

Abstract
FAK mediated tumour cell migration, invasion, survival, proliferation and regulation of tumour stem cells through its kinase-dependent enzymatic functions and kinase-independent scaffolding functions. At present, the development of FAK PROTACs has become one of the hotspots in current pharmaceutical research to solve above problems. Herein, we designed and synthesised a series of FAK-targeting PROTACs consisted of PF-562271 derivative 1 and Pomalidomide. All compounds showed significant in vitro FAK kinase inhibitory activity, the IC50 value of the optimised PROTAC A13 was 26.4 nM. Further, A13 exhibited optimal protein degradation (85% degradation at 10 nM). Meantime, compared with PF-562271, PROTAC A13 exhibited better antiproliferative activity and anti-invasion ability in A549 cells. More, A13 had excellent plasma stability with T1/2 >194.8 min. There are various signs that PROTAC A13 could be useful as expand tool for studying functions of FAK in biological system and as potential therapeutic agents.
Keywords: FAK, PROTACs, PF-562271, A549 cells, protein degradation
1. Introduction
Focal adhesion kinase (FAK), also named protein tyrosine kinase 2 (PTK2), was first discovered in 1992 by Schaller et al1. As an intracellular non-receptor tyrosine kinase, FAK can be divided into four parts: the middle kinase catalytic domain, the amino-terminal FERM (4.1-ezrin-radixin-moesin) domain, the carboxy-terminal focal adhesion targeting (FAT) domain and proline-rich regions (PRRs). FAK protein contains at least 6 tyrosine phosphorylation sites (Tyr397, Tyr407, Tyr576, Tyr577, Tyr861, and Tyr925), which are distributed in kinase catalytic domain and other scaffolding domain of FAK. The phosphorylation of these tyrosine residues provided a binding site for recruiting effector protein, playing an pivotal role in regulating the biological functions of FAK2. The kinase domain was mainly responsible for regulating the kinase activity of FAK, while the FERM, FAT and PRR domains regulated protein-protein interactions, followed by the activation of downstream signalling. Hence, FAK exerted catalytic and scaffolding functions, both of them were essential in early embryonic development, reproduction, cancer development, etc3–6. Overexpression of FAK had been detected in multiple tumour types, and biological studies revealed that FAK was essential for the development and progression of various human cancers7–10. Thus, the FAK has emerged as a promising target for anti-cancer therapy.
To date, several potent FAK inhibitors have been developed, and some of them were conducted clinical evaluation. GSK-2256098, VS-4718, PF-562271 and Defactinib are ATP-competitive inhibitors of kinase domain. GSK-2256098 is a highly selective inhibitor of FAK and has been shown to efficiently inhibit the phosphorylation of Tyr39711. VS-4718 is used to treat metastatic non-hematological malignancies, advanced non-hematological malignancies or advanced pancreatic cancer of patients in combination with gemcitabine or nabutaxel12. PF-562271 is a highly active and highly selective FAK inhibitor developed by Pfizer13. Defactinib is evaluated to treat ovarian cancer, pancreatic cancer, non-small cell lung cancer and mesothelioma in combination with checkpoint inhibitors14. As we said above, FAK exerts kinase-dependent enzymatic functions and kinase-independent scaffolding functions. The development of the small-molecule inhibitors can inhibit the enzymatic functions of FAK, but can’t prevent the kinase-independent scaffolding functions. In addition, small molecule drugs are likely to cause off-target toxicity and even drug resistance at high concentrations. In summary, it is necessary to develop a strategy against both the enzymatic functions and scaffolding functions of FAK.
Proteolysis targeting chimaeras (PROTACs) are a class of heterobifunctional molecules containing three moieties: the ligand of the protein of interest (POI), the ligand of E3 ubiquitin ligase (E3 ligase) and the linker15. The PROTAC molecules formed a POI-PROTAC-E3 ligase ternary complexes after entering the cell, resulting in ubiquitination of the target protein and consequently proteasome-mediated degradation16. PROTACs are not required to occupy binding sites for prolonged periods of time during degradation and can function multiple times in the cell, which means that only a catalytic amount of the PROTAC molecule is needed to degrade the target protein. More importantly, PROTAC can inhibit both kinase-dependent enzymatic functions and the essential functions mediated by the FAK scaffolding moieties (Figure 1).
Figure 1.

Structures of representative FAK degraders.
Several targeting-FAK PROTACs have been successfully developed (Figure 1). In 2018, Crew’s group reported PROTAC-3 as a selective and potent Fak degrader, which can potentially induce the degradation of FAK in human prostate cancer cell line PC3 cells with a DC50 value of 3.0 nM17. Using FAK inhibitor BI-4644 and VHL E3 ligase binder, PROTAC BI-3663 was designed by Boehringer Ingelheim, which can effectively degrade FAK protein in 12 cell lines. However, BI-3663 did not display more potent antiproliferative activity than BI-464418. Rao et al. identified that the degradation of FAK induced by FC-11 can be restored if PROTAC molecules were removed19,20. GlaxoSmithKline reported GSK215 as a potent and selective FAK-degrading PROTAC, which induced rapid and prolonged FAK degradation with a single dose (8 mg/kg) in mice21,22. FAK PROTACs with different E3 ligase ligands and different FAK inhibitors are valuable to enhance activities, drug-like properties, and exploration of structure-activity relationships (SARs). Therefore, the research of novel PROTAC molecules targeting FAK kinase still has important academic significance and practical application requirements.
PF-562271, a highly active and highly selective FAK inhibitor, was tolerated well in phase I clinical trials. In this study, we presented a series of FAK PROTACs based on PF-562271 and CRBN E3 ligase ligand Pomalidomide. The co-crystal of PF-562271 and FAK was analysed (PDB ID: 3BZ3), as shown in Figure 2, the 2,4-diaminopyrimidine core formed two donor-acceptor interactions with Cys502 in the hinge region, N-methyl sulphonamide fragment formed a hydrogen bond interaction with Asp564 in the DFG-motif, the trifluoromethyl group contained a hydrophobic interaction with the gatekeeper residue Met499, the lactam fragment extended to the solvent region and could be modified to bind to the linkers.
Figure 2.

Three-dimensional space matching diagram and detailed interactions of PF-562271 in the ATP-binding site of FAK (PDB ID: 3BZ3). The interactions are illustrated with yellow dashed lines.
The lactam fragment of PF-562271 was replaced with a piperazine ring, leaving a binding site for the linkers. At the same time, the pyridine ring was replaced with a benzene ring to obtain compound 1. In vitro enzymatic activity of compound 1 remained comparable to that of PF-562271. A docking study was conducted to analyse the binding mode of compound 1 and FAK. The results of molecular docking were shown in Figure 3(A). The 2,4-diaminopyrimidine core formed two donor-acceptor interactions with Cys502 in the hinge region, N-methyl sulphonamide fragment formed a hydrogen bond interaction with Asp564 in the DFG-motif, the trifluoromethyl group contained a hydrophobic interaction with the gatekeeper residue Met499, the piperazine ring of compound 1 extended to the solvent region to connect with the linkers. Pomalidomide was an inhibitor of CRBN developed by Celgene for the treatment of multiple myeloma, and had been widely used in the design of PROTACs because of low molecular weight. The co-crystal of pomalidomide (PDB ID: 4CI3) with CRBN protein was analysed, as shown in Figure 3(A), piperidine-2,6-dione formed three donor-acceptor interactions with His380 and Trp382, the amino-substituted benzene ring moiety was exposed in the solvent region, serving as the attachment site for the linkers. Based on the above strategies, we designed 15 FAK-targeting PROTACs based on PF-562271 and Pomalidomide, and alkyl-chains or PEG-chains of different lengths and compositions as the linkers (Figure 3(B)). Further, the synthesis and biological evaluation of the designed FAK PROTACs were exhibited in this paper.
Figure 3.

