

Abstract
Genes encoding glutamine phosphoribosylpyrophosphate amidotransferase (GPAT) and glycinamide ribonucleotide synthetase (GARS) from Aquifex aeolicus were expressed in Escherichia coli, and the enzymes were purified to near homogeneity. Both enzymes were maximally active at a temperature of at least 90°C, with half-lives of 65 min for GPAT and 60 h for GARS at 80°C. GPAT activity is known to depend upon channeling of NH3 from a site in an N-terminal glutaminase domain to a distal phosphoribosylpyrophosphate site in a C-terminal domain where synthesis of phosphoribosylamine (PRA) takes place. The efficiency of channeling of NH3 for synthesis of PRA was found to increase from 34% at 37°C to a maximum of 84% at 80°C. The mechanism for transfer of PRA to GARS is not established, but diffusion between enzymes as a free intermediate appears unlikely based on a calculated PRA half-life of approximately 0.6 s at 90°C. Evidence was obtained for coupling between GPAT and GARS for PRA transfer. The coupling was temperature dependent, exhibiting a transition between 37 and 50°C, and remained relatively constant up to 90°C. The calculated PRA chemical half-life, however, decreased by a factor of 20 over this temperature range. These results provide evidence that coupling involves direct PRA transfer through GPAT-GARS interaction rather than free diffusion.
The first two steps in de novo purine nucleotide synthesis involve reactions in which labile enzyme intermediates are transferred between distal, noncontiguous sites. In the first reaction, catalyzed by glutamine phosphoribosylpyrophosphate (PRPP) amidotransferase (GPAT), NH3, generated by hydrolysis of glutamine at an N-terminal glutaminase domain, is sequestered from H2O by transfer through a 20-Å tunnel to a C-terminal PRPP site where synthesis of phosphoribosylamine (PRA) takes place (11). Channeling of NH3 through a hydrophobic tunnel prevents its release from the enzyme and also avoids the likelihood of protonation of NH3 to NH4+, which is not a substrate (13). These steps are shown by equations 1 and 2, and the synthesis of glycinamide ribonucleotide (GAR) by the second enzyme, GAR synthetase (GARS), is given in equation 3.
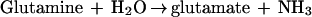 |
1 |
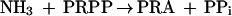 |
2 |
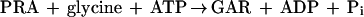 |
3 |
The GPAT half reactions are coupled, and NH3 is channeled by transient forms of the enzyme, in which closing of a flexible loop upon PRPP binding activates the glutaminase domain and forms an NH3 channel between the active sites (1, 2, 4, 11).
PRA, the product of the second GPAT half reaction, is susceptible to hydrolysis to ribose 5-phosphate, with a half-life of 5 s under physiological conditions at 37°C (17). The kinetics for the synthesis of GAR by Escherichia coli GPAT and GARS were found to be inconsistent with free diffusion of PRA between enzymes and, as a result, a direct-transfer mechanism was proposed (17). Since evidence for a GPAT-GARS physical interaction was not obtained, direct PRA transfer was assumed to involve a transient channeling interaction between GPAT and GARS. The transient NH3 channel in E. coli GPAT may be a precedent for a similar type of GPAT-GARS channel.
As a first step to assess how labile intermediates of purine biosynthesis are utilized at elevated temperatures in hyperthermophilic bacteria, we have expressed Aquifex aeolicus purF and purD in E. coli and purified the GPAT and GARS encoded by their respective genes. With a maximum growth temperature near 95°C, A. aeolicus is one of the most thermophilic bacteria known (6). Here we report an initial characterization of the two enzymes, the function of the GPAT NH3 channel, and evidence for coupling between GPAT and GARS.
MATERIALS AND METHODS
Cloning of A. aeolicus purF and purD.
The A. aeolicus purF and purD genes were amplified by PCR from chromosomal DNA, generously supplied by Robert Huber and Karl Stetter, Universität Regensburg, Regensburg, Germany. The purF and purD genes, with appropriate flanking restriction sites, were digested with NdeI/HindIII and NdeI/XhoI, respectively, and then ligated into the corresponding restriction sites in pET24a vector (Novagen). The complete nucleotide sequence of each cloned gene, determined by the DNA sequencing facility of Iowa State University, was identical with that previously determined (6).
Enzyme purification.
