

Abstract

Cobalt-catalyzed C–H amination via M-nitrenoid species is spiking the interest of the research community. Understanding this process at a molecular level is a challenging task, and here we report a well-defined macrocyclic system featuring a pseudo-Oh aryl-CoIII species that reacts with aliphatic azides to effect intramolecular Csp2–N bond formation. Strikingly, a putative aryl-Co=NR nitrenoid intermediate species is formed and is rapidly trapped by a carboxylate ligand to form a carboxylate masked-nitrene, which functions as a shortcut to stabilize and guide the reaction to productive intramolecular Csp2–N bond formation. On one hand, several intermediate species featuring the Csp2–N bond formed have been isolated and structurally characterized, and the essential role of the carboxylate ligand has been proven. Complementarily, a thorough density functional theory study of the Csp2–N bond formation mechanism explains at the molecular level the key role of the carboxylate-masked nitrene species, which is essential to tame the metastability of the putative aryl-CoIII=NR nitrene species to effectively yield the Csp2–N products. The solid molecular mechanistic scheme determined for the Csp2–N bond forming reaction is fully supported by both experimental and computation complementary studies.
Short abstract
A well-defined pseudo-Oh aryl-CoIII species reacts with aliphatic azides to effect intramolecular Csp2−N bond formation via a carboxylate masked-CoIII-nitrene, which serves as a shortcut to guide the reaction to productive Csp2−N bond formation.
Introduction
The introduction of nitrogen functionalities into organic frameworks has attracted considerable interest in the development of new methodologies, given their ubiquitous occurrence in pharmaceuticals and natural products.1 A powerful strategy to achieve the construction of C–N bonds is based on the direct functionalization of C–H bonds, which has been widely studied in the last few decades.2−8 This field has been mainly dominated by the use of noble metal catalysis; yet, the development of more sustainable methodologies using M-nitrenoid species with first-row transition metals has recently become a hot topic.9
M-Nitrenoid species are rare and unstable species for late transition metals. For Group 8 M-nitrenoids, a prominent example is the relatively stable octahedral iron(IV) terminal imido complex [FeIV(N4Py)(NTs)]2+ reported by Que and co-workers,10 with S = 1 and a half-life of 3 h at room temperature.11,12 For transition metals in Group 9 and beyond, the common instability of Oh M-nitrenoid species may be overcome by changing the spin state or the geometry of the complex. In particular, the isolation of Group 9 Co-nitrene species has been achieved by lowering the symmetry and coordination number of the complex,13−15 highlighting four-coordinated complexes featuring tetrahedral geometry.16,17 Among all aminating reagents used to forge C–N bonds with cobalt catalysis, organic azides constitute an attractive N-source due to its 2e-oxidant character with concomitant extrusion of inert N2.9,18,19 The latter, together with the low-symmetry requirement, forces the design of low oxidation state CoI species that form isolable CoIII-imido multiple-bonded species upon reaction with N3-R.16,17 Although this chemistry is dominated by the use of low valent cobalt systems, few examples are reported on direct Csp2–N bond formation through C–H activation involving putative high-valent Co platforms.20,21 Indeed, highly unstable octahedral high valent MV=NR species with Group 9 metals are proposed as key intermediate species in Csp2–N bond forming processes. Remarkably, their relevance has been clearly pointed out formally in Oh MV Group 9 complexes bearing a Cp* ligand.9,22 Reaction of cyclometalated Cp*RhIII and Cp*IrIII complexes with N3-R render the proposed Cp*MV=NR intermediate species,23,24 which are essential for the inner-sphere Csp2–N bond forming step with the cyclometalated ligand (Figure 1a). Regarding the analogous cobalt chemistry bearing a Cp* ligand, Matsunaga and Kanai demonstrated the ability of Cp*CoIII catalysts to perform the Csp2–H amidation of indoles using sulfonyl azides and phosphoryl azides.25,26 The Csp2–H amidation of indoles using acyl azides has also been reported using Cp*CoIII by Punniyamurthy and co-workers (Figure 1b)27 and using phenyl azidoformates by Chang and co-workers.28 Also, isoquinolone synthesis via Cp*CoV cobaltacycles using N-chloroamides was reported by Zhu and co-workers.29 Despite these incipient reports, the high valent approach in Co-catalyzed Csp2–H amination is still in its infancy.
Figure 1.

(a, b) Cp*-M-nitrenoid species proposed for the C–H amination reactions (M = Ir, Rh, Co); (c) this work.
The examination of structure and electronic properties of key intermediate species is foremost for unveiling the mechanistic intricacies of inner-sphere N atom transfer chemistry. To this end, our group has been interested in the elucidation of transient intermediates involved in several Csp2–H functionalization reactions. In 2016, we reported a set of aryl-CoIII complexes synthesized through Csp2–H activation which were catalytically competent in alkyne and diazoacetate annulation reactions.30−34 Thanks to the stability offered by the 12-membered macrocyclic model substrate employed, we were able to isolate an unprecedented C-metalated cis-aryl-CoIII-alkyl enolate complex, i.e., a masked-carbene species, which was demonstrated to be an on-cycle intermediate in the catalytic formation of the final Csp2–C products.30,31 Because of the extra stabilization offered by these model platforms, we hypothesized that they could offer a suitable electronic and geometric environment for studying the reactivity of pseudo-Oh aryl-CoIII organometallic complexes toward organic azides.
