

Abstract
The efficacy and specificity of conventional monoclonal antibody (mAb) drugs in the clinic require further improvement. Currently, the development and application of novel antibody formats for improving cancer immunotherapy have attracted much attention. Variable region‐retaining antibody fragments, such as antigen‐binding fragment (Fab), single‐chain variable fragment (scFv), bispecific antibody, and bi/trispecific cell engagers, are engineered with humanization, multivalent antibody construction, affinity optimization and antibody masking for targeting tumor cells and killer cells to improve antibody‐based therapy potency, efficacy and specificity. In this review, we summarize the application of antibody variable region engineering and discuss the future direction of antibody engineering for improving cancer therapies.
Keywords: antibody engineering, bi/trispecific killer engager, cancer immunotherapy, chimeric antigen receptor‐T (CAR‐T) cell, nanobody, natural killer (NK) cell, scFv
Abbreviations
- ADCC
Antibody‐dependent cellular cytotoxicity
- ADCP
Antibody‐dependent cellular phagocytosis
- Akt
v‐akt murine thymoma viral oncogene homolog; protein kinase B
- ALL
Acute lymphoblastic leukemia
- AML
Acute myelogenous leukemia
- B‐ALL
B cell acute lymphoblastic leukemia
- BCMA
B cell maturation antigen
- C
Constant region
- CAR‐T
Chimeric antigen receptor T cell
- CDC
Complement‐dependent cytotoxicity
- CDR3
Complementarity‐determining region 3
- CRC
Colorectal cancers
- CT83
Cancer/testis antigen 83
- EGFR
Epidermal growth factor receptor
- EpCAM
Epithelial cell adhesion molecule
- Fab
antigen‐binding fragment
- FcRn
Neonatal Fc receptor
- ForCE
Format chain exchange
- FR2
Framework region 2
- HER2
erbB‐2
- HGF
Hepatocyte growth factor
- HSA
Human serum albumin
- ICAM1
Intercellular adhesion molecule 1
- LALA
234 and 235 leucine‐to‐alanine mutation
- mAb
Monoclonal antibody
- MAPK
Mitogen‐activated protein kinase
- MHC
Major histocompatibility complex
- MM
Multiple myeloma
- MUC1
Mucin 1
- NK cell
Natural killer cell
- NSCLC
Non‐small cell lung cancer
- PI3K
Phosphoinositide 3‐kinase
- PLAC1
Placenta‐enriched 1
- PRAME
PRAME nuclear receptor transcriptional regulator
- PSMA
Prostate‐specific membrane antigen
- scFv
Single‐chain variable fragment
- SLCO6A1
Solute carrier organic anion transporter family member 6A1
- SP17
Sperm autoantigenic protein 17
- STAT
Signal transducer and activator of transcription
- SVM
Support vector machine
- TCR
T cell receptor
- TIL
Tumor‐infiltrating T cells
- Treg
Regulatory T cells
- US FDA
US Food and Drug Administration
- V
Variable region
- VEGF
Vascular endothelial growth factor
1. BACKGROUND
An antibody is composed of a variable region that binds to the antigen and a constant region, which can be separated and expressed as smaller antibody fragments such as antigen‐binding fragment (Fab) and single‐chain variable fragment (scFv). The first anti‐cancer monoclonal antibody (mAb) rituximab was approved in 1997 after extensive engineering and optimization [1, 2]. However, full‐length antibodies still face limited tumor penetration, prolonged retention in circulation, unspecific binding to non‐tumor tissues, and high production costs. There have been unprecedented advances in antibody fragment construct engineering for cancer treatment since the approval of the first bispecific antibody blinatumomab in 2018 [3] and the nanobody caplacizumab in 2019 [4]. More advanced techniques in molecular cloning, genetic engineering, yeast/phage display platforms and high‐throughput antibody screening systems have facilitated the engineering of antibody fragment formats. Currently, there are several hundred antibody‐derived modular formats, such as scFv, Fab, diabodies, triabodies, minibodies and nanobodies, all with the aim of improving antibody‐based cancer immunotherapy [5, 6, 7]. These modular formats offer a range of advantages over mAbs: alternative routes of administration, ability to bind antigens that are inaccessible to mAbs, ability to cross the blood‐brain barrier, reduced Fc region‐related adverse effects, economically friendly for mass production, increased stability, high flexibility for modification and engineering, and reduced immunogenicity. Although these antibody fragment formats may hold great potential for improving drug efficacy and safety compared with conventional anti‐cancer antibodies, few antibody fragment‐based therapies have been approved for clinical use. In this review, we introduce and summarize the most recent advances in anti‐cancer antibody fragment formats and the future direction of the field.
2. ANTIBODY FRAGMENT FORMATS
Variable region engineering has led to an explosion of choices for cancer therapy, which renders the screening and validation of antibody fragment candidates exponentially more difficult than that for full immunoglobulin G (IgG). A range of engineering approaches have been developed to aid antibody therapy optimization, with the potential to assist the development of antibody fragment optimization (Figure 1). A deeper understanding of the function of the wide range of antibody fragments is required to accelerate their application in cancer therapy.
FIGURE 1.
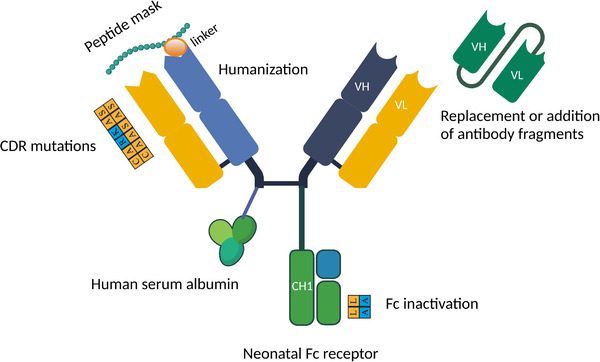
Graphic summary of antibody format engineering, including variable region humanization, peptide masking, CDR region mutagenesis, replacement of antibody variable region, and Fc inactivation by mutagenesis.Abbreviations: CDR, complementarity‐determining region; Fc, fragment crystallizable region; VH, heavy variable region; VL, light variable region; CH1, 2, 3, heavy constant region 1, 2, 3; VHH, single variable domain on a heavy chain
2.1. scFv and nanobodies
The scFv is composed of a heavy‐chain variable (VH) domain connected to a light‐chain variable (VL) domain. The most common linker is (G4S)3/(SG4)3, which is sufficiently flexible to orientate the two domains [8]. In addition to Gly and Ser, other amino acids such as Thr and Ala are introduced to increase flexibility. Longer G4S linkers have been introduced to scFv, subsequently demonstrating stronger binding [9, 10]. With the development of computational tools, linkers are now designed with computational algorithms to optimize scFv flexibility, solubility and stability based on the VH and VL amino acid sequences [11]. The modular VH‐(G4S)3/(SG4)3‐VL construction provides more freedom for modification than a full immunoglobulin (IgG, IgA, IgE and IgM). Due to the simplicity of the design, this format can be easily engineered to introduce additional modules such as drug conjugates, imaging tracers, stabilizing agents, or another binding region. However, the simple format is also subject to drawbacks because of the lack of the Fc region. Unlike the full immunoglobulin, scFv are comparably unstable and tend to aggregate and misfold. Therefore, such antibody fragments require extensive modification and engineering to achieve satisfactory quality for in vivo studies.
Nanobodies, also termed VHH, are derived from camelids and express natural VH‐only antibodies with CH2 and CH3 domains. Nanobodies are the smallest antibody fragments (12‐15 kDa), only half the size of an scFv (25 kDa), and are composed of the VH variable domain onl y. Although nanobodies interact with antigens mainly via the complementarity‐determining region 3 (CDR3), similar to full IgG, their CDR3 region can form a stretched convex paratope with concave epitopes [12, 13]. This feature allows nanobodies to access clefts or hidden and cryptic epitopes inaccessible to conventional mAbs [14]. On the other hand, the shorter region of interaction with the antigen increases the difficulty of isolating high‐affinity nanobodies [15]. The development of several nanobody screening and isolation platforms illustrate the advantages of the selected nanobodies over conventional mAbs in terms of physical and chemical robustness. Nanobodies remain stable under extreme conditions, including high temperatures, high pressures, low pH and low protease concentrations, reducing manufacturing costs [16, 17]. Recent findings have suggested that the high stability allows intracellular nanobody expression, which may present new avenues for targeting intracellular cancer antigens [18]. In addition, the stretched CDR3 region, substitutions of conserved hydrophobic residues to hydrophilic amino acids in the VH framework region 2 (FR2), and additional disulphide bond render nanobodies conformationally stable and less prone to aggregation [16, 17, 19, 20].
2.2. scFv and nanobody format engineering
Nanobodies and scFv present a few advantages over the full immunoglobulin. The solid tumor is relatively inaccessible to whole antibodies because of several biological barriers [21]. In contrast, scFv and nanobodies have superior tissue penetration properties compared with mAbs: they demonstrate a higher perfusion rate across endothelial and epithelial barriers and diffuse faster through the tumor stroma because of their smaller size [22, 23, 24, 25]. Nevertheless, the small size of scFv and nanobodies also has its drawbacks. For example, the molecular weight of nanobodies (15 kDa) is below the renal cut‐off, rendering their circulating half‐life extremely short (0.1–0.3 h) [26, 27]. This short half‐life greatly limits the efficacy and potency of nanobody therapies, especially for chronic diseases. Consequently, many strategies have been developed for improving the half‐life of scFv and nanobodies, including the conjugation of a human Fc domain to the C terminal, linking the variable region of an anti‐albumin antibody to scFv or nanobodies, polyethylene glycol (PEG)ylation, coupling to human serum albumin (HSA), or more recently, polyglutamic acid and PASylation [28, 29, 30]. The coupling to HSA extends the half‐life of nanobodies by 5–10‐fold at the expense of a marginally higher molecular weight of 50 kDa [31, 32]. Although the conjugated anti‐epidermal growth factor receptor (EGFR) nanobodies demonstrate comparable tumor penetration and uptake speed to cetuximab, the ability of conjugated nanobodies to inhibit tumor growth remains much weaker [33]. Some more complicated designs also take advantage of the small size and assemble nanobodies by binding with scFv or Fab to form multivalent antibody formats, which prolongs the half‐life by engaging the neonatal Fc receptor (FcRn)‐mediated IgG recycling pathway [34]. Although the short plasma half‐life and lack of an Fc region may result in lower bioavailability and immune response than mAbs, these features render scFv/nanobodies more specific with fewer adverse effects. In addition, the short half‐life renders them ideal for medical imaging with minimal background [35, 36, 37].
2.3. Multivalent antibody fragments
Monovalent antibody fragments such as scFv and single‐domain antibodies can be further engineered into multivalent antibody fragments such as diabodies, triabodies, tetrabodies, minibodies, and more intricate formats (Figure 2). Diabodies are formed via either non‐covalent or covalent bonding between two scFv with linkers containing less than 11 amino acids, while triabodies and tetrabodies are formed when the linker contains less than 3 amino acids. These formats demonstrate higher specificity and affinity than monovalent fragments [38]. Moreover, two scFv can be linked by the CH3 domain to form a minibody, which is still relatively small but more stable than other multivalent formats, rendering it ideal as an imaging agent and for drug conjugation [39]. For example, a radio‐labeled minibody targeting CD8 exhibits good tolerance and sensitive detection of CD8‐positive T cells in tissue; therefore, it is being evaluated for monitoring the CD8+ T‐cell response after immunotherapy [40]. The α‐particle‐emitting astatine‐211 (211At)‐labeled anti‐prostate stem cell antigen (PSCA) A11 minibody demonstrated potent tumor growth inhibition in vivo, but radiotoxicity was also present, possibly due to the free 211At accumulation in the thyroid, stomach, salivary gland and spleen [41].
FIGURE 2.
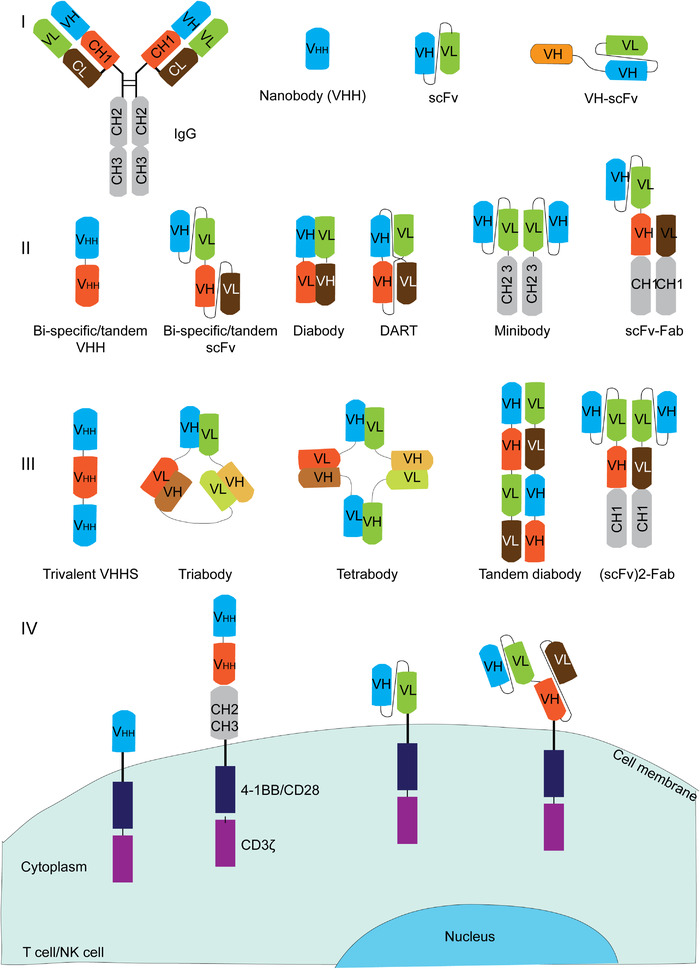
Illustration of four classes of antibody formats. I: mono‐specific antibody, which includes full IgG, nanobody, scFv and VH‐scFv; II: bispecific antibodies, which includes bi‐specific VHH, bi‐specific scFv, diabody, DART, minibody and scFv‐Fab; III: multi‐specific antibodies, which includes trivalent VHHs, triabody, tetrabody, tandem diabody, (scFv)2‐Fab; IV: antibody format applied on CAR, which includes nanobody‐based CAR, bi‐specific nanobody‐based CAR, scFv CAR, and bi‐specific scFv‐based CAR.Abbreviations: IgG, immunoglobulin G; VH, heavy variable region; VL, light variable region; VHH: single variable domain on a heavy chain; CH1, 2, 3, heavy constant region 1, 2, 3; scFv: single‐chain variable fragment; DART, Dual‐Affinity Re‐Targeting; Fab, fragment antigen‐binding
The immunogenicity of nanobodies is meant to be lower than that of non‐human full antibodies because of the lack of an Fc and the smaller size. Nevertheless, nanobodies induce anti‐nanobody autoantibodies and cytokine release syndrome even after humanization [42, 43]. Nanobody humanization has been suggested to be redundant, where a non‐humanized gallium‐68 (68Ga)‐labeled anti‐erbB‐2 (HER2) nanobody is in a phase II clinical trial for positron emission tomography (PET) of breast cancer [26]. Non‐humanized nanobodies have a relatively short half‐life, which induces minimal immunogenicity. Another study found that the heat‐induced aggregates of anti‐EGFR were not immunogenic, while the insoluble aggregates were immunogenic [44]. Modifications for reducing nanobody immunogenicity remain an active field of research. Further structural and screening platforms will aid our understanding of the factors contributing to nanobody immunogenicity.