E. coli strain B834 (DE3) (7) was used for enzyme production. Plasmid-bearing cells were grown at 37°C in Luria broth medium containing 50 μg of kanamycin/ml for 15 to 20 h. The cells were collected by centrifugation and washed with phosphate-buffered saline as described previously (4). All steps, except as noted, were carried out at approximately 4°C. The cells in buffer containing 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 0.1 mM EDTA, and 100 μg of phenylmethane sulfonylfluoride/ml were disrupted with a French press, and the soluble extract was obtained following centrifugation (4). The extract (35 ml; 19 mg of protein/ml) was incubated at 75°C for 5 min and then cooled in ice. The heat-denatured proteins were removed by centrifugation at 18,000 × g for 10 min. The heat treatment and centrifugation steps were repeated a second time, and the supernatant was then fractionated by precipitation with 40% saturated (NH4)2SO4 for GPAT and 50% (NH4)2SO4 for GARS. The soluble protein solutions, obtained after centrifugation at 18,000 × g for 10 min, were loaded onto a 2.5- by 8-cm column of butyl Sepharose equilibrated with column buffer (50 mM Tris-HCl [pH 7.5], 10 mM MgCl2) and 40% (NH4)2SO4. GPAT was eluted from the column with 100 ml of a 20 to 0% linear gradient of (NH4)2SO4 in column buffer and GARS was eluted with 100 ml of a 25 to 0% linear gradient of (NH4)2SO4 in column buffer. For GPAT, the brown-colored fractions with an absorbance maximum at 415 nm were pooled and concentrated by ultrafiltration using a Centricon-30 ultrafilter. Fractions containing GARS were identified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and were pooled and concentrated as for GPAT. E. coli GARS was overproduced and purified as described previously (4).
Enzyme assays.
Two assays were used for GPAT. The production of glutamate was used to assay glutaminase activity (equation 1). Each reaction mixture contained 50 mM Tris-HCl (pH 7.5 at the temperature of assay), 10 mM PRPP, 30 mM glutamine, 10 mM MgCl2, 50 μg of bovine serum albumin, and 0.2 to 12 μg of enzyme in a final volume of 50 μl. Incubation was for 5 min at various temperatures. Reactions were quenched by the addition of EDTA to a final concentration of 20 mM, and the tubes were placed in ice. Glutamate was determined by the glutamate dehydrogenase method (13). The overall reaction, production of PRA from glutamine and PRPP (equations 1 and 2), was assayed by measuring PPi formation. This activity is referred to as Gln-PRA. The reaction mixture and incubation time were exactly the same as for determination of glutaminase activity. PPi was measured (23) with a kit supplied by Molecular Probes, Inc. (Eugene, Oreg.). The 200-μl reaction mixture contained 20 mM Tris-HCl [pH 7.5], 0.4 mM 2-amino-6-mercapto-7-methylpurine ribonucleoside, 5 mM MgCl2, 0.2 U of purine nucleoside phosphorylase, 0.2 U of inorganic pyrophosphatase, and 5 to 10 μl of the Gln-PRA reaction mixture. Incubation was for 10 min at room temperature, after which formation of 2-amino-6-mercapto-7-methylpurine was measured at 360 nm.
The coupling of PRA formation to synthesis of GAR (the sum of equations 1 to 3) was determined in a 50-μl reaction mixture containing 50 mM Tris-HCl (pH 7.5 at each temperature), 30 mM glutamine, 10 mM PRPP, 10 mM MgCl2, 50 μg of bovine serum albumin, 10 mM ATP, 5 mM [14C]glycine (1,330 cpm/nmol), and a 10-fold excess of GARS enzyme units relative to GPAT enzyme units at each temperature of assay. Incubation was for 5 min at the desired temperature, and reactions were quenched by addition of trichloroacetic acid to a final concentration of 8%. GAR was measured as described by Schendel et al. (18). This coupled reaction is referred to as Gln-GAR.
GAR synthetase was assayed by two procedures differing in the method used to continuously generate the unstable substrate PRA. In one method, PRA was generated enzymatically using 10-fold excess units of GPAT compared to GARS. Otherwise, the assay mixture was the same as for Gln-GAR. In a second method, PRA was generated chemically (19) in a reaction mixture containing 50 mM Tris-HCl (pH 9.2), 500 mM NH4Cl, and 250 mM ribose-5-phosphate incubated at 37°C for 15 min. An aliquot of the PRA mixture was diluted fivefold into a 50-μl GARS reaction mixture that also contained 50 mM Tris-HCl, 50 μg of bovine serum albumin, 10 mM ATP, 5 mM [14C]glycine (1,330 cpm/nmol), and 10 mM CaCl2. The temperature-sensitive Tris buffer resulted in an assay pH of 9.0 at 37°C, 8.7 at 50°C, 8.5 at 60°C, 8.3 at 70°C, 8.1 at 80°C, and 7.9 at 90°C. GARS activity determined by chemical generation of PRA was approximately three- to fivefold higher than by the enzymatic method, due mainly to the inclusion of CaCl2. CaCl2 inhibited the enzymatic generation of PRA and therefore was not used with GPAT. For the Gln-GAR assay, the amount of GAR synthetase was based on enzymatic generation of PRA. For A. aeolicus GARS, the assay temperature was varied between 37 and 90°C. E. coli GARS was assayed at 37 and 50°C.
For all enzyme assays, 1.0 U of activity is defined as the amount of enzyme needed to produce 1 μmol of product per min. Specific activity is expressed in units per milligram of protein. To improve quantitation, a larger number of repeated measurements was taken for assay conditions that produced lower specific activities. Each reported specific activity includes multiple measurements for which the signal was 5 to 15 times greater than the background absorbance or radioactivity. Protein was measured by the Bradford method (3) using bovine serum albumin as a standard.
Inhibition by nucleotides.