Herein, we report the N atom transfer reactivity of organic azides with well-defined aryl-CoIII complexes (Figure 1c), focusing on the step-by-step reactivity of intermediate species to unravel key mechanistic details of the Csp2–N bond formation. Aliphatic azides were found to efficiently effect the Csp2–N bond products. With a combination of experimental and density functional theory (DFT) studies, the full reconstruction of the N atom transfer process was revealed. Several intermediate species featuring the Csp2–N bond formed have been isolated and structurally characterized. The essential role of carboxylate-masked nitrenoid species to tame the metastability of the putative Co-nitrenoid was confirmed both experimentally and theoretically, affording a solid mechanistic picture of the Csp2-N bond forming process. The Co-nitrenoid is clearly described as an pseudo-Oh aryl-CoIII-nitrene based on molecular orbital and electron density analyses, in contrast to the previously reported Oh Cp*CoV=NR imido species (Figure 1a,b).25,26
Results and Discussion
The reactivity of the well-defined aryl-CoIII complex (1-OAc) with organic azides as nitrene precursors started by examining its reaction with p-NO2-phenyl azide. Unfortunately, the use of aromatic azides led to decomposition and formation of unidentified products. On the other hand, positive results were obtained with aliphatic azides. We started with the addition of benzyl azide (a) to 1-OAc complex using fluorinated alcohols as solvent (TFE or HFIP) at 50 °C, affording the aryl-amine coupling complex 2a-OAc in 46% yield (Scheme 2). This complex was structurally characterized by 2D NMR studies, where a diagnostic HMBC peak between the benzylic −CH2 of the formal azide and the quaternary carbon of the aryl moiety was observed, proving the formation of a new Csp2-NH bond. The coordinatively saturated complex 2a-OAc slowly evolved at room temperature to a more stable dinuclear species, 3a-OAc, in quantitative yield (see 1H NMR time-evolution in Figure S1). Crystals of 3a-OAc were obtained from slow evaporation from a CH2Cl2 solution (DCM/pentane) at −4 °C, allowing for an unambiguous characterization of this dimeric species. Compound 3a-OAc features the new Csp2–NH bond, and each CoIII center presents a distorted octahedral geometry, with coordination to Npy, NHL, and NHazide as well as one OAc and two μ-hydroxo bridging ligands. Independent blank experiments exposing 2a-OAc crude mixture to H2O or O2 clearly suggested that the origin of the hydroxo groups in 3a-OAc is O2.
Scheme 2. Thermal Decomposition and Acidic Work-up Affording the Corresponding Aminated Product P-a, P-b, P-c, and P-a-cyc.

Isolated yields shown.
Encouraged by these results, we explored the reactivity of 1-OAc with (2-azidoethyl)benzene (b) and (3-azidopropyl)benzene (c) (Scheme 1). Using an excess of the azide b and c (6 equiv), the corresponding inserted complex (2b-OAc and 2c-OAc) was obtained in 26% and 25% yield, respectively. Both complexes led to the quantitative formation of the corresponding dimer 3b-OAc and 3c-OAc, analogous to complex 3a-OAc (vide supra).
Scheme 1. Reactivity of the Aryl-CoIII (1-OAc) with Organic Azides to Afford Complexes 2x-OAc and 3x-OAc.

NMR yields of 3x-OAc are based on 2x-OAc. Selected bond distances for [Å] and angles [deg]: C(19)–N(27) 1.441(16), N(27)–Co(1) 2.009(11), C(19)–N(27)–Co(1) 110.3(9), Co(1)–O(5) 1.924(9), Co(1)–N(16) 1.955(12), Co(1)–O(3) 1.932(9), Co(1)–O(4) 1.930 (9). Hydrogen atoms, anions, and solvents molecules have been omitted for clarity.
The better yields observed for 2a-OAc after treatment of 1-OAc with benzyl azide prompted us to scrutinize the demetalation step. On the basis of previous reports,24 the protodemetalation step to render the aminated product was predicted to be kinetically and thermodynamically disfavored. Thus, to favor this step, we designed alternative strategies based on the use of strong acids and thermolysis (Scheme 2). First, HCl (2 M) was added to a solution of 2a-OAc in CHCl3, and after 16 h the crude mixture was basified and extracted, affording the aminated product P-a in 44% isolated yield. The analogous reaction using 2b-OAc and 2c-OAc afforded the corresponding aminated product P-b and P-c in 40% and 42% respectively. On the other hand, heating 2a-OAc to 100 °C in HFIP furnished the cyclized product P-a-cyc in 27% yield. The analogous cyclic products using 2b-OAc and 2c-OAc were not formed under the same conditions, which highlights the importance of the benzylic position for the formation of cyclized product (see mechanistic proposal for P-a-cyc formation in Scheme S7).
The absence of an analogous cyclic product from 2b-OAc and 2c-OAc led us to investigate in depth the reactivity of these azides under different thermal conditions (Scheme 3). When 1-OAc was mixed with an excess of azide b at 100 °C in TFE, a new paramagnetic species appeared and was stable under inert atmosphere. XRD analysis showed a CoII complex with distorted octahedral geometry bearing the phenylethan-1-amine moiety inserted (4b-OAc), which under acid conditions forms the product P-b in 41% NMR yield with respect to the 4b-OAc complex.
Scheme 3. (A) Reactivity of Aryl-CoIII (1-OAc) with Organic Azide (b) at 100 °C and (B) Crystal Structure of 4b-OAc Complex.

NMR yield of P-b is based on 4b-OAc.
Selected bond distances for [Å] and angles [deg]: C(12)–N(20) 1.439(5), N(20)–Co(1) 2.176(4), C(12)–N(20)–Co(1) 109.7(2), Co(1)–N(18) 2.127(4), Co(1)–N(9) 2.259(4), Co(1)–N(2) 2.255(4), Co(1)–O(33) 2.075(3), Co(1)–O(29) 2.056(3). Hydrogen atoms and solvent molecules have been omitted for clarity. NH(L) refers to the coordination of the Co(II) center to another ligand moiety (depicted as N9 in the crystal structure).
