

Abstract
Despite the promise of immunotherapy such as the immune checkpoint inhibitors (ICIs) anti-PD-1 and anti-CTLA-4 for advanced melanoma, only 26%–52% of patients respond, and many experience grade III/IV immune-related adverse events. Motivated by the need for an effective therapy for patients non-responsive to clinically approved ICIs, we have developed a novel nanoimmunotherapy that combines locally administered Prussian blue nanoparticle-based photothermal therapy (PBNP-PTT) with systemically administered agonistic anti-CD137 monoclonal antibody therapy (aCD137). PBNP-PTT was administered at various thermal doses to melanoma cells in vitro, and was combined with aCD137 in vivo to test treatment effects on melanoma tumor progression, animal survival, immunological protection against tumor rechallenge, and hepatotoxicity. When administered at a melanoma-specific thermal dose, PBNP-PTT elicits immunogenic cell death (ICD) in melanoma cells and upregulates markers associated with antigen presentation and immune cell co-stimulation in vitro. Consequently, PBNP-PTT eliminates primary melanoma tumors in vivo, yielding long-term tumor-free survival. However, the antitumor immune effects generated by PBNP-PTT cannot eliminate secondary tumors, despite significantly slowing their growth. The addition of aCD137 enables significant abscopal efficacy and improvement of survival, functioning through activated dendritic cells and tumor-infiltrating CD8+ T cells, and generates CD4+ and CD8+ T cell memory that manifests in the rejection of tumor rechallenge, with no long-term hepatotoxicity. This study describes for the first time a novel and effective nanoimmunotherapy combination of PBNP-PTT with aCD137 mAb therapy for melanoma.
Keywords: photothermal therapy, Prussian blue nanoparticles, anti-CD137, melanoma, nanoimmunotherapy
1. Introduction
Melanoma is a prevalent cutaneous malignancy estimated to account for over 7,000 deaths in 2021 in the United States alone [1]. Although highly curable when detected early, the 5-year survival rates decline precipitously for patients with regional (68.0%) and metastatic (29.8%) melanoma, indicating a lack of efficacy of conventional therapies, and a strong need for novel therapies for this patient population [1]. Immune checkpoint inhibitors (ICIs) represent a class of monoclonal antibody immunotherapy (mAbs) that targets key immune checkpoints on a variety of cells, including T cells (e.g., programmed death 1 (PD-1), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)), which have resulted in dramatic treatment responses in previously untreatable cancers, particularly metastatic melanoma [2–8]. Anti-PD-1 (aPD-1) mAb therapy has become a new gold standard for advanced melanoma [7]. Despite this immense promise, responses to ICIs are limited to small subsets of patients, with a 26%–52% five-year survival, depending on the ICI(s) administered and previous treatment received [9]. Additionally, ICIs are associated with grade III/IV immune-related adverse events (irAEs) in 23%–59% of these patients [8–13]. Thus, there is a great need for novel therapies for this patient population.
Responding to this need, we have engineered a novel nanoimmunotherapy, which combines Prussian blue nanoparticle-based photothermal therapy (PBNP-PTT) with agonistic anti-CD137 mAb therapy (aCD137) for melanoma. Instead of targeting immune checkpoints such as PD-1 and CTLA-4 to reverse T cell immunosuppression, we utilize aCD137, which targets and activates CD137 (4-1BB), and represents a promising new target for immunomodulation [14, 15]. The engagement of this co-stimulatory molecule expressed on activated T cells, among other immune cells, can result in CD4+ and CD8+ T cell activation that could elicit antitumor immune responses and facilitate tumor clearance through T cell cytotoxicity and IFN-ɣ and other pro-inflammatory cytokine secretion [16]. Because of these antitumor properties of CD137 signaling, aCD137 has been recently investigated for use in cancer immunotherapy to co-stimulate T cells in the context of tumor cells [17]. Urelumab and utomilumab are two aCD137 antibodies in clinical development that function through activating CD137 on immune effector cells [18]. However, while aCD137 has been explored in early clinical trials, its widespread use has been limited, suggesting that a strategy relying solely on the activation of T cells, without providing antigen targets for activated T cells, is insufficient for tumor clearance. Further, aCD137 has been shown to generate dose-limiting hepatotoxicity [19, 20].
Therefore, in this nanoimmunotherapy, we utilize the effects of aCD137 to complement the effects of PBNP-PTT. To our knowledge, this is the first evidence of a combination nanoimmunotherapy comprising PTT and a co-stimulatory molecule targeted agonistic mAb. Our rationale for combining aCD137 with PBNP-PTT lies in the fact that PBNP-PTT is immunostimulatory [21]. PBNP-PTT comprises intratumoral (i. t.) administration of light-absorbing PBNPs followed by near-infrared (NIR) laser illumination, which heats the nanoparticles by converting absorbed NIR light into thermal energy, thereby ablating surrounding tumor cells. In this proposed combination nanoimmunotherapy, PBNP-PTT serves the dual purpose of ablating the primary tumor to decrease tumor burden and releasing tumor antigens to provide a “multi-antigen vaccination effect [22–25].” PTT and other ablation methods have been demonstrated to generate immunogenic cell death (ICD) in cancer [21, 24, 26, 27], a cell death mechanism characterized by the engagement and stimulation of dendritic cells (DCs) and T cells against cancer cells [28–30]. We have shown that this phenomenon is maximally engaged only when administered at a tumor-specific thermal dose [24]. In a different cancer model, we have shown that PBNP-PTT administered at the neuroblastoma-specific thermal dose window is immunogenic and significantly improves the survival of neuroblastoma-bearing mice [22–24, 31, 32]. However, these effects are not robust enough for complete abscopal eradication of distal neuroblastoma tumors or metastases [31]. Therefore, herein we complement the immunogenic effects of PBNP-PTT by the administration of aCD137, which may further activate infiltrating T cells against the tumor by activating a co-stimulatory pathway, and generate systemic antitumor immunological memory [33]. We hypothesize that the interplay between the local and regional immunomodulatory effects of PBNP-PTT coupled with the systemic activation of a T cell response elicited by the aCD137 mAbs will result in complete response and long-term survival in models of advanced melanoma. The combination of PBNP-PTT and aCD137 mAbs represents a new therapeutic modality, as this combination has not been previously investigated.
In this study, we utilize the SM1 murine melanoma cell line that harbors the BRAFV600E mutation [34]. SM1 melanoma is predicted to be non-responsive to aPD-1 therapy by its mutation status [35], and has been shown to be non-responsive to both aPD-1 and aCTLA-4 in mouse models at the doses tested [36]. Additionally, aCD137 has shown improved preclinical efficacy over ICIs in SM1 cells [36]. We first determine the thermal dose of PBNP-PTT that elicits SM1 tumor cell-specific ICD and upregulates markers associated with antigen presentation and immune cell co-stimulation in vitro, and validate these findings in two human melanoma cell lines with the BRAFV600E mutation, similar to SM1 cells. We evaluate whether PBNP-PTT can eliminate primary melanoma tumors in vivo, if the immune response generated is robust enough to eliminate the secondary tumor, and whether aPD-1 can improve the efficacy of PBNP-PTT in the SM1 melanoma model. We then test how the addition of aCD137 impacts the abscopal efficacy of PBNP-PTT and/or improvement of survival, and the underlying immunological mechanisms driving the observed effects by measuring cellular and macromolecular subsets. We also assess both short- and long-term hepatotoxicity in response to the combination treatment. Finally, we test the ability of PBNP-PTT + aCD137 to generate immunological memory that enables rejection of tumor rechallenge. Through these studies, we seek to demonstrate for the first time the efficacy of this novel combination nanoimmunotherapy for melanoma.
2. Methods
2.1. Cells
Murine SM1 melanoma cells (BRAFV600E mutation) were obtained through a Materials Transfer Agreement with the American Type Culture Collection (ATCC). Human WM9 and WM793 melanoma cells (BRAFV600E mutation) were obtained from the laboratory of Dr. Meenhard Herlyn at The Wistar Institute. SM1 [34] and WM9 were cultured in Roswell Park Memorial Institute medium (RPMI 1640, Gibco, Carlsbad, CA) containing 1% L-glutamine (pre-supplemented), 10% fetal bovine serum (FBS, Gibco, Carlsbad, CA), and 1% penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO). WM793 was cultured in Dulbecco’s modified Eagle medium (DMEM, Gibco, Carlsbad, CA) containing 1% L-glutamine (pre-supplemented), 10% fetal bovine serum, and 1% penicillin/streptomycin.
2.2. PBNP synthesis
Potassium hexacyanoferrate(II) trihydrate (K4[Fe(CN)6]·3H2O) and iron(III) chloride hexahydrate (FeCl3·6H2O) were purchased from Sigma-Aldrich (St. Louis, MO). All synthetic procedures were conducted using ultrapure water obtained from a Milli-Q system (Millipore Corporation, Billerica, MA). Prussian blue nanoparticles were synthesized using a scheme as described previously [37] and characterized for uniformity (Fig. S1 in the Electronic Supplementary Material (ESM)). Briefly, an aqueous solution of 54.06 mg of FeCl3·6H2O (10 mM) in 20 mL of Milli-Q water was added under vigorous stirring to an aqueous solution containing 84.5 mg of K4Fe(CN)6·3H2O (10 mM) in 20 mL of Milli-Q water. After stirring for 15 min, the precipitate was isolated by centrifugation in equal parts water and acetone (10,000g for 15 min) and rinsed by sonication (30 s, 40% amplitude) in Milli-Q water. The isolation and rinsing steps were repeated three times before the particles were resuspended by sonication in Milli-Q water.
2.3. In vitro PBNP-PTT
5 million SM1 cells were suspended in 500 μL of 1× phosphate buffered saline (PBS). When used, PBNPs were added to the samples at 0.15 mg/mL. The samples were then illuminated by the NIR laser (808 nm; Laserglow Technologies, Toronto, Canada) for 10 min at varied power (0.75, 1.0, 1.5, and 2.0 W) to administer PBNP-PTT at various thermal doses/temperature ranges. The maximum temperature of the cell suspension was measured using a thermal camera (forward-looking infrared (FLIR), Arlington, VA), and temperatures were recorded every minute for 10 min. Thermal doses were calculated using the formula established previously in the Ref. [38] ; where ti is the ith time interval, R = 0.25 when T < 43 °C and 0.5 when T > 43 °C, and T is the average temperature during ti.
2.4. In vitro cell analysis
Following in vitro PBNP-PTT (as described above), cell suspensions were centrifuged and the cell pellet was re-plated in RPMI media in 6-well plates at 37 °C. Antibodies were purchased from Biolegend (San Diego, CA), denoted with “#” prior to the catalog number, and Abcam (Cambridge, UK), denoted with “ab” prior to the catalog number. After 24 h, cells were harvested and stained with Zombie Violet/Green™ Fixable viability dye (#423114 or #423112), fluorescent antibodies against calreticulin (ab209577), CD80 (#104718), CD86 (#105008), MHC-1 (#116510), CD137L (#107105), PD-L1 (#124315) (surface stains), and high mobility group box-1 protein (HMGB1) (ab195011), and Melan-A/MART1 (ab225500) (intracellular stains). Flow cytometry was performed using the BD Biosciences Celesta Cell Analyzer (Franklin Lakes, NJ), and cytometric analysis was done using FlowJo software (Ashland, OR). For estimation of intracellular ATP, cells were harvested 24 h after in vitro PBNP-PTT, washed with 1× PBS, and mixed with ATP reagent from the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI); luminescence was measured using a SpectraMax microplate reader (Molecular Devices, San Jose, CA).
2.5. Animals
Five-week-old female C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME). The animals were acclimated for 7 days prior to handling. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of George Washington University (GWU), Washington, DC (Protocol #s A396 and A2021-001), and were in accordance with the humane care of research animals. Tumor volumes (mm3) were monitored every other day by caliper measurement and calculated using the formula (long diameter × short diameter2)/2. A tumor size of 20 mm in diameter in any dimension for single tumors, or 15 mm in any dimension if two tumors were present, was designated as the endpoint, and mice were euthanized at that time (if not earlier as designated in study design). Euthanasia was achieved through cervical dislocation after CO2 narcosis. If tumors impaired the mobility of the animal, became ulcerated, or appeared infected, or if the mice displayed signs of distress exhibiting a sick mouse posture, the mouse was euthanized.
2.6. Ex vivo co-culture analysis
To assess the effects of PBNP-PTT treatment of SM1 cells on direct T cell activation, spleens were harvested from tumor-free C57BL/6 mice after euthanasia. Splenic T cells were magnetically isolated by negative selection using Pan T cell isolation Kit II mouse (Miltenyi Biotec, Bergisch Gladbach, Germany), and subsequently co-cultured with SM1 cells pre-treated with PBNP-PTT at varied laser power (1, 1.5, and 2 W) for 24 or 48 h in TexMACS™ medium (Miltenyi Biotec). Cells were then harvested and stained with Zombie Red™ Fixable viability dye (#423109) and fluorescent antibodies to detect CD3 (#100236), CD4 (#100443), CD8a (#100723), CD25 (#101917), and CD69 (#104539) (Biolegend).
2.7. Tumor inoculation
The backs of five-week-old female C57BL/6 mice were shaved prior to tumor cell inoculation. For the single tumor model, 1 million SM1 cells were inoculated into the backs of C57BL/6 mice. For two-tumor models, two injections of 1 million SM1 cells were inoculated simultaneously (synchronous model) or four days apart (metachronous model). Long-term surviving animals were rechallenged by inoculating 1 million SM1 cells into the mice 66 days post-treatment. Tumor growth was regularly monitored and measured by calipers.