Design of FAK-targeting PROTACs. (A) Molecular docking model of compound 1 with FAK protein (PDB ID: 3BZ3) and the co-crystal binding modes of Pomalidomide (PDB ID: 4CI3). Piperazine fragment of 1 with CRBN E3 ligase ligand to develop FAK PROTACs by linkers. (B) FAK-targeting PROTACs including three parts: FAK Ligand, Linker, and CRBN Ligand.
2. Experimental section
The starting materials, reagents and solvents were commercially available products without further purification unless otherwise. The organic solvents used are purified and preserved by conventional methods. The reaction was monitored by thin-layer chromatography (TLC) on HSGF-254 (10–40 μm) silica gel plates and visualised with UV light. Column chromatography was performed on silica gel (200–300 mesh ASTM). Nuclear magnetic resonance (NMR) data were recorded in DMSO-d6 or CDCl3 on Bruker ARX-600 NMR or Bruker ARX-400 NMR spectrometers with TMS as an internal standard. LC-MS analysis was performed using an Agilent 1200 liquid phase mass spectrometer (ESI mode). High-resolution accurate mass spectrometry (HRMS) determinations for all final target compounds were obtained on a Bruker micromass time of flight mass spectrometer equipped with an electrospray ionisation (ESI) detector.
2.1. Synthesis
2.1.1. 2–(2,6-Dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione (a2)
4-Fluoroisobenzofuran-1,3-dione (1.0 equiv) was added to a stirred solution of 3-aminopiperidine-2,6-dione (1.1 equiv) and sodium acetate (1.3 equiv) in AcOH. The resulted mixture was heated to 140 °C and stirred for 8 h before being cooled to room temperature. The acetic acid was directly removed under vacuum, and the residue was purified by column chromatography (5% petroleum ether/ethyl acetate) to afford intermediate a2. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 7.97 − 7.92 (m, 1H), 7.79 (d, J = 7.3 Hz, 1H), 7.73 (t, J = 8.9 Hz, 1H), 5.16 (dd, J = 12.8, 5.4 Hz, 1H), 2.89 (ddd, J = 17.1, 13.9, 5.5 Hz, 1H), 2.66 − 2.51 (m, 2H), 2.11 − 2.02 (m, 1H). MS (ESI) m/z (%): 277.1 [M + H]+.
2.1.2. Tert-Butyl [2–(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl]glycinate (a3)
To a solution of intermediate a2 (1.0 equiv) in DMF, tert-butylglycine (1.2 equiv) and DIPEA (1.3 equiv) were added. The mixtures were heated to 90 °C for 5 h. After being cooled to room temperature, the solution was quenched with water and extracted with CH2Cl2. The organic layer was dried over Na2SO4 and filtered. The filtrate was concentrated and purified by column chromatography (20% petroleum ether/ethyl acetate) to obtain intermediate a3. 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.51 (dd, J = 8.4, 7.2 Hz, 1H), 7.15 (d, J = 6.8 Hz, 1H), 6.76 (d, J = 8.4 Hz, 1H), 6.71 (s, 1H), 4.96 − 4.90 (m, 1H), 3.94 (s, 2H), 2.89 − 2.74 (m, 3H), 2.14 − 2.10 (m, 1H), 1.50 (s, 9H). MS (ESI) m/z (%): 410.2 [M + Na]+.
2.1.3. Synthesis of intermediates a4 and a6
TFA (2 ml) was added to a solution of intermediates a3 and a5 in CH2Cl2 (10 mL), respectively, the reaction was stirred at 25 °C for 8 h. The solvent was evaporated to dryness to obtain intermediates a4 and a6, respectively.
2.1.4. Synthesis of intermediates a7–a11
To a solution of intermediate a6 (1.0 equiv) in CH2Cl2, bromine substituted carboxylic acid chain (1.1 equiv), HATU (1.2 equiv) and DIPEA (1.5 equiv) were added. The mixtures were stirred at 25 °C. After the reaction was completed, the solution was diluted with CH2Cl2, washed with H2O and brine, and dried over Na2SO4 overnight. The organic phase was concentrated under vacuum and purified through column chromatography (20% petroleum ether/ethyl acetate) to obtain intermediates a7–a11.
2.1.5. Synthesis of intermediates a13–a14
TEA (1.3 equiv) and p-toluenesulfonyl chloride (1.1 equiv) were added to a solution of intermediate a11–a12 (1.0 equiv) in CH2Cl2. The mixtures were stirred at 30 °C until completion of the reaction. The mixture was diluted with CH2Cl2, the organic layer was washed with brine, dried over Na2SO4 and filtered. The filtrate was concentrated and purified by column chromatography (20% petroleum ether/ethyl acetate) to give intermediates a13–a14.
2.1.6. Synthesis of intermediates a16–a21
SOCl2 (5 mL) was added to the corresponding bromocarboxylic acid chain (1.2 equiv), and the mixture was stirred at 80 °C for 5 h. After being cooled to room temperature, the solvent was evaporated to dryness. The residue was dissolved with dry THF, to the solution was added with commercially available a15 (1.0 equiv). The mixture was stirred at 50 °C until completion of the reaction. After cooling to room temperature, the crude product of intermediates a16–a21 can be obtained by filtered.
2.1.7. 2-[2–(2-Azidoethoxy)ethoxy]ethan-1-ol (a23)
2-[2–(2-Chloroethoxy)ethoxy]ethan-1-ol (1.0 equiv) and sodium azide (2.0 equiv) were added to DMF and reacted at 100 °C 12 h. After cooling to room temperature, the reaction was quenched with H2O and extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4 and filtered. The organic phase was concentrated under vacuum and purified by column chromatography (1% Methanol/Dichloromethane) to obtain intermediate a23. MS (ESI) m/z (%): 176.3 [M + H]+.
2.1.8. 1-Azido-2-[2–(2-bromoethoxy)ethoxy]ethane (a24)
Intermediate a23 (1.0 equiv), carbon tetrabromide (1.2 equiv), triphenylphosphine (1.2 equiv) were added to CH2Cl2, and reacted at 25 °C for 12 h. The reaction was quenched with H2O and extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4 and filtered. The organic phase was concentrated under vacuum and purified by column chromatography (15% petroleum ether/ethyl acetate) to obtain intermediate a24. MS (ESI) m/z (%): 238.1 [M + H]+.
2.1.9. Tert-Butyl 3–(3-hydroxypropoxy)propanoate (a26)
tert-Butyl acrylate (1.0 equiv), 1,3-propanediol (5.0 equiv) and 40% benzyltrimethylammonium hydroxide (0.1 equiv) were added to acetonitrile, and the mixtures were heated to 25 °C and stirred for 3 d. The reaction was quenched with H2O and extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4 and concentrated under vacuum. The residue was purified by column chromatography (1% Methanol/Dichloromethane) to obtain intermediate a26. MS (ESI) m/z (%): 227.0 [M + Na]+.
2.1.10. Tert-Butyl 3–(3-iodopropoxy)propanoate (a27)
Triphenylphosphine (1.2 equiv), imidazole (1.2 equiv) and iodine (1.5 equiv) were dissolved in dry THF. Intermediate a26 (1.0 equiv) in THF was added dropwise to the reaction system, and the reaction was stirred at 25 °C for 3 h under Argon. The mixture was filtered to remove the white precipitate. The filtrate was concentrated under vacuum and purified by column chromatography (15% petroleum ether/ethyl acetate) to obtain intermediate a27.
2.1.11. N-[2-(aminomethyl)phenyl]-N-methylmethanesulfonamide (a30)
2-Fluoro-benzonitrile (1.0 equiv), N-methyl-methanesulfonamide (1.0 equiv) and caesium carbonate (1.2 equiv) were added to acetonitrile and the mixture was stirred at 80 °C for 16 h. After cooling to room temperature, the mixture was filtered. The filtrate was concentrated to obtain the crude product of intermediate a29. A 2 M solution of BH3 in THF (1.5 equiv) was added to a solution of intermediate a29 (1.0 equiv) in THF. The reaction was carried out at 60 °C for 12 h under Argon. And H2O was added to quench the reaction. The mixture was extracted with CH2Cl2, the organic layer was washed with brine and dried over Na2SO4. The organic phase was concentrated and purified by column chromatography (1% Methanol/Dichloromethane) to obtain intermediate a30.
2.1.12. Tert-butyl 4–(4-{[4-chloro-5-(trifluoromethyl)pyrimidin-2-yl]amino}phenyl)piperazine-1-carboxylate (a32)
2,4-Dichloro-5-(trifluoromethyl)pyrimidine (1.0 equiv) was added to a mixed solvent of tert-butanol and 1,2-dichloroethane, and zinc bromide (1.2 equiv) was added after cooling to 0 °C. The mixture was stirred for 30 min. Then, tert-butyl 4–(4-aminophenyl)piperazine-1-carboxylate (1.0 equiv) and TEA (2.0 equiv) were added, stirring at 0 °C for 3 h. The reaction was quenched with H2O and extracted with CH2Cl2. The organic layer was washed with brine and dried over Na2SO4. The organic layer was concentrated under reduced pressure and purified by column chromatography (20% petroleum ether/ethyl acetate) to give intermediate a32. 1H NMR (400 MHz, CDCl3) δ 8.51 (s, 1H), 7.54 (s, 1H), 7.44 (d, J = 8.8 Hz, 2H), 6.94 (d, J = 8.7 Hz, 2H), 3.64 − 3.55 (m, 4H), 3.16 − 3.08 (m, 4H), 1.49 (s, 10H).
2.1.13. Tert-butyl 4-{4-[(4-{[2-(N-methylmethylsulfonamido)benzyl]amino}-5-(trifluoromethyl)pyrimidin-2-yl)amino]phenyl}piperazine-1-carboxylate (a33)
Intermediate a32 (1.0 equiv), intermediate a30 (1.0 equiv) and DIPEA (1.0 equiv) were added to 1,4-dioxane, the mixture was heated to 100 °C for 6 h. After cooling to room temperature, the reaction was quenched with H2O, extracted with EtOAc, the organic layer was washed with brine and dried over Na2SO4. The organic phase was concentrated under reduced pressure and purified by column chromatography (1% Methanol/Dichloromethane) to obtain intermediate a33. TFA (2 mL) was added to a solution of intermediate a33 in CH2Cl2 (10 mL), the mixture was stirred at 25 °C for 5 h. The solvent was removed under reduced pressure, the residue was dissolved with saturated aqueous NaHCO3 and extracted with CH2Cl2. The organic layer was concentrated under reduced pressure to give compound 1.
2.1.14. N-Methyl-N-(2-{[(2-{[4-(piperazin-1-yl)phenyl]amino}-5-(trifluoromethyl)pyrimidin-4-yl)amino]methyl}phenyl)methanesulfonamide (1)
TFA (2 mL) was added to a solution of intermediate a33 in CH2Cl2 (10 mL), the mixture was stirred at 25 °C for 5 h. The solvent was removed under reduced pressure, the residue was dissolved with saturated aqueous NaHCO3 and extracted with CH2Cl2. The organic layer was concentrated under reduced pressure to give compound 1.
2.1.15. N-(2-{[(2-{[4–(4-{2-[2–(2-azidoethoxy)ethoxy]ethyl}piperazin-1-yl)phenyl]amino}-5-(trifluoromethyl)pyrimidin-4-yl)amino]methyl}phenyl)-N-methylmethanesulfonamide (a34)
Compound 1 (1.0 equiv) and intermediate a24 (0.12 equiv) were dissolved in acetonitrile, potassium carbonate (1.6 equiv) was added, and the mixture was heated to 80 °C and stirred for 6 h. After cooling to room temperature, H2O was added to the solution and extracted with CH2Cl2. The organic phase was washed with brine, dried over Na2SO4 overnight. The organic layer was concentrated under reduced pressure and purified by column chromatography (1% Methanol/Dichloromethane) to acquire intermediate a34. 1H NMR (400 MHz, CDCl3) δ 8.13 (s, 1H), 7.54 − 7.44 (m, 2H), 7.40 (d, J = 9.0 Hz, 2H), 7.37 − 7.30 (m, 2H), 7.28 − 7.26 (m, 1H), 6.87 (d, J = 9.0 Hz, 2H), 5.98 (s, 1H), 5.13 (s, 1H), 4.65 (s, 1H), 3.72 − 3.64 (m, 8H), 3.40 − 3.36 (m, 2H), 3.24 (s, 3H), 3.20 − 3.16 (m, 4H), 2.97 (s, 3H), 2.77 − 2.69 (m, 6H).
2.1.16. N-(2-{[(2-{[4–(4-{2-[2–(2-aminoethoxy)ethoxy]ethyl}piperazin-1-yl)phenyl]amino}-5-(trifluoromethyl)pyrimidin-4-yl)amino]methyl}phenyl)-N-methylmethanesulfonamide (a35)
Intermediate a34 (1.0 equiv) and triphenylphosphine (3.0 equiv) were added to a mixed solvent of THF (10 ml) and H2O (3 mL), and the mixture was stirred at 80 °C until completion of the reaction. The solution was concentrated under reduced pressure and extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4 and filtered. The filtrate was concentrated under reduced pressure to give intermediate a35.
2.1.17. Synthesis of compounds A1-A2
Intermediate a4 (1.1 equiv), HATU (1.3 equiv) and DIPEA (1.5 equiv) were added to a solution of intermediate a35 (1.0 equiv) in CH2Cl2, the mixture was stirred at 25 °C until completion of the reaction. The solution was diluted with H2O and extracted with CH2Cl2, the organic layer with brine, dried over Na2SO4 and filtered. The organic layer was concentrated under reduced pressure and purified by column chromatography (6% Methanol/Dichloromethane) to obtain compound A1.
Intermediate a6 (1.1 equiv), HATU (1.3 equiv) and DIPEA (1.5 equiv) were added to a solution of intermediate a37 (1.0 equiv) in CH2Cl2, the mixture was stirred at 25 °C until completion of the reaction. The solution was diluted with H2O and extracted with CH2Cl2, the organic layer with brine, dried over Na2SO4 and filtered. The organic layer was concentrated under reduced pressure and purified by column chromatography (6% Methanol/Dichloromethane) to obtain compound A2.
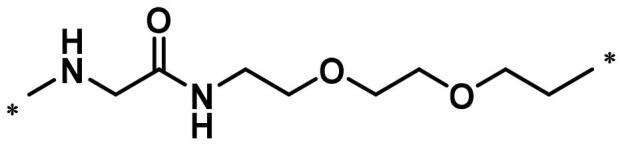
2.1.17.1. 2-{[2–(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl]amino}-N-(2-{2-[2–(4-{4-[(4-{[2-(N-methylmethylsulfonamido)benzyl]amino}-5-(trifluoromethyl)pyrimidin-2-yl)amino]phenyl}piperazin-1-yl)ethoxy]ethoxy}ethyl)acetamide (A1)
Yellow solid; yield: 36.8%; 1H NMR (600 MHz, CDCl3) δ 10.00 (s, 1H), 8.14 (s, 1H), 7.51 − 7.48 (m, 1H), 7.44 (d, J = 5.8 Hz, 1H), 7.39 (d, J = 8.7 Hz, 2H), 7.35 − 7.27 (m, 3H), 7.18 (d, J = 7.1 Hz, 1H), 6.97 (s, 1H), 6.84 (d, J = 8.7 Hz, 2H), 6.80 (d, J = 8.5 Hz, 1H), 6.70 (t, J = 5.7 Hz, 1H), 5.96 (s, 1H), 5.13 (s, 1H), 4.90 (dd, J = 11.4, 4.4 Hz, 1H), 4.66 (s, 1H), 3.94 (d, J = 5.8 Hz, 2H), 3.62 − 3.52 (m, 9H), 3.45 − 3.42 (m, 1H), 3.24 (s, 3H), 3.15 (s, 4H), 2.97 (s, 4H), 2.86 − 2.77 (m, 2H), 2.72 − 2.64 (m, 6H), 2.11 (dd, J = 8.8, 3.9 Hz, 1H). 13 C NMR (150 MHz, DMSO-d6) δ 172.81, 170.05, 168.70, 168.58, 167.31, 160.93, 158.34, 154.70, 146.25, 145.80, 139.57, 139.23, 136.22, 132.05, 131.79, 128.37, 127.83, 127.56, 127.07, 125.20 (q, J = 269.2 Hz), 120.75 (2 C), 117.46, 115.68 (2 C), 110.98, 109.86, 69.62, 69.52, 68.94, 67.98, 57.03, 52.99 (2 C), 48.57 (3 C), 45.17, 38.66, 38.57, 36.00, 30.98, 22.16. HRMS calcd for C45H52F3N11O9S, [M + H]+, 980.3695; found 980.3712.
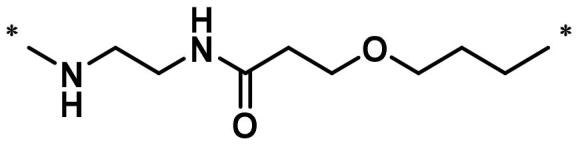
2.1.17.2. N-(2-{[2–(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl]amino}ethyl)-3-[3–(4-{4-[(4-{[2-(N-methylmethylsulfonamido)benzyl]amino}-5-(trifluoromethyl)pyrimidin-2-yl)amino]phenyl}piperazin-1-yl)propoxy]propanamide (A2)
