

Abstract
A stable dimethyl(thiodimethyl)sulfonium tetrafluoroborate salt was employed for the electrophilic cyclization reaction of o-alkynyl thioanisoles for the synthesis of 2,3-disubstituted benzo[b]thiophenes. The reaction described herein works well with various substituted alkynes in excellent yields, and a valuable thiomethyl group was introduced with ease. The reaction utilizes moderate reaction conditions and ambient temperature while tolerating various functionalities. To elucidate the mechanism, electrophilic addition reactions using the dimethyl(thiodimethyl)sulfonium tetrafluoroborate salt with diphenylacetylene was demonstrated.
Graphical Abstract
Benzo[b]thiophenes are considered a valuable class of heterocycles in the realm of medicinal chemistry1 because of possessing diverse anticancer,2 antioxidant,3 anti-inflammatory,4 antitubercular,5 antimalarial,6 and antimicrobial activities.7 The use of benzo[b]thiophenes has also been of considerable interest in materials chemistry as they have found applications in organic solar cells,8 organic light-emitting diodes,9 and semiconductors.10 The privileged benzo[b]thiophene scaffold is also a part of several FDA-approved drug such as raloxifene, brexpiprazole, zileuton, and sertaconazole (Figure 1).1b
Figure 1.

Some of the recent methods for the synthesis of the benzo[b]thiophene core structure include metal-mediated annulation of 2-halo alkynyl benzenes,11 photocatalytic radical annulation of o-methylthioarenediazonium salts,12 a cascade of alkene bond thiolation, aromatic sulfur-denitration of 2-nitrochalcones with elemental sulfur,13 iodine-catalyzed cascade reactions of substituted thiophenols with alkynes,14 and C–H bond functionalization and aromatic nucleophilic substitution of various 2′-nitrochalcones using elemental sulfur.15
An attractive approach for gaining direct access to the benzo[b]thiophene core is the electrophilic cyclization reaction of alkynyl thioanisoles. In the past, several electrophiles have been employed as the cyclizing agents including I2, ICl, NIS, Br2, NBS, PhSCl, and PhSeBr.16 Recently, our group has reported copper-catalyzed chloro-, bromo-, and iodocyclization reactions using sodium halides as the source of electrophiles.17 Other examples for the synthesis of benzo[b]thiophene via alkynyl thioanisole includes gold(I)–NHC-catalyzed cyclization,18 iodocyclization via in situ generated molecular iodine,19 electrochemical sulfonylation/cyclization using sodium sulfinates,20 photoredox-catalyzed cascade radical annulation, aryl sulfide and NCS,21 and S2Cl2 mediated electrophilic cyclization followed by reduction and alkylation of the sulfur moiety.22
The use of sulfur electrophiles for the cyclization of 2-alkynylthioanisole to the corresponding benzo[b]thiophenes was first demonstrated by Larock and co-workers.16b However, their method was limited to the use of the electrophile p-NO2C6H4SCl, containing an electron-deficient thioaryl group. Almost a decade later Zeni and others reported an Fe-promoted electrophilic cyclization employing dialkyldisulfides and dialkyldiselenides as the source of electrophilic sulfur for the synthesis of thiophenes and selenophenes; however, they did not demonstrate the synthesis of benzo[b]thiophene. In addition, they obtained poor yields with sulfur electrophiles.23
Very recently, Flynn and co-workers reported the use of methanesulfenyl chloride as a highly effective electrophile. Methanesulfenyl chloride is a low boiling liquid usually generated through the reaction of methyl disulfide and sulfuryl chloride at low temperature.24 Even though their methods are very attractive for the synthesis of thiomethyl substituted benzo[b]thiophene, the use of toxic, corrosive, and lachrymatory sulfuryl chloride limits the applicability of their process. In addition, their method also employs flammable methyl disulfide. Recently Huang and co-workers reported thiotrifluoromethyl radical-mediated cyclization reaction of o-alkynyl thioanisole. Their method requires an equivalent amount of silver(I) trifluoromethanethiolate along with potassium persulfate as an oxidant.25 In another recent work Zhao and co-workers demonstrated the synthesis of indoles, benzofuran, and benzo[b]thiophene via in situ generated organosulfenyl chloride via an interrupted Pummerer reaction of PhICl2 and sulfoxides.26 However, in the study they only demonstrated one example of a benzo[b]thiophene derivative. Some of the above-mentioned methodologies are summarized in Scheme 1, along with our newly developed process. With only a handful of methods known for electrophilic sulfur mediated cyclization, our method filled in the gap for the desired methodology that employs nontoxic reagents, requires milder reaction conditions, and results in high yields of products, as described below. In this study, we report a high yielding synthesis of benzo[b]thiophene using commercially available and stable dimethyl(thiodimethyl)sulfonium tetrafluoroborate salt (2) as the source of electrophilic thiomethyl for the cyclization of 2-alkynylthioanisoles.
Scheme 1.
Synthetic Routes for the Construction of 3-Sulfenyl Benzo[b]thiophenes Derivatives from o-Alkynylthioanisole
We determined that the reaction of phenyl substituted alkynyl thioanisole 1 with tetrafluoroborate salt (2) furnishes the corresponding cyclized product 3-(methylthio)-2-phenylbenzo[b]thiophene 3 in 99% yield at room temperature (Table 1, entry 1). We established that the reaction worked best when dichloromethane was employed as the solvent. We also found that the highest yields were obtained when an excess (2 equiv) of tetrafluoroborate salt (2) was employed, and the reaction was allowed to run for 24 h at room temperature. To study the scope of this reaction we employed various substituted alkynyl thioanisoles 4–25 (Table 1). We first investigated the electronic effect by employing electron-rich and electron-deficient groups on the remote phenyl group. The mildly activating methyl group, when present in the ortho or para positions, resulted in lower yields of the cyclized product when compared with the unsubstituted phenyl group (see entries 2, 3). The presence of the methyl in the meta position did not seem to affect the yield and resulted in the quantitative yield of the product 28.
Table 1.
Synthesis of Benzo[b]thiophenes via Electrophilic Thiomethyl Mediated Cyclization Reaction

| |||||
|---|---|---|---|---|---|
| Entry | Alkyne (R1) | Product | Yield (%)a | ||
| 1 |

|

|
|||
| R3 = H | 1 | 3 | 99 | ||
| 2 | R3 = p-Me | 4 | 26 | 84 | |
| 3 | R3 = o-Me | 5 | 27 | 92 | |
| 4 | R3 = m-Me | 6 | 28 | 100b | |
| 5 | R3 = p-OMe | 7 | 29 | 79 | |
| 6 | R3 = o-OMe | 8 | 30 | 80 | |
| 7 | R3 = m-OMe | 9 | 31 | 87 | |
| 8 | R3 = p-Cl | 10 | 32 | 100 | |
| 9 | R3 = o-Cl | 11 | 33 | 85 | |
| 10 | R3 = m-Cl | 12 | 34 | 93 | |
| 11 | R3 = p-CN | 13 | 35 | 92 | |
| 12 | R3 = o-CN | 14 | 36 | 100* | |
| 13 | R3 = m-CN | 15 | 37 | 88 | |
| 14 |