3. MONOVALENT FORMAT
The monovalent format of the antibody fragment is more focused on the antagonistic effects because of the lack of an Fc region. Nanobodies and scFv cannot initiate immune responses, including antibody‐dependent cellular cytotoxicity (ADCC), antibody‐dependent cellular phagocytosis (ADCP), or complement‐dependent cytotoxicity (CDC). Beginning in the late 1990s, scFv and nanobodies have been developed against well‐studied cancer cell surface proteins that were successfully targeted by mAbs such as cetuximab, panitumumab, trastuzumab, rituximab and obinutuzumab [6, 7, 45]. Compared with conventional IgG, antibody fragments are miniature antagonists with deeper penetration into solid tumors, fewer adverse effects, and lower manufacturing costs [15, 38]. Currently, there are approximately 700 active mAb drug projects in clinical and preclinical development, with 106 mAb drugs approved by the US Food and Drug Administration (FDA) for cancer therapy, which provide targets for alternative antibody format research as proof of concept [46, 47].
3.1. EGFR family
EGFR is considered one of the main antigens for therapeutic intervention of cancer as it is overexpressed in various epithelial cell tumors, including breast cancer, non‐small cell lung cancer (NSCLC) and colorectal cancer (CRC), and is associated with poor prognosis [48]. Ligand binding to EGFR leads to activation of the downstream signaling pathways, including that for phosphoinositide 3‐kinase (PI3K), v‐akt murine thymoma viral oncogene homolog 1 or protein kinase B (Akt), mitogen‐activated protein kinase (MAPK) and signal transducer and activator of transcription (STAT), which promote tumor cell growth, migration and survival [49]. The US FDA has approved four anti‐EGFR mAbs (cetuximab, panitumumab, necitumumab and amivantamab) for treating CRC and NSCLC, paving the way for research on anti‐EGFR scFv and nanobodies [50]. With similar downstream signaling pathways, HER2 is closely related to EGFR and is overexpressed mainly in breast and ovarian cancers. Its high expression level renders HER2 the ideal target for breast cancer treatment [51]. Indeed, anti‐HER2 trastuzumab is arguably one of the most well‐established mAb drugs, improving the survival of patients with metastatic breast cancer from 20.3 months to 25.1 months and reducing the risk of relapse by around 10% within 3 years [52].
Despite the success of full IgG anti‐EGFR/HER2 drugs, improvements can still be made with antibody fragment engineering, such as by joining monospecific antibody fragments. The Fab arm‐exchanged bispecific antibody amivantamab was approved recently by US FDA for treating NSCLC by targeting EGFR and tyrosine‐protein kinase Met (cMET) simultaneously [53]. Anti‐HER2 nanobodies and scFv have also been studied to improve the penetration of anti‐HER2 therapies. Due to the lack of an Fc, antibody fragments cannot exert ADCC or ADCP. Approaches have been developed for conjugating drugs to antibody fragments for anti‐tumor toxicity. CAM‐H2 is an anti‐HER2 131‐iodine (131I)‐conjugated nanobody that is currently tested in a phase I/II clinical trial for the treatment of advanced and progressive HER2‐positive malignancy [54]. Given its small size, the nanobody was able to cross the blood‐brain barrier to target metastatic HER2‐positive tumors, which are inaccessible to conventional antibodies [36, 55]. HER3 expression was higher in 83% of gastrointestinal tumors and 20% of breast, ovarian and bladder cancers than in normal tissues [56, 57]. HER3 confers resistance to HER2 inhibition by maintaining the RAS‐Akt signaling pathway through a negative feedback mechanism [56]. In addition, HER3 may form heterodimers to promote tumor proliferation, rendering it a critical target for cancer immunotherapy [57]. Lumretuzumab was developed to target HER3 and inhibit its phosphorylation and ligand binding. In a phase I clinical trial, monotherapy treatment with lumretuzumab and other anti‐HER3 mAbs only had a control rate of 21% without ADCC effects [58, 59]. Single‐domain antibodies have subsequently been studied for targeting alternative HER3 domains, thereby mediating the anti‐proliferative effect via distinctive mechanisms [60]. Although antibody fragments against HER2 and HER3 cannot exert ADCC or ADCP, they can act as building blocks for multi‐specific antibody complexes that target distinctive epitopes on HER3 and HER2 to combat tumor resistance against monotherapy.
3.2. Vascular endothelial growth factor (VEGF) and hepatocyte growth factor (HGF)
VEGF and VEGF receptor (VEGFR) are highly expressed during angiogenesis and vasculogenesis, promoting the development of new blood vessels in various cancer types, including colon, breast, and lung cancers [61]. Inhibiting VEGF and VEGFR were shown to extend the survival of patients with cancer when administered with chemotherapy [62]. The US FDA approved bevacizumab (Avastin) in 2004 for treating metastatic CRC by neutralizing VEGF [63]. Avastin blocks VEGF‐VEGFR interaction, thereby promoting chemotherapeutic drug delivery to the tumor via blood vessels [64]. Currently, few nanobodies against VEGF or VEGFR2 have been developed for inhibiting tumor angiogenesis, and anti‐VEGF nanobodies inhibited human umbilical vein endothelial cell proliferation and tube formation in vitro [65, 66]. However, there is currently no evidence of any anti‐tumor effect of anti‐VEGF/VEGFR nanobody in vivo, possibly due to the short half‐life of the nanobodies in the blood. Some studies have attempted to fuse the IgG1 Fc to anti‐VEGFR nanobody, but the in vivo effect remained unclear despite the Fc‐mediated function in vitro [67, 68]. Future studies on the humanization and affinity maturation of anti‐VEGF/VEGFR nanobodies may facilitate the development of functional monovalent nanobodies in vivo.
HGF is a plasminogen‐like protein that interacts with the cMET receptor. The engagement results in the activation of tyrosine kinase, which promotes tumor outgrowth and metastasis [69]. Currently, five mAbs are in phase I‐III clinical trials for CRC, renal cell carcinoma, glioma, gastric cancer and oesophageal cancer [70]. Unfortunately, onartuzumab (anti‐cMET) and rilotumumab (anti‐HGF) failed to demonstrate significant improvement even in patients with high cMET expression in phase III clinical trials [71, 72]. Although it has been argued that cMET may not be the best biomarker for enrolling patients, cMET and HGF may have kinase‐independent pathways in tumorigenesis that render the kinase region targeted by anti‐HGF/cMET mAbs and anti‐HGF nanobodies obsolete [73]. To address this issue, nanobodies against the whole cMET ectodomain were developed and demonstrated higher uptake by tumor tissues compared to that by normal tissues and delayed tumor growth compared with saline control [19, 74].
The success of conventional antibodies for cancer therapy is a double‐edged sword for research on antibody fragment formats. It is difficult to overlook the issues regarding the emerging antibody formats: lack of evidence for efficacy and potency, safety concerns and a short half‐life. Nevertheless, antibody fragments demonstrated an unparalleled advantage over conventional antibodies as building blocks in multi‐specific antibody constructs and chimeric antigen receptor T (CAR‐T)/CAR‐natural killer (NK) cell therapy, given their flexible modular feature.
4. MULTI‐SPECIFIC FORMAT
The multi‐specific antibody format is composed of more than 2 antibody variable fragments, either a VH domain alone or a VH plus VL domain, that bind to multiple antigens (Figure 2). By engaging multiple antigens sequentially or simultaneously, the multi‐specific antibody formats can achieve novel functionalities via various mechanisms.
4.1. Bispecific T‐cell engager (BiTE) antibody
Conventional mAbs do not recruit T cells because of their lack of an FcR. On the other hand, bispecific antibodies can be designed to tether T cells by anti‐CD3 single‐chain antibodies. BiTEs recruit T cells to target tumor cells, leading to T cell activation via CD3 binding in the T cell receptor (TCR) complex [75, 76, 77]. T cells are activated regardless of major histocompatibility complex (MHC) peptide restriction, leading to the destruction of target cells in a TCR specificity‐independent manner [75]. Currently, more than 15 BiTEs against solid tumors are under clinical study, which targets well‐established antigens, including EGFR, HER2, prostate‐specific membrane antigen (PSMA) and epithelial cell adhesion molecule (EpCAM) (Table 1). The development of mAbs targeting solid tumors is more complicated than that for hematological tumors because of the poor delivery rate [76, 78, 79]. BiTEs are formed by scFv or nanobodies; therefore, they potentially offer higher tumor penetration due to their smaller size than full size IgG. For example, a bispecific nanobody against CD3 and EGFR bound specifically with EGFR‐overexpressing tumor cells, thereby mediating lysis via both T cell activation and EGFR signaling blockade [80]. In 2009, catumaxomab, an anti‐CD3 and EpCAM‐bispecific antibody targeting EpCAM‐positive tumors for T cell‐mediated lysis, became the first US FDA‐approved BiTE for intraperitoneal treatment of patients with malignant ascites [48, 81]. Despite its stability and longer half‐life compared to scFv, it caused severe adverse effects. The Fc of catumaxomab bound to the FcγR of tissue Kupffer cells that initiate local cytokine release, resulting in the death of the patient [82]. Thus, catumaxomab was voluntarily withdrawn from the US market in 2013 and the European Union (EU) market in 2017.
TABLE 1.
Antibody fragments for cancer immunotherapy in clinical trials
| Drug | Monovalent | Cancer type | Clinical stage |
|---|---|---|---|
| LCAR‐B38M (Legend/Janssen) | BCMA nanobodies incorporated into CAR‐T cells | MM | Phase II |
| TXB4 (Ossianix) | TfR1 variable new antigen receptor with mAb payload | Primary central nervous system lymphoma | Preclinical (mouse) |
| CAM‐H2 (Precirix) | HER2 monomer 131I‐conjugated nanobody | Solid malignancies (breast and gastric) | Phase I (NCT04467515) |
| L‐DOS47 (Helix BioPharma and Theradex) | HER2 nanobody monomer combined with doxorubicin | Pancreatic cancer | Phase Ib/II (NCT04203641) |
| TAS266 (Novartis) | DR5 tetrameric nanobody | Advanced solid malignancies (Pancreatic) | Phase I, suspended (NCT01529307) |
| Multivalent | |||
| A‐319 (Generon) | CD3 + CD19 scFv‐Fab (II, 1+1) | Hematological malignancies (ALL and B cell ALL [B‐ALL]) | Investigational new drug (IND), active |
| AFM11 (Affimed) | CD3 + CD19 tandem diabodies (III, 2+2) | Hematological malignancies (NHL and ALL) | Phase I, suspended (NCT02106091 and NCT02848911) |
| Blinatumomab (Amgen) | CD3 + CD19 tandem scFv (II, 1+1) | Hematological malignancies (ALL and B‐ALL) | Marketed |
| MGD011 (MacroGenics and Janssen) | CD3 + CD19 Dual‐Affinity Re‐Targeting (DART) (II, 1+1) | B cell lymphoma | Phase I, terminated (NCT02454270) |
| AMG562 (Amgen) | CD3 + CD19 tandem scFv (II, 1+1) | NHL | Phase I (NCT03571828) |
| A319 (Generon and EVIVE Biotechnology) | CD3 + CD19 (scFv)2‐Fab (III, 1+2) | B cell lymphoma | Phase I (NCT04056975) |
| AMG330 (Amgen) | CD3 + CD33 tandem scFv (II, 1+1) | Hematological malignancies (AML) | Phase I (NCT02520427) |
| AMV‐564 (Amphivena Therapeutics) | CD3 + CD33 tandem diabodies (III, 2+2) | Hematological malignancies (AML and myelodysplastic syndrome [MDS]) | Phase I (NCT03144245 and NCT03516591) |
| GEM333 (GEMoaB Monoclonals) | CD3 + CD33 single‐chain diabody (scDb) (II, 1+1) | Hematological malignancies (AML) | Phase I (NCT03516760) |
| AMG673 (Amgen) | CD3 + CD33 Fc fused tandem scFv (II, 1+1) | AML | Phase I (NCT03224819) |
| RG7828 (Roche) | CD3 + CD20 full‐length bispecific (1+1) | Hematological malignancies | Phase I/II (NCT02500407) |
| REGN1979 (Regeneron) | CD3 + CD20 Fc modified full‐length bispecific (1+1) | Hematological malignancies | Phase II (NCT03888105) |
| RG6026 (Roche) | CD3 + CD20 Fc modified full‐length bispecific (1+2) | NHL | Phase I (NCT03075696) |
| GEN3013 (Genmab) | CD3 + CD20 DuoBody (1+1) | Hematological malignancies | Phase I/II (NCT03625037) |
| FBTA05 (Trion) | CD3 + CD20 full‐length bispecific (1+1) | B cell lymphoma | Phase I/II (NCT01138579) |
| Plamotamab (Xencor) | CD3 + CD20 full‐length bispecific (1+2) | Hematological malignancies | Phase I (NCT02924402) |
| AMG424 (Amgen) | CD3 + CD38 bispecific hetero‐Fc (1+1) | MM | Phase I, terminated (NCT03445663) |
| AMG420, BI 836909 (Boehringer Ingelheim) | CD3 + BCMA tandem scFv (II, 1+1) | Hematological malignancies (MM) |
Phase I (NCT02514239 and NCT03836053) |
| AMG701 (Amgen) | CD3 + BCMA tandem scFv (II, 1+1) | MM | Phase I (NCT04998747) |
| JNJ‐64007957 (Janssen) | CD3 + BCMA full‐length bispecific (1+1) | MM | Phase I (NCT03145181) |
| PF‐6863135 (Pfizer) | CD3 + BCMA full‐length bispecific (1+2) | MM | Phase I (NCT03269136) |
| REGN5458 (Regeneron) | CD3 + BCMA full‐length bispecific (1+1) | MM | Phase I (NCT03761108) |
| TNB383B (Abbvie) | CD3 + BCMA full‐length bispecific (1+2) | MM | Phase I (NCT03933735) |
| Xmab14045 (Xencor) | CD3 + CD123 scFv‐Fab (II, 1+1) | Hematological malignancies (AML, BLL and chronic myeloid leukaemia) | Phase I (NCT02730312) |
| Flotetuzumab, MGD006 (Macrogenics) | CD3 + CD123 DART (II, 1+1) | Hematological malignancies (AML, MDS and CML) | Phase II, pending (NCT02152956 and NCT03739606) |
| JNJ‐63709178 (Janssen) | CD3 + CD123 full‐length bispecific (1+1) | Hematological malignancies (AML and CML) | Phase I (NCT02715011) |
| AMG111 (Amgen) | CD3 + CEA BiTE (II, 1+1) | Gastrointestinal adenocarcinoma | Phase I (NCT02291614) |
| RG7802 (Roche) | CD3 + CEA full‐length bispecific (1+2) | Solid malignancies | Phase I (NCT02324257) |
| HPN‐424 (Harpoon) | CD3 + PSMA VH‐scFv (I, 1+1) | Solid malignancies (prostate cancer) | Phase I/II (NCT03577028) |
| AMG160 (Amgen) | CD3 + PSMA Fc fused bispecific/tandem scFv (II, 1+1) | Solid malignancies (prostate cancer) | Phase I (NCT03792841) |
| MOR209 (Aptevo Therapeutics) | CD3 + PSMA (scFv)2‐Fc (II, 1+1) | Solid malignancies (prostate cancer) | Phase I (NCT02262910) |
| Pasotuxizumab A212 | CD3 + PSMA tandem scFv (II, 1+1) | Solid malignancies (prostate cancer) | Phase I (NCT01723475) |
