

Abstract
The Arf GAPs are a family of proteins with a common catalytic function of hydrolyzing GTP bound to ADP-ribosylation factors (Arf) with proposed cellular functions that are diverse (Inoue and Randazzo, 2007; Kahn et al., 2008). Understanding the biochemistry of the Arf GAPs is valuable for designing and interpreting experiments using standard cell biology techniques described elsewhere. The following briefly reviews some common approaches for in vivo studies of Arf GAPs and discusses the use of isolated cellular organelles to complement in vivo experiments. Detailed protocols for examining the activity of Arf GAPs in whole cell lysates and in association with isolated focal adhesions are provided.
Keywords: ADP-ribosylation factor, GTPase-activating protein, guanine nucleotide binding protein, Arf1, Arf6
INTRODUCTION
The Arf GTPase-activating proteins (GAPs) are a family of proteins encoded by 31 genes in humans. The proteins are subdivided into ten groups on the basis of domain structure and phylogenetic analysis (Kahn et al., 2008). The Arf GAPs have a common catalytic domain that induces the hydrolysis of GTP bound to the small GTP binding protein Arf, converting Arf-GTP to Arf-GDP. Despite the common catalytic function, the Arf GAPs have diverse cellular functions.
The substrates for Arf GAPs are Arf proteins (Kahn et al., 2006) bound to GTP. Humans have five Arf proteins: Arf1 and Arf3 are in class 1; Arf4 and Arf5 are in class 2; and Arf6 is in class 3. The Arf proteins regulate membrane traffic and actin remodeling. The best studied Arfs, Arf1 and Arf6, function in distinct cellular sites with specific effector proteins. The Arf GAPs have been found to affect membrane and actin remodeling in part through regulation of specific Arfs (Nie and Randazzo, 2006; Inoue and Randazzo, 2007). The GAPs, which outnumber the Arfs, may have greater site specificity than the substrate Arfs. At least six Arf GAPs (GIT1/2, ASAP1/3, ARAP2, and AGAP2) have been found to associate with cellular adhesions to the extracellular matrix, but not with the Golgi apparatus (Randazzo et al., 2007; Campa and Randazzo, 2008). Other Arf GAPs (e.g., ArfGAP1/2/3, ARAP1) associate with Golgi membranes but have not been detected in cellular adhesive structures (Cukierman et al., 1995; Miura et al., 2002; Frigerio et al., 2007). The function of the Arf GAPs may extend beyond simple regulation of Arf, including functioning as Arf effectors, and the GAPs themselves are likely regulated by signals other than Arfs.
Exploration of the cellular roles of Arf GAPs has centered on two related questions: (1) what is the site of action of a given GAP and (2) what is the Arf specificity for a given GAP? Given that many cellular functions of Arfs are known (Kahn et al., 2006; Gillingham and Munro, 2007; Donaldson and Jackson, 2011), particularly for Arf1 and Arf6, identification of the Arf specificity of an Arf GAP would provide some information about potential functions of the GAP. Some common approaches to address these questions are described and some discussion of the potential pitfalls of the approaches is also included. Using cellular fractions for in vitro GAP assays as a complementary technique to overcome some of the pitfalls of the in vivo methods is recommended. Detailed protocols for measuring GAP activity in cell lysates and in isolated focal adhesions (FAs) are provided.
Common Approaches for Studying Arf GAPs in Cells
Some progress has been made in understanding the cellular function of Arf GAPs, but the following two critical and related questions either require further clarification or need to be determined for most Arf GAPs: (1) what is the cellular event regulated by the Arf GAP and (2) what is the particular Arf with which the GAP interacts for a specific cellular event.
Identifying specific cellular events or sites of action depends, to a great extent, on localization by microscopy. This approach provides the best information when endogenous proteins can be visualized, usually by immunofluorescence (IF). For example, the function of ASAP1 in cell migration was identified by finding that ASAP1 is located in FAs (Randazzo et al., 2000; Liu et al., 2002, 2005). Unfortunately, it has been difficult to obtain antibodies reliable for IF for most of the Arf GAPs. This approach may further be complicated by low concentrations of the Arf GAPs. Typically, cultured cells for Arf GAP expression levels are screened using immunoblots, and determination of localization of endogenous proteins in cells with the highest expression using immunofluorescence is attempted. However, functioning as enzymes, they may be present at nanomolar concentrations or less, in which case this approach may not be feasible.
Epitope tags or GFP fused to recombinant proteins can sometimes provide information about localization; however, the epitope tags and GFP are not always innocuous. For example, it has been found that a GFP-tagged ARAP2 was toxic to cells and did not target FAs in the same way that the endogenous ARAP2 (Fig. 17.13.1) does. Part of the mislocalization could be due to misfolded recombinant proteins or limited binding sites (see below). The ability to rescue a phenotype generated by reduced expression of endogenous protein is not sufficient evidence to support the use of the epitope-tagged protein for localization studies, particularly when expression levels are greater than physiological expression levels. Presuming, for example, that 10% of the expressed Arf GAP functions like the endogenous Arf GAP, which may be adequate to rescue the phenotype of the reduced expression of endogenous protein, the imaging signal from 90% may be in a location that is not physiologically relevant.
Figure 17.13.1.
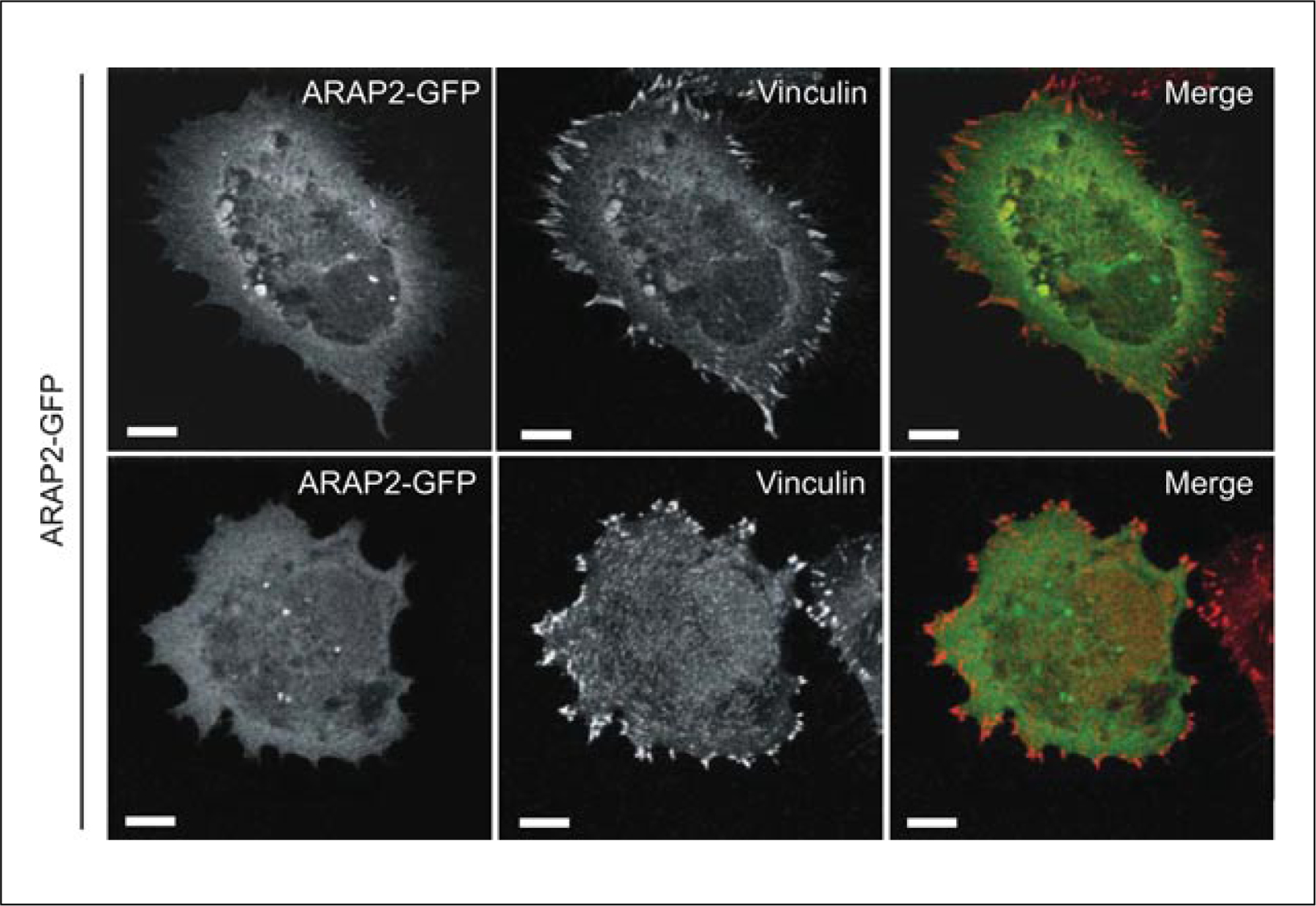
Subcellular localization of ARAP2-GFP. HeLa cells transfected with GFP-tagged ARAP2 were plated on fibronectin, fixed, and stained with anti-vinculin antibody. ARAP2-GFP did not co-localize with the focal adhesion (FA) marker, vinculin, as shown in two representative images. Overexpression of ARAP2-GFP was toxic to cells as cells often became multinucleated and/or were vacuolated. Scale bars, 10 μm.