|
16 |

|
38 | 100 |
| 15 |

|
17 |

|
39 | 83 |
| 16 |

|
18 |

|
40 | 100 |
| 17 |

|
19 |

|
41 | 65 |
| + | + | ||||
| 42 | 24 | ||||
| 18 |
|
20 |

|
43 | 72 |
| 19 |
|
21 |

|
44 | - |
| 20 | 21 | 44 | 69c | ||
| 21 |
|
22 |

|
45 | - |
| 22 |

|
23 |

|
46 | - |
| 23 |

|
24 |

|
47 | 23 |
| + | + | ||||
| 48 | trace | ||||
| 24 |
|
25 |

|
49 | 95 |
Unless otherwise mentioned all the reactions were performed in using 1.0 equiv of alkyne (0.3 mmol) and 2.0 equiv of 2 in 3 mL of DCM for 24 h.
Reaction was allowed to run for 48 h.
Reaction was run for 1 h using 1.1 equiv of 2.
In these cyclization reactions, a strong activating group such as methoxy, when present on the remote phenyl ring, could activate the triple bond favorably for these cyclization reactions. The methoxy group also showed a similar trend to the methyl group (entries 5–7). The ortho and para methoxy substituted phenyl ring furnished the products 29 and 30 in slightly lower yields of 79 and 80%, respectively, whereas the meta substituted product 31 was obtained in a higher yield of 87%.
We also employed the mild electron-withdrawing chloro group in all three possible positions of the remote phenyl ring. The reaction proceeded with ease in all three cases. We obtained the cyclized product 32, 33, and 34 in excellent yields (entries 8–10). p-Chloro substituted product 32 was obtained in quantitative yield, whereas the ortho and meta substituted products 33 and 34 were obtained in 85 and 93% yield, respectively.
In a study reported by Larock and co-workers for the synthesis of benzofuran, strong electron-withdrawing groups, such as nitro or nitrile, could affect the electrophilic cyclization reaction adversely as they decrease the electron density on the alkyne carbon, impeding the approach of an electrophile.27 Later Flynn and co-workers reported a detailed mechanistic study of iodocyclization reaction where they reported the mitigation of the unfavorable electronic effects on alkyne due to the substituents.28 It appears that the electrophilic cyclization with dimethyl(thiodimethyl)sulfonium tetrafluoroborate salt is also not affected by the electronic effects of its substituents. When we introduced a nitrile functionality on the remote phenyl group, the reaction proceeded with ease, resulting in excellent yields of the product irrespective of the position of the nitrile (entries 11–13). The ortho nitrile substrate resulted in a quantitative yield of the product 36; however, it required a longer reaction time of 48 h. The para position resulted in 92% yield of the desired benzo[b]thiophene 35, and the meta substitution furnished 88% yield of 37.
While the reaction appears to have a minimal electronic effect, we decided to investigate if steric effects altered the reactivity. When we employed the linear n-butyl group, the resulting benzo[b]thiophene 38 was attained in quantitative yield (entry 14). Changing the linear n-butyl group to the linear 3-cyanopropyl group resulted in the cyclized compound 39 in an only slightly lower yield of 88% (entry 15). The sterically bulky tert-butyl group did not affect the cyclization reaction, as product 40 was obtained an excellent 100% yield (entry 16).
The bulky trimethylsilyl (TMS) group resulted in a moderate yield of 65% of the desired product 41 along with 24% of the side product 42 (entry 17). After the first hour, the product ratio did not appear to change with time, suggesting that a desilylation reaction takes place before the cyclization. We believe that initially there is competition between the cyclization reaction and the replacement of the alkynyl TMS group with a thiomethyl group. Replacing the relatively more labile TMS group with a bulkier and stable triisopropyl resulted in a desired cyclized product 43 with an improved yield of 72% yield, and no desilylated side product was obtained (entry 18).
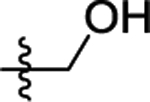
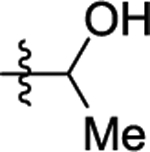
Substitution of the alkynyl thioanisole with a primary alcohol resulted in a complex reaction mixture. Reducing the amount of electrophile used and shortening the reaction time to 1 h resulted in cyclized product 44 in moderate 69% yield (entry 20). Secondary and tertiary propargyl alcohols 22 and 23 did not result in the desired cyclized product 45 and 46 with our standard or modified reaction conditions (entries 21 and 22).
We also placed a vinyl moiety, and the starting alkyne 24 was subjected to our standard reaction conditions (entry 23). This reaction resulted in a complex mixture, and the desired product 47 was isolated in a poor 23% yield. Altering the equivalents of salt 2 and changing the reaction time did not result in a favorable outcome. We also observed trace amounts of a product by GC-MS with m/z = 244, which we believe is the dicyclized product 48; however, we could not isolate or fully characterize it.
Finally, alkynyl thioanisole 25 containing a bromine functionality in the aromatic ring bearing the thiomethyl group was synthesized. The cyclization of compound 25 resulted in an excellent 95% yield of the 6-bromo benzo[b]thiophene 49 (entry 24). The presence of halogens such as bromine could be very convenient in the further functionalization of the cyclized product 49.
We believe that the mechanism of this reaction begins with the alkyne attacking the electrophilic sulfur of the salt 2, followed by the removal of dimethyl sulfide, as shown in Scheme 2. The intermediate 50 formed thereby could undergo cyclization by intramolecular nucleophilic ring-opening of the three-membered sulfonium ring. Finally, the dimethyl sulfide could act as a nucleophile and demethylate the cationic intermediate 51 via an SN2 displacement reaction, thereby forming the desired 3-thiomethylbenzo[b]thiophene 3 and trimethyl sulfonium cation.
Scheme 2.

Proposed Mechanism for Cyclization Reaction
We postulated that if the salt 2 reacted with an alkyne without a tethered nucleophilic thiomethyl group capable of intramolecular cyclization, the addition reaction of Scheme 3 should occur. We thought that the reaction of diphenyl alkyne 52 and the salt 2 would form intermediate 53, after which the dimethyl sulfide generated in the first step could open up the cationic 3-membered sulfonium intermediate 53 to form salt 54 via an SN2 attack. This salt could lose methyl tetrafluoroborate to generate trans-1,2-bis(methylthio)-1,2-diphenylethene 55.
Scheme 3.
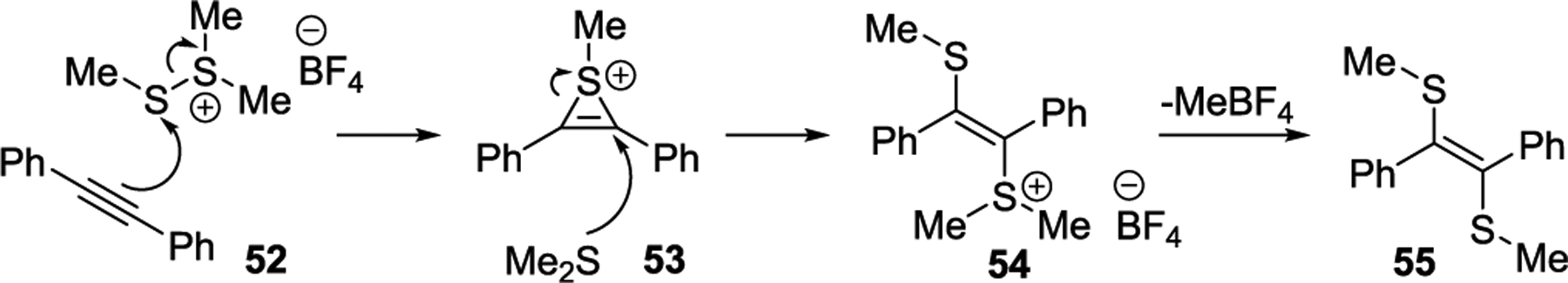
Proposed Mechanism for the Addition Reaction
To test our hypothesis, we subjected diphenylacetylene to our standard reaction conditions (Scheme 4). The reaction was monitored via GC, and we observed 1,2-bis(methylthio)-1,2-diphenylethene as the minor product. However, TLC suggested a very polar compound as the major product. Upon column chromatography, we isolated the salt 54 (71%) as the major product instead of the anticipated addition product 55 (eq 1).
Scheme 4.

Electrophilic Addition Reactions of Diphenyl Alkyne
To further evaluate our assumption, we used 2 equiv of TBAB along with the slightly lower quantity (1.5 equiv) of salt 2. As expected, we obtained the alkyne addition products 56 and 57 where thiomethyl and bromine were added to the two alkynyl carbons (eq 2). However, to our disappointment we obtained an inseparable mixture of E and Z isomers suggesting that the second step of our proposed mechanism may be more subtle, as depicted in Scheme 5.
Scheme 5.