Yellow solid; yield: 31.7%; 1H NMR (600 MHz, CDCl3) δ 10.76 (s, 1H), 8.14 (s, 1H), 7.72 (s, 1H), 7.50 − 7.46 (m, 1H), 7.45 − 7.41 (m, 3H), 7.34 − 7.27 (m, 3H), 7.08 (d, J = 7.1 Hz, 1H), 6.97 (d, J = 8.6 Hz, 1H), 6.82 (d, J = 8.7 Hz, 3H), 6.41 (t, J = 4.9 Hz, 1H), 5.99 (s, 1H), 5.12 (s, 1H), 4.88 (dd, J = 12.0, 5.4 Hz, 1H), 4.65 (d, J = 11.3 Hz, 1H), 3.66 (t, J = 5.6 Hz, 2H), 3.51 − 3.44 (m, 6H), 3.22 (s, 3H), 3.15 (s, 4H), 2.96 (s, 3H), 2.80 − 2.65 (m, 7H), 2.50 − 2.46 (m, 4H), 2.09 (dd, J = 7.8, 5.3 Hz, 1H), 1.82 − 1.78 (m, 2H). 13 C NMR (150 MHz, CDCl3) δ 172.53, 172.47, 169.60, 169.56, 167.70, 161.05, 158.88, 154.69, 147.17, 146.82, 139.93, 139.11, 136.36, 132.60, 132.01, 130.67, 129.32, 128.87, 126.42, 125.00 (q, J = 269.3 Hz), 121.89 (2 C), 116.92 (2 C), 116.88, 111.94, 110.40, 69.42, 66.88, 55.33, 52.95 (2 C), 49.23 (2 C), 49.00, 42.17, 40.65, 39.32, 39.02, 37.11, 35.63, 31.56, 26.37, 22.98. HRMS calcd for C45H52F3N11O8S, [M + H]+, 964.3746; found 964.3788.
2.1.18. Synthesis of compounds A3-A15
Potassium iodide (1.1 equiv) and potassium carbonate (1.3 equiv) were added to a stirred solution of compound 1 (1.0 equiv) and the corresponding int ermediates a13–a14, a16–a21 and a7–a11 (1.1 equiv) in acetonitrile. The mixture was stirred at 80 °C until completion of the reaction. The solution was diluted with H2O and extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4 and filtered. The filtrate was concentrated under reduced pressure and purified by column chromatography (6% Methanol/Dichloromethane) to obtain compounds A3–A4, A5–A10 and A11–A15.
2.1.18.1. N-(2-{[(2-{[4–(4-{2-[2–(2-{[2–(2,6-Dioxopiperidin-3-yl)]-1,3- Dioxyisoindolin-4-acyl}amino)ethoxy]ethoxy}ethylpiperazin-1-yl)phenyl]amino}-5-(trifluoromethyl)pyrimidin-4-yl)amino]methyl}phenyl)-N-methylmethanesulfonamide (A3)
Yellow solid; yield: 37.7%; 1H NMR (600 MHz, CDCl3) δ 10.40 (s, 1H), 8.15 (s, 1H), 7.71 (s, 1H), 7.49 − 7.45 (m, 1H), 7.44 (d, J = 6.6 Hz, 1H), 7.40 (d, J = 9.0 Hz, 2H), 7.34 − 7.27 (m, 3H), 7.09 (d, J = 7.1 Hz, 1H), 6.90 (d, J = 8.6 Hz, 1H), 6.85 (d, J = 8.9 Hz, 2H), 6.52 (t, J = 5.5 Hz, 1H), 6.00 (s, 1H), 5.13 (s, 1H), 4.87 (dd, J = 11.5, 4.1 Hz, 1H), 4.64 (d, J = 11.5 Hz, 1H), 3.73 − 3.69 (m, 4H), 3.67 − 3.64 (m, 4H), 3.47 − 3.44 (m, 2H), 3.22 (s, 3H), 3.17 (s, 4H), 2.96 (s, 3H), 2.80 − 2.65 (m, 9H), 2.10 − 2.06 (m, 1H). 13 C NMR (150 MHz, CDCl3) δ 172.28, 169.44, 169.30, 167.75, 161.07, 158.89, 154.69, 147.33, 146.89, 139.91, 139.14, 136.14, 132.66, 132.01, 130.70, 129.31, 128.84, 126.41, 125.00 (q, J = 269.3 Hz), 121.98 (2 C), 116.99 (2 C), 116.83, 111.75, 110.48, 70.78, 70.48, 69.51, 68.77, 57.70, 53.45 (2 C), 49.54 (2 C), 48.96, 42.48, 40.67, 39.30, 35.64, 31.50, 23.00. HRMS calcd for C43H49F3N10O8S, [M + H]+, 923.3480; found 923.3513.
2.1.18.2. N-{2-[({[2-({4-[4–(2-{2-[2–(2-{[2–(2,6-Dioxopiperidin-3-yl)]- 1,3-Dioxyisoindoline-4-acyl}amino)ethoxy]ethoxy}ethoxy)ethylpiperazin-1-yl]phenyl}amino)-5-(trifluoromethyl) yl]pyrimidin-4-yl}amino)methyl]phenyl}-N-methylmethanesulfonamide (A4)
Yellow solid; yield: 29.6%; 1H NMR (600 MHz, CDCl3) δ 10.79 (s, 1H), 8.15 (s, 1H), 7.61 (s, 1H), 7.49 − 7.43 (m, 2H), 7.39 (d, J = 8.9 Hz, 2H), 7.35 − 7.27 (m, 3H), 7.09 (d, J = 7.1 Hz, 1H), 6.89 (d, J = 8.5 Hz, 1H), 6.83 (d, J = 8.9 Hz, 2H), 6.50 (t, J = 5.4 Hz, 1H), 5.99 (s, 1H), 5.13 (s, 1H), 4.88 (dd, J = 12.3, 5.4 Hz, 1H), 4.64 (d, J = 11.8 Hz, 1H), 3.72 − 3.64 (m, 12H), 3.46 − 3.42 (m, 2H), 3.23 (s, 3H), 3.16 (s, 4H), 2.96 (s, 3H), 2.84 − 2.67 (m, 9H), 2.12 − 2.08 (m, 1H). 13 C NMR (150 MHz, CDCl3) δ 172.27, 169.46, 169.44, 167.80, 161.13, 158.90, 154.79, 147.45, 146.88, 139.93, 139.17, 136.12, 132.69, 131.82, 130.71, 129.32, 128.85, 126.42, 125.02 (q, J = 269.7 Hz), 121.99 (2 C), 116.85 (2 C), 116.81, 111.73, 110.49, 70.96, 70.60 (2 C), 70.43, 69.44, 68.48, 57.53, 53.44 (2 C), 49.40 (2 C), 49.02, 42.43, 40.65, 39.32, 35.66, 31.64, 23.05. HRMS calcd for C45H53F3N10O9S, [M + H]+, 967.3743; found 967.3778.
2.1.18.3. N-[2–(2,6-Dioxopiperidin-3-yl)1,3-dioxoisoindol-4-yl]-5–(4-{4-[(4-{[2 -(N-Methylmethylsulfonamido)benzyl]amino)}5-(trifluoromethyl)pyrimidin-2-yl)amino]phenyl}piperazin-1-yl)pentanamide (A5)
Yellow solid; yield: 32.5%; 1H NMR (600 MHz, CDCl3) δ 10.04 (s, 1H), 9.43 (s, 1H), 8.81 (d, J = 8.5 Hz, 1H), 8.15 (s, 1H), 7.83 − 7.68 (m, 2H), 7.54 (d, J = 7.3 Hz, 1H), 7.44 (d, J = 7.3 Hz, 1H), 7.41 (d, J = 8.8 Hz, 2H), 7.35 − 7.27 (m, 3H), 6.85 (d, J = 8.8 Hz, 2H), 6.00 (s, 1H), 5.14 (s, 1H), 4.92 (dd, J = 12.4, 5.3 Hz, 1H), 4.64 (d, J = 12.3 Hz, 1H), 3.23 (s, 3H), 3.14 (s, 4H), 2.96 (s, 3H), 2.87 (d, J = 15.8 Hz, 1H), 2.79 − 2.71 (m, 2H), 2.61 (s, 4H), 2.50 (t, J = 7.3 Hz, 2H), 2.47 − 2.44 (m, 2H), 2.13 − 2.10 (m, 1H), 1.82 − 1.77 (m, 2H), 1.66 − 1.60 (m, 2H). 13 C NMR (150 MHz, CDCl3) δ 172.25, 171.83, 169.34, 168.67, 166.84, 161.06, 158.91, 154.67, 147.57, 139.94, 139.17, 137.92, 136.55, 131.79, 131.25, 130.77, 129.33, 128.87, 126.40, 125.37, 125.00 (q, J = 269.4 Hz), 121.95 (2 C), 118.59, 116.82 (2 C), 115.45, 58.01, 53.23 (2 C), 49.70 (2 C), 49.39, 40.68, 39.34, 37.78, 35.65, 31.51, 26.10, 23.30, 22.82. HRMS calcd for C42H45F3N10O7S, [M + H]+, 891.3218; found 891.3225.
2.1.18.4. N-(2–(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindol-4-yl)-6–(4-{4-[(4- {[2-(N-Methylmethylsulfonamido)benzyl]amino}-5-(trifluoromethyl)pyrimidin-2-yl)amino]phenyl}piperazin-1-yl)hexanamide (A6)
Yellow solid; yield: 41.9%; 1H NMR (600 MHz, CDCl3) δ 9.90 (s, 1H), 9.43 (s, 1H), 8.81 (d, J = 8.5 Hz, 1H), 8.15 (s, 1H), 7.78 − 7.64 (m, 2H), 7.54 (d, J = 7.2 Hz, 1H), 7.45 − 7.41 (m, 3H), 7.36 − 7.27 (m, 3H), 6.86 (d, J = 8.8 Hz, 2H), 6.01 (s, 1H), 5.13 (s, 1H), 4.94 (dd, J = 12.3, 5.3 Hz, 1H), 4.65 (d, J = 11.6 Hz, 1H), 3.24 (s, 3H), 3.21 (s, 4H), 2.97 (s, 3H), 2.89 (d, J = 15.7 Hz, 1H), 2.81 − 2.70 (m, 6H), 2.53 − 2.46 (m, 4H), 2.1 − 2.14 (m, 1H), 1.82 − 1.76 (m, 2H), 1.67 − 1.61 (m, 2H), 1.46 − 1.41 (m, 2H). 13 C NMR (150 MHz, CDCl3) δ 172.24, 171.69, 169.35, 168.60, 166.83, 161.02, 158.91, 154.64, 147.22, 139.95, 139.15, 137.91, 136.57, 132.11, 131.25, 130.74, 129.35, 128.89, 126.41, 125.38, 124.98 (q, J = 269.6 Hz), 121.94 (2 C), 118.61, 117.12 (2 C), 115.48, 58.19, 53.02 (2 C), 49.40 (2 C), 49.37, 40.69, 39.35, 37.87, 35.65, 31.51, 26.98, 25.90, 25.11, 22.88. HRMS calcd for C43H47F3N10O7S, [M + H]+, 905.3375; found 905.3394.
2.1.18.5. N-[2–(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindol-4-yl]-7–(4-{4-[(4-{[2-(N-Methylmethylsulfonamido)benzyl]amino}-5-(trifluoromethyl)pyrimidin-2-yl)amino]phenyl}piperazin-1-yl)heptamide (A7)
Yellow solid; yield: 36.1%; 1H NMR (600 MHz, CDCl3) δ 10.10 (s, 1H), 9.44 (s, 1H), 8.80 (d, J = 8.5 Hz, 1H), 8.14 (s, 1H), 7.70 (t, J = 7.9 Hz, 1H), 7.54 (d, J = 7.3 Hz, 1H), 7.44 − 7.40 (m, 3H), 7.35 − 7.27 (m, 3H), 6.87 (d, J = 8.7 Hz, 2H), 6.00 (s, 1H), 5.13 (s, 1H), 4.93 (s, 1H), 4.65 (d, J = 11.3 Hz, 1H), 3.23 (s, 3H), 3.18 (s, 4H), 2.97 (s, 3H), 2.88 (d, J = 15.6 Hz, 1H), 2.79 − 2.72 (m, 2H), 2.68 (s, 4H), 2.46 (t, J = 7.1 Hz, 4H), 2.17 − 2.14 (m, 1H), 1.79 − 1.74 (m, 2H), 1.60 − 1.57 (m, 2H), 1.44 − 1.38 (m, 4H). 13 C NMR (150 MHz, CDCl3) δ 172.41, 171.72, 169.41, 168.70, 166.84, 161.12, 158.90, 154.77, 147.40, 139.95, 139.17, 137.93, 136.56, 131.97, 131.27, 130.76, 129.34, 128.87, 126.42, 125.40, 125.00 (q, J = 269.4 Hz), 122.04 (2 C), 118.59, 117.04 (2 C), 115.48, 58.50, 53.04 (2 C), 49.55 (2 C), 49.37, 40.68, 39.34, 38.01, 35.66, 31.48, 28.99, 27.23, 26.12, 25.28, 22.92. HRMS calcd for C44H49F3N10O7S, [M + H]+, 919.3531; found 919.3543.
2.1.18.6. N-[2–(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindol-4-yl]-8–(4-{4-[(4-{[2-(N-Methylmethylsulfonamido)benzyl]amino}-5-(trifluoromethyl)pyrimidin-2-yl)amino]phenyl}piperazin-1-yl)octamide (A8)
Yellow solid; yield: 38.2%; 1H NMR (600 MHz, CDCl3) δ 10.73 (s, 1H), 9.44 (s, 1H), 8.79 (d, J = 8.5 Hz, 1H), 8.13 (s, 1H), 7.98 (s, 1H), 7.69 (t, J = 7.9 Hz, 1H), 7.53 (d, J = 7.3 Hz, 1H), 7.45 − 7.41 (m, 3H), 7.35 − 7.27 (m, 3H), 6.84 (d, J = 8.8 Hz, 2H), 6.04 (s, 1H), 5.12 (s, 1H), 4.93 (dd, J = 11.9, 5.4 Hz, 1H), 4.64 (d, J = 11.2 Hz, 1H), 3.28 − 3.20 (m, 7H), 2.96 (s, 3H), 2.87 (d, J = 13.6 Hz, 1H), 2.76 − 2.72 (m, 6H), 2.53 − 2.49 (m, 2H), 2.44 (t, J = 7.3 Hz, 2H), 2.17 − 2.15 (m, 1H), 1.74 (dt, J = 13.3, 6.7 Hz, 2H), 1.62 − 1.58 (m, 2H), 1.41 − 1.30 (m, 6H). 13 C NMR (150 MHz, CDCl3) δ 172.49, 172.06, 169.40, 168.94, 166.86, 160.88, 158.90, 154.36, 147.09 (2 C), 139.92, 139.07, 137.88, 136.52, 132.16, 131.27, 130.64, 129.33, 128.88, 126.43, 125.36, 124.92 (q, J = 269.4 Hz), 121.89 (2 C), 118.55 (2 C), 117.13, 115.49, 58.47, 52.85 (2 C), 49.39, 49.04 (2 C), 40.71, 39.34, 37.90, 35.65, 31.52, 29.14, 28.91, 27.45, 25.87, 25.34, 22.93. HRMS calcd for C45H51F3N10O7S, [M + H]+, 933.3688; found 933.3702.
2.1.18.7. N-[2–(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindol-4-yl]-9–(4-{4-[(4-{[2-(N-methylmethylsulfonamido)benzyl]amino}-5-(trifluoromethyl)pyrimidin-2-yl)amino]phenyl}piperazin-1-yl)nonanamide (A9)
Yellow solid; yield: 29.7%; 1H NMR (600 MHz, CDCl3) δ 10.97 (s, 1H), 9.47 (s, 1H), 8.80 (d, J = 8.5 Hz, 1H), 8.14 (s, 1H), 7.77 − 7.64 (m, 2H), 7.53 (d, J = 7.3 Hz, 1H), 7.45 − 7.40 (m, 3H), 7.35 − 7.26 (m, 3H), 6.84 (d, J = 8.9 Hz, 2H), 6.02 (s, 1H), 5.12 (s, 1H), 4.93 (dd, J = 12.2, 5.4 Hz, 1H), 4.64 (d, J = 11.6 Hz, 1H), 3.25 − 3.21 (m, 7H), 2.96 (s, 3H), 2.86 (dd, J = 13.2, 2.4 Hz, 1H), 2.80 − 2.71 (m, 6H), 2.52 (s, 2H), 2.45 (dd, J = 14.3, 7.3 Hz, 2H), 2.17 − 2.12 (m, 1H), 1.77 − 1.71 (m, 2H), 1.63 − 1.58 (m, 2H), 1.40 − 1.30 (m, 8H). 13 C NMR (150 MHz, CDCl3) δ 172.51, 172.17, 169.36, 168.99, 166.88, 161.02, 158.89, 154.65, 147.06, 139.91, 139.10, 137.91, 136.49, 132.15, 131.25, 130.61, 129.31, 128.85, 126.42, 125.30, 124.97 (q, J = 270.5 Hz), 121.94 (2 C), 118.49, 117.12 (2 C), 115.46, 58.39, 52.78 (2 C), 49.37, 49.05 (2 C), 40.67, 39.31, 38.04, 35.64, 31.55, 29.17, 29.06, 28.91, 27.38, 25.84, 25.24, 22.89. HRMS calcd for C46H53F3N10O7S, [M + H]+, 947.3844; found 947.3866.
2.1.18.8. N-[2–(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindol-4-yl]-10–(4-{4-[(4-{[2-(N-methylmethylsulfonamido)benzyl]amino}-5-(trifluoromethyl)pyrimidin-2-yl)amino]phenyl}piperazin-1-yl)decanoamide (A10)
Yellow solid; yield: 39.1%; 1H NMR (600 MHz, CDCl3) δ 11.02 (s, 1H), 9.44 (s, 1H), 8.81 (d, J = 8.3 Hz, 1H), 8.14 (s, 1H), 7.76 (s, 1H), 7.69 (t, J = 7.7 Hz, 1H), 7.53 (d, J = 7.0 Hz, 1H), 7.44 − 7.39 (m, 3H), 7.34 − 7.27 (m, 3H), 6.84 (d, J = 8.1 Hz, 2H), 6.01 (s, 1H), 5.13 (s, 1H), 4.93 (dd, J = 11.8, 4.8 Hz, 1H), 4.64 (d, J = 8.0 Hz, 1H), 3.23 (s, 3H), 3.18 (s, 4H), 2.96 (s, 3H), 2.87 (d, J = 15.6 Hz, 1H), 2.81 − 2.69 (m, 6H), 2.46 − 2.43 (m, 4H), 2.16 − 2.13 (m, 1H), 1.76 − 1.72 (m, 2H), 1.55 (s, 2H), 1.40 − 1.30 (m, 10H). 13 C NMR (150 MHz, CDCl3) δ 172.58, 172.18, 169.35, 168.92, 166.90, 161.03, 158.90, 154.61, 147.34 (2 C), 139.92, 139.12, 137.93, 136.48, 131.89, 131.24, 130.69, 129.32, 128.85, 126.42, 125.32, 124.97 (q, J = 269.5 Hz), 121.94 (2 C), 118.48, 116.90 (2 C), 115.45, 58.65, 52.97 (2 C), 49.41, 49.30 (2 C), 40.67, 39.32, 38.15, 35.65, 31.61, 29.55, 29.21, 29.17, 29.02, 27.61, 26.21, 25.33, 22.86. HRMS calcd for C47H55F3N10O7S, [M + H]+, 961.4001; found 961.4022.
2.1.18.9. N-(2-{[2–(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl]amino}ethyl)-5–(4 –{4-[(4-{[2-(N-Methylmethylsulfonamido)benzyl]amino}-5-(trifluoromethyl)pyrimidin-2-yl)amino]phenyl}piperazine -1-yl)pentanamide (A11)
Yellow solid; yield: 40.3%; 1H NMR (600 MHz, CDCl3) δ 10.57 (s, 1H), 8.14 (s, 1H), 7.78 (s, 1H), 7.46 (dd, J = 17.1, 9.0 Hz, 2H), 7.41 (d, J = 8.6 Hz, 2H), 7.34 − 7.26 (m, 3H), 7.07 (d, J = 7.1 Hz, 1H), 6.96 (d, J = 8.6 Hz, 1H), 6.82 (d, J = 8.6 Hz, 2H), 6.48 (s, 1H), 6.41 (s, 1H), 6.00 (s, 1H), 5.12 (s, 1H), 4.89 (dd, J = 12.0, 5.3 Hz, 1H), 4.65 (d, J = 11.5 Hz, 1H), 3.44 (s, 4H), 3.22 (s, 3H), 3.13 (s, 4H), 2.96 (s, 3H), 2.82 − 2.70 (m, 3H), 2.63 (s, 4H), 2.44 (s, 2H), 2.20 (t, J = 7.0 Hz, 2H), 2.09 − 2.05 (m, 1H), 1.66 − 1.62 (m, 2H), 1.58 − 1.54 (m, 2H). 13 C NMR (150 MHz, CDCl3) δ 173.89, 172.44, 169.58 (2 C), 167.69, 161.05, 158.88, 154.66, 147.24, 146.86, 139.92, 139.11, 136.36, 132.58, 131.94, 130.67, 129.32, 128.87, 126.42, 125.00 (q, J = 269.3 Hz), 121.91 (2 C), 116.93, 116.87 (2 C), 111.96, 110.41, 57.83, 52.98 (2 C), 49.34 (2 C), 49.02, 42.25, 40.66, 39.31, 39.01, 36.10, 35.64, 31.56, 25.82, 23.46, 22.91. HRMS calcd for C44H50F3N11O7S, [M + H]+, 934.3640; found 934.3689.
2.1.18.10. N-(2-{[2–(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl]amino}ethyl)-6–(4-{4-[(4-{[2-(N-methylmethylsulfonamido)benzyl]amino}-5-(trifluoromethyl)pyrimidin-2-yl)amino]phenyl}piperazine-1-yl)hexanamide (A12)
Yellow solid; yield: 32.9%; 1H NMR (600 MHz, CDCl3) δ 11.03 (s, 1H), 8.14 (s, 1H), 7.65 (s, 1H), 7.47 (t, J = 7.7 Hz, 1H), 7.43 (d, J = 6.9 Hz, 1H), 7.39 (d, J = 8.3 Hz, 2H), 7.35 − 7.27 (m, 3H), 7.09 (d, J = 7.0 Hz, 1H), 6.95 (d, J = 8.4 Hz, 1H), 6.82 (d, J = 8.4 Hz, 2H), 6.40 (s, 1H), 6.24 (s, 1H), 5.98 (s, 1H), 5.12 (s, 1H), 4.88 (dd, J = 11.6, 5.1 Hz, 1H), 4.68 − 4.59 (m, 1H), 3.53 − 3.41 (m, 4H), 3.22 (s, 3H), 3.16 (s, 4H), 2.96 (s, 3H), 2.84 − 2.66 (m, 7H), 2.43 (d, J = 6.3 Hz, 2H), 2.18 (s, 2H), 2.11 − 2.07 (m, 1H), 1.68–1.63 (m, 2H), 1.58 − 1.54 (m, 2H), 1.35 − 1.31 (m, 2H). 13 C NMR (150 MHz, CDCl3) δ 174.04, 172.46, 169.74, 169.60, 167.70, 161.13, 158.90, 154.78, 147.30, 146.81, 139.95, 139.14, 136.33, 132.66, 131.95, 130.66, 129.33, 128.87, 126.45, 125.03 (q, J = 269.5 Hz), 121.96 (2 C), 116.93 (3 C), 112.01, 110.60, 58.07, 52.89 (2 C), 49.25 (2 C), 49.05, 42.01, 40.68, 39.32, 38.82, 36.49, 35.69, 31.61, 26.97, 25.74, 25.67, 22.99. HRMS calcd for C45H52F3N11O7S, [M + H]+, 948.3797; found 948.3851.
2.1.18.11. N-(2-{[2–(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl]amino}ethyl)-7–(4 -{4-[(4-{[2-(N-methylmethylsulfonamido)benzyl]amino}-5-(trifluoromethyl)pyrimidin-2-yl)amino]phenyl}piperazine -1-yl)heptamide (A13)
Yellow solid; yield: 31.4%; 1H NMR (600 MHz, CDCl3) δ 11.07 (s, 1H), 8.14 (s, 1H), 7.60 (s, 1H), 7.49 − 7.46 (m, 1H), 7.44 (d, J = 7.1 Hz, 1H), 7.39 (d, J = 8.6 Hz, 2H), 7.35 − 7.27 (m, 3H), 7.09 (d, J = 7.1 Hz, 1H), 6.93 (d, J = 8.5 Hz, 1H), 6.83 (d, J = 8.7 Hz, 2H), 6.41 (s, 1H), 6.11 (s, 1H), 5.98 (s, 1H), 5.12 (s, 1H), 4.88 (dd, J = 12.2, 5.4 Hz, 1H), 4.64 (d, J = 9.0 Hz, 1H), 3.59 − 3.56 (m, 1H), 3.47 − 3.40 (m, 3H), 3.22 (s, 3H), 3.17 (s, 4H), 2.96 (s, 3H), 2.83 − 2.67 (m, 7H), 2.49 − 2.40 (m, 2H), 2.22 − 2.13 (m, 2H), 2.09 − 2.07 (m, 1H), 1.65 (t, J = 11.2 Hz, 2H), 1.54 (s, 2H), 1.36 − 1.29 (m, 4H). 13 C NMR (150 MHz, CDCl3) δ 174.11, 172.45, 169.62, 169.50, 167.73, 161.14, 158.90, 154.79, 147.33, 146.77, 139.95, 139.15, 136.28, 132.66, 131.89, 130.67, 129.33, 128.87, 126.46, 125.03 (q, J = 269.6 Hz), 121.98 (2 C), 116.90 (2 C), 116.78, 111.93, 110.57, 58.46, 52.90 (2 C), 49.17 (2 C), 49.05, 42.28, 40.67, 39.33, 38.89, 36.68, 35.70, 31.65, 29.19, 27.36, 25.91, 25.58, 22.97. HRMS calcd for C46H54F3N11O7S, [M + H]+, 962.3953; found 962.4011.