Moreover, X-ray absorption spectroscopy (XAS) was conducted for 4b-OAc, clearly confirming the CoII oxidation state for the metal center (Table S2, Figure S22, Panels S1–S3) compared to CoIII species 1-OAc and the newly synthesized aryl-CoIII-benzylamine complex (5-OAc, see Scheme S13). The Co-ligand bond distances in the crystal structure of 4b-OAc (>2.1 Å) suggest a high spin CoII–d7 electronic configuration, which was supported by the μeff calculated using Evans method in CD2Cl2. The obtained value of μeff = 4.22 MB is in agreement with the presence of three unpaired electrons. We hypothesized that the CoII complex 4b-OAc stemmed from reductive elimination of an in situ aryl-CoIV-imido, although more investigations are needed to shed some light on the detailed mechanism of the formation of 4b-OAc.
The nature of the carboxylate ligand was also investigated, and we prepared the analogous 1-(OOCR) complex bearing a substituted benzoate instead of the initial acetate (see Figures S23–S24 for the XRD of 1-(OBz-CF3) and 1-(OBz-OMe)). The use of EWG and EDG substituents did not affect the formation of the inserted 2a-OBz-X complex (Scheme S9). Subjecting the mixture to acidic conditions led the formation of P-a product in similar yields. Moreover, the direct formation of P-a-cyc product was achieved by reacting several 1-(OBz-X) with benzyl azide (a) under thermal conditions. The most coordinating p-OMe-benzoate affords a 42% yield, whereas the least coordinating p-NO2-benzoate affords only 16% yield, thus following the expected trend (see Scheme S11). However, the coordinating p-Me-benzoate drops to 17%, and the p-Cl-benzoate affords 49%. Therefore, the use of EWG and EDG substituents did not affect the formation of either complex 2a-OBz-X complex or the final organic product P-a-cyc.
Mechanistic Investigations
To gain more mechanistic insights of the Csp2–N bond formation, additional tests were performed. By adding TEMPO radical at 50 °C, MS peaks matching with a CoII complex bearing the formed Csp2–N bond were detected, whereas at 100 °C the yield of P-a-cyc dropped from 27% to 7%. These results are not conclusive for either a radical or nonradical pathway since Csp2–N coupling is occurring, although in lower yields. Therefore, on the basis of all experimental evidence, a thorough computational DFT study was mandatory to unravel the precise mechanism for the intriguing Csp2–N bond forming step using benzyl azide and 1-OAc (Figure 2 and Figure 3). The calculations were performed at the revTPSS-D3BJ/Def2TZVP//BP86-D3BJ/Def2SVP level of theory (see Supporting Information for full computational details and benchmark study). The rate-determining step of the reaction corresponds to the N2 extrusion from 1-OAc·N3Bz to yield the short-lived species INT-N. Wave function analysis of INT-N indicated without a doubt that INT-N is best described as an aryl-CoIII=N-R (R = −CH2Ph) nitrene species (Fischer-type) with σ and π bonds between the Co and the N atoms (bond order of 1.51 and bond length of 1.71 Å). Effective oxidation state (EOS) analysis in INT-N dissects the N–Co σ and π bonds into two contributions from the ligand and the metal, as shown in Figure 2 (see also Supporting Information). Considering the corresponding occupations of the effective fragment orbitals (EFOs), the EOS analysis assigns the two electrons of the N–Co σ bond to the nitrene, whereas the two electrons of the N–Co π bond are assigned to the Co. Therefore, INT-N may be described as an aryl CoIII-nitrene with significant back-donation from Co to N. Qualitative analysis of the relative contributions of N and Co to the π and π* canonic molecular orbitals also characterize INT-N as a aryl CoIII-nitrene (Figure S33 and Table S10), ruling out an aryl-CoV-imido species (Schrock-type).35
Figure 2.
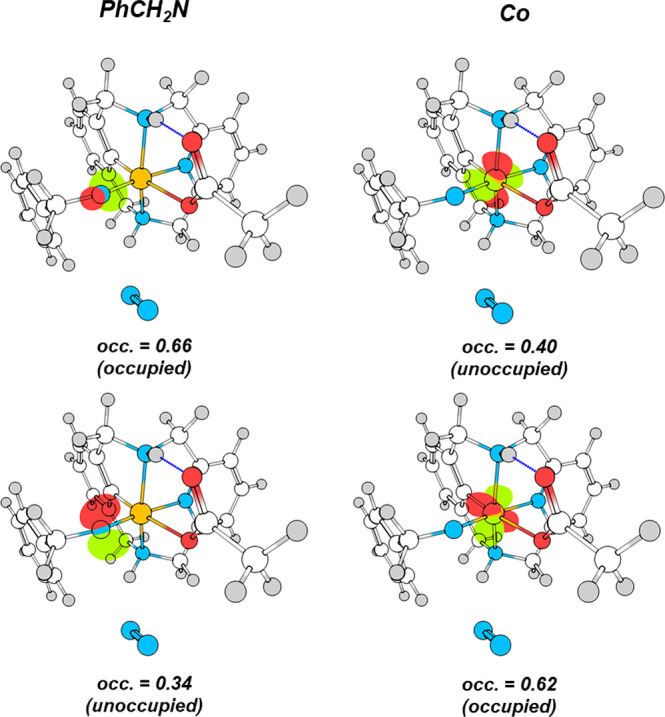
Effective fragment orbitals (EFOs) graphical representation and occupations—in the [0,1] range—associated with the σ (top) and π (bottom) interaction between the PhCH2N ligand and the Co center.
Figure 3.

RevTPSS-D3BJ/Def2TZVP//BP86-D3BJ/Def2SVP free energy profile for the studied reaction mechanism. Gibbs free energies (G, in kcal·mol–1) are relative to 1-OAc·N3Bz. The pathway in black corresponds to the singlet species (S = 0), while the blue pathway corresponds to the triplet species (S = 1). Geometries for all S = 0 intermediates and transition states are shown (nitrogen atoms are represented in blue, oxygens in red, cobalt in orange, carbon in white, and hydrogens in gray. Note that hydrogens bonded to carbon have been hidden for clarity). Relevant distances have also been included (in Å).