2.8. In vivo PBNP-PTT and mAb treatments
When tumors became palpable (~ 60 mm3), mice were randomly divided into treatment groups. Mice were anesthetized prior to and during treatment using 2%–5% isoflurane. Tumors receiving PBNP-PTT were intratumorally injected with 2.5 mg/kg PBNPs. Following injection, tumors were irradiated with the NIR laser (808 nm; Laserglow Technologies, Toronto, ON, Canada) for 10 min, and temperatures were measured by thermal camera at one minute intervals (FLIR, Arlington, VA). Eyes were covered with opaque black cardboard during treatment to avoid eye damage by the laser. Mice receiving mAb treatments were intraperitoneally injected with 5, 10, or 15 mg/kg anti-CTLA-4 (clone 9D9), 15 mg/kg anti-PD-1 (clone RMP1–14), or 15 mg/kg anti-CD137 (clone 3H3) (all mAbs were purchased from BioXCell, West Lebanon, NH) every three days for two weeks (i.e., on days 1, 4, 7, 10, 13, 16, totaling 6 doses). After administering the individual treatments, the mice were monitored for tumor progression and survival.
2.9. Ex vivo immunological analysis
All antibodies were purchased from Biolegend unless otherwise stated. Synchronous tumor-bearing mice were euthanized 14 days after initial treatment, and their secondary tumors, spleens, lymph nodes, blood, and livers were harvested for further analysis. Secondary tumors were cut into smaller sections and digested using a tissue digestion mixture (1 g of Collagenese I (Sigma-Aldrich, C0130), 1 g of Collagenase IV (Sigma-Aldrich, C5138), 0.5 g of Hyaluronidase V (Sigma-Aldrich, H6254), 0.2 g of DNAase I, and Pierce Protease Inhibitor, ethylenediaminetetraacetic acid (EDTA) free (ThermoFisher, Waltham, MA; A32965) in 200 mL of Hank’s Balanced Salts Solution (HBSS) (Sigma-Aldrich, 55021C)), followed by preparation of single cell suspensions by passing the digested tumors through a series of 100 and 80 μm cell strainers. 1 million cells from each tumor sample were then stained with Zombie Aqua™ Fixable viability dye (#423102) and antibodies against CD3 (#147501 kit), CD4 (#147501 kit), and CD8 (#100737). Single cell suspensions of splenic cells were prepared by passing the isolated spleens through a 100 μm cell strainer using a plunger. Cells were centrifuged, and the cell pellet was suspended in ammonium–chloride–potassium (ACK) lysis buffer (ThermoFisher) to lyse red blood cells. Splenic T cells were magnetically isolated by negative selection using a mouse Pan T cell isolation Kit II (Miltenyi Biotec). They were then stained with a Zombie Aqua™ Fixable viability dye and antibodies against CD3 (#147501 kit), CD4 (#147501 kit), CD8 (#100737), CD69 (#104508), CD25 (#102020), PD-L1 (#135214), CD44 (#147501 kit), and CD62L (#147501 kit). Lymph nodes were cut into small sections and single cell suspensions were prepared by passing the isolated lymph nodes through 100 μm cell strainers using a plunger. The cells were then counted and stained using a Zombie Aqua™ Fixable viability dye and antibodies against CD11c (#117320), CD80 (#104714), and CD86 (#105007). Samples were then analyzed by flow cytometry using the FlowJo software. Blood was collected post-euthanasia and allowed to clot (first on ice, then at room temperature for 30 min) and then centrifuged to separate cells from serum. Serum was then collected and analyzed for cytokine/chemokine content using a custom magnetic bead-based Multiplexing Assay kit from R&D Systems and the Luminex MAGPIX Instrument with xPOTENT 4.3 software. When analyzed, values that fell below the detectable range of the Luminex Instrument were recorded as 0 pg/mL.
2.10. Liver histology
Harvested livers were fixed in 4% paraformaldehyde (PFA) and embedded in paraffin wax, followed by sectioning using a microtome, and staining with hematoxylin and eosin (H&E). Tissues were analyzed by a trained medical pathologist. The number of tumor metastatic infiltrates was recorded as clusters of greater than 10 neoplastic cells. Individual cells or smaller clusters in sinusoids were not included. Inflammatory scoring was evaluated adapted from previous literature [39]. Hepatic inflammation was evaluated in five liver tissue samples per animal, encompassing multiple lobes. Three H&E-stained tissue sections were evaluated for each animal. The slide with the greatest evidence of inflammation for each mouse was selected for more detailed analysis. All tissue sections on the slide were scanned for clusters of inflammatory cells (foci). The number of inflammatory cells was counted in each focus within three areas of the liver, including portal tracts, central veins, and lobular parenchyma (lobules). Each of the three areas of liver was analyzed for the number of inflammatory foci. Each inflammatory focus was assigned an inflammatory score of 0–4, dependent on the number of inflammatory cells in the focus (1: minimal (< 20 cells); 2: mild (21–50 cells); 3: moderate (51–150 cells); 4: severe (> 150 cells)). The average score and standard deviation were calculated of the 10 most affected foci were determined for each animal. One mouse in the age-matched naïve control group appeared to have signs of fatty liver disease.
2.11. Liver enzyme analysis
Blood from long-term surviving PBNP-PTT + aCD137-treated mice (n = 2) and naïve tumor-free age-matched C57BL/6 mice (n = 3) was obtained post-euthanasia and allowed to clot (first on ice, then at room temperature for 30 min) and then centrifuged to separate cells from serum. Serum was then carefully collected and analyzed for alanine aminotransferase (ALT) (GTP Assay Kit (Colorimetric); Catalog #KA1294, Novus Biologicals LLC, Centennial, CO, USA) and aspartate aminotransferase (AST) (Mouse AST SimpleStep ELISA Kit, ab263882, Abcam, Cambridge, UK) using protocols provided by the manufacturers.
2.12. Statistics
Statistical significance was determined by one-way ANOVA using Tukey’s multiple comparison test. Statistics on tumor growth curves were determined by two-way ANOVA using Tukey’s multiple comparison test and multiple t test, comparing averaged tumor volumes between each individual time point. Survival results were analyzed according to a Kaplan-Meier curve. The log-rank (Mantel-Cox) test was used to determine statistically significant differences in survival between groups. Values were considered statistically significantly different when p values were less than 0.05.
3. Results
3.1. PBNP-PTT increases the immunogenicity of melanoma cells, which directly activates T cells in a thermal dose-dependent manner in vitro
To uncover the immunogenic effects of PBNP-PTT on melanoma cells, we first established metrics for measuring immunogenicity in vitro. Specifically, we measured melanoma cell death, the upregulation of markers defining ICD, the induction of molecular patterns associated with immune cell engagement, and the direct activation of T cells after co-culture with PBNP-PTT-treated melanoma cells (Fig. 1(a)). To perform these analyses, PBNPs were synthesized and characterized for uniform size distributions, stability, and photothermal heating properties (Fig. S1 in the ESM) using methods previously described by our group [23, 37]. Using a constant PBNP concentration (0.15 mg/mL PBNPs), SM1 cells were subjected to PBNP-PTT at varied laser powers (0.75–2.0 W) for ten min in vitro. SM1 cells heated in a laser power-dependent manner, reaching the highest temperature (81.5 °C) at the highest laser power (2.0 W) (Fig. 1(b) and Fig. S2 in the ESM). The time-temperature plots were converted to cumulative equivalent minutes at 43 °C (∑CEM43), a parameter used to quantify the thermal dose [38], which illustrated that the thermal doses administered to the SM1 cells were also laser power-dependent (Fig. 1(c)).
Figure 1.
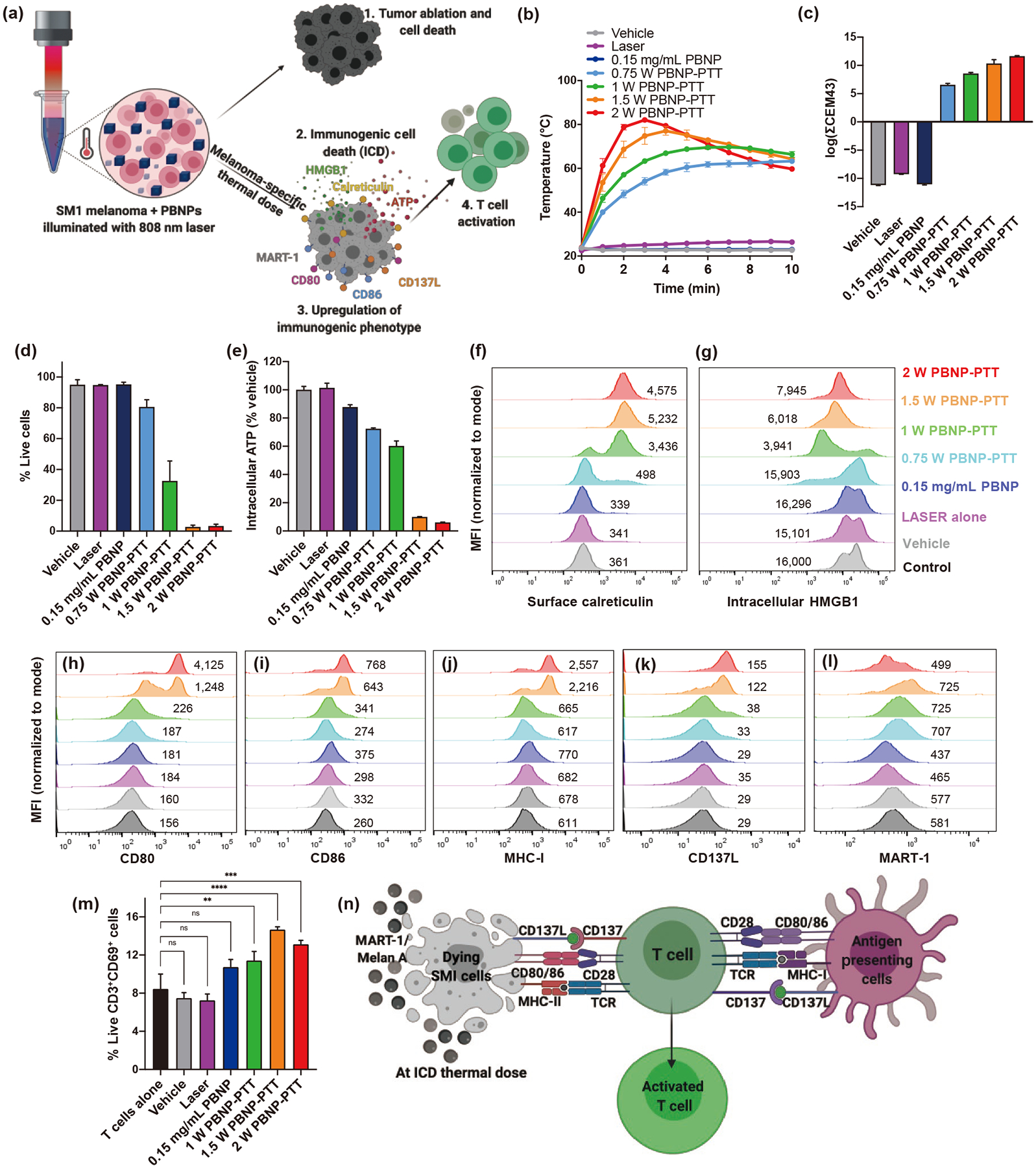
PBNP-PTT generates immunogenicity of SM1 cells in vitro. SM1 cells (10 million cells/mL) were treated with varied doses of PBNP-PTT or controls. (a) Effect of varying thermal doses of PBNP-PTT on immunogenicity was studied, as measured by tumor cell viability, ICD correlates, cellular markers, and activation of T cells. (b) Cell suspension temperature was measured every minute for ten min using a thermal camera. (c) Thermal dose (∑CEM43) values. (d) Viability, (e) intracellular ATP, (f) calreticulin, (g) intracellular HMGB1, (h) CD80, (i) CD86, (j) MHC1, (k) CD137L, and (l) MART-1 were visualized on SM1 cells 24 h after treatment. Values indicate MFI. (m) SM1 cells were left untreated (CTRL) or treated in vitro with Vehicle (PBS), PBNPs, or PBNP-PTT, and then co-cultured with ex vivo T cells isolated from spleens of naïve mice at an E:T ratio of 5:1. After 48 h, CD69 expression was measured on live T cells. (n) PBNP-PTT-treated SM1 cells express APC-like phenotype. Values represent means ± standard deviation (SD), n = 3/group; ns: not significant, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To determine whether the heat generated during PBNP-PTT elicited cytotoxicity and ICD in SM1 cells in vitro, cell death and the consensus biochemical correlates for ICD (i.e., release of intracellular ATP and HMGB1, and upregulation of cell surface calreticulin) were measured [28]. As expected, SM1 cells were killed in a thermal dose-dependent manner, with < 10% live cells remaining after treatment with ≥ 1.5 W laser power (Fig. 1(d) and Fig. S2 in the ESM). PBNP-PTT generated a laser power- and thermal dose-dependent decrease in intracellular ATP of SM1 cells, suggesting its release (Fig. 1(e)), with < 10% intracellular ATP remaining after treatment with ≥ 1.5 W laser power. Concurrently, PBNP-PTT generated an increase in SM1 cell surface calreticulin (Fig. 1(f)) and decreased intracellular HMGB1 (Fig. 1(g)) as measured by flow cytometry, suggesting the upregulation of cell surface calreticulin and the release of HMGB1 at laser powers ≥ 1 W. The maximum expression of the ICD correlates was attained at a thermal dose of 8.5–10.5 log(∑CEM43) (Fig. S3 in the ESM).