2.1.18.12. N-(2-{[2–(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl]amino}ethyl)-8–(4-{4-[(4-{[2-(N-methylmethylsulfonamido)benzyl]amino}-5-(trifluoromethyl)pyrimidin-2-yl)amino]phenyl}piperazine-1-yl) octanamide (A14)
Yellow solid; yield: 39.7%; 1H NMR (600 MHz, CDCl3) δ 10.46 (s, 1H), 8.14 (s, 1H), 7.62 (s, 1H), 7.50 − 7.47 (m, 1H), 7.44 (d, J = 7.5 Hz, 1H), 7.40 (d, J = 8.9 Hz, 2H), 7.35 − 7.26 (m, 3H), 7.09 (d, J = 7.1 Hz, 1H), 6.98 (d, J = 8.6 Hz, 1H), 6.83 (d, J = 8.9 Hz, 2H), 6.40 (d, J = 5.0 Hz, 1H), 6.13 (s, 1H), 5.99 (s, 1H), 5.12 (s, 1H), 4.90 (dd, J = 12.3, 5.4 Hz, 1H), 4.64 (d, J = 13.1 Hz, 1H), 3.49 − 3.43 (m, 4H), 3.23 (s, 3H), 3.20 (s, 4H), 2.97 (s, 3H), 2.86 − 2.67 (m, 7H), 2.46 (d, J = 7.7 Hz, 2H), 2.16 (t, J = 7.5 Hz, 2H), 2.11 − 2.08 (m, 1H), 1.63 − 1.59 (m, 2H), 1.56 − 1.53 (m, 2H), 1.33 − 1.28 (m, 6H). 13 C NMR (150 MHz, CDCl3) δ 174.12, 172.23, 169.58, 169.45, 167.71, 161.11, 158.90, 154.75, 147.23, 146.84, 139.94, 139.15, 136.37, 132.62, 131.99, 130.69, 129.35, 128.88, 126.43, 125.01 (q, J = 269.9 Hz), 121.96 (2 C), 117.00 (2 C), 116.88, 112.01, 110.50, 58.32, 52.91 (2 C), 49.20 (2 C), 49.08, 42.12, 40.67, 39.34, 39.04, 36.62, 35.67, 31.67, 29.03, 28.98, 27.10, 26.04, 25.61, 22.93. HRMS calcd for C47H56F3N11O7S, [M + H]+, 976.4110; found 976.4171.
2.1.18.13. N-(2-{[2–(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl]amino}ethyl)-10–(4 -{4-[(4-{[2-(N-methylmethylsulfonamido)benzyl]amino}-5-(trifluoromethyl)pyrimidin-2-yl)amino]phenyl}piperazine -1-yl)decanoamide (A15)
Yellow solid; yield: 34.8%; 1H NMR (600 MHz, CDCl3) δ 9.94 (s, 1H), 8.15 (s, 1H), 7.57 − 7.44 (m, 3H), 7.41 (d, J = 8.9 Hz, 2H), 7.35 − 7.27 (m, 3H), 7.10 (d, J = 7.1 Hz, 1H), 7.00 (d, J = 8.6 Hz, 1H), 6.85 (d, J = 8.9 Hz, 2H), 6.40 (d, J = 5.2 Hz, 1H), 5.99 (d, J = 15.2 Hz, 2H), 5.14 (s, 1H), 4.91 (dd, J = 12.4, 5.4 Hz, 1H), 4.66 (s, 1H), 3.49 − 3.44 (m, 4H), 3.24 (s, 3H), 3.19 (s, 4H), 2.97 (s, 3H), 2.87 − 2.66 (m, 7H), 2.44 (s, 2H), 2.16 (t, J = 7.5 Hz, 2H), 2.12 − 2.08 (m, 1H), 1.63 − 1.60 (m, 2H), 1.57 − 1.53 (m, 2H), 1.33–1.26 (m, 8H). 13 C NMR (150 MHz, CDCl3) δ 174.09, 171.95, 169.59, 169.19, 167.68, 161.19, 158.92, 154.85, 147.43, 146.91, 139.98, 139.20, 136.39, 132.64, 131.89, 130.76, 129.35, 128.88, 126.45, 125.05 (q, J = 269.4 Hz), 122.02 (2 C), 116.97 (2 C), 116.88, 112.07, 110.54, 58.60, 53.08 (2 C), 49.40 (2 C), 49.11, 42.22, 40.68, 39.36, 39.19, 36.71, 35.72, 31.67, 29.25 (2 C), 29.19, 27.45, 26.31, 25.70, 22.94. HRMS calcd for C49H60F3N11O7S, [M + H]+, 1004.4423; found 1004.4482.
2.2. Fak HTRF assay
The FAK kinase assay was performed using the HTRF® KinEASE™-TK kit (Cisbio Bioassays, France) in white 384-well small volume plates with a total working volume of 20 μL. Purified FAK enzyme was purchased from Carna Biosciences (Japan). Compounds were diluted step-by-step from a concentrated stock of 8 mM in 100% DMSO and with serial kinase reaction buffer dilutions. The IC50 measurements were performed in replicates. For each assay, 4 μL of dispensed compounds, 4 μL of mix 1 (ATP + Substrate TK) and 2 μL of kinase (0.111 ng/mL) were added to the assay wells. The assay plates were incubated at 30 °C for 50 min and reactions were terminated by adding 10 μL of mix 2 (Sa-XL665 + TK-Antibody-Cryptate). After a final incubation (60 min at room temperature), HTRF signals were obtained by reading plates using an Infinite® F500 microplate reader (Tecan, Switzerland). The fluorescence was measured at 620 nM (Cryptate) and 665 nM (XL665). A ratio was calculated (665/620) for each well. For IC50 measurements, values were normalised and fitted with Graphpad prism 8.3.0.
2.3. Cell proliferation assay
A549 cells were cultured in a 96-well plate at a density of 4000 cells/well and were maintained at 37 °C in a humidified atmosphere of 5% CO2 for 24 h. The tested compounds were added to the culture medium at the indicated final concentrations and incubated for 72 h. Fresh MTT was added to each well at a final concentration of 5 mg/mL in phosphate-buffered saline (PBS), and the cells were then incubated at 37 °C for 4 h. Removing culture solution and the formazan crystals in each well were dissolved in 150 μL DMSO, and the absorbance of each test was measured at λ490nm using Infinite® F500 microplate reader (Tecan, Switzerland). The IC50 values were calculated by concentration-response curve fitting using Graphpad prism 8.3.0.
2.4. Western blotting
Antibodies used in this study are: Anti-FAK (CST-3285S) was obtained from Cell Signalling Technology (MA, USA), anti-GAPDH (Milipore-MAB374, 1:5000 for Western Blotting) was purchased from Milipore (MA, USA). A549 cells were cultured in F12K medium containing 10% foetal bovine serum and 1% pen/strep in an incubator containing 5% CO2 at 37 °C. The drug was prepared with DMSO to a concentration of 5 mM; After the cells were plated for 24 h, the drug-containing medium was changed, and then the drugs were added to co-culture for 8 h, and then the cells were collected. Add 10 μL PMSF (100 mM) to 1 mL lysate, shake well and put it on ice (PMSF can be mixed with lysate only after shaking well until there is no crystallization). According to the number of cells, add 100–500 μL lysis solution containing PMSF to each tube of cells, and lyse on ice for 30 min. Centrifuge at 12,000 rpm for 10 min at 4 °C. The centrifuged supernatant was packaged and transferred to a 1.5 mL centrifuge tube for protein quantitative detection. The protein was pre-treated using BCA method. The protein solution was mixed with 5× loading buffer according to the volume ratio of 4:1, boiled in boiling water for 10 min, cooled and then subjected to SDS-PAGE electrophoresis.
According to the molecular weight of the target protein, 12% separation gel and 5% concentrated gel were prepared. Calculate the volume of solution containing 50 μg protein, which is the loading amount. Use 1× loading buffer to make up the total volume of each sample loading hole consistent; The voltage of concentrated glue is 80 V for 30 min, and the voltage of separated glue is 120 V for 30–50 min. Bromophenol blue can stop electrophoresis and transfer film when it reaches the glue bottom. PVDF membrane with constant pressure of 100 V, 60 min and 0.45 μm; Immerse PVDF membrane completely in 5% BSA-PBST, and gently shake at room temperature for 60 min. The primary antibody was diluted with 5% BSA-PBST and incubated overnight at 4 °C. The PVDF membrane was taken out the next day, and PBST washed the membrane five times, each time for 6 min. PBST diluted secondary antibody and incubated at room temperature for 60 min. PBST membrane washing 5 times and each time is 6 min. After mixing ECL A and B solution according to the volume of 1:1, drop them evenly on the film, set the exposure time and type as required, start the exposure, save the picture and export the picture after the exposure, and use image analysis software Image J for grey Analysis.
2.5. Wound healing assay
A549 cells (6 × 105 cells) were seeded in six-well plates and grown to approximately 100% confluence in culture medium. Subsequently, a cell-free line was manually created by scratching the confluent cell monolayers with a 200 μL pipette tip. The wounded cell monolayers were washed three times with phosphate-buffered saline (PBS) and incubated with serum-free medium. Then, cells were treated with different concentrations of compound A13, incubated for 72 h, and photographed at 0, 24 and 72 h with an inverted microscope.
2.6. Transwell invasion assay
On the first day, 0.2× basement membrane extract (BME) working solution was prepared by diluting 5× BME stock solution in 1× Travigen Inc. coating buffer. Briefly, 100 μL of 10× coating buffer was diluted in 900 μL of sterile water to make 1× coating buffer. Then 960 μL of 1× coating buffer was mixed with 40 μL of 5× BME to make a working 0.2× BME solution. Corning Transwell permeable inserts (Costar Transwell chambers, Corning) were placed on a 24-well plate, and 100 μL of 0.2× BME solution was added to each Transwell insert and incubated for 16 h. The following day, A549 cells were trypsinized and cells were suspended in serum-free medium. Approximately 100 μL from the cell suspension (∼3 × 105 cells) was added to each Transwell insert followed by another 100 μL of PROTAC or control containing serum-free RPMI medium. The lower chamber was filled with 10% FBS containing RPMI medium, and the whole setup was incubated at 37 °C and 5% CO2 for 24 h. After 24 h, the cell culture medium was removed from both lower and upper chambers and the Transwell inserts were washed three times with PBS. Non-invasive cells were removed using a cotton swab, and the bottom side of the membrane of the Transwell inserts was fixed with 4% formaldehyde for 10 min at room temperature followed by permeabilization with PBST (pH-7.4, 50 mM Tris-HCl, 150 mM NaCl, 0.1% Triton-X100) for another 10 min. Inserts were washed once with PBS and stained with 0.2% (w/v) crystal violet solution for 20 min at room temperature. Inserts were then extensively washed with PBS and once with water to remove all excess dye and salts. Cells migrated through the membrane were captured using a camera attached to a light microscope. Images were then analysed and the number of cells on the bottom side of the membrane were counted by ImageJ software.
2.7. Plasma stability assay
The pooled frozen human plasma (BioreclamationIVT, batch No. BRH1569252) was thawed in a water bath at 37 °C prior to experiment. Plasma was centrifuged at 4000 rpm for 5 min and the clots were removed if any. The pH will be adjusted to 7.4 ± 0.1 if required. 100 μM propantheline bromide solution was prepared by diluting 5 µL of 10 mM stock solution with 495 µL H2O as a positive control. The other detected compound solution was prepared by the same method. 98 µL of blank plasma was spiked with 2 μL of dosing solution (100 μM) to achieve 2 μM of the final concentration in duplicate and samples were incubated at 37 °C in a water bath. At each time point (0, 10, 30, 60 and 120 min), 400 μL of stop solution (200 ng/mL tolbutamide and 200 ng/mL Labetalol in ACN) was added to precipitate protein and mixed thoroughly. Centrifuged sample plates at 4000 rpm for 15 min. An aliquot of supernatant (50 μL) was transfered from each well and mixed with 100 μL ultra-pure water. The samples were shaked at 800 rpm for about 10 min before submitting to LC-MS/MS analysis.
2.8. The membrane permeability assay
After 21–28 days, the cells seeded on transwell filters were used for permeability experiments. The monolayers were rinsed once with a solution of 0.02% EDTA in DPBS on the apical and basolateral side, and three times with HBSS buffered at pH 7.4 with 25 mM HEPES buffer (N-2-hydroxyethylpiperazine-N’-2-ethane sulphonic acid). The cells were then left to equilibrate at 37 °C for 30 min in HBSS/HEPES solution. TEER values were then measured. All cell monolayers used had TEER values between 2.5 and 5 kΩ/cm2 before the experiments.
At the start of the experiment (time = 0 min), 600 μL of buffered HBSS had been placed on the basolateral side of the monolayer and 100 μL of 200 μM solution of the test compound prepared in HBSS/HEPES was added to the apical side of the monolayer. At time 30, 90, 120 and 150 min, 400 μL were sampled from the basolateral side. The volume sampled (400 μL) was replaced with 400 μL of HBSS/HEPES buffer solution. Each compound was tested on 4 wells/experiment and at least three experiments were conducted using different passage number cells. LC–MS/MS analysis was performed on each sample and compared to a standard curve (0.05–100 μM) to determine the concentration of each respective peptide derivative. Apparent permeability values (Papp) were then calculated using the following formula.
where dC/dt is the steady state rate of the change in the chemical concentration (M/s); Vr is the volume of the receiver chamber (cm3); A is the surface area of the cell monolayer (cm2); C0 is the initial concentration in the donor chamber (M).
2.9. Molecular docking study
Ligand structures were prepared by using Maestro 9.0 within the Schrodinger package. The crystal structures of FAK (PDB ID: 3BZ3) were retrieved from RCSB Protein Data Bank (http://www. pdb.org), and prepared for molecular docking by using Protein Preparation Wizard. The ligand structures were optimised by Maestro’s Ligprep module, regulated to a protonated state of pH 7.4, and minimised with OPLS_2005 force field to produce low-energy conformers. Compounds were docked into binding sites with the Glide module within the Schrodinger package on account of united-atom scoring function. The docking program was implemented twenty times, providing sufficient constellation groups and the output was characterised by the favourable binding affinity value. In addition, the figure was prepared using PyMOL.
3. Experimental section
3.1. Chemistry
The synthesis of intermediates a4, a7–a11, a13–a14 and a16–a21 was followed as Scheme 1. Commercially available 4-fluoroisobenzofuran-1,3-dione (a1) gave intermediate a2 via cyclisation reaction with 3-aminopiperidine-2,6-dione. The F atom of intermediate a2 was substituted by tert-butylglycine, followed by hydrolysis reaction to obtain intermediate a4. The F atom of intermediate a2 was substituted by Boc-protected ethylenediamine to generate intermediate a5. After removing the Boc protecting group, intermediate a5 was condensed with the corresponding long chain of carboxylic acid to acquire intermediates a7–a11. Nucleophilic substitution of intermediate a2 with amino-substituted PEG-chains gave intermediates a11–a12. And intermediates a11–a12 were protected by TsCl to afford intermediates a13–a14. Commercially available a15 was condensed with the corresponding long chain of carboxylic acid to obtain intermediates a16–a21.
Scheme 1.