The EOS analysis of the S = 1 spin state of INT-N indicates that the triplet INT-N can also be described as a CoIII-nitrene with a Co–N bond length of 1.73 Å. As it can be seen by the occupation and shape of the effective fragment orbitals (EFOs) depicted in Figure S36, in the singlet–triplet transition, the S = 0 beta electron of the lone-pair of the N is transferred to a p-type EFO of the N, resulting in a triplet state with two alpha p-type nonbonding electrons on the N. In addition, the remaining two beta electrons form two Co–N one-electron π bonds polarized toward the Co (see Figure S37). This analysis agrees with the fact that the major contribution of the spin density (i.e., electron density of alpha electrons minus the electron density of the beta electrons, which indicates the localization of the unpaired electrons) of the S = 1 state of INT-N is localized in the N (see Figure S34 and Table S12) and that the singlet → triplet spin-crossing does not cause significant change in the Co–N bond distance or in the formal oxidation state of the Co.
This intermediate species rapidly evolves overcoming a very low barrier (<2 kcal/mol, TS2) to a 14 kcal/mol more stable INT-MaskN species by formation of a five-member acetoxy(benzyl)amide ring via carboxylate attack to the N atom, formally defined as a masked aryl-Co-nitrene. Wave function analysis describes INT-MaskN as a masked CoIII nitrene with a single σ bond between the Co and the N. The lack of a Co=N π bond is also evidenced by the increased bond distance of 1.91 Å (1.71 Å for INT-N) and a decreased bond order of 0.78.
The masked aryl-CoIII-nitrene (or “nitrenoid”)36 is not sufficiently stabilized to be experimentally trapped since it allows the formal nucleophilic attack of the aryl moiety to the N atom of the masked nitrene to finally achieve the Csp2–N coupling through a barrier lower than 9 kcal/mol, TS3. The partial atomic charge of the N atom in INT-MaskN (−0.51) vs INT-N (−0.74) explains the enhanced electrophilic character of the former, induced by the formation of the acetoxy(benzyl)amide. Moreover, EOS analysis also reveals a larger occupation of the aryl sigma contribution in INT-MaskN compared to INT-N, which favors the SN2 attack, and is an indication of the enhanced nucleophilic character of the aryl in INT-MaskN (see Figure S32).
The reaction profile has also been evaluated for S = 2 and S = 1 spin states. The energies of the S = 2 state of all intermediates and transition states involved in the reaction mechanism are far higher than the singlet, and therefore the quintuplet states play no role in the studied reaction mechanism (see Table S11). The triplet states does not play a key role either. The S = 1 state of the initial complex interacting with the benzyl azide is 22.1 kcal/mol above the singlet (Figure 3). The Gibbs energy of S = 1 states for TS1, TS2, INT-MaskN, TS3, and 2a are also clearly higher than their S = 0 counterparts. The only exception is Co-nitrene intermediate INT-N, for which the triplet state is only 0.5 kcal/mol more stable than the singlet. However, because of the important electron reorganization that takes places on the N in the singlet–triplet transition, the probability of a spin-crossing between the singlet and the triplet Gibbs energy surfaces is strongly reduced. Thus, all of the computational evidence indicates that the reaction profile undergoes a singlet species.
We have performed several DFT relaxed PES scans to explore the stability of aryl-CoIII-nitrene complex INT-N upon distortion or disconnection of one of the coordinating N.17 However, all of the calculations confirm that the tight coordination environment imposed by the macrocyclic ligand in the aryl-CoIII-nitrene complex INT-N is mandatory for its stabilization. In addition, we have tried to locate the transition state that corresponds to the direct formation of the C–N bond (species 2a) from intermediate INT-N. However, all of our attempts lead to transition state TS2 or intermediate INT-MaskN.
The role of carboxylate anions was experimentally confirmed by using the acetate-free organometallic [aryl-CoIII-(CH3CN)2]2+ complex (1-CH3CN) (Scheme S14). Applying reaction conditions at 50 and 100 °C using benzyl azide (a), neither the aryl-amine coupling complex (analogous to 2a-OAc) (50 °C) nor final organic product P-a-cyc (100 °C) was detected. Furthermore, the presence of benzylaldehyde as a side product suggested the degradation of benzyl azide, pointing out the importance of the formation of the masked-aryl-Co-nitrene species INT-MaskN toward the Csp2–N coupling.
Previously, some of us studied the mechanism of Csp2–H functionalization with diazo esters catalyzed with a aryl-CoIII-carboxylate compound,31 in which the key role of a carboxylate-masked aryl-CoIII-carbene was proven. Analogously, the formation of the aryl-Co-nitrene, the facile evolution of the nitrene through a low-lying transition state to form a five-member acetoxy(benzyl)amide ring, as well as the final SN2-type substitution of the aryl-Co to the masked nitrene are reminiscent of the mechanism of the cobalt-catalyzed Csp2-H functionalization with diazo esters. The key difference between both mechanisms is the stability of the masked carbene and the masked nitrene. Whereas the masked-carbene could be isolated and fully experimentally characterized because the final nucleophilic attack is the rate-determining step of the reaction, the analogous masked nitrene could not be isolated due to the barrier to form the coupling product being much smaller than the barrier for the formation of the aryl-CoIII-nitrene. Experimentally, we conducted UV–vis monitoring analyses at variable temperature, which allows us to determine that the release of N2 toward the formation of the nitrene species INT-N is indeed the rate-determining step (rds) of the reaction (ΔG‡ = 23.9 kcal/mol, Eyring plot in Figure S20), which nicely agrees with the DFT Gibbs energy profile of the reaction mechanism presented in Figure 3. MS analysis after mixing time agrees with the accumulation of 1-OAc·N3Bz species (Figures S19 and S20).
The proposed general mechanism is shown in Scheme 4. This study demonstrates the stabilizing masking effect of the carboxylate group to the Co-nitrene moiety, to tame the extraordinary reactivity and elusiveness of Co-nitrene species.