Proposed Mechanism for the Addition Reaction in the Presence and Absence of TBAB
We believe that the sulfonium intermediate 53 as well as the vinyl cationic intermediate 58 could form in these addition reactions. We propose that in absence of TBAB the formation of only one isomer of the salt 50 is favored via intermediate 53, whereas in the presence of TBAB the reaction could proceed through intermediate 58 or both intermediates 53 and 58 to give the addition product as a mixture of E and Z isomers.
To further expand the scope of our thiomethyl cyclization strategy, we first demonstrated that the reaction can easily be scaled up without significant loss in the reaction yield. A gram scale cyclization of alkyne 1 was performed and 4.6 mmol of 1 was allowed to react with 9.2 mmol of dimethyl(thiodimethyl)sulfonium tetrafluoroborate salt under standard conditions. The cyclized product 3 was isolated in an excellent yield of 87% (Scheme 6). The sulfoxide functional group is an integral part of many drugs, such as esomeprazole, modafinil, armodafinil, and sulindac. Therefore, to expand the application of our cyclization methodology, compound 3 was transformed into the corresponding sulfoxide by employing m-CPBA mediated sulfoxidation reaction. The corresponding oxidized product 58 was obtained in 82% yield.
Scheme 6.
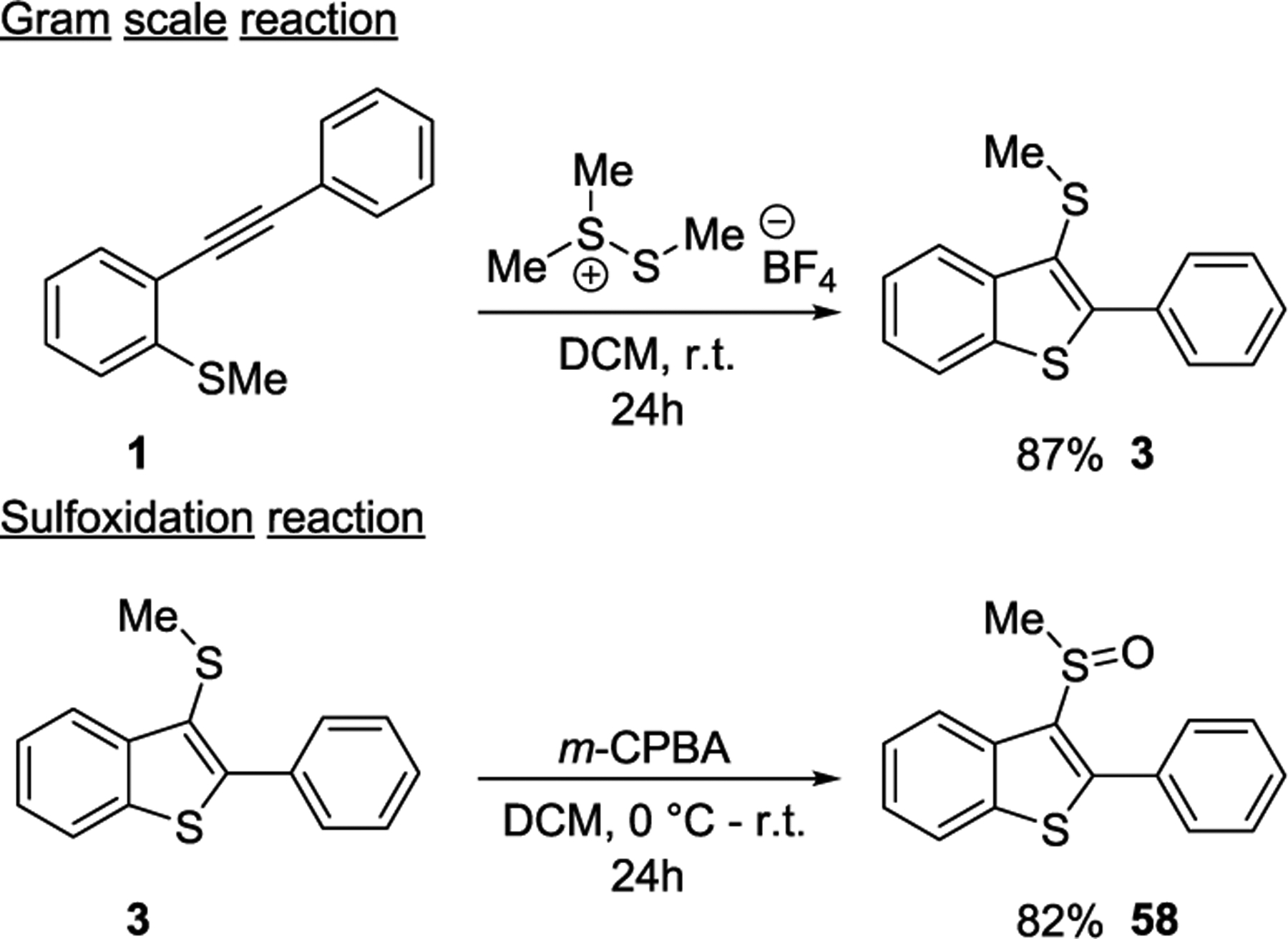
Scale-up Reaction and Further Elaboration
In summary, we have demonstrated the first electrophilic cyclization reaction of an alkyne involving stable dimethyl(thiodimethyl)sulfonium tetrafluoroborate salt. The reaction not only proceeds under ambient reaction conditions, but also furnishes the desired cyclized products in excellent yields. The reaction yield does not seem to be influenced by steric or electronic factors when various groups were employed in the remote alkyne. We have also demonstrated electrophilic addition reactions using the tetrafluoroborate salt.
EXPERIMENTAL SECTION
General Methods.
The 1H and 13C NMR spectra were recorded at 400 and 100 MHz Bruker NMR, respectively. High-resolution mass spectra (HRMS) were recorded on a VG-70S magnetic sector mass spectrometer using direct probe sample introduction and electron ionization (EI). Thin-layer chromatography was performed using commercially obtained 60-mesh silica gel plates, employing short wavelength UV light (254 nm) for the visualization. All obtained melting points are uncorrected.
Reagents.
Unless otherwise noted all reagents were obtained commercially and were used without further purification. Alkynes 1, 4–13, 16–25 were prepared by previously reported methods.16b,17,29,30
General Procedure for the Synthesis of 14 and 15.
2-Ethynylthioanisole (2.00 mmol, 1.2 equiv), Pd(PPh3)2Cl2 (0.16 mmol, 0.1 equiv), and iodobenzonitrile (1.66 mmol, 1.0 equiv) were added to a 50 mL round-bottomed flask (RBF). Et3N (10 mL) was added to the reaction mixture. CuI (0.08 mmol, 0.05 equiv) was added, and the reaction mixture was flushed with nitrogen. The reaction was allowed to run at room temperature, and the reaction progress was monitored by thin-layer chromatography (TLC). After the reaction was completed, the mixture was filtered and absorbed in silica gel before purification via column chromatography using hexanes and ethyl acetate as the eluent.
2-((2-(Methylthio)phenyl)ethynyl)benzonitrile (14).
Column chromatography was performed using hexanes and ethyl acetate (20:1) as the eluent. Light orange-yellow solid; 0.206 g, 83%; mp 78.4–79.5 °C; 1H NMR (400 MHz, CDCl3) δ 2.53 (s, 3H), 7.13 (td, J = 7.6, 0.8 Hz, 1H), 7.20 (d, J = 8.0 Hz, 1H), 7.35 (td, J = 8.0, 1.6 Hz, 1H), 7.41 (td, J = 8.0, 0.8 Hz, 1H), 7.56 (td, J = 8.0, 1.2 Hz, 1H), 7.58 (dd, J = 7.6, 1.2 Hz, 1H), 7.67 (d, J = 7.2 Hz, 1H), 7.68 (d, J = 7.6 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 15.4, 91.6, 93.4, 115.0, 117.7, 120.4, 124.5, 127.2, 128.5, 129.8, 132.5, 132.6, 132.8, 133.3, 142.3; FTIR (ATR) cm−1 2925, 2854, 2227, 2216, 1580, 1563, 1485, 1436, 1318, 1287, 1268, 1167, 1093, 1067, 1038, 953, 751, 746, 716, 701; HRMS (EI-ion trap) m/z [M]+ calcd for (C16H11NS)+ 249.0612, found 249.0623.
3-((2-(Methylthio)phenyl)ethynyl)benzonitrile (15).
Column chromatography was performed using hexanes and ethyl acetate (20:1) as the eluent. Light yellow solid; 0.194 g, 78%; mp 73.4–74.1 °C; 1H NMR (400 MHz, CDCl3) δ 2.53 (s, 3H), 7.13 (td, J = 7.6, 0.8 Hz, 1H), 7.20 (d, J = 8.0 Hz, 1H), 7.35 (td, J = 8.0, 1.6 Hz, 1H), 7.48 (t, J = 8.4, 1H), 7.49 (d, J = 7.6 Hz, 1H), 7.61 (dt, J = 8.0, 1.2 Hz, 1H), 7.78 (dt, J = 8.0, 1.6 Hz, 1H), 7.85 (s, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 15.2, 89.5, 93.4, 113.0, 118.2, 120.4, 124.3, 124.5, 125.0, 129.4, 129.6, 131.6, 132.5, 135.0, 135.7, 142.4; FTIR (ATR) cm−1 3051, 2924, 2856, 2232, 1602, 1574, 1479, 1462, 1434, 1322, 1281, 1257, 1198, 1071, 1042, 884, 792, 753, 719, 677; HRMS (EI-ion trap) m/z [M]+ calcd for (C16H11NS)+ 249.0612, found 249.0615.
Gram Scale Reaction for the Synthesis of 3.
2-Methyl(2-(phenylethynyl)phenyl)sulfane 1 (4.45 mmol, 1.000 g, 1.0 equiv) and DCM (40 mL) were added to a 100 mL RBF equipped with a magnetic stirring bar at room temperature. To this solution dimethyl(methylthio)sulfonium tetrafluoroborate (8.90 mmol, 1.750 g, 2.0 equiv) was added portion-wise over the period of 5 min. The reaction mixture was allowed to stir for 24 h. Upon completion, the reaction mixture was filtered, concentrated under a vacuum, absorbed in silica gel, and purified via column chromatography using hexanes as the eluent. Product 3 was obtained as a white solid in 87% yield (0.995 g).
General Procedure for the Cyclization Reaction.
Alkynylthioanisole (0.3 mmol, 1.0 equiv) and CH2Cl2 (3 mL) was added to a 6-dram vial followed by dimethyl(methylthio)sulfonium tetrafluoroborate (0.6 mmol, 2.0 equiv). The reaction was stirred for 24 h at room temperature and was monitored using TLC. The reaction mixture was filtered, concentrated under a vacuum, absorbed in silica gel, and purified via column chromatography using hexanes and ethyl acetate as the eluent.
3-(Methylthio)-2-phenylbenzo[b]thiophene (3).
Column chromatography was performed using hexanes as the eluent. White solid; Yield 0.076 g, 99%; mp 93.5–94.9 °C; 1H NMR (400 MHz, CDCl3) δ 2.24 (s, 3H), 7.34–7.43 (m, 2H), 7.43–7.52 (m, 3H), 7.73–7.80 (m, 2H), 7.82 (d, J = 8.0 Hz, 1H), 8.03 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 19.1, 122.4, 123.6, 124.9, 125.0, 128.5, 128.7, 130.0, 134.0, 138.5, 141.4, 146.2; FTIR (ATR) cm−1 3064, 3048, 2922, 2852, 1477, 1456, 1442, 1431, 1319, 1217, 1077, 1032, 1018, 968, 901, 753, 729, 696; HRMS (EI-ion trap) m/z [M]+ calcd for (C15H12S2)+ 256.0380, found 256.0386.