Synthesis of intermediates a4, a7–a11, a13–a14 and a16–a21. Reagents and conditions: (a) 3-aminopiperidine-2,6-dione, NaOAc, AcOH, 140 °C, 58.3–68.7% yield; (b) DIPEA, DMF, 90 °C,53.6–65.2% yield; (c) CF3COOH, CH2Cl2, 25 °C, 86.3–90.2% yield; (d) HATU, DIPEA, 25 °C, 67.4–75.3% yield; (e) TsCl, Et3N, CH2Cl2, 30 °C, 72.6–79.3% yield; (f) corresponding bromocarboxylic acid, SOCl2, CH2Cl2, 40 °C, 61.5–85.8% yield.
The synthetic routes for intermediates a24 and a27 were shown in Scheme 2. Using 2-[2–(2-chloroethoxy)ethoxy]ethan-1-ol (a22) as the starting material, the intermediate a24 was generated by azide and bromination. The addition reaction of tert-butyl acrylate with 1,3-propanediol gave intermediate a26, which was then substituted by iodine to afford intermediate a27.
Scheme 2.

Synthesis of intermediates a24 and a27. Reagents and conditions: (a) NaN3, DMF, 100 °C, 47.5% yield; (b) CBr4, PPh3, CH2Cl2, 25 °C, 53.7% yield; (c) propane-1,3-diol, tritionB, CH3CN, 25 °C, 36.2% yield; (d) I2, PPh3, imidazole, THF, 25 °C, 31.8% yield.
Compound 1 was synthesised as depicted in Scheme 3. Commercially available o-fluorobenzonitrile was substituted by N-methyl-methanesulfonamide to give intermediate a29, which was then subjected to a reduction to obtain intermediate a30. Commercially available 2,4-dichloro-5-(trifluoromethyl)pyrimidine a31 afforded the intermediate a33 through a two-step substitution reaction, and was followed by a deprotection to produce compound 1.
Scheme 3.