Scheme 4. Proposed Mechanism for the Reaction of 1-OAc with Benzylazide (a).

Intermolecular Nitrene-Transfer Attempts
Additionally, nitrene transfer was attempted by adding xanthene (2 equiv) to the mixture of 1-OAc and benzyl azide (a), but no intermolecular Csp2–N coupling product with xanthene was detected, and intramolecular 2a-OAc (20%) was formed (see Scheme S15).
Comparing the Reactivity of Organometallic CoIII and RhIII Complexes
To gain more insight into the mechanism, we explored the reactivity of benzyl azide using an analogous aryl-RhIII complex. However, attempts to isolate the aryl-RhIII analogous to 1-OAc using L-H were unsuccessful. Thus, we attempted the formation of P-a-cyc by reacting the L-H ligand with benzyl azide (a) and stoichiometric amounts of Rh(OAc)3 in TFE at 100 °C. In contrast to the aryl-CoIII complex, P-a-cyc cyclic product was not formed, and we only detected the formation of hydrazine. Therefore, since the aryl-RhIII complex was not isolable with L-H, we attempted successfully the comparison of both aryl-CoIII (1Me-OAc) and aryl-RhIII (6Me-OAc) synthesized with the L-Me ligand. CoIII complex 1Me-OAc was mixed with benzylazide, but no reaction was observed. By reacting complex 6Me-OAc with benzylazide, only aryl-RhIII-imine species (7xMe-OAc) was observed, with no trace of Csp2-N bond-formed species (Scheme S18). The contrasting reactivity of Co versus Rh analogues highlights the uniqueness of Co reactivity, its versatility to stabilize metastable species via carboxylate masking, and the value of studying in depth the role of first row metals in C–N formation.
Conclusions
In summary, we have studied the reactivity of well-defined pseudo-Oh aryl-CoIII species (1-OAc) with different azides, with successful intramolecular Csp2–N bond formation with aliphatic azides. In brief, when benzyl azide is used, we are able to trap the just-formed Csp2–N bond species, consisting of a CoIII complex (2a-OAc) that tends to dimerize to form complex 3a-OAc. Analogous reactivity is found for (2-azidoethyl)benzene (b) and (3-azidopropyl)benzene (c). However, only 2a-OAc evolves to an intramolecular cyclization to obtain the organic product P-a-cyc, whereas 1-OAc reacts with b under thermal treatment to afford a well-defined CoII complex featuring the already formed Csp2–N bond (4b-OAc). The thorough DFT study performed demonstrates the stabilizing masking effect of the carboxylate group to tame the extraordinary reactivity and elusiveness of an aryl-CoIII=N-R nitrene species (INT-N). First, the Gibbs energy barrier of the rate-determining step of the reaction, which corresponds to the N2 extrusion, is in agreement with the Gibbs energy barrier extracted from the Eyring plot and the mild experimental conditions applied (50 °C). More importantly, detailed wave function analysis of the masked aryl-CoIII-nitrene species INT-MaskN clearly shows an increase of electrophilicity on N and an increase of nucleophilicity on the Csp2-aryl compared to INT-N, thus promoting the facile SN2-like attack to effect the Csp2–N coupling. This is in line with the fact that the SN2-like barrier is far lower than the barrier for the formation of the aryl-CoIII-nitrene and with the fact that these masked species could not be trapped as in the case of the masked carbene.31 The key role of carboxylate anions in the formation of masked aryl-CoIII-nitrene species, fully supported by both experimental and computation studies, culminated in a solid mechanistic picture of the Csp2–N bond forming amination process, which is thought to be valuable for the future development of catalytic Csp2–N methodologies via Co=NR species. Indeed, CoIII masked nitrene species have been crystallographically isolated very recently,37 further supporting the validity of our study. Moreover, the occurrence of other transition metal masked nitrene species (Ru) is also proposed in chiral α-amino acid synthesis using carbamate derivatives.38 Interestingly, this in situ masking strategy is a straightforward alternative to the use of stabilized nitrene sources such as dioxazolones (CO2-evolving reagent), which focus the scope on amidation reactions.39−41
Experimental Section
Formation of 2x-OAc and 3x-OAc Complexes
1-OAc (0.048 mmol) and organic azides (a–c) with 1 mL of HFIP were mixed in a 2 mL vial and sealed. The mixture was heated at 50 °C overnight. Then, the crude mixture was concentrated under a vacuum line until the initial volume was reduced to two-thirds observing the formation of 2x-OAc intermediate complex by 1H NMR (CDCl3) and HRMS. The corresponding dimeric species 3x-OAc were slowly formed by recrystallization with CHCl3 layered with pentane under air.
Synthesis of P-x Products
Once the 2x-OAc were formed, each crude mixture was dissolved in CHCl3, and HCl (3 mmol, 2 M) was added and stirred overnight. The crude was basified until pH 14 and extracted with CHCl3. The products were purified by column chromatography using neutral alumina (CHCl3, then CHCl3/MeOH 8:2), giving the corresponding Csp2–N coupling products (P-a, P-b, and P-c).
Synthesis of P-a-cyc Product
1-OAc (0.048 mmol) and benzyl azide (a) (2.1 equiv) were mixed in HFIP (1 mL) in a 2 mL vial and sealed. The crude was heated at 100 °C overnight in the absence of light. The solvent was then removed, and the cyclic product was purified by column chromatography using neutral alumina (CHCl3, then CHCl3/MeOH. 8:2).
Formation of 4b-OAc
1-OAc (0.048 mmol) and (2-azidoethyl)benzene (b) (6.0 equiv) were mixed in TFE (1 mL) in a 2 mL vial and sealed. The crude was heated at 100 °C, and after 16 h the solvent was removed. Pentane diffusion in a concentrated solution of CH2Cl2 anhydrous under inert atmosphere yields the 4b-OAc complex.