| Ertumaxomab (Trion) | CD3 + HER2 scFv‐IgG (1+1) | Breast cancer | Phase II (NCT00522457) |
| GBR1302 (Glenmark Pharmaceuticals) | CD3 + HER2 scFv‐IgG (1+1) | Solid tumor (HER2‐positive) | Phase I, terminated (NCT02829372) |
| M802 (YZY Bio) | CD3 + HER2 scFv‐IgG (1+1) | Solid tumor (HER2‐positive) | Phase I (NCT04501770) |
| BTRC4017A (Genetech) | CD3 + HER2 | Solid tumor (HER2‐positive) | Phase I (NCT03448042) |
| MGD007 (MacroGenics) | CD3 + gpA33 DART (II, 1+1) | CRC | Phase II (NCT02248805) |
| MGD009 (MacroGenics) | CD3 + B7H3 DART (II, 1+1) | Solid tumor | Phase I (NCT02628535) |
| REGN4018 (Regeneron) | CD3 + MUC16 full‐length bispecific (1+1) | Solid tumor | Phase I (NCT03564340) |
| AMG596 (Amgen) | CD3 + EGFRvIII tandem scFv (II, 1+1) | Solid malignancies (EGFRvIII‐positive glioblastoma) | Phase I (NCT03296696) |
| A‐337 (Generon) | CD3 + EpCAM bispecific minibody (II, 1+2) | Solid malignancies (NSCLC) | Phase I |
| BFCR4350A (Genentech) | CD3 + CD307 tandem scFv (II, 1+1) | Hematological malignancies (MM) | Phase I (NCT03275103) |
| AFM13 (Affimed) | CD16 + CD30 tetravalent homodimer (III, 2+2) | Hodgkin Lymphoma | Phase II (NCT04101331) |
| GTB‐3550 (GT Biopharma) | CD16 + CD33 + IL‐15 TriKE (III, 1+1+1) | Hematological malignancies | Phase I/II (NCT03214666) |
| SAR442257 (Sanofi) | CD3 + CD28 + CD38 trispecific full IgG (III, 1+1+1) | MM and NHL | Phase I (NCT04401020) |
| NKP46 NKCE (AstraZeneca) | NKP46 + CD16 + undisclosed | Preclinical | |
| GNC‐038 (Sichuan Baili) | CD3 + CD19 + 41BB + PD‐L1 tetraspecific (III, 1+1+1+1) | NHL | Phase I (NCT04606433) |
| RO5520985 Vanucizumab (Roche) | EGFR + Ang2 full‐length bispecific (1+1) | Solid tumor | Phase I (NCT01688206) |
| BI‐836880 (Ablynx) | EGFR + Ang2 bispecific nanobody (II, 1+1) | NSCLC | Phase I (NCT03468426) |
| Amivantamab (Genmab and Janssen) | EGFR + cMET full‐length bispecific (1+1) | NSCLC | Phase I (NCT02609776) |
| EMBO1 (Epimab Biotherapeutics) | EGFR + cMET | Solid tumor | Phase I (NCT03797391) |
| Zenocutuzumab MCLA128 (Merus) | HER2 + HER3 full‐length bispecific (1+1) | Breast cancer | Phase II (NCT02912949) |
| KN026 (Alphamab) | HER2 + HER2 Fc‐fused (scFv)2 (1+1) | Solid tumor | Phase II (NCT04521179) |
| MBS301 (Beijing Mabworks Biotech) | HER2 + HER2 full‐length bispecific (1+1) | Solid tumor | Phase I (NCT03842085) |
| Zanidatamab ZW25 (Zymeworks) | HER2 + HER2 scFv‐Fab‐Fc (II, 1+1) | HER2‐amplified biliary tract cancers | Phase II (NCT04466891) |
| MP0274 (Molecular Partners AG) | HER2 + HER2 natural ankyrin repeat proteins (1+1) | Solid tumor (HER2‐positive) | Phase I (NCT03084926) |
| OXS‐1550, DT2219ARL (GT Biopharma) | CD19 + CD22 tandem scFv fusion protein (II, fused to modified diphtheria toxin, 1+1) | Hematological malignancies (B cell lymphoma and leukemia) | Phase I/II (NCT02370160) |
| BI 836880 (Ablynx/Boehringer Ingelheim) | Ang2 + VEGF tandem nanobody (I, nanobody, 1+1, anti‐HSA for half‐life extension) | Solid malignancies (NSCLC) | Phase I (NCT02689505) |
| Chinese PLA General Hospital | CD19 + CD22 scFv incorporated into CAR‐T therapy | Relapse or refractory B cell lymphoma | Phase I (NCT03185494) |
4.2. Modifications for improving BiTE efficacy
Due to the modular features of antibody fragments, there is much room for engineering to optimize BiTE specificity, potency, and affinity while reducing its toxicity. Following the withdrawal of catumaxomab, the engineering shifted to the variable regions of BiTEs. To prevent adverse/off‐target effects, the next‐generation BiTE currently undergoing clinical trials feature silenced Fc or Fc‐free constructs composed of modular antibody fragments [67, 68]. The US FDA approved blinatumomab (CD3+CD19) in 2014 for treating acute lymphoblastic leukemia (ALL); it is a bispecific antibody that recognizes CD3 and CD19 with two scFv joined by a linker [4, 83‐85]. In doses as low as 0.005 mg/m2 daily, it eliminated CD19+ cells in patients with non‐Hodgkin lymphoma (NHL) [86]. The phase II clinical trial confirmed the sustained depletion of peripheral tumor cells [86]. All patients exhibited ongoing regression for >6 months with manageable adverse effects such as lymphopenia, leukopenia, cytokine release and chills. The lack of autoimmune or cytokine storm syndrome in treated patients encouraged further follow‐up research on BiTEs. Subsequent clinical trials on relapse and refractory ALL confirmed the effectiveness (43% remission rate) and safety (2% death) of blinatumomab. The current BiTE development also focuses on the characterization of novel anti‐CD3 domains. A low‐affinity anti‐CD3 scFv was incorporated to reduce the peripheral cytokine concentration and tissue cytotoxicity [87, 88]. Several groups have utilized the high‐throughput platform to test numerous anti‐CD3 fragments to maximize T cell cytotoxicity and minimize adverse effects [89].
To increase specificity and avidity, the format can be engineered to contain at least 2 binding sites for the target antigen in combination with anti‐CD3 (2+1, 3+1, 4+1). For example, tandem diabody CD3+CD19 AFM11 (2+2) and CD3+CD123 (2+2) are currently in development and are composed of tetravalent anti‐CD3 and CD19/CD123 fragments. This tetravalent antibody can target T cells, B cells and myeloid cells simultaneously [90]. An attractive feature of the tetravalent antibody format is the ability to achieve the same efficacy independent of the T cell: target cell ratio. The multivalent format can potentially reduce the effective dosing in therapeutic applications, thereby improving the safety of multi‐specific antibody therapy. Although the tetravalent format achieved >10‐fold potency compared with the bivalent format in vivo [91], clinical trial data supporting the superiority of this format in terms of efficacy are lacking. Trivalent antibodies such as RG6026, which is bivalent for CD20 and monovalent for CD3 (2+1), achieved an overall 42% complete remission rate in a phase I clinical trial, but 8% of the patients experienced grade ≥3 cytokine release syndrome [92, 93]. scFv and nanobody combinations have also been formulated by flanking anti‐CD3 scFv with anti‐EGFR and anti‐EpCAM nanobodies. The trispecific T‐cell engager induced T cell‐dependent killing of EGFR‐EpCAM double‐positive cells in vitro [94]. These formats are expected to deliver higher tumor antigen specificity, T cell lysis and fewer adverse effects than the bivalent format. Despite the success of BiTEs in lymphoma, there remains room for improvement in terms of higher specificity and potency in cancer immunotherapy.
4.3. Modifications for improving BiTE specificity
To improve the specificity of effector cell activation, specific effector cell markers for improving the precision of T cell activation are being evaluated. Anti‐CD3 BiTEs activate CD3 cells of all lineages, including regulatory T cells (Tregs) [95]. Therefore, the unspecific engagement of T cells may reduce the efficacy and potency of BiTEs via Treg‐mediated perforin‐dependent cytotoxicity against CD8+ T cells [96, 97, 98]. γδ T cells are enriched in the tumor microenvironment and are associated with tumor regression. Accordingly, bispecific anti‐γδ T nanobodies targeting HER2 and EGFR have been constructed for treating solid malignancies [99]. The latter molecule induced the lysis of patient‐derived CRC cells and exhibited minimal activity against primary EGFR+ keratinocytes, thereby holding promise for an extended therapeutic window.
The critical limiting factor for anti‐CD3 or anti‐γδ T fragments is the binding to T cells in normal tissues, which limits BiTE efficacy and safety. Hence, approaches for targeting tumor‐infiltrating T cells (TILs) have been achieved by masking anti‐CD3/γδ T binding by linking inhibitors [e.g., mimotope or epitope mimetic peptide, laminin‐mimetic peptide (LAP), IgG hinge region and affinity peptide] to the N terminal of the antibody variable region [100]. The most widely used approach, i.e., mimotopes, is linked to the antibody fragment N terminal via a protease‐prone linker, which is only cleaved in a microenvironment with protease overexpression. The resultant probody strategy has been applied to several traditional targets such as EGFR, HER2 and CD3, leading to a 100‐ to 300,000‐fold reduction in affinity [101, 102]. Other than mimotopes, masking peptides that bind to the conserved region of the variable region has been engineered to extend to the paratope region, thereby preventing antibody fragment from binding to the antigen. LAP was modified to mask the framework region of the anti‐EGFR variable region, which was readily cleaved in the tumor microenvironment [103]. Compared with mimotopes, LAP had the advantage of antigen‐independent masking of antibody fragments, but the masking only reduced the affinity by <3 folds [104]. These modifications were aimed at reducing off‐tumor targeting that often causes cytokine release syndrome. For example, coiled‐coil inhibition of anti‐mouse CD3 was recently demonstrated to reduce off‐target effects and cytokine release syndrome in Balb/c mice [105]. It maintained a high percentage of antibody masking in the non‐tumor environment and a high release rate at the tumor site. The coiled‐coil has also been applied to other antibodies, including anti‐HER2, anti‐CD20 and anti‐CD19, exhibiting wide application in anti‐cancer mAb engineering [106, 107, 108]. Despite the progress on protease‐activating masking moieties, designing peptide sequences specific to tumor proteases remains challenging. An alternative approach is format chain exchange (ForCE) technology to the CH3 domain of bispecific antibodies, which allows the activation and arm exchange of two inactive probodies only when they are in close proximity in vitro [109]. Drug‐conjugated probodies against CD71 have entered phase I clinical trials, demonstrating the capability of probodies for pinpointing previously undruggable antigens.
4.4. Bi/trispecific killer engagers (BiKE/TriKE)
In addition to BiTE, bispecific and trispecific NK cell engagers have been developed to engage the activating receptors on NK cells and tumor‐associated antigens [110]. NK cells are safer than T cells as NK cell infusion does not induce graft‐versus‐host disease [111]. CD16 expressed on NK cells is the basis for inducing ADCC by docking the Fc of tumor‐binding antibodies. Therefore, bispecific scFv binds both CD16 and cancer antigens, including CD19, CD20, CD33, CD30, B‐cell maturation antigen (BCMA), and more. BiKE/TriKE promote NK cell degranulation, resulting in enhanced cancer cell lysis independent of antibody Fc [112]. AFM13 is a tetravalent bispecific tandem antibody against CD16A and CD30 with a median inhibitory concentration (IC50) of 35.8 nmol/L for CD30 antigen [113, 114]. It was well‐tolerated in two phase I trials, with a high response rate (77%–88%) at the highest dose in patients with Hodgkin lymphoma [114]. AFM13 efficacy is now undergoing a phase II clinical trial as monotherapy for CD30‐positive T cell lymphoma (NCT04101331). To optimize the specificity, anti‐CD16A‐based bispecific antibodies have also been developed to engage NK cells and macrophages to target cancer cells without unspecific binding to other Fc receptors [101]. BiKEs have also been engineered to incorporate additional modules, forming TriKE with enhanced specificity and/or NK cell activity and survival. Vallera et al. [115] cross‐linked interleukin (IL)‐15 between anti‐CD16 and anti‐CD33 scFv, constructing a TriKE that promoted NK cell survival and tumor‐suppressing effects in vivo. TriKE that targets two NK cell‐activating receptors (e.g., NKp46 and CD16) and tumor antigen have also been developed, as the full activation of NK cells requires the engagement of a combination of receptors [116]. Currently, the CD16/IL‐15/CD33 TriKE is undergoing a phase II clinical trial for acute myelogenous leukemia (AML) (NCT03214666).
4.5. Other bispecific antibodies
Bispecific scFv against multiple signaling pathways in tumor cells have also been developed to reduce cancer resistance to single antibody drugs by targeting multiple cancer antigens on the same cell [117, 118]. For example, EGFR and HER2 mutations were found in 15%–30% of NSCLC samples and 1.6% of HER2‐positive breast cancer cases, respectively [119]. EGFR, HER2 and HER3 share similar downstream pathways, and anti‐EGFR treatment induces HER2 overexpression, which confers tumor resistance against anti‐EGFR treatment [117, 118]. Although anti‐HER2 and anti‐EGFR mAbs achieved clinical benefit for HER2/EGFR‐positive patients, only 10%‐25% of HER2‐positive patients responded to trastuzumab treatment [119]. To achieve a high response rate and low tumor resistance, multi‐specific antibody therapies have been studied for treating patients who do not respond to monotherapy. Bispecific antibodies such as KN026 (trastuzumab combined with pertuzumab) against two HER2 epitopes have been constructed for targeting cancers with low HER2 expression levels [120]. KN026 contains an engineered CH3 region for heterodimerization of the trastuzumab Fab Fc on the knob and the pertuzumab Fab Fc on the hole. In a phase I study, it exhibited similar efficacy to trastuzumab combined with pertuzumab [121]. Dual targeting of EGFR + HER2, HER2 + HER3, or EGFR + cMET has been developed, and some of them have entered phase I clinical trials (Table 1) [121, 122, 123].
Another main hurdle in cancer therapy is the immunosuppressive tumor microenvironment. Attempts have been made to combat the immunosuppressive microenvironment and tumor angiogenesis by treating NSCLC with bispecific nanobodies against VEGF and angiopoietin‐2 (Ang2) in combination with anti‐programmed death‐ligand 1 (PD‐L1). In the most recent phase Ib clinical trial, two of 12 patients exhibited partial response while the adverse effects were manageable [124]. These bispecific antibody formats demonstrate potent cytotoxicity and reduced resistance. However, their antitumor effect against cancers with low target antigen expression remains quite poor. This may result from the narrow epitope coverage by each antibody domain and compromised binding affinity when combined with a bispecific antibody.
