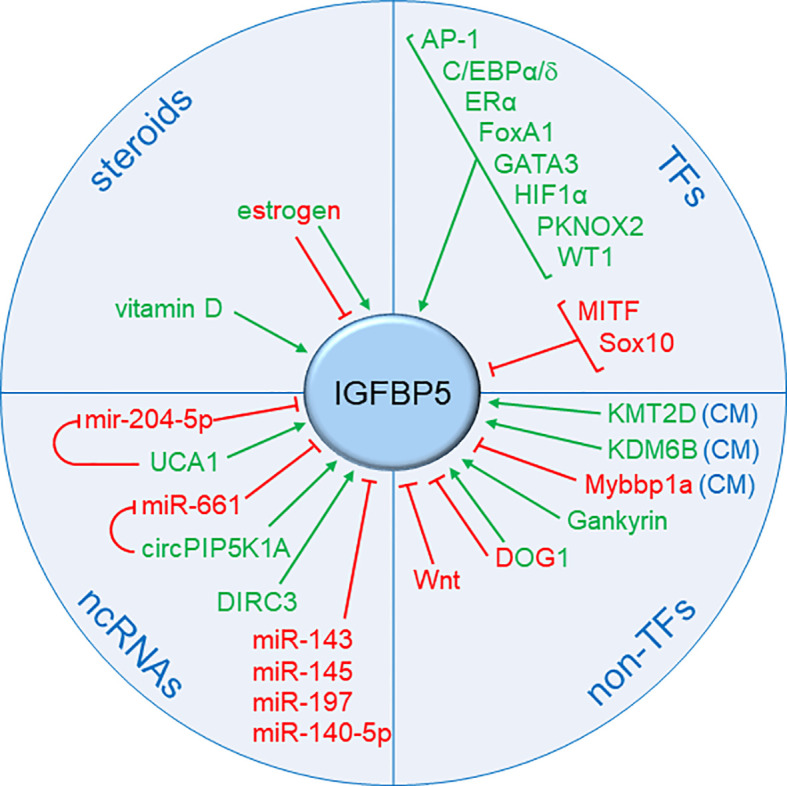
Summary of known factors regulating IGFBP5 expression. Besides proteins that regulate IGFBP5 transcription, namely transcription factors (TFs) (see also Figure 5 ) and chromatin modifiers (CM), other factors have been identified that interfere with IGFBP5 expression. These include other proteins (Wnt, DOG1, Gankyrin), steroids and non-coding RNAs (ncRNAs), namely microRNAs (miRs) and long noncoding RNAs (lncRNAs). Factors that stimulate IGFBP5 expression are denoted in green, those which have a negative effect are shown in red. Factors, which can have either effect dependent on the cellular context, are bicolored.
Transcriptional regulation
The IGFBP5 gene is located on the q-arm of chromosome 2 and is tightly linked to the IGFBP2 gene in a tail-to-tail orientation (146). Besides proximal promoter elements, sequences far upstream (telomeric) and downstream (centromeric) of the transcriptional start site (TSS) regulate IGFBP5 transcription ( Figure 5 ). The far upstream regulatory elements are located in the 2q35 protein-coding gene desert (23–27). This region has been recognized as a BC susceptibility locus, where several single nucleotide polymorphism (SNP) sites and a deletion variant (called enCNV or esv3594306) have been identified, potentially linking IGFBP5 to BC risk (25–27, 129).
Figure 5.

Overview of chromosome 2q regions relevant to the transcriptional regulation of the human IGFBP5 gene. These regions include the proximal IGFBP5 promoter, several enhancer elements in the protein-coding gene desert 2q35 and an HIF1α-binding downstream regulatory element. Numbers above or below the horizontal line indicate the genomic positions or the nucleotide positions relative to the transcriptional start site (+1) of the IGFBP5 gene, respectively. Green ovals indicate stimulatory transcription factors, red ones repressors. The relative position of the gene DIRC3, whose RNA product interferes with MITF and Sox10 activities, and the SNP site rs4442975, which interferes with FoxA1 binding activity, are indicated. In the lower panel, the transcription factor configuration in the deletion variant esv3594306 and its consequences for the estrogen (E2) response, as measured by promoter reporter or by allele-specific expression assays, are shown. PRE, putative regulatory element.
Factors binding to the breast cancer susceptibility locus 2q35
ERα is an important regulator of IGFBP5 transcription in ERα-positive BCs. Three independent studies on primary BCs revealed that ERα-positive BCs express higher IGFBP5 mRNA levels than ERα-negative BCs (119, 147, 148). Additionally, in vitro studies showed that estrogen modulates IGFBP5 expression in ERα-positive BC cell lines, whereby the mode of its action varies between cell lines (99, 147). In T47D and BT474 cells, estrogen upregulates (24, 147), whereas, in MCF-7 cells and in a TAM-resistant MCF-7 subline, it downregulates IGFBP5 expression (99, 149). In the latter cells, anti-estrogens TAM and FULV upregulate IGFBP5 expression instead. Additionally, MCF-7 cells show much lower IGFBP5 expression than T47D and BT474 cells (100) and harbor a monoallelic variant (esv3594306) within the chromosomal region 2q35, which is not found in the other two cell lines (see below). This variant may be responsible for the repressive effect of estrogen on IGFBP5 expression in MCF-7 cells (26). Of note, a repressive effect of estrogen on IGFBP5 was also found with the estrogen-sensitive PE04 ovarian cancer cell line (150).
Genome-wide association studies (GWASs) are used to identify low-penetrance genetic loci which are associated with diseases, such as cancer. Such studies have identified a BC susceptibility locus at 2q35 (151), which is hundreds of kilobase pairs (kbps) away from the IGFBP5 gene (23). Capture Hi-C, a method performed to map genomic contacts, revealed that proteins binding to 2q35 (~217,913 kbp- ~218,262 kbp) interact with proteins binding to the IGFBP5 gene (217,536,828 bp-217,560,272 bp) (23) suggesting a DNA loop being formed between 2q35 and the IGFBP5 gene. Subsequent studies identified several regulatory elements within the 2q35 region that interfere with the transcription of the IGFBP5 gene (23, 24, 26, 27, 152). One regulatory element, located ~345 kbp telomeric to the IGFBP5 promoter, is an enhancer that harbors a binding site for FoxA1 (23, 24). A SNP site (rs4442975, genomic position: 217,920,769 bp) has been identified at an important position of the FoxA1 recognition element. The replacement of the G at this site by a T significantly increases FoxA1 DNA binding. G/T heterozygous BC cell lines show higher IGFBP5 mRNA levels than G/G homozygous ones suggesting that the remote FoxA1-binding enhancer plays a role in IGFBP5 transcription. Studies on normal breast tissue confirmed the link between the T-allele of rs4442975 and higher IGFBP5 expression. Interestingly, estrogen responsiveness is also increased in the G/T heterozygous BT474 cells as compared to G/G homozygous MCF-7 cells. This finding may relate to the strong link between FoxA1 and ERα activities (34). Given this link, it is striking that the T-allele of rs4442975 is associated with a reduced risk of women to develop ERα-positive BC (27, 129). Moreover, the protective effect of the T-allele was stronger, when women were under current treatment with estrogen and progesterone, a therapy that increases the risk of developing ERα-positive BC by approximately two-fold.
In another 2q35 variant (esv3594306), a stretch of ~ 1.4 kbp between 217,955,891 and 217,957,273 is deleted, which has a profound effect on IGFBP5 transcription (26, 27). The deleted sequence is part of the putative regulatory element PRE2, located ~400kbp telomeric to the IGFBP5 gene. On both sites of the deleted sequence, functional binding sites for ERα and FoxA1 as well as for GATA binding protein 3 (GATA3), another ERα-cooperating TF (153), are found (27). Luciferase-based promoter assays showed that, by bringing the two composite enhancer elements in close proximity, the deletion potentiates the PRE2-dependent enhancer activity. Additionally, in an allele-specific chromosomal approach by using PRE2-heterozygous MCF-7 cells, a stronger binding activity of ERα to the deletion variant allele was found as compared to the WT allele (26). Interestingly, in the absence of estrogen, the variant allele was found to be the major driver of ERα-dependent IGFBP5 expression, whereas, in the presence of estrogen, IGFBP5 transcription on the variant allele was suppressed. This may explain the inhibitory effect of estrogen on IGFBP5 expression in MCF-7 cells (99, 149). Of note, in the luciferase-based promoter assay, estrogen did not suppress, but rather promote the activity of the deletion variant enhancer (27) suggesting that the inhibitory effect of estrogen on the variant allele is dependent upon the chromatin structure.
The esv3594306 deletion on one or both alleles is found in ~9% of European Americans and ~27% of African Americans (26). In both groups, carriers of this deletion have a reduced risk of developing BC. This association was stronger for ERα-positive than for ERα-negative BC supporting the notion that the ERα activity is important for the effect of esv3594306 on BC risk. Interestingly, evidence has been provided that this deletion does not affect the expression of IGFBP5’s neighboring genes IGFBP2 and RPL37A (27). This may suggest that changes in IGFBP5 expression is the major, if not only reason for the esv3594306-dependent reduction in BC risk. However, interchromosomal interactions of the 2q35 region have to be considered as well. A major interaction event in trans has been found between 2q35 and chromosome 1q42.12, which includes enable homolog gene (ENAH) (23). ENAH is frequently highly expressed in BC and shows an inverse expression to ERα suggesting that it is regulated by estrogen (154).
Even further upstream of the IGFBP5 gene an additional putatitive enhancer element, PRE3, has been identified (218,305,944- 218,306,443) (27). It contains functional GATA3 and FoxA1 sites and enhances IGFBP5 promoter activity in luciferase-based promoter assays with estrogen amplifying this effect.
Telomeric to PRE2 and including the PRE3 sequence lies a gene that codes for the long non-coding RNA (lncRNA) disrupted in renal carcinoma 3 (DIRC3) (152). DIRC3, which contains 3384 nucleotides, belongs to a subset of nuclear lncRNAs that control transcription of protein-coding genes. One target of DIRC3 is its neighbor IGFBP5. Two DNA looping interactions connect the DIRC3 locus to the IGFBP5 promoter. In melanoma cells, where DIRC3 is highly expressed, DIRC3 increases IGFBP5 transcription by preventing the TFs SRY-Box transcription factor 10 (Sox10) and melanocyte inducing transcription factor (MITF) from inhibiting IGFBP5 transcription. Sox10 is highly expressed in basal-like BCs (155) and might negatively regulate IGFBP5 transcription in this mostly ERα-negative subtype. Interestingly, Sox10 plays an important role in mammary development and a link between Sox10 and Wnt signaling has been suggested (156). It is therefore possible that Sox10 is involved in the inverse relationship between IGFBP5 expression and Wnt activity, as discussed above.