It may not be possible to determine the localization of some Arf GAPs using immunomicroscopy for the reasons described above. In that case, it may be useful to determine localization by immunoblotting cell fractions enriched in particular organelles, as described in this unit. However, if antibodies are not available, in vitro GAP assays may provide information about the Arf GAP subtype. Properties that can be used to discern between some of the GAPs are listed (see Table 17.13.1). The protocols described for examining GAP activity in isolated focal adhesions allow for this type of approach. The GAP assay can be performed under a set of conditions that satisfy the lipid requirement of particular Arf GAPs, e.g., activation by PIP3 but not PIP2 would be consistent with an ARAP subtype. Arf6 specificity within the PIP3-dependent Arf GAPs is consistent with ARAP2, and Arf1 or Arf5 specificity is consistent with ARAP1 or ARAP3.
Table 17.13.1.
Distinguishing Features of Some Arf GAPs
| Arf GAP | Substrate (Arf1 vs.Arf5 vs. Arf6) | Phosphoinositidea dependence | Other distinguishing features |
|---|---|---|---|
| ArfGAP1 | Arf1 ≥ Arf5 ≥ Arf6 | None | Not determined |
| ASAP1/2/3 | Arf5 ≥ Arf1≫Arf6 | PI(4,5)P2 ≥ PI(3,4,5)P3 | Not determined |
| ACAP1/2 | Arf6>Arf1 = Arf5 | PI(3,5)P2 = PI(4,5)P2 = PIP3 | Not determined |
| ARAP1 | Arf5>Arf1>Arf6 | PI(3,4,5)P3>PIP2 | Not determined |
| ARAP2 | Arf6>Arf5 = Arf1 | PI(3,4,5)P3>PIP2 | Not determined |
| AGAP1/2 | Arf1>Arf5>Arf6 | PIP3=PIP2 | Activation by C-terminal peptide of RhoA |
PI (4,5) P2 and PIP2, phosphatidylinositol 4,5-biphosphate; PI (3,5) P2, phosphatidylinositol 3,5-bisphosphate; PI (3,4,5) P3 and PIP3, phosphatidylinositol 3,4,5-triphosphate.
Substrate specificity has also been determined in vivo using the assay described in detail in UNIT 14.12 (Cohen and Donaldson, 2010). To understand the potential pitfalls of the in vivo experiments, a description of the experiments is useful. An Arf GAP is either overexpressed or its expression is reduced in cells that also express Arf-HA. The cells are lysed and the Arf-HA-GTP is precipitated using GST fused to a fragment of the Arf effector GGA, which binds selectively to the GTP-bound form of Arf. The amount of Arf detected in the precipitates is proportional to the Arf-HA-GTP in the cells (Cohen and Donaldson, 2010; Yoon et al., 2006). If overexpression of the Arf GAP reduces the signal from a particular Arf-HA or reduced expression increases the signal, the Arf is considered the substrate. Factors considered critical to interpreting the experiments are described in Critical Parameters.
BASIC PROTOCOL 1
PREPARATION OF CELL LYSATES FOR GAP ASSAY
This protocol utilizes whole cell lysates from cells overexpressing an Arf GAP. This method provides a way to assay the GAP in the context of cellular proteins. Interpretation could be affected if the enzyme is not fully active as described above, which can be partly addressed by appropriate titrations of enzyme and substrate. There are also potentially confounding factors arising from the use of a “dirty” enzyme preparation, e.g., presence of exchange factors, effectors that can sequester the substrate, and other GAPs. By controlling the conditions, the influence of these factors can be minimized.
Materials
HeLa cells maintained at 37°C in Dulbecco’s modified Eagle’s medium supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum (FBS)
Plasmids expressing Arf GAP or mutants of Arf GAP (e.g., ASAP1 plasmids, Randazzo et al., 2000)
PBS (Quality Biological)
Lysis buffer for HeLa cells (see recipe)
BioRad protein assay reagent
Dilution buffer (lysis buffer without 1% Triton X-100)
1% Triton X-100 (APPENDIX 2A)
6-well tissue culture plate
37°C, 5% CO2 incubator
Cell scraper (SARSTEDT)
1.5-ml microcentrifuge tubes
Plate HeLa cells at 2.63 × 105 cells/well in 6-well tissue culture plates. Incubate overnight in a 37°C, 5% CO2 incubator.
- On the following day, cells should be ~90% confluent. Transfect cells with plasmids expressing an Arf GAP or mutants of the Arf GAP.Transfections are typically performed using a transfection reagent such as Lipofectamine (Invitrogen) or Fugene 6 (promega) according to manufacturer’s instructions.Cells can also be transfected with siRNA or plasmids expressing proteins to determine if these treatments affect GAP activity.
- Harvest whole cell lysates. Pre-cool PBS and lysis buffer to 4°C before starting the experiment. Place tissue culture plate on ice. Rinse cell monolayer two times with 2 ml ice-cold PBS. Remove PBS as much as possible and immediately add 250 μl ice-cold lysis buffer to each well. Use cell scrapers to scrape off all the materials from the plates and transfer to 1.5-ml microcentrifuge tubes (one tube/well). Incubate lysates 15 min on ice. Clear lysates by centrifuging 15 min at 21,000 × g, 4°C. Transfer the supernatants to new 1.5-ml microcentrifuge tubes.The volume of lysis buffer used should be such that the protein concentration will be >2 mg/ml. If this initial lysate concentration is too low, it will be difficult to saturate GAP reaction and determine the C50.
- Determine protein concentration using BioRad protein assay reagent according to manufacturer’s instructions.The whole cell lysates can be dispensed into 10- to 100-μl aliquots, flash frozen using either a dry ice/ethanol bath or liquid nitrogen, and stored at −80°C.
Before starting the GAP assay, dilute cell lysates with dilution buffer to decrease Triton X-100 concentration to 0.2% (1 vol lysates plus 4 vol dilution buffer). Determine protein concentration again as in step 4 and adjust lysate volume with dilution buffer plus 0.2% Triton X-100 so that every sample has the same protein and detergent concentrations.
BASIC PROTOCOL 2
PREPARATION OF FA FRACTION
Different Arf GAPs have different sites of action and their activity may be regulated by changes in the local environment like lipid composition and interacting partners. Subcellular fractionation is one way to assess GAP activity in particular compartments. This protocol describes the use of a fractionated sample for GAP assay. It describes how to measure Arf GAP activity in isolated focal adhesions (FAs; to date, six Arf GAPs, including ASAP1/3, GIT1/2, ARAP2, and AGAP2, are found to localize to FAs). An FA fraction was prepared following methods described previously with some modifications (Tong et al., 2010; Kuo et al., 2011). The principle of this isolation method is to expose cells to a pH-balanced, low ionic strength buffer so that cells swell under the osmotic pressure. Once the plasma membrane is weakened, a hydrodynamic force is applied to shear the cell bodies including nuclei, internal membrane-bound organelles, soluble cytosolic proteins, and most of the cytoskeleton (Fig. 17.13.2) from the cellular ventral membranes. In contrast to previously published methods optimized for proteomic studies, this method is designed for measuring enzymatic activity after fractionation. This protocol is scaled down for practical amounts of materials (purified substrate, [α32P]GTP, and LUVs) needed for the GAP assay. Accordingly, a repetitive pipettor is used rather than a dental water jet apparatus because it is easier to maneuver inside a small well, while keeping the ability to produce a consistent force. This method is not designed to produce pure FA fractions but is useful for the relatively fast and reproducible method of fractionating cells for the GAP assay. The time required for preparing an FA fraction is ∼10 to 15 min.
Figure 17.13.2.