3-(Methylthio)-2-(p-tolyl)benzo[b]thiophene (26).
Column chromatography was performed using hexanes and ethyl acetate (80:1) as the eluent. White solid; Yield 0.068 g, 84%; mp 65.5–66.5 °C; 1H NMR (400 MHz, CDCl3) δ 2.24 (s, 3H), 2.41 (s, 3H), 2.26 (d, J = 8.0 Hz, 2H), 7.34 (td, J = 8.0, 1.2 Hz, 1H), 7.44 (td, J = 8.0, 0.8, 1H), 7.66 (d, J = 8.0 Hz, 2H), 7.80 (d, J = 8.0 Hz, 1H), 8.02 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 19.1, 21.5, 122.4, 123.2, 123.5, 124.9, 124.9, 129.3, 129.8, 131.1, 138.4, 138.8, 141.4, 146.5; FTIR (ATR) cm−1 3059, 3022, 2922, 2856, 1742, 1490, 1456, 1434, 1377, 1322, 1217, 801, 755, 730; HRMS (EI-ion trap) m/z [M]+ calcd for (C16H14S2)+ 270.0537, found 270.0543.
3-(Methylthio)-2-(o-tolyl)benzo[b]thiophene (27).
Column chromatography was performed using hexanes and ethyl acetate (80:1) as the eluent. Yellow solid; 0.074 g, 92%; mp 76.6–77.8 °C; 1H NMR (400 MHz, CDCl3) δ 2.17 (s, 3H), 2.27 (s, 3H), 7.21–7.35 (m, 4H), 7.39 (td, J = 8.0, 0.8 Hz, 1H), 7.47 (td, J = 8.0, 0.8 Hz, 1H), 7.84 (d, J = 8.0 Hz, 1H), 8.01 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 18.6, 20.4, 122.5, 123.3, 124.8, 124.9, 125.5, 125.6, 129.1, 130.3, 131.1, 133.5, 137.8, 139.3, 140.3, 145.7; FTIR (ATR) cm−1 3068, 2992, 2920, 2858, 1740, 1475, 1451, 1433, 1377, 1257, 1224, 1195, 1115, 1071, 954, 905, 756, 745, 734; HRMS (EI-ion trap) m/z [M]+ calcd for (C16H14S2)+ 270.0537, found 270.0545.
3-(Methylthio)-2-(m-tolyl)benzo[b]thiophene (28).
Column chromatography was performed using hexanes and ethyl acetate (80:1) as the eluent. Yellow oil; 0.081 g, 100%; 1H NMR (400 MHz, CDCl3) δ 2.24 (s, 3H), 2.42 (s, 3H), 7.21 (d, J = 8.8 Hz, 1H), 7.31–7.40 (m, 2H), 7.45 (td, J = 8.0, 1.2 Hz, 1H), 7.53–7.62 (m, 2H), 7.81 (d, J = 8.0 Hz, 1H), 8.02 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 19.1, 21.6, 122.4, 123.6, 124.9, 124.9, 127.1, 128.4, 129.5, 130.6, 133.9, 138.2, 138.5, 141.3, 146.4; FTIR (Salt Plate) cm−1 3055, 2919, 2855, 1604, 1454, 1434, 1319, 1294, 1241, 1159, 1095, 1074, 1018, 970, 856, 787, 756, 731, 695; HRMS (EI-ion trap) m/z [M]+ calcd for (C16H14S2)+ 270.0537, found 270.0544.
2-(4-Methoxyphenyl)-3-(methylthio)benzo[b]thiophene (29).
Column chromatography was performed using hexanes and ethyl acetate (20:1) as the eluent. Light brown solid; 0.067 g, 79%; mp 85.7–86.3 °C; 1H NMR (400 MHz, CDCl3) δ 2.24 (s, 3H), 3.85 (s, 3H), 6.99 (dt, J = 8.8, 3.2 Hz, 2H), 7.35 (td, J = 8.0, 1.2 Hz, 1H), 7.44 (td, J = 8.0, 0.8 Hz, 1H), 7.71 (dt, J = 8.8, 2.8 Hz, 2H), 7.80 (d, J = 8.0 Hz, 1H), 8.01 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 19.0, 55.5, 114.0, 122.3, 122.7, 123.4, 124.8, 124.8, 126.4, 131.2, 138.2, 141.5, 146.2, 160.1; FTIR (ATR) cm−1 3059, 3007, 2963, 2917, 2854, 1742, 1608, 1534, 1491, 1459, 1432, 1364, 1301, 1287, 1251, 1177, 1160, 1025, 832, 810, 759, 730; HRMS (EI-ion trap) m/z [M]+ calcd for (C16H14OS2)+ 286.0486, found 286.0476.
2-(2-Methoxyphenyl)-3-(methylthio)benzo[b]thiophene (30).
Column chromatography was performed using hexanes and ethyl acetate (20:1) as the eluent. Yellow solid; 0.068 g, 80%; mp 70.8–73.2 °C; 1H NMR (400 MHz, CDCl3) δ 2.22 (s, 3H), 3.80 (s, 3H), 7.00 (d, J = 8.4 Hz, 1H), 7.04 (t, J = 7.6 Hz, 1H), 7.36 (td, J = 8.0, 1.2 Hz, 1H), 7.39–7.47 (m, 3H), 7.82 (d, J = 8.0 Hz, 1H), 8.00 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 18.7, 55.8, 111.3, 120.5, 122.5, 122.9, 123.3, 124.6, 124.8, 125.9, 130.5, 132.5, 139.5, 140.4, 142.7, 157.5; FTIR (ATR) cm−1 3005, 2970, 2924, 2836, 1738, 1602, 1580, 1484, 1435, 1377, 1278, 1247, 1184, 1117, 1048, 1025, 903, 762, 739, 714; HRMS (EI-ion trap) m/z [M]+ calcd for (C16H14OS2)+ 286.0486, found 286.0489.
2-(3-Methoxyphenyl)-3-(methylthio)benzo[b]thiophene (31).
Column chromatography was performed using hexanes and ethyl acetate (40:1) as the eluent. Yellow liquid; 0.075 g, 87%; 1H NMR (400 MHz, CDCl3) δ 2.14 (s, 3H), 3.74 (s, 3H), 6.84 (dt, J = 7.2, 2.0 Hz, 1H), 7.19–7.29 (m, 4H), 7.34 (td, J = 8.0, 0.8 Hz, 1H), 7.700 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 19.1, 55.4, 114.3, 115.5, 122.4, 123.6, 123.7, 124.9, 125.1, 129.5, 135.2, 138.4, 141.3, 145.9, 159.5; FTIR (Salt Plate) cm−1 3020, 2925, 2837, 1599, 1579, 1466, 1435, 1288, 1216, 1176, 1167, 1051, 928, 755, 669; HRMS (EI-ion trap) m/z [M]+ calcd for (C16H14OS2)+ 286.0486, found 286.0497.
2-(4-Chlorophenyl)-3-(methylthio)benzo[b]thiophene (32).
Column chromatography was performed using hexanes and ethyl acetate (80:1) as the eluent. Yellow solid; 0.087 g, 100%; mp 81.8–82.9 °C; 1H NMR (400 MHz, CDCl3) δ 2.25 (s, 3H), 7.39 (td, J = 8.0, 1.2 Hz, 1H), 7.42–7.52 (m, 3H), 7.71 (dt, J = 8.4, 2.4 Hz, 2H), 7.83 (d, J = 8.0 Hz, 1H), 8.03 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 19.1, 122.5, 123.7, 124.2, 125.1, 125.3, 128.8, 131.3, 132.5, 134.8, 138.4, 141.3, 144.8; FTIR (ATR) cm−1 3053, 2924, 2858, 1519, 1474, 1433, 1397, 1252, 1217, 1178, 1091, 1013, 991, 903, 831, 799, 757, 732, 707; HRMS (EI-ion trap) m/z [M]+ calcd for (C15H11ClS2)+ 289.9991, found 289.9997.
2-(2-Chlorophenyl)-3-(methylthio)benzo[b]thiophene (33).
Column chromatography was performed using hexanes and ethyl acetate (80:1) as the eluent. Light yellow solid; 0.074 g, 85%; mp 103.1–104.3 °C; 1H NMR (400 MHz, CDCl3) δ 2.23 (s, 3H), 7.31–7.57 (m, 6H), 7.85 (d, J = 8.0 Hz, 1H), 8.03 (d, J = 7.6 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 18.6, 122.6, 123.6, 124.9, 125.3, 126.6, 126.9, 129.9, 130.3, 132.8, 133.1, 134.7, 139.4, 140, 143; FTIR (ATR) cm−1 3072, 2994, 2922, 2856, 1619, 1593, 1461, 1430, 1318, 1250, 1219, 1126, 1077, 1061, 1029, 992, 911, 761, 746, 736, 705; HRMS (EI-ion trap) m/z [M]+ calcd for (C15H11ClS2)+ 289.9991, found 289.9991.
2-(3-Chlorophenyl)-3-(methylthio)benzo[b]thiophene (34).
Column chromatography was performed using hexanes and ethyl acetate (80:1) as the eluent. yellow viscous liquid; 0.093 g, 81%; 1H NMR (400 MHz, CDCl3) δ 2.25 (s, 3H), 7.34–7.43 (m, 3H), 7.47 (t, J = 8.0 Hz, 1H), 7.65 (t, J = 4.0 Hz, 1H), 7.77 (s, 1H), 7.83 (d, J = 8.0 Hz, 1H), 8.04 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 19.1, 122.5, 123.8, 124.7, 125.1, 125.4, 128.2, 128.7, 129.8, 129.9, 134.4, 135.8, 138.5, 141.9, 144.4; FTIR (Salt Plate) cm−1 3059, 2985, 2920, 2852, 1792, 1594, 1564, 1463, 1433, 1319, 1294, 1249, 1217, 1150, 1095, 1082, 998, 970, 913, 880, 781, 756, 734, 687; HRMS (EI-ion trap) m/z [M]+ calcd for (C15H11ClS2)+ 289.9991, found 289.9992.
