

Abstract
A wide range of functionalized pyridine ligands have been employed to synthesize a variety of Pd(II) complexes of the general formulas [PdL4](NO3)2 and [PdL2Y2], where L = 4-X-py and Y = Cl– or NO3–. Their structures have been unambiguously established via analytical and spectroscopic methods in solution (NMR spectroscopy and mass spectrometry) as well as in the solid state (X-ray diffraction). This in-depth characterization has shown that the functionalization of ligand molecules with groups of either electron-withdrawing or -donating nature (EWG and EDG) results in significant changes in the physicochemical properties of the desired coordination compounds. Downfield shifts of signals in the 1H NMR spectra were observed upon coordination within and across the complex families, clearly indicating the relationship between NMR chemical shifts and the ligand basicity as estimated from pKa values. A detailed crystallographic study has revealed the operation of a variety of weak interactions, which may be factors explaining aspects of the solution chemistry of the complexes. The Pd(II) complexes have been found to be efficient and versatile precatalysts in Suzuki–Miyaura and Heck cross-coupling reactions within a scope of structurally distinct substrates, and factors have been identified that have contributed to efficiency improvement in both processes.
Short abstract
A wide range of pyridine derivatives have been employed to synthesize a variety of di- and tetrasubstituted Pd(II) complexes of square-planar geometry. This in-depth characterization has shown that the functionalization of ligand molecules with groups of either electron-withdrawing or -donating nature results in significant changes in the physicochemical properties of the coordination compounds. Moreover, the complexes have been found to be of practical utility as efficient precatalysts for both Suzuki−Miyaura and Heck cross-coupling reactions.
Introduction
Since its discovery by Thomas Anderson in 1849, pyridine has been one of the most popular heterocyclic compounds used in chemistry. Despite the many similarities between pyridine and benzene, the introduction of an electronegative N atom to the aromatic ring significantly differentiates their physicochemical properties.1 The presence of a pair of nonbonding electrons in the valence shell of the N atom enables pyridine derivatives to act as Lewis bases toward a wide variety of metal ions.2 Both pyridine and polypyridine ligands are good neutral donors of mono- or multidentate nature, and their coordination properties can be relatively easily altered by substitution with electron-donating or -withdrawing groups.3 The structural and electronic modifications achieved by the functionalization of heterocyclic rings enable modulation of the metal coordination sphere, which can lead to improvement of the desired properties and potential applicability.4 Furthermore, pyridine and its derivatives can be utilized as model units for research on important biomolecules such as nicotine, pyridoxine, or nicotinamide adenine dinucleotide phosphate (NADP).4a,5 A wide variety of transition-metal complexes with pyridine-based ligands having both academic and industrial importance have been successfully generated, as reflected in the rich literature in this field.6 Notably, coordination structures based on pyridyl units have shown real application potential as catalysts,7 compounds of cytotoxic activity,8 and molecule magnets.9
Where direct complexation equilibrium measurements of the Lewis basicity of a ligand are unavailable, the Brønsted basicity, measured as pKa values, usually rather readily obtained for pyridine derivatives, has been widely applied as a measure of the effect of any substituent on pyridine donor behavior.10 In a recent study of Pt(II) complexes of 4-substituted pyridines having some parallels with the present study of Pd(II) species,11 it was found that the 1H NMR chemical shifts of the 2/6 protons showed a same linear dependence on the pKa for the coordinated as well as free ligands, consistent with protonation being a useful guide to coordination behavior. Pd(II) complexes with pyridine derivatives have been used as efficient catalysts in reactions such as the carbonylation of nitro compounds,12 reduction of nitro compounds to amines,13 or carbonylation of aniline derivatives by CO/O2.14 A successful example of the correlation between the catalytic efficiency and ligand basicity is provided in the conversion of nitrobenzene to ethyl N-phenylcarbamate catalyzed by a series of [PdL2Cl2] complexes, where L = various di- and monosubstituted pyridines.12a An increase in the reaction yield was observed when Pd(II) complexes based on more basic ligands were used as catalysts, although steric effects were also apparent in species with 2/6 substituents, leading to the conclusion that 3 or 4 substitution provided the best correlation with basicity.
In this work, we have employed a series of 4-substituted pyridine ligands, L1–L12 (Scheme 1), to generate an array of coordination compounds with Pd(II) cations of a square-planar geometry. The particular reaction conditions employed provided di- and tetrasubstituted complexes diversified in terms of their charge and composition. We anticipated that the properties of such complexes could be tuned by modifying the nature of the ligand substituents, and our efforts to prove this are presented below. Detailed analyses have been made of the structures and 1H NMR spectra of the complexes in relation to substituent effects, and we have extended this investigation to that of functionality by employing the complexes as catalyst precursors in Suzuki–Miyaura and Heck cross-coupling reactions involving a range of organic reagents.
Scheme 1. Synthetic Routes for the Pd(II) Complexes Based on the Pyridine Ligands L1–L12.
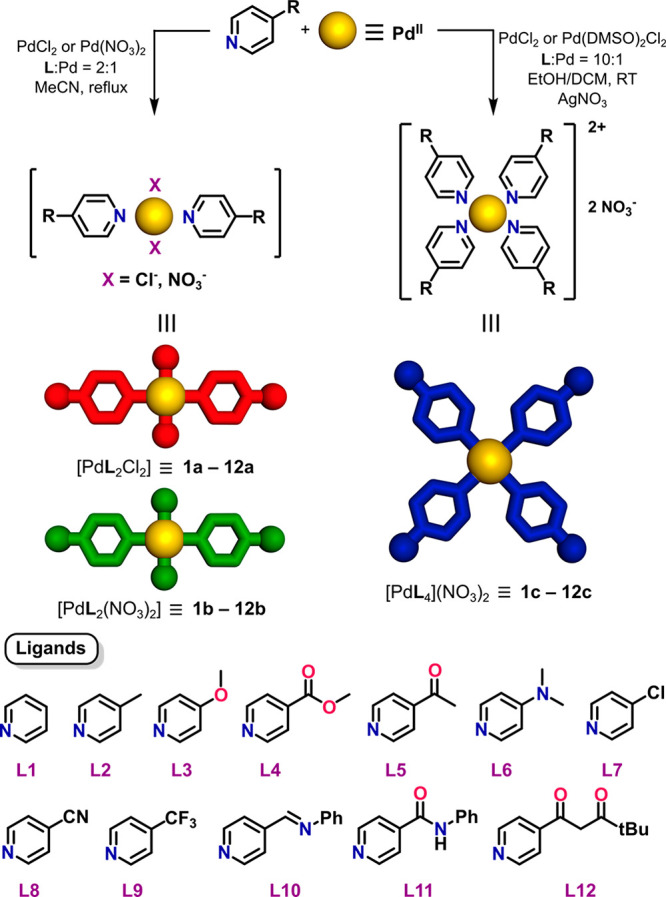
Results and Discussion
Synthesis of Complexes
The substituents on the 4 position of ligands L1–L12 span a range of both electron-withdrawing and -donating groups, but pyridine-N-bound complexes of all of the ligands can be isolated under the appropriate reaction conditions. While, in principle, mono, bis, tris, and tetrakis species are possible, the use of an exact 2:1 L/Pd(II) reaction stoichiometry enabled the isolation of neutral [PdL2Y2], where Y = Cl– or NO3–, whereas the use of a large excess of the ligand [10:1 L/Pd(II)] was required to shift the reaction equilibrium toward the exclusive generation of tetra-substituted compounds, allowing for the ready isolation of cationic complexes [PdL4]2+ as their nitrate salts. The synthetic procedures are outlined in Scheme 1 and described in detail in the SI. On the basis of their 1H NMR spectra [see the Supporting Information (SI) and Figure 1b] allied to the X-ray structural results (see below), all of the [PdL2Cl2] products (1a–12a) appeared to contain only the trans isomer, whereas the [PdL2(NO3)2] products (1b–12b) contained minor but detectable amounts of the cis isomer. This preference for the trans configuration mimics that known for various Pt(II) analogues.11 The use of PdCl2 or [Pd(DMSO)2Cl2] (DMSO = dimethyl sulfoxide) as reactants for the formation of [PdL4]2+ cations (1c–12c) was efficient but required the elimination of chloride from the reaction mixtures by the addition of AgNO3. The ease of formation of the [PdL4]2+ cations appeared to increase with the basicity of the ligand, and this may explain why a pure species 6b could not be isolated with the most strongly basic unit L6. The substituents of ligands L10–L12 involve good coordinating sites that appeared, probably through competition with nitrate, to divert the formation of products 10b–12b into that of inseparable mixtures.
Figure 1.
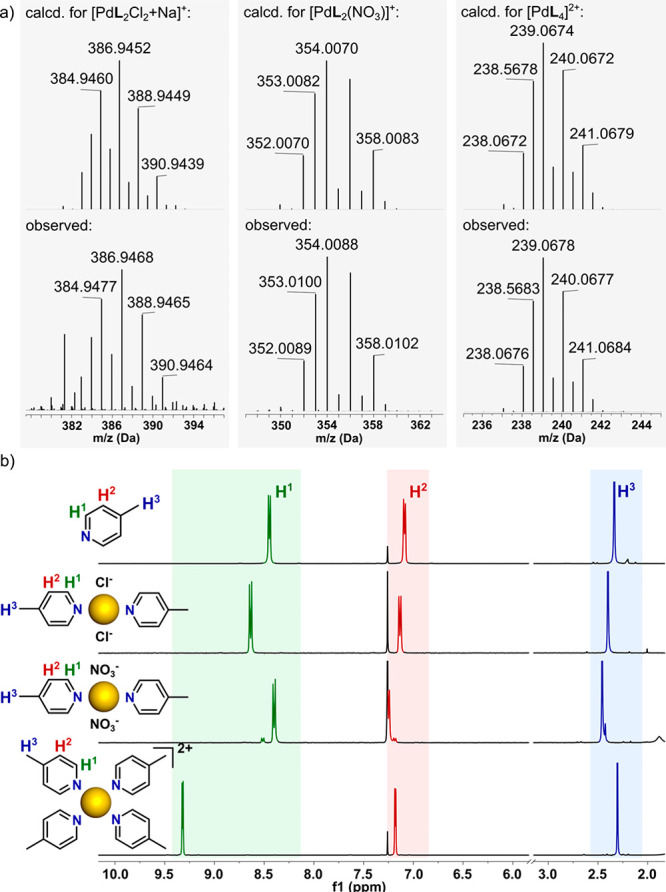
(a) ESI-MS spectra of Pd(II) complexes 2a–2c, showing the calculated isotope model (top) and observed data (bottom). (b) 1H NMR spectra (300 MHz, CDCl3) of compounds 2a–2c.