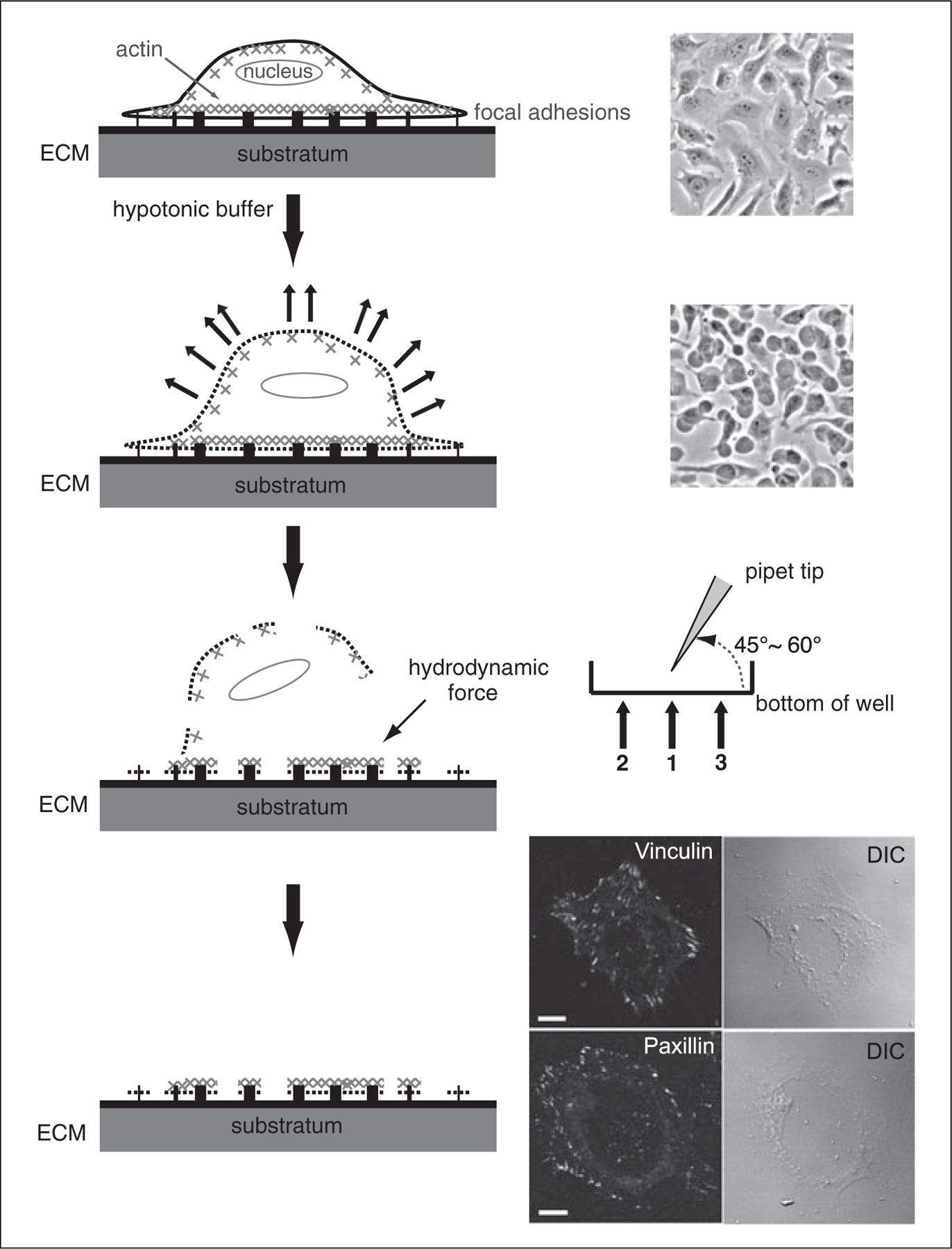
The procedure of FA isolation. Left panel, schematic diagram of the FA isolation procedure. Right panel, representative images of cells at each step (top to bottom). Phase contrast images of HeLa cell morphology before and after incubation with hypotonic lysis buffer. Cells appear swollen but do not detach from the substratum after 5 min of treatment. The position of the repetitive pipettor tip for each wash labeled as 1, 2, 3. The FA fraction is validated by the loss of nuclei and cell bodies (DIC) and the preservation of FAs shown by vinculin and paxillin immunofluorescence staining. Scale bar, 10 μm.
Materials
Fibronectin (from bovine plasma; Sigma)
Phosphate buffered saline (PBS; Quality Biological Inc.)
HeLa cells maintained at 37°C in Dulbecco’s modified Eagle’s medium supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum (FBS)
Plasmids expressing Arf GAP or mutants of Arf GAP
Hypotonic lysis buffer, ice cold (see recipe)
12-well tissue culture plates (Corning) or microscope coverslips (no. 1.5, 12-mm circle; Fisher)
37°C, 5% CO2 incubator
Repetitive pipettor (Eppendorf)
Prepare tissue culture plates/coverslips and cells
-
1Coat 12-well tissue culture plates or coverslips for immunofluorescence with fibronectin for growth of cells by incubating in 10 μg/ml bovine plasma fibronectin diluted in PBS overnight at 4°C. Before use, rinse each well or coverslip once with PBS.HeLa cells form FAs on tissue culture plates even without the fibronectin coating. FA fractions are obtained using cells cultured on surfaces with or without fibronectin coating. FA fractions isolated from cells grown on fibronectin contain more GAP activity than those collected from cells grown on surfaces without fibronectin. Different concentrations of fibronectin and other ECM proteins can also be used in this step.
-
2Plate HeLa cells into the 12-well plates at 1.2 × 105 cells/well (place coverslips into the wells). Set up triplicate wells for each experimental condition. Incubate 16 to 24 hr (cells should be ~80% confluent) in a 37°C, 5% CO2 incubator. Transfect cells with plasmids expressing an Arf GAP or mutants of the Arf GAP (as described in Basic Protocol 1).For studies involving other cell types or siRNA, depending on the time required for a maximum knockdown, plating density and time should be planned accordingly. The authors obtain significant reduction of protein levels after 72 hr of siRNA treatment and usually transfect cells with siRNA in a bigger dish (10-cm plate), then transfer the cells into the 12-well plate 48 hr after siRNA transfection and perform the GAP assay on the next day (72 hr of siRNA treatment). Other perturbations, like addition of growth factors or pharmacological inhibitors, can be employed in this step to study effects on GAP activity.
Isolate FAs
-
3Apply hypotonic shock to cells to weaken the plasma membrane on the following day when cells are ~85% to 90% confluent. Pre-cool PBS and hypotonic lysis buffer before starting the experiment. Place the tissue culture plate on ice. Rinse cells three times with 1 ml ice-cold PBS. Rinse cells once with 0.5 ml ice-cold hypotonic lysis buffer. Incubate cells in 1 ml hypotonic lysis buffer 5 min at 4°C.To preserve the FA composition and structure, incubation time should be as short as possible. Optimal conditions can be determined by microscopic examination of the cell morphology during the hypotonic shock. Choose the shortest time that allows most cells to swell but not burst.
-
4Remove cell bodies with hydrodynamic force. Immediately after the cells have swelled, flush cells three times with 0.75 ml hypotonic lysis buffer using a repetitive pipettor to remove the cell bodies including nuclei, other internal organelles, bulk of the cytoskeleton, and soluble materials of the cytoplasm.A repeating pipettor is used because it generates consistent force and it is maneuverable inside the well of the tissue culture plates. During trituration, the tip of the repeating pipettor is held at a 45° to 60° angle and ~0.5 cm from the surface of the bottom of the well, and is placed at different areas inside the well for each flush (Fig. 17.13.2). Perform the GAP assay immediately after FA isolation is completed, as it has not been determined whether the isolated FAs can be stored and retain its activity.These recommendations should be experimentally determined and adjusted to the specific system, since optimal conditions vary depending on the cell type and pipet being used. Validation of the cell body removal and FA preparation can be done by confirming the presence and organization of known FA proteins such as paxillin, vinculin, focal adhesion kinase, etc. using a standard immunofluorescence staining protocol (see Fig. 17.13.2 for typical preparation).
BASIC PROTOCOL 3
EXPRESSION OF HUMAN MYRISTOYLATED Arf1, Arf5, AND Arf6 IN E. coli
In this protocol, recombinant Arf1, Arf5, and Arf6 are myristoylated by co-expression with yeast N-myristoyltransferase (NMT) in BL21(DE3) bacteria. Bacteria are transformed with a plasmid constructed of cDNA for an Arf inserted into pET3, which is then selected with ampicillin, and a plasmid containing the cDNA for yeast n-myristoyltransferase selected using kanamycin. The co-transformants are stored as glycerol stocks. The preparation of the transformed bacteria is described in detail in Randazzo and Kahn (1995).
Materials
Bacteria co-transformed with expression plasmids for Arf and yeast N-myristoyltransferase glycerol stocks
LB agar plate containing 100 μg/ml ampicillin and 50 μg/ml kanamycin
LB broth containing 100 μg/ml ampicillin and 50 μg/ml kanamycin
50 μM sodium myristate (mol. wt. 250.4, 12.52 mg/1 liter)
100 mM isopropyl β-D-1-thiogalactopyranoside (IPTG)
37°C incubator and bacteria shaker
Spectrophotometer
Refrigerated centrifuge
Room temperature bacteria shaker
Streak the glycerol stock for expression of the desired Arf on LB agar plates containing 100 μg/ml ampicillin and 50 μg/ml kanamycin, then incubate overnight at 37°C.
Inoculate 100 ml LB broth containing 100 μg/ml ampicillin and 50 μg/ml kanamycin with a single colony of co-transformants, then grow at 37°C until OD600 ~0.6 (2 to 6 hr), and leave overnight at 4°C.
- Centrifuge culture 10 min at 1500 × g, 4°C, collect the pellet, and resuspend the pellet in 2 liters of LB containing 100 μg/ml ampicillin and 50 μg/ml kanamycin. Grow 3 to4 hr at 37°C until OD600 ∼0.6.An OD600 of 0.7 to 1.0 is acceptable.