4-(3-(Methylthio)benzo[b]thiophen-2-yl)benzonitrile (35).
Column chromatography was performed using hexanes and ethyl acetate (40:1) as the eluent. Light brown solid; 0.077 g, 92%; mp 74.2–75.4°°C; 1H NMR (400 MHz, CDCl3) δ 2.27 (s, 3H), 7.44 (td, J = 8.0, 12 Hz, 1H), 7.51 (td, J = 8.0, 0.8 Hz, 1H), 7.76 (d, J = 8.4 Hz, 2H), 7.87 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 8.4 Hz, 2H), 8.07 (d, J = 7.6 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 19.2, 112.1, 118.7, 122.6, 124.0, 125.3, 125.8, 125.8, 130.6, 132.2, 138.6, 138.6, 141.1, 143.5; FTIR (ATR) cm−1 2992, 2920, 2852, 2228, 1742, 1602, 1523, 1433, 1366, 1217, 1021, 992, 953, 848, 837, 806, 762, 734; HRMS (EI-ion trap) m/z [M]+ calcd for (C16H11NS2)+ 281.0333, found 281.0331.
2-(3-(Methylthio)benzo[b]thiophen-2-yl)benzonitrile (36).
Column chromatography was performed using hexanes and ethyl acetate (40:1) as the eluent. Light pink solid; 0.084 g, 100%; mp 106.6–107.3 °C °C; Product was isolated as a light pink solid: mp 106.6–107.3 °C; 1H NMR (400 MHz, CDCl3) δ 2.32 (s, 3H), 7.44 (td, J = 7.2, 1.2 Hz, 1H), 7.47–7.60 (m, 3H), 7.66 (td, J = 7.6, 1.2 Hz, 1H), 7.81 (dd, J = 7.6, 0.8 Hz, 1H), 7.86 (d, J = 8.0 Hz, 1H), 8.05 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 18.8, 114.2, 118.0, 122.7, 123.9, 125.2, 125.7, 127.8, 129.1, 132.2, 132.4, 133.3, 137.7, 139.4, 140.1, 141.3; FTIR (ATR) cm−1 3070, 3003, 2926, 2854, 2229, 1740, 1595, 1447, 1427, 1377, 1294, 1220, 1169, 1021, 970, 908, 879, 768, 759, 735; HRMS (EI-ion trap) m/z [M]+ calcd for (C16H11NS2)+ 281.0333, found 281.0323.
3-(3-(Methylthio)benzo[b]thiophen-2-yl)benzonitrile (37).
Column chromatography was performed using hexanes and ethyl acetate (40:1) as the eluent. Light brown liquid; 0.074 g, 88%; Product was isolated as a light brown liquid; 1H NMR (400 MHz, CDCl3) δ 2.25 (s, 3H), 7.41 (td, J = 7.2, 1.2 Hz, 1H), 7.48 (td, J = 8.4, 1.2 Hz, 1H), 7.55 (t, J = 8.0 Hz, 1H), 7.67 (dt, J = 8.0, 1.2 Hz, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.97 (dt, J = 8.0, 1.2 Hz, 1H), 8.04 (d, J = 8.0 Hz, 1H), 8.06 (t, J = 1.6 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 19.2, 112.9, 118.6, 122.6, 123.9, 125.3, 125.4, 125.8, 129.4, 131.9, 133.4, 134.3, 135.3, 138.5, 141, 143.2; FTIR (Salt Plate) cm−1 3064, 3018, 2921, 2852, 2230, 1599, 1580, 1471, 1454, 1433, 1421, 1318, 1224, 1215, 1161, 1018, 971, 795, 757, 732, 686; HRMS (EI-ion trap) m/z [M]+ calcd for (C16H11NS2)+ 281.0333, found 281.0331.
2-Butyl-3-(methylthio)benzo[b]thiophene (38).
Column chromatography was performed using hexanes as the eluent. light yellow liquid; 0.071 g, 100%; Product was isolated as a light yellow liquid; 1H NMR (400 MHz, CDCl3) δ 0.99 (t, J = 7.2 Hz, 3H), 1.44 (sextet, J = 3.6 Hz, 2H), 1.72 (quintet, J = 7.6 Hz, 2H), 2.29 (s, 3H), 3.14 (t, J = 7.6 Hz, 2H), 7.31 (td, J = 7.2, 0.8 Hz, 1H), 7.41 (t, J = 7.2 Hz, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 14.0, 19.2, 22.5, 29.3, 33.8, 122.5, 122.7, 123.5, 124.3, 124.6, 138.0, 140.7, 150.9; FTIR (Salt Plate) cm−1 3059, 2955, 2923, 2871, 2857, 1455, 1434, 1378, 1314, 1252, 1152, 1018, 971, 942, 755, 731, 715; HRMS (EI-ion trap) m/z [M]+ calcd for (C13H16S2)+ 236.0693, found 236.0700.
4-(3-(Methylthio)benzo[b]thiophen-2-yl)butanenitrile (39).
Column chromatography was performed using hexanes and ethyl acetate (80:1) as the eluent. light orange liquid; 0.062 g, 83%; 1H NMR (400 MHz, CDCl3) δ 2.11 (quintet, J = 7.6 Hz, 2H), 2.31 (s, 3H), 2.43 (t, J = 7.2 Hz, 2H), 3.29 (t, J = 7.6 Hz, 2H), 7.35 (t, J = 8.0 Hz, 1H), 7.44 (t, J = 7.6 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.93 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 16.7, 19.2, 27.2, 28.2, 119.3, 122.7, 123.0, 124.9, 125.0, 125.3, 138.0, 140.5, 146.8; FTIR (Salt Plate) cm−1 3059, 2922, 2852, 2247, 1454, 1434, 1315, 1252, 1187, 1158, 1134, 1068, 1018, 971, 942, 758, 733; HRMS (EI-ion trap) m/z [M]+ calcd for (C13H13NS2)+ 247.0489, found 247.0485.
2-(tert-Butyl)-3-(methylthio)benzo[b]thiophene (40).
Column chromatography was performed using hexanes as the eluent. light orange viscous liquid; 0.071 g, 100%; 1H NMR (400 MHz, CDCl3) δ 1.62 (s, 9H), 2.30 (s, 3H), 7.31 (td, J = 8.0, 1.2 Hz, 1H), 7.40 (td, J = 8.0, 0.8 Hz, 1H), 7.76 (d, J = 8.0 Hz, 1H), 7.93 (d, J = 8.4 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 19.7, 31.1, 36.3, 121.8, 122.3, 122.3, 124.2, 124.6, 136.6, 142.3, 159.7; FTIR (Salt Plate) cm−1 3058, 2960, 2921, 2855, 1454, 1428, 1391, 1361, 1255, 1236, 1211, 1047, 1018, 969, 917, 831, 756, 732, 650; HRMS (EI-ion trap) m/z [M]+ calcd for (C13H16S2)+ 236.0693, found 236.0682.
Trimethyl(3-(methylthio)benzo[b]thiophen-2-yl)silane (41).
Column chromatography was performed using hexanes (80:1) as the eluent. light yellow liquid; 0.049 g, 65%; 1H NMR (400 MHz, CDCl3) δ 0.47 (s, 9H), 2.33 (s, 3H), 7.35 (td, J = 8.0, 0.8 Hz, 1H), 7.43 (td, J = 8.0, 0.8 Hz, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.99 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 0.1, 19.9, 122.7, 124.5, 124.7, 134.6, 141.9, 142.9, 146.6; FTIR (Salt Plate) cm−1 3052, 2955, 2921, 2897, 2854, 1456, 1450, 1415, 1320, 1285, 1246, 1160, 1076, 1010, 969, 900, 841, 756, 733, 712, 635; HRMS (EI-ion trap) m/z [M]+ calcd for (C12H16S2Si)+ 252.0463, found 252.0472.
2,3-Bis(methylthio)benzo[b]thiophene (42).
Column chromatography was performed using hexanes as the eluent. Light orange viscous liquid; 0.016 g, 24%; Product was isolated as a light orange viscous liquid; 1H NMR (400 MHz, CDCl3) δ 2.35 (s, 3H), 2.64 (s, 3H), 7.30 (td, J = 8.0, 0.8 Hz, 1H), 7.40 (td, J = 8.0, 0.8 Hz, 1H), 7.74 (d, J = 8.0 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 18.3, 18.6, 122.1, 122.3, 124.2, 125.1, 138.7, 140.9, 145.2; FTIR (Salt Plate) cm−1 3057, 2957, 2920, 2851, 1589, 1466, 1452, 1416, 1377, 1314, 1296, 1248, 1077, 1015, 972, 918, 753, 729; HRMS (EI-ion trap) m/z [M]+ calcd for (C10H10S3)+ 225.9945, found 225.9941.
Triisopropyl(3-(methylthio)benzo[b]thiophen-2-yl)silane (43).
Column chromatography was performed using hexanes as the eluent. Colorless viscous liquid; 0.073 g, 73%; Product was isolated as a colorless viscous liquid; 1H NMR (400 MHz, CDCl3) δ 1.16 (d, J = 7.6 Hz, 18H), 1.65–1.80 (m, 3H), 2.38 (s, 3H), 7.36 (td, J = 1.2, 7.2 Hz, 1H), 7.43 (td, J = 1.2, 8.0 Hz, 1H), 7.86 (d, J = 8.0 Hz, 1H), 8.00 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 13.0, 19.2, 19.3, 122.5, 123.1, 124.3, 124.5, 134.7, 141.5, 141.7, 143.3 FTIR (Salt Plate) cm−1 2944, 2925, 2865, 1460, 1449, 1243, 1076, 1019, 1001, 882, 756, 732, 714, 672, 647; HRMS (EI-ion trap) m/z [M]+ calcd for (C18H28S2Si)+ 336.1402, found 336.1405.