Computational Details
All DFT calculations were carried out using Gaussian16 program. Geometry optimizations have been performed without any symmetry restrictions, considering the effect of the HFIP solvent via the Self-Consistent Reaction Field method using the SMD solvation model42 and taking into account dispersion effects with Grimme and co-workers DFT-D3BJ correction,43,44 at the BP86-D3BJ(SMD)/Def2SVP level of theory.45−48 The HFIP solvent is not implemented in GAUSSIAN16, so we performed those calculations using the Solvent = Generic,Read options for the SCRF keyword (see Supporting Information for further details). All geometry optimized structures were characterized by analytical frequency calculations, which also afforded enthalpy and entropy corrections at 298.15 K. All points in the reaction pathway were connected via IRC calculations. Single point calculations on the equilibrium geometries, including the solvent and dispersion effects (Esp), were carried out at the revTPSS-D3BJ(SMD)/Def2TZVP level of theory.49 Then, the total Gibbs energy values (G) are given by
| 1 |
where the Gibbs energy correction (Gcorr.) was obtained from the thermodynamical analysis at the optimization level of theory but corrected using the GoodVibes code50 so that frequencies below 100 are not treated with the Harmonic Approximation, but rather with the Quasi-Harmonic Approximation as described by Grimme.51 Finally, the additional correction term ΔG°/* accounts for the transition from the standard state concentration (gas phase, pressure of 1 atm) to the concentrations used experimentally.
Metal and ligands oxidation states (OS) were assigned with the effective oxidation states (EOS) analysis, which relies on Mayer’s effective fragment orbitals (EFOs) and their occupations. The EFOs are sorted by decreasing occupation number, and individual electrons (or pairs for closed-shell singlets) are assigned to those EFOs with higher occupations. This leads to an effective configuration of the atoms/ligands within the molecule, which directly determines their OS. EOS analysis was performed at the revTPSS-D3BJ/Def2TZVP level of theory with the in-house developed program APOST-3D,52 using the Topological Fuzzy Voronoi Cells (TFVC) atomic definition and a 40 × 146 atomic grid for the required numerical integrations.
Acknowledgments
This work was financially supported by MICINN (CTQ2016-77989-P and PID2019-104498GB-I00 to X.R., PGC2018-098212-B-C22 to J.M.L. and PGC2018-095808-B-I00 to T.P.) and Generalitat de Catalunya (2017SGR264 to X.R. and 2017SGR39 to J.M.L.). X.R. is thankful for an ICREA Academia award. X-ray absorption experiments were performed at the CLAESS beamline at ALBA Synchrotron with the collaboration of ALBA staff (V.M.-D.) as part of the in-house experiment 2019093969. We thank Dr. A. Company for fruitful discussions and STR-UdG for technical support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.2c02111.
Additional experimental details, materials, instrumentation, spectroscopic characterization of all compounds, computational details and X-ray crystallography details of 1-OBzCF3 (CCDC 2097542), 1-OBzOMe (CCDC 2097543), 3a-OAc (CCDC 2097546), 4b-OAc (CCDC 2097544), and 5-OAc (CCDC 2097545) (PDF)
Author Present Address
# Queen Mary University of London, Mile End Road, London E1 4NS, UK
Author Contributions
⊥ L.C. and M.M. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Hili R.; Yudin A. K. Making carbon-nitrogen bonds in biological and chemical synthesis. Nat. Chem. Biol. 2006, 2, 284–287. 10.1038/nchembio0606-284. [DOI] [PubMed] [Google Scholar]
- Davies H. M. L.; Long M. S. Recent Advances in Catalytic Intramolecular C-H Aminations. Angew. Chem., Int. Ed. 2005, 44, 3518–3520. 10.1002/anie.200500554. [DOI] [PubMed] [Google Scholar]
- Halfen J. A. Recent Advances in Metal-Mediated Carbon-Nitrogen Bond Formation Reactions: Aziridination and Amidation. Curr. Org. Chem. 2005, 9, 657–669. 10.2174/1385272053765024. [DOI] [Google Scholar]
- Davies H. M. L.; Manning J. R. Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collet F.; Dodd R. H.; Dauban P. Catalytic C-H amination: recent progress and future directions. Chem. Commun. 2009, 5061–5074. 10.1039/b905820f. [DOI] [PubMed] [Google Scholar]
- Zalatan D. N.; Bois J. D.. Metal-Catalyzed Oxidations of C-H to C-N Bonds. In C-H Activation; Yu J.-Q.; Shi Z., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2010; pp 347–378. [DOI] [PubMed] [Google Scholar]
- Müller P.; Fruit C. Enantioselective Catalytic Aziridinations and Asymmetric Nitrene Insertions into CH Bonds. Chem. Rev. 2003, 103, 2905–2920. 10.1021/cr020043t. [DOI] [PubMed] [Google Scholar]
- Louillat M.-L.; Patureau F. W. Oxidative C-H amination reactions. Chem. Soc. Rev. 2014, 43, 901–910. 10.1039/C3CS60318K. [DOI] [PubMed] [Google Scholar]