Inhibition by nucleotides was carried out as described previously (5). Synergism refers to the increased inhibition by one nucleotide, expressed as the concentration required for 50% inhibition (I0.5), at a fixed concentration of a second nucleotide. The concentration of the fixed nucleotide was set to approximately 0.1 or 0.5 times its I0.5 value. Inhibition was carried out at 80°C.
Calculation of channeling efficiency.
The fraction of the NH3 produced by the GPAT glutaminase domain (Table 1, glutaminase) that is channeled to the PRTase domain for synthesis of PRA (Table 1, Gln-PRA) is referred to as channeling efficiency (2). Channeling efficiency is greater than 90% for E. coli GPAT at 37°C in vitro (2). It is not known whether the experimental value is lower than 100% because of limitations in the assay or whether channeling of NH3 is truly less than 100% efficient.
TABLE 1.
Ammonia channel function and GPAT-GARS coupling
| GPATa | GARSa | Temp (°C) | Activity (U/mg)
|
Channel efficiencyb | Coupling efficiencyc | ||
|---|---|---|---|---|---|---|---|
| Glutaminase | Gln-PRA | Gln-GAR | |||||
| Ec | Ec | 37 | 54.6 ± 1.85 (3)d | 52.4 ± 2.67 (3) | 51.0 ± 2.27 (3) | 0.96 | 0.97 |
| Aa | Ec | 37 | 4.24 ± 0.20 (8) | 1.42 ± 0.15 (8) | 0.41 ± 0.05 (3) | 0.34 | 0.29 |
| Aa | Aa | 37 | 4.24 ± 0.20 (8) | 1.42 ± 0.15 (8) | 0.26 ± 0.04 (12) | 0.34 | 0.18 |
| Aa | Ec | 50 | 7.36 ± 0.60 (9) | 3.90 ± 0.39 (6) | 3.21 ± 0.2 (3) | 0.53 | 0.82 |
| Aa | Aa | 50 | 7.36 ± 0.60 (9) | 3.90 ± 0.39 (6) | 2.63 ± 0.30 (8) | 0.53 | 0.76 |
| Aa | Aa | 60 | 11.8 ± 0.80 (3) | 7.63 ± 0.24 (3) | 5.84 ± 0.14 (3) | 0.65 | 0.76 |
| Aa | Aa | 70 | 30.4 ± 1.40 (6) | 22.6 ± 1.00 (3) | 15.1 ± 0.4 (3) | 0.74 | 0.67 |
| Aa | Aa | 80 | 40.5 ± 1.30 (10) | 35.4 ± 1.50 (3) | 22.4 ± 1.1 (3) | 0.87 | 0.63 |
| Aa | Aa | 90 | 53.2 ± 1.00 (5) | 44.7 ± 1.20 (4) | 27.9 ± 1.1 (5) | 0.84 | 0.62 |
Ec, E. coli; Aa, A. aeolicus.
Fraction of NH3 derived from glutamine hydrolysis (glutaminase) utilized for PRA (Gln-PRA).
Fraction of PRA (Gln-PRA) converted to GAR (Gln-GAR).
Number of repeated measurements.
Calculation of coupling efficiency.
Coupling efficiency is defined as the fraction of PRA produced by GPAT (Table 1, Gln-PRA) that is converted to GAR by an excess of GARS (Table 1, Gln-GAR).
Estimation of chemical half-lives.
The spontaneous rate of PRPP hydrolysis was determined at 90°C in 50 mM Tris-HCl (pH 7.5). Aliquots were removed as a function of time, and the PRPP remaining was determined using E. coli GPAT. Glutamate production was measured, and a first-order rate constant and half-life were calculated. The rate of PRA decay at pH 7.5 and 90°C was estimated by extrapolation of rate constants determined at 24°C (0.31 min−1) and 37°C (1.09 min−1) (18). These conditions are those used for in vitro enzyme assay. They differ from conditions with an E. coli extract (17) and were designed to mimic an in vivo environment. For the latter case, a rate constant of 8.4 min−1 was determined for PRA decay at 37°C (17).
RESULTS
Isolation and characterization of A. aeolicus GPAT.
The A. aeolicus purF gene was amplified by PCR from chromosomal DNA and inserted into a T7 expression vector for enzyme production in E. coli. The A. aeolicus enzyme overproduced in E. coli was purified to near homogeneity as indicated by SDS-PAGE (Fig. 1). The brown color of the protein with an absorption maximum at 415 nm (data not shown) is indicative of the presence of an Fe-S cluster, perhaps similar to the 4Fe-4S cluster in the Bacillus subtilis enzyme (20). Four cysteine ligands to the Fe-S cluster in B. subtilis GPAT are conserved in the A. aeolicus sequence. The amino acid sequences of the A. aeolicus and B. subtilis GPAT enzymes are 44% identical.
FIG. 1.

Purification of A. aeolicus GPAT and GARS from an E. coli expression system. SDS-PAGE of GPAT (lanes 1 to 4) and GARS (lanes 5 to 8) is shown. Lanes 1 and 5, soluble cell extracts; lanes 2 and 6, soluble protein following two heat denaturation steps; lanes 3 and 7, soluble protein following (NH4)2SO4 precipitation; lanes 4 and 8, purified proteins; lane 9, molecular mass markers (Bio-Rad). The SDS–10% PAGE gel was stained with Coomassie brilliant blue R. The arrows show the positions of GPAT and GARS.