(3-(Methylthio)benzo[b]thiophen-2-yl)methanol (44).
Column was performed using hexanes and ethyl acetate (10:1) as the eluent. White solid; 0.043 g, 69%; mp 70.6–71.5 °C; 1H NMR (400 MHz, CDCl3) δ 2.32 (s, 3H), 2.44 (s, 1H), 5.10 (s, 2H), 7.36 (t, J = 7.2 Hz, 1H), 7.43 (t, J = 7.6 Hz, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 19.4, 59.5, 122.9, 122.9, 124.5, 124.8, 125.1, 138.7, 140.5, 148.2; FTIR (ATR) cm−1 3055, 2922, 2861, 1744, 1519, 1454, 1421, 1368, 1311, 1298, 1228, 1167, 1152, 1037, 1025, 995, 973, 937, 758, 732; HRMS (EI-ion trap) m/z [M]+ calcd for (C10H10OS2)+ 210.0173, found 210.0177.
2-(Cyclohex-1-en-1-yl)-3-(methylthio)benzo[b]thiophene (47).
Column was performed using hexanes as the eluent. Yellow liquid; 0.018 g, 20%; 1H NMR (400 MHz, CDCl3) δ 1.69–1.76 (m, 2H), 1.78–1.84 (m, 2H), 2.25–2.30 (m, 2H), 2.32 (s, 3H), 2.57–2.63 (m, 2H), 6.21–6.26 (m, 1H), 7.32 (td, J = 8.0, 1.2 Hz, 1H), 7.41 (td, J = 8.0, 1.2 Hz, 1H), 7.76 (d, J = 8.0 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 19.3, 21.9, 23.1, 26.0, 29.9, 121.9, 122.3, 123.0, 124.5, 124.6, 131.7, 131.9, 137.7, 141.4, 149.6; FTIR (Salt Plate) cm−1 3059, 3020, 2923, 2856, 2830, 1452, 1432, 1348, 1320, 1292, 1250, 1176, 1158, 1136, 1073, 1018, 970, 920, 755, 731; HRMS (EI-ion trap) m/z [M]+ calcd for (C15H16S2)+ 260.0693, found 260.0688.
6-Bromo-3-(methylthio)-2-phenylbenzo[b]thiophene (49).
Column was performed using hexanes and ethyl acetate (80:1) as the eluent. Pale yellow liquid; 0.096 g, 95%; 1H NMR (400 MHz, CDCl3) δ 2.21 (s, 3H), 7.38–7.52 (m, 3H), 7.55 (d, J = 8.4 Hz, 1H), 7.75 (d, J = 7.6 Hz, 2H), 7.87 (d, J = 7.6 Hz, 1H), 7.96 (s, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 19.3, 119.1, 123.8, 125.0, 125.1, 128.6, 128.9, 129.2, 130.1, 133.7, 140.0, 140.5, 146.9; FTIR (Salt Plate) cm−1 3061, 3022, 2919, 2852, 1580, 1542, 1473, 1443, 1385, 1310, 1263, 1217, 1090, 895, 813, 763, 748, 693; HRMS (EI-ion trap) m/z [M]+ calcd for (C15H11BrS2)+ 333.9486, found 333.9493.
Synthesis of (E)-Dimethyl(2-(methylthio)-1,2-diphenylvinyl)sulfonium tetrafluoroborate (54).
Diphenylacetylene (0.6 mmol, 1.0 equiv) and dimethyl(methylthio)sulfonium tetrafluoroborate (1.2 mmol, 2.0 equiv) were added to a 6-dram vial. Methylene chloride (3 mL) was added to the vial, and the reaction was allowed to stir at room temperature. The progress of the reaction was monitored by TLC. After the reaction was completed, the reaction mixture was filtered, concentrated under a vacuum, and absorbed in silica gel before purification via column chromatography using hexanes as the eluent. The purification was performed via column chromatography, first using hexanes and ethyl acetate (10:1) to remove the nonpolar impurities and then with CH2Cl2 and methanol (98:2) as the eluent. Light yellow solid; 0.174 g, 71%; mp 183.4–185.1 °C; 1H NMR (400 MHz, CDCl3) δ 1.79 (s, 3H), 2.61 (s, 6H), 7.38–7.40 (m, 4H), 7.54 (t, J = 7.6 Hz, 2H), 7.58–7.62 (m, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 16.4, 27.4, 114.2, 127.8, 128.5, 129.9, 130.2, 130.6, 131.1, 131.8, 133.2, 166.8.; FTIR (ATR) cm−1 3020, 2968, 2930, 1737, 1606, 1576, 1490, 1431, 1372, 1327, 1287, 1200, 1180, 1055, 1005, 813, 772, 743, 710; HRMS (ESI-ion trap) m/z [M]+ calcd for (C17H19S2)+ 287.0923, found 287.0922.
Synthesis of (2-Bromo-1,2-diphenylvinyl)(methyl)sulfane (56 and 57).
Diphenylacetylene (0.6 mmol, 1.0 equiv), TBAB (1.8 mmol, 3.0 equiv), and dimethyl(methylthio)sulfonium tetrafluoroborate (0.9 mmol, 1.5 equiv) were added to a 6-dram vial. Methylene chloride (3 mL) was added to the vial and the progress of the reaction was monitored by TLC. After the reaction was completed, the reaction mixture was filtered, concentrated under a vacuum, and absorbed in silica gel before purification via column chromatography using hexanes as the eluent. Column was performed using hexanes and ethyl acetate (80:1) as the eluent. Light-yellow solid; 0.174 g, 95%; mp 64.0–67.0 °C; 1H NMR (400 MHz, CDCl3) δ 1.79 (s, 0.87H), 1.89 (s, 2.2H), 7.05–7.13 (m, 5H), 7.15–7.20 (m, 2H), 7.34–7.53 (m, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 16.4, 17.2, 116.5, 117.4, 127.5, 127.8, 128.2, 128.3, 128.4, 128.5, 128.7, 129.5, 130.1, 130.2, 136.8, 137.5, 139.3, 140.5, 140.5, 140.7; FTIR (ATR) cm−1 3081, 3059, 3027, 2926, 1746, 1598, 1565, 1486, 1442, 1425, 1366, 1324, 1283, 1215, 1073, 1029, 986, 951, 911, 847, 791, 763, 726, 693, 616; HRMS (EI-ion trap) m/z [M]+ calcd for (C15H13BrS)+ 303.9921, found 303.9918.
Synthesis of 3-(Methylsulfinyl)-2-phenylbenzo[b]thiophene (58).
m-CPBA (0.47 mmol, 0.95 equiv) was added to a stirred solution of 3-(methylthio)-2-phenylbenzo[b]thiophene (0.5 mmol, 1.0 equiv) and 3 mL of DCM at 0 °C. The reaction was stirred overnight. The reaction mixture was diluted with 30 mL of DCM and washed with saturated NaHCO3 solution (2 × 10 mL). The resulting organic solution was dried with Na2SO4 and concentrated under a vacuum. The obtained crude mixture was purified using column chromatography using hexanes and ethyl acetate (3:1) as the eluent. Colorless liquid; 0.106 g, 82%; 1H NMR (400 MHz, CDCl3) δ 3.08 (s, 3H), 7.42–7.52 (m, 7H), 7.89 (d, J = 7.6 Hz, 1H), 7.67 (d, J = 7.2 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 40.2, 122.9, 124.0, 125.6, 125.8, 129.1, 129.9, 130.4, 131.6, 132.1, 136.7, 139.6, 147.8; FTIR (ATR) cm−1 3061, 2920, 2853, 1580, 1542, 1473, 1443, 1385, 1310, 1263, 1217, 1090, 1032, 966, 895, 813, 763, 748, 693; HRMS (EI-ion trap) m/z [M]+ calcd for (C15H12OS2)+ 272.0330, found 272.0332.
Supplementary Material
ACKNOWLEDGMENTS
The authors are thankful to the University of West Florida (UWF), and UWF’s Office of Research and Sponsored Programs and Office of Undergraduate Research for supporting this research. ZA is grateful to NIH for support through the Institute of General Medical Sciences of the National Institutes of Health under Grant Number 1T34GM110517-01. This content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors are also grateful to Dr. Tim Royappa for his help throughout the project.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c02606.