- Shin K.; Kim H.; Chang S. Transition-Metal-Catalyzed C-N Bond Forming Reactions Using Organic Azides as the Nitrogen Source: A Journey for the Mild and Versatile C-H Amination. Acc. Chem. Res. 2015, 48, 1040–1052. 10.1021/acs.accounts.5b00020. [DOI] [PubMed] [Google Scholar]
- Klinker E. J.; Jackson T. A.; Jensen M. P.; Stubna A.; Juhász G.; Bominaar E. L.; Münck E.; Que Jr L. A Tosylimido Analogue of a Nonheme Oxoiron(IV) Complex. Angew. Chem., Int. Ed. 2006, 45, 7394–7397. 10.1002/anie.200602799. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Faponle A. S.; Barman P.; Vardhaman A. K.; Sastri C. V.; Kumar D.; de Visser S. P. Long-Range Electron Transfer Triggers Mechanistic Differences between Iron(IV)-Oxo and Iron(IV)-Imido Oxidants. J. Am. Chem. Soc. 2014, 136, 17102–17115. 10.1021/ja508403w. [DOI] [PubMed] [Google Scholar]
- Coin G.; Patra R.; Rana S.; Biswas J. P.; Dubourdeaux P.; Clémancey M.; de Visser S. P.; Maiti D.; Maldivi P.; Latour J.-M. Fe-Catalyzed Aziridination Is Governed by the Electron Affinity of the Active Imido-Iron Species. ACS Catal. 2020, 10, 10010–10020. 10.1021/acscatal.0c01427. [DOI] [Google Scholar]
- Zhang L.; Liu Y.; Deng L. Three-Coordinate Cobalt(IV) and Cobalt(V) Imido Complexes with N-Heterocyclic Carbene Ligation: Synthesis, Structure, and Their Distinct Reactivity in C-H Bond Amination. J. Am. Chem. Soc. 2014, 136, 15525–15528. 10.1021/ja509731z. [DOI] [PubMed] [Google Scholar]
- Reckziegel A.; Pietzonka C.; Kraus F.; Werncke C. G. C-H Bond Activation by an Imido Cobalt(III) and the Resulting Amido Cobalt(II) Complex. Angew. Chem., Int. Ed. 2020, 59, 8527–8531. 10.1002/anie.201914718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünwald A.; Anjana S. S.; Munz D. Terminal Imido Complexes of the Groups 9–11: Electronic Structure and Developments in the Last Decade. Eur. J. Inorg. Chem. 2021, 2021, 4147–4166. 10.1002/ejic.202100410. [DOI] [Google Scholar]
- Baek Y.; Das A.; Zheng S.-L.; Reibenspies J. H.; Powers D. C.; Betley T. A. C-H Amination Mediated by Cobalt Organoazide Adducts and the Corresponding Cobalt Nitrenoid Intermediates. J. Am. Chem. Soc. 2020, 142, 11232–11243. 10.1021/jacs.0c04252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao W.; Fehn D.; Heinemann F. W.; Scheurer A.; Munz D.; Meyer K. A Pair of Cobalt(III/IV) Terminal Imido Complexes. Angew. Chem., Int. Ed. 2021, 60, 16480–16486. 10.1002/anie.202103170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dequirez G.; Pons V.; Dauban P. Nitrene Chemistry in Organic Synthesis: Still in Its Infancy?. Angew. Chem., Int. Ed. 2012, 51, 7384–7395. 10.1002/anie.201201945. [DOI] [PubMed] [Google Scholar]
- Park Y.; Semproni S. P.; Zhong H.; Chirik P. J. Synthesis, Electronic Structure, and Reactivity of a Planar Four-Coordinate, Cobalt-Imido Complex. Angew. Chem., Int. Ed. 2021, 60, 14376–14380. 10.1002/anie.202104320. [DOI] [PubMed] [Google Scholar]
- Du C.; Li P.-X.; Zhu X.; Han J.-N.; Niu J.-L.; Song M.-P. Cobalt-Catalyzed Oxidative C-H/N-H Cross-Coupling: Selective and Facile Access to Triarylamines. ACS Catal. 2017, 7, 2810–2814. 10.1021/acscatal.7b00262. [DOI] [Google Scholar]
- Jia Q.; Kong L.; Li X. Cobalt(iii)-catalyzed C-H amidation of weakly coordinating sulfoxonium ylides and α-benzoylketene dithioacetals. Org. Chem. Front. 2019, 6, 741–745. 10.1039/C8QO01270A. [DOI] [Google Scholar]
- Sau Y.-K.; Yi X.-Y.; Chan K.-W.; Lai C.-S.; Williams I. D.; Leung W.-H. Insertion of nitrene and chalcogenolate groups into the Ir-C σ bond in a cyclometalated iridium(III) complex. J. Organomet. Chem. 2010, 695, 1399–1404. 10.1016/j.jorganchem.2010.02.002. [DOI] [Google Scholar]
- Park S. H.; Kwak J.; Shin K.; Ryu J.; Park Y.; Chang S. Mechanistic Studies of the Rhodium-Catalyzed Direct C-H Amination Reaction Using Azides as the Nitrogen Source. J. Am. Chem. Soc. 2014, 136, 2492–2502. 10.1021/ja411072a. [DOI] [PubMed] [Google Scholar]
- Figg T. M.; Park S.; Park J.; Chang S.; Musaev D. G. Comparative Investigations of Cp*-Based Group 9 Metal-Catalyzed Direct C-H Amination of Benzamides. Organometallics 2014, 33, 4076–4085. 10.1021/om5005868. [DOI] [Google Scholar]
- Sun B.; Yoshino T.; Matsunaga S.; Kanai M. Air-Stable Carbonyl(pentamethylcyclopentadienyl)cobalt Diiodide Complex as a Precursor for Cationic (Pentamethylcyclopentadienyl)cobalt(III) Catalysis: Application for Directed C-2 Selective C-H Amidation of Indoles. Adv. Synth. Catal. 2014, 356, 1491–1495. 10.1002/adsc.201301110. [DOI] [Google Scholar]
- Sun B.; Yoshino T.; Matsunaga S.; Kanai M. A Cp*CoI2-dimer as a precursor for cationic Co(iii)-catalysis: application to C-H phosphoramidation of indoles. Chem. Commun. 2015, 51, 4659–4661. 10.1039/C4CC10284C. [DOI] [PubMed] [Google Scholar]
- Shah T. A.; De P. B.; Pradhan S.; Banerjee S.; Punniyamurthy T. Cp*Co(III)-Catalyzed Regioselective C2 Amidation of Indoles Using Acyl Azides. J. Org. Chem. 2019, 84, 16278–16285. 10.1021/acs.joc.9b02244. [DOI] [PubMed] [Google Scholar]