Synthesis of compound 1. Reagents and conditions: (a) N-methylmethanesulfonamide, Cs2CO3, CH3CN, 80 °C, 82.6% yield; (b) BH3 (2 M in THF), anhydrous THF, 60 °C, 47.9% yield; (c) tert-butyl 4–(4-aminophenyl)piperazine-1-carboxylate, ZnBr2, TEA, t-BuOH/DCE, 0 °C, 72.3% yield; (d) a30, DIPEA, 1,4-dioxane, 100 °C, 81.3%; e) CF3COOH, CH2Cl2, 25 °C, 93.6% yield.
The synthetic routes of compounds A1-A2 are shown in Scheme 4. Compound 1 was substituted by intermediate a24 to give intermediate a34. Intermediate a34 was reduced to a primary amine and condensed with intermediate a4 to obtain compound A1. Compound 1 was substituted by intermediate a27 to give intermediate a36, which was hydrolysed and condensed with intermediate a6 to obtain compound A2.
Scheme 4.

Synthesis of compounds A1-A2. Reagents and conditions: (a) 81 or 84, K2CO3, CH3CN, 80 °C, 58.6–63.3% yield; (b) PPh3, THF/H20, 70 °C, 76.3–81.2% yield; (c) 65 or 67, HATU, DIPEA, CH2Cl2, 25 °C, 31.7–36.8% yield; (d) TFA, DCM, 30 °C, 95% yield.
The compounds A3–A15 were synthesised as shown in Scheme 5. Compound 1 was substituted by intermediates a13–a14, a16–a21 and a7–a11 to afford compounds A3–A4, A5–A10 and A11–A15.
Scheme 5.