Factors binding to proximal promoter elements
Four functional TF recognition elements have been localized in the proximal human IGFBP5 promoter. These include binding sites for CCAAT-enhancer-binding proteins/C/EBPs), Wilms tumor gene 1 (WT1), activating protein-1 (AP-1) and PBX/Knotted Homeobox 2 (PKNOX2).
A C/EBP binding site has been identified in the proximal IGFBP5 promoter at position -71 relative to the transcriptional start site (TSS) (157, 158). In HNSCC, it allows C/EBPα to upregulate IGFBP5 expression (157). In primary rat osteoblasts, it mediates responsiveness to C/EBPδ and confers reactivity of these cells to prostaglandin E2 (158). C/EBPα is considered to act as a tumor suppressor in a number of solid cancers, including BC (159). In contrast, C/EBPδ shows tumor-promoting activities in BC, as evidenced by its ability to increase BC stem cell activity and to support BC growth and metastasis (160). Interestingly, in BC, expression of C/EBPα is upregulated by ERα and downregulated by HIF1α. On the other hand, C/EBPδ upregulates HIF1α expression in BC cells and, hence, might additionally promote IGFBP5 transcription indirectly through HIF1α (see below). Whether and to which extent C/EBP proteins contribute IGFBP5 expression in BC remains to be seen.
Further upstream of the C/EBP recognition element are several potential binding sites for WT1 between positions -848 and -190 relative to TSS (161). WT is a zinc finger TF, which may act as a tumor suppressor or promoter depending on the type of tumor (162). A short sequence (KTS) between the third and fourth zinc finger is important for its affinity to DNA. The splicing variant WT1(-KTS), which misses this sequence, was found to bind to and activate the IGFBP5 promoter in osteosarcoma cells (161). Interestingly, WT1 acts repressive on the IGF2 P3 promoter (163) suggesting that WT1 interferes with the balance between IGF2 and IGFBP5 expression. In BC, both isoforms, WT1 and WT1(KTS), are expressed and may play a role in ERα and Her2 expression (164).
A binding site for AP-1, hetero- or homodimeric TFs consisting of members of the Jun, Fos, ATF or Maf families (165), has been identified at position -1199 relative to IGFBP5 TSS (166). A SNP at -1195C>T, which reduced binding of AP-1 to the IGFBP5 promoter coincided with lower promoter activity in HNSCC cells. The T-allele was found to be associated with higher tumor grade in non-oropharyngeal HNSCC. In BC, AP-1 is well recognized as being involved in tumor progression (167). Additionally, it can serve as a docking protein for ERα. By tethering to AP-1, ERα is able to drive transcription through AP-1 binding sites (29). The AP-1 binding site within the proximal IGFBP5 promoter might therefore contribute to ERα-dependent IGFBP5 transcription.
Several recognition elements for PKNOX2, a homodomain TF, have been identified between -800 and -1800 relative to IGFBP5 TSS by using chromatin immunoprecipitation (ChiP) assays. PKNOX2 is able to upregulate IGFBP5 transcription in gastric cancer cells (168). Being a pro-apoptotic and anti-mitogenic protein, PKNOX2 acts tumor-suppressive on gastric cancer. There is evidence that IGFBP5 partially mediates this tumor-suppressive effect. Whether PKNOX2 plays a role in IGFBP5 expression in BC is not known.
HIF1α binding to a downstream recognition element
IGFBP5 was found to be a target of hypoxia-inducible factor 1α (HIF1α) (169). Expression of HIF1α is commonly regulated by hypoxia, but, particularly in cancer, can also be modulated by other factors, such as inhibitors of mammalian target of rapamycin (mTOR) (170). This important downstream target of the PI3K/AKT pathway forms two complexes, mTORC1 and mTORC2, of which mTORC1 negatively feeds back on this signaling pathway to limit the pathway’s activity. Part of this feedback mechanism includes secreted IGFBP5 (169). By stimulating the translation of HIF1α mTORC1 increases HIF1α protein abundance. In turn, HIF1α binds to multiple sites at a sequence approximately 3 kbp centromeric to the IGFBP5 promoter and increases its activity. Consequently, more IGFBP5 is secreted which then reduces IGF1R-dependent PI3K/AKT activity. Identified by using tuberous sclerosis complex (TSC)-/- mouse embryonic fibroblasts, the IGFBP5-dependent feed back regulation of the PI3K/AKT pathway was also demonstrated to take place in cancer cells, including MCF-7 cells. Hence, the mTORC1/HIF1α/IGFBP5 axis may play a general role in restricting IGF1R-driven PI3K/AKT activity in (breast) cancer cells.
Chromatin modifiers
Chromatin accessibility is vital to active transcription. Post-transcriptional modifications of histones are key events in regulating chromatin accessibility (171). Certain histone modifications, such as H3K4me1, indicate open chromatin. In melanoma cells, the histone methyltransferase KMT2D, which can methylate Lys-4 in histone 3, has been found to keep proximal and distal IGFBP5-regulating enhancers in an open state, thereby contributing to high IGFBP5 levels (172). Both KMT2D and IGFBP5 act as tumor suppressors in melanoma cells and, in metastatic melanoma, the IGFBP5 level correlates with that of KMT2D. It is possible that KMT2D plays also an important role in ERα-dependent transcription of IGFBP5 in BC, as KMT2D is important for ERα/FoxA1-depending transcription (173).
Myb binding protein 1a (Mybbp1a) is involved in chromatin remodelling (174) and interferes with the function of certain nuclear factors (175). It acts primarily as a tumor suppressor. In Huh-7 human hepatocellular carcinoma (HCC) cells, Mybbp1a promotes the methylation of CpG islands within the IGFBP5 gene by binding to DNA methyltransferase 1 (DNMT1), thereby blocking IGFBP5 transcription (176). Expressed as well in BCs, Mybbp1a might regulate IGFBP5 gene methylation in BCs (177). Additionally, it might interfere with ERα-dependent transcription of IGFBP5. Mybbp1a has been shown to be an activating co-factor of estrogen-related receptor α (ERRα) (178), a TF whose recognition site overlaps with that of ERα (179). Interestingly, BC cells may rise the ERRα to ERα expression ratio in response to the ERα inhibitor TAM (37).
Translational regulation by non-coding RNAs
Noncoding RNAs, including microRNAs (miRs) and lncRNAs, have an impact on IGF/IGF1R pathway activity (reviewed in (180)). MiRs are double-stranded short RNAs that are binding to the 3’-upstream region (UTR) of mRNAs to block their translation or to induce their degradation (181). A subset of lnc-RNAs, non-protein coding transcripts of sizes of more than 200 bases, are able to counteract the actions of miRs (182). Several miRs and counteracting lncRNAs were also found to be able to regulate IGFBP5 translation.
In breast tissues, miR-140-5p may be an important regulator of IGFBP5 translation (183). MiR-140-5p is more highly expressed in breast cancer lesions compared to normal breast tissue.
In papillary thyroid cancer (PTC), mir-204-5p was shown to suppress IGFBP5 expression and to inhibit cellular growth (184). Growth could be restored by overexpression of IGFBP5 or expression of the lncRNA Urothelial Carcinoma Associated 1 (UCA1) that limits the activity of miR-204-5p (185). In BC, miR-204-5p is often downregulated and considered to be a tumor suppressor (186), whereas UCA1 is overpressed and shows cancer-promoting effects (187, 188). Whether these activities in BC are linked to IGFBP5 has not been analyzed yet.
In ovarian cancer, the ratio between miR-661 and the circular lnc-RNA circPIP5K1A plays a role in regulating IGFBP5 translation (189). In the cell lines SKOV3 and A2780, circPIP5K1A and IGFBP5 promote cellular growth. In BC, miR-661 acts tumor-suppressive and higher miR-661 expression correlates with better outcome (190, 191). If miR-661 regulates IGFBP5 expression in BC is not known.
In meningioma cells, miR-197 was found to inhibit the expression of IGFBP5 (192). In BC, miR-197 seems to be upregulated compared to healthy breast tissue (193).
Other miRs that can potentially interfere with IGFBP5 translation are miR-143 and miR-145. In the intestine, these two miRs play a role in regulating IGFBP5 in injury-activated myofibroblasts (113). Like miR-204-5p in thyroid cancer cells (113), miR-143 was found to be counteracted by lncRNA UCA1 in colon cancer cells (194). A significant correlation between miR-143 and miR-145 expression was found in BC (195). Both were found to act as tumor-suppressors.
In sum, there are a number of miRs that potentially could interfere with IGFBP5 expression in BC. Of these, most act as tumor suppressors in BC. Only miR-197 and miR-140-5p might have tumor-promoting functions. It remains to be shown to what extent IGFBP5 contributes to their tumor-suppressing or tumor-promoting activities in BC.
Regulation of IGFBP expression by other factors
The oncoprotein gankyrin is a subunit of the 26S proteasome and contributes to the degradation of proteins, such as p53 and Rb (196). Gankyrin has been shown to upregulate IGFBP5 expression in Huh-7 human hepatocellular carcinoma (HCC) and U-2 OS osteosarcoma cell lines (197). IGFBP5 knock-down in these cells resulted in growth inhibition. Gankyrin is also expressed in BCs, particularly in Her2-positive and TAM-resistant BCs (198, 199).
A calcium-dependent chloride channel protein called Discovered On Gastrointestinal stromal tumors 1 (DOG1) or anoctamin 1 (ANO1) (200) has a strong impact on IGFBP5 expression in gastrointestinal stromal tumor cell lines (201). In BC, DOG1 acts as a growth- and invasion-promoting factor, whose expression is associated with poorer survival (202).
Vitamin D-related compounds, which act through the vitamin D receptor, were also found to regulate IGFBP5 expression. Shown for MCF-7 cells, vitamin D increases the secretion of IGFBP5 and inhibit IGF-1-mediated cellular growth, an effect that was dependent upon the IGFBP binding domain of IGF-1 (203).