Add 50 μM sodium myristate for 20 min prior to adding 0.1 mM IPTG, grow overnight at room temperature for myrArf1 and myrArf5, or add 1 mM IPTG grow 3 hr at 37°C for myrArf6.
Harvest bacteria by centrifuging 10 min at 6000 × g, 4°C. If not used immediately, store pellets at –80°C.
BASIC PROTOCOL 4
ISOLATION OF MyrArf1 OR MyrArf5 PROTEIN
Myristoylated Arf1 and Arf5 are extracted from the bacteria used for expression and then purified with a three-column strategy. Anion exchange (Q column) is used as a first step. Arf does not adhere to the column under the conditions used, but many proteins and nucleotides do adhere. MyrArf is separated from non-myristoylated Arf and other proteins by hydrophobic interaction chromatography using phenyl Sepharose. Sodium chloride, rather than ammonium sulfate, is used for salting the protein onto the column because the latter can cause nucleotide dissociation, resulting in denatured protein. Myristate imparts hydrophobicity sufficient for myrArf to adhere to the column. Non-myristoylated Arf does not. Size exclusion on Superdex 75 is the final step.
Materials
2-liter bacterial culture
T20M100M1D1 (see recipe)
Complete protease inhibitor tablets (Roche)
5M NaCl (APPENDIX 2A)
HiLoad 16/10 phenyl Sepharose HP column (GE Healthcare Life Sciences)
SDS-PAGE apparatus (UNIT 6.1)
Coomassie blue dye
Hiload 26/60 Superdex 75 column (GE Healthcare Life Sciences)
Buffer for phenylsepharose column (T50N3000M1D1; see recipe)
French press or cell disruptor (e.g., Microfluidics M-110P)
Refrigerated ultracentrifuge with Ti45 fixed-angle rotor (Beckman)
5-ml HiTrap Q HP column (GE Healthcare Life Sciences)
AKTA FPLC (GE Healthcare Life Sciences) or similar protein chromatography system
50-ml Amicon stirred ultrafiltration cell attached to nitrogen gas tank or 50-ml Amicon centrifugal filters (Ultracel 10K)
Amicon 43-mm YM-10 filters (if using the ultrafiltration cell)
- Suspend bacteria from a 2-liter culture in 30 ml T20M100M1D1 with one protease inhibitor tablet if using a French press for lysis.If using a cell disruptor, such as a Microfluidics M-110P, suspend in 50 ml T20M100M1D1 with one protease inhibitor tablet.
Lyse cells using a French press or cell disruptor. In the latter case, pass the cells three times and add 20 ml at the end of the lysis to push the remaining lysate from the loop.
Clarify cell lysate by centrifuging in a fixed-angle rotor (e.g., Ti45, Beckman) 40 to 60 min at 100,000 × g, 4°C.
- Pass supernatant, which contains soluble Arf, through a 5-ml HiTrap Q HP column, which has been pre-equilibrated in T20N100M1D1 per manufacturer’s instructions.The column is isocratically developed in the same buffer. Arf does not adhere to the column and is recovered in the eluate that passes through the column. Protein recovery in the eluate is confirmed using the Bio-Rad protein assay (or equivalent).
- Adjust eluate from the HiTrap Q flow-through containing Arf to 3 M NaCl by adding 5 M NaCl and apply to a HiLoad 16/10 phenyl Sepharose HP column.The column is developed in a 75-ml gradient of NaCl from 3000 mM T50N3000M1D1 to 20 mM T20N20M1D1 using an AKTA FPLC or equivalent chromatography system. Non-myristoylated Arf1 and Arf5 are not adsorbed onto the column in 3 M NaCl. MyrArf1 and Arf5 elute with ∼1.5 M NaCl (i.e., middle of the gradient).
- Analyze 10-μl samples of the fractions by SDS-PAGE (UNIT 6.1) and stain with Coomassie blue dye. Pool fractions containing Arf.The presence of a band at ∼20 KDa confirms the presence of Arf in the fractions.
- Fractionate pool from the phenylsepharose column by size exclusion using a Hiload 26/60 Superdex 75 column, which is equilibrated and run in T20M100M1D1. Set the buffer flow rate to be 0.3 ml/min at 4°C and collect 3.6-ml fractions per tube.The column requires use of medium pressure, achieved with an FPLC or HPLC pump. MyrArf1 or myrArf5 elute ~200 ml in 330 ml bed volume of the Hiload 26/60 Superdex 75 column.
Analyze 10-μl samples of the fractions by SDS-PAGE and pool fractions containing Arf.
- Concentrate the pool, using either conventional ultrafiltration through an Amicon YM-10 membrane driven by nitrogen gas or centrifugal filtration with an Amicon Ultracel 10K to ~1.0 ml (protein concentration should be 0.5 to 2.0 mg/ml). Dispense the purified protein into 50-ml aliquots and snap freeze in dry ice/ethanol for 1 year at −80°C.The Arf can be stored for 1 to 2 weeks at 4°C. Do not store the protein at −20°C.
BASIC PROTOCOL 5
ISOLATION OF MyrArf6-GDP
MyrArf6 is extracted from bacteria with both GDP and GTP bound. Both forms are very hydrophobic. Protein purification begins with protein extraction. First, bacteria are lysed in the absence of detergent, and then the pellet is collected and washed. MyrArf6 is extracted from the pellet into a solution containing the detergent Triton X-100. The protein is precipitated from the detergent extract using ammonium sulfate, dissolved, and then incubated with GDP and an Arf exchange factor so that only myrArf6-GDP is present. After removing the exchange factor, the myrArf6-GDP is further purified by size exclusion.
Materials
Bacterial culture pellet
T20N25M1D1G10 plus protease inhibitor (see recipe)
T20N100M1D1 (see recipe)
Ammonium sulfate
0.5 M EDTA (APPENDIX 2A)
GDP, disodium salt (Sigma)
GST-GEP100sec7-PH (protein preparation described in Sakurai et al., 2011)
Glutathione Sepharose 4B beads (GE Healthcare)
Hiload 16/60 Superdex 75 column
SDS-PAGE
Microfluidics M-110P cell disruptor
Refrigerated centrifuge
Resuspend bacterial pellets from 1 liter of culture in 25 ml of T20N100M1D1 plus protease inhibitor.
Lyse cells with Microfluidics M-110P cell disruptor (18,000 psi) three times. Add 20 ml T20N100M1D1 at the end of lysis to push the remaining lysate out of the loop (total volume is ∼45 ml).
Clarify cell lysates by centrifuging 40 to 60 min at 100,000 × g (Ti45 rotor), 4°C.
Resuspend pellet in 30 ml T20N100M1D1 and centrifuge 40 min at 100,000 × g, 4°C.
Resuspend pellet in 25 ml T20N25M1D1G10 plus 1% Triton X-100 to extract Arf6-GTP.
Centrifuge 1 hr at 100,000 × g, 4°C.
To the supernatant, gradually add ammonium sulfate to 35% on ice (add 201 g to 1 liter at 4°C).
- Centrifuge 30 min at 25,000 × g (Ti45 rotor), 4°C.After centrifugation, there is no tight pellet, only a thin layer of yellowish membrane.
Remove the supernatant and resuspend the yellowish membrane in 5 ml of T20N25M1D1G10 plus 1% Triton X-100 (this contains myrArf6-GTP).
To convert myrArf6-GTP to myrArf6-GDP, add 2 mM EDTA (20 μl of 0.5 M EDTA) and 10 mM GDP (100 μl of 0.5 M GDP) to the 5 ml myrArf6-GTP sample. In addition, add 1 μM of GST-GEP100sec7-PH to facilitate the conversion. Incubate sample 100 min at 30°C.
- Pass treated myrArf6 sample through glutathione Sepharose 4B beads (200 μl slurry, pre-equilibrated in T20N25M1D1G10 plus 0.1% Triton X-100 and 50 μM GDP) five times to remove GST-GEP100sec7-PH.Total flow-through volume is ∼5.8 ml.
Equilibrate a Hiload 16/60 Superdex 75 column with T20N25M1D1G10 plus 0.1% Triton X-100 and 50 μM GDP. Apply half of the flow-through from step 11 to the column. Run column two times.
- Confirm Arf by analyzing with SDS-PAGE as described in Basic Protocol 5. Pool fractions containing Arf6 and dispense into 50- to 100-μl aliquots. Flash freeze the protein in an ethanol/dry-ice bath for 1 year at −80°C.The protein can also be stored for 1 week at 4°C. Do not store protein−20°C.
BASIC PROTOCOL 6
LOADING Arf WITH [α32P]GTP TO FORM SUBSTRATE FOR Arf GAPs
Arf is loaded with radiolabeled nucleotide by exchange, accelerated by reducing Mg2+ concentrations to 1 to 10 μM (achieved with an EDTA buffer). Large unilamellar vesicles (LUVs) are necessary to stabilize the product of the exchange reaction, myrArf-GTP.