In addition to ICD, we investigated the ability of PBNP-PTT to upregulate molecules involved in antigen presentation and immune cell co-stimulation on SM1 cells in vitro. Interestingly, PBNP-PTT generated laser power- and thermal dose-dependent increases in the expression of CD80 (maximum MFI (mMFI) at 2 W: 4,125; Fig. 1(h)), CD86 (mMFI: 768; Fig. 1(i)), MHC-I (mMFI: 2557; Fig. 1(j)), and CD137 ligand (CD137L) (mMFI: 155; Fig. 1(k)) on SM1 cells. Melanoma antigen recognized by T cells (MART-1/Melan-A) was also increased with higher PBNP-PTT thermal dose (mMFI: 725 at 1.5 W), suggesting an increase in the antigenicity and immunogenicity potential of the cells in response to treatment (Fig. 1(l); see the gating strategy in Fig. S4 in the ESM).
To establish reproducibility of these effects in human cells, we administered PBNP-PTT to WM9 (Fig. S5 in the ESM) and WM793 (Fig. S6 in the ESM) human melanoma cell lines harboring the BRAFV600E mutation and predicted to be unresponsive to aPD-1 therapy [40–42], and illustrated consistent results. Briefly, both WM9 and WM793 tumor cells also generated thermal dose-dependent increases in ICD biochemical correlates and molecules involved in antigen presentation and immune cell co-stimulation. Maximum cell death and immunogenicity, as measured by the aforementioned markers, was elicited with PBNP-PTT using a 2 W laser power (12.8 log(∑CEM43)) for WM9 cells and 1.5 W laser power (10.6 log(∑CEM43)) for WM793 cells, correlating with the effects observed in murine SM1 cells.
Because PBNP-PTT upregulated molecules involved in antigen presentation and immune cell co-stimulation, we sought to uncover whether SM1 cells treated with PBNP-PTT could directly activate T cells. For this purpose, T cells were isolated from the spleens of naïve mice (not exposed to SM1 cells) and co-cultured ex vivo at a 5:1 effector to target cell (E:T) ratio with SM1 cells treated in vitro with either vehicle (water), PBNPs (0.15 mg/mL), or PBNP-PTT (0.15 mg/mL PBNP + 10 min laser illumination at 1, 1.5, or 2 W). After 48 h, expression of the activation marker CD69 was significantly increased on T cells after co-culture with SM1 cells treated with PBNP-PTT at all thermal doses tested (Fig. 1(m)). The highest T cell activation was observed with 1.5 W PBNP-PTT (14.7% CD3+/CD69+). Expression of CD69 and CD25, another T cell activation marker, was also measured in response to PBNP-PTT-treated SM1 cells at 5:1 and 10:1 E:T ratios after 24 and 48 h of co-culture; no other tested condition elicited direct T cell activation as measured by increased CD69 or CD25 expression (Fig. S7 in the ESM). Co-culture with SM1 cells (after vehicle and PBNP-PTT treatments) generated small but stastistically significant decreases in CD25 expression (Fig. S7 in the ESM). These data suggest that activation of T cells by surviving PBNP-PTT-treated cells, in a similar manner to antigen-presenting cells (APCs), can be achieved under specific thermal dose conditions (Fig. 1(n)).
The overall goal of these described in vitro studies was to establish the bounds of PBNP-PTT thermal dose that elicits immunogenicity in melanoma cells. Based on these studies, we have observed that the optimal PBNP-PTT is generated in vitro using a laser power of 1.5–2 W, which corresponds to 10.3–10.5 log(∑CEM43) for the tested melanoma cells. These thermal doses enable PBNP-PTT to generate optimal effects, that is, high percentage of tumor cell killing, induction of ICD, upregulation of immunostimulatory molecules, and direct T cell activation. Thus, PBNP-PTT should be administered at a thermal dose above 10.3 log(∑CEM43) for optimal cell death and immunogenicity.
3.2. PBNP-PTT eliminates local SM1 tumors but cannot eliminate a distant tumor, and is not improved with the addition of aPD-1 ICI therapy
To determine the efficacy of ICI therapy on SM1 melanoma, we separately investigated the effects of aPD-1 and aCTLA-4 mAb immunotherapy in the SM1 melanoma mouse model. As predicted based on its mutation status (BRAFV600E), as well as previous descriptions in the literature [35, 36], we verified that, using the specific dosing scheme tested herein, neither aPD-1 nor aCTLA-4 ICI therapy was able to significantly slow the growth of SM1 tumors nor improve the survival of SM1 tumor-bearing mice (Fig. S8 in the ESM).
Having established that SM1 melanoma did not respond to ICI therapy at the doses tested, and that PBNP-PTT enabled SM1 cell death and immunogenicity in vitro, we next sought to understand the effect of PBNP-PTT on SM1 melanoma in vivo. SM1 tumor-bearing mice were treated with PBNP-PTT or left untreated. PBNP-PTT eliminated 100% of treated tumors, while tumors of mice left untreated (CTRL) progressed (Fig. 2(a)), resulting in a significant improvement in survival for mice treated with PBNP-PTT (median survival (MS): not achieved in the study) versus CTRL (MS: 16 days) (Fig. 2(b)). Long-term surviving PBNP-PTT-treated mice (n = 3, 75%) were rechallenged with SM1 tumors ~ 70 days after initial treatment. 0% of the rechallenged mice rejected the tumor, and 100% succumbed to tumor burden within 32 days after rechallenge (Fig. 2(b)). These data suggest that although PBNP-PTT is effective for local tumor control in vivo, it does not generate an effective immunological memory response.
Figure 2.
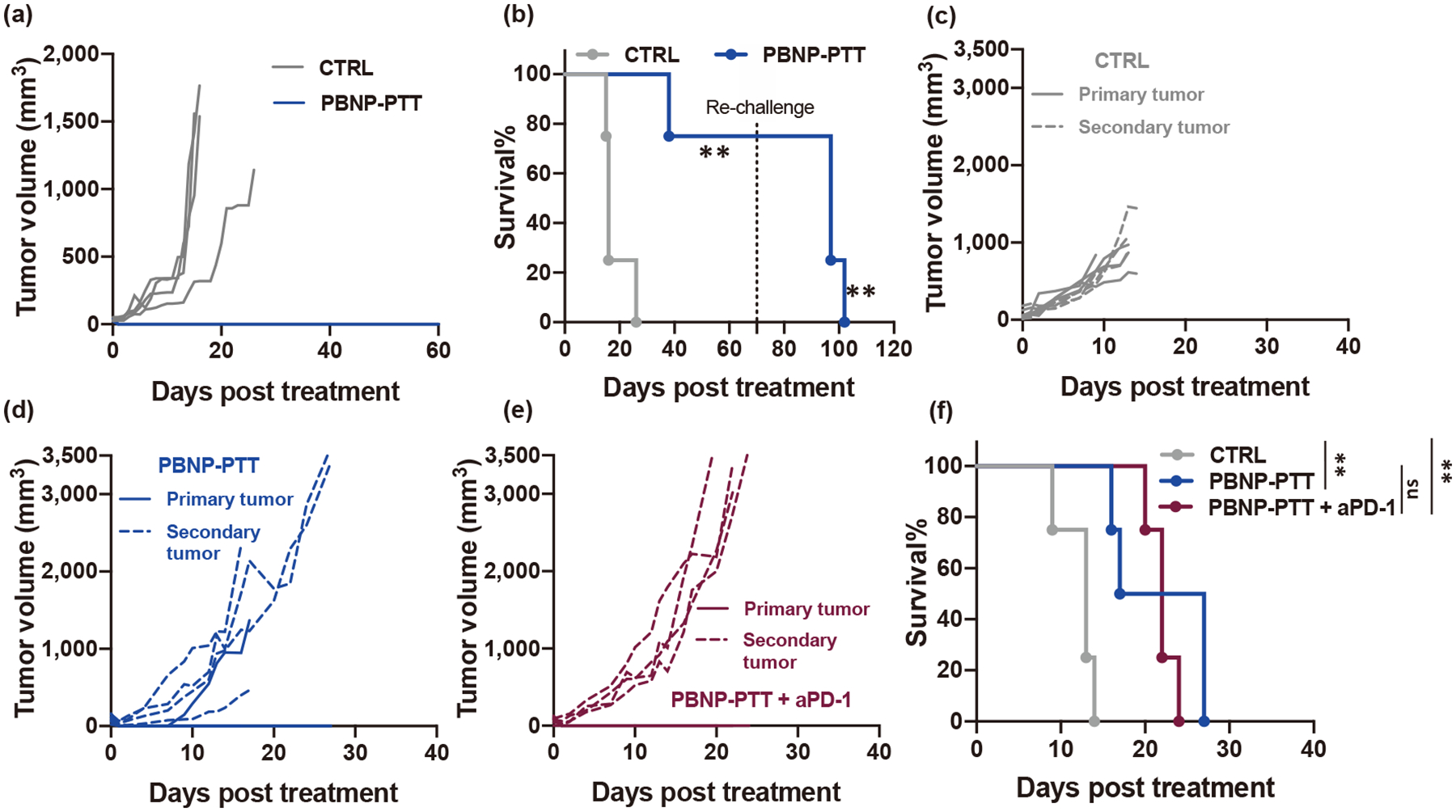
PBNP-PTT effectively treats local but not distal SM1 melanoma in vivo. (a)–(b) Effect of PBNP-PTT on tumor growth and survival. Tumor-bearing mice were left untreated (CTRL) or treated with PBNP-PTT. (a) Tumor growth observed in the treatment groups. (b) Survival of tumor-bearing animals post-treatment and rechallenge. n = 4/group, **p < 0.01. (c)–(f) Effect of PBNP-PTT on tumor growth and survival in a two tumor model. Tumor-bearing mice were left untreated (CTRL) or treated with PBNP-PTT or PBNP-PTT + aPD-1. When used, PBNP-PTT was administered to only one (“primary”) tumor, and the other (“secondary”) tumor was left untreated. Tumor growth observed in mice treated with (c) CTRL, (d) PBNP-PTT, and (e) PBNP-PTT + aPD-1. Solid lines represent the primary (treated) tumor, dashed lines represent the secondary (untreated) tumor. (f) Survival of tumor-bearing animals post-treatment. n = 4/group; **p < 0.01, ns: not significant.
To determine whether local treatment with PBNP-PTT generated a systemic immune response able to impact the growth of a secondary tumor, SM1 cells were simultaneously inoculated into contralateral flanks of mice to generate two synchronous tumors. PBNP-PTT was administered to one tumor (designated “primary” tumor), and the other tumor was left untreated (designated “secondary” tumor). While PBNP-PTT-treated primary tumors were eliminated, the secondary untreated tumor growth was unchanged compared to CTRL (Figs. 2(c) and 2(d)). The tumor burden endpoints for each individual tumor in the CTRL group were lower than in the groups receiving PBNP-PTT because mice in the CTRL group had two tumors (thus lowering the endpoint criteria) whereas those in the PBNP-PTT-treated groups had only one tumor. These results confirm our previous observations and suggest that although PBNP-PTT is effective in treating single tumors, the immunogenic effects cannot eliminate established distal disease, and thus may benefit from combination with immunotherapy. However, mice treated with the combination treatment of PBNP-PTT + aPD-1 did not exhibit any benefit over PBNP-PTT alone in this two-tumor model (MS: 22) (Figs. 2(e) and 2(f)). Although survival was marginally improved for mice treated with PBNP-PTT (MS: 22 days) over CTRL (MS: 13.5 days), all PBNP-PTT and PBNP-PTT + aPD-1-treated mice succumbed to tumor burden and there were no long-term survivors (Fig. 2(f)). These data suggest that PBNP-PTT may require a different complementary immunotherapy intervention to exert improved therapeutic benefit for this melanoma model.
To uncover whether the lack of responsiveness to PBNP-PTT + aPD-1 therapy was due to a lack of expression of PD-L1, a known ligand for PD-1, we treated SM1 cells with varying doses of PBNP-PTT and found that PD-L1 was unchanged or decreased from basal levels in response to all tested conditions of PBNP-PTT in SM1 melanoma cells and in both human melanoma cell lines (Fig. S9 in the ESM), suggesting that the effects of PBNP-PTT are not improved by combining with aPD-1 therapy.