Conclusions
On the one hand, higher IGFBP5 expression seems to predict lower risk of developing BC. On the other hand, higher IGFBP5 expression is linked to poorer outcome of BC patients. This apparent contradiction can be explained by the different functions of IGFBP5 in normal breast as compared to BC. In normal breast, it reduces the risk of BC initiation by counteracting mitogenic IGF and inducing apoptosis, while, in BC, it can have multiple functions, depending on BC subtype, the individual cellular context, the tumor microenvironment and on whether it acts IGF-dependent or -independent. The BC subtype is important, because IGF is a mitogen particularly in ERα-positive BCs and because ERα is a major driver of IGFBP5 transcription.
Given that IGFBP5 is a target of ERα, IGFBP5 could be expected to play a more specific role in ERα-positive BCs. However, no clear function has been defined yet even in this BC subtype. Data obtained from clinical studies are inconsistent as to whether IGFBP5 expression in ERα-positive BCs is of any value for predicting patients’ outcome as long as patients with any kind of ERα-positive BCs and treatment are evaluated together. Further specification by molecular taxonomy and/or by treatment may be necessary to define IGFBP5’s role(s) in ERα-positive BCs. So far, such more specific analyses has revealed a link between high IGFBP5 expression and poor survival of patients with luminal A, but not luminal B tumors, and an association between high IGFBP5 levels and more favorable outcome in TAM-treated patients with mostly lobular cancers. Only in the latter case are the clinical data backed up by in vitro experiments showing a sensitizing effect of IGFBP5 to anti-estrogens.
Given the mostly poor survival linked to higher IGFBP5 expression, it is puzzling that, in both ERα-positive and -negative BC cell lines, IGFBP5 shows pro-apoptotic activities. Hence, there must be additional, tumor-promoting IGFBP5 activities, which have yet to be uncovered. For instance, IGFBP5 might play a promoting role in BC cell dissemination and/or metastasis. In this case, IGFBP5 would have a dual effect in BC, suppressive in the initial phase by its pro-apoptotic activity and promoting in the late phase of tumor development by supporting the metastatic activity of the tumor. Such a behaviour would be reminiscent of the dual role of transforming growth factor β, which is known to act as a tumor suppressor in pre-malignant and as a tumor promoter in malignant cells (204).
Overall, it is not clear yet whether and how IGFBP5 is involved in BC progression. IGFBP5 might just be a surrogate marker and, as such, not contribute itself to BC progression. Clearly, more research is necessary to better understand the function of IGFBP5 in BC. Particularly, research focussing on IGFBP5’s role in BC dissemination, dormancy and metastatic outgrowth may advance our knowledge on the mechanism(s) by which IGFBP5 interferes with BC progression.
Author contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Acknowledgments
The author thanks Angela Dittmer for critically reading the manuscript.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- 1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin (2015) 65:87–108. doi: 10.3322/caac.21262 [DOI] [PubMed] [Google Scholar]
- 2. Hüsemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, et al. Systemic spread is an early step in breast cancer. Cancer Cell (2008) 13:58–68. doi: 10.1016/j.ccr.2007.12.003 [DOI] [PubMed] [Google Scholar]
- 3. Dittmer J. Mechanisms governing metastatic dormancy in breast cancer. Semin Cancer Biol (2017) 44:72–82. doi: 10.1016/j.semcancer.2017.03.006 [DOI] [PubMed] [Google Scholar]
- 4. Risson E, Nobre AR, Maguer-Satta V, Aguirre-Ghiso JA. The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nat Cancer (2020) 1:672–80. doi: 10.1038/s43018-020-0088-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peitzsch C, Tyutyunnykova A, Pantel K, Dubrovska A. Cancer stem cells: The root of tumor recurrence and metastases. Semin Cancer Biol (2017) 44:10–24. doi: 10.1016/j.semcancer.2017.02.011 [DOI] [PubMed] [Google Scholar]
- 6. Dittmer J. Breast cancer stem cells: Features, key drivers and treatment options. Semin Cancer Biol (2018) 53:59–74. doi: 10.1016/j.semcancer.2018.07.007 [DOI] [PubMed] [Google Scholar]
- 7. Brooks MD, Burness ML, Wicha MS. Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell (2015) 17:260–71. doi: 10.1016/j.stem.2015.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lambert AW, Weinberg RA. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer (2021) 21:325–38. doi: 10.1038/s41568-021-00332-6 [DOI] [PubMed] [Google Scholar]
- 9. Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer (2009) 9:576–86. doi: 10.1038/nrc2683 [DOI] [PubMed] [Google Scholar]
- 10. Rocca A, Maltoni R, Bravaccini S, Donati C, Andreis D. Clinical utility of fulvestrant in the treatment of breast cancer: a report on the emerging clinical evidence. Cancer Manag Res (2018) 10:3083–99. doi: 10.2147/CMAR.S137772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dixon JM. Prospects of neoadjuvant aromatase inhibitor therapy in breast cancer. Expert Rev Anticancer Ther (2008) 8:453–63. doi: 10.1586/14737140.8.3.453 [DOI] [PubMed] [Google Scholar]
- 12. Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene (2007) 26:6469–87. doi: 10.1038/sj.onc.1210477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mathew A, Romond EH. Systemic therapy for HER2-positive early-stage breast cancer. Curr Probl Cancer (2016) 40:106–16. doi: 10.1016/j.currproblcancer.2016.09.002 [DOI] [PubMed] [Google Scholar]
- 14. Bou Zerdan M, Ghorayeb T, Saliba F, Allam S, Bou Zerdan M, Yaghi M, et al. Triple negative breast cancer: Updates on classification and treatment in 2021. Cancers (Basel) (2022) 14:1253. doi: 10.3390/cancers14051253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol (2011) 5:5–23. doi: 10.1016/j.molonc.2010.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harbeck N, Gnant M. Breast cancer. Lancet (2017) 389:1134–50. doi: 10.1016/s0140-6736(16)31891-8 [DOI] [PubMed] [Google Scholar]
- 17. Kim H, Abd Elmageed ZY, Ju J, Naura AS, Abdel-Mageed AB, Varughese S, et al. PDZK1 is a novel factor in breast cancer that is indirectly regulated by estrogen through IGF-1R and promotes estrogen-mediated growth. Mol Med (2013) 19:253–62. doi: 10.2119/molmed.2011.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Song RX, Chen Y, Zhang Z, Bao Y, Yue W, Wang JP, et al. Estrogen utilization of IGF-1-R and EGF-r to signal in breast cancer cells. J Steroid Biochem Mol Biol (2010) 118:219–30. doi: 10.1016/j.jsbmb.2009.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Farabaugh SM, Boone DN, Lee AV. Role of IGF1R in breast cancer subtypes, stemness, and lineage differentiation. Front Endocrinol (Lausanne) (2015) 6:59. doi: 10.3389/fendo.2015.00059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Clemmons DR. Role of insulin-like growth factor binding proteins in controlling IGF actions. Mol Cell Endocrinol (1998) 140:19–24. doi: 10.1016/s0303-7207(1098)00024-00020 [DOI] [PubMed] [Google Scholar]
- 21. Kalus W, Zweckstetter M, Renner C, Sanchez Y, Georgescu J, Grol M, et al. Structure of the IGF-binding domain of the insulin-like growth factor-binding protein-5 (IGFBP-5): implications for IGF and IGF-I receptor interactions. EMBO J (1998) 17:6558–72. doi: 10.1093/emboj/6517.6522.6558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baxter RC. IGF binding proteins in cancer: mechanistic and clinical insights. Nat Rev Cancer (2014) 14:329–41. doi: 10.1038/nrc3720 [DOI] [PubMed] [Google Scholar]
- 23. Dryden NH, Broome LR, Dudbridge F, Johnson N, Orr N, Schoenfelder S, et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by capture Hi-c. Genome Res (2014) 24:1854–68. doi: 10.1101/gr.175034.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ghoussaini M, Edwards SL, Michailidou K, Nord S, Cowper-Sal Lari R, Desai K, et al. Evidence that breast cancer risk at the 2q35 locus is mediated through IGFBP5 regulation. Nat Commun (2014) 4:4999. doi: 10.1038/ncomms5999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garner CP, Ding YC, John EM, Ingles SA, Olopade OI, Huo D, et al. Genetic variation in IGFBP2 and IGFBP5 is associated with breast cancer in populations of African descent. Hum Genet (2008) 123:247–55. doi: 10.1007/s00439-008-0468-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wyszynski A, Hong CC, Lam K, Michailidou K, Lytle C, Yao S, et al. An intergenic risk locus containing an enhancer deletion in 2q35 modulates breast cancer risk by deregulating IGFBP5 expression. Hum Mol Genet (2016) 25:3863–76. doi: 10.1093/hmg/ddw223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baxter JS, Johnson N, Tomczyk K, Gillespie A, Maguire S, Brough R, et al. Functional annotation of the 2q35 breast cancer risk locus implicates a structural variant in influencing activity of a long-range enhancer element. Am J Hum Genet (2021) 108:1190–203. doi: 10.1016/j.ajhg.2021.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fox EM, Davis RJ, Shupnik MA. ERbeta in breast cancer–onlooker, passive player, or active protector? Steroids (2008) 73:1039–51. doi: 10.1016/j.steroids.2008.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Johnson AB, O'Malley BW. Steroid receptor coactivators 1, 2, and 3: critical regulators of nuclear receptor activity and steroid receptor modulator (SRM)-based cancer therapy. Mol Cell Endocrinol (2012) 348:430–9. doi: 10.1016/j.mce.2011.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gibson DA, Saunders PT. Estrogen dependent signaling in reproductive tissues - a role for estrogen receptors and estrogen related receptors. Mol Cell Endocrinol (2012) 348:361–72. doi: 10.1016/j.mce.2011.09.026 [DOI] [PubMed] [Google Scholar]