Mass Spectrometry (MS) Analysis
The successful generation of the desired mononuclear Pd(II) compounds was confirmed via electrospray ionization mass spectrometry (ESI-MS). As shown in Figure 1a, with the example of complexes with L2, isotopically resolved peaks were generally found for [M + Na+]+, [M – NO3–]+, and [M – 2NO3–]2+, where M represents the intact assembly for units of the general formulas [PdL2Cl2] (2a), [PdL2(NO3)2] (2b), and [PdL4](NO3)2 (2c), respectively. All of the peaks were in good agreement with their calculated distribution, allowing the molecularity to be unambiguously established and to distinguish the specific types of complexes. The MS data for all of the units are available in the SI.
NMR Spectroscopy
Apart from signals due to different substituents, the 1H NMR spectra were all very similar, with the 2-fold symmetry of the free ligands retained in all of the complexes and all ligand units in any particular complex being equivalent. In general, a greater sensitivity to the composition and structure of the complexes was seen in the chemical shifts of the H1 protons (on C adjacent to N) rather than in those of the H2 protons, and the presence of small amounts of cis isomer in the products 1b–12b, <10% in all cases, was readily discerned on this basis. Spectra typical of the whole group are shown for the complexes of L2 in Figure 1b, with the results for all other species being included in the SI. A comparison of the various trans-[PdL2(NO3)2] (1b–12b) and trans-[PdL2Cl2] (1a–12a) pairs shows that the H1 chemical shifts are sensitive to the nature of the adjacent donor atom, and this is presumably a contributor to the very large downfield shifts (∼0.5–1 ppm relative to those of the free ligands) for the H1 proton signals of complexes 1c–12c, although the dominant effect here may be that of ion pairing involving C–H1···ONO2 bonding, as proposed to explain similar observations on Pt(II) analogues.11 Again, as observed for Pt(II), the H1 chemical shifts (Table 1) show a close-to-linear dependence on the pKa values of the protonated ligands (Figure 2), indicating that the substituent effects remain operative along with any effects of Pd(II) coordination.
Table 1. 1H NMR Chemical Shifts (δ, ppm) in CDCl3 of H1 Protons for Pd(II) Complexes Based on Ligands L1–L12.
| δ(H1) [ppm] |
|||||
|---|---|---|---|---|---|
| pKab | L | PdL2Cl2 (1a–12a) | PdL2(NO3)2 (1b–12b) | PdL4(NO3)2 (1c–12c) | |
| L1 | 5.23 | 8.62 | 8.84 | 8.61 | 9.63 |
| L2 | 5.98 | 8.45 | 8.63 | 8.40 | 9.32 |
| L3 | 6.47 | 8.41 | 8.59 | 8.33 | 9.21 |
| L4 | 3.49 | 8.79 | 9.01 | 8.78 | 9.80 |
| L5 | 3.57 | 8.79 | 9.05 | 8.82 | 9.87 |
| L6 | 9.61 | 8.21 | 8.25 | 8.71 | |
| L7 | 3.83 | 8.49 | 8.75 | 8.50 | 9.52 |
| L8 | 2.10 | 8.79 | 9.08 | 8.83 | |
| L9 | 2.46 | 8.82 | 9.09 | 8.86 | |
| L10 | 3.07 | 8.77 | 8.97 | 9.79 | |
| L11a | 3.12 | 8.79 | 8.99 | 9.44 | |
| L12 | 2.86 | 8.75 | 8.95 | 9.75 | |
Figure 2.
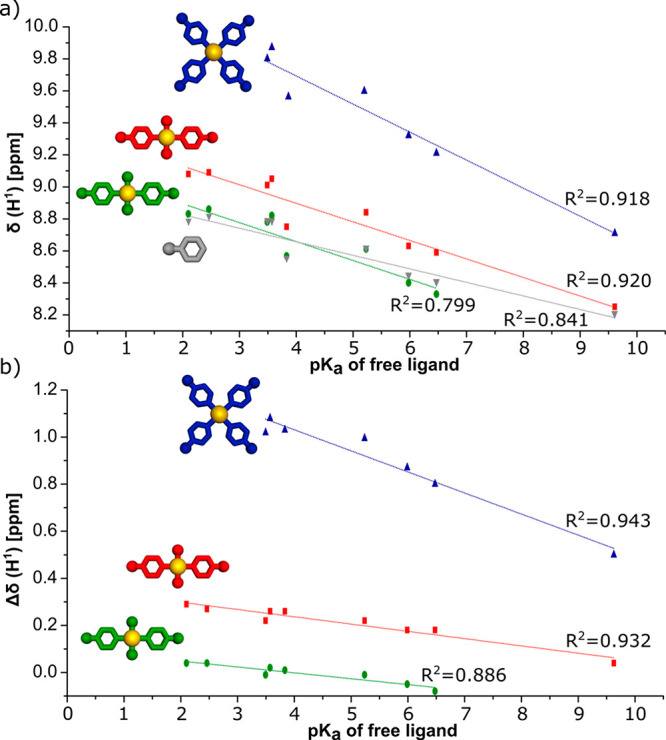
(a) Relationships between the chemical shifts (δ, ppm) of the signal H1 in the 1H NMR spectra (CDCl3, 25 °C) and pKa values of free ligands for the Pd(II) complexes. (b) Relationships between the chemical shift changes (Δδ, ppm) of the signal H1 in the 1H NMR spectra (CDCl3, 25 °C) and pKa values of free ligands for the Pd(II) complexes. Only ligands of known pKa values are included in the graphs.
Solution Complexation Equilibria
In regard to ligand substitution processes, Pd(II) is classified as a labile metal ion and its substitution reaction rates are typically orders of magnitude faster than those of Pt(II).17 This lability was readily observed for complexes 2a–2c by using 1H NMR spectroscopy to follow titrations with acid (methanesulfonic acid, MSA) and base (triethylamine, Et3N). Results typical of what was observed generally are shown in Figure 3. Thus, the equilibrium mixture of cis- and trans-2b reacted with Et3N to give ultimately some 2c, while no intermediates such as [PdL3Y]+ were detected via NMR. Because of the ligand deficiency after the altered L/Pd(II) complex stoichiometry, species 2c were necessarily accompanied by unidentifiable Pd(II) species to which L2 was not coordinated. Neutralization of the reaction mixture with MSA did not return the original complex, retaining the structure of tetrakis(pyridine) units (Figures 3a and S33). In another experiment, the 1H NMR titration of complex 2c with sequential portions of MSA led to changes indicative of the dissociation and protonation of L2 and probably some substitution of nitrate by methanesulfonate, which resulted in the complete disappearance of signals from 2c. Although the bis(ligand) units were identified as one of the decomposition products, their content decreased with increasing acid concentration. Neutralization of the mixture allowed regeneration of the tetrakis ions 2c (Figures 3b and S36).
Figure 3.
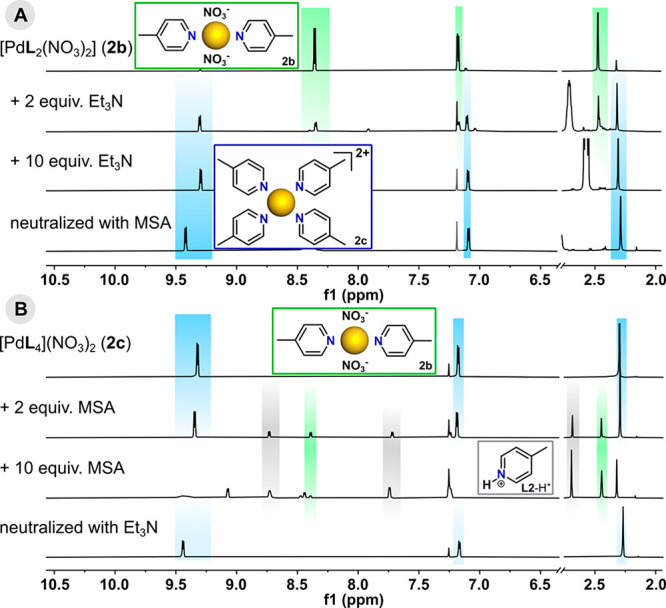
Parts of the 1H NMR spectra (600 MHz, CDCl3) showing the transformations of (a) 2b upon the addition of Et3N and (b) 2c upon the addition of MSA.
Conversely, no significant changes were observed during the 1H NMR titrations of 2b and 2c with MSA and Et3N, respectively (Figures S34 and S35). Moreover, 2a turned out to be completely insensitive in both the basic and acidic environments (Figures S31 and S32).
In contrast to the series of [PtL4]Cl2 units obtained by Marzilli et al.,11 the Pd(II) analogues have not been isolated despite many synthetic attempts. All experiments led to the formation of disubstituted species 1a–12a even if a significant excess of ligand was used. To explain the distinct behavior of Pd(II) and Pt(II) complexes, we investigated the influence of Cl– anions on the stability of the tetrakis(ligand) unit 2c. During 1H NMR titration with triethylamine hydrochloride (Et3N·HCl) as the chloride source, complete disappearance of the signals from 2c was noticed just after the addition of 2 equiv of the organic salt (Figures 4a and S29). Thus, in the presence of chloride, complete decomposition of 2c and finally conversion to 2a was observed along with the release of noncoordinated ligand molecules, as evidenced by the full consistency of NMR chemical shifts. Additionally, 2a was titrated with sequential portions of ligand in an attempt to form the tetrakis species. Its structure remained initially intact, but unusually high downfield signals [Δδ(H1) = ∼1.8 ppm compared to that of free ligand] were found to appear with increasing ligand concentration (Figures 4b and S30). These signals could signify the replacement of chloride anions by L2 and generation of the target [PdL4]Cl2 (2c″), but the bis(pyridine) complex 2a was still the dominant form even with a large excess of ligand (10 equiv). All attempts to isolate tetrakis units were unsuccessful.