The myrArfs are purified bound to GDP. The substrate for GAPs is myrArf-GTP. Therefore, for the GAP assay, the GDP is exchanged for GTP. Radioisotopically labeled GTP is used following the hydrolysis reaction, i.e., the conversion of [α32P]GTP to [α32P]GDP. In this preparatory reaction, nucleotide exchange is accelerated by buffering Mg2+ to micromolar levels with EDTA. MyrArf-GTP is not stable without a hydrophobic binding site.
The GAP assay reactions described in this protocol contain LUVs composed of 40% (mole fraction) PC, 25% PE, 15% PS, 8% PI, 1% PIP2, 1% PIP3, and 10% cholesterol. LUVs are prepared by extrusion using a lipid extruder (Avanti Polar Lipids).
Materials
Lipids (Avanti Polar Lipids)
Phosphatidylcholine (PC, chicken egg)
Phosphatidylethanolamine (PE, bovine liver)
Phosphatidylserine (PS, porcine brain)
Phosphatidylinositol (PI, bovine liver)
Phosphatidylinositol 4,5-bisphosphate (PIP2, porcine brain)
Cholesterol
Phosphatidylinositol 3,4,5-trisphosphate (PIP3)
Chloroform
Nitrogen source
Lipid hydration buffer (see recipe)
Ethanol/dry ice bath
5× exchange buffer (see recipe)
[α32P]GTP (Perkin Elmer)
100 μM GTP
2.5 mM MgCl2 (APPENDIX 2A)
Arf
12 ×75–mm siliconized glass tube
Lyophilizer
Vortexer
37°C water bath
Lipid extruder (Avanti Polar Lipids)
Whatman Nuclepore Track Etched membrane with 1.0-μm pore size
Prepare large unilamellar vesicles (LUVs)
-
1Mix lipids in chloroform from manufacturer in a siliconized glass tube in the desired molar ratios.The authors have used 25% PC, 25% PE, 15% PS, 8% PI, 1% PIP2, 1% PIP3, and 10% cholesterol.
-
2
Evaporate chloroform under a stream of nitrogen for 15 to 20 min at room temperature in a fume hood.
-
3
Remove residual chloroform using a lyophilizer with a pressure of <100 μm of mercury for at least 1 hr.
-
4
Add lipid hydration buffer to the dried lipids to achieve a total phospholipid concentration of 5 mM. Allow lipids to hydrate 5 to 10 min at room temperature, and then vortex to obtain a suspension that appears homogeneous by eye. If necessary, dislodge dry lipids from the wall of the glass tube with buffer rapidly expelled from a pipet.
-
5
Freeze and thaw the suspension five times using an ethanol/dry ice bath and a 37°C water bath (suspension will appear white and opaque).
-
6Using the lipid extruder, pass the lipid suspension eleven times through a Whatman Nuclepore Track Etched membrane with pore size of 1.0 μm. The suspension will become opalescent.The LUVs prepared as described provide the hydrophobic surface.
Load myrArf protein with [α32P]GTP by exchange
-
7
To prepare 100 μl of ~0.25 μM Arf-GTP, add 20 μl of 5× exchange buffer, 10 μl LUV suspension, 2.5 μl radiolabeled GTP with 5 μl of 10 μM GTP (specific activity 25,000 to 100,000 cpm/pmol), and 52.5 μl water. Mix and add 10 μl of 5 μM Arf (0.1 mg/ml). Incubate for 30 to 60 min at 30°C.
-
8
Add 1.5 μl of 100 mM MgCl2 to adjust Mg2+ to final concentration of 2 mM and place on ice. The [α32P]GTP-Arf is stable for hours, but should be used the same day it is prepared.
BASIC PROTOCOL 7
MEASURING GAP ACTIVITY IN CELL LYSATES OR ISOLATED FAS
In this GAP assay, the conversion of Arf bound [α32P]GTP to [α32P]GDP is described. Briefly, myrArf-[α32P]GTP is incubated with a source of GAP, whereupon after a specified period of time the reaction is stopped by cooling and diluting the reaction. The radiolabeled GTP attached to Arf is trapped on nitrocellulose, separating the protein-bound GTP and GDP from free GTP and GDP. The radiolabeled nucleotides are extracted from the nitrocellulose and separated by thin layer chromatography, from which the radiolabel can be quantified.
Materials
myrArf protein labeled with [α32P]GTP (see Basic Protocol 6)
Cell lysates
5× GAP reaction buffer (see recipe) LUVs
T25N100M10D1 wash buffer (see recipe)
2 N formic acid
1N LiCl
Focal adhesion preparations (FAs)
Nitrocellulose membranes
Vacuum manifold
Polyethyleneimine-cellulose (TLC) plate
Phosphorimager (e.g., StormImager, GE Healthcare Lifesciences) with screens
12-well plates
GAP assay using cell lysate
1a. Load myrArf protein with [α32P]GTP according to Basic Protocol 6.
2a. Prepare serial dilutions of cell lysate (e.g., 1:0, 1:3, 1:9, 1:27) all containing 0.2% Triton X-100.
3a. Set up a 25-μl reaction on ice as follows, add 5 μl of 5× GAP reaction buffer, 2.5 μl LUV, 2.5 μl of diluted cell lysate (usually use entire dilution series), 2.5 μl of [α32P]GTP-labeled myrArf, and 12.5 μl water. The final concentration of Triton X-100 in the reaction is 0.02%.
4a. Incubate 3 min at 30°C.
5a. Stop the reaction by adding 2 ml of ice-cold T25N100M10D1 wash buffer.
6a. Trap protein-bound nucleotides on nitrocellulose membrane using a vacuum manifold. Wash filters four times with 2 ml, each time, of ice-cold T25N100M10D1.
7a. Submerge the nitrocellulose membrane in 0.75 ml of 2 N formic acid to elute the protein-bound nucleotides.
8a. Spot 20 to 30 μl of the nucleotide-containing formic acid solution on a polyethyleneimine-cellulose (TLC) plate.
9a. Separate GDP from GTP on the TLC plate by thin layer chromatography using a solution containing 1 N formic acid and 1 N LiCl.
10a.Expose TLC plate to a phosphorimager screen, then read after an appropriate period of time on a phosphorimager (e.g., StormImager). Determine the relative mass of radiolabeled GDP and GTP (Randazzo et al., 2001).
GAP assay using FA
1b. Preload myrArf protein with [α32P]GTP according to Basic Protocol 6.
2b. For GAP assay on focal adhesions in a 12-well plate, prepare a reaction mixture containing 5 μl of 5× GAP reaction buffer, 2.5 μl LUV, 2.5 μl of [α32P]GTP-labeled myrArf, and 15 μl water for each well. Keep mixture on ice.
3b. Immediately after FA preparation, add 25 μl of the reaction mixture to each well.
4b. Incubate 5 min at room temperature.
5b. Stop the reaction with 2 ml of ice-cold T25N100M10D1 wash buffer and proceed with steps 6a through 10a.
SUPPORT PROTOCOL
DETERMINING BINDING STOICHIOMETRY OF myrArf PROTEIN FOR GTP
The efficiency of nucleotide exchange on Arf should be determined to assess the quality of the protein preparation. Suitable preparations bind >0.3 mol GTPγS/mol Arf, when the mass of Arf is measured using a dye binding assay like the BioRad assay or the amido black assay (Schaffne and Weissman, 1973), use bovine serum albumin as a standard.
Materials
5× exchange buffer (see Basic Protocol 6, except with the addition of 50 μM GTPγS)
LUVs
myrArf proteins
[35S]GTPγS (PerkinElmer)
T25N100M10D1 (see recipe)
Nitrocellulose filters
Liquid scintillation counter
Set up a 100-μl reaction on ice, add 20 μl of 5× exchange buffer, 10 μl LUVs, 10 μl of 10 μM myrArf protein, 0.5 μl [35S]GTPγS, and 59.5 μl water (final concentration for myrArf is 1 μM and GTPγS is 10 μM).
Remove 5 μl of the sample, determine total radioactivity using a scintillation counter directly to calculate specific activity.
Incubate reaction tube for different time points at 30°C.
Remove 5 μl of the sample at different time points (e.g., 5, 10, 20, 30, 45, 60, 75, 90 min) and add to 2 ml of ice-cold T25N100M10D1 wash buffer.
Trap Arf-bound nucleotides on nitrocellulose filters by rapid filtration. Apply samples to nitrocellulose filters on a vacuum manifold, which is used to draw the solution through the filter. Then, wash filters four times while on the vacuum manifold with 2 ml, each wash, of ice-cold T25N100M10D1. Wash filters four times with 2 ml each of ice-cold T25N100M10D1.
Determine the amount of GTPγS bound to myrArf from each sample by liquid scintillation (assuming [35S]GTPγS and GTPγS bind to myrArf equally well).
Fit data to a first-order rate equation. The moles of GTPγS in the 5-μl sample are calculated from the amount of radioactivity and the specific activity. The moles of Arf in the samples is calculated from the concentration and volume (i.e., in this case, 5 × 10–6 liters × 1 × 10–6 mol Arf/liter = 5 pmol Arf).