- 31. Gu F, Hsu HK, Hsu PY, Wu J, Ma Y, Parvin J, et al. Inference of hierarchical regulatory network of estrogen-dependent breast cancer through ChIP-based data. BMC Syst Biol (2010) 4:170. doi: 10.1186/1752-0509-4-170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou Y, Gerrard DL, Wang J, Li T, Yang Y, Fritz AJ, et al. Temporal dynamic reorganization of 3D chromatin architecture in hormone-induced breast cancer and endocrine resistance. Nat Commun (2019) 10:1522. doi: 10.1038/s41467-019-09320-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Le Romancer M, Poulard C, Cohen P, Sentis S, Renoir JM, Corbo L. Cracking the estrogen receptor's posttranslational code in breast tumors. Endocr Rev (2011) 32:597–622. doi: 10.1210/er.2010-0016 [DOI] [PubMed] [Google Scholar]
- 34. Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet (2011) 43:27–33. doi: 10.1038/ng.730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fox EM, Arteaga CL, Miller TW. Abrogating endocrine resistance by targeting ERalpha and PI3K in breast cancer. Front Oncol (2012) 2:145. doi: 10.3389/fonc.2012.00145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nass N, Kalinski T. Tamoxifen resistance: from cell culture experiments towards novel biomarkers. Pathol Res Pract (2015) 211:189–97. doi: 10.1016/j.prp.2015.01.004 [DOI] [PubMed] [Google Scholar]
- 37. Dittmer J. Nuclear mechanisms involved in endocrine resistance. Front Oncol (2021) 11:736597. doi: 10.3389/fonc.2021.736597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hawsawi Y, El-Gendy R, Twelves C, Speirs V, Beattie J. Insulin-like growth factor - oestradiol crosstalk and mammary gland tumourigenesis. Biochim Biophys Acta (2013) 1836:345–53. doi: 10.1016/j.bbcan.2013.10.005 [DOI] [PubMed] [Google Scholar]
- 39. Ekyalongo RC, Yee D. Revisiting the IGF-1R as a breast cancer target. NPJ Precis Oncol (2017) 1:14. doi: 10.1038/s41698-017-0017-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jin Q, Esteva FJ. Cross-talk between the ErbB/HER family and the type I insulin-like growth factor receptor signaling pathway in breast cancer. J Mammary Gland Biol Neoplasia (2008) 13:485–98. doi: 10.1007/s10911-008-9107-3 [DOI] [PubMed] [Google Scholar]
- 41. Li H, Batth IS, Qu X, Xu L, Song N, Wang R, et al. IGF-IR signaling in epithelial to mesenchymal transition and targeting IGF-IR therapy: overview and new insights. Mol Cancer (2017) 16:6. doi: 10.1186/s12943-016-0576-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gaben AM, Sabbah M, Redeuilh G, Bedin M, Mester J. Ligand-free estrogen receptor activity complements IGF1R to induce the proliferation of the MCF-7 breast cancer cells. BMC Cancer (2012) 12:291. doi: 10.1186/1471-2407-12-291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Becker MA, Ibrahim YH, Cui X, Lee AV, Yee D. The IGF pathway regulates ERalpha through a S6K1-dependent mechanism in breast cancer cells. Mol Endocrinol (2011) 25:516–28. doi: 10.1210/me.2010-0373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maor S, Mayer D, Yarden RI, Lee AV, Sarfstein R, Werner H, et al. Estrogen receptor regulates insulin-like growth factor-I receptor gene expression in breast tumor cells: involvement of transcription factor Sp1. J Endocrinol (2006) 191:605–12. doi: 10.1677/joe.1.07016 [DOI] [PubMed] [Google Scholar]
- 45. Iida M, Tsuboi K, Niwa T, Ishida T, Hayashi SI. Compensatory role of insulin-like growth factor 1 receptor in estrogen receptor signaling pathway and possible therapeutic target for hormone therapy-resistant breast cancer. Breast Cancer (2019) 26:272–81. doi: 10.1007/s12282-018-0922-0 [DOI] [PubMed] [Google Scholar]
- 46. Morelli C, Garofalo C, Bartucci M, Surmacz E. Estrogen receptor-alpha regulates the degradation of insulin receptor substrates 1 and 2 in breast cancer cells. Oncogene (2003) 22:4007–16. doi: 10.1038/sj.onc.1206436 [DOI] [PubMed] [Google Scholar]
- 47. Luo J, Liu D. Does GPER really function as a G protein-coupled estrogen receptor in vivo ? Front Endocrinol (Lausanne) (2020) 11:148. doi: 10.3389/fendo.2020.00148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ignatov A, Ignatov T, Roessner A, Costa SD, Kalinski T. Role of GPR30 in the mechanisms of tamoxifen resistance in breast cancer MCF-7 cells. Breast Cancer Res Treat (2010) 123:87–96. doi: 10.1007/s10549-009-0624-6 [DOI] [PubMed] [Google Scholar]
- 49. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science (2005) 307:1625–30. doi: 10.1126/science.1106943 [DOI] [PubMed] [Google Scholar]
- 50. Gonzalez de Valdivia E, Broselid S, Kahn R, Olde B, Leeb-Lundberg LMF. G Protein-coupled estrogen receptor 1 (GPER1)/GPR30 increases ERK1/2 activity through PDZ motif-dependent and -independent mechanisms. J Biol Chem (2017) 292:9932–43. doi: 10.1074/jbc.M116.765875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. De Marco P, Bartella V, Vivacqua A, Lappano R, Santolla MF, Morcavallo A, et al. Insulin-like growth factor-I regulates GPER expression and function in cancer cells. Oncogene (2012) 32:678–88. doi: 10.1038/onc.2012.97 [DOI] [PubMed] [Google Scholar]
- 52. De Marco P, Romeo E, Vivacqua A, Malaguarnera R, Abonante S, Romeo F, et al. GPER1 is regulated by insulin in cancer cells and cancer-associated fibroblasts. Endocr Relat Cancer (2014) 21:739–53. doi: 10.1530/ERC-14-0245 [DOI] [PubMed] [Google Scholar]
- 53. Yerushalmi R, Gelmon KA, Leung S, Gao D, Cheang M, Pollak M, et al. Insulin-like growth factor receptor (IGF-1R) in breast cancer subtypes. Breast Cancer Res Treat (2012) 132:131–42. doi: 10.1007/s10549-011-1529-8 [DOI] [PubMed] [Google Scholar]
- 54. Christopoulos PF, Corthay A, Koutsilieris M. Aiming for the insulin-like growth factor-1 system in breast cancer therapeutics. Cancer Treat Rev (2018) 63:79–95. doi: 10.1016/j.ctrv.2017.11.010 [DOI] [PubMed] [Google Scholar]
- 55. Nili M, Mukherjee A, Shinde U, David L, Rotwein P. Defining the disulfide bonds of insulin-like growth factor-binding protein-5 by tandem mass spectrometry with electron transfer dissociation and collision-induced dissociation. J Biol Chem (2012) 287:1510–9. doi: 10.1074/jbc.M111.285528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shimasaki S, Shimonaka M, Zhang HP, Ling N. Identification of five different insulin-like growth factor binding proteins (IGFBPs) from adult rat serum and molecular cloning of a novel IGFBP-5 in rat and human. J Biol Chem (1991) 266:10646–53. [PubMed] [Google Scholar]
- 57. Kiefer MC, Masiarz FR, Bauer DM, Zapf J. Identification and molecular cloning of two new 30-kDa insulin-like growth factor binding proteins isolated from adult human serum. J Biol Chem (1991) 266:9043–9. [PubMed] [Google Scholar]
- 58. McGuire SE, Hilsenbeck SG, Figueroa JA, Jackson JG, Yee D. Detection of insulin-like growth factor binding proteins (IGFBPs) by ligand blotting in breast cancer tissues. Cancer Lett (1994) 77:25–32. doi: 10.1016/0304-3835(1094)90343-90343 [DOI] [PubMed] [Google Scholar]
- 59. Tripathi G, Salih DA, Drozd AC, Cosgrove RA, Cobb LJ, Pell JM. IGF-independent effects of insulin-like growth factor binding protein-5 (Igfbp5) in vivo . FASEB J (2009) 23:2616–26. doi: 10.1096/fj.08-114124 [DOI] [PubMed] [Google Scholar]
- 60. Zesławski W, Beisel HG, Kamionka M, Kalus W, Engh RA, Huber R, et al. The interaction of insulin-like growth factor-I with the n-terminal domain of IGFBP-5. EMBO J (2001) 20:3638–44. doi: 10.1093/emboj/3620.3614.3638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Luther GA, Lamplot J, Chen X, Rames R, Wagner ER, Liu X, et al. IGFBP5 domains exert distinct inhibitory effects on the tumorigenicity and metastasis of human osteosarcoma. Cancer Lett (2013) 336:222–30. doi: 10.1016/j.canlet.2013.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Coverley JA, Baxter RC. Phosphorylation of insulin-like growth factor binding proteins. Mol Cell Endocrinol (1997) 128:1–5. doi: 10.1016/s0303-7207(97)04032-x [DOI] [PubMed] [Google Scholar]
- 63. Mukherjee A, Rotwein P. Insulin-like growth factor binding protein-5 in osteogenesis: facilitator or inhibitor? Growth Horm IGF Res (2007) 17:179–85. doi: 10.1016/j.ghir.2007.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Maeda H, Yonou H, Yano K, Ishii G, Saito S, Ochiai A. Prostate-specific antigen enhances bioavailability of insulin-like growth factor by degrading insulin-like growth factor binding protein 5. Biochem Biophys Res Commun (2009) 381:311–6. doi: 10.1016/j.bbrc.2009.01.096 [DOI] [PubMed] [Google Scholar]
- 65. Zheng B, Clarke JB, Busby WH, Duan C, Clemmons DR. Insulin-like growth factor-binding protein-5 is cleaved by physiological concentrations of thrombin. Endocrinology (1998) 139:1708–14. doi: 10.1210/endo.1139.1704.5945 [DOI] [PubMed] [Google Scholar]