Figure 4.
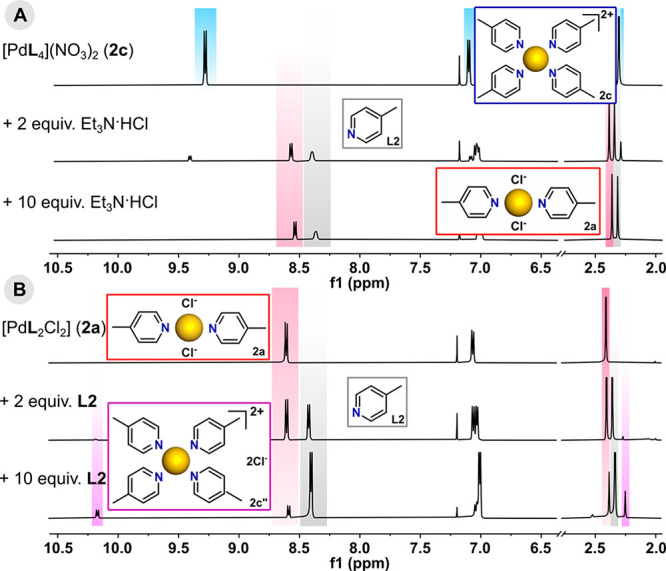
Parts of the 1H NMR spectra (600 MHz, CDCl3) showing the transformations of (a) 2c into 2a with Et3N·HCl and (b) 2a upon the addition of L2.
X-ray Crystallography
Slow diffusion of n-hexane vapor into saturated solutions of the complexes in chloroform afforded a number of crystals of Pd(II) units from three different families. Single-crystal X-ray structure determinations have been performed on 13 complexes: 2b, 2c, 3a, 3b, 4a–4c, 5b, 6a, 6c, and 7a–7c. Separation of the trans isomers of 2b, 3b, 4b, 5b, and 7b was achieved by selective crystallization from the mixture of geometrical isomers. All of the ORTEP representations with atom-labeling schemes are presented in Figures S40–S52. Selected geometric parameters are summarized in Table S18. These structure determinations establish the trans configuration of all of the [PdL2Y2] (Y = Cl– or NO3–) species and the unidentate N coordination of the pyridine ligands in all cases, basic features that, along with the bond lengths and bond angles, are important but in no way exceptional (Tables S14–S17). What a single-crystal X-ray structure determination can add to this information is a definition of the weak interactions that occur within the crystal, and one convenient method to achieve this is to consider the Hirshfeld surfaces of components involving primary bonding interactions, as defined through the use of the program CrystalExplorer.18
The crystal structure of 6a·2CHCl3 contains by far the most strongly basic ligand L6 in the present series, and thus the complex provides a reference point of one extreme of the bis(ligand) species. In the crystals of metal-ion complexes of aza-aromatic ligands, it is common to find that the aza-aromatic units lie in parallel planes, forming arrays described as involving “π–π stacking”,19 although this may be a misleading or at least inadequate term as a description of the actual interactions occurring20 and they may well be only part of a panoply of weak associative effects.21 Indeed, the L6 units in the crystal of 6a do form stacks, but the Hirshfeld surface shows that the interactions involved are purely dispersive and that the only interactions that exceed dispersion are those involving the solvent molecules. These interactions involve both C–H···Cl and Cl···Cl (halogen bonding22) contacts and provide a model for solvation of the complex by chloroform as well as possibly explaining why the pyridine units are tilted with respect to the PdN2Cl2 plane (Figure 5a).
Figure 5.
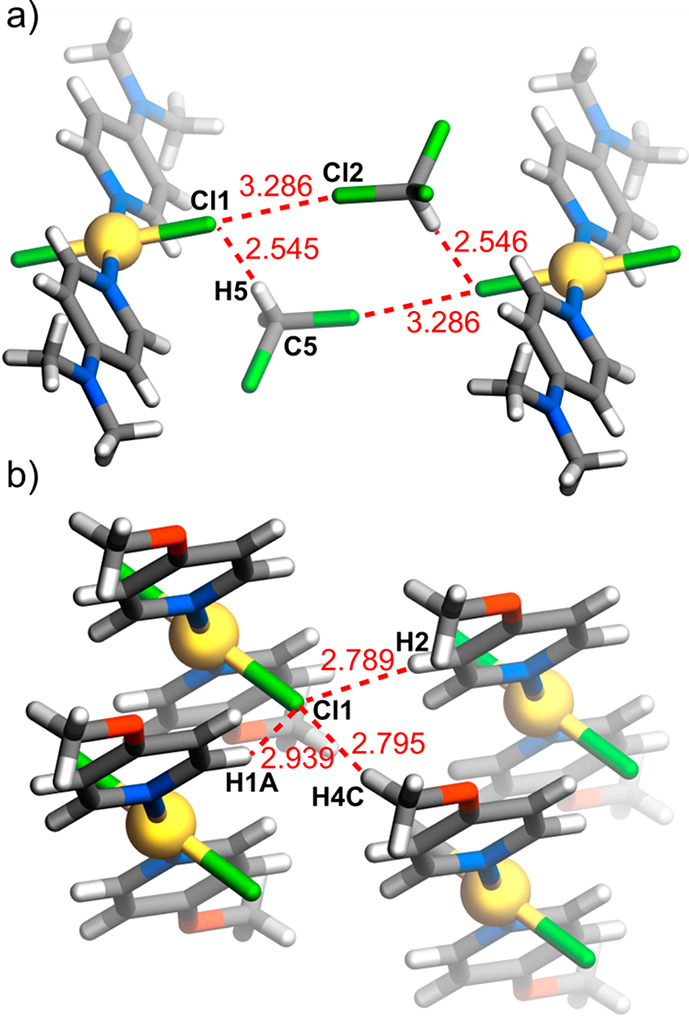
Weak interactions within the crystal structures of (a) 6a·2CHCl3 and (b) 3a.
Passage to a complex of a much less basic ligand L2 and the replacement of chloride by nitrate in 2b lead to a much more complicated array of interactions exceeding dispersion. They derive exclusively, however, from the nitrate ligands and involve both O···H–C and O···C(aromatic) bonding, here perhaps indicating how an association between molecules might occur in solution, with the absence of solvent in the crystal indicating that solvation involves weaker interactions. Note that the L2 units do lie in parallel planes but with a centroid···centroid separation of 4.78 Å and no overlap in the projection perpendicular to the planes, so that they do not constitute a “stacked” array (Figure S53).
A more direct comparison of the consequences of replacing chloride by nitrate is possible through examination of the structures of 3a and 3b. Ligand L3 is again much less basic than L6, although slightly more basic than L2, and the methoxyl O is now an important point of interaction in both structures. In both, it is possible to find stacked arrays of the pyridine units, but again any interaction does not exceed dispersion in either case. In complex 3a, where the molecular unit has 2-fold symmetry, the chloride ligands are involved in interactions exceeding dispersion with both aromatic and aliphatic H, and these are complemented by O(methoxyl)···H–C(methoxyl), O(methoxyl)···H–C(pyridine) and C(methoxyl)–H···Cl interactions (Figures 5b and S54). In the crystal of complex 3b, the two pyridine ligands of each molecule are not equivalent and only one methoxyl group is involved in interactions exceeding dispersion. The polyatomic nature of the nitrate ligands means that they have multiple sites for interaction, but just like the chloride ligands of 3a, they serve to link molecules through interactions with both aromatic and aliphatic HC (Figure S55).
Pyridine (L1) itself is, of course, the parent ligand of all of the derivatives considered here, so that the nature of its complexes provides another reference point for the present series. Its consideration at this stage is appropriate in that it is less basic than L2, L3, or L6 but more so than any of the other ligands presently employed. Complex 1a has particular significance in that it has been structurally characterized in three different polymorphs, space groups C2/c,23P1̅,24 and P21/n.25 This polymorphism can be understood in that the Hirshfeld surfaces show dispersion interactions to be dominant, completely for the P1̅ polymorph and in association with limited reciprocal C–H···Cl interactions for the other two. Only in the P21/n polymorph can it be said that there is an approach to a stacked array of pyridine units, but the centroid···centroid separation is 3.9159(2) Å and no indication of ring atom interactions beyond dispersion are apparent. Given the nondirectional nature of dispersion interactions, it is understandable that subtle differences in the conditions of crystallization might well give rise to the occupation of different local energy minima.
Another direct comparison of the consequences of replacing chloride by nitrate is provided in the structures of 7a and 7b. In complex 7a, each coordinated chloride has two interactions with pyridine-CH units and one barely discernible interaction with a pyridine-4-Cl unit (Figure 6a) While in complex 7b each bound nitrate is involved in O···H–C(pyridine) interactions analogous to the Cl···H–C interactions in 7a, one is also bound (through separate O atoms) to aromatic C and, as is clearly evident, to pyridine-4-Cl, and the other has just an additional O···Cl(pyridine) interaction (Figure 6b; in complex 7a, the two chloride ligands are equivalent). In both complexes, these local interactions are associated with limited stacking of the pyridine units, but these involve centroid···centroid separations near 4.8 Å, with no evidence of any interaction outside dispersion. As a different polymorph (but also P1̅), complex 7a has been structurally characterized previously as part of an investigation of halogen bonding within crystals of complexes of the [M(X-py)2(halogen)2] type.26 The Hirshfeld surface for this polymorph is very similar to that of complex 7a, although the Cl···Cl interactions are somewhat more prominent. The centroid···centroid separation of the closest parallel pyridine ring pairs is also shorter at 3.9039(8) Å, although still with no indication of interactions exceeding dispersion.
Figure 6.