- Lee J.; Lee J.; Jung H.; Kim D.; Park J.; Chang S. Versatile Cp*Co(III)(LX) Catalyst System for Selective Intramolecular C-H Amidation Reactions. J. Am. Chem. Soc. 2020, 142, 12324–12332. 10.1021/jacs.0c04448. [DOI] [PubMed] [Google Scholar]
- Yu X.; Chen K.; Guo S.; Shi P.; Song C.; Zhu J. Direct Access to Cobaltacycles via C-H Activation: N-Chloroamide-Enabled Room-Temperature Synthesis of Heterocycles. Org. Lett. 2017, 19, 5348–5351. 10.1021/acs.orglett.7b02632. [DOI] [PubMed] [Google Scholar]
- Planas O.; Roldán-Gómez S.; Martin-Diaconescu V.; Luis J. M.; Company A.; Ribas X. Mechanistic insights into the SN2-type reactivity of aryl-Co(iii) masked-carbenes for C-C bond forming transformations. Chem. Sci. 2018, 9, 5736–5746. 10.1039/C8SC00851E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas O.; Roldán-Gómez S.; Martin-Diaconescu V.; Parella T.; Luis J. M.; Company A.; Ribas X. Carboxylate-Assisted Formation of Aryl-Co(III) Masked-Carbenes in Cobalt-Catalyzed C-H Functionalization with Diazo Esters. J. Am. Chem. Soc. 2017, 139, 14649–14655. 10.1021/jacs.7b07880. [DOI] [PubMed] [Google Scholar]
- Planas O.; Whiteoak C. J.; Martin-Diaconescu V.; Gamba I.; Luis J. M.; Parella T.; Company A.; Ribas X. Isolation of Key Organometallic Aryl-Co(III) Intermediates in Cobalt-Catalyzed C(sp2)-H Functionalizations and New Insights into Alkyne Annulation Reaction Mechanisms. J. Am. Chem. Soc. 2016, 138, 14388–14397. 10.1021/jacs.6b08593. [DOI] [PubMed] [Google Scholar]
- Planas O.; Whiteoak C. J.; Ribas X.. Recent Advances in Cobalt-Catalyzed Cross-coupling Reactions. In Non-Noble Metal Catalysis; Gebbink R. J. M. K.; Moret M. E., Eds.; Wiley: 2019; pp 297–328. [Google Scholar]
- Planas O.; Chirila P. G.; Whiteoak C. J.; Ribas X.. Chapter Four - Current Mechanistic Understanding of Cobalt-Catalyzed C-H Functionalization. In Adv. Organomet. Chem.; Pérez P. J., Ed.; Academic Press: 2018; Vol. 69, pp 209–282. [Google Scholar]
- Kuijpers P. F.; van der Vlugt J. I.; Schneider S.; de Bruin B. Nitrene Radical Intermediates in Catalytic Synthesis. Chem.—Eur. J. 2017, 23, 13819–13829. 10.1002/chem.201702537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero A.; Pérez P. J. Dimensioning the Term Carbenoid. Chem.—Eur. J. 2017, 23, 14389–14393. 10.1002/chem.201702392. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Sun M.-C.; Yang D.; Li T.; Song M.-P.; Niu J.-L. Cobalt(II)-Catalyzed Activation of C(sp3)-H Bonds: Organic Oxidant Enabled Selective Functionalization. ACS Catal. 2022, 12, 1650–1656. 10.1021/acscatal.1c05250. [DOI] [Google Scholar]
- Ye C.-X.; Shen X.; Chen S.; Meggers E. Stereocontrolled 1,3-nitrogen migration to access chiral α-amino acids. Nat. Chem. 2022, 14, 566–573. 10.1038/s41557-022-00895-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S. Y.; Hwang Y.; Lee M.; Chang S. Mechanism-Guided Development of Transition-Metal-Catalyzed C-N Bond-Forming Reactions Using Dioxazolones as the Versatile Amidating Source. Acc. Chem. Res. 2021, 54, 2683–2700. 10.1021/acs.accounts.1c00198. [DOI] [PubMed] [Google Scholar]
- Lee S.; Rovis T. Rh(III)-Catalyzed Three-Component Syn-Carboamination of Alkenes Using Arylboronic Acids and Dioxazolones. ACS Catal. 2021, 11, 8585–8590. 10.1021/acscatal.1c02406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunny S.; Karvembu R. Recent Advances in Cobalt-Catalyzed, Directing-Group-Assisted C-H Bond Amidation Reactions. Adv. Synth. Catal. 2021, 363, 4309–4331. 10.1002/adsc.202100558. [DOI] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Perdew J. P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. 10.1103/PhysRevB.33.8822. [DOI] [PubMed] [Google Scholar]
- Schäfer A.; Huber C.; Ahlrichs R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. 10.1063/1.467146. [DOI] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Ruzsinszky A.; Csonka G. I.; Constantin L. A.; Sun J. Workhorse Semilocal Density Functional for Condensed Matter Physics and Quantum Chemistry. Phys. Rev. Lett. 2009, 103, 026403. 10.1103/PhysRevLett.103.026403. [DOI] [PubMed] [Google Scholar]
- Luchini G.; Alegre-Requena J.; Funes-Ardoiz I.; Paton R. GoodVibes: automated thermochemistry for heterogeneous computational chemistry data [version 1; peer review: 2 approved with reservations]. F1000Research 2020, 9, 291. 10.12688/f1000research.22758.1. [DOI] [Google Scholar]
- Grimme S. Supramolecular Binding Thermodynamics by Dispersion-Corrected Density Functional Theory. Chem.—Eur. J. 2012, 18, 9955–9964. 10.1002/chem.201200497. [DOI] [PubMed] [Google Scholar]
- Ramos-Cordoba E.; Postils V.; Salvador P. Oxidation States from Wave Function Analysis. J. Chem. Theory Computat. 2015, 11, 1501–1508. 10.1021/ct501088v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.