- 66. Overgaard MT, Boldt HB, Laursen LS, Sottrup-Jensen L, Conover CA, Oxvig C. Pregnancy-associated plasma protein-A2 (PAPP-A2), a novel insulin-like growth factor-binding protein-5 proteinase. J Biol Chem (2001) 276:21849–53. doi: 10.21074/jbc.M102191200 [DOI] [PubMed] [Google Scholar]
- 67. Imai Y, Moralez A, Andag U, Clarke JB, Busby WH, Jr., Clemmons DR. Substitutions for hydrophobic amino acids in the n-terminal domains of IGFBP-3 and -5 markedly reduce IGF-I binding and alter their biologic actions. J Biol Chem (2000) 275:18188–94. doi: 10.11074/jbc.M000070200 [DOI] [PubMed] [Google Scholar]
- 68. Andress DL, Birnbaum RS. Human osteoblast-derived insulin-like growth factor (IGF) binding protein-5 stimulates osteoblast mitogenesis and potentiates IGF action. J Biol Chem (1992) 267:22467–72. [PubMed] [Google Scholar]
- 69. Andress DL, Loop SM, Zapf J, Kiefer MC. Carboxy-truncated insulin-like growth factor binding protein-5 stimulates mitogenesis in osteoblast-like cells. Biochem Biophys Res Commun (1993) 195:25–30. doi: 10.1006/bbrc.1993.2004 [DOI] [PubMed] [Google Scholar]
- 70. Shand JH, Beattie J, Song H, Phillips K, Kelly SM, Flint DJ, et al. Specific amino acid substitutions determine the differential contribution of the n- and c-terminal domains of insulin-like growth factor (IGF)-binding protein-5 in binding IGF-I. J Biol Chem (2003) 278:17859–66. doi: 10.1074/jbc.M300526200 [DOI] [PubMed] [Google Scholar]
- 71. Allan GJ, Tonner E, Szymanowska M, Shand JH, Kelly SM, Phillips K, et al. Cumulative mutagenesis of the basic residues in the 201-218 region of insulin-like growth factor (IGF)-binding protein-5 results in progressive loss of both IGF-I binding and inhibition of IGF-I biological action. Endocrinology (2006) 147:338–49. doi: 10.1210/en.2005-0582 [DOI] [PubMed] [Google Scholar]
- 72. Duan C, Allard JB. Insulin-like growth factor binding protein-5 in physiology and disease. Front Endocrinol (Lausanne) (2020) 11:100. doi: 10.3389/fendo.2020.00100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Perks CM, McCaig C, Clarke JB, Clemmons DR, Holly JM. Effects of a non-IGF binding mutant of IGFBP-5 on cell death in human breast cancer cells. Biochem Biophys Res Commun (2002) 294:995–1000. doi: 10.1016/S0006-1291X(1002)00570-00573 [DOI] [PubMed] [Google Scholar]
- 74. Bushman TL, Kuemmerle JF. IGFBP-3 and IGFBP-5 production by human intestinal muscle: reciprocal regulation by endogenous TGF-beta1. Am J Physiol (1998) 275:G1282–1290. doi: 10.1152/ajpgi.1998.1275.1286.G1282 [DOI] [PubMed] [Google Scholar]
- 75. Xu C, Graf LF, Fazli L, Coleman IM, Mauldin DE, Li D, et al. Regulation of global gene expression in the bone marrow microenvironment by androgen: androgen ablation increases insulin-like growth factor binding protein-5 expression. Prostate (2007) 67:1621–9. doi: 10.1002/pros.20655 [DOI] [PubMed] [Google Scholar]
- 76. Thomas LN, Wright AS, Lazier CB, Cohen P, Rittmaster RS. Prostatic involution in men taking finasteride is associated with elevated levels of insulin-like growth factor-binding proteins (IGFBPs)-2, -4, and -5. Prostate (2000) 42:203–10. doi: [DOI] [PubMed] [Google Scholar]
- 77. Miyake H, Pollak M, Gleave ME. Castration-induced up-regulation of insulin-like growth factor binding protein-5 potentiates insulin-like growth factor-I activity and accelerates progression to androgen independence in prostate cancer models. Cancer Res (2000) 60:3058–64. [PubMed] [Google Scholar]
- 78. Miyake H, Nelson C, Rennie PS, Gleave ME. Overexpression of insulin-like growth factor binding protein-5 helps accelerate progression to androgen-independence in the human prostate LNCaP tumor model through activation of phosphatidylinositol 3'-kinase pathway. Endocrinology (2000) 141:2257–65. doi: 10.1210/endo.2141.2256.7520 [DOI] [PubMed] [Google Scholar]
- 79. Kuemmerle JF, Zhou H. Insulin-like growth factor-binding protein-5 (IGFBP-5) stimulates growth and IGF-I secretion in human intestinal smooth muscle by ras-dependent activation of p38 MAP kinase and Erk1/2 pathways. J Biol Chem (2002) 277:20563–71. doi: 10.21074/jbc.M200885200 [DOI] [PubMed] [Google Scholar]
- 80. Abrass CK, Berfield AK, Andress DL. Heparin binding domain of insulin-like growth factor binding protein-5 stimulates mesangial cell migration. Am J Physiol (1997) 273:F899–906. doi: 10.1152/ajprenal.1997.1273.1156.F1899 [DOI] [PubMed] [Google Scholar]
- 81. Mohan S, Nakao Y, Honda Y, Landale E, Leser U, Dony C, et al. Studies on the mechanisms by which insulin-like growth factor (IGF) binding protein-4 (IGFBP-4) and IGFBP-5 modulate IGF actions in bone cells. J Biol Chem (1995) 270:20424–31. doi: 10.21074/jbc.20270.20435.20424 [DOI] [PubMed] [Google Scholar]
- 82. Berfield AK, Andress DL, Abrass CK. IGFBP-5(201-218) stimulates Cdc42GAP aggregation and filopodia formationin migrating mesangial cells. Kidney Int (2000) 57:1991–2003. doi: 10.1046/j.1523-1755.2000.00049.x [DOI] [PubMed] [Google Scholar]
- 83. Schedlich LJ, Young TF, Firth SM, Baxter RC. Insulin-like growth factor-binding protein (IGFBP)-3 and IGFBP-5 share a common nuclear transport pathway in T47D human breast carcinoma cells. J Biol Chem (1998) 273:18347–52. doi: 10.11074/jbc.18273.18329.18347 [DOI] [PubMed] [Google Scholar]
- 84. Schedlich LJ, Le Page SL, Firth SM, Briggs LJ, Jans DA, Baxter RC. Nuclear import of insulin-like growth factor-binding protein-3 and -5 is mediated by the importin beta subunit. J Biol Chem (2000) 275:23462–70. doi: 10.21074/jbc.M002208200 [DOI] [PubMed] [Google Scholar]
- 85. Akkiprik M, Hu L, Sahin A, Hao X, Zhang W. The subcellular localization of IGFBP5 affects its cell growth and migration functions in breast cancer. BMC Cancer (2009) 9:103. doi: 10.1186/1471-2407-9-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Su Y, Nishimoto T, Feghali-Bostwick C. IGFBP-5 promotes fibrosis independently of its translocation to the nucleus and its interaction with nucleolin and IGF. PLoS One (2015) 10:e0130546. doi: 10.1371/journal.pone.0130546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Amaar YG, Baylink DJ, Mohan S. Ras-association domain family 1 protein, RASSF1C, is an IGFBP-5 binding partner and a potential regulator of osteoblast cell proliferation. J Bone Miner Res (2005) 20:1430–9. doi: 10.1359/JBMR.050311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Malpeli G, Innamorati G, Decimo I, Bencivenga M, Nwabo Kamdje AH, Perris R, et al. Methylation dynamics of RASSF1A and its impact on cancer. Cancers (Basel) (2019) 11:959. doi: 10.3390/cancers11070959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tognoli ML, Vlahov N, Steenbeek S, Grawenda AM, Eyres M, Cano-Rodriguez D, et al. RASSF1C oncogene elicits amoeboid invasion, cancer stemness, and extracellular vesicle release via a SRC/Rho axis. EMBO J (2021) 40:e107680. doi: 10.15252/embj.2021107680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wang W, Lim KG, Feng M, Bao Y, Lee PL, Cai Y, et al. KDM6B counteracts EZH2-mediated suppression of IGFBP5 to confer resistance to PI3K/AKT inhibitor treatment in breast cancer. Mol Cancer Ther (2018) 17:1973–83. doi: 10.1158/1535-7163.MCT-17-0802 [DOI] [PubMed] [Google Scholar]
- 91. Reeves ME, Firek M, Chen ST, Amaar YG. Evidence that RASSF1C stimulation of lung cancer cell proliferation depends on IGFBP-5 and PIWIL1 expression levels. PLoS One (2014) 9:e101679. doi: 10.1371/journal.pone.0101679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Schedlich LJ, Muthukaruppan A, O'Han MK, Baxter RC. Insulin-like growth factor binding protein-5 interacts with the vitamin d receptor and modulates the vitamin d response in osteoblasts. Mol Endocrinol (2007) 21:2378–90. doi: 10.1210/me.2006-0558 [DOI] [PubMed] [Google Scholar]
- 93. Amaar YG, Thompson GR, Linkhart TA, Chen ST, Baylink DJ, Mohan S. Insulin-like growth factor-binding protein 5 (IGFBP-5) interacts with a four and a half LIM protein 2 (FHL2). J Biol Chem (2002) 277:12053–60. doi: 10.1074/jbc.M110872200 [DOI] [PubMed] [Google Scholar]
- 94. Hwang JR, Huh JH, Lee Y, Lee SI, Rho SB, Lee JH. Insulin-like growth factor-binding protein-5 (IGFBP-5) inhibits TNF-alpha-induced NF-kappaB activity by binding to TNFR1. Biochem Biophys Res Commun (2011) 405:545–51. doi: 10.1016/j.bbrc.2011.01.064 [DOI] [PubMed] [Google Scholar]
- 95. Sureshbabu A, Okajima H, Yamanaka D, Tonner E, Shastri S, Maycock J, et al. IGFBP5 induces cell adhesion, increases cell survival and inhibits cell migration in MCF-7 human breast cancer cells. J Cell Sci (2012) 125:1693–705. doi: 10.1242/jcs.092882 [DOI] [PubMed] [Google Scholar]
- 96. Andress DL. Insulin-like growth factor-binding protein-5 (IGFBP-5) stimulates phosphorylation of the IGFBP-5 receptor. Am J Physiol (1998) 274:E744–750. doi: 10.1152/ajpendo.1998.1274.1154.E1744 [DOI] [PubMed] [Google Scholar]
- 97. Meadows KA, Holly JM, Stewart CE. Tumor necrosis factor-alpha-induced apoptosis is associated with suppression of insulin-like growth factor binding protein-5 secretion in differentiating murine skeletal myoblasts. J Cell Physiol (2000) 183:330–7. doi: [DOI] [PubMed] [Google Scholar]
- 98. Butt AJ, Dickson KA, Jambazov S, Baxter RC. Enhancement of tumor necrosis factor-alpha-induced growth inhibition by insulin-like growth factor-binding protein-5 (IGFBP-5), but not IGFBP-3 in human breast cancer cells. Endocrinology (2005) 146:3113–22. doi: 10.1210/en.2004-1408 [DOI] [PubMed] [Google Scholar]