REAGENTS AND SOLUTIONS
Use deionized, distilled water in all recipes and protocol steps. For common stock solutions, see APPENDIX 2A; for suppliers, see SUPPLIERS APPENDIX.
Exchange buffer, 5×
125 mM HEPES, pH 7.4
5 mM EDTA, pH 8.0 (APPENDIX 2A)
2.5 mM MgCl2 (APPENDIX 2A)
500 mM NaCl (APPENDIX 2A)
5 mM ATP (APPENDIX 2A)
5 mM DTT (APPENDIX 2A)
Store indefinitely at –20°C
GAP reaction buffer, 5×
125 mM HEPES, pH 7.4
500 mM NaCl (APPENDIX 2A)
10 mM MgCl2 (APPENDIX 2A)
5 mM DTT (APPENDIX 2A)
5 mM GTP
Store indefinitely at −20°C
Hypotonic lysis buffer
2.5 mM Tris Cl, pH 8 (APPENDIX 2A)
2 mM MgCl2 (APPENDIX 2A)
0.5 mM CaCl2 (APPENDIX 2A)
1 mM DTT
1 mM Na3VO4
1 mM NaF
Complete protease inhibitor tablets (Roche)
Prepare fresh immediately prior to use
Lipid hydration buffer
25 mM HEPES, pH 7.4
100 mM NaCl (APPENDIX 2A)
1mM dithiothreitol (APPENDIX 2A)
Store for 1 to 2 weeks at 4°C
Lysis buffer for HeLa cells
50 mM Tris Cl, pH 7.5 (APPENDIX 2A)
100 mM NaCl (APPENDIX 2A)
2 mM MgCl2 (APPENDIX 2A)
1 mM DTT (APPENDIX 2A)
1% Triton X-100 (APPENDIX 2A)
Add the Halt protease and phosphatase inhibitor cocktail (ThermoScientific) just before use
Store buffer without inhibitor cocktail 1 month at 4°C
T20N25M1D1G10 plus 0.1% Triton X-100
20 mM Tris Cl, pH 8.0 (APPENDIX 2A)
25 mM NaCl (APPENDIX 2A)
1 mM MgCl2 (APPENDIX 2A)
1 mM DTT (APPENDIX 2A)
10% glycerol
0.1% Triton X-100 (APPENDIX 2A)
Store up to 1 month at 4°C
T25N100M10D1 wash buffer
25 mM Tris Cl, pH 8.0 (APPENDIX 2A)
100 mM NaCl (APPENDIX 2A)
10 mM MgCl2 (APPENDIX 2A)
1 mM DTT (APPENDIX 2A)
Store up to1 to 2 weeks at 4°C
T20N100M1D1
20 mM Tris Cl, pH 8.0 (APPENDIX 2A)
100 mM NaCl (APPENDIX 2A)
1 mM MgCl2 (APPENDIX 2A)
1 mM DTT (APPENDIX 2A)
Store 1 to 2 weeks at 4°C
T50N3000M1D1
50 mM Tris Cl, pH 8.0 (APPENDIX 2A)
3000 mM NaCl (APPENDIX 2A)
1 mM MgCl2 (APPENDIX 2A)
1 mM DTT (APPENDIX 2A)
Store 1 to 2 weeks at 4°C
COMMENTARY
Background Information
The assays for Arf GAPs depend on one of two types of measurements. One measurement is relative amounts of GTP and GDP bound to Arf, which is usually determined using a radioactive tracer. Other assays are based on the difference in tryptophan fluorescence of Arf-GTP and Arf-GDP, first noted by Richard Kahn (Kahn and Gilman, 1986). In either case, Arf-GTP is treated as the substrate when analyzing the data, which is possible because of the very low intrinsic exchange rates of Arf. The reaction can be treated as a single substrate reaction. The dependence of initial velocity on substrate concentration and the dependence of kobs on GAP often fit simple hyperbolic equations. Enzyme parameters can be determined to help understand the biochemistry related to the biologic process, in addition to understanding the reaction mechanism, and is useful for comparison to other Arf GAPs as described below.
The substrate for Arf GAPs, myrArf-GTP, is restricted to hydrophobic surfaces. The particular surface does have a role, and the appropriate lipid environment for optimal activity should be established as part of the analysis of enzymatic properties. Keeping the lipid environment constant is important for any comparative analysis (see Critical Parameters). The lipid environment is also important to consider because of the potential effect of surface dilution phenomena (Dennis, 1973a,b; Deems et al., 1975). Comparative analysis requires a sufficient and constant surface. Holding the Arf/surface area constant should also be suitable, although the authors have never tried this. An Arf GAP that is maximally active without a surface has not been found. In the protocol described in this unit, the surface is the ventral membrane for the GAP assay using FAs and the surface is the LUVs for the GAP assay using cell lysates. However, lipids are needed to load the Arf with [α32P]GTP. Once formed, Arf-GTP is not stable without the lipids. When adding Arf to the reaction, the amount of lipid in each condition, e.g., at different Arf concentrations, must be kept constant.
Critical Parameters
The most critical parameters are the quality of the substrates and the amount of lipid surface available.
The substrate of the reaction is Arf-GTP. To determine most enzymatic parameters, the Arf must bind GTP efficiently to stoichiometries of >0.3 mol GTP/mol Arf. Myristoylated Arf binds GTP much more efficiently than the non-myristoylated Arf. Both myrArf and non-myrArf require hydrophobic surfaces, in the form of a detergent micelle, LUV, or other phospholipid membrane, to bind GTP. A C-terminal histidine tag on Arf has been found to be relatively innocuous, although preparation of the native protein is simple so that the tag may not be warranted. Other modifications of Arf often have a deleterious effect on either GTP binding and/or interaction with GAP. For example, GST-fused Arf binds <0.005 mol GTP/mol Arf and is poorly recognized by GAPs (Jian et al., 2010). Truncating the N-terminal 17 amino acids from Arf, e.g., [∆17]Arf1, results in a protein that binds nucleotide with high stoichiometry but is not used by some Arf GAPs (Randazzo et al., 1994; Yoon et al., 2004; Luo et al., 2005). In general, modifying Arf has not been found to be useful except when performing mutational analysis to determine the regions of Arf involved in GAP recognition.
Arthur Kornberg said, “Thou shall not waste clean thinking on dirty enzymes.” (Kornberg, 2000, 2003). The data from the assays described are easiest to interpret when using purified GAPs; however, the purified GAPs are not always stable and/or may lack factors for maximal activity. These considerations justify the use of cell fractions; however, confounding factors must be considered. First, the assay could fail because of contaminating exchange factor activity (signal is lost due to dissociation of [α32P]GDP from Arf). This is easily assessed by determining the recovery of Arf-bound radioisotope. The authors have found that the amount of [α32P]GXP (GXP = GTP + GDP) bound to Arf to be constant under the conditions of this assay, indicating that exchange is not an important confounding factor. Another confounding factor is effectors sequestering Arf-GTP. In the latter case, even if the reaction goes to completion, it does not exclude the presence of Arf effectors that may slow the reaction or influence kinetics. Ways to address this issue are being considered, but have yet to find a robust approach at this time. The other confounding factor, multiple Arf GAPs, can be addressed in several ways. One is using standard kinetic approaches, described by Segel (1975). The other approach is to use subcellular fractions from cells in which the GAP of interest has either been overexpressed or eliminated using siRNA or by immunodepletion if a suitable antibody is available.
As previously described, ensuring that lipids are constant is important for making comparisons. The FA preparations have been consistent in the authors’ hands, presumably with a constant amount of lipid surface. The only other lipid comes from the preparation of Arf-GTP, which can easily be controlled and held constant.
The following parameters are critical in interpreting experimental results.
First, it is useful to know that the GAP is fully active under the conditions of the experiment. Epitope tags can disrupt GAP activity, especially those on the N-terminus of Arf GAPs with the Arf GAP domain at the extreme N-terminus of the protein. Epitope tags on Arf also disrupt interaction with the GAP (Jian et al., 2010). Some Arf GAPs are autoinhibited in the full-length form (Jian et al., 2009). In the autoinhibited state, the Arf GAPs have some activity, which could be observed at high concentrations of the proteins used in overexpression experiments, but they are not specific for a particular Arf. Activation, either by addition of the appropriate ligand or by removal of an autoinhibitory motif, results in greater activity for the physiologically relevant Arf. Unfortunately, the physiologically relevant pathways leading to activation of the Arf GAPs in cells are not well described.
Second, experiments in which the Arf and/or GAP are overexpressed can also be difficult to interpret for reasons related to the properties of the enzymatic reaction. The particular Arf used as substrate is determined by the relative concentration of the substrate Arf and the GAP relative to the Km for the reaction. At high concentrations of the GAP, non-substrate Arfs may be used. The concentration of Arf-HA is also relevant, as the nonphysiologically relevant Arf may be used if present in excess of the physiologically relevant Arf. For example, if the Km for Arf1 is 0.1 μM and the Km for Arf6 is 1 μM, little difference in activity would be observed if the GAP and/or Arfs were present at concentrations approaching 1 μM (Segel, 1975).