3.3. PBNP-PTT combined with aCD137 generates an abscopal effect that significantly improves the long-term survival of metachronous tumor-bearing mice
In light of the findings illustrating the increase of CD137L on SM1 melanoma cells in response to PBNP-PTT in vitro, we utilized aCD137, a T cell agonistic mAb, as a second component of our nanoimmunotherapy in combination with PBNP-PTT. We sought to uncover if the antitumor effects of the combination treatment could be effective in a model of distal melanoma. To test this hypothesis, we utilized bilateral metachronous SM1 tumors with four days between tumor cell inoculations. The treatment groups were: CTRL (untreated), PBNP-PTT, aCD137, and PBNP-PTT + aCD137. In CTRL animals, both tumors were untreated. When used, PBNP-PTT was administered once locally to only the first established (“primary”) tumor, while the secondary tumor was left untreated (Fig. S10 in the ESM). When used, aCD137 was systemically delivered via i.p. administration. This setup enabled us to observe a potential abscopal effect (Fig. 3(a)). Primary and secondary tumors of CTRL mice rapidly progressed (Fig. 3(b)). Interestingly, aCD137 treatment did not affect primary tumor growth compared to CTRL, but prevented 100% of secondary tumors in the metachronous two-tumor model (Fig. 3(c)), suggesting that it could mount an effective antitumor immune response to attack a nascent tumor, but did not have the cytotoxic capability to eliminate an established tumor. 100% of PBNP-PTT-treated primary tumors were eliminated, and secondary tumors exhibited significantly delayed growth kinetics compared to CTRL (Fig. 3(d)). This pattern suggests that PBNP-PTT exerts both a cytotoxic effect to the primary tumor and a suboptimal abscopal antitumor immune response that was able to slow untreated secondary tumor growth, although unable to completely prevent its eventual growth. Strikingly, 60% of mice treated with PBNP-PTT + aCD137 exhibited complete primary tumor elimination and prevention of secondary tumor growth (Fig. 3(e)), suggesting the synergistic advantage of combining the two treatment modalities. Comparative tumor growth rates were illustrated in superimposed curves (Fig. 3(f)). These effects on tumor growth resulted in significantly improved survival benefit for mice treated with PBNP-PTT (MS: 40 days) or PBNP-PTT + aCD137 (MS: not achieved during the study) compared to no treatment (MS: 17 days) and aCD137 treatment (MS: 17 days) (Fig. 3(g)). These data suggest that combining local PBNP-PTT with systemic aCD137 administration enables robust abscopal efficacy against SM1 tumors in vivo.
Figure 3.
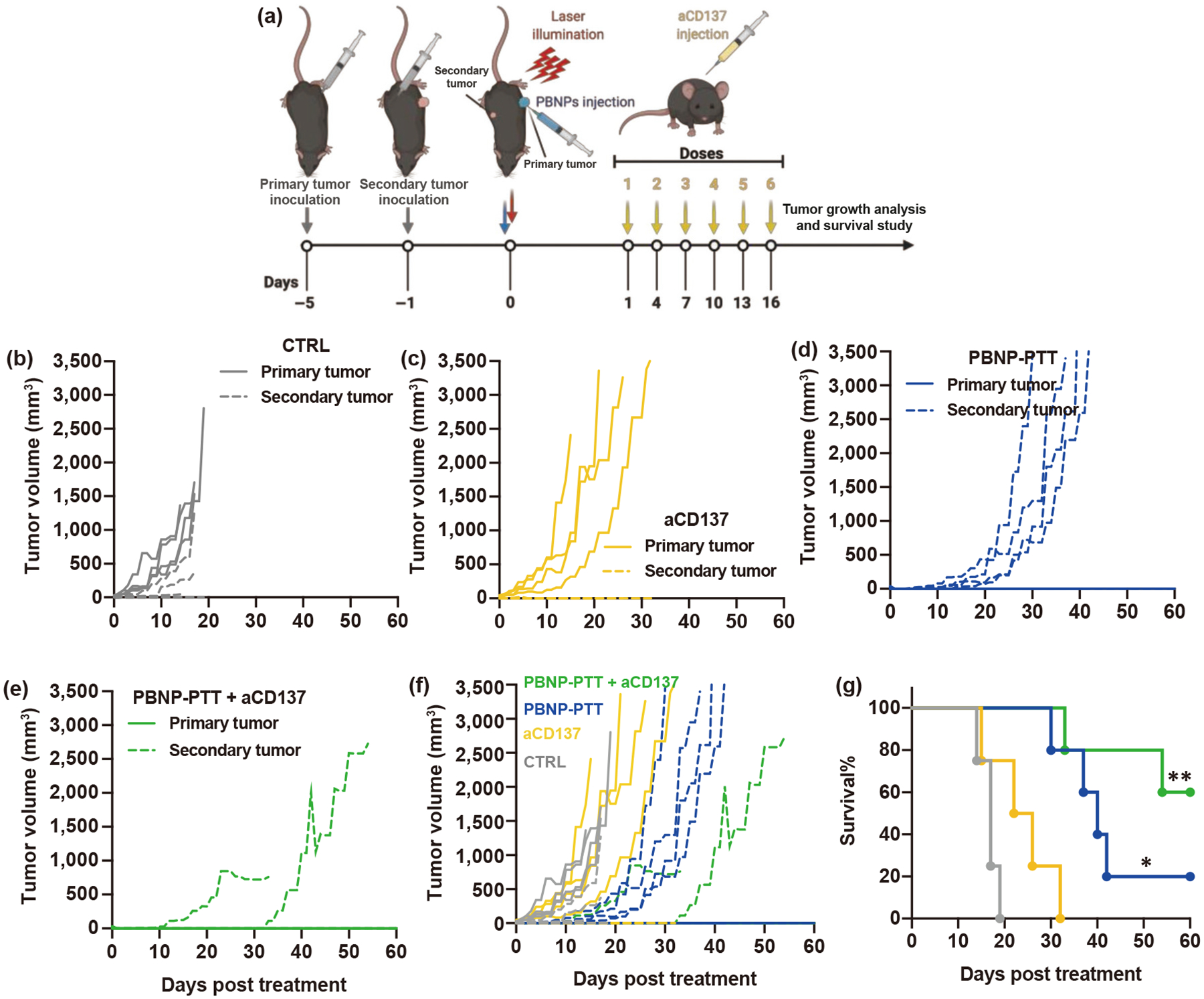
PBNP-PTT + aCD137 improves the survival of metachronous tumor-bearing mice through abscopal effect. (a) Schematic overview of the study. Two SM1 tumors were inoculated into opposite sides of C57BL/6 mice four days apart. Secondary tumors were left untreated. Tumor growth observed in mice treated with (b) CTRL, (c) aCD137, (d) PBNP-PTT, and (e) PBNP-PTT + aCD137, with (f) tumor growth curves superimposed for comparison. Each line represents an individual mouse; solid lines represent the primary tumors and dashed lines represent the secondary tumors. (g) Survival of tumor-bearing animals post-treatment. n = 4–5/group; *p < 0.05, **p < 0.01 compared with CTRL and aCD137.
3.4. PBNP-PTT combined with aCD137 generates an abscopal antitumor effect via tumor-infiltrating CD8+ T cells and systemic activation of DCs and T cells
Having illustrated the treatment benefit of PBNP-PTT + aCD137 on bilateral SM1 melanoma tumors in vivo, we sought to identify the key mechanistic components of immunity driving the observed abscopal efficacy. In order to analyze infiltrating immune cells in an established secondary tumor, a synchronous, rather than metachronous, bilateral tumor model was generated. This model enabled the collection of secondary tumors not present in all treatment groups in the metachronous model. The treatment groups were CTRL (untreated), PBNP-PTT, aCD137, and PBNP-PTT + aCD137. When used, PBNP-PTT was administered once locally to only one tumor denoted as “primary,” while “secondary” tumors were left untreated. When used, aCD137 was systemically delivered via i.p. administration (Fig. 4(a)). CTRL mice rapidly grew both primary and secondary tumors (Figs. 4(b) and 4(c)). Unlike the metachronous model, aCD137 was unable to impact the growth of either tumors and all aCD137-treated mice rapdily grew both primary and secondary tumors, likely due to the higher tumor burden. PBNP-PTT, alone and in combination with aCD137, eliminated 100% of primary tumors (Figs. 4(b) and 4(c)). PBNP-PTT + aCD137 significantly slowed secondary tumor growth (1.68-fold reduction in tumor volume compared to CTRL at the day of euthanasia), confirming the abscopal effect (Fig. 4(c)). Secondary tumors were not eliminated, as they were in mice treated with PBNP-PTT + aCD137 in the metachronous model, since the synchronous model comprised two established tumors, thus representing a higher initial tumor burden. After 14 days (a timepoint established before treatment), mice were euthanized, and tumors, lymph nodes, spleens, blood, and livers were harvested for analysis. To illustrate the abscopal effect on metastasis, livers were harvested and analyzed for the presence of metastatic foci. 60% of livers from CTRL and aCD137-treated mice harbored tumor foci (>10 tumor cells) (Fig. S11 in the ESM), as compared to mice treated with PBNP-PTT (20%) or PBNP PTT + aCD137 (20%), suggesting that PBNP-PTT prevented the occurrence of hepatic metastases via abscopal immune effects.
Figure 4.
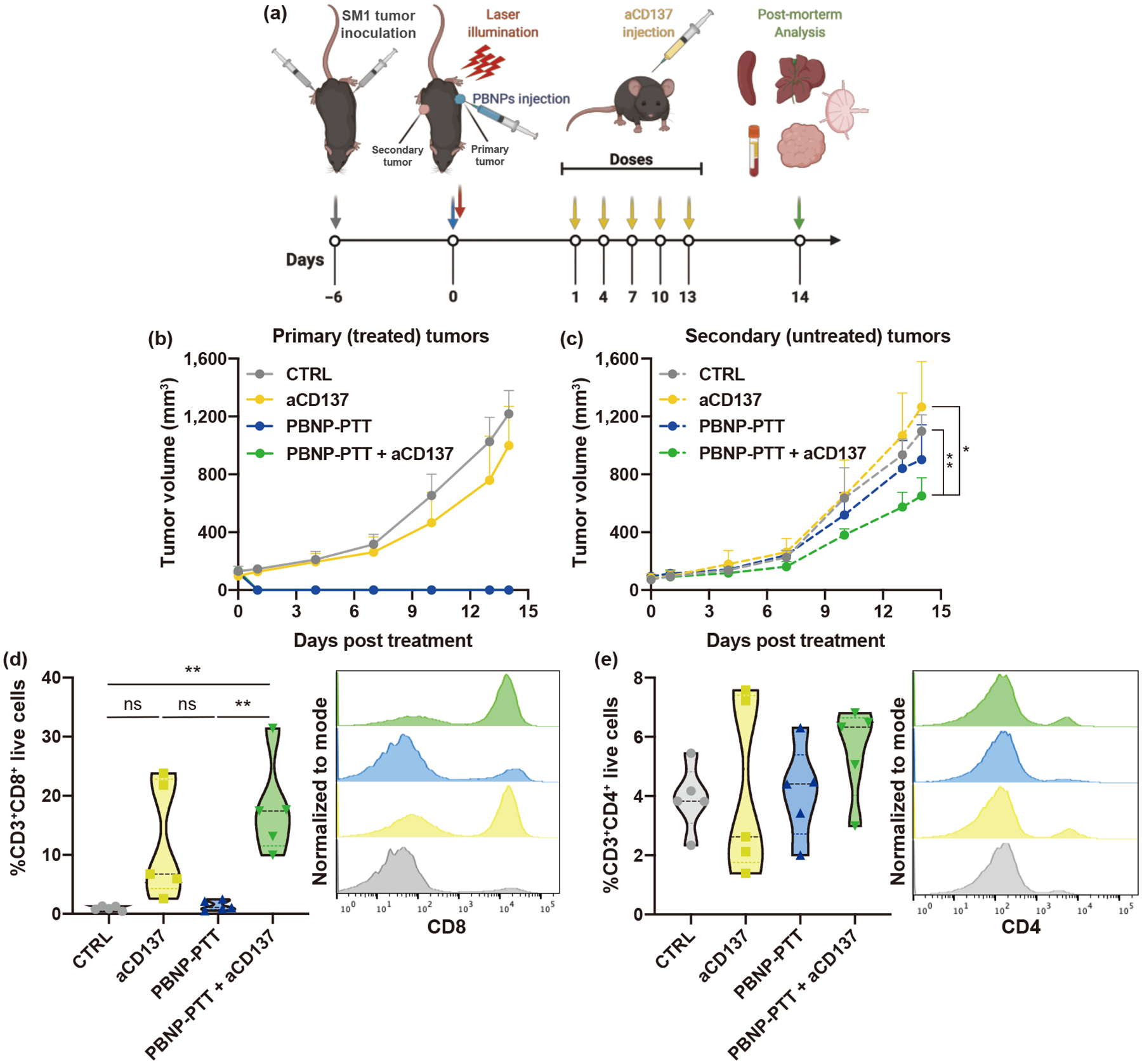
PBNP-PTT + aCD137 generates abscopal antitumor effect via infiltrating cytotoxic T cells. (a) Schematic overview of the study. Two SM1 tumors were inoculated into opposite sides of C57BL/6 mice simultaneously. Secondary tumors were left untreated. (b) Primary and (c) secondary tumor growth. Secondary tumor-infiltrating (d) CD8+ T cells and (e) CD4+ T cells after 14 days. Values represent means ± SD, n = 5/group; ns: not significant, *p < 0.05, **p < 0.01.
To understand the mechanism through which PBNP-PTT + aCD137 exerted robust abscopal effect, the presence of tumor-infiltrating lymphocytes (TILs) in secondary tumors was investigated by flow cytometry. The percentage of CD8+ T cells observed in tumors was significantly higher in PBNP-PTT + aCD137-treated mice (17%; Fig. 4(d) and Fig. S12 in the ESM) compared with CTRL (< 1%) and PBNP-PTT-treated mice (< 1%), suggesting that CD8+ T cell infiltration may drive the responses seen in the secondary tumors. Although not statistically significant, aCD137 monotherapy also marginally increased CD8+ T cell infiltration into secondary tumors (Fig. 4(d) and Fig. S12 in the ESM). The content of CD4+ T cells in tumors was statistically identical across treatment groups (2%–7%; Fig. 4(e) and Fig. S12 in the ESM).