4.6. Minibody engineering
Unlike full IgG, antibody fragments do not necessarily contain CH1, CH2, or CH3 domains, as the constant region will increase the size and reduce the penetration of the antibody fragments. Therefore, single CH2 or CH3 domains are used instead of the full CH1, CH2, or CH3 to link monovalent antibody fragments (e.g., minibodies) to minimize the size while maintaining adequate stability. One effective strategy for enhancing the stability of the single CH2 or CH3 domain is introducing additional disulfide bonds, either inter‐ or intra‐domain. The disulfide bonds are the most critical covalent bond in IgG for thermostability [49]. R292C/N297G/V302C mutations in CH2 introduced additional intra‐domain disulfide bonds that facilitated the improved stability and slower rate of clearance in the circulation [125]. The CH3 domain has also been engineered to add intra‐domain and inter‐domain disulfide bonds by mutating P445, G446 and K44 at the C terminal by G, E and C, respectively [126, 127].
Glycosylation modifications have also been applied to the CH2 domain to alter Fc‐fused antibody fragment binding to FcγR and complement. The introduction of an N297 mutation effectively rendered the antibody glycosylated, which is being used to treat autoimmune diseases because of the reduced binding to FcγR and complement. Glycoform engineering in CH2 was also important for the safety of Fc‐fused antibody fragment therapy because common cell lines such as CHO will introduce immunogenic glycosylation in the CH2 domain. Lonza and Roche developed genetically modified CHO cells to produce non‐immunogenic and homogenously glycosylated CH2 [128]. A glycoengineered anti‐CD20 (obinutuzumab [GA101]), which demonstrates improved effector function through higher affinity against FcγRIII, has been approved for treating chronic lymphocytic leukemia (NCT01300247) [129].
Antibody fragments with an Fc domain have improved stability and serum half‐life, but the Fc region creates safety issues because of the binding to Fc receptors and complement. Patients who received monoclonal anti‐CD3 therapy (OKT3) or bispecific anti‐CD3X EpCAM antibodies experienced systemic cytokine storm due to T cell activation through Fc receptor binding on immune cells [130]. The Fc receptor and complement binding was disabled using a common Fc domain engineering termed LALA (234 and 235 leucine to alanine mutation), which significantly improved the safety of anti‐CD3 therapies [131]. Subsequently, more sophisticated Fc mutations such as glycosylated IgG1, IgG2m4 (H268Q/V309L/A330S/P331S, changes to IgG4), hIgG1‐P329G LALA, IgG4 ProAlaAla (S228P/L234A/L235A) and IgG2 (V234A/G237A/P238S/H268A/V309L/A330S/P331S) have been introduced to completely inactivate Fc effector functions [132, 133].
4.7. Antibody fragment multimerization
scFv and nanobody multimerization has become popular in recent years and has improved their avidity [134, 135]. Trispecific and tetraspecific scFv or nanobodies have been formulated by combining antibody formats against the same antigen. Theoretically, the multivalent format can yield antibody formats that target multiple epitopes on the same antigen, thereby boosting potency. A multivalent nanobody (DR5Nb1 by Ablynx) against death receptor 5 (DR5) demonstrated greater tumor‐killing potency than conventional mAb therapy in vitro [136]. DR5Nb1 demonstrated a serum half‐life of 5‐9 h, while the half‐life of the full‐length antibody was >1 week, which largely reduced the efficacy of DR5Nb1 in vivo. Following the successful correlation of valency and potency, the tetravalent anti‐GD2 scFv was developed and was PEGylated to increase stability and half‐life. PEGylated GD2‐specific tetravalent scFv achieved better tumor retention and lower off‐target binding compared with full‐length anti‐GD2 mAb (dinutuximab). Nevertheless, the tumor cell cytotoxicity and tumor growth inhibitory ability of the tetravalent scFv in vivo remained less optimal [34]. More complicated designs such as liposomal drugs coated with trispecific antibodies have also been tested with anti‐HER2 + anti‐FAP + anti‐modified (m)PEG antibody, which demonstrated heightened cytotoxicity than bispecific antibody alone in vitro [137]. Compromised binding affinity and instability are commonly observed with the multivalent format [74]. Future studies on affinity and stability optimizsation combined with high‐throughput screening and deep learning are critical for the multivalent antibody format to shine (Figure 3).
FIGURE 3.
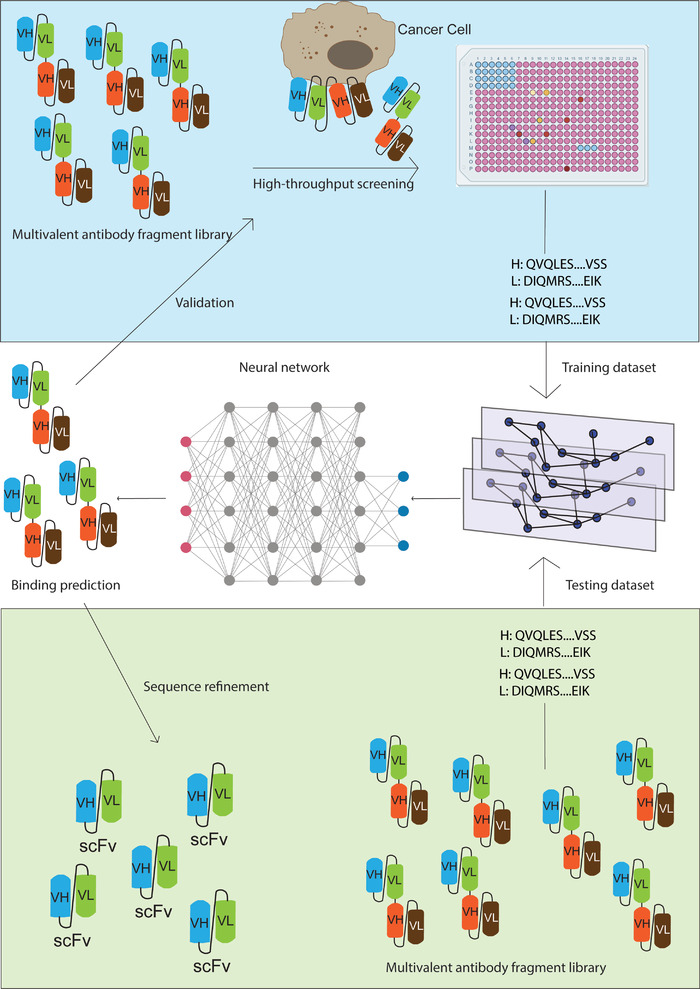
Neural network‐aided high‐throughput bispecific antibody screening. The multivalent antibody fragment library is screened for their binding to target cancer cells in a high‐throughput manner. The sequences of target cancer cell binders are fed into the neural network as training dataset. The sequences of the larger multivalent antibody fragment library are fed into the neural network as testing dataset. The neural network can then predict the multivalent antibodies that bind to target cancer cells, which can be validated by high‐throughput screening.Abbreviations: H, heavy chain sequences; L, light chain sequences
5. ScFv AND NANOBODIES IN CAR‐T/NK CELLS
CAR is a fusion protein composed of an intracellular signaling domain, transmembrane domain hinge region, and an antigen‐binding domain [138]. CAR‐engineered T/NK cells have accomplished inspiring achievements in treating hematological malignancies.
5.1. ScFv/nanobodies in CAR‐T/NK cells
CAR‐T cells have yielded exciting clinical results in certain cancer types such as B‐cell leukemia and multiple myeloma (MM). The antigen‐binding domain is conventionally designed to be an scFv or nanobody that recognizes cancer cell surface antigens or soluble ligands to induce T cell activation in an MHC‐independent manner [139, 140]. TCR mimicking CAR has also been incorporated to bind intracellular tumor‐associated antigens, leading to MHC‐dependent T cell activation [141]. The US FDA has approved five CAR‐T therapies: Abecma, Breyanzi, Kymriah, Tecartus and Yescarta, four of which target lymphoma/leukemia [142]. Kymriah, Breyanzi and Yescarta used murine‐derived anti‐CD19 FMC63 for CAR, raising the concern of a potential anti‐CAR immune response in patients [143]. In addition to treatment for hematological disease, CAR‐T therapy against solid tumors has also been developed. BioNTech Cell and Gene Therapies GmbH have developed a combination therapy that utilizes anti‐claudin 6 CAR‐T cells boosted by liposome‐mRNA complexes to treat solid tumors, including ovarian, testicular, uterine, lung and gastric cancers [144]. The liposome‐mRNA complexes encoding claudin 6 were introduced to dendritic cells to promote the survival and expansion of the infused CAR‐T cells, which induced satisfactory tumor regression in vivo [145]. The safety and efficacy of the treatment against multiple solid tumors are now being evaluated in a phase I/II clinical trial (NCT04503278) [144].
CAR‐NK therapies have also gained more attention in recent years as they present several advantages compared to CAR‐T cells. NK cells are innate lymphoid cells that do not have antigen‐specific receptors such as TCR; therefore, they can kill cancer cells or infected cells with an intrinsic CAR‐independent mechanism [146]. The human leukocyte antigen‐independent killing of NK cells also renders CAR‐NK therapy safer as it does not induce graft‐versus‐host complications [147]. However, only 20 CAR‐NK clinical trials are being conducted worldwide, whereas clinicaltrials.gov lists more than 500 CAR‐T clinical trials, possibly due to the slow ex vivo expansion of primary NK cells by good manufacturing practice standards and suboptimal viral transduction success rates [148, 149]. To address the problem, the NK‐92 cell line with anti‐HER2 CAR is being evaluated in a clinical trial (NCT03383978) [150, 151]. Another study used primary NK cells from core blood for anti‐CD19 CAR incorporation, resulting in a 73% response rate without adverse effects such as cytokine storm or graft‐versus‐host syndrome [152]. Active research into CAR engineering has progressed greatly in the last 5 years, with innovative scFv and nanobody formats incorporated into CAR‐T therapies entering early‐phase clinical trials.
The antigen‐binding domain (scFv or nanobody) is arguably the most critical component of CAR, determining the antigen‐binding affinity and specificity of CAR‐T cells. Single amino acid mutation in the scFv against ganglioside GD2 completely abolished the therapeutic effect of anti‐GD2 CAR‐T cells in vivo [153]. The development of CAR‐T cells targeting antigens with ideal affinity and specificity is complicated, as the affinity needs to be adequate for CAR‐T cells to recognize the tumor antigen, which in turn induces T cell lysis of target cells; high affinity (receptor affinity [Kd] in the nanomolar range) could also result in binding to healthy tissues [154, 155]. Moreover, high‐affinity CAR‐T cells demonstrated low persistence of duration in vivo due to excessive activation‐mediated T cell death [156]. This on‐target, off‐tumor toxicity was observed in clinical trials with anti‐CD19 CAR‐T cells in B cell lymphoma as bone marrow CD19 was also depleted, leading to hypogammaglobulinemia [157]. Although antibody infusion could ameliorate this phenomenon, the more severe off‐tumor toxicities in other CAR‐T agents limit their clinical use. For example, off‐tumor toxicities were associated with anti‐HER2 CAR‐T cells bound to non‐malignant lung tissue, which led to the death of one patient with multi‐organ failure [158]. An anti‐EGFRvIII CAR‐T phase I pilot study also reported one patient death from pulmonary edema out of the 33 patients enrolled. At high doses, the patient developed pulmonary vasculature, possibly due to the off‐tumor toxicity of the activated CAR‐T cells [159]. These alarming consequences should spur more research into CAR engineering, especially scFv, for achieving minimum off‐tumor toxicity.
5.2. ScFv/nanobody engineering for reducing CAR‐T off‐tumor toxicity
Similar to the engineering of bispecific antibody fragments, the success of parental mAbs does not predict CAR functionality. A trastuzumab variable region was adopted for anti‐HER2 CAR‐T cells, yet consequential off‐tumor toxicities occurred due to the HER2 expression on normal tissues [158, 160, 161]. Some strategies, such as affinity tuning, exploit the difference in antigen expression level on tumor and non‐tumor tissue to generate CAR that is more tumor‐specific. TCR affinity against peptide MHC ranges from 0.8 to 100 μmol/L, which allows the lysis of target cells without collateral damage [161, 162]. On the contrary, scFv converted from affinity‐matured mAb typically exhibits affinity in the nanomolar range. The high affinity of scFv CAR demonstrated rapid exhaustion and suboptimal persistence in circulation [163]. To optimize the safety and therapeutic window of CAR‐T cells, low‐affinity scFv at micromolar range Kd were selected to target antigens that are highly expressed on tumors while leaving the low‐expression tissue unbound. The strategy generated low‐affinity scFv anti‐CD19 and anti‐EGFR CAR‐T cells, demonstrating longer persistence because of the antigen‐induced expansion of CAR‐T cells and superior anti‐tumor effect with higher specificity in vivo [154, 164]. Affinity engineering by directed evolution isolated an anti‐intercellular adhesion molecule 1 (ICAM1) CAR with affinity of 10 μmol/L. The medium affinity anti‐ICAM1 CAR‐T cells demonstrated potent cancer killing in vivo and more potent therapeutic effects compared with high‐affinity (1 nmol/L) anti‐ICAM1 CAR‐T cells. Most importantly, the high‐affinity CAR‐T cells targeted non‐cancer cells regardless of antigen expression density, while the low‐affinity CAR‐T cells spared them [165]. The scFv structure may also influence CAR function, as scFv with similar affinity against BCMA gave rise to CAR‐T cells with strikingly different expansion rates and anti‐tumor effects [166].
In addition to affinity, the format of the antibody fragment on the CAR also influences CAR‐T therapy potency and efficacy. Alternatives to scFv were investigated for reducing scFv‐based CAR immunogenicity, aggregation and misfolding [167]. As discussed earlier, nanobodies exhibit lower immunogenicity, smaller size and outstanding stability under harsh conditions, rendering them ideal for constructing CAR. With the first US FDA‐approved nanobody monotherapy in 2019, considerably more effort has been invested in the feasibility of nanobody‐based CAR‐T therapy. Bispecific nanobody anti‐CD19+CD20 CAR‐T cells were constructed and killed B cell lines in vitro with high specificity and enhanced proliferation [168].
Despite the success of scFv‐based CAR‐T therapy for hematological tumors, treatment for solid tumors remains challenging. One of the main obstacles is antigen accessibility to the binding domain of CAR, which is even worse when scFv with sub‐micromolar affinity is incorporated into the CAR. On the other hand, nanobody‐based CAR‐T cells, including anti‐CD105, anti‐EIIIB and anti‐BCMA, could access hidden epitopes because of the long CDR3 region and demonstrated effective tumor cell line killing in vitro and tumor size reduction in vivo [169, 170, 171]. An attractive feature of nanobodies is the ability to target >1 epitope on the antigen. The anti‐BCMA CAR‐T therapy LCAR‐B38M utilizes a nanobody that recognizes two epitopes on BCMA, boosting treatment specificity and inducing less toxicity. Although it is difficult to directly compare the results between clinical trials, LCAR‐B38M demonstrated a higher overall response rate (88%) compared with the scFv‐based anti‐BCMA CAR‐T therapy bb‐2121. Moreover, the LCAR‐B38M dose used in clinical trials (median total CAR‐T cells = 32.6 × 106 cells) was significantly lower than that of the scFv‐based therapies (range = 150‐450 × 106 cells) [172, 173]. These promising early‐stage clinical trials raise hopes for other nanobody‐based CAR‐T therapies in solid malignancies. Compared with antibody‐based therapy, CAR‐T therapy faces even more difficulty when the target antigen is expressed at low concentrations. The problem is even more profound when CAR‐T cells are used for solid tumors due to the antigen inaccessibility and immunosuppressive microenvironment [170, 174, 175].