Third is a factor that affects interpretation of experiments in which expression of a particular Arf GAP is reduced. Multiple Arf GAPs are expressed in cells. Unless a condition is found under which the Arf GAP in question is the dominant active Arf GAP, one might not observe an effect over background. Even if the Arf GAP is active at a particular site, Arf associated with the site may represent only a small fraction of total cellular Arf, and therefore a change would not be detected.
A fourth consideration for interpreting the in vivo results is that the Arfs are also regulated by GEFs and bind to effectors. Any change caused by perturbing the GAP could be complicated by changes in GEF activity. For instance, Arf1 exchange factors are activated by Arf6-GTP (Cohen et al., 2007; DiNitto et al., 2010), so Arf6GAPs could change Arf1-GTP levels secondary to its direct effect on Arf6-GTP.
One approach the authors have taken to complement the in vivo determination of Arf GAP specificity is to assay GAP activity in cellular fractions of interest. Although these have the shortcomings of in vitro experiments in that the reaction is removed from the cellular environment, it has the following advantages: Native Arf is used as substrate. Native Arf is the covalently modified with myristate Arf lacking epitope tags. The protein can be expressed and purified from bacteria as described.
Arf concentration is controlled, allowing determination of relative activity towards different isoforms.
The quantity of GAP is limited to the amount that can associate with the compartment of interest.
The isolated GAP reaction is examined without exchange factor activity confounding results.
Dirty enzymes, which have potential confounding factors, have been used (Kornberg, 2000, 2003). Some of these shortcomings, e.g., presence of more than one GAP, can be addressed, at least in part, as described in the protocols.
Troubleshooting
Lack of signal is often related to the preparation of the substrate Arf-GTP. This problem could result from nucleotidases that frequently contaminate Arf preparations, limiting the amount of GTP binding due to hydrolysis before the GAP reaction. One virtue of assays using [α32P]GTP is that the problem is easily identified by the large amount of Arf1-α32P]GDP in the substrate preparation after loading. Nucleotidase can be inhibited with high concentrations of ATP and pyrophosphate if the contamination is minor. The nucleotidases can often be removed by an additional purification step. Hydroxylapaptite has been the most useful column, but others such as a cation exchange column are also effective.
Some Arf preparations may load poorly with nucleotide. If Arf is not myristoylated, nucleotide binding stoichiometries of ≤5% are typical. One way to check myristoylation is to compare migration of the protein with Arf that was expressed in bacteria without NMT, known to not have myristate by analysis on SDS-PAGE. Myristoylated Arf migrates faster than non-myristoylated Arf. Mutations in switch regions of Arf can also affect relative affinities for GTP and GDP, and may either decrease or increase GTP binding.
Impure preparations of Arf GAPs can have confounding factors, e.g., inhibitors or Arf effectors that can sequester Arf-GTP. Sometimes simply diluting the Arf GAP preparation can reduce the effect of proteins that sequester Arf-GTP. Adding saturating amounts of Arf-GTP can also eliminate problems associated with sequestration, although interpretation could be complicated for other reasons, as described in the introduction. It is worthwhile to titrate the substrate to help in interpretation when these confounding factors may be present.
Curve fitting of the time dependence of the signals can be useful for identifying artifacts. For substrate saturation kinetics, the time dependence can be linear or, if a significant amount of substrate is consumed, the time dependence can be modeled by a first or second order reaction. The authors have not encountered a reaction that cannot be reasonably well fit to one of these equations. While it is true that there may be better equations with linear and exponential elements, first and second order equations will give a good fit of almost every data set with substrate in excess. If they do not, suspect an artifact. For single turnover, or when enzyme concentration is high relative to substrate, burst kinetics may be observed, which will fit a linear and exponential equation (Gutfreund, 1972). However, if the enzyme is limiting, the presence of a burst of activity consuming more substrate than there is enzyme would raise concerns of an artifact.
Anticipated Results
In Figure 17.13.3, thin layer chromatographic separation of GDP from GTP for an experiment in which C50 was determined is shown. In the sample in which GAP was not added, >90% of the guanine nucleotide recovered was GTP. In the sample with GAP, >90% of the nucleotide was GDP.
Figure 17.13.3.
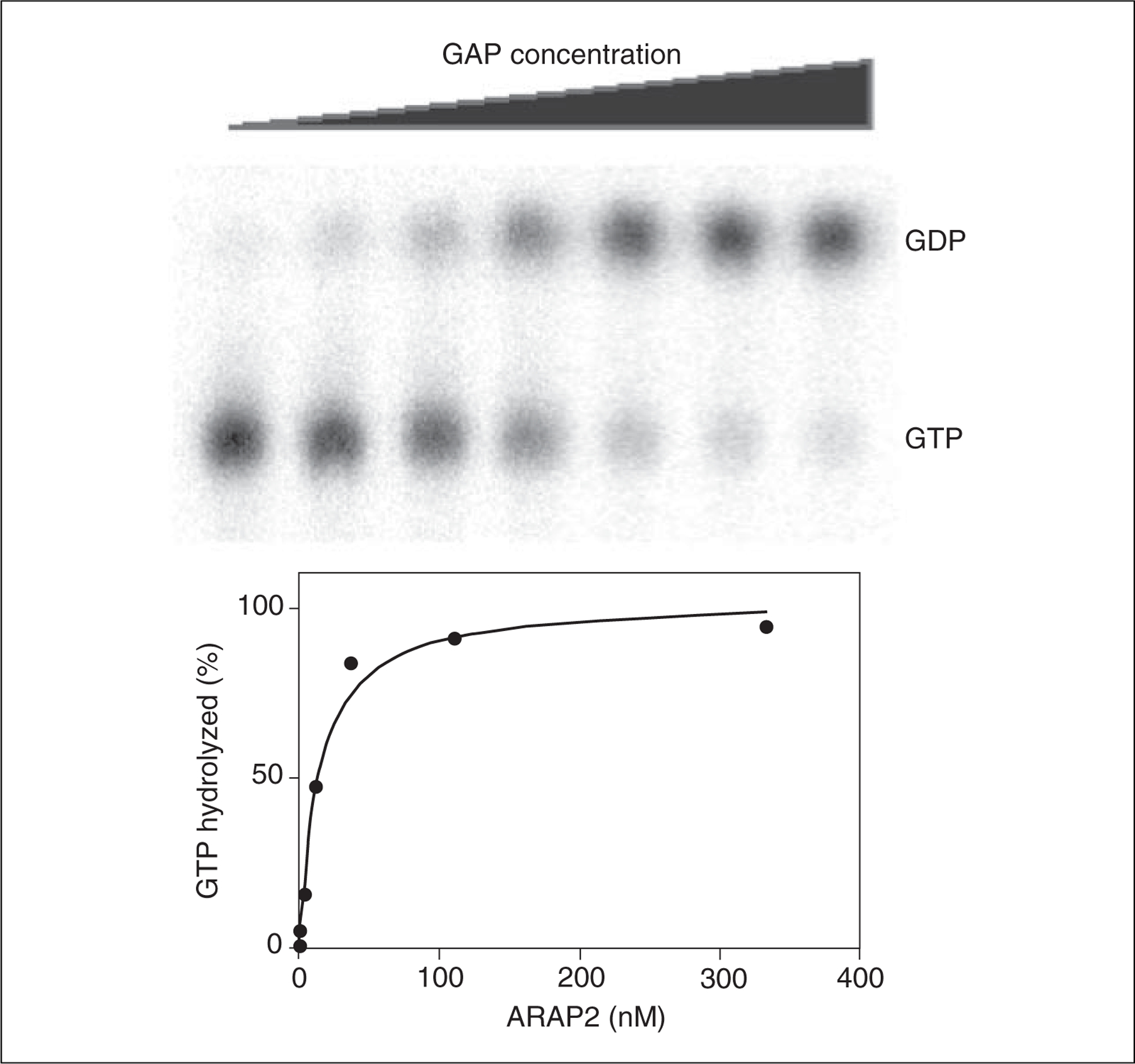
Determination of C50. Top panel, phosphoimager image from an experiment in which ARAP2 partially purified from cell lysates was used to catalyze GTP hydrolysis on myrArf6. Lower panel, the quantified GTP hydrolysis is fitted to hyperbolic equation to determine C50.
Time Considerations
In the preparation of myrArf, the bacteria are first recovered from the glycerol stock with an overnight incubation. A culture is started on day 2. On day 3, the bacteria are induced to express protein. Lysis and extraction of the protein from the bacteria typically takes 3 hr, including clarifying centrifugation, and each of the chromatographic steps is about half of a day of work. Together, plan on 1 week to prepare the purified protein. Enough protein is recovered for multiple experiments.