PBNP-PTT + aCD137-treated mice also exhibited significantly higher DC maturation and activation in the inguinal lymph nodes as measured by a significantly increased proportion of CD11c+ cells (21.6%, Fig. 5(a) and Fig. S13 in the ESM) and CD11c+/CD80+ cells (20.4%, Fig. 5(b) and Fig. S13 in the ESM) compared to all other groups. CD11c+/CD86+ cells were not statistically different across treatment groups compared to CTRL, likely due to the high variance of CD11c+/CD86+ DCs in the lymph nodes of CTRL mice (24%–66%) (Fig. 5(c)). Treatment with aCD137 also generated significantly increased CD11c+ (12.2%) and CD11c+/CD80+ (13.3%) cells in the lymph nodes compared to CTRL and PBNP-PTT treatments, but these populations were significantly lower than those found in mice treated with PBNP-PTT + aCD137. These observations suggested that PBNP-PTT + aCD137 elicited systemic antitumor immune responses driven by DCs. Because these analyses were performed 14 days after the administration of PBNP-PTT, the acute effects of PBNP-PTT on DC activation and maturation were not measured. Based on our prior data [22], we predicted that PBNP-PTT would generate short-term increases in CD80+ and CD86+ DC populations in the lymph nodes.
Figure 5.
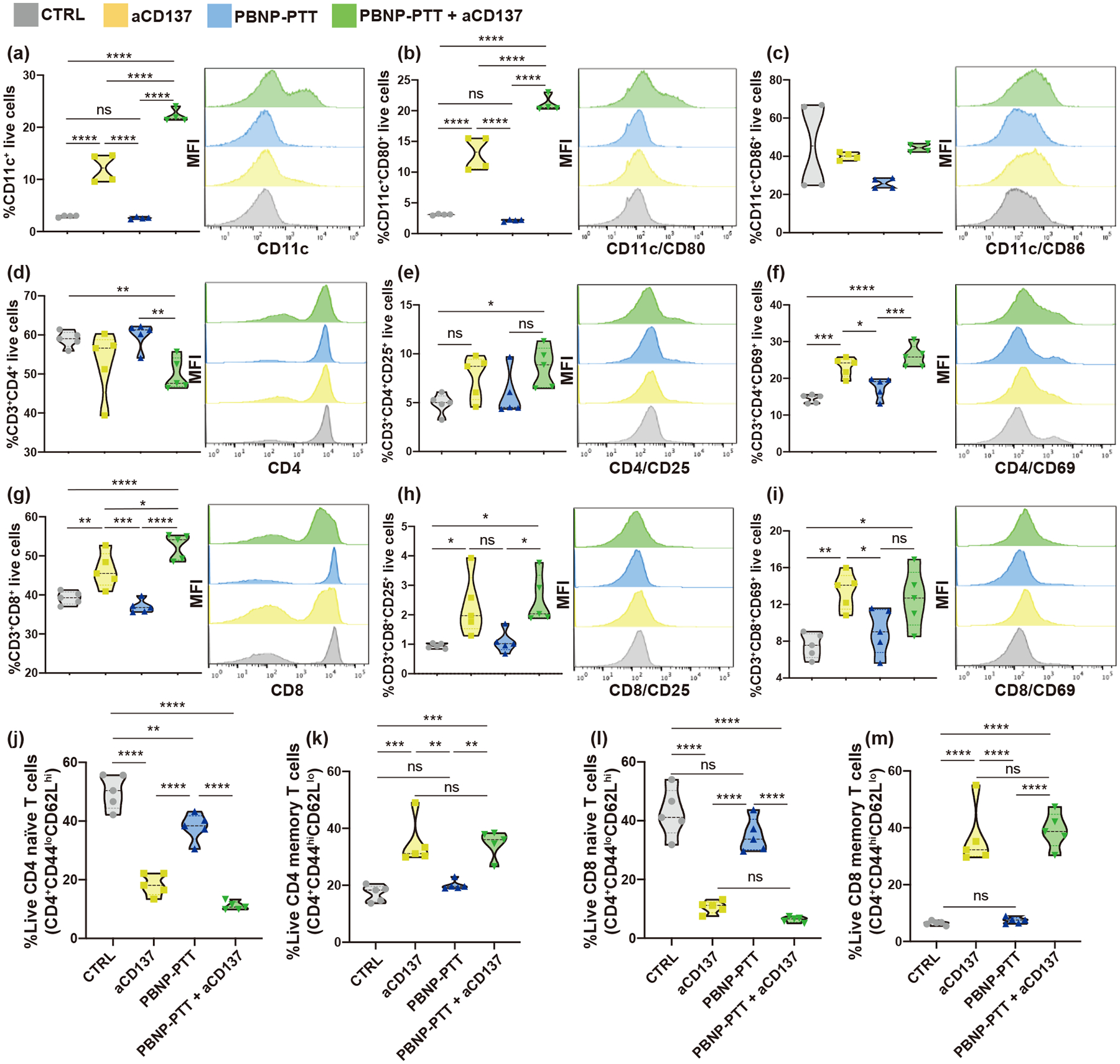
PBNP-PTT + aCD137 enhances DC activation in lymph nodes and generates systemic T cell activation and memory. Synchronous SM1 tumors were established and treated as described. Lymph nodes were harvested and analyzed for (a) CD11c, (b) CD80, and (c) CD86 expression. Spleens were harvested and analyzed for (d)–(f) CD4, (g)–(i) CD8, ((e) and (h)) CD25, ((f) and (i)) CD69, and ((j)–(m)) CD62 and CD44 expression levels. Values represent means ± SD, n = 5/group; ns: not significant, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
To complement the observations in the secondary tumors and lymph nodes, spleens were analyzed for T cell activation by flow cytometry 14 days after treatment initiation (24 h after final aCD137 administration). PBNP-PTT + aCD137 treatment generated a significant decrease in CD4+ splenic T cells (47%) compared to CTRL (59%) and PBNP-PTT (61%) treatment (Fig. 5(d) and Fig. S14 in the ESM). Interestingly, despite the decrease in CD4+ T cell populations in PBNP-PTT + aCD137-treated mice, the CD4+ T cells appeared to be significantly activated, as seen by their increased expression of CD25 (9% vs. 5% in CTRL; Fig. 5(e) and Fig. S14 in the ESM) and CD69 (25.7% vs. 14% in CTRL; Fig. 5(f) and Fig. S14 in the ESM). aCD137-treated mice also exhibited significantly increased CD4+/CD69+ cells (24.2%) compared to CTRL and PBNP-PTT treatments. PBNP-PTT + aCD137 treatment generated a significant increase in splenic CD8+ T cells (64%) compared to CTRL (40%), aCD137 (45%), and PBNP-PTT (37%) (Fig. 5(g) and Fig. S15 in the ESM), complementing the observed increase in tumor-infiltrating CD8+ T cells. Importantly, CD25 (2%; Fig. 5(h) and Fig. S15 in the ESM) and CD69 (13%; Fig. 5(i) and Fig. S15 in the ESM) expression were both significantly increased in the CD8+ T cells in the spleens of mice treated with PBNP-PTT + aCD137, suggesting that the treatment caused a systemic T cell activation. Splenic T cells of treated mice also showed no change in T cell exhaustion marker, PD-1, compared to CTRL (Fig. S16 in the ESM). To further elucidate the T cell response driving abscopal efficacy in SM1 melanoma-bearing mice, splenic T cells were analyzed for CD4+ and CD8+ naïve (CD62Lhi/CD44lo) versus memory (CD62Llo/CD44hi) markers. Strikingly, PBNP-PTT + aCD137 caused a significant decrease in CD4+ naïve (10%; Fig. 5(j)) and a significant increase in CD4+ memory T cells (36%, Fig. 5(k)). The same trend was observed in the CD8+ T cell subset (6.4% naïve; 39% memory) (Figs. 5(l) and 5(m); see the gating strategy in Fig. S17 in the ESM). These data suggested that not only did the combination nanoimmunotherapy cause DC and T cell activation and infiltration, but also generated a T cell-mediated immunological memory response against SM1 melanoma. A similar pattern of CD4+ and CD8+ memory T cell generation was observed in mice treated with aCD137 monotherapy, although this treatment did not slow tumor growth nor increase survival benefit. These findings suggested the importance of the localized PBNP-PTT in initiating the antitumor response that was then potentiated by aCD137 in our combination nanoimmunotherapy.
To further characterize the systemic immune response generated by PBNP-PTT + aCD137, serum was harvested from mice 14 days after treatments were initiated, and analyzed for cytokines and chemokines associated with T cell-mediated immunity. TNFα, IL-10, Fas ligand, MIP-1a, and RANTES were chosen as representative cytokines/chemokines involved in an inflammatory immune response [43–46]. IL-5 was investigated as it has shown different immunological functions dependent on its context [47–49]. Measured by magnetic multiplexing, PBNP-PTT + aCD137 significantly increased serum concentrations of TNFα (4.5 pg/mL), IL-5 (23.5 pg/mL), IL-10 (14.1 pg/mL), Fas ligand (73.4 pg/mL), MIP-1a (7.1 pg/mL), and RANTES (26.8 pg/mL) compared to CTRL (0, 13, 2.8, 0, 0, 0 pg/mL, respectively) and PBNP-PTT treatment (0, 15, 4.3, 0, 0.4, 0 pg/mL, respectively) (Figs. 6(a)–6(f)). TNFα (2.1 pg/mL), IL-10 (14.4 pg/mL), Fas ligand (122.6 pg/mL), MIP-1a (5.6 pg/mL), and RANTES (24.8 pg/mL) were also significantly increased by aCD137 treatment alone. However, PBNP-PTT + aCD137 significantly increased serum IL-5 expression compared to all other groups (Fig. 6(b)). These changes in serum cytokines and chemokines suggested the engagement of a systemic antitumor immune effect driving the observed responses. It is important to note that these measurements represented a snapshot in time, based on when the serum was harvested from the animal (14 days after PBNP-PTT and 24 h after the final aCD137 dose). As such, these results were limited in scope and did not represent the dynamic cytokine landscape likely to adapt over time.
Figure 6.