- 99. Huynh H, Yang XF, Pollak M. A role for insulin-like growth factor binding protein 5 in the antiproliferative action of the antiestrogen ICI 182780. Cell Growth Differ (1996) 7:1501–6. [PubMed] [Google Scholar]
- 100. Sheikh MS, Shao ZM, Clemmons DR, LeRoith D, Roberts CT, Jr., Fontana JA. Identification of the insulin-like growth factor binding proteins 5 and 6 (IGFBP-5 and 6) in human breast cancer cells. Biochem Biophys Res Commun (1992) 183:1003–10. doi: 10.1016/s0006-1291x(1005)80290-80296 [DOI] [PubMed] [Google Scholar]
- 101. Hermani A, Shukla A, Medunjanin S, Werner H, Mayer D. Insulin-like growth factor binding protein-4 and -5 modulate ligand-dependent estrogen receptor-alpha activation in breast cancer cells in an IGF-independent manner. Cell Signal (2013) 25:1395–402. doi: 10.1016/j.cellsig.2013.02.018 [DOI] [PubMed] [Google Scholar]
- 102. Wang H, Arun BK, Wang H, Fuller GN, Zhang W, Middleton LP, et al. IGFBP2 and IGFBP5 overexpression correlates with the lymph node metastasis in T1 breast carcinomas. Breast J (2008) 14:261–7. doi: 10.1111/j.1524-4741.2008.00572.x [DOI] [PubMed] [Google Scholar]
- 103. Vijayan A, Guha D, Ameer F, Kaziri I, Mooney CC, Bennett L, et al. IGFBP-5 enhances epithelial cell adhesion and protects epithelial cells from TGFbeta1-induced mesenchymal invasion. Int J Biochem Cell Biol (2013) 45:2774–85. doi: 10.1016/j.biocel.2013.10.001 [DOI] [PubMed] [Google Scholar]
- 104. Becker MA, Hou X, Harrington SC, Weroha SJ, Gonzalez SE, Jacob KA, et al. IGFBP ratio confers resistance to IGF targeting and correlates with increased invasion and poor outcome in breast tumors. Clin Cancer Res (2012) 18:1808–17. doi: 10.1158/1078-0432.CCR-11-1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wu CC, Chang SC, Zeng GY, Chu HW, Huang Y, Liu HP. Proteome analyses reveal positive association of COL2A1, MPO, TYMS, and IGFBP5 with canine mammary gland malignancy. Proteomics Clin Appl (2019) 13:e1800151. doi: 10.1002/prca.201800151 [DOI] [PubMed] [Google Scholar]
- 106. Li X, Cao X, Li X, Zhang W, Feng Y. Expression level of insulin-like growth factor binding protein 5 mRNA is a prognostic factor for breast cancer. Cancer Sci (2007) 98:1592–6. doi: 10.1111/j.1349-7006.2007.00565.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer (2016) 16:582–98. doi: 10.1038/nrc.2016.73 [DOI] [PubMed] [Google Scholar]
- 108. Dittmer J, Leyh B. The impact of tumor stroma on drug response in breast cancer. Semin Cancer Biol (2015) 31:3–15. doi: 10.1016/j.semcancer.2014.05.006 [DOI] [PubMed] [Google Scholar]
- 109. Nguyen XX, Muhammad L, Nietert PJ, Feghali-Bostwick C. IGFBP-5 promotes fibrosis via increasing its own expression and that of other pro-fibrotic mediators. Front Endocrinol (Lausanne) (2018) 9:601. doi: 10.3389/fendo.2018.00601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Arai T, Arai A, Busby WH, Jr., Clemmons DR. Glycosaminoglycans inhibit degradation of insulin-like growth factor-binding protein-5. Endocrinology (1994) 135:2358–63. doi: 10.1210/endo.2135.2356.7527332 [DOI] [PubMed] [Google Scholar]
- 111. Jones JI, Gockerman A, Busby WH, Jr., Camacho-Hubner C, Clemmons DR. Extracellular matrix contains insulin-like growth factor binding protein-5: potentiation of the effects of IGF-I. J Cell Biol (1993) 121:679–87. doi: 10.1083/jcb.1121.1083.1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Akkiprik M, Peker I, Ozmen T, Amuran GG, Gulluoglu BM, Kaya H, et al. Identification of differentially expressed IGFBP5-related genes in breast cancer tumor tissues using cDNA microarray experiments. Genes (Basel) (2015) 6:1201–14. doi: 10.3390/genes6041201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Chivukula RR, Shi G, Acharya A, Mills EW, Zeitels LR, Anandam JL, et al. An essential mesenchymal function for miR-143/145 in intestinal epithelial regeneration. Cell (2014) 157:1104–16. doi: 10.1016/j.cell.2014.03.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kojima H, Kunimoto H, Inoue T, Nakajima K. The STAT3-IGFBP5 axis is critical for IL-6/gp130-induced premature senescence in human fibroblasts. Cell Cycle (2012) 11:730–9. doi: 10.4161/cc.11.4.19172 [DOI] [PubMed] [Google Scholar]
- 115. Li X, Zhou Q, Wang S, Wang P, Li J, Xie Z, et al. Prolonged treatment with y-27632 promotes the senescence of primary human dermal fibroblasts by increasing the expression of IGFBP-5 and transforming them into a CAF-like phenotype. Aging (Albany NY) (2020) 12:16621–46. doi: 10.18632/aging.103910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Gregory CW, Kim D, Ye P, D'Ercole AJ, Pretlow TG, Mohler JL, et al. Androgen receptor up-regulates insulin-like growth factor binding protein-5 (IGFBP-5) expression in a human prostate cancer xenograft. Endocrinology (1999) 140:2372–81. doi: 10.1210/endo.2140.2375.6702 [DOI] [PubMed] [Google Scholar]
- 118. Tennant MK, Thrasher JB, Twomey PA, Birnbaum RS, Plymate SR. Insulin-like growth factor-binding proteins (IGFBP)-4, -5, and -6 in the benign and malignant human prostate: IGFBP-5 messenger ribonucleic acid localization differs from IGFBP-5 protein localization. J Clin Endocrinol Metab (1996) 81:3783–92. doi: 10.1210/jcem.3781.3710.8855838 [DOI] [PubMed] [Google Scholar]
- 119. Mita K, Zhang Z, Ando Y, Toyama T, Hamaguchi M, Kobayashi S, et al. Prognostic significance of insulin-like growth factor binding protein (IGFBP)-4 and IGFBP-5 expression in breast cancer. Jpn J Clin Oncol (2007) 37:575–82. doi: 10.1093/jjco/hym066 [DOI] [PubMed] [Google Scholar]
- 120. Ahn BY, Elwi AN, Lee B, Trinh DL, Klimowicz AC, Yau A, et al. Genetic screen identifies insulin-like growth factor binding protein 5 as a modulator of tamoxifen resistance in breast cancer. Cancer Res (2010) 70:3013–9. doi: 10.1158/0008-5472.CAN-09-3108 [DOI] [PubMed] [Google Scholar]
- 121. Yu QC, Verheyen EM, Zeng YA. Mammary development and breast cancer: A wnt perspective. Cancers (Basel) (2016) 8:65. doi: 10.3390/cancers8070065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Liu BY, Soloviev I, Huang X, Chang P, Ernst JA, Polakis P, et al. Mammary tumor regression elicited by wnt signaling inhibitor requires IGFBP5. Cancer Res (2012) 72:1568–78. doi: 10.1158/0008-5472.CAN-11-3668 [DOI] [PubMed] [Google Scholar]
- 123. Tonner E, Barber MC, Travers MT, Logan A, Flint DJ. Hormonal control of insulin-like growth factor-binding protein-5 production in the involuting mammary gland of the rat. Endocrinology (1997) 138:5101–7. doi: 10.1210/endo.5138.5112.5619 [DOI] [PubMed] [Google Scholar]
- 124. Chapman RS, Lourenco PC, Tonner E, Flint DJ, Selbert S, Takeda K, et al. Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of Stat3. Genes Dev (1999) 13:2604–16. doi: 10.1101/gad.2613.2619.2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Tonner E, Barber MC, Allan GJ, Beattie J, Webster J, Whitelaw CB, et al. Insulin-like growth factor binding protein-5 (IGFBP-5) induces premature cell death in the mammary glands of transgenic mice. Development (2002) 129:4547–57. doi: 10.1242/dev.4129.4519.4547 [DOI] [PubMed] [Google Scholar]
- 126. Allan GJ, Beattie J, Flint DJ. The role of IGFBP-5 in mammary gland development and involution. Domest Anim Endocrinol (2004) 27:257–66. doi: 10.1016/j.domaniend.2004.06.009 [DOI] [PubMed] [Google Scholar]
- 127. Ning Y, Hoang B, Schuller AG, Cominski TP, Hsu MS, Wood TL, et al. Delayed mammary gland involution in mice with mutation of the insulin-like growth factor binding protein 5 gene. Endocrinology (2007) 148:2138–47. doi: 10.1210/en.2006-0041 [DOI] [PubMed] [Google Scholar]
- 128. Francis SM, Bergsied J, Isaac CE, Coschi CH, Martens AL, Hojilla CV, et al. A functional connection between pRB and transforming growth factor beta in growth inhibition and mammary gland development. Mol Cell Biol (2009) 29:4455–66. doi: 10.1128/MCB.00473-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kapoor PM, Lindstrom S, Behrens S, Wang X, Michailidou K, Bolla MK, et al. Assessment of interactions between 205 breast cancer susceptibility loci and 13 established risk factors in relation to breast cancer risk, in the breast cancer association consortium. Int J Epidemiol (2020) 49:216–32. doi: 10.1093/ije/dyz193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Butt AJ, Dickson KA, McDougall F, Baxter RC. Insulin-like growth factor-binding protein-5 inhibits the growth of human breast cancer cells in vitro and in vivo . J Biol Chem (2003) 278:29676–85. doi: 10.1074/jbc.M301965200 [DOI] [PubMed] [Google Scholar]
- 131. McCaig C, Perks CM, Holly JM. Intrinsic actions of IGFBP-3 and IGFBP-5 on Hs578T breast cancer epithelial cells: inhibition or accentuation of attachment and survival is dependent upon the presence of fibronectin. J Cell Sci (2002) 115:4293–303. doi: 10.1242/jcs.00097 [DOI] [PubMed] [Google Scholar]
- 132. McCaig C, Perks CM, Holly JM. Signalling pathways involved in the direct effects of IGFBP-5 on breast epithelial cell attachment and survival. J Cell Biochem (2002) 84:784–94. doi: 10.1002/jcb.10093 [DOI] [PubMed] [Google Scholar]