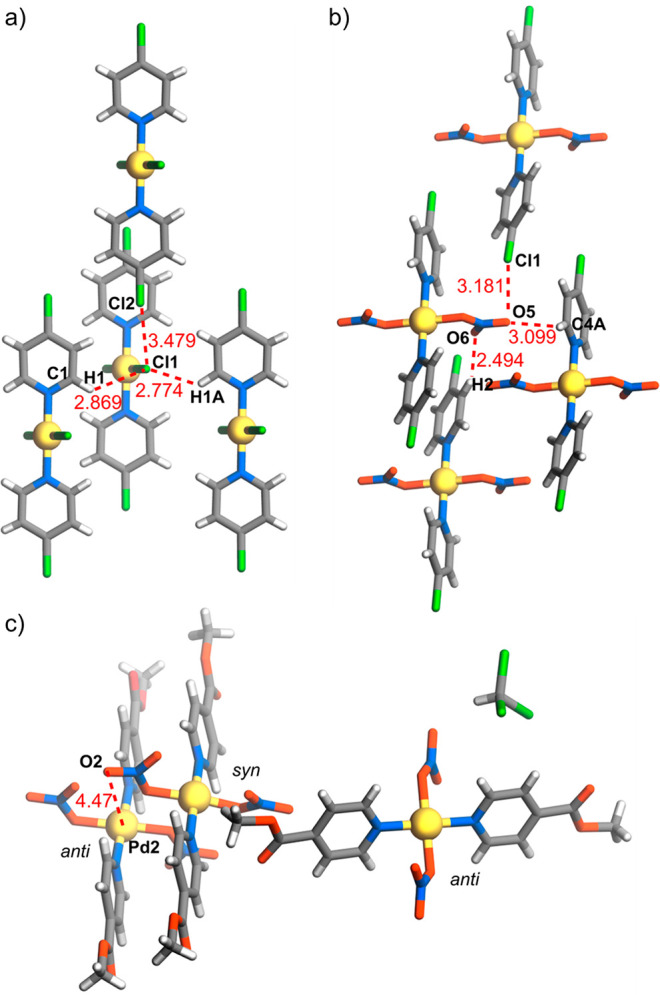
Weak interactions within the crystal structures of (a) 7a and (b) 7b. (c) Syn and anti orientations of nitrate ligands in the structure of 4b.
Ligand L4 is appreciably less basic than pyridine, implying a significant electron-withdrawing effect of the methoxycarbonyl group on the pyridine ring, but in the structure of both complexes 4a and 4b, there is no indication on the Hirshfeld surfaces of a change in face-to-face pyridine ring interactions from that of dispersion. Instead, as observed in the structures already described, it is the coordinated anions and pyridine substituent that are involved in all interactions that exceed dispersion. The chloride ligands of the centrosymmetric complex 4a are involved in interactions with both pyridine and ester methyl CH atoms of adjacent molecules of the ester substituent interacting with pyridine C, again a reciprocated case (Figure S56). Complex 4b was in fact crystallized as a hemisolvate, 4b2·CHCl3, and the presence of three inequivalent Pd sites as well as the presence of the solvent makes a description of the weak interactions in the crystal rather complicated. What is particularly interesting here, though, is the fact that while two of the three inequivalent Pd centers can be considered to have a square-planar coordination sphere, the third (Pd1) is square-pyramidal because of axial binding to nitrate O. Axial binding of a reaction substrate can, of course, be one of the initial steps in a catalytic mechanism, and while five coordination of Pd(II) is a well-understood occurrence,27 it does not appear to be particularly favored in the present systems. The Pd1 environment in complex 4b is unique in the present series in that, perhaps in order to accommodate the axial interaction, the two nitrate ligands have a syn orientation relative to the PdN2O2 plane, while in all other cases, it is anti (Figure 6c).
Ligand L5 has Brønsted basicity very similar to that of L4, but the Hirshfeld surface for the centrosymmetric complex 5b indicates that the acetyl substituent produces more significant charge relocalization in the pyridine ring than does the methyl ester group. Thus, nitrate O is involved in interactions not only with both aromatic and aliphatic H, as in complexes 2b, 3b, 4b, and 7b, but also with the carbonyl C of the substituent and the pyridine C adjacent to it (Figure S57). Unlike complex 4b, complex 5b shows no evidence of an axial interaction with Pd exceeding dispersion, but as for Pd2 and Pd3 in complex 4b, two O atoms are located, here, 3.776(6) Å above and below the PdN2O2 plane in a line with the Pd, indicating again that an axial approach could be a minimum energy pathway to binding an extra ligand. (In complex 4b, the O atoms are 3.141(3) Å from Pd2 and 3.252(3) Å from Pd3.)
The application of Hirshfeld surface analysis to complexes 2c, 4c, 6c, and 7c is limited by the disorder present in the structures of 4c and 7c, so that a detailed analysis has been applied to the structures of complexes 2c and 6c only. All four complexes do, however, have a structure in which all four pyridine units lie close to perpendicular to the PdN4 plane, a feature well-known in various tetrakis(pyridine) complexes and commonly ascribed to its enabling of the minimization of repulsion between the ligands,28 with such a repulsion also being considered the reason for the difficulty in obtaining hexakis(pyridine) complexes of octahedral metal ions.29 For [PtL4]2+ cations, where the same conformation is observed, an alternative explanation based on the observation of specific interactions of cations with counteranions has, however, been offered.11 In the structure of complex 6c, it is possible to discern a degree of interlocking of the cations with a resemblance to what is found in instances of the “terpyridine embrace”,19b but as is seen in the bis(ligand) species 6a, the Hirshfeld surface provides no evidence for interactions exceeding dispersion between L6 units. What is evident on the Hirshfeld surface is the versatility of nitrate in forming O···H–C bonds involving both aromatic and aliphatic H atoms. One result of this is that nitrate anions do form “caps” to each cation, as seen with the Pt(II) analogues,11 by the interaction of O1 with three aromatic CH atoms (H1A, H6, and H6A) of adjacent ligands (Figure 7). The additional interactions of O2 with methyl CH (H9B and H9AB) and aromatic CH (H2) atoms serve to link cations into sheets parallel to (010) from which L6 units project so that the sheets are linked through dispersion interactions. In the structure of complex 2c, there is disorder of the anions, which complicates the interpretation of their interactions, but the Hirshfeld surface of the cations shows that the chains of cations running along [001] are, in fact, linked by C(aromatic)–H···C(aromatic) interactions, providing an example of where changing the substituent on pyridine results in the generation of pyridine···pyridine interactions exceeding dispersion, a feature not apparent in any of the other comparisons of the present work. Regardless of their disorder, the nitrate anions do appear to occupy capping regions of the cations, as seen in complex 6c, and this is true also for the nondisordered anions associated with the disordered cations in complexes 4c and 7c.
Figure 7.
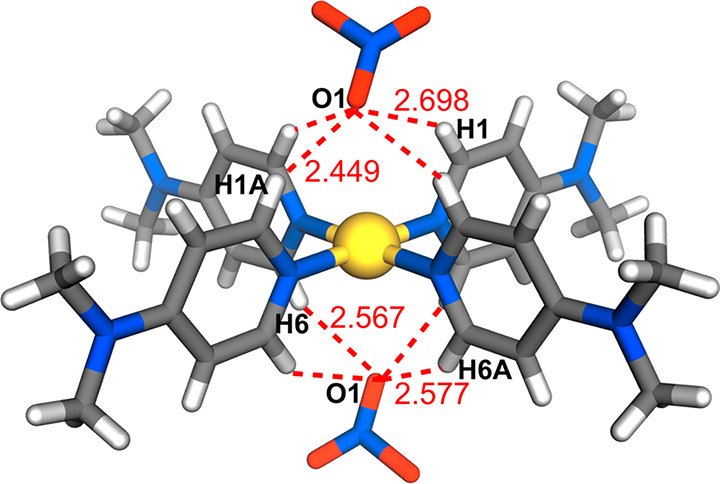
Nonbonding C–H···O interactions between [PdL4]2+ and nitrate counterions in the structure of complex 6c.
What is observed in the solid state through crystal structure determinations does not necessarily apply to solutions, but the rarity of solvent incorporation in the structures presently described indicates that solvation interactions can be in competition with a variety of other forces determined by the particular nature of the solute. What has not been overtly considered in the discussion above of the tetrakis(pyridine) complexes is the fact that they are considered to be ionic species and thus that there should be an electrostatic factor to be allowed for in the cation···anion interactions. The calculation of Hirshfeld surfaces with neutral-atom wave functions may therefore be misleading in regard to the intensity of interactions but not their directionality, so that the cation capping by nitrate seen in the structures of complexes 2c, 4c, 6c, and 7c can still be seen as a consequence of O···H–C interactions. As argued in the case of Pt(II) analogues,11 the preservation of such interactions in solution could explain why strong downfield shifts are also observed in the 1H NMR spectra of complexes 2c, 4c, 6c, and 7c, although it is also important to note that the environment of the pyridine protons in the bis(pyridine) complexes is quite varied and quite different from that in the tetrakis species. In regard to catalysis by [PdLnYm], the interactions of different substituents and counteranions indicate possible structural features of a substrate that might enhance its binding to the catalyst, but this, of course, is one step in what must be a more complicated process.
Catalytic Studies
Because of the structural differences between Pd(II) complexes with pyridine ligands, catalytic studies were undertaken in order to investigate their activity in Pd-catalyzed cross-coupling reactions and explore their diversity in functionality as well. Thus, their catalytic properties were tested and compared in both the Suzuki–Miyaura and Heck reactions.
Suzuki–Miyaura Coupling
Complex 2c was selected as a model catalyst precursor for which the reaction conditions were optimized in the coupling between 4′-bromoacetophenone and phenylboronic acid (Table S22). Among the tested bases (K2CO3, K3PO4, NaOH, and Et3N) and solvents (chloroform, toluene, 1,4-dioxane, and N,N-dimethylformamide), the combination of K3PO4 and toluene allowed formation of the expected 4-acetylbiphenyl in the highest gas chromatography (GC) yield. The catalytic reactions were performed at 80 °C and, importantly, without the need to exclude air or water. Taking economic and environmental considerations into account, the optimal catalyst concentration was 0.1 mol %, which resulted in an almost quantitative conversion just after 2 h.