Synthesis of PRA takes place in two steps at separated sites. First, a glutaminase activity hydrolizes glutamine to glutamate plus NH3 (equation 1). Second, NH3 derived from glutamine hydrolysis is transferred through a 20-Å channel for reaction with PRPP to yield PRA. The first half reaction is catalyzed by a glutaminase activity, and the overall reaction is designated Gln-PRA. There was a broad pH optimum between pHs 6.5 and 8.5 for glutaminase activity, determined at 80°C (data not shown). B. subtilis GPAT has a similarly broad pH optimum at 37°C (14). Accordingly, pH 7.5 was used for assays of glutaminase and Gln-PRA activities and for the coupled synthesis of GAR by GPAT and GARS. The data in Table 1 show the temperature dependence of the glutaminase and Gln-PRA activities. The optimal assay temperature for both activities was at least 90°C. At 90°C, the two activities of the A. aeolicus GPAT, glutaminase and Gln-PRA, were 97 and 85%, respectively, of the corresponding activities of the E. coli enzyme at 37°C. The stability of the A. aeolicus GPAT was tested at 37 and 80°C. The glutaminase half-life was 25 h at 37°C and 65 min at 80°C. Loss of activity was accompanied by loss of brown color.
The Km values for PRPP and glutamine at 90°C were 0.87 ± 0.22 and 3.0 ± 0.4 mM, respectively, and were not significantly different at 60°C. In contrast to E. coli GPAT (9), the A. aeolicus enzyme has essentially no basal glutaminase activity at any of the assay temperatures. That is, the glutaminase activity is completely dependent on binding of PRPP.
Inhibition of GPAT by nucleotides.
Synergistic end product inhibition by adenine and guanine nucleotides, a hallmark of microbial and mammalian GPAT (5, 13, 14, 24), is a mechanism for the regulatory enzyme of the purine pathway to sense output from both branches of the pathway. Synergistic inhibition of B. subtilis GPAT results from the interaction between the β-phosphate of a nucleoside diphosphate (ADP) bound to an allosteric site and the ribose 2′ OH of a nucleoside monophosphate (GMP) bound to the catalytic site (5). A limited survey for inhibition of A. aeolicus GPAT by adenine and guanine nucleotides at 80°C indicated similar inhibition by AMP and ADP (Table 2). GDP and GMP were inhibitory at slightly higher concentrations than the adenine nucleotides. AMP and GDP were the most inhibitory pair, exhibiting five- to sixfold synergism under conditions that produce 188-fold synergism for inhibition of B. subtilis GPAT by ADP and GMP (5).
TABLE 2.
Synergistic inhibition of A. aeolicus GPAT by nucleotides
| Nucleotide
|
I0.5b | Synergismc | |
|---|---|---|---|
| Varied | Fixeda | ||
| AMP | 6.12 ± 1.2 | ||
| ADP | 4.10 ± 0.8 | ||
| GMP | 17.9 ± 2.2 | ||
| GDP | 8.50 ± 1.2 | ||
| AMP | GDP (0.85) | 5.80 ± 1.7 | 1.1 |
| AMP | GDP (4.2) | 1.15 ± 0.2 | 5.3 |
| AMP | GMP (8.0) | 4.77 ± 1.4 | 1.3 |
| ADP | GMP (8.0) | 3.22 ± 0.9 | 1.3 |
| GMP | ADP (2.0) | 9.80 ± 2.0 | 1.8 |
| GDP | AMP (0.6) | 3.30 ± 0.7 | 2.6 |
| GDP | AMP (3.0) | 1.39 ± 0.4 | 6.1 |
The millimolar concentration of fixed nucleotide is given in parentheses.
The millimolar concentration of varied nucleotide required for 10 or 50% inhibition ± standard deviation.
Decreased I0.5 due to the second nucleotide.
Isolation and purification of A. aeolicus GARS.
A thermophilic GARS was required to investigate coupling with GPAT at elevated temperatures. The purD gene, encoding GARS, was cloned by PCR from A. aeolicus DNA and inserted into an expression vector in a manner similar to that used for purF. The enzyme produced in E. coli was purified and, similar to GPAT, gave a single major band upon SDS-PAGE (Fig. 1). The optimal temperature for GARS was at least 90°C. Enzyme activity at 90°C was eight times higher than at 50°C. In the coupled assay with A. aeolicus GPAT, the specific activity was 4.01 ± 0.43 U/mg at 90°C. The Km values were 1.01 ± 0.30 mM for ATP and 0.59 ± 0.10 mM for glycine at 90°C. The Km values were slightly temperature dependent and were approximately twofold lower at 50°C. Higher specific activities were obtained in the GARS assay with chemically generated PRA by substitution of Ca2+ for Mg2+. Ca2+ inhibits GPAT activity and could not be used in the coupled assay. Ca2+-dependent A. aeolicus GARS specific activity was 22.8 ± 1.5 U/mg at 90°C and pH 7.9, comparable to the specific activity of E. coli GARS under optimal conditions (29.5 ± 2.0 U/mg). The thermal stability of this enzyme is remarkable. The enzyme half-life at 80°C was 60 h.