- 133. Dittmer A, Lange T, Leyh B, Dittmer J. Protein- and growth-modulatory effects of carcinoma-associated fibroblasts on breast cancer cells: Role of interleukin6. Int J Oncol (2020) 56:258–72. doi: 10.3892/ijo.2019.4918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Leyh B, Dittmer A, Lange T, Martens JW, Dittmer J. Stromal cells promote anti-estrogen resistance of breast cancer cells through an insulin-like growth factor binding protein 5 (IGFBP5)/B-cell leukemia/lymphoma 3 (Bcl-3) axis. Oncotarget (2015) 6:39307–28. doi: 10.18632/oncotarget.5624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Dittmer A, Dittmer J. Long-term exposure to carcinoma-associated fibroblasts makes breast cancer cells addictive to integrin β1. Oncotarget (2018) 9:22079–94. doi: 10.18632/oncotarget.25183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res (2008) 68:826–33. doi: 10.1158/0008-5472.CAN-07-2707 [DOI] [PubMed] [Google Scholar]
- 137. Beelen K, Opdam M, Severson TM, Koornstra RH, Vincent AD, Wesseling J, et al. PIK3CA mutations, phosphatase and tensin homolog, human epidermal growth factor receptor 2, and insulin-like growth factor 1 receptor and adjuvant tamoxifen resistance in postmenopausal breast cancer patients. Breast Cancer Res (2014) 16:R13. doi: 10.1186/bcr3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Robertson JFR, Ferrero J-M, Bourgeois H, Kennecke H, de Boer RH, Jacot W, et al. Ganitumab with either exemestane or fulvestrant for postmenopausal women with advanced, hormone-receptor-positive breast cancer: a randomised, controlled, double-blind, phase 2 trial. Lancet Oncol (2013) 14:228–35. doi: 10.1016/s1470-2045(13)70026-3 [DOI] [PubMed] [Google Scholar]
- 139. Sarwar N, Kim JS, Jiang J, Peston D, Sinnett HD, Madden P, et al. Phosphorylation of ERalpha at serine 118 in primary breast cancer and in tamoxifen-resistant tumours is indicative of a complex role for ERalpha phosphorylation in breast cancer progression. Endocr Relat Cancer (2006) 13:851–61. doi: 10.1677/erc.1.01123 [DOI] [PubMed] [Google Scholar]
- 140. Wang VY, Li Y, Kim D, Zhong X, Du Q, Ghassemian M, et al. Bcl3 phosphorylation by akt, Erk2, and IKK is required for its transcriptional activity. Mol Cell (2017) 67:484–497.e485. doi: 10.1016/j.molcel.2017.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Czapiewski P, Cornelius M, Hartig R, Kalinski T, Haybaeck J, Dittmer A, et al. BCL3 expression is strongly associated with the occurrence of breast cancer relapse under tamoxifen treatment in a retrospective cohort study. Virchows Arch (2022) 480:529–41. doi: 10.1007/s00428-00021-03238-00428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Hawsawi Y, Humphries MP, Wright A, Berwick A, Shires M, Al-Kharobi H, et al. Deregulation of IGF-binding proteins -2 and -5 contributes to the development of endocrine resistant breast cancer in vitro . Oncotarget (2016) 7:32129–43. doi: 10.18632/oncotarget.38534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Verret B, Cortes J, Bachelot T, Andre F, Arnedos M. Efficacy of PI3K inhibitors in advanced breast cancer. Ann Oncol (2019) 30:x12–20. doi: 10.1093/annonc/mdz381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Pavlicek A, Lira ME, Lee NV, Ching KA, Ye J, Cao J, et al. Molecular predictors of sensitivity to the insulin-like growth factor 1 receptor inhibitor figitumumab (CP-751,871). Mol Cancer Ther (2013) 12:2929–39. doi: 10.1158/1535-7163.MCT-13-0442-T [DOI] [PubMed] [Google Scholar]
- 145. Neuzillet Y, Chapeaublanc E, Krucker C, De Koning L, Lebret T, Radvanyi F, et al. IGF1R activation and the in vitro antiproliferative efficacy of IGF1R inhibitor are inversely correlated with IGFBP5 expression in bladder cancer. BMC Cancer (2017) 17:636. doi: 10.1186/s12885-017-3618-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Allander SV, Larsson C, Ehrenborg E, Suwanichkul A, Weber G, Morris SL, et al. Characterization of the chromosomal gene and promoter for human insulin-like growth factor binding protein-5. J Biol Chem (1994) 269:10891–8. doi: 10.1016/s0021-9258(17)34142-x [DOI] [PubMed] [Google Scholar]
- 147. Figueroa JA, Jackson JG, McGuire WL, Krywicki RF, Yee D. Expression of insulin-like growth factor binding proteins in human breast cancer correlates with estrogen receptor status. J Cell Biochem (1993) 52:196–205. doi: 10.1002/jcb.240520211 [DOI] [PubMed] [Google Scholar]
- 148. Yee D, Sharma J, Hilsenbeck SG. Prognostic significance of insulin-like growth factor-binding protein expression in axillary lymph node-negative breast cancer. J Natl Cancer Inst (1994) 86:1785–9. doi: 10.1093/jnci/1786.1723.1785 [DOI] [PubMed] [Google Scholar]
- 149. Parisot JP, Leeding KS, Hu XF, DeLuise M, Zalcberg JR, Bach LA. Induction of insulin-like growth factor binding protein expression by ICI 182,780 in a tamoxifen-resistant human breast cancer cell line. Breast Cancer Res Treat (1999) 55:231–42. doi: 10.1023/a:1006274712664 [DOI] [PubMed] [Google Scholar]
- 150. Walker G, MacLeod K, Williams AR, Cameron DA, Smyth JF, Langdon SP. Insulin-like growth factor binding proteins IGFBP3, IGFBP4, and IGFBP5 predict endocrine responsiveness in patients with ovarian cancer. Clin Cancer Res (2007) 13:1438–44. doi: 10.1158/1078-0432.CCR-06-2245 [DOI] [PubMed] [Google Scholar]
- 151. Milne RL, Benitez J, Nevanlinna H, Heikkinen T, Aittomaki K, Blomqvist C, et al. Risk of estrogen receptor-positive and -negative breast cancer and single-nucleotide polymorphism 2q35-rs13387042. J Natl Cancer Inst (2009) 101:1012–8. doi: 10.1093/jnci/djp167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Coe EA, Tan JY, Shapiro M, Louphrasitthiphol P, Bassett AR, Marques AC, et al. The MITF-SOX10 regulated long non-coding RNA DIRC3 is a melanoma tumour suppressor. PLoS Genet (2019) 15:e1008501. doi: 10.1371/journal.pgen.1008501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Miyoshi Y, Murase K, Saito M, Imamura M, Oh K. Mechanisms of estrogen receptor-alpha upregulation in breast cancers. Med Mol Morphol (2010) 43:193–6. doi: 10.1007/s00795-010-0514-3 [DOI] [PubMed] [Google Scholar]
- 154. Di Modugno F, Mottolese M, Di Benedetto A, Conidi A, Novelli F, Perracchio L, et al. The cytoskeleton regulatory protein hMena (ENAH) is overexpressed in human benign breast lesions with high risk of transformation and human epidermal growth factor receptor-2-positive/hormonal receptor-negative tumors. Clin Cancer Res (2006) 12:1470–8. doi: 10.1158/1078-0432.CCR-05-2027 [DOI] [PubMed] [Google Scholar]
- 155. Klaric KA, Riaz N, Asleh K, Wang XQ, Atalla T, Strickland S, et al. SRY-box transcription factor 10 is a highly specific biomarker of basal-like breast cancer. Histopathology (2022) 80:589–97. doi: 10.1111/his.14592 [DOI] [PubMed] [Google Scholar]
- 156. Mertelmeyer S, Weider M, Baroti T, Reiprich S, Frob F, Stolt CC, et al. The transcription factor Sox10 is an essential determinant of branching morphogenesis and involution in the mouse mammary gland. Sci Rep (2020) 10:17807. doi: 10.1038/s41598-020-74664-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Chang KW, Hung PS, Lin IY, Hou CP, Chen LK, Tsai YM, et al. Curcumin upregulates insulin-like growth factor binding protein-5 (IGFBP-5) and C/EBPalpha during oral cancer suppression. Int J Cancer (2010) 127:9–20. doi: 10.1002/ijc.25220 [DOI] [PubMed] [Google Scholar]
- 158. Ji C, Chen Y, Centrella M, McCarthy TL. Activation of the insulin-like growth factor-binding protein-5 promoter in osteoblasts by cooperative e box, CCAAT enhancer-binding protein, and nuclear factor-1 deoxyribonucleic acid-binding sequences. Endocrinology (1999) 140:4564–72. doi: 10.1210/endo.4140.4510.7061 [DOI] [PubMed] [Google Scholar]
- 159. Lourenco AR, Coffer PJ. A tumor suppressor role for C/EBPalpha in solid tumors: more than fat and blood. Oncogene (2017) 36:5221–30. doi: 10.1038/onc.2017.151 [DOI] [PubMed] [Google Scholar]
- 160. Balamurugan K, Mendoza-Villanueva D, Sharan S, Summers GH, Dobrolecki LE, Lewis MT, et al. C/EBPdelta links IL-6 and HIF-1 signaling to promote breast cancer stem cell-associated phenotypes. Oncogene (2019) 38:3765–80. doi: 10.1038/s41388-018-0516-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Muller M, Persson AB, Krueger K, Kirschner KM, Scholz H. The wilms tumor protein WT1 stimulates transcription of the gene encoding insulin-like growth factor binding protein 5 (IGFBP5). Gene (2017) 619:21–9. doi: 10.1016/j.gene.2017.03.037 [DOI] [PubMed] [Google Scholar]
- 162. Ullmark T, Montano G, Gullberg U. DNA And RNA binding by the wilms' tumour gene 1 (WT1) protein +KTS and -KTS isoforms-from initial observations to recent global genomic analyses. Eur J Haematol (2018) 100:229–40. doi: 10.1111/ejh.13010 [DOI] [PubMed] [Google Scholar]
- 163. Drummond IA, Rupprecht HD, Rohwer-Nutter P, Lopez-Guisa JM, Madden SL, Rauscher FJ, 3rd, et al. DNA Recognition by splicing variants of the wilms' tumor suppressor, WT1. Mol Cell Biol (1994) 14:3800–9. doi: 10.1128/mcb.3814.3806.3800-3809.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Nasomyon T, Samphao S, Sangkhathat S, Mahattanobon S, Graidist P. Correlation of wilms' tumor 1 isoforms with HER2 and ER-α and its oncogenic role in breast cancer. Anticancer Res (2014) 34:1333–42. [PubMed] [Google Scholar]
- 165. Bejjani F, Evanno E, Zibara K, Piechaczyk M, Jariel-Encontre I. The AP-1 transcriptional complex: Local switch or remote command? Biochim Biophys Acta Rev Cancer (2019) 1872:11–23. doi: 10.1016/j.bbcan.2019.04.003 [DOI] [PubMed] [Google Scholar]