Subsequently, the catalytic activity of the full range of structurally diversified Pd(II) complexes with pyridine ligands was tested under the same optimized reaction conditions. The majority of the catalyst precursors provided the cross-coupling product in excellent yields of >90% (Table 2). Only minor differences were observed between bis and tetrakis complexes of a given ligand, so it appears that the nature of the complex and the different counterions does not directly influence the effectiveness of the catalyzed reaction. Nevertheless, some differences could be noted depending on the ring substituent. The lowest GC yields were observed for the complexes based on L4 (64–78%). Better GC yields (>70%) were achieved for the complexes based on L5, L7–L9, and L11, while those of L1–L3, L6, and L10 showed the highest activity in Suzuki–Miyaura coupling. Although no simple correlation was observed between GC yields and pKa values of the ligands (Figure S93), Pd(II) complexes with more basic pyridine ligands generally showed slightly greater catalytic effectiveness.
Table 2. GC Yields [%]a in Suzuki–Miyaura and Heck Cross-Coupling Reactions Catalyzed by Pd(II) Complexes Based on Ligands L1–L12.
| GC yield [%] in Suzuki–Miyaura couplingb |
GC yield
[%] in Heck couplingc |
||||||
|---|---|---|---|---|---|---|---|
| pKa of L | PdL2Cl2 (1a–12a) | PdL2(NO3)2 (1b–12b) | PdL4(NO3)2 (1c–12c) | PdL2Cl2 (1a–12a) | PdL2(NO3)2 (1b–12b) | PdL4(NO3)2 (1c–12c) | |
| L1 | 5.23 | 97 | 93 | 95 | 85 | 88 | 90 |
| L2 | 5.98 | 93 | 92 | 98 | 90 | 91 | 94 |
| L3 | 6.47 | 93 | 91 | 91 | 86 | 82 | 76 |
| L4 | 3.49 | 78 | 72 | 64 | 89 | 92 | 79 |
| L5 | 3.57 | 86 | 87 | 88 | 80 | 92 | 75 |
| L6 | 9.61 | 93 | 90 | 86 | 83 | ||
| L7 | 3.83 | 82 | 74 | 75 | 90 | 92 | 80 |
| L8 | 2.10 | 88 | 66 | 91 | 93 | ||
| L9 | 2.46 | 87 | 70 | 81 | 91 | ||
| L10 | 3.07 | 98 | 90 | 93 | 88 | ||
| L11 | 3.12 | 86 | 79 | 88 | 90 | ||
| L12 | 2.86 | 83 | 92 | 92 | 77 | ||
Reaction yields were determined by GC–MS measurement of 4′-bromoacetophenone or iodobenzene decay as the average of three results.
Reaction conditions: 4′-bromoacetophenone (0.2 mmol, 1 equiv), phenylboronic acid (0.24 mmol, 1.2 equiv), K3PO4 (0.4 mmol, 2 equiv), and Pd(II) complex (0.1 mol %) were stirred in toluene (2 mL) at 80 °C for 2 h.
Reaction conditions: iodobenzene (0.2 mmol, 1 equiv), styrene (0.24 mmol, 1.2 equiv), Et3N (1.0 mmol, 5 equiv), and Pd(II) complex (0.1 mol %) were stirred in DMSO (2 mL) at 120 °C for 2 h.
Complex 2c, as one of the most effective systems, was selected to explore the capabilities in the Suzuki–Miyaura cross-coupling in terms of functional-group tolerance. Under the optimized reaction conditions, a set of functionalized aryl bromides and arylboronic acids were reacted together. The 2c unit enabled the synthesis of scope of structurally distinct biphenyl derivatives 3aa–3cc in high-to-excellent yields (74–100%; Scheme 2). The high efficiency was observed regardless of the presence of electron-donating (−Me and −OMe) or electron-withdrawing (−CF3 and −COMe) substituents in the substrate molecules, highlighting the catalyst precursor versatility.
Scheme 2. Scope of the Suzuki–Miyaura Cross-Coupling Reaction between Aryl Bromides and Arylboronic Acids.
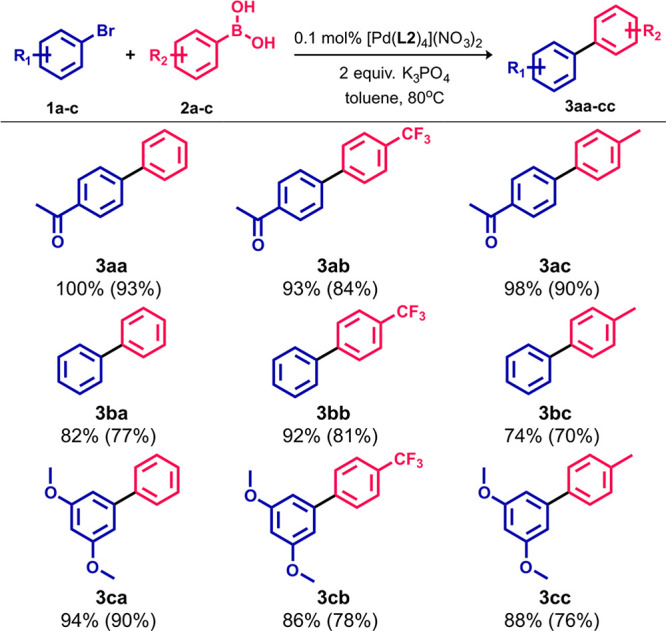
The GC yields were determined by GC–MS measurement of aryl bromide decay. The yields in parentheses are for the isolated compounds.
Heck Coupling
For an initial assessment of the efficacy of the complexes as catalyst precursors for the Heck reaction, the cross-coupling of iodobenzene with styrene catalyzed by 2c was chosen as a model reaction to develop the reaction conditions (Table S24). Under the conditions optimized for the Suzuki–Miyaura reaction, only traces of the Heck coupling product were observed. For this reason, different variations in terms of solvents and bases were tested using a 1 mol % Pd(II) complex. The reaction did not proceed successfully in the presence of inorganic bases (K3PO4 and K2CO3) or nonpolar solvent (toluene). The pair of Et3N and DMSO represented the best combination to reach high yields because almost quantitative conversion was achieved at 120 °C just after 2 h. Additional experiments showed that the catalyst loading could be reduced to 0.1 mol %. This concentration was sufficient to guarantee good conversion at the same time, whereas using 0.01 mol % significantly extended the reaction time. With these results in hand, subsequent catalytic reactions were performed in DMSO at 120 °C using Et3N as a base and 0.1 mol % Pd(II) complex. Note that the Pd(II) complexes essentially retain their structure under the reaction conditions, as indicated by the 1H NMR spectra recorded after heating in DMSO at 120 °C (Figures S37–S39).
A comparison of the catalytic activities for a number of the other Pd(II) complexes was performed for the Heck reaction as well. As with the Suzuki–Miyaura cross-coupling, potential catalyst precursors were examined to evaluate the substituent effect on the efficiency in catalyzed reactions. Under the same conditions, very high GC yields (>90%) were obtained in most of the reactions, and yields of <80% were observed in only a few cases (Table 2). Overall, the tetrakis(pyridine) complexes, especially with ligands L3–L5 and L12, provided lower GC yields (75–79%) in comparison to neutral bis(ligand) species. Any ring substituent effect was negligible, and no clear relationship between the ligand basicity and catalytic activity of the Pd(II) complexes was apparent (Figure S95). In all cases, the selectivity in the (E)-stilbene formation was very high, ranging from 89% to 99%, and was completely independent of the catalyst precursor structure.
To investigate the scope of the Heck cross-coupling reaction, the catalytic properties of complex 2c were further studied by using a set of functionalized substrates under the conditions described above. As shown in Scheme 3, 2c showed good catalytic activity and selectivity in the reactions between aryl iodides and olefins, giving GC yields in the range of 61–100%. It is noteworthy that an excellent conversion was accomplished for acrylate derivatives (97–100%). The reaction system exhibited also great chemoselectivity toward iodoarenes because no cross-coupling involving bromoarene moieties, as either olefin or haloarene coupling partners, was observed.
Scheme 3. Scope of the Heck Cross-Coupling Reaction between Aryl Iodides and Olefins.
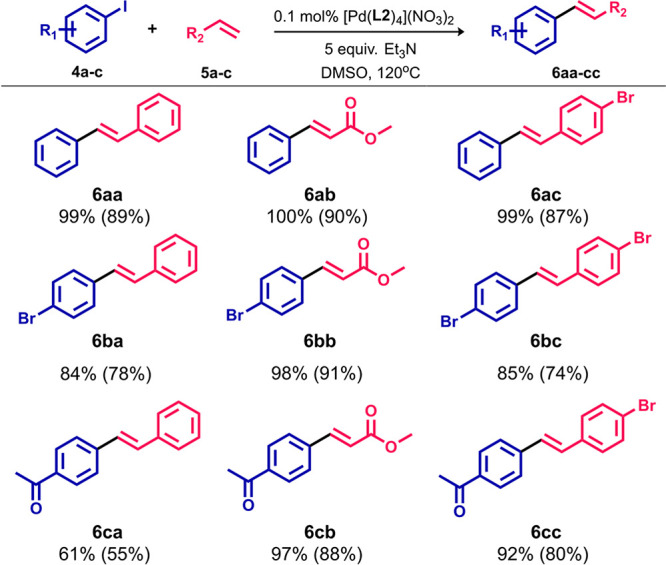
The GC yields were determined by GC–MS measurement of aryl iodide decay. The yields in parentheses are for the isolated compounds.
Complex 2c as a representative of the multiple family of Pd(II) complexes with pyridyl ligands has been extensively investigated with respect to catalytic properties that demonstrated high catalytic activity in the Suzuki–Miyaura and Heck cross-coupling reactions. On the basis of the experiments carried out, it can be concluded that all of the units presented herein constitute a group of versatile precatalysts that can be successfully applied in Pd-catalyzed reactions.
Because of the multitude of literature reports on the mechanism of both Suzuki–Miyaura and Heck cross-coupling, profound studies have not been conducted in this area. We assume that the bis- and tetrakis(pyridine) complexes considered in this paper play the precatalyst role. According to the generally accepted mechanism, the reduction of Pd(II) to Pd(0) occurs at the beginning of the catalytic cycle, leading to the generation of active species. The process then proceeds in a typical manner for Pd-catalyzed transformations, through the sequence of three consecutive stages involving oxidative addition, transmetalation or carbometalation, and reductive elimination, as described in numerous works.30 The precatalyst was degraded during the cycle that was observed as precipitation of metallic Pd; therefore, it could not be regenerated and then reused.