5.3. Multivalent scFv in CAR‐T therapy for improving specificity
Monovalent CAR‐T therapy has achieved success in treating hematological malignancy but nevertheless faces limitations such as a high relapse rate after 6 months, potentially due to the low persistence of CAR‐T cells or downregulation of the antigen target on tumor cells via distinct mechanisms [158, 159]. Most importantly, solid tumor targeting by monovalent CAR‐T cells demonstrates limited efficacy because of target inaccessibility and low expression levels [176]. Few constructs have been designed for targeting multiple antigens on tumor or non‐malignant tissue to improve persistence and inhibit off‐tumor activation of CAR‐T cells. For example, the synNotch system created an AND logic that requires binding two tumor‐associated antigens to fully activate T cells [177]. Other approaches incorporate scFv against multiple tumor antigens connected with an activation module and inhibitory scFv against non‐tumor antigens. However, these approaches are limited by the relatively chunky design and slow activation loop. Instead of engineering two separate scFv with intracellular domains, some recent studies have tested bispecific scFv CAR and reported satisfactory prevention of antigen escape by dual targeting [178, 179, 180]. CAR avidity and functional avidity are vital for CAR‐T cell cytotoxicity, especially in low‐antigen density scenarios. Several studies have reported that elevated CAR avidity improved the CAR‐T cell response [181, 182]. By design, bispecific antibodies exhibit higher avidity than monospecific antibodies as they can bind two antigens on the same target. CD19 and CD20 dual‐targeting CAR‐T cells demonstrate signs of benefiting patients with relapsed B cell lymphoma with manageable grade 1 cytokine release syndrome [183]. Theoretically, dual targeting can prevent tumor relapse caused by the downregulation of a single tumor antigen [184, 185]. In addition to anti‐CD20, anti‐CD22, anti‐CD123 and BAFF‐R (TNF receptor superfamily member 13C) have been combined with anti‐CD19 as bispecific CAR [186]. In a pilot study of six patients with refractory lymphoblastic leukemia, bispecific anti‐CD19+CD22 CAR‐T cells exhibited a long persistence duration of 100 days and high expansion after 2 weeks of infusion, raising hopes for the following phase I clinical trial [187].
In addition to lymphoma, a single‐center clinical study on MM tested bispecific antibody formats with anti‐BCMA and anti‐CD38 CAR‐T cells and demonstrated a 1‐year progression‐free survival rate of 68% [188]. Although it is too early to draw conclusions based on phase I and pilot clinical studies, significantly milder cytokine release syndrome and other adverse effects have been observed in bispecific CAR‐T/CAR‐NK treatment compared with conventional mono‐scFv CAR‐T/CAR‐NK therapy. Despite the high expectations for bispecific CAR‐T/CAR‐NK cells to enhance the therapeutic effects of CAR‐T/CAR‐NK therapies, the structure of multi‐specific CAR requires extensive optimization, such as the linker length and sequence and the orientation of the variable regions [178, 179, 189]. Furthermore, tumor resistance to CAR‐T/CAR‐NK therapies via impaired interferon‐gamma (IFN‐γ) signaling or Fas expression has also been reported, which raises concerns about their efficacy on solid tumors [190].
Cancer/testis antigens (CTAs) are expressed in various cancers, rendering them ideal targets for immunotherapy [191]. Compared with conventional targets such as EGFR, HER2, VEGF and mucin 1 (MUC1), which are also expressed in normal tissues, CTAs hold great potential for minimizing off‐tumor toxicity [192]. Although most CTAs are expressed intracellularly, recent bioinformatics studies indicated the cytomembrane expression of CTAs and the subsequent finding of melanoma‐associated antigen 1 (MAGE‐A1) expression on the cytomembrane of lung adenocarcinoma cell lines [193]. Anti‐MAGE‐A1 CAR‐T cells demonstrated tumor growth inhibition in a xenograft model by binding to MAGE‐A1‐positive cells, which validated the hypothesis for targeting CTAs [194]. Other CTAs that have been suggested to have surface expression include PRAME nuclear receptor transcriptional regulator (PRAME), CTA 83 (CT83), sperm autoantigenic protein 17 (SP17), solute carrier organic anion transporter family member 6A1 (SLCO6A1) and placenta‐enriched 1 (PLAC1), whose surface binding as CAR‐T treatments have not been proven [195, 196]. The advances in deep learning and high‐throughput screening may facilitate the development of next‐generation bispecific CAR against novel cancer antigens.
6. CONCLUSIONS AND FUTURE PERSPECTIVE
The current antibody fragments for cancer therapies can be classified into three main categories: mono‐specific, multi‐specific and incorporated as CAR. Antibody fragment engineering is opening up new avenues for cancer treatment. The US FDA has approved one fragment‐based bispecific antibody and five fragment‐based CAR‐T therapies for cancer treatment. More than 60 antibody fragments alone and 150 antibody fragment‐based CAR‐T therapies are being evaluated in clinical trials for both hematological and solid tumors. Antibody fragments are considerably smaller than conventional antibodies, making them easier to diffuse into tumor sites and potentially more effective for targeting solid tumors. Recurrence is often reported after treatment with single mAb therapy. To combat cancer resistance, bispecific and multivalent bindings have been developed to target >1 tumor antigen. The drawbacks of the lack of Fc have also been addressed by different engineering strategies, such as fusing anti‐CD3 scFv to antibody fragments to engage effector T cells for tumor cell killing or fusing adaptor proteins such as HSA to antibody fragments to extend the half‐life.
Currently, more than 30 antibody fragment engineering platforms are generating novel antibody fragment formats for cancer therapy. The growing number of engineering strategies and formats facilitate the development of novel antibody drugs; therefore, careful selection of a suitable strategy is required. The binding affinity, avidity, valency, epitope interaction/accessibility, stability and flexibility, and half‐life of the format all require optimization to generate the ideal drug for clinical benefits. Therefore, a deeper understanding of the engineering strategies is necessary for designing a suitable format. We should expect an explosion of novel antibody fragment candidates for cancer immunotherapy via the joint efforts of deep learning and high‐throughput screening platforms. Deep learning and machine learning algorithms have been applied for modeling a range of antibody optimization processes, such as predicting antibody structure, antibody solubility and stability, and antibody‐antigen binding and affinity. With the introduction of alpha‐folding, similar algorithms have been built for modeling antibody structure [197]. Antibody structure prediction could also be considered a special case for general protein structure prediction. Deep learning methods such as DeepAb have been developed to simulate antibody structures by predicting the inter‐residue distances and orientation [198]. The key question in antibody function is antibody‐antigen binding. An antibody‐antigen prediction has also been the most popular task for machine learning. For example, “PEPITO” [199] and “DiscoTope” [200] have been developed for predicting epitopes, while Paratome and support vector machine (SVM)‐based algorithms have been built for predicting paratopes [201, 202]. In addition to the antibody‐antigen interface prediction algorithms, convolutional neural networks have also been used for predicting whether antibodies bind to specific antigens and the developability of specific antibodies [203, 204, 205].
Currently, more than 100 multivalent antibody fragment formats have been validated experimentally, illustrating the robustness of the modular domains. The antibody domains can be assembled like Lego bricks to form a novel antibody derivative, targeting antigen combinations. The modular antibody fragment format design is determined by a complex interplay of parameters, including scFv/nanobody binding affinity, specificity, valency, antigen topology, density and expression site. The design is complicated by the structural transformation when two antibody fragments are linked. For example, the parental anti‐HER3 mAb does not inhibit the HER3 signaling pathway in combination with anti‐HER2 mAb. However, the bispecific anti‐HER2+HER3 antibody derived from the parental mAbs can inhibit both the HER2 and HER3 signaling pathways and tumor growth [196, 206]. As it is difficult to predict the function of the domain antibody format from parental full antibodies, high‐throughput screening such as phage/yeast/Escherichia coli display systems have been adapted to aid the identification of functional multivalent antibody formats from several thousand candidates. For example, unbiased phenotypic screening of more than 500 bispecific IgG yielded a novel HER2‐HER3 bispecific IgG1 that reduced tumor volume in vivo by a “dock and block” mechanism that is not feasible with the two parental mAbs [123]. In addition to the antibody‐antigen interaction and antigen selection, it has been acknowledged that the multivalent domain antibody format impacts the function of multivalent domain antibodies. The future panels of unbiased screening should include several thousand antibody fragments in a range of formats to yield the best fit for the purpose.
Although it was believed that the complicated interplay of design parameters rendered it almost impossible to predict functional multivalent antibody fragments from parental mAbs, novel methods have been developed to tackle the issue. The rational design of therapeutic antibodies depends on the accurate epitope, paratope and antibody structure. In addition to the conventional methods, including alanine scanning, crystallography and cryogenic electron microscopy, machine learning and deep learning have currently been applied to assist in therapeutic antibody design optimization. Compared with conventional structural biology methods, which are mostly time‐ and cost‐consuming, machine/deep learning can achieve ultra‐high‐throughput prediction and optimization within hours to a few days depending on the size and complexity of the database. Neural networks can extract the key features of a range of properties (such as VH gene usage, CDRs, paratopes, epitopes and even 3D structures) and calculate each layer in latent spaces, which is not feasible with traditional prediction algorithms such as clonotyping.
The binding and function data from the high‐throughput multivalent antibody platform can be collected to train novel neural networks for predicting untested combinations of modular antibody fragments, thereby accelerating the discovery of functional multivalent antibody fragments. Validation of the neural network prediction will be used to optimize the algorithm, in turn boosting prediction accuracy. The evolution of the neural network will hopefully form a positive feedback loop to enhance the efficiency of multivalent antibody format engineering for cancer therapy.
With the recent advances in antibody variable region engineering, considerably more antibody‐based cancer therapies have been approved by the US FDA in the past decade. The engineering of the variable region that are described above can improve the safety, stability, specificity and potency of the antibody‐based cancer therapy, which could provide guidance for future cancer immunotherapy development.
DECLARATIONS
COMPETING INTERESTS
The authors declare that they have no competing interests.
FUNDING
This work was supported by grants from the CAMS Innovation Fund for Medical Sciences (2021‐I2M‐1‐017).
AUTHOR CONTRIBUTIONS
Lou H wrote the manuscript and drafted the figures. Cao X revised the manuscript. All authors approved the final manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
ACKNOWLEDGMENTS
We thank Drs. Nan Li, Jiannan Feng and Bingjing Wang for revising the manuscript.