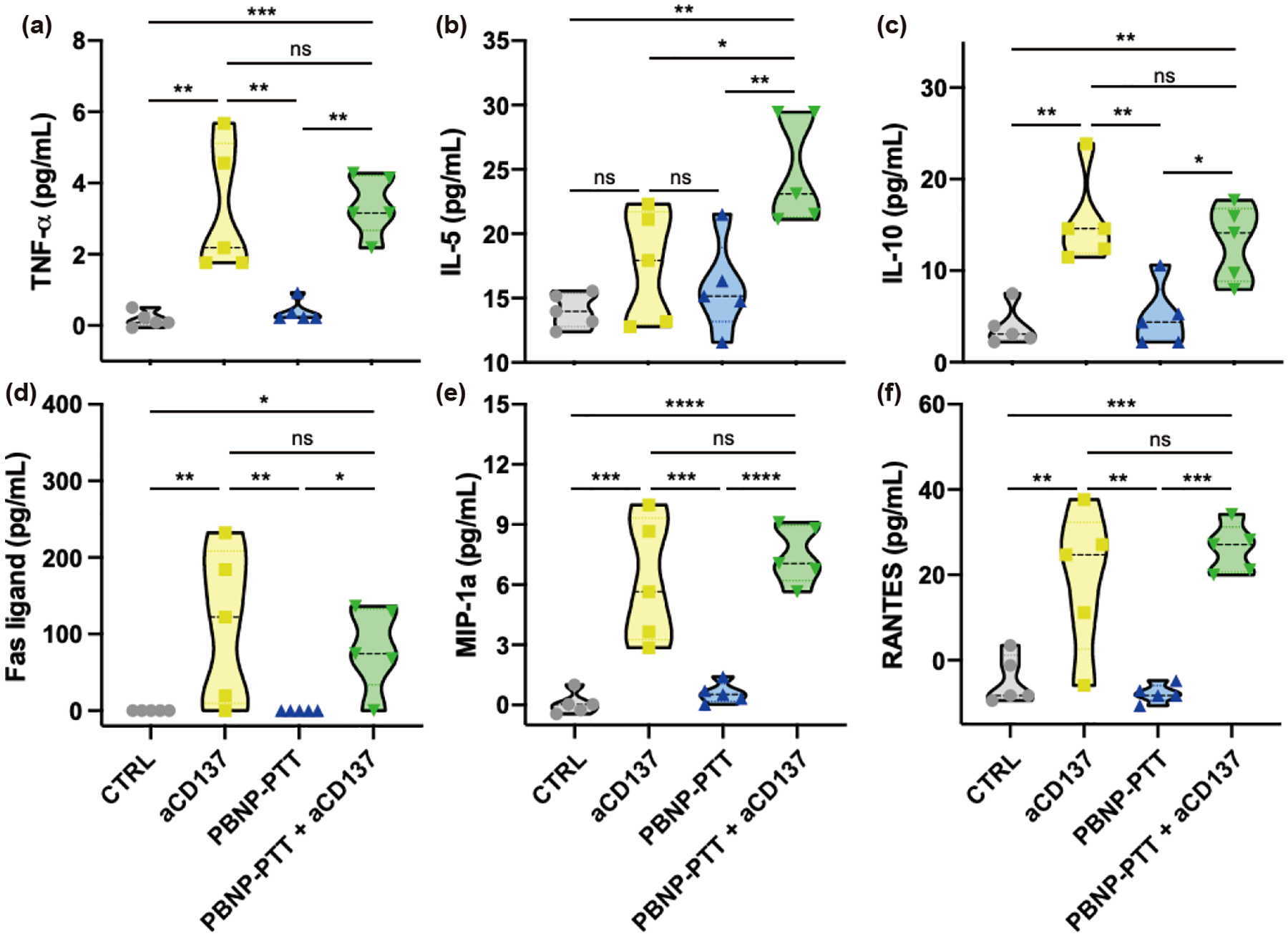
PBNP-PTT + aCD137 generates an increase in serum cytokines/chemokines associated with T cell activation. Synchronous SM1 tumors were established and treated as previously described. On Day 14, blood was harvested and serum was analyzed for (a) TNFα, (b) IL-5, (c) IL-10, (d) Fas ligand, (e) MIP-1a, and (f) RANTES expression. Normalized values in the negative range are listed as “0” pg/mL. Values represent mean ± SD, n = 5/group; ns: not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
3.5. PBNP-PTT combined with aCD137 generates T cell-mediated immunological memory
Having established the therapeutic efficacy of PBNP-PTT + aCD137 to treat SM1 melanoma tumors in vivo and generate both CD4+ and CD8+ memory T cells, we sought to demonstrate the ability of the memory T cells to prevent melanoma recurrence in vivo. To this end, we utilized a single SM1 tumor model in C57BL/6 mice. The nanoimmunotherapy or control treatments were administered to tumor-bearing mice, and the mice were monitored for tumor growth and long-term survival. After completion of the treatments, all long-term surviving animals were rechallenged with SM1 cells (Fig. 7(a)). aCD137 monotherapy was unable to significantly slow tumor growth compared to CTRL, and thus generated no significant survival benefit (Figs. 7(b) and 7(c)). Consistent with previous studies, PBNP-PTT eliminated 100% of treated tumors and generated a significant increase in survival. However, 60% of treated mice succumbed to their disease (Fig. 7(d)). Importantly, PBNP-PTT + aCD137 enabled complete tumor cure and long-term survival in 60% of treated mice (Fig. 7(e)). Long-term surviving mice treated with PBNP-PTT (n = 2) and PBNP-PTT + aCD137 (n = 3) were rechallenged with SM1 cells 66 days after the initial treatment to examine immunological memory. Strikingly, 66% of mice treated with PBNP-PTT + aCD137 rejected tumor rechallenge (Figs. 7(e) and 7(f)) and exhibited complete tumor-free survival (Fig. 7(g)), suggesting that immunological memory was established. Naïve age-matched mice and rechallenged PBNP-PTT-treated mice rapidly succumbed to tumor burden (Fig. S18 in the ESM). Comparative tumor growth kinetics are illustrated in averaged curves (Fig. 7(f)). These effects in tumor growth resulted in significantly improved survival benefit for mice treated with PBNP-PTT (MS: 44 days) and PBNP-PTT + aCD137 (MS: not achieved by day of rechallenge) compared to CTRL (MS: 19 days) and aCD137 treatment (MS: 19 days) (Fig. 7(g)). These data complemented the observations of increased CD4+ and CD8+ memory T cells in the spleens of PBNP-PTT + aCD137-treated mice, and illustrated that antitumor immune memory was generated, manifesting in the rejection of melanoma rechallenge.
Figure 7.
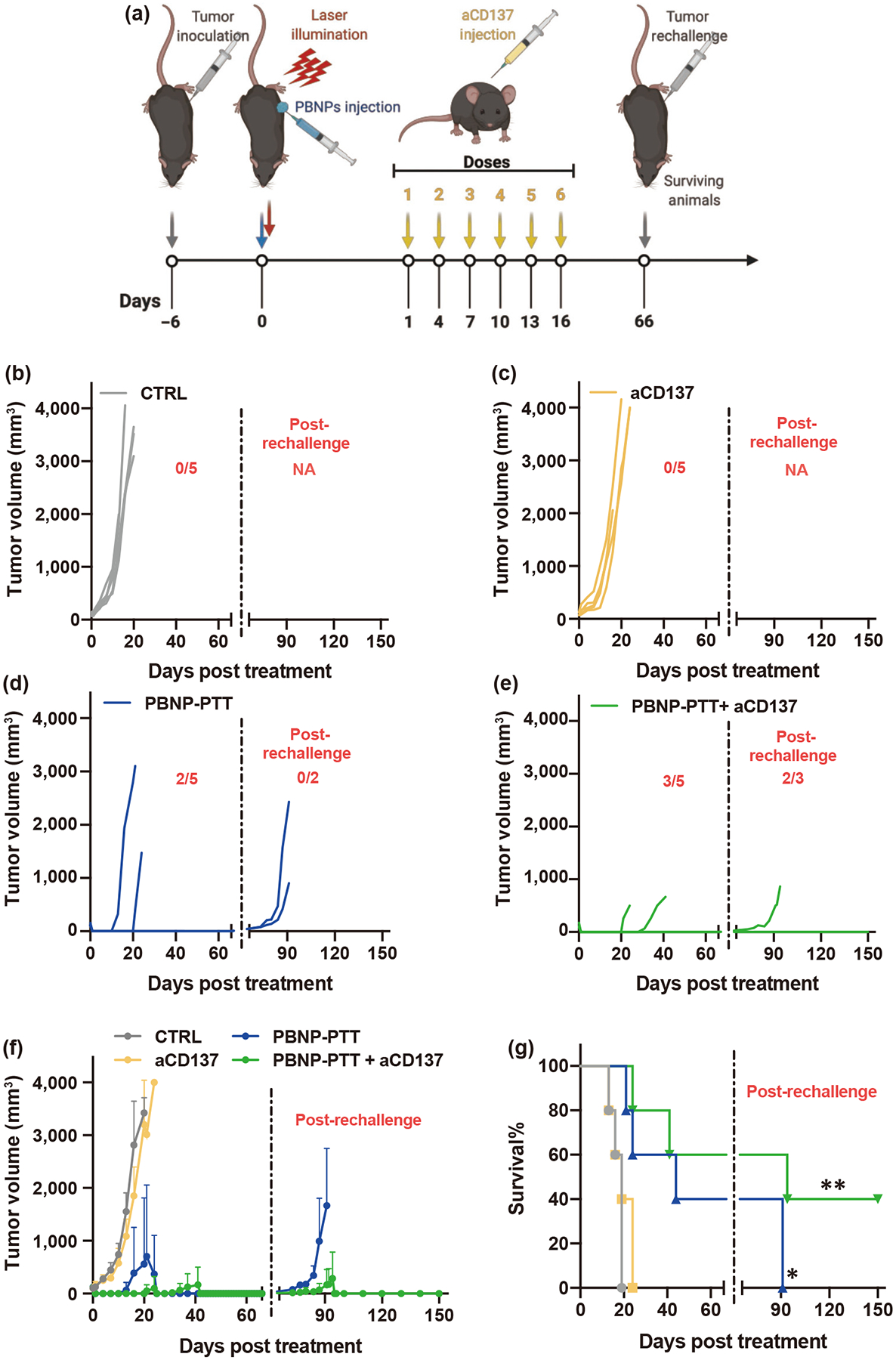
PBNP-PTT + aCD137 improves the survival of single tumor-bearing mice and generates immunological memory. (a) SM1 tumor-bearing mice were left untreated (CTRL) or treated with PBNP-PTT, aCD137, or PBNP-PTT + aCD137. Long-term surviving mice were rechallenged with SM1 cells. (b)–(f) Tumor growth observed in the treatment groups. (g) Survival of tumor-bearing animals post-treatment. n = 5/group; *p < 0.05, **p < 0.01. The values given in red represent number of surviving animals before and post-rechallenge.
3.6. PBNP-PTT combined with aCD137 generates acute hepatotoxicity similar to aCD137 monotherapy, but appears to recover over time
Critical to the success of aCD137 therapy is the reduction of hepatotoxicity [15, 39, 50]. We therefore examined inflammation in harvested livers to quantify acute toxicity 14 days after PBNP-PTT and 24 h after the final aCD137 i.p. injection. Livers from CTRL and PBNP-PTT-treated animals appeared healthy and non-inflamed (Fig. S19 in the ESM). As predicted, aCD137 monotherapy generated acute hepatitis and significant increases in inflammatory foci (90 foci), primarily around the central veins, compared to CTRL (8 foci) and PBNP-PTT-treated animals (12 foci; Fig. S19 in the ESM). Livers from PBNP-PTT + aCD137-treated mice also showed increased liver inflammation (142 foci; Fig. S19 in the ESM) compared to controls (Fig. S19 in the ESM). However, we predicted that the synergy generated by the combination therapy may enable a lower effective aCD137 dose, thereby mitigating aCD137-mediated hepatotoxicity. It is also noteworthy that this assessment was made 24 h after injection of aCD137, and therefore it may represent short-term hepatic inflammation.
Thus, to investigate the long-term effect of PBNP-PTT + aCD137 on hepatotoxicity, we harvested livers from the long-term surviving PBNP-PTT + aCD137-treated mice that had recovered from both the primary SM1 tumor and rechallenged SM1 tumor (226 days post-treatment). Although livers from PBNP-PTT + aCD137-treated mice exhibited significantly increased inflammation in the portal tracts compared to age-matched controls, this inflammation was scored as mild, and there was no increased inflammation in the lobules, central veins, or overall (Fig. S20 in the ESM). Additionally, serum levels of ALT and AST were measured from these mice at the same timepoint as an indication of liver damage. Long-term surviving PBNP-PTT + aCD137-treated mice had statistically identical serum ALT and AST (Fig. S20 in the ESM) levels to their age-matched controls, suggesting normal liver health. These findings indicated that although PBNP-PTT + aCD137 induced acute hepatotoxicity, there were no long-term liver health concerns above normal aging effects.
4. Discussion
Our work illustrates the efficacy of PBNP-PTT combined with agonistic aCD137 mAbs, a novel nanoimmunotherapy for the treatment of melanoma. As monotherapies, both PBNP-PTT and aCD137 have limitations. PBNP-PTT provides a strong cytotoxic effect to treated tumors [22–24, 51–54], and while it appears to generate tumor immunogenicity and a mild abscopal effect [22–24]. It is ineffective at the cytotoxic thermal dose to elicit a robust antitumor immune response capable of eliminating distal or metastatic tumors [31]. Alternatively, aCD137 generates T cell-mediated antitumor immune effects [33, 55–58], but is incapable of debulking established tumors [50] and generates hepatotoxicity [39, 59, 60]. Thus, the complementary effects of PBNP-PTT and CD137 agonism are combined in our study to generate beneficial synergistic responses in the SM1 model of melanoma.
We demonstrate herein the ability of PBNP-PTT to generate efficient cytotoxicity and elimination of primary tumors (Fig. 2) in addition to eliciting immunogenicity of the treated tumor cells (Fig. 1) for surveillance by circulating and infiltrating immune cells. The upregulation of molecules involved in T cell co-stimulation and antigen presentation suggests that PBNP-PTT-treated tumor cells can function in conjunction with APCs to activate T cells. This effect is also confirmed in two human melanoma cell lines (Figs. S5 and S6 in the ESM). Although we demonstrate T cell activation by PBNP-PTT-treated SM1 cells in the absence of DCs (Fig. 1(m)), our in vivo data (Figs. 3, 4, and 7) suggests that this effect is made more potent in the presence of DCs as would be expected to occur in the animals.
We have established that PBNP-PTT + aCD137 enables a robust abscopal antitumor immune response, as demonstrated by treating one tumor with PBNP-PTT and leaving a second tumor untreated (Figs. 3 and 4). Analysis of the infiltrating immune cells in the secondary tumors suggests the abscopal effect is driven primarily via infiltrating cytotoxic CD8+ T cells (Fig. 4(d)). Analysis of the immune cells in the spleen and lymph nodes of treated mice reveales that PBNP-PTT + aCD137 generates a significant increase in the presence and maturation of DCs in the lymph node, and systemic increases of total CD8+ and activated CD8+ T cells, as measured in the spleen (Fig. 5). Importantly, we observe that naïve subsets of both CD4+ and CD8+ T cells are significantly decreased after nanoimmunotherapy treatment, while memory populations are significantly increased (Figs. 5(j)–5(m)), suggesting that the rejection of tumor rechallenge (Fig. 7) is mediated via both CD4+ and CD8+ memory T cells. These observations are made 14 days after the administration of PBNP-PTT and 24 h after the final administration of aCD137. As such, the measured immunological effects are likely skewed toward revealing the acute effects of aCD137, as prior literature suggests that the short-term effects of PBNP-PTT would occur much earlier [61, 62]. However, these studies illuminate the complementary immunological effects of the nanoimmunotherapy at 14 days post-treatment initiation.