GPAT NH3 channel efficiency.
Binding of PRPP to a site in the E. coli GPAT PRTase domain results in ordering of a PRTase “flexible loop” leading to activation of the glutaminase site and formation of a 20-Å NH3 channel that connects the sites for glutamine hydrolysis and PRA synthesis (11). In E. coli GPAT, essentially all of the NH3 derived from glutamine hydrolysis is channeled to the PRPP site for synthesis of PRA (2). The data in Table 1, line 1, show that channeling efficiency, determined from the ratio of Gln-PRA to glutaminase, is near unity, in agreement with recent work in which Gln-PRA was measured by a different method (2). Channeling efficiency for A. aeolicus GPAT, on the other hand, was only 0.34 at 37°C. Although the glutaminase activity at this temperature was less than 10% of the maximal activity, most of the NH3 produced was not used for synthesis of PRA. The channeling efficiency increased to nearly 90% at 80°C. Given the chemical lability of PRPP (half-life, 20 min at 90°C), it is uncertain whether channeling of NH3 approaches 100% at high temperature. Nevertheless, it is clear that interdomain signaling (activation of glutaminase by PRPP) and channeling of NH3 are strongly temperature dependent.
GPAT-GARS coupling.
PRA, the product of the GPAT reaction, is a labile intermediate (17, 18). Based on PRA half-lives at 24 and 37°C (18), we estimated a half-life of approximately 0.6 s at 90°C, an optimal growth temperature for A. aeolicus (6). Since such a labile metabolite is unlikely to be free and diffusible in the cell, we investigated GPAT-GARS coupling efficiency, defined as the fraction of PRA produced that can be converted to GAR by excess GARS. For these experiments, we used a 10-fold excess of GARS relative to GPAT. Line 1 in Table 1 shows a control reaction with E. coli GPAT and GARS at 37°C. With the E. coli enzymes, the coupling efficiency approached 1.0, indicating that essentially all of the PRA generated was converted to GAR. When PRA was generated by A. aeolicus GPAT, the fractional conversion to GAR by excess E. coli GARS at 37°C was decreased to 0.29 (Table 1, line 2). The coupling efficiency for the A. aeolicus GPAT-E. coli GARS heterologous pair was increased threefold at 50°C (Table 1, line 4). Since E. coli GARS is not stable at higher temperatures, we could not test coupling of this heterologous enzyme pair at 60°C. For reactions with GPAT and GARS, both from A. aeolicus, coupling efficiency was less than 20% at 37°C but was markedly improved at 50°C. Further temperature increases did not afford better coupling.
DISCUSSION
There are multiple steps in de novo purine nucleotide synthesis where pathway function likely depends upon metabolite channeling. In the first reaction, catalyzed by GPAT, NH3 derived from glutamine hydrolysis at an N-terminal glutaminase site must be sequestered from water and channeled to a distal phosphoribosyltransferase site for reaction with PRPP to yield PRA. Crystallographic (11) and biochemical (1, 2, 4) studies have characterized structural changes that result in the transient formation of a 20-Å NH3 channel that connects the two sites in E. coli GPAT. For the next step of the pathway, it appears unlikely that PRA, having a half-life of 5 s under physiological conditions at 37°C and an estimated half-life of 0.5 s at 90°C, could dissociate from GPAT, equilibrate with the solvent, and then bind to GARS. Rudolph and Stubbe (17) reported kinetic evidence supporting direct transfer of PRA between the two E. coli enzymes at 37°C, although a stable GPAT-GARS interaction was not detected. However, at least two transient conformations exist during each turnover of E. coli GPAT (4, 11). Any GPAT-GARS complex would also be transient and may involve a transient form of GPAT. The objective for our initial characterization of GPAT and GARS was to examine channeling in GPAT and GARS from an extreme thermophile where substrate and product lability may be more problematic than at 37°C.
A. aeolicus GPAT resembles the B. subtilis enzyme somewhat more closely than the enzyme from E. coli. A. aeolicus and B. subtilis GPAT share 44% amino acid identity, compared to 35% for A. aeolicus and E. coli. A. aeolicus GPAT contains an as-yet-uncharacterized Fe-S cluster that is likely a 4Fe-4S cluster based on conservation of the four cysteinyl ligands in the B. subtilis enzyme. The 4Fe-4S cluster imparts air sensitivity to the B. subtilis GPAT. However, the Fe-S cluster and the A. aeolicus enzyme are extremely stable at 37°C, retaining 50% activity after 25 h in air. Nevertheless, enzyme inactivation at 37 and 80°C was accompanied by loss of brown color, indicating the importance of the Fe-S cluster for structural integrity. A. aeolicus GPAT retains the synergistic end product inhibition by nucleotides that is the hallmark of GPAT regulation from other sources (5, 13, 14, 24).