For the preparation of cell lysates and FA fractions, the total time taken for preparing whole cell lysates and FA fractions depends on the type of treatments on the cells. Growth factors or pharmacological inhibitor–treated cells can be lysed in minutes or hours. For overexpression types of studies, cells are plated on day 1, transfected on day 2, and lysed on day 3. siRNA-mediated knockdown experiments take longer depending on the time required to reduce protein expression (for 72 hr of siRNA treatment, the total time until harvest lysates is 5 days). For FA fractions, allow an additional 1 day for ECM coating before setting up cells. Following different treatments, it takes ~45 to 60 min to harvest total cell lysates and determine protein concentration. Cell lysates can be prepared in advance, frozen in small aliquots, and stored at −80°C until used. On the other hand, FA fractions have to be prepared fresh right before the GAP assay. It takes ~10 to 15 min from intact cells on the plates to the FA fraction ready for GAP assay. It is critical to keep the time between the end of FA preparation and the addition of Arf-[α32P]GTP as short as possible. Therefore, allow enough time to purify myrArf and to load [α32P]GTP (30 min to 1 hr depending on which Arf is used) onto myrArf before start of FA preparation.
Acknowledgment
This work was supported by the intramural program of the National Cancer Institute, National Institutes of Health.
Literature Cited
- Campa F and Randazzo PA 2008. Arf GTPase-activating proteins and their potential role in cell migration and invasion. Cell Adh. Migr 2:258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LA and Donaldson JG 2010. Analysis of Arf GTP-binding protein function in cells. Curr. Protoc. Cell Bio 48:14.12.1–14.12.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LA, Honda A, Varnai P, Brown FD, Balla T, and Donaldson JG 2007. Active Arf6 recruits ARNO/cytohesin GEFs to the PM by binding their PH domain. Mol. Biol. Cell 18:2244–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukierman E, Huber I, Rotman M, and Cassel D 1995. The Arf1 GTPase-activating protein— Zinc-finger motif and Golgi complex localization. Science 270:1999–2002. [DOI] [PubMed] [Google Scholar]
- Deems RA, Eaton BR, and Dennis EA 1975. Kinetic-analysis of phospholipase-A2 activity toward mixed micelles and its implications for study of lipolytic enzymes. J. Biol. Chem 250:9013–9020. [PubMed] [Google Scholar]
- Dennis EA 1973a. Phospholipase A2 activity towards phosphatidylcholine in mixed micelles— Surface dilution kinetics and effect of thermotropic phase-transitions. Arch. Biochem. Biophys 158:485–493. [DOI] [PubMed] [Google Scholar]
- Dennis EA 1973b. Kinetic dependence of phospholipase A2 activity on detergent triton X-100. J. Lipid Res 14:152–159. [PubMed] [Google Scholar]
- DiNitto JP, Lee MT, Malaby AW, and Lambright DG 2010. Specificity and membrane partitioning of Grsp1 signaling complexes with Grp1 family Arf exchange factors. Biochemistry 49:6083–6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG and Jackson CL 2011. ARF family G proteins and their regulators: Roles in membrane transport, development and disease. Nat. Rev. Mol. Cell Biol 12:362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigerio G, Grimsey N, Dale M, Majoul I, and Duden R 2007. Two human ARFGAPs associated with COP-I-coated vesicles. Traffic 8:1644–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingham AK and Munro S 2007. The small G proteins of the Arf family and their regulators. Ann. Rev. Cell Dev. Biol 23:579–611. [DOI] [PubMed] [Google Scholar]
- Gutfreund H 1972. Enzymes: Physical Principles John Wiley and Sons, New York. [Google Scholar]
- Inoue H and Randazzo PA 2007. Arf GAPs and their interacting proteins. Traffic 8:1465–1475. [DOI] [PubMed] [Google Scholar]
- Jian XY, Brown P, Schuck P, Gruschus JM, Balbo A, Hinshaw JE, and Randazzo PA 2009. Autoinhibition of Arf GTPase-activating protein activity by the BAR domain in ASAP1. J. Biol. Chem 284:1652–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian XY, Cavenagh M, Gruschus J, Randazzo PA, and Kahn RA 2010. Modifications to the C-terminus of Arf1 alter cell functions and protein interactions. Traffic 11:732–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn RA and Gilman AG 1986. The protein cofactor necessary for ADP-ribosylation of Gs by cholera-toxin is itself a GTP binding-protein. J. Biol. Chem 261:7906–7911. [PubMed] [Google Scholar]
- Kahn RA, Cherfils J, Elias M, Lovering RC, Munro S, and Schurmann A 2006. Nomenclature for the human Arf family of GTP-binding proteins: ARF, ARL, and SAR proteins. J. Cell Biol 172:645–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn RA, Bruford E, Inoue H, Logsdon JM, Nie Z, Premont RT, Randazzo PA, Satake M, Theibert AB, Zapp ML, and Cassel D 2008. Consensus nomenclature for the human ArfGAP domain containing proteins. J. Cell Biol 182:1039–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg A 2000. Ten commandments: Lessons from the enzymology of DNA replication. J. Bacteriol 182:3613–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg A 2003. Ten commandments of enzymology, amended. Trends Biochem. Sci 28:515–517. [DOI] [PubMed] [Google Scholar]
- Kuo JC, Han XM, Hsiao CT, Yates JR, and Waterman CM 2011. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for beta-Pix in negative regulation of focal adhesion maturation. Nat. Cell Biol 13:383–U109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YH, Loijens JC, Martin KH, Karginov AV, and Parsons JT 2002. The association of ASAP1, an ADP ribosylation factor-GTPase activating protein, with focal adhesion kinase contributes to the process of focal adhesion assembly. Mol. Biol. Cell 13:2147–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Yerushalmi GM, Grigera PR, and Parsons JT 2005. Mislocalization or reduced expression of Arf GTPase-activating protein ASAP1 inhibits cell spreading and migration by influencing Arf1 GTPase cycling. J. Biol. Chem 280:8884–8892. [DOI] [PubMed] [Google Scholar]
- Luo RB, Jacques K, Ahvazi B, Stauffer S, Premont RT, and Randazzo PA 2005. Mutational analysis of the Arf1 center dot GTP/Arf GAP interface reveals an Arf1 mutant that selectively affects the Arf GAP ASAP1. Curr. Biol 15:2164–2169. [DOI] [PubMed] [Google Scholar]
- Miura K, Jacques KM, Stauffer S, Kubosaki A, Zhu KJ, Hirsch DS, Resau J, Zheng Y, and Randazzo PA 2002. ARAP1: A point of convergence for Arf and Rho signaling. Mol. Cell 9:109–119. [DOI] [PubMed] [Google Scholar]
- Nie ZZ and Randazzo PA 2006. Arf GAPs and membrane traffic. J. Cell Sci 119:1203–1211. [DOI] [PubMed] [Google Scholar]
- Randazzo PA and Kahn RA 1995. Myristoylation and ADP-ribosylation factor function. Method Enzymol 250:394–405. [DOI] [PubMed] [Google Scholar]
- Randazzo PA, Terui T, Sturch S, and Kahn RA 1994. The amino-terminus of ADP-ribosylation factor (Arf)-1 is essential for interaction with G(s) and Arf GTPase-activating protein. J. Biol. Chem 269:29490–29494. [PubMed] [Google Scholar]
- Randazzo PA, Andrade J, Miura K, Brown MT, Long YQ, Stauffer S, Roller P, and Cooper JA 2000. The Arf GTPase-activating protein ASAP1 regulates the actin cytoskeleton. Proc. Natl. Acad. Sci. U.S.A 97:4011–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randazzo PA, Miura K, and Jackson TR 2001. Assay and purification of phosphoinositide-dependent ADP-ribosylation factor (ARF). Methods Enzymol 329:343–354. [DOI] [PubMed] [Google Scholar]
- Randazzo PA, Inoue H, and Bharti S 2007. Arf GAPs as regulators of the actin cytoskeleton. Biol. Cell 99:583–600. [DOI] [PubMed] [Google Scholar]
- Sakurai A, Jian X, Lee CJ, Manavski Y, Chavakis E, Donaldson J, Randazzo PA, and Gutkind JS 2011. Phosphatidylinositol-4-phosphate 5-kinase and GEP100/Brag2 protein mediate antiangiogenic signaling by semaphorin 3E-plexin-D1 through Arf6 protein. J. Biol. Chem 286:34335–34345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffne W and Weissman C 1973. Rapid, sensitive, and specific method for determination of protein in dilute-solution. Anal. Biochem 56:502–514. [DOI] [PubMed] [Google Scholar]
- Segel I 1975. Enzyme Kinetics John Wiley and Sons, Inc., New York. [Google Scholar]
- Tong WY, Liang M, Tam V, Yip HK, Kao YT, Cheung KMC, Yeung KWK, and Lam YW 2010. Biochemical characterization of the cell-biomaterial interface by quantitative proteomics. Mol. Cell Proteomics 9:2089–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon H-Y, Jacques K, Nealon B, Stauffer S, Premont RT, and Randazzo PA 2004. Differences between AGAP1, ASAP1 and Arf GAP1 in substrate recognition: Interaction with the N-terminus of Arf1. Cell. Signal 16:1033–1044. [DOI] [PubMed] [Google Scholar]
- Yoon H-Y, Bonifacino JS, and Randazzo PA 2006. In vitro assays of Arf1 interaction with GGA proteins. Methods Enzymol 404:316–332. [DOI] [PubMed] [Google Scholar]