Conclusions
In summary, a series of Pd(II) complexes based on a wide range of functionalized pyridine derivatives have been successfully generated and analyzed in solution via NMR spectroscopy and MS as well as in the solid state via X-ray diffraction. This work has been based on two sets of complexes of the general formulas [PdL4](NO3)2 and [PdL2Y2], where Y = Cl– or NO3–. Their properties have been examined in light of the ligand basicity as a factor of influence, although the results obtained have shown that this is just one of several factors that may be important. The complexes have been found to be of practical utility as simple and efficient catalyst precursors for both the Suzuki–Miyaura and Heck cross-coupling reactions for a scope of substrates under relatively mild conditions.
Experimental Section
General Procedures
All reagents were purchased from commercial suppliers (mainly Merck or Fluorochem) and used without further purification. High-purity solvents were purchased from VWR. NMR solvents were purchased from Deutero GmbH (Germany) and used as received. NMR spectra were acquired on Bruker Fourier 300 MHz, Bruker Avance IIIHD 400 MHz, and Bruker Avance IIIHD 600 MHz spectrometers at 25 °C and referenced to a tetramethylsilane signal or solvent residual peaks. All NMR data were processed with Mestrelab Research MNova software. ESI-MS spectra were recorded on Bruker HD Impact and ABSciex QTOF 5600 spectrometers in positive-ion mode. Theoretical MS spectra were predicted using Mestrelab Research MNova software. GC–MS analyses were performed on a Bruker 450-GC spectrometer with a 30 m Varian DB-5 0.25 mm capillary column and a Scion SQ-MS detector.
X-ray Crystallography
X-ray measurements were performed using an Oxford Diffraction SuperNova diffractometer with monochromatic Cu Kα radiation for 2c, 3a, 5b, and 6a. The diffraction data were collected on a Rigaku XtaLAB Synergy diffractometer equipped with a rotating anode as a Cu Kα radiation source for 4a. The remaining compounds were subjected to X-ray measurements on an Oxford Diffraction Xcalibur diffractometer with Mo Kα radiation. Data collection and data reduction for all Pd(II) complexes were carried out using the CrysAlisPRO software.31 Using OLEX2, the intrinsic phasing method (ShelXT) was used for crystal structure solution.32 The exception is the 4a structure, which was solved with direct methods (ShelXS).33 The refinement process was performed with anisotropic displacement parameters for non-H atoms with the full-matrix least-squares method based on F2 (ShelXL).32b In all structures, except for 7a and 7b, the H atoms were placed in calculated positions and refined using a riding model. The high quality of the obtained single crystals of complexes 7a and 7b made it possible to carry out high-resolution X-ray measurements. Therefore, the H atoms have been derived from the difference Fourier map and refined without constraints. Crystallographic data, details on the refinement, twin structures, and disordered fragments in the crystal structures are included in the SI.
Synthesis of Ligands
Ligands L1–L9 were purchased from commercial suppliers and used as received. Ligands L10–L12 were prepared according to the previously described procedures.7e,34
Synthesis of [PdL2Cl2] Complexes (1a–12a)
One of the ligands L1–L12 (∼0.2 mmol, 2 equiv) was added to an acetonitrile (MeCN) solution of PdCl2 (∼0.1 mmol, 1 equiv in 5 mL of MeCN). Then the resulting mixture was heated under reflux for 12 h. The precipitate that formed was centrifuged off, washed with MeCN (10 mL) and diethyl ether (Et2O; 2 × 10 mL), and dried under vacuum. Specific details on the synthetic procedures and analytical data (quantities used, yields, NMR and MS data, etc.) can be found in the SI.
Synthesis of [PdL2(NO3)2] Complexes (1b–12b)
One of the ligands L1–L12 (0.2 mmol, 2 equiv) was added to an MeCN solution of Pd(NO3)2·2H2O (0.1 mmol, 1 equiv in 5 mL of MeCN). Then, the resulting mixture was heated under reflux for 12 h. The solvent was then evaporated under reduced pressure. The crude product was redissolved in MeCN (1 mL) and reprecipitated by the addition of Et2O (10 mL). The precipitate was centrifuged off, washed with Et2O (2 × 10 mL), and dried under a vacuum. Specific details on the synthetic procedures and analytical data (quantities used, yields, NMR and MS data, etc.) can be found in the SI.
Synthesis of [PdL4](NO3)2 Complexes (1c–12c)
To a suspension of PdCl2 or Pd(DMSO)2Cl2 (∼0.1 mmol, 1 equiv) in ethanol (5 mL) was added a solution of one of the ligands L1–L12 (∼1.0 mmol, 10 equiv) in dichloromethane (DCM; 5 mL), and the resulting mixture was stirred at room temperature for 1 h. Then, AgNO3 (∼0.2 mmol, 2 equiv) in 0.5 mL of H2O was added, and the resulting suspension was stirred for an additional 12 h excluding light. The reaction mixture was filtered to remove AgCl, and then the filtrate was evaporated under reduced pressure. The crude product was redissolved in DCM (1 mL) and reprecipitated by the addition of n-hexane (10 mL). The precipitate was centrifuged off, washed with n-hexane (2 × 10 mL), and dried under a vacuum. Specific details on the synthetic procedures and analytical data (quantities used, yields, NMR and MS data, etc.) can be found in the SI.
Suzuki–Miyaura Coupling
A reaction vessel equipped with a stirring bar was charged with aryl bromide (1.0 mmol, 1.0 equiv) and arylboronic acid (1.2 mmol, 1.2 equiv) dissolved in toluene (10 mL). Then, the Pd(II) precatalyst (0.001 mmol, 0.001 equiv) as a solution in chloroform (0.05 mL) and solid K3PO4 (2.0 mmol, 2.0 equiv) was added. The vial was sealed, and the reaction mixture was heated for 2 h at 80 °C. Then, the resulting solution was cooled to room temperature, diluted with DCM (50 mL), and washed with distilled water (40 mL). The collected aqueous phase was extracted with DCM (2 × 50 mL). The organic layers were gathered, dried over Na2SO4, and filtered, and the solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel to obtain the desired products 3aa–3cc. The full characterization of the coupling products is available in the SI.
Heck Reaction
A reaction vessel equipped with a stirring bar was charged with aryl iodide (1.0 mmol, 1.0 equiv) and olefin (1.2 mmol, 1.2 equiv) dissolved in DMSO (10 mL). Then, the Pd(II) precatalyst (0.001 mmol, 0.001 equiv) as a solution in DMSO (0.05 mL) and Et3N (5.0 mmol, 5.0 equiv) was added. The vial was sealed, and the reaction mixture was heated for 2 h at 120 °C. Then, the resulting solution was cooled to room temperature, diluted with ethyl acetate (50 mL), and washed with icy distilled water (40 mL). The collected aqueous phase was extracted with ethyl acetate (2 × 50 mL). The organic layers were gathered, dried over Na2SO4, and filtered, and the solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel to obtain the desired products 6aa–6cc. The full characterization of the coupling products is available in the SI.
Acknowledgments
We thank the National Science Center (Grant SONATA BIS 2018/30/E/ST5/00032 to A.R.S.) for financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.2c01996.
