

Learning Points
Skin-limited Langerhans cell histiocytosis (LCH) during the neonatal period with spontaneous clearing has been termed congenital self-healing reticulohistiocytosis.
As the course of this disease cannot be predicted and extracutaneous organ involvement can manifest later on, some authors prefer avoiding the use of this designation and instead refer to it as neonatal LCH with cutaneous involvement.
There are no clinical or morphological patterns that can distinguish between skin-limited and multisystem LCH (MS-LCH).
Classical neonatal LHC with cutaneous involvement are multiple, red-brown papules, nodules, or vesicles resembling a varioliform rash that may be crusted or ulcerated. However, a variety of cutaneous lesions have been described, including solitary nodules.
To determine the extent of organ involvement, a series of laboratory and imaging evaluations must be done, and there is a need for continued long-term follow-up for signs of MS-LCH.
Case Presentation
A 1-day-old female patient presented with a congenital, ulcerated nodule on her left palm. She was born by vaginal delivery after an unremarkable pregnancy. There was no history of trauma.
Physical examination revealed a 1.2 cm × 1.0 cm violaceous, indurated nodule with central ulceration and peripheral yellow desquamation on the hypothenar eminence of the left hand [Figure 1]. Lymph nodes were not palpable. The rest of the physical exam was unremarkable.
Figure 1.
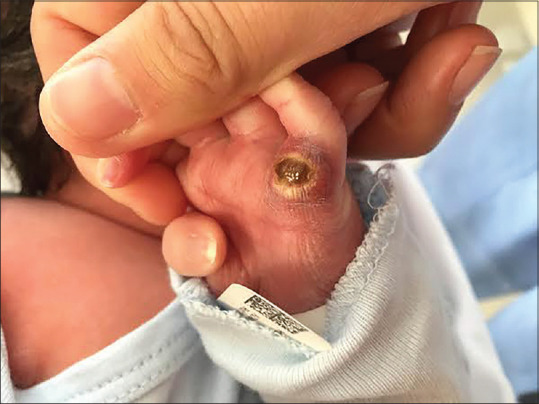
Physical examination at birth shows a solitary violaceus nodule on the hypothenar eminence of the left hand, measuring 1.2 cm × 1.0 cm. The nodule is firm in consistency and presents central ulceration and peripheral yellow desquamation.
Biopsy showed histiocytes on the hematoxylin-eosin stain [Figure 2], and immunohistochemistry was positive for S-100 and CD1a [Figure 3], CD8, and langerin cells.
Figure 2.

Sample tissue of a nodule on the hypothenar eminence of the left hand
Figure 3.

Sample tissue of a nodule on the hypothenar eminence of the left hand
Blood tests, including complete blood cell count, liver enzymes, and albumin, were normal. No abnormalities were detected on abdominal ultrasound and brain magnetic resonance imaging. Radiographs of several body parts were taken, including hands, forearms, femur, and skull.
What is Your Diagnosis?
Answer: Neonatal Langerhans cell histiocytosis with cutaneous involvement.
Discussion
The diagnosis was neonatal Langerhans cell histiocytosis with cutaneous involvement. The differential diagnoses include pyogenic granuloma, hemangioma, cyst, and malignancy, including Langerhans cell histiocytosis (LCH).
The lesion did not require any specific treatment, and it resolved gradually following the biopsy. She was followed up by us every 6 months during the first year and then once every year. After 3 years of follow-up, there have been no recurrence signs.
LCH is a neoplastic histiocytic disorder driven by activating mutations in the mitogen-activated protein kinase (MAPK) pathway. The BRAF V600E mutation, which leads to the activation of the MAPK pathway, is present in over half of cases.[1]
The dendritic histiocytes accumulate in one or many organs. LCH is classified based on the number of organ systems involved as single-system or multisystem (MS-LCH).
The incidence of LCH in children under one year of age is 9.9 cases per million per year,[2] while the estimated incidence of neonatal LCH (LCH diagnosed within 28 days after birth) is 1–2 cases per million per year.[3]
Reports on LCH in the neonatal period describe a skin-limited disease form with a high tendency to spontaneous regression called congenital self-healing reticulohistiocytosis.[3] Although our patient had a solitary skin lesion that spontaneously involuted, we did not use the terms “self-healing” or “skin-limited” as upon diagnosis, the course of the disease cannot be predicted.[3]
There are no clinical or morphological patterns that can distinguish between skin-limited LCH and MS-LCH.[3] The detection of the BRAF-V600E mutation is often seen in MS-LCH but rarely in skin-limited LCH.[1] However, the ability of the BRAF-V600E mutation to predict MS-LCH still requires validation.
The diagnosis of cutaneous LCH is based on clinical signs, histological finds, and immunophenotypic stains.
Classical cutaneous neonatal LHC are multiple, red-brown papules, nodules, or vesicles resembling a varioliform rash that may be crusted or ulcerated. Other common manifestations are scaly, eczematous patches, similar to seborrheic dermatitis, more prominent in the intertriginous areas and on the scalp. Solitary nodules are also documented as a single red-brown firm nodule that tends to become ulcerated, necrotic, or crusted. Blue nodular skin infiltrates resembling “blueberry-muffin baby” have also been reported.[4]
Positivity for Langerhans cells can be confirmed with the microscopy (presence of Birbeck granules) and immunohistochemical demonstration of dendritic cells (CD1, S100 protein, and CD68 positivity).[5]
Once the diagnosis is made, baseline laboratory and radiographic evaluation must be done to determine the extent of organ involvement, such as complete blood count, liver enzymes, coagulation studies, a skeletal survey, and chest radiography.[4]
There are no specific treatments for cutaneous neonatal LHC, but if lesions persist, topical corticosteroids, tacrolimus, or nitrogen mustard can be used, or localized lesions may be excised.[4]
Although skin-limited LCH generally has a good prognosis, the cases of recurrence and complications suggest that there is a need for continued long-term, and perhaps indefinite, follow-up for signs of MS-LCH.[5]
Ethics committee/Institutional review board's permission
There was no need to be assessed by an ethics committee because this is a Quiz.
Patient's consent
We have the patient's consent to use photographs that may reveal the identity of the patient is enclosed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Héritier S, Emile JF, Barkaoui MA, Thomas C, Fraitag S, Boudjemaa S, et al. BRAF mutation correlates with high-risk Langerhans cell histiocytosis and increased resistance to first-line therapy. J Clin Oncol. 2016;34:3023–30. doi: 10.1200/JCO.2015.65.9508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salotti JA, Nanduri V, Pearce MS, Parker L, Lynn R, Windebank KP. Incidence and clinical features of Langerhans cell histiocytosis in the UK and Ireland. Arch Dis Child. 2009;94:376–80. doi: 10.1136/adc.2008.144527. [DOI] [PubMed] [Google Scholar]
- 3.Minkov M, Prosch H, Steiner M, Grois N, Pötschger U, Kaatsch P, et al. Langerhans cell histiocytosis in neonates. Pediatr Blood Cancer. 2005;45:802–7. doi: 10.1002/pbc.20362. [DOI] [PubMed] [Google Scholar]
- 4.Lee YH, Talekar MK, Chung CG, Bell MD, Zaenglein AL. Congenital self-healing reticulohistiocytosis. J Clin Aesthet Dermatol. 2014;7:49–53. [PMC free article] [PubMed] [Google Scholar]
- 5.Wheller L, Carman N, Butler G. Unilesional self-limited Langerhans cell histiocytosis: A case report and review of the literature. J Cutan Pathol. 2013;40:595–9. doi: 10.1111/cup.12121. [DOI] [PubMed] [Google Scholar]