Synthesis of compounds A3–A15. Reagents and conditions: (a) a13–a14, DIPEA, DMF, 90 °C, 29.6–37.7%; (b) a16–a21 or a7–a11, KI, K2CO3, CH3CN, 80 °C, 29.4–41.9%.
3.2. In vitro activity against FAK kinase and SAR analyses
Compounds A1–A15 were evaluated for their activities against FAK kinases using homogeneous time-resolved technology (HTRF) assays at concentrations of 0.5 μM and 0.1 μM. PF-562271 was served as a positive control to confirm the experimental condition. PF-562271 showed a strong inhibitory activity against FAK with an IC50 value of 2.8 nM (Figure 4), which was similar to the reported data. As shown in Table 1, the results from the assay showed that inhibitory activities of most of PROTACs were more than 90% at the concentration of 0.5 μM. Furthermore, at the concentration of 0.1 μM, all PROTACs inhibited FAK protein by more than 50%, indicating that all the PROTACs had good affinities to FAK protein. In addition, compounds A1, A6 and A13 containing alkyl-linker or PEG-linker were selected to test their IC50 values, both of them exhibited good inhibitory activities with IC50 values of 22.3, 24.4 and 26.4 nM, respectively, which were slightly lower than PF-562271 (Figure 4).
Figure 4.

The enzyme inhibition activity curve of compound A1, A6, A13 and PF-562271.
Table 1.
FAK inhibition of compound 1 based PROTACs A1–A15 with different linkers.
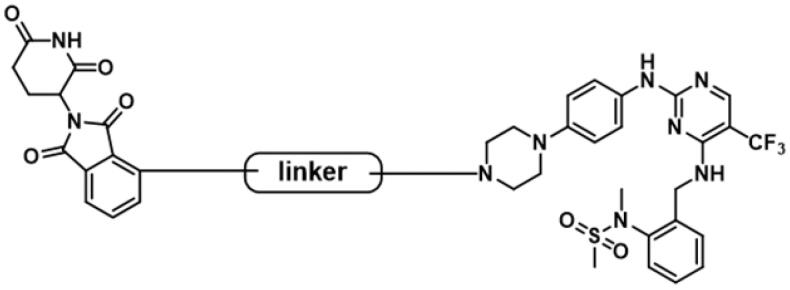
| Compd. | linker | FAK inhibition (%)a |
|
|---|---|---|---|
| 0.5 μM | 0.1 μM | ||
| A1 |
|
98.0 | 94.9 |
| A2 |
|
98.8 | 89.0 |
| A3 |
|
97.5 | 87.8 |
| A4 |
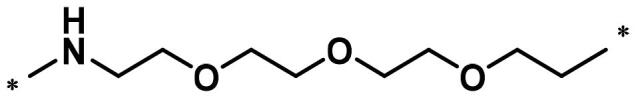
|
97.0 | 90.8 |
| A5 |
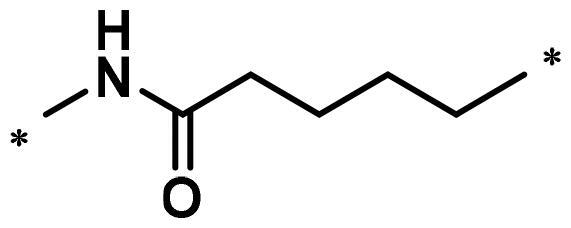
|
98.0 | 91.2 |
| A6 |
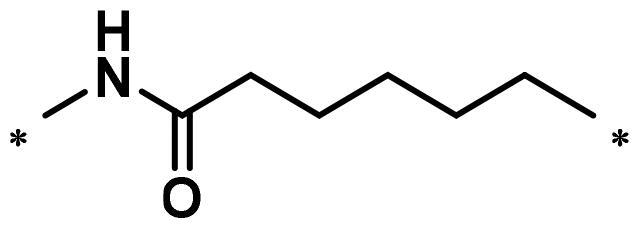
|
96.8 | 88.2 |
| A7 |
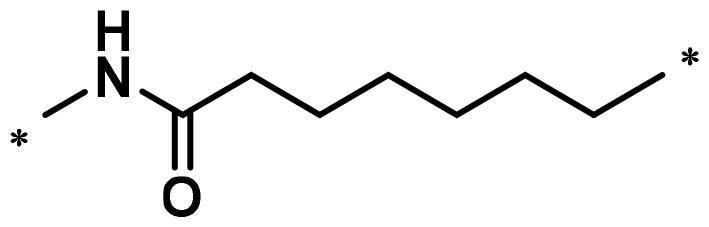
|
95.4 | 87.5 |
| A8 |
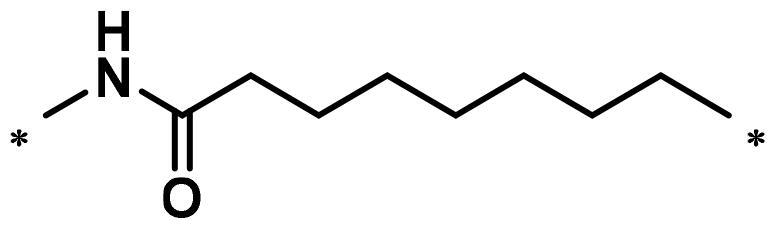
|
83.0 | 67.7 |
| A9 |
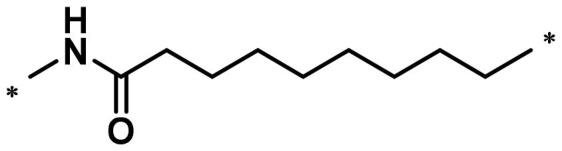
|
92.3 | 79.0 |
| A10 |
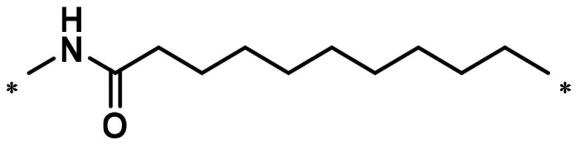
|
90.0 | 66.3 |
| A11 |
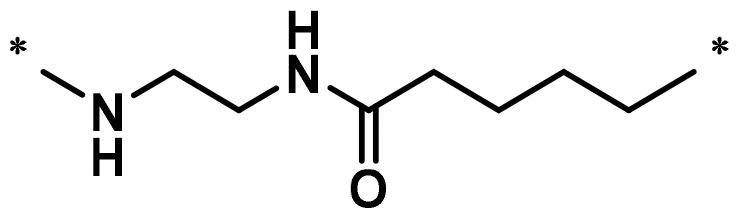
|
98.6 | 93.5 |
| A12 |
|
98.0 | 86.1 |
| A13 |
|
96.9 | 84.4 |
| A14 |

|
92.8 | 84.0 |
| A15 |
|
95.8 | 74.9 |
| PF-562271 | 95.6 | 91.2 | |
aThe inhibition ratio are shown as the mean values from two separate experiments.
Next, the degradation efficiency of FAK-PROTAC was evaluated in A549 cells. A549 was a human non-small cell lung cancer cell with high expression of FAK. We selected two concentration gradients (10 nM, 100 nM) of PROTACs to incubate with A549 cells, and then the cells were lysed and performed Western blot experiments to observe the changes of FAK protein content in A549 cells. As shown in Figure 5, the results of Western blot test showed that most of the compounds had potent degradation activities on FAK protein in A549 cells. Compounds A4–A15 had stronger degradation activities, the degradation rate of FAK protein was more than 50% at concentration of 10 nM. Among them, compound A13 exhibited optimal protein degradation (85% degradation at 10 nM).
Figure 5.