- 166. Niu J, Huang YJ, Wei S, Liu Z, Wang LE, Chang S, et al. Association between a functional polymorphism (-1195T>C) in the IGFBP5 promoter and head and neck cancer risk. Head Neck (2011) 33:650–60. doi: 10.1002/hed.21514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol (2015) 17:1218–27. doi: 10.1038/ncb3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Zhang L, Li W, Cao L, Xu J, Qian Y, Chen H, et al. PKNOX2 suppresses gastric cancer through the transcriptional activation of IGFBP5 and p53. Oncogene (2019) 38:4590–604. doi: 10.1038/s41388-019-0743-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Ding M, Bruick RK, Yu Y. Secreted IGFBP5 mediates mTORC1-dependent feedback inhibition of IGF-1 signalling. Nat Cell Biol (2016) 18:319–27. doi: 10.1038/ncb3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci (2012) 33:207–14. doi: 10.1016/j.tips.2012.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications. Biochim Biophys Acta (2014) 1839:627–43. doi: 10.1016/j.bbagrm.2014.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Maitituoheti M, Keung EZ, Tang M, Yan L, Alam H, Han G, et al. Enhancer reprogramming confers dependence on glycolysis and IGF signaling in KMT2D mutant melanoma. Cell Rep (2020) 33:108293. doi: 10.1016/j.celrep.2020.108293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science (2017) 355:1324–30. doi: 10.1126/science.aah6893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Cavellan E, Asp P, Percipalle P, Farrants AK. The WSTF-SNF2h chromatin remodeling complex interacts with several nuclear proteins in transcription. J Biol Chem (2006) 281:16264–71. doi: 10.1074/jbc.M600233200 [DOI] [PubMed] [Google Scholar]
- 175. Felipe-Abrio B, Carnero A. The tumor suppressor roles of MYBBP1A, a major contributor to metabolism plasticity and stemness. Cancers (Basel) (2020) 12:254. doi: 10.3390/cancers12010254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Weng X, Wu J, Lv Z, Peng C, Chen J, Zhang C, et al. Targeting Mybbp1a suppresses HCC progression via inhibiting IGF1/AKT pathway by CpG islands hypo-methylation dependent promotion of IGFBP5. EBioMedicine (2019) 44:225–36. doi: 10.1016/j.ebiom.2019.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Akaogi K, Ono W, Hayashi Y, Kishimoto H, Yanagisawa J. MYBBP1A suppresses breast cancer tumorigenesis by enhancing the p53 dependent anoikis. BMC Cancer (2013) 13:65. doi: 10.1186/1471-2407-1113-1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Cerutti C, Zhang L, Tribollet V, Shi JR, Brillet R, Gillet B, et al. Computational identification of new potential transcriptional partners of ERRalpha in breast cancer cells: specific partners for specific targets. Sci Rep (2022) 12:3826. doi: 10.1038/s41598-022-07744-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Stein RA, McDonnell DP. Estrogen-related receptor alpha as a therapeutic target in cancer. Endocr Relat Cancer (2006) 13:S25–32. doi: 10.1677/erc.1671.01292 [DOI] [PubMed] [Google Scholar]
- 180. Kerr A, Baxter RC. Noncoding RNA actions through IGFs and IGF binding proteins in cancer. Oncogene (2022) 41:3385–93. doi: 10.1038/s41388-022-02353-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med (2014) 20:460–9. doi: 10.1016/j.molmed.2014.06.005 [DOI] [PubMed] [Google Scholar]
- 182. Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell (2018) 172:393–407. doi: 10.1016/j.cell.2018.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Gullu G, Peker I, Haholu A, Eren F, Kucukodaci Z, Gulec B, et al. Clinical significance of miR-140-5p and miR-193b expression in patients with breast cancer and relationship to IGFBP5. Genet Mol Biol (2015) 38:21–9. doi: 10.1590/S1415-475738120140167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Liu L, Wang J, Li X, Ma J, Shi C, Zhu H, et al. MiR-204-5p suppresses cell proliferation by inhibiting IGFBP5 in papillary thyroid carcinoma. Biochem Biophys Res Commun (2015) 457:621–6. doi: 10.1016/j.bbrc.2015.01.037 [DOI] [PubMed] [Google Scholar]
- 185. Liu H, Li R, Guan L, Jiang T. Knockdown of lncRNA UCA1 inhibits proliferation and invasion of papillary thyroid carcinoma through regulating miR-204/IGFBP5 axis. Onco Targets Ther (2018) 11:7197–204. doi: 10.2147/OTT.S175467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Hong BS, Ryu HS, Kim N, Kim J, Lee E, Moon H, et al. Tumor suppressor miRNA-204-5p regulates growth, metastasis, and immune microenvironment remodeling in breast cancer. Cancer Res (2019) 79:1520–34. doi: 10.1158/0008-5472.CAN-18-0891 [DOI] [PubMed] [Google Scholar]
- 187. Alkhathami AG, Hadi A, Alfaifi M, Alshahrani MY, Verma AK, Beg MMA. Serum-based lncRNA ANRIL, TUG1, UCA1, and HIT expressions in breast cancer patients. Dis Markers (2022) 2022:9997212. doi: 10.1155/2022/9997212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Yao F, Wang Q, Wu Q. The prognostic value and mechanisms of lncRNA UCA1 in human cancer. Cancer Manag Res (2019) 11:7685–96. doi: 10.2147/CMAR.S200436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Sun Y, Li X, Chen A, Shi W, Wang L, Yi R, et al. circPIP5K1A serves as a competitive endogenous RNA contributing to ovarian cancer progression via regulation of miR-661/IGFBP5 signaling. J Cell Biochem (2019) 120:19406–14. doi: 10.1002/jcb.29055 [DOI] [PubMed] [Google Scholar]
- 190. Reddy SD, Pakala SB, Ohshiro K, Rayala SK, Kumar R. MicroRNA-661, a c/EBPalpha target, inhibits metastatic tumor antigen 1 and regulates its functions. Cancer Res (2009) 69:5639–42. doi: 10.1158/0008-5472.CAN-09-0898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Hoffman Y, Bublik DR, Pilpel Y, Oren M. miR-661 downregulates both Mdm2 and Mdm4 to activate p53. Cell Death Differ (2014) 21:302–9. doi: 10.1038/cdd.2013.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Hu SA, Cheng J, Zhao WH, Zhao HY. Quercetin induces apoptosis in meningioma cells through the miR-197/IGFBP5 cascade. Environ Toxicol Pharmacol (2020) 80:103439. doi: 10.1016/j.etap.2020.103439 [DOI] [PubMed] [Google Scholar]
- 193. Shaker O, Maher M, Nassar Y, Morcos G, Gad Z. Role of microRNAs -29b-2, -155, -197 and -205 as diagnostic biomarkers in serum of breast cancer females. Gene (2015) 560:77–82. doi: 10.1016/j.gene.2015.01.062 [DOI] [PubMed] [Google Scholar]
- 194. Jahangiri B, Khalaj-Kondori M, Asadollahi E, Sadeghizadeh M. Cancer-associated fibroblasts enhance cell proliferation and metastasis of colorectal cancer SW480 cells by provoking long noncoding RNA UCA1. J Cell Commun Signal (2019) 13:53–64. doi: 10.1007/s12079-018-0471-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Johannessen C, Moi L, Kiselev Y, Pedersen MI, Dalen SM, Braaten T, et al. Expression and function of the miR-143/145 cluster in vitro and in vivo in human breast cancer. PLoS One (2017) 12:e0186658. doi: 10.1371/journal.pone.0186658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Wang X, Jiang B, Zhang Y. Gankyrin regulates cell signaling network. Tumour Biol (2016) 37:5675–82. doi: 10.1007/s13277-016-4854-z [DOI] [PubMed] [Google Scholar]
- 197. Umemura A, Itoh Y, Itoh K, Yamaguchi K, Nakajima T, Higashitsuji H, et al. Association of gankyrin protein expression with early clinical stages and insulin-like growth factor-binding protein 5 expression in human hepatocellular carcinoma. Hepatology (2008) 47:493–502. doi: 10.1002/hep.22027 [DOI] [PubMed] [Google Scholar]
- 198. Kim YH, Kim JH, Choi YW, Lim SK, Yim H, Kang SY, et al. Gankyrin is frequently overexpressed in breast cancer and is associated with ErbB2 expression. Exp Mol Pathol (2013) 94:360–5. doi: 10.1016/j.yexmp.2012.12.002 [DOI] [PubMed] [Google Scholar]
- 199. Jahangiri R, Mosaffa F, EmamiRazavi A, Gharib M, Jamialahmadi K. Increased expression of gankyrin and stemness factor Oct-4 are associated with unfavorable clinical outcomes and poor benefit of tamoxifen in breast carcinoma patients. Pathol Oncol Res (2020) 26:1921–34. doi: 10.1007/s12253-019-00766-2 [DOI] [PubMed] [Google Scholar]
- 200. West RB, Corless CL, Chen X, Rubin BP, Subramanian S, Montgomery K, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol (2004) 165:107–13. doi: 10.1016/s0002-9440(10)63279-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Simon S, Grabellus F, Ferrera L, Galietta L, Schwindenhammer B, Muhlenberg T, et al. DOG1 regulates growth and IGFBP5 in gastrointestinal stromal tumors. Cancer Res (2013) 73:3661–70. doi: 10.1158/0008-5472.CAN-12-3839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Bae JS, Park JY, Park SH, Ha SH, An AR, Noh SJ, et al. Expression of ANO1/DOG1 is associated with shorter survival and progression of breast carcinomas. Oncotarget (2018) 9:607–21. doi: 10.18632/oncotarget.23078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Rozen F, Yang XF, Huynh H, Pollak M. Antiproliferative action of vitamin d-related compounds and insulin-like growth factor-binding protein 5 accumulation. J Natl Cancer Inst (1997) 89:652–6. doi: 10.1093/jnci/1089.1099.1652 [DOI] [PubMed] [Google Scholar]
- 204. David CJ, Massague J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat Rev Mol Cell Biol (2018) 19:419–35. doi: 10.1038/s41580-018-0007-0 [DOI] [PMC free article] [PubMed] [Google Scholar]