The A. aeolicus GPAT glutaminase and Gln-PRA activities are highly temperature dependent, with optima at 90°C or higher. An important property of the glutaminase activity is its strict dependence upon PRPP. In the absence of PRPP, no “basal” glutaminase activity was detected at 37°C or at higher temperatures, in contrast to E. coli GPAT (9). This suggests less mobility of the A. aeolicus structure than occurs with the E. coli enzyme. In the ligand-free E. coli enzyme, the 30-residue PRTase flexible loop is essentially unfolded. PRPP binding results in folding of the flexible loop, which activates glutaminase and also forms the NH3 channel. Inherent flexibility of the glutaminase active site, perhaps in concert with motion of the flexible loop, is thought to be responsible for basal glutaminase activity. In contrast, the ligand-free A. aeolicus enzyme is inferred to be folded in a stable, inactive conformation that requires PRPP binding to equilibrate with a conformation competent for glutaminase activity, a step referred to as interdomain signaling (1, 4), and to form the NH3 channel. The two steps, interdomain signaling and channel formation, are not as tightly linked as in the E. coli enzyme. The data in Table 1 show that at 37 and 50°C, about 50% or less of the NH3 derived from glutamine hydrolysis was channeled to the PRPP site. Elevated temperatures increased channeling efficiency from 34% at 37°C to 87% at 80°C. It is reasonable to imagine that elevated temperatures are required to disrupt stable interactions of the PRTase flexible loop structure to allow the reorganization that is needed to form the NH3 channel. Initial steps in loop reorganization that take place at temperatures below the optimum for activity appear to permit activation of the glutamine site by PRPP even though the structure of the NH3 channel has not been optimized.
Purification of A. aeolicus GARS permitted experiments to investigate coupling of PRA production with synthesis of GAR. Given the short half-life of PRA at 37°C, and particularly at 90°C, free diffusion of PRA seems unlikely. We found two types of evidence for coupling: species specificity at low temperature and improved coupling at higher temperatures. By comparison with the highly efficient coupling of the two E. coli enzymes, the relatively low capacity of A. aeolicus GPAT to couple with A. aeolicus or E. coli GARS at 37°C was surprising. In our experiments, a 10-fold excess of E. coli GARS converted essentially all of the PRA produced by E. coli GPAT to GAR. In contrast, there was only 29% coupling of E. coli GARS and A. aeolicus GPAT at 37°C. The observed species specificity at 37°C suggests that enzyme-enzyme interactions are involved in coupling. The temperature dependence of coupling with A. aeolicus GPAT further supports the importance of enzyme-enzyme interaction. At 50°C, coupling between the heterologous enzyme pair was improved to 82% and that between the two A. aeolicus enzymes was improved to 76% (from 18% at 37°C). If not for GPAT-GARS interactions, a temperature increase from 37 to 50°C should decrease the synthesis of GAR because of a reduced PRA chemical half-life at 50°C. It is noteworthy that further increases in temperature, which increased Gln-PRA activity 12-fold and GARS activity 11-fold, had no further effect on coupling efficiency. However, in the absence of direct transfer of PRA between the two A. aeolicus enzymes, coupling efficiency at 90°C should be substantially less than at 50°C because of the difference in PRA half-life (12 s at 50°C and 0.6 s at 90°C). It appears unlikely that a 60% coupling efficiency could suffice in vivo. If true, some additional component not available in vitro or some feature of the physiological condition may contribute to high-efficiency GPAT-GARS coupling.
We conclude that a structural change in A. aeolicus GPAT is an essential component of coupling, based on the cross-species data at 37 and 50°C. Regions of A. aeolicus GPAT needed to interact with E. coli GARS were not available at 37°C (29% coupling) but became accessible at 50°C (82% coupling). The deficiency at 37°C is not due to E. coli GARS, which coupled efficiently with E. coli GPAT at both 37 and 50°C. The increase in coupling efficiency with the two A. aeolicus enzymes (from 18% at 37°C to 76% at 50°C) is also attributed to structural changes in GPAT. Indeed, the increase in all three GPAT activities with temperature attests to some type of GPAT structural reorganization. This is consistent with the structural changes known to occur in E. coli GPAT during its catalytic cycle, in which flexible regions of the free enzyme adopt specific conformations upon substrate binding. In its 95°C habitat, A. aeolicus experiences no selective pressure for structural flexibility at 37°C. Regions of A. aeolicus GPAT that are flexible at 95°C may adopt more stable, inhibitory conformations at 37°C. The increase in activity with temperature would then result from “unfolding” inhibitory conformations. Consistent with this idea, we found a 15% increase in all A. aeolicus GPAT activities when assayed in 100 mM urea with no change in channeling or coupling efficiency (data not shown).