Reaction optimization table and compound characterization data (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.joc.1c02606
The authors declare no competing financial interest.
Contributor Information
Zahra Alikhani, Department of Chemistry, University of West Florida, Pensacola, Florida 32514, United States.
Alyssa G. Albertson, Department of Chemistry, University of West Florida, Pensacola, Florida 32514, United States
Christopher A. Walter, Department of Chemistry, University of West Florida, Pensacola, Florida 32514, United States
Prerna J. Masih, Department of Biology, University of West Florida, Pensacola, Florida 32514, United States
Tanay Kesharwani, Department of Chemistry, University of West Florida, Pensacola, Florida 32514, United States.
REFERENCES
- (1).(a) Keri RS; Chand K; Budagumpi S; Balappa Somappa S; Patil SA; Nagaraja BM An Overview of Benzo[b]thiophene-Based Medicinal Chemistry. Eur. J. Med. Chem 2017, 138, 1002–1033. [DOI] [PubMed] [Google Scholar]; (b) Wishart DS; Feunang YD; Guo AC; Lo EJ; Marcu A; Grant JR; Sajed T; Johnson D; Li C; Sayeeda Z; Assempour N; Iynkkaran I; Liu Y; Maciejewski A; Gale N; Wilson A; Chin L; Cummings R; Le D; Pon A; Knox C; Wilson M DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46 (D1), D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Sweidan K; Engelmann J; Rayyan W; Sabbah D; Zarga M; Al-Qirim T; Al-Hiari Y; Sheikha G; Shattat G Synthesis and Preliminary Biological Evaluation of New Heterocyclic Carboxamide Models. Lett. Drug Des. Discovery 2015, 12 (5), 417–429. [Google Scholar]; (b) Martorana A; Gentile C; Perricone U; Piccionello AP; Bartolotta R; Terenzi A; Pace A; Mingoia F; Almerico AM; Lauria A Synthesis, Antiproliferative Activity, and in Silico Insights of New 3-Benzoylamino-Benzo[b]thiophene Derivatives. Eur. J. Med. Chem 2015, 90, 537–546. [DOI] [PubMed] [Google Scholar]
- (3).Rosada B; Bekier A; Cytarska J; Płaziński W; Zavyalova O; Sikora A; Dzitko K; Łączkowski KZ Benzo[b]thiophene-Thiazoles as Potent Anti-Toxoplasma Gondii Agents: Design, Synthesis, Tyrosinase/tyrosine Hydroxylase Inhibitors, Molecular Docking Study, and Antioxidant Activity. Eur. J. Med. Chem 2019, 184, 111765. [DOI] [PubMed] [Google Scholar]
- (4).(a) Fakhr IMI; Radwan MAA; El-Batran S; Abd El-Salam OME; El-Shenawy SM Synthesis and Pharmacological Evaluation of 2-Substituted Benzo[b]thiophenes as Anti-Inflammatory and Analgesic Agents. Eur. J. Med. Chem 2009, 44 (4), 1718–1725. [DOI] [PubMed] [Google Scholar]; (b) Isloor AM; Kalluraya B; Sridhar Pai K Synthesis, Characterization and Biological Activities of Some New Benzo[b]thiophene Derivatives. Eur. J. Med. Chem 2010, 45 (2), 825–830. [DOI] [PubMed] [Google Scholar]
- (5).(a) Rao GK Synthesis, Antitubercular and Antibacterial Activities of Some Quinazolinone Analogs Substituted with Benzothiophene. Chem. Sci. J 2015, DOI: 10.4172/2150-3494.100092. [DOI] [Google Scholar]; (b) Mahajan PS; Nikam MD; Nawale LU; Khedkar VM; Sarkar D; Gill CH Synthesis and Antitubercular Activity of New Benzo[b]thiophenes. ACS Med. Chem. Lett 2016, 7 (8), 751–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Rackham MD; Brannigan JA; Moss DK; Yu Z; Wilkinson AJ; Holder AA; Tate EW; Leatherbarrow RJ Discovery of Novel and Ligand-Efficient Inhibitors of Plasmodium falciparum and Plasmodium vivax N-Myristoyltransferase. J. Med. Chem 2013, 56 (1), 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Banerjee T; Kapoor N; Surolia N; Surolia A Benzothiophene Carboxamide Derivatives as Novel Antimalarials. IUBMB Life 2011, 63 (12), 1111–1115. [DOI] [PubMed] [Google Scholar]
- (7).(a) Shafieex A; Vossoghi M; Wossooghi J; Yazdani S Synthesis and Antibacterial and Antifungal Activities of Alkyl and Polyhalophenyl Esters of Benzo[b]thiophene-3-Carbamic Acid. J. Pharm. Sci 1981, 70 (5), 566–568. [DOI] [PubMed] [Google Scholar]; (b) Naganagowda G; Padmashali B Synthesis, Antimicrobial, and Anthelmintic Activities of Some New 3-Chlorobenzothiophene-2-Carbonylchloride Derivatives. Phosphorus Sulfur Silicon Relat. Elem 2010, 185 (8), 1691–1700. [Google Scholar]; (c) Jagtap VA; Agasimundin YS Synthesis and Preliminary Evaluation of Some 2-Amino-N’-[substituted]-4, 5, 6, 7-Tetrahydro-1-Benzothiophene-3-Carbohydrazide as Antimicrobial Agents. J. Pharm. Res 2015, 9 (1), 10–14. [Google Scholar]
- (8).(a) Chang S-L; Hung K-E; Cao F-Y; Huang K-H; Hsu C-S; Liao C-Y; Lee C-H; Cheng Y-J Isomerically Pure Benzothiophene-Incorporated Acceptor: Achieving Improved VOC and JSC of Nonfullerene Organic Solar Cells via End Group Manipulation. ACS Appl. Mater. Interfaces 2019, 11 (36), 33179–33187. [DOI] [PubMed] [Google Scholar]; (b) Kim J; Cho N; Min Ko H; Kim C; Kwan Lee J; Ko J Push-Pull Organic Semiconductors Comprising of Bis-Dimethylfluorenyl Amino Benzo[b]thiophene Donor and Various Acceptors for Solution Processed Small Molecule Organic Solar Cells. Sol. Energy Mater. Sol. Cells 2012, 102, 159–166. [Google Scholar]
- (9).(a) Kim M-S; Choi B-K; Lee T-W; Shin D; Kang SK; Kim JM; Tamura S; Noh T A Stable Blue Host Material for Organic Light-Emitting Diodes. Appl. Phys. Lett 2007, 91 (25), 251111. [Google Scholar]; (b) Najare MS; Patil MK; Mantur S; Nadaf AA; Inamdar SR; Khazi IAM Highly Conjugated D-π-A-π-D Form of Novel Benzo[b]thiophene Substituted 1,3,4-oxadiazole Derivatives; Thermal, Optical Properties, Solvatochromism and DFT Studies. J. Mol. Liq 2018, 272, 507–519. [Google Scholar]
- (10).Kim J; Ko HM; Cho N; Paek S; Lee JK; Ko J Efficient Small Molecule Organic Semiconductor Containing Bis-Dimethylfluorenyl Amino Benzo[b]thiophene for High Open Circuit Voltage in High Efficiency Solution Processed Organic Solar Cell. RSC Adv. 2012, 2 (7), 2692–2695. [Google Scholar]
- (11).(a) Sun L-L; Deng C-L; Tang R-Y; Zhang X-G CuI/TMEDA-Catalyzed Annulation of 2-Bromo Alkynylbenzenes with Na2S: Synthesis of Benzo[b]thiophenes. J. Org. Chem 2011, 76 (18), 7546–7550. [DOI] [PubMed] [Google Scholar]; (b) Kuhn M; Falk FC; Paradies J Palladium-Catalyzed C-S Coupling: Access to Thioethers, Benzo[b]thiophenes, and thieno[3,2-b]thiophenes. Org. Lett 2011, 13 (15), 4100–4103. [DOI] [PubMed] [Google Scholar]
- (12).Hari DP; Hering T; König B Visible Light Photocatalytic Synthesis of Benzothiophenes. Org. Lett 2012, 14 (20), 5334–5337. [DOI] [PubMed] [Google Scholar]
- (13).Nguyen TB; Retailleau P DIPEA-Promoted Reaction of 2-Nitrochalcones with Elemental Sulfur: An Unusual Approach to 2-Benzoylbenzothiophenes. Org. Lett 2017, 19 (18), 4858–4860. [DOI] [PubMed] [Google Scholar]