Additional experimental details, materials and methods, NMR and ESI-MS spectra for all compounds, NMR titration experiments, crystal data and structure refinement for Pd(II) complexes, reaction development for catalytic tests, and characterization of cross-coupling products (PDF)
Author Contributions
All authors have approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- a Pal S.Pyridine: A Useful Ligand in Transition Metal Complexes; IntechOpen, 2018. [Google Scholar]; b Joule J. A.; Mills K.; Smith G. F.. Heterocyclic Chemistry, 3rd ed.; CRC Press, 1995. [Google Scholar]
- Jensen W. B. The Lewis acid-base definitions: a status report. Chem. Rev. 1978, 78 (1), 1–22. 10.1021/cr60311a002. [DOI] [Google Scholar]
- a Patroniak V.; Kubicki M.; Stefankiewicz A. R.; Grochowska A. M. Preparation of new heterotopic ligands. Tetrahedron 2005, 61 (23), 5475–5480. 10.1016/j.tet.2005.03.131. [DOI] [Google Scholar]; b Stefankiewicz A. R.; Wałȩsa-Chorab M.; Harrowfield J.; Kubicki M.; Hnatejko Z.; Korabik M.; Patroniak V. Self-assembly of transition metal ion complexes of a hybrid pyrazine–terpyridine ligand. Dalton Trans. 2013, 42 (5), 1743–1751. 10.1039/C2DT31982A. [DOI] [PubMed] [Google Scholar]; c Gorczyński A.; Harrowfield J. M.; Patroniak V.; Stefankiewicz A. R. Quaterpyridines as Scaffolds for Functional Metallosupramolecular Materials. Chem. Rev. 2016, 116 (23), 14620–14674. 10.1021/acs.chemrev.6b00377. [DOI] [PubMed] [Google Scholar]; d Brzechwa-Chodzyńska A.; Zieliński M.; Gilski M.; Harrowfield J. M.; Stefankiewicz A. R. Dynamer and Metallodynamer Interconversion: An Alternative View to Metal Ion Complexation. Inorg. Chem. 2020, 59 (12), 8552–8561. 10.1021/acs.inorgchem.0c01019. [DOI] [PubMed] [Google Scholar]; e Čonková M.; Drożdż W.; Miłosz Z.; Cecot P.; Harrowfield J.; Lewandowski M.; Stefankiewicz A. R. Influencing prototropy by metal ion coordination: supramolecular transformation of a dynamer into a Zn-based toroidal species. J. Mater. Chem. C 2021, 9 (9), 3065–3069. 10.1039/D0TC05598K. [DOI] [Google Scholar]
- a Krogul A.; Cedrowski J.; Wiktorska K.; Ozimiński W. P.; Skupińska J.; Litwinienko G. Crystal structure, electronic properties and cytotoxic activity of palladium chloride complexes with monosubstituted pyridines. Dalton Trans. 2012, 41 (2), 658–666. 10.1039/C1DT11412C. [DOI] [PubMed] [Google Scholar]; b Bugarčić Ž. D.; Petrović B.; Zangrando E. Kinetics and mechanism of the complex formation of [Pd(NNN)Cl]+ with pyridines in methanol: synthesis and crystal structure of [Pd(terpy)(py)](ClO4)2. Inorg. Chim. Acta 2004, 357 (9), 2650–2656. 10.1016/j.ica.2004.02.015. [DOI] [Google Scholar]
- a Schlosser M.; Mongin F. Pyridine elaboration through organometallic intermediates: regiochemical control and completeness. Chem. Soc. Rev. 2007, 36 (7), 1161–1172. 10.1039/b706241a. [DOI] [PubMed] [Google Scholar]; b Dell’arciprete M. L.; Cobos C. J.; Furlong J. P.; Mártire D. O.; Gonzalez M. C. Reactions of sulphate radicals with substituted pyridines: a structure-reactivity correlation analysis. ChemPhysChem 2007, 8 (17), 2498–505. 10.1002/cphc.200700456. [DOI] [PubMed] [Google Scholar]
- a Debata N. B.; Tripathy D.; Chand D. K. Self-assembled coordination complexes from various palladium(II) components and bidentate or polydentate ligands. Coord. Chem. Rev. 2012, 256 (17), 1831–1945. 10.1016/j.ccr.2012.04.001. [DOI] [Google Scholar]; b Peloquin D. M.; Schmedake T. A. Recent advances in hexacoordinate silicon with pyridine-containing ligands: Chemistry and emerging applications. Coord. Chem. Rev. 2016, 323, 107–119. 10.1016/j.ccr.2016.02.005. [DOI] [Google Scholar]; c Pazderski L.; Szłyk E.; Sitkowski J.; Kamieński B.; Kozerski L.; Toušek J.; Marek R. Experimental and quantum-chemical studies of 15N NMR coordination shifts in palladium and platinum chloride complexes with pyridine, 2,2′-bipyridine and 1,10-phenanthroline. Magn. Reson. Chem. 2006, 44 (2), 163–170. 10.1002/mrc.1740. [DOI] [PubMed] [Google Scholar]; d Happ B.; Winter A.; Hager M. D.; Schubert U. S. Photogenerated avenues in macromolecules containing Re(i), Ru(ii), Os(ii), and Ir(iii) metal complexes of pyridine-based ligands. Chem. Soc. Rev. 2012, 41 (6), 2222–2255. 10.1039/C1CS15154A. [DOI] [PubMed] [Google Scholar]; e Kristiansson O. Bis(4-aminopyridine)silver(I) nitrate and tris(2,6-diaminopyridine)silver(I) nitrate. Acta Crystallogr. C 2000, 56 (2), 165–167. 10.1107/S0108270199014432. [DOI] [PubMed] [Google Scholar]; f Webb M. I.; Wu B.; Jang T.; Chard R. A.; Wong E. W. Y.; Wong M. Q.; Yapp D. T. T.; Walsby C. J. Increasing the Bioavailability of RuIII Anticancer Complexes through Hydrophobic Albumin Interactions. Chem. Eur. J. 2013, 19 (50), 17031–17042. 10.1002/chem.201302671. [DOI] [PubMed] [Google Scholar]
- a Pardey A. J.; Longo C. Catalysis by rhodium complexes bearing pyridine ligands: Mechanistic aspects. Coord. Chem. Rev. 2010, 254 (3), 254–272. 10.1016/j.ccr.2009.08.004. [DOI] [Google Scholar]; b Togni A.; Venanzi L. M. Nitrogen Donors in Organometallic Chemistry and Homogeneous Catalysis. Angew. Chem., Int. Ed. 1994, 33 (5), 497–526. 10.1002/anie.199404971. [DOI] [Google Scholar]; c Walczak A.; Stachowiak H.; Kurpik G.; Kaźmierczak J.; Hreczycho G.; Stefankiewicz A. R. High catalytic activity and selectivity in hydrosilylation of new Pt(II) metallosupramolecular complexes based on ambidentate ligands. J. Catal. 2019, 373, 139–146. 10.1016/j.jcat.2019.03.041. [DOI] [Google Scholar]; d Abdine R. A. A.; Kurpik G.; Walczak A.; Aeash S. A. A.; Stefankiewicz A. R.; Monnier F.; Taillefer M. Mild temperature amination of aryl iodides and aryl bromides with aqueous ammonia in the presence of CuBr and pyridyldiketone ligands. J. Catal. 2019, 376, 119–122. 10.1016/j.jcat.2019.06.020. [DOI] [Google Scholar]; e Walczak A.; Stefankiewicz A. R. pH-Induced Linkage Isomerism of Pd(II) Complexes: A Pathway to Air- and Water-Stable Suzuki–Miyaura-Reaction Catalysts. Inorg. Chem. 2018, 57 (1), 471–477. 10.1021/acs.inorgchem.7b02711. [DOI] [PubMed] [Google Scholar]
- a Rakić G. M.; Grgurić-Šipka S.; Kaluđerović G. N.; Gómez-Ruiz S.; Bjelogrlić S. K.; Radulović S. S.; Tešić Ž. L. Novel trans-dichloridoplatinum(II) complexes with 3- and 4-acetylpyridine: Synthesis, characterization, DFT calculations and cytotoxicity. Eur. J. Med. Chem. 2009, 44 (5), 1921–1925. 10.1016/j.ejmech.2008.11.004. [DOI] [PubMed] [Google Scholar]; b Holford J.; Sharp S. Y.; Murrer B. A.; Abrams M.; Kelland L. R. In vitro circumvention of cisplatin resistance by the novel sterically hindered platinum complex AMD473. Br. J. Cancer 1998, 77 (3), 366–373. 10.1038/bjc.1998.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lannes A.; Intissar M.; Suffren Y.; Reber C.; Luneau D. Terbium(III) and Yttrium(III) Complexes with Pyridine-Substituted Nitronyl Nitroxide Radical and Different β-Diketonate Ligands. Crystal Structures and Magnetic and Luminescence Properties. Inorg. Chem. 2014, 53 (18), 9548–9560. 10.1021/ic500779y. [DOI] [PubMed] [Google Scholar]; b Alexandropoulos D. I.; Cunha-Silva L.; Pham L.; Bekiari V.; Christou G.; Stamatatos T. C. Tetranuclear Lanthanide(III) Complexes with a Zigzag Topology from the Use of Pyridine-2,6-dimethanol: Synthetic, Structural, Spectroscopic, Magnetic and Photoluminescence Studies. Inorg. Chem. 2014, 53 (6), 3220–3229. 10.1021/ic5001045. [DOI] [PubMed] [Google Scholar]
- a Palusiak M. Substituent effect in para substituted Cr(CO)5–pyridine complexes. J. Organomet. Chem. 2007, 692 (18), 3866–3873. 10.1016/j.jorganchem.2007.05.029. [DOI] [Google Scholar]; b Nakano K.; Suemura N.; Yoneda K.; Kawata S.; Kaizaki S. Substituent effect of the coordinated pyridine in a series of pyrazolato bridged dinuclear diiron(ii) complexes on the spin-crossover behavior. Dalton Trans. 2005, (4), 740–743. 10.1039/b416986g. [DOI] [PubMed] [Google Scholar]; c Jaju K.; Pal D.; Chakraborty A.; Chakraborty S. Electronic substituent effect on Se-H···N hydrogen bond: A computational study of para-substituted pyridine-SeH2 complexes. Chem. Phys. Lett. 2019, 737, 100031. 10.1016/j.cpletx.2019.100031. [DOI] [Google Scholar]; d Kimura A.; Ishida T. Pybox-Iron(II) Spin-Crossover Complexes with Substituent Effects from the 4-Position of the Pyridine Ring (Pybox = 2,6-Bis(oxazolin-2-yl)pyridine). Inorganics 2017, 5 (3), 52. 10.3390/inorganics5030052. [DOI] [Google Scholar]
- Lewis N. A.; Pakhomova S.; Marzilli P. A.; Marzilli L. G. Synthesis and Characterization of Pt(II) Complexes with Pyridyl Ligands: Elongated Octahedral Ion Pairs and Other Factors Influencing 1H NMR Spectra. Inorg. Chem. 2017, 56 (16), 9781–9793. 10.1021/acs.inorgchem.7b01294. [DOI] [PubMed] [Google Scholar]
- a Krogul A.; Skupińska J.; Litwinienko G. Tuning of the catalytic properties of PdCl2(XnPy)2 complexes by variation of the basicity of aromatic ligands. J. Mol. Catal. A Chem. 2014, 385, 141–148. 10.1016/j.molcata.2014.01.020. [DOI] [Google Scholar]; b Krogul A.; Skupińska J.; Litwinienko G. Catalytic activity of PdCl2 complexes with pyridines in nitrobenzene carbonylation. J. Mol. Catal. A Chem. 2011, 337 (1), 9–16. 10.1016/j.molcata.2011.01.007. [DOI] [Google Scholar]
- Krogul A.; Litwinienko G. Application of Pd(II) Complexes with Pyridines as Catalysts for the Reduction of Aromatic Nitro Compounds by CO/H2O. Org. Process Res. Dev. 2015, 19 (12), 2017–2021. 10.1021/acs.oprd.5b00273. [DOI] [Google Scholar]
- Krogul A.; Litwinienko G. One pot synthesis of ureas and carbamates via oxidative carbonylation of aniline-type substrates by CO/O2 mixture catalyzed by Pd-complexes. J. Mol. Catal. A Chem. 2015, 407, 204–211. 10.1016/j.molcata.2015.06.027. [DOI] [Google Scholar]
- a Tehan B. G.; Lloyd E. J.; Wong M. G.; Pitt W. R.; Gancia E.; Manallack D. T. Estimation of pKa Using Semiempirical Molecular Orbital Methods. Part 2: Application to Amines, Anilines and Various Nitrogen Containing Heterocyclic Compounds. Quant. Struct.-Act. Rel. 2002, 21 (5), 473–485. . [DOI] [Google Scholar]; b Brivio M.; Schlosrich J.; Ahmad M.; Tolond C.; Bugg T. D. Investigation of acid-base catalysis in the extradiol and intradiol catechol dioxygenase reactions using a broad specificity mutant enzyme and model chemistry. Org. Biomol. Chem. 2009, 7 (7), 1368–73. 10.1039/b821559f. [DOI] [PubMed] [Google Scholar]; c Iyehara Ogawa H.; Liu S.-Y.; Sakata K.; Niyitani Y.; Tsuruta S.; Kato Y. Inverse correlation between combined mutagenicity in Salmonella typhimurium and strength of coordinate bond in mixtures of cobalt(II) chloride and 4-substituted pyridines. Mutat. Res. Genet. Toxicol. 1988, 204 (2), 117–121. 10.1016/0165-1218(88)90081-X. [DOI] [PubMed] [Google Scholar]; d Uwai K.; Konno N.; Kitamura S.; Ohta S.; Takeshita M. Purification and characterization of rat liver enzyme catalyzing stereoselective reduction of acetylpyridines. Chirality 2005, 17 (8), 494–500. 10.1002/chir.20197. [DOI] [PubMed] [Google Scholar]; e Honda H. 1H-MAS-NMR Chemical Shifts in Hydrogen-Bonded Complexes of Chlorophenols (Pentachlorophenol, 2,4,6-Trichlorophenol, 2,6-Dichlorophenol, 3,5-Dichlorophenol, and p-Chlorophenol) and Amine, and H/D Isotope Effects on 1H-MAS-NMR Spectra. Molecules 2013, 18 (4), 4786. 10.3390/molecules18044786. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Chatzopoulou M.; Kotsampasakou E.; Demopoulos V. J. Clauson–Kaas-Type Synthesis of Pyrrolyl-phenols, from the Hydrochlorides of Aminophenols, in the Presence of Nicotinamide. Synth. Commun. 2013, 43 (21), 2949–2954. 10.1080/00397911.2012.753460. [DOI] [Google Scholar]; g Stilinović V.; Kaitner B. Salts and Co-Crystals of Gentisic Acid with Pyridine Derivatives: The Effect of Proton Transfer on the Crystal Packing (and Vice Versa). Cryst. Growth Des. 2012, 12 (11), 5763–5772. 10.1021/cg301267h. [DOI] [Google Scholar]
- https://scifinder.cas.orgCalculated using Advanced Chemistry Development (ACD/Labs) Software V11.02 (© 1994–2022 ACD/Labs).