The question of how labile intermediates are utilized in organisms that live at elevated temperatures has been examined previously for carbamoyl phosphate (16). In Pyrococcus abyssi, a hyperthermophile with an optimal growth temperature of 96°C, carbamoyl phosphate having a half-life of 2 to 3 s at 96°C is utilized in pathways for the synthesis of arginine and pyrimidines. Evidence was obtained for channeling of carbamoyl phosphate from carbamoyl phosphate synthetase to ornithine transcarbamoylase (arginine pathway) and to aspartate transcarbamoylase (pyrimidine pathway). The data were consistent with partial channeling at 37°C that was increased substantially at elevated temperature. The kinetics for the formation of carbamoyl aspartate by carbamoyl phosphate synthetase and aspartate transcarbamoylase exhibit lags before reaching the steady-state rate at 37 and 70°C, consistent with a partial channeling of carbamoyl phosphate at these temperatures. At 90°C, however, a lag was not detected for carbamoyl aspartate synthesis, suggesting essentially complete channeling between carbamoyl phosphate synthetase and aspartate transcarbamoylase at 90°C. Since evidence was not obtained for physical association of enzymes, channeling was proposed to occur during transient enzyme interactions. The similarities are apparent for the utilization of carbamoyl phosphate and PRA by enzymes from hyperthermophiles.
Thus, while convincing support for intermediate channeling between enzymes has been garnered from biochemical studies, evidence of a physical complex of two enzymes is still lacking for any system. Clues about the nature of interenzyme channels may come from intraenzyme channels, which have been observed in crystal structures of several enzymes having multiple active sites. In addition to the NH3 channel of GPAT (11), these include the indole channel of tryptophan synthase (8), the NH3 and carbamate channels of carbamoyl phosphate synthetase (22), and the NH3 channel of asparagine synthetase (12). The NH3 channel is a general feature of glutamine amidotransferases, which produce nitrogen from glutamine for a variety of biosynthetic pathways (25). These channels come in two varieties—permanent and transient. In tryptophan synthase, carbamoyl phosphate synthetase, and asparagine synthetase, the channels appear to be permanent fixtures of the enzymes, in that the “walls” are always present in channel form. However, GPAT (15), GMP synthetase (21), and anthranilate synthase (10) all have NH3 channels that are not present or are incompletely formed in the resting enzyme. GPAT is the only system for which both channel-formed and unformed states have been visualized in crystal structures. Structural data from GPAT, GMP synthetase, and anthranilate synthase are consistent with the idea that transient channels are formed as a result of rearrangements of floppy structures, initiated by binding of a substrate to one of the interconnected catalytic sites. Such signaling and channel construction may also be a feature of the channeling of labile intermediates between enzymes.
ACKNOWLEDGMENTS
We thank Robert Huber and Karl Stetter for a gift of A. aeolicus DNA.
This work was supported by Public Health Service grants GM24658 (to H.Z.) and DK42303 (to J.L.S.).
Footnotes
Journal paper 16255 from the Purdue University Agricultural Experiment Station.
REFERENCES
- 1.Bera A K, Chen S, Smith J L, Zalkin H. Interdomain signaling in glutamine phosphoribosylpyrophosphate amidotransferase. J Biol Chem. 1999;274:36498–36504. doi: 10.1074/jbc.274.51.36498. [DOI] [PubMed] [Google Scholar]
- 2.Bera A K, Smith J L, Zalkin H. Dual role for the glutamine phosphoribosylpyrophosphate amidotransferase ammonia channel. Interdomain signaling and intermediate channeling. J Biol Chem. 2000;275:7975–7979. doi: 10.1074/jbc.275.11.7975. [DOI] [PubMed] [Google Scholar]
- 3.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 4.Chen S, Burgner J W, Krahn J M, Smith J L, Zalkin H. Tryptophan fluorescence monitors multiple conformational changes required for glutamine phosphoribosylpyrophosphate amidotransferase interdomain signaling and catalysis. Biochemistry. 1999;38:11659–11669. doi: 10.1021/bi991060o. [DOI] [PubMed] [Google Scholar]
- 5.Chen S, Tomchick D R, Wolle D, Hu P, Smith J L, Switzer R L, Zalkin H. Mechanism of the synergistic end-product regulation of Bacillus subtilis glutamine phosphoribosylpyrophosphate amidotransferase by nucleotides. Biochemistry. 1997;36:10718–10726. doi: 10.1021/bi9711893. [DOI] [PubMed] [Google Scholar]
- 6.Deckert G, Warren P V, Gaasterland T, Young W G, Lenox A L, Graham D E, Overbeek R, Snead M A, Keller M, Aujay M, Huber R, Feldman R A, Short J M, Olsen G J, Swanson R V. The complete genome of the hyperthermophilic bacterium A. aeolicus. Nature. 1998;392:353–358. doi: 10.1038/32831. [DOI] [PubMed] [Google Scholar]