- (14).Yan K; Yang D; Zhang M; Wei W; Liu Y; Tian L; Wang H Facile Access to Benzothiophenes through Metal-Free Iodine-Catalyzed Intermolecular Cyclization of Thiophenols and Alkynes. Synlett 2015, 26 (13), 1890–1894. [Google Scholar]
- (15).Nguyen TB; Retailleau P Cooperative Activating Effect of Tertiary Amine/DMSO on Elemental Sulfur: Direct Access to Thioaurones from 2′-Nitrochalcones under Mild Conditions. Org. Lett 2018, 20, 186–189. [DOI] [PubMed] [Google Scholar]
- (16).(a) Flynn BL; Verdier-Pinard P; Hamel E A Novel Palladium-Mediated Coupling Approach to 2,3-Disubstituted Benzo[b]thiophenes and Its Application to the Synthesis of Tubulin Binding Agents. Org. Lett 2001, 3 (5), 651. [DOI] [PubMed] [Google Scholar]; (b) Yue D; Larock RC Synthesis of 2,3-Disubstituted Benzo[b]thiophenes via Palladium-Catalyzed Coupling and Electrophilic Cyclization of Terminal Acetylenes. J. Org. Chem 2002, 67 (6), 1905–1909. [DOI] [PubMed] [Google Scholar]; (c) Mehta S; Waldo JP; Larock RC Competition Studies in Alkyne Electrophilic Cyclization Reactions. J. Org. Chem 2009, 74 (3), 1141–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen C-C; Chen C-M; Wu M-J Transition Metal-Catalyzed Cascade Cyclization of Aryldiynes to Halogenated benzo[b]naphtho[2,1-D]thiophene Derivatives. J. Org. Chem 2014, 79, 4704–4711. [DOI] [PubMed] [Google Scholar]; (e) Sanz R; Guilarte V; Hernando E; Sanjuán AM Synthesis of Regioselectively Functionalized Benzo[b]thiophenes by Combined Ortho-Lithiation-Halocyclization Strategies. J. Org. Chem 2010, 75 (21), 7443–7446. [DOI] [PubMed] [Google Scholar]; (f) Cunningham C; Cloyd M; Phillips A; Khan S; Whalen K; Kesharwani A One-pot Successive Cyclization-Alkylation Strategy for the Synthesis of 2,3-Disubstituted Benzo[b]thiophenes. Org. Biomol. Chem 2021, 19, 4107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Mori T; Nishimura T; Yamamoto T; Doi I; Miyazaki E; Osaka I; Takimiya K Consecutive Thiophene-Annulation Approach to π-Extended Thienoacene-Based Organic Semiconductors with [1]-Benzothieno[3,2-b][1]benzothiophene (BTBT) Substructure. J. Am. Chem. Soc 2013, 135, 13900–13913. [DOI] [PubMed] [Google Scholar]
- (17).(a) Kesharwani T; Giraudy KA; Morgan JL; Kornman C; Olaitan AD Green Synthesis of Halogen Containing Thiophene, Selenophene and Benzo[b]selenophene Derivatives Using Sodium Salts as the Source of Halide. Tetrahedron Lett. 2017, 58 (7), 638–641. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kesharwani T; Kornman C; Tonnaer A; Hayes A; Kim S; Dahal N; Romero R; Royappa A Sodium Halides as the Source of Electrophilic Halogens in Green Synthesis of 3-Halo- and 3,n-Dihalobenzo[b]thiophenes. Tetrahedron 2018, 74 (24), 2973–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Walter C; Fallows N; Kesharwani T Copper-Catalyzed Electrophilic Chlorocyclization Reaction Using Sodium Chloride as the Source of Electrophilic Chlorine. ACS Omega 2019, 4 (4), 6538–6545. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kim S; Dahal N; Kesharwani T Environmentally benign process for the synthesis of 2,3-disubstituted benzo[b]thiophenes using electrophilic cyclization. Tetrahedron Lett. 2013, 54, 4373–4376. [Google Scholar]
- (18).Dillon CC; Keophimphone B; Sanchez M; Kaur P; Muchalski H Org. Biomol. Chem 2018, 16, 9279–9284. [DOI] [PubMed] [Google Scholar]
- (19).Kesharwani T; Kornman CT; Tonnaer AL; Royappa AD Green synthesis of benzo[b]thiophenes via iron(III) mediated 5-endo-dig iodocyclization of 2-alkynylthioanisoles. Tetrahedron Lett. 2016, 57 (3), 411–414. [Google Scholar]
- (20).Zhang M-M; Sun Y; Wang W-W; Chen K-K; Yang W-C; Wang L Electrochemical synthesis of sulfonated benzothiophenes using 2-alkynylthioanisoles and sodium sulfinates. Org. Biomol. Chem 2021, 19, 3844–3849. [DOI] [PubMed] [Google Scholar]
- (21).Tambe SD; Jadhav MS; Rohokale RS; Kshirsagar UA Metal-Free Synthesis of 3-Thiocyanatobenzothiophenes by Eosin Y Photoredox-Catalyzed Cascade Radical Annulation of 2-Alkynylthioanisoles. Eur. J. Org. Chem 2018, 2018, 4867–4873. [Google Scholar]
- (22).Wang C; Nakamura H; Sugino H; Takimiya K Methylthionated benzo[1,2-b:4,5-b’] dithiophenes: a model study to control packing structures and molecular orientation in thienacene based organic semiconductors. Chem. Commun 2017, 53, 9594–9597. [DOI] [PubMed] [Google Scholar]
- (23).(a) Gai BM; Stein AL; Roehrs JA; Bilheri FN; Nogueira CW; Zeni G Synthesis and Antidepressant-like Activity of Selenophenes Obtained via Iron(III)-PhSeSePh-Mediated Cyclization of Z-Selenoenynes. Org. Biomol. Chem 2012, 10 (4), 798–807. [DOI] [PubMed] [Google Scholar]; (b) Sonawane AD; Shimozuma A; Udagawa T; Ninomiya M; Koketsu M Synthesis and Photophysical Properties of Selenopheno-[2,3-b]quinoxaline and Selenopheno[2,3-b]pyrazine Heteroacenes. Org. Biomol. Chem 2020, 18 (21), 4063–4070. [DOI] [PubMed] [Google Scholar]
- (24).Dillon AS; Flynn BL Polyynes to Polycycles: Domino Reactions Forming Polyfused Chalcogenophenes. Org. Lett 2020, 22 (8), 2987–2990. [DOI] [PubMed] [Google Scholar]
- (25).Wang L; Wang H; Meng W; Xu X-H; Huang Y Facile Syntheses of 3-Trifluoromethylthio Substituted Thioflavones and Benzothiophenes via the Radical Cyclization. Chin. Chem. Lett 2021, 32, 389–392. [Google Scholar]
- (26).Zhang B; Li X; Li X; Yu Z; Zhao B; Wang X; Du Y; Zhao K An Interrupted Pummerer Reaction Mediated by a Hypervalent Iodine(III) Reagent: In Situ Formation of RSCl and Its Application for the Synthesis of 3-Sulfenylated Indoles. J. Org. Chem 2021, 86, 17274–17281. [DOI] [PubMed] [Google Scholar]
- (27).Yue D; Yao T; Larock RC Synthesis of 2,3-Disubstituted Benzo[b]furans by the Palladium-Catalyzed Coupling of o-Iodoanisoles and Terminal Alkynes, Followed by Electrophilic Cyclization. J. Org. Chem 2005, 70 (25), 10292–10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Volpe R; Aurelio L; Gillin MG; Krenske EH; Flynn BL Mapping the Interactions of I2, I., I−, and I+ with Alkynes and Their Roles in Iodocyclizations. Chem. Eur. J 2015, 21, 10191–10199. [DOI] [PubMed] [Google Scholar]
- (29).Lu WD; Wu MJ Halocyclization of 2-Alkynylthioanisoles by Cupric Halides: Synthesis of 2-Substituted 3-Halobenzo[b]thiophenes. Tetrahedron 2007, 63, 356–362. [Google Scholar]
- (30).Yamauchi T; Shibahara F; Murai T Pd/phenanthrolinecatalyzed arylative cyclization of o-(1-alkynyl)thioanisoles: synthesis of 3-arylated benzo[b]thiophenes. Tetrahedron Lett. 2016, 57, 2945–2948. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.