Lou H, Cao X. Antibody variable region engineering for improving cancer immunotherapy. Cancer Commun. 2022;42:804–827. 10.1002/cac2.12330
REFERENCES
- 1. Leavy O. The birth of monoclonal antibodies. Nat Immunol. 2016;17(1):S13. [Google Scholar]
- 2. Grillo‐Lopez AJ, White CA, Dallaire BK, Varns CL, Shen CD, Wei A, et al. Rituximab the first monoclonal antibody approved for the treatment of lymphoma. Curr Pharm Biotechnol. 2000;1(1):1‐9. [DOI] [PubMed] [Google Scholar]
- 3. Duggan S. Caplacizumab: first global approval. Drugs. 2018;78(15):1639‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jen EY, Xu Q, Schetter A, Przepiorka D, Shen YL, Roscoe D, et al. FDA approval: blinatumomab for patients with B‐cell precursor acute lymphoblastic leukemia in morphologic remission with minimal residual disease. Clin Cancer Res. 2019;25(2):473‐7. [DOI] [PubMed] [Google Scholar]
- 5. Wang Y, Wang M, Wu HX, Xu RH. Advancing to the era of cancer immunotherapy. Cancer Commun (Lond). 2021;41(9):803‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nelson AL, Reichert JM. Development trends for therapeutic antibody fragments. Nat Biotechnol. 2009;27(4):331‐7. [DOI] [PubMed] [Google Scholar]
- 7. Nelson AL. Antibody fragments: hope and hype. MAbs 2: 77‐83. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen X, Zaro JL, Shen W‐C. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65(10):1357‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shen Z, Yan H, Zhang Y, Mernaugh RL, Zeng X. Engineering peptide linkers for scFv immunosensors. Anal Chem. 2008;80(6):1910‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yusakul G, Sakamoto S, Pongkitwitoon B, Tanaka H, Morimoto S. Effect of linker length between variable domains of single chain variable fragment antibody against daidzin on its reactivity. Biosci Biotechnol Biochem. 2016;80(7):1306‐12. [DOI] [PubMed] [Google Scholar]
- 11. Krokhotin A, Du H, Hirabayashi K, Popov K, Kurokawa T, Wan X, et al. Computationally guided design of single‐chain variable fragment improves specificity of chimeric antigen receptors. Molecular Therapy‐Oncolytics. 2019;15:30‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Muyldermans S, Baral TN, Retamozzo VC, De Baetselier P, De Genst E, Kinne J, et al. Camelid immunoglobulins and nanobody technology. Vet Immunol Immunopathol. 2009;128(1‐3):178‐83. [DOI] [PubMed] [Google Scholar]
- 13. Pardon E, Laeremans T, Triest S, Rasmussen SGF, Wohlknig A, Ruf A, et al. A general protocol for the generation of Nanobodies for structural biology. Nat Protoc. 2014;9(3):674‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De Genst E, Silence K, Decanniere K, Conrath K, Loris R, Kinne J, et al. Molecular basis for the preferential cleft recognition by dromedary heavy‐chain antibodies. Proc Natl Acad Sci. 2006;103(12):4586‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Debie P, Lafont C, Defrise M, Hansen I, van Willigen DM, van Leeuwen FWB, et al. Size and affinity kinetics of nanobodies influence targeting and penetration of solid tumours. J Controlled Release. 2020;317:34‐42. [DOI] [PubMed] [Google Scholar]
- 16. Kunz P, Zinner K, Mcke N, Bartoschik T, Muyldermans S, Hoheisel JD. The structural basis of nanobody unfolding reversibility and thermoresistance. Sci Rep. 2018;8(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kunz P, Ortale A, Mucke N, Zinner K, Hoheisel JD. Nanobody stability engineering by employing the DeltaTm shift; a comparison with apparent rate constants of heat‐induced aggregation. Protein Eng Des Sel. 2019;32(5):241‐9. [DOI] [PubMed] [Google Scholar]
- 18. Dingus J, Tang JCY, Cepko C. A general approach for stabilizing nanobodies for intracellular expression. bioRxiv. 2021:2021.04.06.438746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vosjan MJWD, Vercammen J, Kolkman JA, Stigter‐van Walsum M, Revets H, van Dongen GAMS. Nanobodies targeting the hepatocyte growth factor: potential new drugs for molecular cancer therapy. Mol Cancer Ther. 2012;11(4):1017‐25. [DOI] [PubMed] [Google Scholar]
- 20. Steeland S, Vandenbroucke RE, Libert C. Nanobodies as therapeutics: big opportunities for small antibodies. Drug Discovery Today. 2016;21(7):1076‐113. [DOI] [PubMed] [Google Scholar]
- 21. Deonarain MP, Xue Q. Tackling solid tumour therapy with small‐format drug conjugates. Antibody Ther. 2020;3(4):237‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Christiansen J, Rajasekaran AK. Biological impediments to monoclonal antibody‐based cancer immunotherapy. Mol Cancer Ther. 2004;3(11):1493‐501. [PubMed] [Google Scholar]
- 23. Teicher BA, Chari RVJ. Antibody conjugate therapeutics: challenges and potential. Clin Cancer Res. 2011;17(20):6389‐97. [DOI] [PubMed] [Google Scholar]
- 24. Bae J, Song Y. Engineering a cell‐penetrating hyperstable antibody scFv(Ras) ‐ An extraordinary approach to cancer therapeutics. Synth Syst Biotechnol. 2021;6(4):343‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Du Y, Lin X, Feng Q, Pan X, Song S, Yang J. Inhibition of human lung cancer cells by anti‐p21Ras scFv mediated by the activatable cell‐penetrating peptide. Anticancer Drugs. 2022;33(1):e562–e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Keyaerts M, Xavier C, Heemskerk J, Devoogdt N, Everaert H, Ackaert C, et al. Phase I Study of 68Ga‐HER2‐Nanobody for PET/CT Assessment of HER2 Expression in Breast Carcinoma. J Nucl Med. 2016;57(1):27‐33. [DOI] [PubMed] [Google Scholar]
- 27. Li Z, Krippendorff B‐F, Sharma S, Walz AC , Lav. Influence of molecular size on tissue distribution of antibody fragments. 2016. p. 113‐9. [DOI] [PMC free article] [PubMed]
- 28. Griffiths K, Binder U, McDowell W, Tommasi R, Frigerio M, Darby WG, et al. Half‐life extension and non‐human primate pharmacokinetic safety studies of i‐body AD‐114 targeting human CXCR4. mAbs. 2019;11(7):1331‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Adams R, Griffin L, Compson JE, Jairaj M, Baker T, Ceska T, et al. Extending the half‐life of a fab fragment through generation of a humanized anti‐human serum albumin Fv domain: An investigation into the correlation between affinity and serum half‐life. mAbs. 2016;8(7):1336‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pan H, Liu J, Deng W, Xing J, Li Q, Wang Z. Site‐specific PEGylation of an anti‐CEA/CD3 bispecific antibody improves its antitumor efficacy. Int J Nanomed. 2018;13:3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nilsen J, Trabjerg E, Grevys A, Azevedo C, Brennan SO, Stensland M, et al. An intact C‐terminal end of albumin is required for its long half‐life in humans. Communications Biology. 2020;3(1):181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shen Z, Xiang Y, Vergara S, Chen A, Xiao Z, Santiago U, et al. A resource of high‐quality and versatile nanobodies for drug delivery. iScience. 2021;24(9):103014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tijink BM, Laeremans T, Budde M, Stigter‐van Walsum M, Dreier T, de Haard HJ, et al. Improved tumor targeting of anti‐epidermal growth factor receptor Nanobodies through albumin binding: taking advantage of modular Nanobody technology. Mol Cancer Ther. 2008;7(8):2288‐97. [DOI] [PubMed] [Google Scholar]
- 34. Kholodenko IV, Kalinovsky DV, Svirshchevskaya EV, Doronin II, Konovalova MV, Kibardin AV, et al. Multimerization through pegylation improves pharmacokinetic properties of scFv fragments of GD2‐specific antibodies. Molecules. 2019;24(21):3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chatalic KL, Veldhoven‐Zweistra J, Bolkestein M, Hoeben S, Koning GA, Boerman OC, et al. A Novel (1)(1)(1)In‐Labeled Anti‐Prostate‐Specific Membrane Antigen Nanobody for Targeted SPECT/CT Imaging of Prostate Cancer. J Nucl Med. 2015;56(7):1094‐9. [DOI] [PubMed] [Google Scholar]
- 36. Hrynchak I, Santos L, Falcao A, Gomes CM, Abrunhosa AJ. Nanobody‐Based Theranostic Agents for HER2‐Positive Breast Cancer: Radiolabeling Strategies. Int J Mol Sci. 2021;22(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xavier C, Blykers A, Vaneycken I, D'Huyvetter M, Heemskerk J, Lahoutte T, et al. (18)F‐nanobody for PET imaging of HER2 overexpressing tumors. Nucl Med Biol. 2016;43(4):247‐52. [DOI] [PubMed] [Google Scholar]
- 38. Alibakhshi A, Kahaki FA, Ahangarzadeh S, Yaghoobi H, Yarian F, Arezumand R, et al. Targeted cancer therapy through antibody fragments‐decorated nanomedicines. J Controlled Release. 2017;268:323‐34. [DOI] [PubMed] [Google Scholar]
- 39. Hu S‐z, Shively L, Raubitschek A, Sherman M, Williams LE, Wong JYC, et al. Minibody: a novel engineered anti‐carcinoembryonic antigen antibody fragment (single‐chain Fv‐CH3) which exhibits rapid, high‐level targeting of xenografts. Cancer Res. 1996;56(13):3055‐61. [PubMed] [Google Scholar]
- 40. Pandit‐Taskar N, Postow MA, Hellmann MD, Harding JJ, Barker CA, O'Donoghue JA, et al. First‐in‐humans imaging with 89Zr‐Df‐IAB22M2C anti‐CD8 minibody in patients with solid malignancies: preliminary pharmacokinetics, biodistribution, and lesion targeting. J Nucl Med. 2020;61(4):512‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bäck TA, Jennbacken K, Hagberg Thulin M, Lindegren S, Jensen H, Olafsen T, et al. Targeted alpha therapy with astatine‐211‐labeled anti‐PSCA A11 minibody shows antitumor efficacy in prostate cancer xenografts and bone microtumors. Ejnmmi Res. 2020;10(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Van Roy M, Ververken C, Beirnaert E, Hoefman S, Kolkman J, Vierboom M, et al. The preclinical pharmacology of the high affinity anti‐IL‐6R Nanobody® ALX‐0061 supports its clinical development in rheumatoid arthritis. Arthritis Res Ther. 2015;17(1):135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Papadopoulos KP, Isaacs R, Bilic S, Kentsch K, Huet HA, Hofmann M, et al. Unexpected hepatotoxicity in a phase I study of TAS266, a novel tetravalent agonistic Nanobody® targeting the DR5 receptor. Cancer Chemother Pharmacol. 2015;75(5):887‐95. [DOI] [PubMed] [Google Scholar]
- 44. Kibria MG, Akazawa‐Ogawa Y, Rahman N, Hagihara Y, Kuroda Y. The immunogenicity of an anti‐EGFR single domain antibody (V(HH)) is enhanced by misfolded amorphous aggregation but not by heat‐induced aggregation. Eur J Pharm Biopharm. 2020;152:164‐74. [DOI] [PubMed] [Google Scholar]
- 45. Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23(9):1126‐36. [DOI] [PubMed] [Google Scholar]
- 46. Mullard A. FDA approves fourth CAR‐T cell therapy. Nat Rev Drug Discovery. 2021;20(3):166. [DOI] [PubMed] [Google Scholar]
- 47. Morrison C. Nanobody approval gives domain antibodies a boost. Nat Rev Drug Discovery. 2019;18(7):485‐8. [DOI] [PubMed] [Google Scholar]
- 48. Xie Y‐H, Chen Y‐X, Fang J‐Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduction and Targeted Therapy. 2020;5(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu L, Jacobsen FW, Everds N, Zhuang Y, Yu YB, Li N, et al. Biological characterization of a stable effector functionless (SEFL) monoclonal antibody scaffold in vitro. J Biol Chem. 2017;292(5):1876‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Garca‐Foncillas J, Sunakawa Y, Aderka D, Wainberg Z, Ronga P, Witzler P, et al. Distinguishing features of cetuximab and panitumumab in colorectal cancer and other solid tumors. Front Oncol. 2019;9:849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. English DP, Roque DM, Santin AD. HER2 expression beyond breast cancer: therapeutic implications for gynecologic malignancies. Molecular Diagnosis & Therapy. 2013;17(2):85‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hudis CA. Trastuzumab—mechanism of action and use in clinical practice. N Engl J Med. 2007;357(1):39‐51. [DOI] [PubMed] [Google Scholar]
- 53. Yun J, Lee S‐H, Kim S‐Y, Jeong S‐Y, Kim J‐H, Pyo K‐H, et al. Antitumor activity of amivantamab (JNJ‐61186372), an EGFR‐MET bispecific antibody, in diverse models of EGFR exon 20 insertion‐driven NSCLC. Cancer Discov. 2020;10(8):1194‐209. [DOI] [PubMed] [Google Scholar]
- 54. Jadot G, Augusto D, Samartzi V, Castela S, Lejeune S, Seront E, et al. Clinical, biological and histological features predictive of response to everolimus in metastatic breast cancer patients. Ann Oncol. 2019;30:iii59‐iii60. [Google Scholar]
- 55. Puttemans J, Dekempeneer Y, Eersels JL, Hanssens H, Debie P, Keyaerts M, et al. Preclinical Targeted alpha‐ and beta(‐)‐Radionuclide Therapy in HER2‐Positive Brain Metastasis Using Camelid Single‐Domain Antibodies. Cancers (Basel). 2020;12(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, et al. Escape from HER‐family tyrosine kinase inhibitor therapy by the kinase‐inactive HER3. Nature. 2007;445(7126):437‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hsieh AC, Moasser MM. Targeting HER proteins in cancer therapy and the role of the non‐target HER3. Br J Cancer. 2007;97(4):453‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Meulendijks D, Jacob W, Martinez‐Garcia M, Taus A, Lolkema MP, Voest EE, et al. First‐in‐human phase I study of lumretuzumab, a glycoengineered humanized anti‐HER3 monoclonal antibody, in patients with metastatic or advanced HER3‐positive solid tumors. Clin Cancer Res. 2016;22(4):877‐85. [DOI] [PubMed] [Google Scholar]
- 59. Mirschberger C, Schiller CB, Schrml M, Dimoudis N, Friess T, Gerdes CA, et al. RG7116, a therapeutic antibody that binds the inactive HER3 receptor and is optimized for immune effector activation. Cancer Res. 2013;73(16):5183‐94. [DOI] [PubMed] [Google Scholar]
- 60. Eliseev IE, Ukrainskaya VM, Yudenko AN, Mikushina AD, Shmakov SV, Afremova AI, et al. Targeting ErbB3 Receptor in Cancer with Inhibitory Antibodies from Llama. Biomedicines. 2021;9(9):1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Murukesh N, Dive C, Jayson GC. Biomarkers of angiogenesis and their role in the development of VEGF inhibitors. Br J Cancer. 2010;102(1):8‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Loges S, Mazzone M, Hohensinner P, Carmeliet P. Silencing or fueling metastasis with VEGF inhibitors: antiangiogenesis revisited. Cancer Cell. 2009;15(3):167‐70. [DOI] [PubMed] [Google Scholar]
- 63. Sekeres MA. The avastin story. N Engl J Med. 2011;365(15):1454‐5. [DOI] [PubMed] [Google Scholar]
- 64. Olsson A‐K, Dimberg A, Kreuger J, Claesson‐Welsh L. VEGF receptor signalling? In control of vascular function. Nat Rev Mol Cell Biol. 2006;7(5):359‐71. [DOI] [PubMed] [Google Scholar]
- 65. Karami E, Sabatier J‐M, Behdani M, Irani S, Kazemi‐Lomedasht F. A nanobody‐derived mimotope against VEGF inhibits cancer angiogenesis. J Enzyme Inhib Med Chem. 2020;35(1):1233‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Arezumand R, Mahdian R, Behdani M, Khanahmad H, Langari J, Namvarasl N, et al. Recombinant expression and purification of human placental growth factor 1 and specific camel heavy chain polyclonal antibody preparation. Saudi J Biol Sci. 2014;21(1):35‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sadeghi A, Behdani M, Muyldermans S, Habibi‐Anbouhi M, Kazemi‐Lomedasht F. Development of a mono‐specific anti‐VEGF bivalent nanobody with extended plasma half‐life for treatment of pathologic neovascularization. Drug Test Anal. 2020;12(1):92‐100. [DOI] [PubMed] [Google Scholar]
- 68. Qasemi M, Behdani M, Shokrgozar MA, Molla‐Kazemiha V, Mohseni‐Kuchesfahani H, Habibi‐Anbouhi M. Construction and expression of an anti‐VEGFR2 Nanobody‐Fc fusionbody in NS0 host cell. Protein Expression Purif. 2016;123:19‐25. [DOI] [PubMed] [Google Scholar]