- 7.Doherty A J, Ashford S R, Brannigan J A, Wigley D B. A superior host strain for the overexpression of cloned genes using the T7 promoter based vectors. Nucleic Acids Res. 1995;23:2074–2075. doi: 10.1093/nar/23.11.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hyde C C, Ahmed S A, Padlan E A, Miles E W, Davies D R. Three-dimensional structure of the tryptophan synthase α2β2 multienzyme complex from Salmonella typhimurium. J Biol Chem. 1988;263:17857–17871. [PubMed] [Google Scholar]
- 9.Kim H H, Krahn J M, Tomchick D R, Smith J L, Zalkin H. Structure and function of the glutamine phosphoribosylpyrophosphate amidotransferase glutamine site and communication with the phosphoribosylpyrophosphate site. J Biol Chem. 1996;271:15549–15557. doi: 10.1074/jbc.271.26.15549. [DOI] [PubMed] [Google Scholar]
- 10.Knöchel T, Ivens A, Hester G, Gonzalez A, Bauerle R, Wilmanns M, Kirschner K, Jansonius J N. The crystal structure of anthranilate synthase from Sulfolobus solfataricus: functional implications. Proc Natl Acad Sci USA. 1999;96:9479–9484. doi: 10.1073/pnas.96.17.9479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krahn J M, Kim J H, Burns M R, Parry R, Zalkin H, Smith J L. Coupled formation of an amidotransferase interdomain ammonia channel and a phosphoribosyltransferase active site. Biochemistry. 1997;36:11061–11068. doi: 10.1021/bi9714114. [DOI] [PubMed] [Google Scholar]
- 12.Larsen T M, Boehlein S K, Schuster S M, Richards N G J, Thoden J B, Holden H M, Rayment I. Three-dimensional structure of Escherichia coli asparagine synthetase B: a short journey from substrate to product. Biochemistry. 1999;38:16146–16157. doi: 10.1021/bi9915768. [DOI] [PubMed] [Google Scholar]
- 13.Messenger L J, Zalkin H. Glutamine phosphoribosylpyrophosphate amidotransferase from Escherichia coli: purification and properties. J Biol Chem. 1979;254:3382–3392. [PubMed] [Google Scholar]
- 14.Meyer E, Switzer R L. Regulation of Bacillus subtilis glutamine phosphoribosylpyrophosphate amidotransferase activity by end products. J Biol Chem. 1978;254:5397–5402. [PubMed] [Google Scholar]
- 15.Muchmore C R A, Krahn J M, Kim J H, Zalkin H, Smith J L. Crystal structure of glutamine phosphoribosylpyrophosphate amidotransferase from Escherichia coli. Protein Sci. 1998;7:39–51. doi: 10.1002/pro.5560070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Purcarea C, Evans D R, Hervé G. Channeling of carbamoyl phosphate to the pyrimidine and arginine biosynthetic pathways in the deep sea hyperthermophilic archaeon Pyrococcus abyssi. J Biol Chem. 1999;274:6122–6129. doi: 10.1074/jbc.274.10.6122. [DOI] [PubMed] [Google Scholar]
- 17.Rudolph J, Stubbe J. Investigation of the mechanism of phosphoribosylamine transfer from glutamine phosphoribosylpyrophosphate amidotransferase to glycinamide ribonucleotide synthetase. Biochemistry. 1995;34:2241–2250. doi: 10.1021/bi00007a019. [DOI] [PubMed] [Google Scholar]
- 18.Schendel F J, Cheng Y S, Otvos J D, Wehrli S, Stubbe J. Characterization and chemical properties of phosphoribosylamine, an unstable intermediate in the de novo purine biosynthetic pathway. Biochemistry. 1988;27:2614–2623. doi: 10.1021/bi00407a052. [DOI] [PubMed] [Google Scholar]
- 19.Schrimsher J L, Schendel F J, Stubbe J. Isolation of a multifunctional protein with aminoimidazole ribonucleotide synthetase, glycinamide ribonucleotide synthetase, and glycinamide ribonucleotide transformylase activities: characterization of aminoimidazole ribonucleotide synthetase. Biochemistry. 1986;25:4356–4365. doi: 10.1021/bi00363a027. [DOI] [PubMed] [Google Scholar]
- 20.Smith J L, Zaluzec E J, Wery J-P, Niu L, Switzer R L, Zalkin H, Satow Y. Structure of the allosteric regulatory enzyme of purine biosynthesis. Science. 1994;264:1427–1433. doi: 10.1126/science.8197456. [DOI] [PubMed] [Google Scholar]
- 21.Tesmer J J G, Klem T J, Deras M L, Davisson V J, Smith J L. The crystal structure of GMP synthetase reveals a novel catalytic triad and is a structural paradigm for two enzyme families. Nat Struct Biol. 1996;3:74–86. doi: 10.1038/nsb0196-74. [DOI] [PubMed] [Google Scholar]
- 22.Thoden J B, Holden H M, Wesenberg G, Raushel F M, Rayment I. Structure of carbamoyl phosphate synthetase: a journey of 96 Å from substrate to product. Biochemistry. 1997;36:6305–6316. doi: 10.1021/bi970503q. [DOI] [PubMed] [Google Scholar]
- 23.Upson R H, Haugland R P, Malekzadeh M N, Haugland R P. A spectrophotometric method to measure enzymatic activity in reactions that generate inorganic pyrophosphate. Anal Biochem. 1996;243:41–45. doi: 10.1006/abio.1996.0479. [DOI] [PubMed] [Google Scholar]
- 24.Zalkin H. The amidotransferases. Adv Enzymol Relat Areas Mol Biol. 1993;38:1–39. [Google Scholar]
- 25.Zalkin H, Smith J L. Enzymes utilizing glutamine as an amide donor. Adv Enzymol Relat Areas Mol Biol. 1998;72:87–144. doi: 10.1002/9780470123188.ch4. [DOI] [PubMed] [Google Scholar]