The serum analysis for the cytokine and chemokine content reveals a systemic immune response mediated via TNFα, MIP-1a, RANTES, Fas ligand, IL-10, and IL-5 in mice treated with PBNP-PTT + aCD137 (Fig. 6). Typical of an inflammatory response, TNFα, MIP-1a, and RANTES would be secreted by immune cells (e.g., activated macrophages, NK cells, T cells) and likely helped drive the observed antitumor T cell responses [44, 45]. IL-10 has been shown to mediate antitumor immunity by activating CD8+ T cells in tumor microenvironments (TME) [63, 64], which correlates with the increased CD8+ T cell infiltration into secondary tumors and decreased tumor burden in mice treated with PBNP-PTT + aCD137. IL-5 is the only measured cytokine significantly increased in mice treated with PBNP-PTT + aCD137 over all other groups (Fig. 6(b)). IL-5 is produced by helper T cells [65] and correlates with the increase in activated CD4+ T cells measured in the secondary tumor and spleen, suggesting that IL-5 may play a pivotal role in enabling systemic antitumor immunity. Further, IL-5 has been measured as upregulated in patients who survived drug-induced liver injury (DILI) compared with patients who succumbed to DILI [66], suggesting that IL-5 may be aiding in the liver recovery observed in mice treated with PBNP-PTT + aCD137 (Fig. S20 in the ESM), and that PBNP-PTT could be key in decreasing the toxicity of aCD137 while maintaining its beneficial agonistic function on T cells. Because serum cytokine and chemokine levels are only observed at one timepoint (14 days after treatment initiation), it will be helpful to measure these molecules in a temporal manner in the future. As a result, we may uncover the short-term effects and kinetics of serum cytokine and chemokine expression in response to PBNP-PTT.
Based on the observed responses in vitro and in vivo, we hypothesized the mechanism through which PBNP-PTT + aCD137 functions (Fig. 8). Locally administered PBNP-PTT causes rapid tumor debulking and elicits mild immunogenicity in the treated TME. Circulating APCs recognize danger-associated molecular patterns (DAMPs) released from PBNP-PTT-treated tumor cells, engulf released tumor antigens, and traffic to lymph nodes for maturation. Simultaneously, T cells recognize co-stimulatory molecules on PBNP-PTT-treated tumor cells, but these immunological effects of PBNP-PTT monotherapy only cause a mild, sup-optimal abscopal effect. Matured DCs in lymph nodes of mice treated with PBNP-PTT + aCD137 present tumor-specific antigens and co-stimulate T cells, mounting an antitumor response in CD8+ T cells, causing CD8+ T cell infiltration in secondary tumors and systemic T cell activation. aCD137 promotes systemic activation and expansion of T cells, and these effects in concert generate significant tumor-specific memory T cells and long-term protection against future tumor recurrence.
Figure 8.
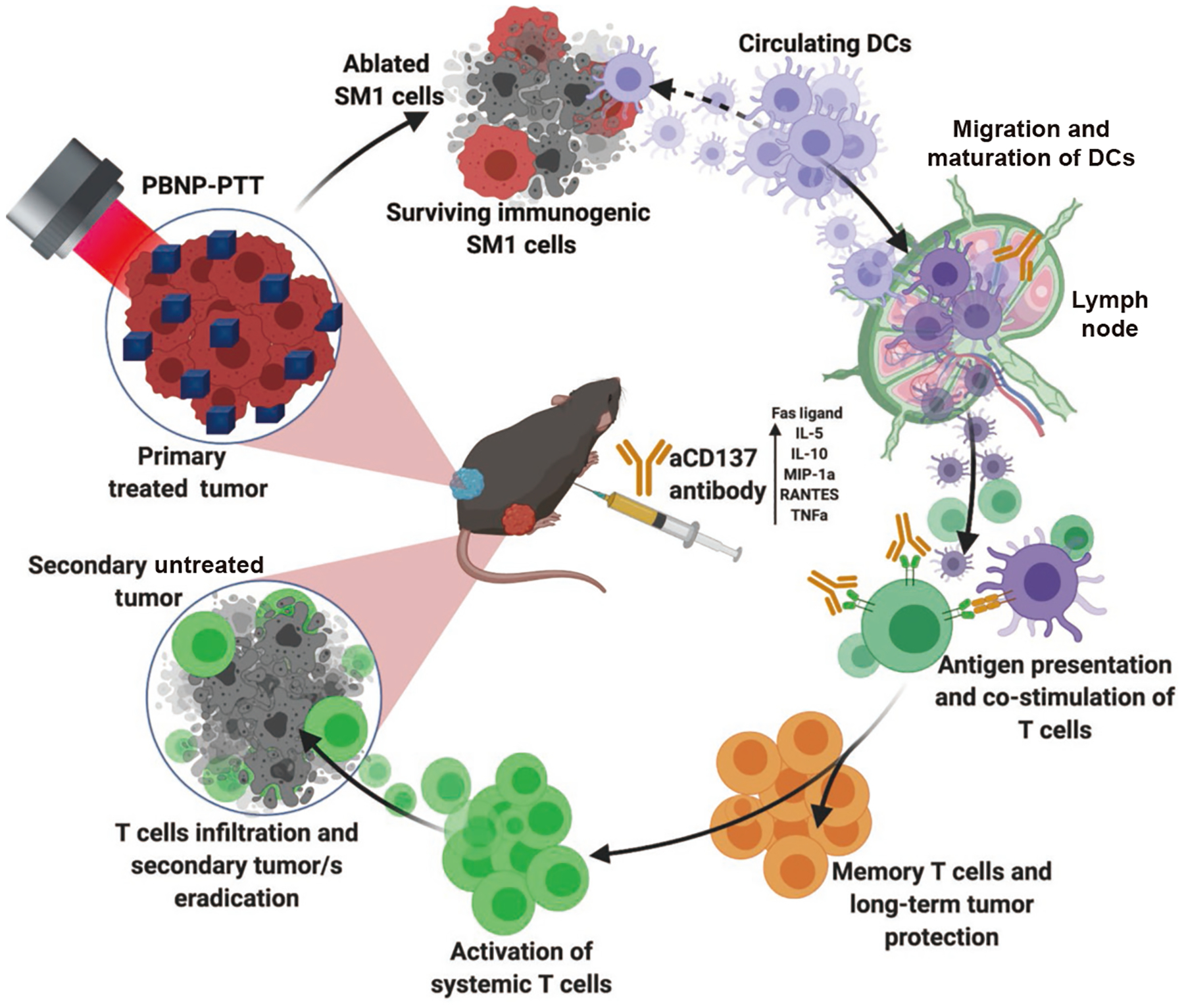
Hypothesized mechanism of action for PBNP-PTT + aCD137. The working theory for efficacy of PBNP-PTT + aCD137 is depicted, illustrating the critical involvement of activated DCs and CD4+ and CD8+ T cells in generating abscopal efficacy against melanoma.
Agonistic mAbs against CD137 have been developed and investigated in Phases I and II clinical trials. Unfortunately, the doses required for efficacy have generated hepatotoxicity in treated patients [20, 33, 67–69]; thus, much clinical development has focused on dosing strategies and mAb modifications to reduce toxicity. Our data illustrates that PBNP-PTT + aCD137 generates a synergistic antitumor response that may enable a lower effective dose of aCD137. We hypothesize that antigens released upon treatment with PBNP-PTT may provide directionality to immune effector cells by exposing specific molecules for immune cell targeting, thereby limiting off-target effects, such that acute hepatotoxicity is mitigated. To realize this effect, we plan to undertake dosing studies to determine the lowest aCD137 concentration needed to effect robust immune cell engagement. Our data also shows that despite the acute hepatotoxicity observed in mice treated with PBNP-PTT + aCD137 (Fig. S19 in the ESM), livers appeared to recover to normal health over time, with no chronic hepatotoxicity (Fig. S20 in the ESM), critical to further development of our combination therapy. Thus, ongoing work focuses on mitigating the acute effects of the treatment.
5. Conclusions
The data presented in this study illustrate a novel nanoimmunotherapy platform for effectively treating melanoma. PBNP-PTT combined with agonistic aCD137 mAb therapy functions at the interface of nanomedicine and immunotherapy, blending complementary and synergistic immunogenic elements and engaging the beneficial consequences of each monotherapy. At an optimal thermal dose, PBNP-PTT provides robust tumor debulking, increased immunogenicity, and release of tumor-specific immune cell targets, which is potentiated by the ability of aCD137 to engage T cell activation in the context of distal or metastasized melanoma. The results described herein provide a foundation for future studies, with the long-term goal of providing an novel treatment option for melanoma patients.
Supplementary Material
Acknowledgements
We would like to acknowledge the George Washington University (GWU) Flow Cytometry Core Facility (Kimberlyn Acklin), the GWU Office of Animal Research (Bethany Rentz), and the GWU Research Pathology Core Lab, for supporting our studies. Research reported in this publication was supported in part by the National Cancer Institute of the National Institutes of Health under Award Nos. R41CA217294 and R37CA226171. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Electronic Supplementary Material: Supplementary material is available in the online version of this article at https://doi.org/10.1007/s12274-021-3813-1.
References
- [1].National Cancer Institute: Surveillance, Epidemiology, and End Results Program. SEER*Explorer [Online]. Washington: National Cancer Institute. https://seer.cancer.gov/explorer/ (accessed Sep 14, 2020). [Google Scholar]
- [2].Hodi FS; O’Day SJ; McDermott DF; Weber RW; Sosman JA; Haanen JB; Gonzalez R; Robert C; Schadendorf D; Hassel JC et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med 2010, 363, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schadendorf D; Hodi FS; Robert C; Weber JS; Margolin K; Hamid O; Patt D; Chen TT; Berman DM; Wolchok JD Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol 2015, 33, 1889–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Robert C; Ribas A; Schachter J; Arance A; Grob JJ; Mortier L; Daud A; Carlino MS; McNeil CM; Lotem M et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol 2019, 20, 1239–1251. [DOI] [PubMed] [Google Scholar]
- [5].Robert C; Schachter J; Long GV; Arance A; Grob JJ; Mortier L; Daud A; Carlino MS; McNeil C; Lotem M et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med 2015, 372, 2521–2532. [DOI] [PubMed] [Google Scholar]
- [6].Weber JS; D’Angelo SP; Minor D; Hodi FS; Gutzmer R; Neyns B; Hoeller C; Khushalani NI; Miller WH Jr.; Lao CD et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015, 16, 375–384. [DOI] [PubMed] [Google Scholar]
- [7].Callahan MK; Kluger H; Postow MA; Segal NH; Lesokhin A; Atkins MB; Kirkwood JM; Krishnan S; Bhore R; Horak C et al. Nivolumab plus ipilimumab in patients with advanced melanoma: Updated survival, response, and safety data in a phase I dose-escalation study. J. Clin. Oncol 2018, 36, 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Postow MA; Chesney J; Pavlick AC; Robert C; Grossmann K; McDermott D; Linette GP; Meyer N; Giguere JK; Agarwala SS et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med 2015, 372, 2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Larkin J; Chiarion–Sileni V; Gonzalez R; Grob JJ; Rutkowski P; Lao CD; Cowey CL; Schadendorf D; Wagstaff J; Dummer R et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med 2019, 381, 1535–1546. [DOI] [PubMed] [Google Scholar]
- [10].Naidoo J; Page DB; Li BT; Connell LC; Schindler K; Lacouture ME; Postow MA; Wolchok JD Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol 2015, 26, 2375–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Michot JM; Bigenwald C; Champiat S; Collins M; Carbonnel F; Postel-Vinay S; Berdelou A; Varga A; Bahleda R; Hollebecque A et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur. J. Cancer 2016, 54, 139–148. [DOI] [PubMed] [Google Scholar]
- [12].Champiat S; Lambotte O; Barreau E; Belkhir R; Berdelou A; Carbonnel F; Cauquil C; Chanson P; Collins M; Durrbach A et al. Management of immune checkpoint blockade dysimmune toxicities: A collaborative position paper. Ann. Oncol 2016, 27, 559–574. [DOI] [PubMed] [Google Scholar]
- [13].Wang DY; Salem JE; Cohen JV; Chandra S; Menzer C; Ye F; Zhao SL; Das S; Beckermann KE; Ha L et al. Fatal toxic effects associated with immune checkpoint inhibitors: A systematic review and meta-analysis. JAMA Oncol 2018, 4, 1721–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mittler RS; Foell J; McCausland M; Strahotin S; Niu LG; Bapat A; Hewes LB Anti-CD137 antibodies in the treatment of autoimmune disease and cancer. Immunol. Res 2004, 29, 197–208. [DOI] [PubMed] [Google Scholar]
- [15].Qi XY; Li FL; Wu Y; Cheng C; Han P; Wang JY; Yang XM Optimization of 4–1BB antibody for cancer immunotherapy by balancing agonistic strength with FcγR affinity. Nat. Commun 2019, 10, 2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shuford WW; Klussman K; Tritchler DD; Loo DT; Chalupny J; Siadak AW; Brown TJ; Emswiler J; Raecho H; Larsen CP et al. 4–1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J. Exp. Med 1997, 186, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chu DT; Bac ND; Nguyen KH; Le Bao Tien N; Van Thanh V; Nga VT; Ngoc VTN; Dao DTA; Hoan LN; Hung NP et al. An update on anti-CD137 antibodies in immunotherapies for cancer. Int. J. Mol. Sci 2019, 20, 1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chester C; Ambulkar S; Kohrt HE 4–1BB agonism: Adding the accelerator to cancer immunotherapy. Cancer Immunol. Immunother 2016, 65, 1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Segal NH; He AR; Doi T; Levy R; Bhatia S; Pishvaian MJ; Cesari R; Chen Y; Davis CB; Huang B et al. Phase I study of single-agent utomilumab (PF-05082566), a 4–1BB/CD137 agonist, in patients with advanced cancer. Clin. Cancer Res 2018, 24, 1816–1823. [DOI] [PubMed] [Google Scholar]
- [20].Segal NH; Logan TF; Hodi FS; McDermott D; Melero I; Hamid O; Schmidt H; Robert C; Chiarion–Sileni V; Ascierto PA et al. Results from an integrated safety analysis of urelumab, an agonist anti-CD137 monoclonal antibody. Clin. Cancer Res 2017, 23, 1929–1936. [DOI] [PubMed] [Google Scholar]
- [21].Chu KF; Dupuy DE Thermal ablation of tumours: Biological mechanisms and advances in therapy. Nat. Rev. Cancer 2014, 14, 199–208. [DOI] [PubMed] [Google Scholar]