- 69. Naldini L, Vigna E, Narsimhan RP, Gaudino G, Zarnegar R, Michalopoulos GK, et al. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto‐oncogene c‐MET. Oncogene. 1991;6(4):501‐4. [PubMed] [Google Scholar]
- 70. Fu J, Su X, Li Z, Deng L, Liu X, Feng X, et al. HGF/c‐MET pathway in cancer: from molecular characterization to clinical evidence. Oncogene. 2021;40(28):4625‐51. [DOI] [PubMed] [Google Scholar]
- 71. Spigel DR, Edelman MJ, O'Byrne K, Paz‐Ares L, Mocci S, Phan S, et al. Results From the Phase III Randomized Trial of Onartuzumab Plus Erlotinib Versus Erlotinib in Previously Treated Stage IIIB or IV Non‐Small‐Cell Lung Cancer: METLung. J Clin Oncol. 2017;35(4):412‐20. [DOI] [PubMed] [Google Scholar]
- 72. Cunningham D, Tebbutt NC, Davidenko I, Murad AM, Al‐Batran S‐E, Ilson DH, et al. Phase III, randomized, double‐blind, multicenter, placebo (P)‐controlled trial of rilotumumab (R) plus epirubicin, cisplatin and capecitabine (ECX) as first‐line therapy in patients (pts) with advanced MET‐positive (pos) gastric or gastroesophageal junction (G/GEJ) cancer: RILOMET‐1 study. 2015. [DOI] [PMC free article] [PubMed]
- 73. Kim K‐H, Kim H. Progress of antibody‐based inhibitors of the HGF‐cMET axis in cancer therapy. Exp Mol Med. 2017;49(3):e307‐e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Su Z, Han Y, Sun Q, Wang X, Xu T, Xie W, et al. Anti‐MET VHH pool overcomes MET‐targeted cancer therapeutic resistance. Mol Cancer Ther. 2019;18(1):100‐11. [DOI] [PubMed] [Google Scholar]
- 75. Stieglmaier J, Benjamin J, Nagorsen D. Utilizing the BiTE (bispecific T‐cell engager) platform for immunotherapy of cancer. Expert Opin Biol Ther. 2015;15(8):1093‐9. [DOI] [PubMed] [Google Scholar]
- 76. Speck T, Heidbuechel JPW, Veinalde R, Jaeger D, Von Kalle C, Ball CR, et al. Targeted BiTE expression by an oncolytic vector augments therapeutic efficacy against solid tumors. Clin Cancer Res. 2018;24(9):2128‐37. [DOI] [PubMed] [Google Scholar]
- 77. Einsele H, Borghaei H, Orlowski RZ, Subklewe M, Roboz GJ, Zugmaier G, et al. The BiTE (bispecific T‐cell engager) platform: development and future potential of a targeted immuno‐oncology therapy across tumor types. Cancer. 2020;126(14):3192‐201. [DOI] [PubMed] [Google Scholar]
- 78. de Bruin RCG, Veluchamy JP, Lougheed SM, Schneiders FL, Lopez‐Lastra S, Lameris R, et al. A bispecific nanobody approach to leverage the potent and widely applicable tumor cytolytic capacity of Vγ9Vσ2‐T cells. Oncoimmunology. 2018;7(1):e1375641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Maeda H, Khatami M. Analyses of repeated failures in cancer therapy for solid tumors: poor tumor‐selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin Transl Med. 2018;7(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mandrup OA, Ong SC, Lykkemark S, Dinesen A, Rudnik‐Jansen I, Dagns‐Hansen NF, et al. Programmable half‐life and anti‐tumour effects of bispecific T‐cell engager‐albumin fusions with tuned FcRn affinity. Communications Biology. 2021;4(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Knödler M, Körfer J, Kunzmann V, Trojan J, Daum S, Schenk M, et al. Randomised phase II trial to investigate catumaxomab (anti‐EpCAM × anti‐CD3) for treatment of peritoneal carcinomatosis in patients with gastric cancer. Br J Cancer. 2018;119(3):296‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Borlak J, Länger F, Spanel R, Schndorfer G, Dittrich C. Immune‐mediated liver injury of the cancer therapeutic antibody catumaxomab targeting EpCAM, CD3 and Fcγ receptors. Oncotarget. 2016;7(19):28059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Romero D. Blinatumomab facilitates complete responses. Nat Rev Clin Oncol. 2018;15(4):200. [DOI] [PubMed] [Google Scholar]
- 84. Newman MJ, Benani DJ. A review of blinatumomab, a novel immunotherapy. J Oncol Pharm Pract. 2016;22(4):639‐45. [DOI] [PubMed] [Google Scholar]
- 85. Gkbuget N, Dombret H, Bonifacio M, Reichle A, Graux C, Faul C, et al. Blinatumomab for minimal residual disease in adults with B‐cell precursor acute lymphoblastic leukemia. Blood, The Journal of the American Society of Hematology. 2018;131(14):1522‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, et al. Immunopharmacologic response of patients with B‐lineage acute lymphoblastic leukemia to continuous infusion of T cell‐engaging CD19/CD3‐bispecific BiTE antibody blinatumomab. Blood, The Journal of the American Society of Hematology. 2012;119(26):6226‐33. [DOI] [PubMed] [Google Scholar]
- 87. Staflin K, de Zafra CLZ, Schutt LK, Clark V, Zhong F, Hristopoulos M, et al. Target arm affinities determine preclinical efficacy and safety of anti‐HER2/CD3 bispecific antibody. JCI Insight. 2020;5(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Slaga D, Ellerman D, Lombana TN, Vij R, Li J, Hristopoulos M, et al. Avidity‐based binding to HER2 results in selective killing of HER2‐overexpressing cells by anti‐HER2/CD3. Sci Transl Med. 2018;10(463). [DOI] [PubMed] [Google Scholar]
- 89. Trinklein ND, Pham D, Schellenberger U, Buelow B, Boudreau A, Choudhry P, et al. Efficient tumor killing and minimal cytokine release with novel T‐cell agonist bispecific antibodies. 2019. p. 639‐52. [DOI] [PMC free article] [PubMed]
- 90. Reusch U, Duell J, Ellwanger K, Herbrecht C, Knackmuss SHJ, Fucek I, et al. A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19+ tumor cells. 2015. p. 584‐604. [DOI] [PMC free article] [PubMed]
- 91. Hoseini SS, Guo H, Wu Z, Hatano MN, Cheung N‐KV. A potent tetravalent T‐cell‐engaging bispecific antibody against CD33 in acute myeloid leukemia. Blood advances. 2018;2(11):1250‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hutchings M, Iacoboni G, Morschhauser F, Offner F, Sureda A, Salles GA, et al. CD20‐Tcb (RG6026), a novel “2: 1” format T‐cell‐engaging bispecific antibody, induces complete remissions in relapsed/refractory B‐cell non‐Hodgkin's lymphoma: preliminary results from a phase I first in human trial. Blood. 2018;132:226. [Google Scholar]
- 93. Morschhauser F, Carlo‐Stella C, Offner F, Salles GA, Hutchings M, Iacoboni G, et al. Dual CD20‐targeted therapy with concurrent CD20‐TCB and obinutuzumab shows highly promising clinical activity and manageable safety in relapsed or refractory B‐cell non‐Hodgkin lymphoma: preliminary results from a phase Ib trial. 2019.
- 94. Tapia‐Galisteo A, i Snchez Rodrguez, Aguilar‐Sopea O, Harwood SL, Narbona J, Ferreras Gutierrez M, et al. Trispecific T‐cell engagers for dual tumor‐targeting of colorectal cancer. OncoImmunology. 2022;11(1):2034355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Duell J, Dittrich M, Bedke T, Mueller T, Eisele F, Rosenwald A, et al. Frequency of regulatory T cells determines the outcome of the T‐cell‐engaging antibody blinatumomab in patients with B‐precursor ALL. Leukemia. 2017;31(10):2181‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21(4):589‐601. [DOI] [PubMed] [Google Scholar]
- 97. Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, et al. Increased regulatory T‐cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66(6):3294‐302. [DOI] [PubMed] [Google Scholar]
- 98. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg‐mediated T cell suppression. Front Immunol. 2012;3:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Peipp M, Kellner C, Sebens S, Rcken C. Novel Bispecific Antibodies Increase T‐Cell Cytotoxicity against Pancreatic Cancer Cells. Cancer Res. 2014; 74(5):1349‐60 [DOI] [PubMed] [Google Scholar]
- 100. Lin W‐W, Lu Y‐C, Chuang C‐H, Cheng T‐L. Ab locks for improving the selectivity and safety of antibody drugs. J Biomed Sci. 2020;27(1):1‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Erster O, Thomas JM, Hamzah J, Jabaiah AM, Getz JA, Schoep TD, et al. Site‐specific targeting of antibody activity in vivo mediated by disease‐associated proteases. J Controlled Release. 2012;161(3):804‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EE, Liang TW, et al. Tumor‐specific activation of an EGFR‐targeting probody enhances therapeutic index. Sci Transl Med. 2013;5(207):207ra144. [DOI] [PubMed] [Google Scholar]
- 103. Chen IJ, Chuang C‐H, Hsieh Y‐C, Lu Y‐C, Lin W‐W, Huang C‐C, et al. Selective antibody activation through protease‐activated pro‐antibodies that mask binding sites with inhibitory domains. Sci Rep. 2017;7(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Lucchi R, Bentanachs J, Oller‐Salvia B. The masking game: design of activatable antibodies and mimetics for selective therapeutics and cell control. ACS Central Science. 2021;7(5):724‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Trang VH, Zhang X, Yumul RC, Zeng W, Stone IJ, Wo SW, et al. A coiled‐coil masking domain for selective activation of therapeutic antibodies. Nat Biotechnol. 2019;37(7):761‐5. [DOI] [PubMed] [Google Scholar]
- 106. Yang J, Shimada Y, Olsthoorn RCL, Snaar‐Jagalska BE, Spaink HP, Kros A. Application of coiled coil peptides in liposomal anticancer drug delivery using a zebrafish xenograft model. Acs Nano. 2016;10(8):7428‐35. [DOI] [PubMed] [Google Scholar]
- 107. Zhang L, Fang Y, Li L, Yang J, Radford DC, Kopeek J. Human Serum Albumin‐Based Drug‐Free Macromolecular Therapeutics: Apoptosis Induction by Coiled‐Coil‐Mediated Cross‐Linking of CD20 Antigens on Lymphoma B Cell Surface. Macromol Biosci. 2018;18(11):1800224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Utterstrm J, Naeimipour S, Selegrd R, Aili D. Coiled coil‐based therapeutics and drug delivery systems. Adv Drug Deliv Rev. 2020;170:26‐43 [DOI] [PubMed] [Google Scholar]
- 109. Dickopf S, Buldun C, Vasic V, Georges G, Hage C, Mayer K, et al. Prodrug‐Activating Chain Exchange (PACE) converts targeted prodrug derivatives to functional bi‐or multispecific antibodies. Biol Chem. 2022;403(56):495‐508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Hu W, Wang G, Huang D, Sui M, Xu Y. Cancer immunotherapy based on natural killer cells: current progress and new opportunities. Front Immunol. 2019;10:1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. de Witte MA, Sarhan D, Davis Z, Felices M, Vallera DA, Hinderlie P, et al. Early reconstitution of NK and γσ T cells and its implication for the design of post‐transplant immunotherapy. Biol Blood Marrow Transplant. 2018;24(6):1152‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Pahl JHW, Koch J, Gtz J‐J, Arnold A, Reusch U, Gantke T, et al. CD16A activation of NK cells promotes NK cell proliferation and memory‐like cytotoxicity against cancer cells. Cancer Immunol Res. 2018;6(5):517‐27. [DOI] [PubMed] [Google Scholar]
- 113. Wu J, Fu J, Zhang M, Liu D. AFM13: a first‐in‐class tetravalent bispecific anti‐CD30/CD16A antibody for NK cell‐mediated immunotherapy. J Hematol Oncol. 2015;8(1):1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Rothe A, Sasse S, Topp MS, Eichenauer DA, Hummel H, Reiners KS, et al. A phase 1 study of the bispecific anti‐CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood, The Journal of the American Society of Hematology. 2015;125(26):4024‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU, et al. IL15 trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin Cancer Res. 2016;22(14):3440‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Gauthier L, Morel A, Anceriz N, Rossi B, Blanchard‐Alvarez A, Grondin G, et al. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell. 2019;177(7):1701‐13. [DOI] [PubMed] [Google Scholar]
- 117. Ciardiello F, Normanno N. HER2 signaling and resistance to the anti‐EGFR monoclonal antibody cetuximab: a further step toward personalized medicine for patients with colorectal cancer. Cancer Discov. 2011;1(6):472‐4. [DOI] [PubMed] [Google Scholar]
- 118. Cai W‐Q, Zeng L‐S, Wang L‐F, Wang Y‐Y, Cheng J‐T, Zhang Y, et al. The latest battles between EGFR monoclonal antibodies and resistant tumor cells. Front Oncol. 2020;10:1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Nahta R, Yu D, Hung M‐C, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2‐targeted therapy in human breast cancer. Nature Clinical Practice Oncology. 2006;3(5):269‐80. [DOI] [PubMed] [Google Scholar]
- 120. Wei H, Cai H, Jin Y, Wang P, Zhang Q, Lin Y, et al. Structural basis of a novel heterodimeric Fc for bispecific antibody production. Oncotarget. 2017;8(31):51037‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Grugan KD, Dorn K, Jarantow SW, Bushey BS, Pardinas JR, Laquerre S, et al. Fc‐mediated activity of EGFR x c‐Met bispecific antibody JNJ‐61186372 enhanced killing of lung cancer cells. 2017. p. 114‐26. [DOI] [PMC free article] [PubMed]
- 122. Cho BC, Lee JS, Han JY, Cho EK, Haura E, Lee KH, et al. JNJ‐61186372 (JNJ‐372), an EGFR‐cMET bispecific antibody, in advanced non‐small cell lung cancer (NSCLC): An update on phase I results. Ann Oncol. 2018;29:viii542. [Google Scholar]
- 123. Geuijen CAW, De Nardis C, Maussang D, Rovers E, Gallenne T, Hendriks LJA, et al. Unbiased combinatorial screening identifies a bispecific IgG1 that potently inhibits HER3 signaling via HER2‐guided ligand blockade. Cancer Cell. 2018;33(5):922‐36. [DOI] [PubMed] [Google Scholar]
- 124. Girard N, Wermke M, Barlesi F, Kim D‐W, Ghiringhelli F, Landsteiner HT, et al. Phase Ib study of BI 836880, a VEGF/Ang2‐blocking nanobody, in combination with BI 754091, an anti‐PD‐1 antibody: Initial results in patients (pts) with advanced non‐small lung cancer (NSCLC). Ann Oncol. 2020;31:S467. [Google Scholar]
- 125. Jacobsen FW, Stevenson R, Li C, Salimi‐Moosavi H, Liu L, Wen J, et al. Engineering an IgG scaffold lacking effector function with optimized developability. J Biol Chem. 2017;292(5):1865‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Yang C, Gao X, Gong R. Engineering of Fc fragments with optimized physicochemical properties implying improvement of clinical potentials for Fc‐based therapeutics. Front Immunol. 2018;8:1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Wozniak‐Knopp G, Stadlmann J, Rker F. Stabilisation of the Fc fragment of human IgG1 by engineered intradomain disulfide bonds. PLoS One. 2012;7(1):e30083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Jefferis R. Glycosylation as a strategy to improve antibody‐based therapeutics. Nat Rev Drug Discovery. 2009;8(3):226‐34. [DOI] [PubMed] [Google Scholar]
- 129. Goede V, Klein C, Stilgenbauer S. Obinutuzumab (GA101) for the treatment of chronic lymphocytic leukemia and other B‐cell non‐hodgkin's lymphomas: a glycoengineered type II CD20 antibody. Oncol Res Treat. 2015;38(4):185‐92. [DOI] [PubMed] [Google Scholar]
- 130. Chatenoud L, Ferran C, Legendre C, Thouard I, Merite S, Reuter A, et al. In vivo cell activation following OKT3 administration. Systemic cytokine release and modulation by corticosteroids. Transplantation. 1990;49(4):697‐702. [DOI] [PubMed] [Google Scholar]
- 131. Woodle ES, Xu D, Zivin RA, Auger J, Charette J, O'Laughlin R, et al. Phase I trial of a humanized, Fc receptor nonbinding OKT3 antibody, huOKT3γ1 (Ala‐Ala) in the treatment of acute renal allograft rejection. Transplantation. 1999;68(5):608‐16. [DOI] [PubMed] [Google Scholar]
- 132. Vafa O, Gilliland GL, Brezski RJ, Strake B, Wilkinson T, Lacy ER, et al. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods. 2014;65(1):114‐26. [DOI] [PubMed] [Google Scholar]
- 133. Schlothauer T, Herter S, Koller CF, Grau‐Richards S, Steinhart V, Spick C, et al. Novel human IgG1 and IgG4 Fc‐engineered antibodies with completely abolished immune effector functions. Protein Eng. Des Sel. 2016;29(10):457‐66. [DOI] [PubMed] [Google Scholar]