FAK-degrading efficiency of PROTACs A1–A15 in A549 cells (A) FAK levels in response to dose escalations of A1–A15 in the A549 cell line after treatment for 12 h, with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) used as the loading control. The experiments were repeated three times, and representative images were selected. (B) FAK protein expression ratio vs control.
3.3. The cell proliferation inhibition potency of PROTACs in A549 cells
To further test the antitumor activities of this class of PROTACs, we selected compound A13 for antiproliferative activity studies against the A549 cell lines23. PF-562271 was used as a positive control. As shown in Figure 6, compound A13 exhibited more significant anti-tumour cell proliferation inhibitory activity than PF-562271. At a concentration of 1.6 μM, the inhibition rate of compound A13 was more than 50%, but the inhibition rate of PF-562271 was less than 20%. And at a concentration of 3.2 μM, the inhibition rate of compound A13 was reached 100%. In contrast, the inhibition rate of PF-562271 was only more than 50% at a concentration of 25.6 μM.
Figure 6.

Antiproliferative activities of compound A13 and PF-562271 against A549 cells. Cell growth inhibition rate was measured by MTT assay. (mean ± SD, n = 3).
3.4. Effects of PROTAC A13 on cell migration
To evaluate the effect of PROTAC A13 on the migration of A549 cells, wound healing assays were performed. For this assay, A549 cells were incubated with different concentrations of A13 (0.24 and 0.48 μM) and DMSO for 72 h, investigating whether compound A13 affects cell lines migration after scratching the cell monolayer (Figure 7). The results from the assay showed that A549 cells filled more than 60% wounded area after scribing 72 h at the concentration of 0.24 μM of PROTAC A13. And at the concentration of 0.48 μM, the migration rate of A549 cells was less than 60%, a concentration-dependent inhibition of A13 was observed in A549 cells.
Figure 7.

(A) The wound healing assay was performed to show the effect of indicated concentrations of PROTAC A13 on A549 lung cancer cell migration. (B) Quantitative analysis of the wound healing assay by PROTAC A13. The migration ratio of A549 cells treated with A13 was measured by wounding healing assay. (mean ± SD, n = 3). *p < 0.05, **p < 0.01 vs CON, #p < 0.05.
3.5. Effects of PROTAC A13 on cell invasion
We further investigated the inhibitory activity of PROTAC A13 on the invasion of A549 cells, transwell assay was performed. PF-562271 was used as a positive control. A549 cells were incubated with DMSO, A13 (0.12, 0.24 and 0.48 μM) and PF-562271 (2.4, 4.8 and 9.6 μM) for 72 h. The results were shown in Figure 8, the number of A549 cells that penetrated the membrane were decreased in a concentration-dependent manner, indicating that compound A13 can dose-dependently inhibit the invasion of A549 cells. In addition, under the concentration of 0.24 μM of A13, the number of A549 cells that penetrated the membrane was lower than that of PF-562271 at the concentration of 2.4 μM, suggesting that the anti-invasion ability against A549 cells of PROTAC A13 was superior than PF-562271.
Figure 8.

(A) Transwell assay was performed to show the effect of indicated concentrations of PROTAC A13 and PF-562271 on A549 lung cancer cell invasion. (B) Quantitative analysis of transwell assay by PROTAC A13 and PF-562271. The Invasion ratio of A549 cells treated with A13 or PF-562271 was measured by transwell assay. (mean ± SD, n = 3). *p < 0.05, **p < 0.01 vs CON; #p < 0.05, ##p < 0.01 vs 1/8 IC50; &p < 0.05, &&p < 0.01 vs 1/4 IC50.
3.6. The plasma stability and membrane permeability of PROTAC A13
Although PROTAC A13 had been shown to degrade FAK, the high molecular weight and multiple hydrogen bond donors (HBDs) and acceptors (HBAs) of A13 consistently limit their physicochemical properties, such as membrane permeability and stability24–26. Hence, the membrane permeability and in vitro plasma stability PROTAC A13 were also evaluated. As shown in Table 2, compound A13 possessed favourable plasma stability with T1/2 > 194.8 min. But it is regrettable that PROTAC A13 exhibited a low membrane permeability with the measured Caco-2 permeability Papp(A−b) < 0.48 × 10−6 cm/s, which is below the standard for “modest” permeability (Papp > 0.5 × 10−6 cm/s) (Table 3).
Table 2.
Plasma stability of PROTAC A13.
| Time Point (min) | 0 | 10 | 30 | 60 | 120 |
|---|---|---|---|---|---|
| Remaining (%) | 100.0 | 98.6 | 84.1 | 79.7 | 65.3 |
| T1/2 (min) | >194.8 | ||||
Table 3.
The membrane permeability of PROTAC A13 in Caco-2 cell lines.
| Compd. | A–B permeability (Papp × 10-6 cm/s)a | B-A permeability (Papp × 10-6 cm/s)a | Efflux ratio (Papp(B−A)/ Papp(A−B)) |
|---|---|---|---|
| B5 | <0.480 | 0.417 | >0.869 |
aDuplicates were performed (n = 2).
4. Conclusion
We designed and synthesised 15 FAK-targeting PROTACs based on FAK inhibitor (PF-562271 derivative 1) and CRBN E3 ligase ligand (Pomalidomide). All PROTACs potently suppressed the enzymatic activities of FAK, the inhibition rate against FAK was more than 50% at the concentration of 0.1 μM. And PROTACs A1, A6 and A13 exhibited potent enzyme inhibition with IC50 values of 22.3, 24.4 and 26.4 nM, respectively. Next, the FAK degradation activities of 15 PROTACs were evaluated, the representative PROTAC A13 had a degradation rate of 85% at the concentration of 10 nM. In addition, the antiproliferative activity by PROTAC A13 exceeded beyond FAK inhibitor PF-562271 at a concentration of 1.6 μM in A549 cells, the inhibition rate of PROTAC A13 was more than 50%. Also, the anti-invasion activity against A549 cells of PROTAC A13 was superior than PF-562271. Further, preliminary drug-like properties evaluations indicate that PROTAC A13 has excellent plasma stability (T1/2 > 194.8 min). In summary, PROTAC A13 can be used as a potent tool to study FAK-related biology, and facilitated the development of new therapeutic agents for the treatment of FAK-related diseases.
Supplementary Material
Funding Statement
We gratefully acknowledge the fnancial support from the Program for Innovative Research Team of the Ministry of Education and Programfor Liaoning Innovative Research Team in University.
Disclosure statement
No potential conflict of interest was reported by the author(s)
References
- 1.Schaller MD, Borgman CA, Cobb BS, et al. Pp125fak a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc Natl Acad Sci U S A 1992;89:5192–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci 2003;116:1409–16. [DOI] [PubMed] [Google Scholar]
- 3.Golubovskaya VM. Focal adhesion kinase as a cancer therapy target. Anticancer Agents Med Chem 2010;10:735–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kessler BE, Sharma V, Zhou Q, et al. Fak expression, not kinase activity, is a key mediator of thyroid tumorigenesis and protumorigenic processes. Mol Cancer Res 2016;14:869–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beraud C, Dormoy V, Danilin S, et al. Targeting fak scaffold functions inhibits human renal cell carcinoma growth. Int J Cancer 2015;137:1549–59. [DOI] [PubMed] [Google Scholar]
- 6.Gogate PN, Kurenova EV, Ethirajan M, et al. Targeting the c-terminal focal adhesion kinase scaffold in pancreatic cancer. Cancer Lett 2014;353:281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sood AK, Coffin JE, Schneider GB, et al. Biological significance of focal adhesion kinase in ovarian cancer. Am J Pathol 2004;165:1087–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beierle EA, Massoll NA, Hartwich J, et al. Focal adhesion kinase expression in human neuroblastoma: immunohistochemical and real-time PCR analyses. Clin Cancer Res 2008;14:3299–305. [DOI] [PubMed] [Google Scholar]
- 9.Furuyama K, Doi R, Mori T, et al. Clinical significance of focal adhesion kinase in resectable pancreatic cancer. World J Surg 2006;30:219–26. [DOI] [PubMed] [Google Scholar]
- 10.van Nimwegen MJ, Verkoeijen S, van Buren L, et al. Requirement for focal adhesion kinase in the early phase of mammary adenocarcinoma lung metastasis formation. Cancer Res 2005;65:4698–706. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J, He DH, Zajac-Kaye M, Hochwald SN.. A small molecule fak kinase inhibitor, gsk2256098, inhibits growth and survival of pancreatic ductal adenocarcinoma cells. Cell Cycle 2014;13:3143–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurmasheva RT, Gorlick R, Kolb EA, et al. Initial testing of vs-4718, a novel inhibitor of focal adhesion kinase (FAK), against pediatric tumor models by the pediatric preclinical testing program. Pediatr Blood Cancer 2017;64:e26304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crompton BD, Carlton AL, Thorner AR, et al. High-throughput tyrosine kinase activity profiling identifies FAK as a candidate therapeutic target in ewing sarcoma. Cancer Res 2013;73:2873–83. [DOI] [PubMed] [Google Scholar]
- 14.Schwock J, Dhani N, Hedley DW.. Targeting focal adhesion kinase signaling in tumor growth and metastasis. Expert Opin Ther Targets 2010;14:77–94. [DOI] [PubMed] [Google Scholar]
- 15.Lai AC, Crews CM.. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov 2017;16:101–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Churcher I. Protac-induced protein degradation in drug discovery: breaking the rules or just making new ones? J Med Chem 2018;61:444–52. [DOI] [PubMed] [Google Scholar]
- 17.Cromm PM, Samarasinghe KTG, Hines J, Crews CM.. Addressing kinase-independent functions of FAK via protac-mediated degradation. J Am Chem Soc 2018;140:17019–26. [DOI] [PubMed] [Google Scholar]
- 18.Popow J, Arnhof H, Bader G, et al. Highly selective PTK2 proteolysis targeting chimeras to probe focal adhesion kinase scaffolding functions. J Med Chem 2019;62:2508–20. [DOI] [PubMed] [Google Scholar]
- 19.Gao H, Wu Y, Sun Y, et al. Design, synthesis, and evaluation of highly potent FAK-targeting protacs. ACS Med Chem Lett 2020;11:1855–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao H, Zheng C, Du J, et al. Fak-targeting protac as a chemical tool for the investigation of non-enzymatic FAK function in mice. Protein Cell 2020;11:534–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Law RP, Nunes J, Chung CW, et al. Discovery and characterisation of highly cooperative FAK-degrading protacs. Angew Chem Int Ed Engl 2021;60:23327–34. [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Xue L, Xu X, et al. FAK-targeting protac demonstrates enhanced antitumor activity against KRAS mutant non-small cell lung cancer. Exp Cell Res 2021;408:112868. [DOI] [PubMed] [Google Scholar]
- 23.Zhou B, Wang GZ, Wen ZS, et al. Somatic mutations and splicing variants of focal adhesion kinase in non-small cell lung cancer. J Natl Cancer Inst 2018;110. [DOI] [PubMed] [Google Scholar]
- 24.Klein VG, Townsend CE, Testa A, et al. Understanding and improving the membrane permeability of vh032-based protacs. ACS Med Chem Lett 2020;11:1732–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edmondson SD, Yang B, Fallan C.. Proteolysis targeting chimeras (protacs) in ‘beyond rule-of-five’ chemical space: recent progress and future challenges. Bioorg Med Chem Lett 2019;29:1555–64. [DOI] [PubMed] [Google Scholar]
- 26.Maple HJ, Clayden N, Baron A, et al. Developing degraders: principles and perspectives on design and chemical space. Medchemcomm 2019;10:1755–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.