- [22].Cano–Mejia J; Bookstaver ML; Sweeney EE; Jewell CM; Fernandes R Prussian blue nanoparticle-based antigenicity and adjuvanticity trigger robust antitumor immune responses against neuroblastoma. Biomater. Sci 2019, 7, 1875–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cano–Mejia J; Burga RA; Sweeney EE; Fisher JP; Bollard CM; Sandler AD; Cruz CRY; Fernandes R Prussian blue nanoparticle-based photothermal therapy combined with checkpoint inhibition for photothermal immunotherapy of neuroblastoma. Nanomed.: Nanotechnol. Biol. Med 2017, 13, 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sweeney EE; Cano–Mejia J; Fernandes R Photothermal therapy generates a thermal window of immunogenic cell death in neuroblastoma. Small 2018, 14, 1800678. [DOI] [PubMed] [Google Scholar]
- [25].den Brok MHMGM; Sutmuller RPM; van der Voort R; Bennink EJ; Figdor CG; Ruers TJM; Adema GJ In situ tumor ablation creates an antigen source for the generation of antitumor immunity. Cancer Res 2004, 64, 4024–4029. [DOI] [PubMed] [Google Scholar]
- [26].Sabel MS; Nehs MA; Su G; Lowler KP; Ferrara JLM; Chang AE Immunologic response to cryoablation of breast cancer. Breast Cancer Res. Treat 2005, 90, 97–104. [DOI] [PubMed] [Google Scholar]
- [27].Wu F; Zhou L; Chen WR Host antitumour immune responses to HIFU ablation. Int. J. Hyperther 2007, 23, 165–171. [DOI] [PubMed] [Google Scholar]
- [28].Kepp O; Senovilla L; Vitale I; Vacchelli E; Adjemian S; Agostinis P; Apetoh L; Aranda F; Barnaba V; Bloy N et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kroemer G; Galluzzi L; Kepp O; Zitvogel L Immunogenic cell death in cancer therapy. Annu. Rev. Immunol 2013, 31, 51–72. [DOI] [PubMed] [Google Scholar]
- [30].Galluzzi L; Vitale I; Warren S; Adjemian S; Agostinis P; Martinez AB; Chan TA; Coukos G; Demaria S; Deutsch E et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cano–Mejia J; Shukla A; Ledezma DK; Palmer E; Villagra A; Fernandes R CpG-coated prussian blue nanoparticles–based photothermal therapy combined with anti-CTLA-4 immune checkpoint blockade triggers a robust abscopal effect against neuroblastoma. Transl. Oncol 2020, 13, 100823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shukla A; Cano-Mejia J; Andricovich J; Burga RA; Sweeney EE; Fernandes R An engineered Prussian blue nanoparticles-based nanoimmunotherapy elicits robust and persistent immunological memory in a TH-MYCN neuroblastoma model. Adv. NanoBiomed Res 2021, 1, 2100021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chester C; Sanmamed MF; Wang J; Melero I Immunotherapy targeting 4–1BB: Mechanistic rationale, clinical results, and future strategies. Blood 2018, 131, 49–57. [DOI] [PubMed] [Google Scholar]
- [34].Koya RC; Mok S; Otte N; Blacketor KJ; Comin–Anduix B; Tumeh PC; Minasyan A; Graham NA; Graeber TG; Chodon T et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res 2012, 72, 3928–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].da Silva IP; Wang KYX; Wilmott JS; Holst J; Carlino MS; Park JJ; Quek C; Wongchenko M; Yan YB; Mann G et al. Distinct molecular profiles and immunotherapy treatment outcomes of V600E and V600K BRAF-mutant melanoma. Clin. Cancer Res 2019, 25, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Knight DA; Ngiow SF; Li M; Parmenter T; Mok S; Cass A; Haynes NM; Kinross K; Yagita H; Koya RC et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J. Clin. Invest 2013, 123, 1371–1381. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [37].Vojtech JM; Cano –Mejia J; Dumont MF; Sze RW; Fernandes R Biofunctionalized prussian blue nanoparticles for multimodal molecular imaging applications. J. Vis. Exp 2015, 28, e52621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sapareto SA; Dewey WC Thermal dose determination in cancer therapy. Int. J. Radiat. Oncol., Biol., Phys 1984, 10, 787–800. [DOI] [PubMed] [Google Scholar]
- [39].Bartkowiak T; Jaiswal AR; Ager CR; Chin R; Chen CH; Budhani P; Ai MD; Reilley MJ; Sebastian MM; Hong DS et al. Activation of 4–1BB on liver myeloid cells triggers hepatitis via an interleukin-27-dependent pathway. Clin. Cancer Res 2018, 24, 1138–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cretella D; Digiacomo G; Giovannetti E; Cavazzoni A PTEN alterations as a potential mechanism for tumor cell escape from PD-1/PD-L1 inhibition. Cancers 2019, 11, 1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Paraiso KHT; Xiang Y; Rebecca VW; Abel EV; Chen YA; Munko AC; Wood E; Fedorenko IV; Sondak VK; Anderson ARA et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res 2011, 71, 2750–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].George S; Miao DN; Demetri GD; Adeegbe D; Rodig SJ; Shukla S; Lipschitz M; Amin-Mansour A; Raut CP; Carter SL et al. Loss of PTEN is associated with resistance to anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity 2017, 46, 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Borsig L; Wolf MJ; Roblek M; Lorentzen A; Heikenwalder M Inflammatory chemokines and metastasis–tracing the accessory. Oncogene 2014, 33, 3217–3224. [DOI] [PubMed] [Google Scholar]
- [44].Zhang JM; An JX Cytokines, inflammation, and pain. Int. Anesthesiol. Clin 2007, 45, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Grivennikov SI; Greten FR; Karin M Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dupont PJ; Warrens AN Fas ligand exerts its pro-inflammatory effects via neutrophil recruitment but not activation. Immunology 2007, 120, 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ikutani M; Yanagibashi T; Ogasawara M; Tsuneyama K; Yamamoto S; Hattori Y; Kouro T; Itakura A; Nagai Y; Takaki S et al. Identification of innate IL-5-producing cells and their role in lung eosinophil regulation and antitumor immunity. J. Immunol 2012, 188, 703–713. [DOI] [PubMed] [Google Scholar]
- [48].Simson L; Ellyard JI; Dent LA; Matthaei KI; Rothenberg ME; Foster PS; Smyth MJ; Parish CR Regulation of carcinogenesis by IL-5 and CCL11: A potential role for eosinophils in tumor immune surveillance. J. Immunol 2007, 178, 4222–4229. [DOI] [PubMed] [Google Scholar]
- [49].Zaynagetdinov R; Sherrill TP; Gleaves LA; McLoed AG; Saxon JA; Habermann AC; Connelly L; Dulek D; Peebles RS Jr.; Fingleton B et al. Interleukin-5 facilitates lung metastasis by modulating the immune microenvironment. Cancer Res 2015, 75, 1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Eskiocak U; Guzman W; Wolf B; Cummings C; Milling L; Wu HJ; Ophir M; Lambden C; Bakhru P; Gilmore DC et al. Differentiated agonistic antibody targeting CD137 eradicates large tumors without hepatotoxicity. JCI Insight 2020, 5, e133647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Fu GL; Liu W; Feng SS; Yue XL Prussian blue nanoparticles operate as a new generation of photothermal ablation agents for cancer therapy. Chem. Commun 2012, 48, 11567–11569. [DOI] [PubMed] [Google Scholar]
- [52].Hoffman HA; Chakrabarti L; Dumont MF; Sandler AD; Fernandes R Prussian blue nanoparticles for laser-induced photothermal therapy of tumors. RSC Adv 2014, 4, 29729–29734. [Google Scholar]
- [53].Sweeney EE; Burga RA; Li CY; Zhu Y; Fernandes R Photothermal therapy improves the efficacy of a MEK inhibitor in neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Sci. Rep 2016, 6, 37035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Cheng L; Gong H; Zhu WW; Liu JJ; Wang XY; Liu G; Liu Z PEGylated Prussian blue nanocubes as a theranostic agent for simultaneous cancer imaging and photothermal therapy. Biomaterials 2014, 35, 9844–9852. [DOI] [PubMed] [Google Scholar]
- [55].Gauttier V; Judor JP; Le Guen V; Cany J; Ferry N; Conchon S Agonistic anti-CD137 antibody treatment leads to antitumor response in mice with liver cancer. Int. J. Cancer 2014, 135, 2857–2867. [DOI] [PubMed] [Google Scholar]
- [56].Murillo O; Arina A; Hervas-Stubbs S; Gupta A; McCluskey B; Dubrot J; Palazón A; Azpilikueta A; Ochoa MC; Alfaro C et al. Therapeutic antitumor efficacy of anti-CD137 agonistic monoclonal antibody in mouse models of myeloma. Clin. Cancer Res 2008, 14, 6895–6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Weigelin B; Bolaños E; Teijeira A; Martinez-Forero I; Labiano S; Azpilikueta A; Morales–Kastresana A; Quetglas JI; Wagena E; Sánchez –Paulete AR et al. Focusing and sustaining the antitumor CTL effector killer response by agonist anti-CD137 mAb. Proc. Natl. Acad. Sci. USA 2015, 112, 7551–7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Palazón A; Teijeira A; Martinez-Forero I; Hervás-Stubbs S; Roncal C; Peñuelas I; Dubrot J; Morales-Kastresana A; Pérez-Gracia JL; Ochoa MC et al. Agonist anti-CD137 mAb act on tumor endothelial cells to enhance recruitment of activated T lymphocytes. Cancer Res 2011, 71, 801–811. [DOI] [PubMed] [Google Scholar]
- [59].Zhang Y; Li N; Suh H; Irvine DJ Nanoparticle anchoring targets immune agonists to tumors enabling anti-cancer immunity without systemic toxicity. Nat. Commun 2018, 9, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Niu LG; Strahotin S; Hewes B; Zhang BY; Zhang YY; Archer D; Spencer T; Dillehay D; Kwon B; Chen LP et al. Cytokine-mediated disruption of lymphocyte trafficking, hemopoiesis, and induction of lymphopenia, anemia, and thrombocytopenia in anti-CD137-treated mice. J. Immunol 2007, 178, 4194–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Huang LP; Li YN; Du YN; Zhang YY; Wang XX; Ding Y; Yang XL; Meng FL; Tu JS; Luo L et al. Mild photothermal therapy potentiates anti-PD-L1 treatment for immunologically cold tumors via an all-in-one and all-in-control strategy. Nat. Commun 2019, 10, 4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chen Q; Xu LG; Liang C; Wang C; Peng R; Liu Z Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Nat. Commun 2016, 7, 13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Oft M IL-10: Master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol. Res 2014, 2, 194–199. [DOI] [PubMed] [Google Scholar]
- [64].Emmerich J; Mumm JB; Chan IH; LaFace D; Truong H; McClanahan T; Gorman DM; Oft M IL-10 directly activates and expands tumor-resident CD8+ T cells without De Novo infiltration from secondary lymphoid organs. Cancer Res 2012, 72, 3570–3581. [DOI] [PubMed] [Google Scholar]
- [65].Hogan SP; Koskinen A; Matthaei KI; Young IG; Foster PS Interleukin-5-producing CD4+ T cells play a pivotal role in aeroallergen-induced eosinophilia, bronchial hyperreactivity, and lung damage in mice. Am. J. Respir. Crit. Care Med 1998, 157, 210–218. [DOI] [PubMed] [Google Scholar]
- [66].Steuerwald NM; Foureau DM; Norton HJ; Zhou J; Parsons JC; Chalasani N; Fontana RJ; Watkins PB; Lee WM; Reddy KR et al. Profiles of serum cytokines in acute drug-induced liver injury and their prognostic significance. PLoS One 2013, 8, e81974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Melero I; Hirschhorn-Cymerman D; Morales-Kastresana A; Sanmamed MF; Wolchok JD Agonist antibodies to TNFR molecules that costimulate T and NK cells. Clin. Cancer Res 2013, 19, 1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Bartkowiak T; Curran MA 4–1BB agonists: Multi-potent potentiators of tumor immunity. Front. Oncol 2015, 5, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Compte M; Harwood SL; Muñoz IG; Navarro R; Zonca M; Perez-Chacon G; Erce-Llamazares A; Merino N; Tapia-Galisteo A; Cuesta AM et al. A tumor-targeted trimeric 4–1BB-agonistic antibody induces potent anti-tumor immunity without systemic toxicity. Nat. Commun 2018, 9, 4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.