- 134. Obeng EM, Dzuvor CKO, Danquah MK. Anti‐SARS‐CoV‐1 and −2 nanobody engineering towards avidity‐inspired therapeutics. Nano Today. 2022;42:101350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Zhang J, Tanha J, Hirama T, Khieu NH, To R, Tong‐Sevinc H, et al. Pentamerization of Single‐domain Antibodies from Phage Libraries: A Novel Strategy for the Rapid Generation of High‐avidity Antibody Reagents. J Mol Biol. 2004;335(1):49‐56. [DOI] [PubMed] [Google Scholar]
- 136. Huet HA, Growney JD, Johnson JA, Li J, Bilic S, Ostrom L, et al. Multivalent nanobodies targeting death receptor 5 elicit superior tumor cell killing through efficient caspase induction. 2014. p. 1560‐70. [DOI] [PMC free article] [PubMed]
- 137. Chen M, Sheu M‐T, Cheng T‐L, Roffler SR, Lin S‐Y, Chen Y‐J, et al. A novel anti‐tumor/anti‐tumor‐associated fibroblast/anti‐mPEG tri‐specific antibody to maximize the efficacy of mPEGylated nanomedicines against fibroblast‐rich solid tumor. Biomaterials Sci. 2022;10(1):202‐15. [DOI] [PubMed] [Google Scholar]
- 138. Guedan S, Calderon H, Posey AD Jr, Maus MV. Engineering and design of chimeric antigen receptors. Molecular Therapy‐Methods and Clinical Development. 2019;12:145‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Awasthi R, Pacaud L, Waldron E, Tam CS, Jger U, Borchmann P, et al. Tisagenlecleucel cellular kinetics, dose, and immunogenicity in relation to clinical factors in relapsed/refractory DLBCL. Blood Adv. 2020;4(3):560‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Mueller KT, Waldron E, Grupp SA, Levine JE, Laetsch TW, Pulsipher MA, et al. Clinical pharmacology of tisagenlecleucel in B‐cell acute lymphoblastic leukemia. Clin Cancer Res. 2018;24(24):6175‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Poorebrahim M, Mohammadkhani N, Mahmoudi R, Gholizadeh M, Fakhr E, Cid‐Arregui A. TCR‐like CARs and TCR‐CARs targeting neoepitopes: an emerging potential. Cancer Gene Ther. 2021;28(6):581‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Sun S, Hao H, Yang G, Zhang Y, Fu Y. Immunotherapy with CAR‐modified T cells: toxicities and overcoming strategies. Journal of Immunology Research. 2018;2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Prasad V. Tisagenlecleucel—the first approved CAR‐T‐cell therapy: implications for payers and policy makers. Nat Rev Clin Oncol. 2018;15(1):11‐2. [DOI] [PubMed] [Google Scholar]
- 144. Reinhard K, Rengstl B, Oehm P, Michel K, Billmeier A, Hayduk N, et al. An RNA vaccine drives expansion and efficacy of claudin‐CAR‐T cells against solid tumors. Science. 2020;367(6476):446‐53. [DOI] [PubMed] [Google Scholar]
- 145. Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534(7607):396‐401. [DOI] [PubMed] [Google Scholar]
- 146. Moretta L, Locatelli F, Pende D, Marcenaro E, Mingari MC, Moretta A. Killer Ig‐like receptor‐mediated control of natural killer cell alloreactivity in haploidentical hematopoietic stem cell transplantation. Blood, The Journal of the American Society of Hematology. 2011;117(3):764‐71. [DOI] [PubMed] [Google Scholar]
- 147. Yoon SR, Lee YS, Yang SH, Ahn KH, Lee J‐H, Lee J‐H, et al. Generation of donor natural killer cells from CD34+ progenitor cells and subsequent infusion after HLA‐mismatched allogeneic hematopoietic cell transplantation: a feasibility study. Bone Marrow Transplant. 2010;45(6):1038‐46. [DOI] [PubMed] [Google Scholar]
- 148. Granzin M, Wagner J, Khl U, Cerwenka A, Huppert V, Ullrich E. Shaping of natural killer cell antitumor activity by ex vivo cultivation. Front Immunol. 2017;8:458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Albinger N, Hartmann J, Ullrich E. Current status and perspective of CAR‐T and CAR‐NK cell therapy trials in Germany. Gene Ther. 2021;28(9):513‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Burger MC, Zhang C, Harter PN, Romanski A, Strassheimer F, Senft C, et al. CAR‐engineered NK cells for the treatment of glioblastoma: turning innate effectors into precision tools for cancer immunotherapy. Front Immunol. 2019:2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2‐specific chimeric antigen receptor‐modified virus‐specific T cells for progressive glioblastoma: a phase 1 dose‐escalation trial. JAMA Oncol. 2017;3(8):1094‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR‐transduced natural killer cells in CD19‐positive lymphoid tumors. N Engl J Med. 2020;382(6):545‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Richman SA, Nunez‐Cruz S, Moghimi B, Li LZ, Gershenson ZT, Mourelatos Z, et al. High‐affinity GD2‐specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol Res. 2018;6(1):36‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low‐affinity CD19 CAR. Nat Med. 2019;25(9):1408‐14. [DOI] [PubMed] [Google Scholar]
- 155. Watanabe K, Kuramitsu S, Posey AD Jr, June CH. Expanding the therapeutic window for CAR T cell therapy in solid tumors: the knowns and unknowns of CAR T cell biology. Front Immunol. 2018;9:2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Greenman R, Pizem Y, Haus‐Cohen M, Goor A, Horev G, Denkberg G, et al. Shaping Functional Avidity of CAR T Cells: Affinity, Avidity, and Antigen Density That Regulate Response. Mol Cancer Ther. 2021;20(5):872‐84. [DOI] [PubMed] [Google Scholar]
- 157. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73‐95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot trial of adoptive transfer of chimeric antigen receptor transduced t cells targeting EGFRvIII in patients with glioblastoma. Journal of immunotherapy (Hagerstown, Md: 1997). 2019;42(4):126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Liu X, Zhang N, Shi H. Driving better and safer HER2‐specific CARs for cancer therapy. Oncotarget. 2017;8(37):62730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Tian S, Maile R, Collins EJ, Frelinger JA. CD8+ T cell activation is governed by TCR‐peptide/MHC affinity, not dissociation rate. J Immunol. 2007;179(5):2952‐60. [DOI] [PubMed] [Google Scholar]
- 162. Zhong S, Malecek K, Johnson LA, Yu Z, de Miera EV‐S, Darvishian F, et al. T‐cell receptor affinity and avidity defines antitumor response and autoimmunity in T‐cell immunotherapy. Proc Natl Acad Sci. 2013;110(17):6973‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 2015;75(17):3505‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity‐tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 2015;75(17):3596‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Park S, Shevlin E, Vedvyas Y, Zaman M, Park S, Hsu Y‐MS, et al. Micromolar affinity CAR T cells to ICAM‐1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep. 2017;7(1):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Smith EL, Staehr M, Masakayan R, Tatake IJ, Purdon TJ, Wang X, et al. Development and evaluation of an optimal human single‐chain variable fragment‐derived BCMA‐targeted CAR T cell vector. Mol Ther. 2018;26(6):1447‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Gorovits B, Koren E. Immunogenicity of chimeric antigen receptor T‐cell therapeutics. BioDrugs. 2019:1‐10. [DOI] [PubMed] [Google Scholar]
- 168. Wang H, Wang L, Li Y, Li G, Zhang X, Jiang D, et al. Nanobody‐armed T cells endow CAR‐T cells with cytotoxicity against lymphoma cells. Cancer Cell Int. 2021;21(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Han L, Zhang J‐S, Zhou J, Zhou K‐S, Xu B‐L, Li L‐L, et al. Single VHH‐directed BCMA CAR‐T cells cause remission of relapsed/refractory multiple myeloma. Leukemia. 2021:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Mo F, Duan S, Jiang X, Yang X, Hou X, Shi W, et al. Nanobody‐based chimeric antigen receptor T cells designed by CRISPR/Cas9 technology for solid tumor immunotherapy. Signal Transduction and Targeted Therapy. 2021;6(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Xie YJ, Dougan M, Jailkhani N, Ingram J, Fang T, Kummer L, et al. Nanobody‐based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc Natl Acad Sci. 2019;116(16):7624‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Zhao W‐H, Liu J, Wang B‐Y, Chen Y‐X, Cao X‐M, Yang Y, et al. A phase 1, open‐label study of LCAR‐B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol. 2018;11(1):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Roex G, Timmers M, Wouters K, Campillo‐Davo D, Flumens D, Schroyens W, et al. Safety and clinical efficacy of BCMA CAR‐T‐cell therapy in multiple myeloma. J Hematol Oncol. 2020;13(1):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK, et al. Regional delivery of chimeric antigen receptor‐engineered T cells effectively targets HER2+ breast cancer metastasis to the brain. Clin Cancer Res. 2018;24(1):95‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART‐19 immunotherapy. Cancer Discov. 2015;5(12):1282‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Maus MV, Grupp SA, Porter DL, June CH. Antibody‐modified T cells: CARs take the front seat for hematologic malignancies. Blood, The Journal of the American Society of Hematology. 2014;123(17):2625‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, et al. SynNotch‐CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. 2021;13(591):eabe7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Zah E, Nam E, Bhuvan V, Tran U, Ji BY, Gosliner SB, et al. Systematically optimized BCMA/CS1 bispecific CAR‐T cells robustly control heterogeneous multiple myeloma. Nat Commun. 2020;11(1):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Zah E, Lin M‐Y, Silva‐Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res. 2016;4(6):498‐508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J Clin Invest. 2016;126(8):3036‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Turatti F, Figini M, Balladore E, Alberti P, Casalini P, Marks JD, et al. Redirected activity of human antitumor chimeric immune receptors is governed by antigen and receptor expression levels and affinity of interaction. J Immunother. 2007;30(7):684‐93. [DOI] [PubMed] [Google Scholar]
- 182. Greenman R, Pizem Y, Haus‐Cohen M, Goor A, Horev G, Denkberg G, et al. Shaping Functional Avidity of CAR T Cells: Affinity, Avidity, and Antigen Density That Regulate Response. Mol Cancer Ther. 2021;20(5):872‐84. [DOI] [PubMed] [Google Scholar]
- 183. Tong C, Zhang Y, Liu Y, Ji X, Zhang W, Guo Y, et al. Optimized tandem CD19/CD20 CAR‐engineered T cells in refractory/relapsed B‐cell lymphoma. Blood. 2020;136(14):1632‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Lemoine J, Ruella M, Houot R. Overcoming intrinsic resistance of cancer cells to CAR T‐cell killing. Clin Cancer Res. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Halford Z, Anderson MK, Bennett LL. Axicabtagene ciloleucel: clinical data for the use of CAR T‐cell therapy in relapsed and refractory large B‐cell lymphoma. Ann Pharmacother. 2021;55(3):390‐405. [DOI] [PubMed] [Google Scholar]
- 186. Wang X, Dong Z, Awuah D, Chang W‐C, Cheng WA, Vyas V, et al. CD19/BAFF‐R dual‐targeted CAR T cells for the treatment of mixed antigen‐negative variants of acute lymphoblastic leukemia. Leukemia. 2022:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Dai H, Wu Z, Jia H, Tong C, Guo Y, Ti D, et al. Bispecific CAR‐T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J Hematol Oncol. 2020;13(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Tang Y, Yin H, Zhao X, Jin D, Liang Y, Xiong T, et al. High efficacy and safety of CD38 and BCMA bispecific CAR‐T in relapsed or refractory multiple myeloma. J Exp Clin Cancer Res. 2022;41(1):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Fry TJ, Shah NN, Orentas RJ, Stetler‐Stevenson M, Yuan CM, Ramakrishna S, et al. CD22‐targeted CAR T cells induce remission in B‐ALL that is naive or resistant to CD19‐targeted CAR immunotherapy. Nat Med. 2018;24(1):20‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Arenas EJ, Martnez‐Sabadell A, Rius Ruiz I, Romn Alonso M, Escorihuela M, Luque A, et al. Acquired cancer cell resistance to T cell bispecific antibodies and CAR T targeting HER2 through JAK2 down‐modulation. Nat Commun. 2021;12(1):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Schutt CA, Mirandola L, Figueroa JA, Nguyen DD, Cordero J, Bumm K, et al. The cancer‐testis antigen, sperm protein 17, a new biomarker and immunological target in head and neck squamous cell carcinoma. Oncotarget. 2017;8(59):100280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Gjerstorff M, Jakobsen M. CAR T cell cancer therapy targeting surface cancer/testis antigens. Front Immunol. 2020;11:1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Da Cunha JPC, Galante PAF, De Souza JE, De Souza RF, Carvalho PM, Ohara DT, et al. Bioinformatics construction of the human cell surfaceome. Proc Natl Acad Sci. 2009;106(39):16752‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. He Q, Jiang X, Zhou X, Weng J. Targeting cancers through TCR‐peptide/MHC interactions. J Hematol Oncol. 2019;12(1):1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Pankov D, Sjstrm L, Kalidindi T, Lee S‐G, Sjstrm K, Gardner R, et al. In vivo immuno‐targeting of an extracellular epitope of membrane bound preferentially expressed antigen in melanoma (PRAME). Oncotarget. 2017;8(39):65917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Harms BD, Kearns JD, Iadevaia S, Lugovskoy AA. Understanding the role of cross‐arm binding efficiency in the activity of monoclonal and multispecific therapeutic antibodies. Methods. 2014;65(1):95‐104. [DOI] [PubMed] [Google Scholar]
- 197. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Ruffolo JA, Sulam J, Gray JJ. Antibody structure prediction using interpretable deep learning. bioRxiv. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Sweredoski MJ, Baldi P. PEPITO: improved discontinuous B‐cell epitope prediction using multiple distance thresholds and half sphere exposure. Bioinformatics. 2008;24(12):1459‐60. [DOI] [PubMed] [Google Scholar]
- 200. Kringelum JV, Lundegaard C, Lund O, Nielsen M. Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLoS Comput Biol. 2012;8(12):e1002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Kunik V, Ashkenazi S, Ofran Y. Paratome: an online tool for systematic identification of antigen‐binding regions in antibodies based on sequence or structure. Nucleic Acids Res. 2012;40(W1):W521‐W4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Daberdaku S, Ferrari C. Antibody interface prediction with 3D Zernike descriptors and SVM. Bioinformatics. 2019;35(11):1870‐6. [DOI] [PubMed] [Google Scholar]
- 203. Mason DM, Friedensohn S, Weber CR, Jordi C, Wagner B, Meng SM, et al. Optimization of therapeutic antibodies by predicting antigen specificity from antibody sequence via deep learning. Nature Biomedical Engineering. 2021;5(6):600‐12. [DOI] [PubMed] [Google Scholar]
- 204. Gainza P, Sverrisson F, Monti F, Rodola E, Boscaini D, Bronstein MM, et al. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat Methods. 2020;17(2):184‐92. [DOI] [PubMed] [Google Scholar]
- 205. Balci AT, Gumeli C, Hakouz A, Yuret D, Keskin O, Gursoy A. DeepInterface: Protein‐protein interface validation using 3D Convolutional Neural Networks. bioRxiv. 2019:617506. [Google Scholar]
- 206. Hausman DF, Hamilton EP, Beeram M, Thimmarayappa J, Ng G, Meric‐Bernstam F. Phase 1 study of ZW25, a bispecific anti‐HER2 antibody, in patients with advanced HER2‐expressing cancers. 2017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.