- Helm L.; Merbach A. E. Inorganic and Bioinorganic Solvent Exchange Mechanisms. Chem. Rev. 2005, 105 (6), 1923–1960. 10.1021/cr030726o. [DOI] [PubMed] [Google Scholar]
- Spackman P. R.; Turner M. J.; McKinnon J. J.; Wolff S. K.; Grimwood D. J.; Jayatilaka D.; Spackman M. A. CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54 (3), 1006–1011. 10.1107/S1600576721002910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Janiak C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc., Dalton Trans. 2000, (21), 3885–3896. 10.1039/b003010o. [DOI] [Google Scholar]; b Scudder M. L.; Goodwin H. A.; Dance I. G. Crystal supramolecular motifs: two-dimensional grids of terpy embraces in [ML2] complexes (L = terpy or aromatic N3-tridentate ligand). New J. Chem. 1999, 23 (7), 695–705. 10.1039/a900564a. [DOI] [Google Scholar]
- a Gavezzotti A. The “sceptical chymist”: intermolecular doubts and paradoxes. CrystEngComm 2013, 15 (20), 4027–4035. 10.1039/c3ce00051f. [DOI] [Google Scholar]; b Martinez C. R.; Iverson B. L. Rethinking the term “pi-stacking. Chem. Sci. 2012, 3 (7), 2191–2201. 10.1039/c2sc20045g. [DOI] [Google Scholar]; c Bloom J. W. G.; Wheeler S. E. Taking the Aromaticity out of Aromatic Interactions. Angew. Chem., Int. Ed. 2011, 50 (34), 7847–7849. 10.1002/anie.201102982. [DOI] [PubMed] [Google Scholar]; d Grimme S. Do Special Noncovalent π–π Stacking Interactions Really Exist?. Angew. Chem., Int. Ed. 2008, 47 (18), 3430–3434. 10.1002/anie.200705157. [DOI] [PubMed] [Google Scholar]; e Ehrlich S.; Moellmann J.; Grimme S. Dispersion-Corrected Density Functional Theory for Aromatic Interactions in Complex Systems. Acc. Chem. Res. 2013, 46 (4), 916–926. 10.1021/ar3000844. [DOI] [PubMed] [Google Scholar]
- a Tiekink E. R. T. Supramolecular assembly based on “emerging” intermolecular interactions of particular interest to coordination chemists. Coord. Chem. Rev. 2017, 345, 209–228. 10.1016/j.ccr.2017.01.009. [DOI] [Google Scholar]; b Meyer E. A.; Castellano R. K.; Diederich F. Interactions with Aromatic Rings in Chemical and Biological Recognition. Angew. Chem., Int. Ed. 2003, 42 (11), 1210–1250. 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- Cavallo G.; Metrangolo P.; Milani R.; Pilati T.; Priimagi A.; Resnati G.; Terraneo G. The Halogen Bond. Chem. Rev. 2016, 116 (4), 2478–2601. 10.1021/acs.chemrev.5b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viossat B.; Dung N.-H.; Robert F. Structure du trans-dichlorobis(pyridine)palladium(II). Acta Crystallogr. C 1993, 49 (1), 84–85. 10.1107/S0108270192004438. [DOI] [Google Scholar]
- Liao C.-Y.; Lee H. M. trans-Dichlorodipyridinepalladium(II). Acta Crystallogr. E 2006, 62 (4), m680–m681. 10.1107/S160053680600688X. [DOI] [Google Scholar]
- Lee H. M.; Liao C.-Y. A new monoclinic polymorph of trans-dichloridodipyridinepalladium(II). Acta Crystallogr. E 2008, 64 (11), m1447. 10.1107/S1600536808033552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zordan F.; Brammer L. M–X···X‘–C Halogen-Bonded Network Formation in MX2(4-halopyridine)2 Complexes (M = Pd, Pt; X = Cl, I; X‘ = Cl, Br, I). Cryst. Growth Des. 2006, 6 (6), 1374–1379. 10.1021/cg050670m. [DOI] [Google Scholar]
- Maresca L.; Natile G. Five-Coordination in Platinum(II) and Palladium(II) Chemistry. Comments Inorg. Chem. 1993, 14 (6), 349–366. 10.1080/02603599308048668. [DOI] [Google Scholar]
- Zhang Y.; Woods T. J.; Rauchfuss T. B. Homoleptic Rhodium Pyridine Complexes for Catalytic Hydrogen Oxidation. J. Am. Chem. Soc. 2021, 143 (27), 10065–10069. 10.1021/jacs.1c05060. [DOI] [PubMed] [Google Scholar]
- Templeton J. L. Hexakis(pyridine)ruthenium(II) tetrafluoroborate. Molecular structure and spectroscopic properties. J. Am. Chem. Soc. 1979, 101 (17), 4906–4917. 10.1021/ja00511a020. [DOI] [Google Scholar]
- a Miyaura N.; Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95 (7), 2457–2483. 10.1021/cr00039a007. [DOI] [Google Scholar]; b Miyaura N.; Yamada K.; Suginome H.; Suzuki A. Novel and convenient method for the stereo-and regiospecific synthesis of conjugated alkadienes and alkenynes via the palladium-catalyzed cross-coupling reaction of 1-alkenylboranes with bromoalkenes and bromoalkynes. J. Am. Chem. Soc. 1985, 107 (4), 972–980. 10.1021/ja00290a037. [DOI] [Google Scholar]; c Knowles J. P.; Whiting A. The Heck–Mizoroki cross-coupling reaction: a mechanistic perspective. Org. Biomol. Chem. 2007, 5 (1), 31–44. 10.1039/B611547K. [DOI] [PubMed] [Google Scholar]; d Matos K.; Soderquist J. A. Alkylboranes in the Suzuki–Miyaura Coupling: Stereochemical and Mechanistic Studies. J. Org. Chem. 1998, 63 (3), 461–470. 10.1021/jo971681s. [DOI] [PubMed] [Google Scholar]; e Braga A. A. C.; Morgon N. H.; Ujaque G.; Lledós A.; Maseras F. Computational study of the transmetalation process in the Suzuki–Miyaura cross-coupling of aryls. J. Organomet. Chem. 2006, 691 (21), 4459–4466. 10.1016/j.jorganchem.2006.02.015. [DOI] [Google Scholar]; f Heck R. F.; Nolley Jr J. Palladium-catalyzed vinylic hydrogen substitution reactions with aryl, benzyl, and styryl halides. J. Org. Chem. 1972, 37 (14), 2320–2322. 10.1021/jo00979a024. [DOI] [Google Scholar]
- CrysAlisPRO; Rigaku Oxford Diffraction Ltd.: Yarnton, Oxfordshire, England, 2019.
- a Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42 (2), 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]; b Sheldrick G. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71 (1), 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Parsons S.; McCall K.; Robertson N. CSD Communication (Private Communication), CCDC, Cambridge, England, 2015.; b Sheldrick G. A short history of SHELX. Acta Crystallogr. A 2008, 64 (1), 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- a Martins F. J.; Mol Lima R.; Alves dos Santos J.; de Almeida Machado P.; Soares Coimbra E.; David da Silva A.; Rezende Barbosa Raposo N. Biological Properties of Heterocyclic Pyridinylimines and Pyridinylhydrazones. Lett. Drug. Des. Discovery 2015, 13, 107–114. 10.2174/1570180812666141216205601. [DOI] [Google Scholar]; b Beck D. E.; Reddy P. V. N.; Lv W.; Abdelmalak M.; Tender G. S.; Lopez S.; Agama K.; Marchand C.; Pommier Y.; Cushman M. Investigation of the Structure–Activity Relationships of Aza-A-Ring Indenoisoquinoline Topoisomerase I Poisons. J. Med. Chem. 2016, 59 (8), 3840–3853. 10.1021/acs.jmedchem.6b00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.