

Abstract
Background
Abnormal expression of protein tyrosine phosphatases (PTPs) has been reported to be a crucial cause of cancer. As a member of PTPs, protein tyrosine phosphatase receptor type O (PTPRO) has been revealed to play tumor suppressive roles in several cancers, while its roles in colorectal cancer (CRC) remains to be elucidated. Hence, we aimed to explore the roles and mechanisms of PTPRO in CRC initiation and progression.
Methods
The influences of PTPRO on the growth and liver metastasis of CRC cells and the expression patterns of different lipid metabolism enzymes were evaluated in vitro and in vivo. Molecular and biological experiments were conducted to uncover the underpinning mechanisms of dysregulated de novo lipogenesis and fatty acid β‐oxidation.
Results
PTPRO expression was notably downregulated in CRC liver metastasis compared to the primary cancer, and such a downregulation was associated with poor prognosis of patients with CRC. PTPRO silencing significantly promoted cell growth and liver metastasis. Compared with PTPRO wild‐type mice, PTPRO‐knockout mice developed more tumors and harbored larger tumor loads under treatment with azoxymethane and dextran sulfate sodium. Gene set enrichment analysis revealed that PTPRO downregulation was significantly associated with the fatty acid metabolism pathways. Blockage of fatty acid synthesis abrogated the effects of PTPRO silencing on cell growth and liver metastasis. Further experiments indicated that PTPRO silencing induced the activation of the AKT serine/threonine kinase (AKT)/mammalian target of rapamycin (mTOR) signaling axis, thus promoting de novo lipogenesis by enhancing the expression of sterol regulatory element‐binding protein 1 (SREBP1) and its target lipogenic enzyme acetyl‐CoA carboxylase alpha (ACC1) by activating the AKT/mTOR signaling pathway. Furthermore, PTPRO attenuation decreased the fatty acid oxidation rate by repressing the expression of peroxisome proliferator‐activated receptor alpha (PPARα) and its downstream enzyme peroxisomal acyl‐coenzyme A oxidase 1 (ACOX1) via activating the p38/extracellular signal‐regulated kinase (ERK) mitogen‐activated protein kinase (MAPK) signaling pathway.
Conclusions
PTPRO could suppress CRC development and metastasis via modulating the AKT/mTOR/SREBP1/ACC1 and MAPK/PPARα/ACOX1 pathways and reprogramming lipid metabolism.
Keywords: AKT, colorectal cancer, fatty acid oxidation, fatty acid synthesis, lipid metabolism, liver metastasis, mTOR, PTPRO, tumorigenesis
Abbreviations
- ACC1
acetyl‐CoA carboxylase
- ACLY
ATP Citrate lyase
- ACOX1
peroxisomal acyl‐coenzyme oxidase 1
- AKT
AKT serine/threonine kinase
- CPT1A
carnitine palmitoyltransferase 1A
- CRC
colorectal cancer
- ERK
extracellular signal‐regulated kinase
- FAO
fatty acid oxidation
- FASN
fatty acid synthetase
- FUSCC
Fudan University Shanghai Cancer Center
- GC‐TOFMS
gas chromatography‐time‐of‐flight mass spectrometry
- GEO
Gene Expression Omnibus
- GSEA
gene set enrichment analysis
- MAPKs
mitogen‐activated protein kinases
- mTOR
mammalian target of rapamycin
- PPARs
peroxisome proliferator‐activated receptors
- PTPRO
protein tyrosine phosphatase receptor type O
- PTPs
protein tyrosine phosphatases
- SCD1
stearoyl‐CoA desaturase
- SPF
specific pathogen‐free
- SREBPs
sterol regulatory element‐binding proteins
- TAG
intracellular triglyceride
- UPLC‐MS/MS
ultra‐performance liquid chromatography‐tandem mass spectrometry
- WT
wild‐type
1. BACKGROUND
Colorectal cancer (CRC) is currently one of the most commonly diagnosed malignant diseases worldwide, with the liver as the most common site for metastasis. The 1‐, 3‐, and 5‐year survival rates of patients with liver metastasis are still far from satisfactory [1, 2]. Therefore, identifying predictive biomarkers and uncovering underpinning mechanisms are urgently warranted for predicting and treating CRC liver metastasis.
Recently, ectopic alteration of energy metabolism has been ubiquitously recognized as a hallmark of cancer during its initiation, progression, therapeutic resistance, and so on [3, 4, 5], of which dysregulation of lipid metabolism has been demonstrated to be a critical metabolic feature of cancer cells [6, 7, 8, 9]. Researches have shown that numerous enzymes involved in fatty acid (FA) uptake, FA synthesis, and FA oxidation (FAO) were abnormally regulated in several cancers, causing reprogramming of FA metabolism and promoting the malignant phenotypes of cancer, indicating the significance of dysregulation of lipid metabolism in cancer [10, 11, 12]. Constant de novo lipogenesis supplies cancer cells with ample lipid molecules for membrane building and signaling transduction, promoting cancer initiation and progression [13, 14]. For example, in response to the inadequate supply of lipids in the brain, breast cancer cells would upregulate the expression of FA synthetase (FASN) to facilitate its brain metastasis [15]. What's more, acyl‐CoA oxidase 1 (ACOX1) is a critical rate‐limiting enzyme involved in FAO. Chen et al. [16] reported that downregulation of deacetylase sirtuin 5 could promote succinylation and activity of ACOX1, enhancing the response of liver cancer cells to oxidative DNA damage. However, the mechanisms underlying the continuous activation of lipogenesis and dysregulation of FAO in CRC have not been completely uncovered.
Abnormal expression of protein tyrosine phosphatases (PTPs) has been shown to be involved in numerous cancers [17, 18, 19, 20]. PTPs could exert tumor suppressive functions via dephosphorylation of oncogenic proteins and promote cancer development and progression by positively regulating signaling pathways [19, 20]. Protein tyrosine phosphatase receptor type O (PTPRO) is a member of PTPs, which phylogenetically belongs to a distinct branch of the tyrosine phosphatases. Previous studies have revealed that it could execute tumor suppressive functions in several cancers [21, 22, 23]. For instance, PTPRO could interact with erb‐b2 receptor tyrosine kinase 2 (ERBB2) and regulate its phosphorylation, inhibiting the signaling pathways downstream of ERBB2 and thus hindering the growth of cancer cells [22]. However, few researches have probed the roles of PTPRO in the development and progression of CRC.
In this study, in vitro and in vivo studies were carried out to explore the biological functions of PTPRO in CRC and whether it can regulate FA metabolism to further execute its functions in CRC.
2. METHODS
2.1. Public dataset and pathway analysis
GSE39582, the largest public microarray dataset of CRC, was downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). The procedure of data processing has been described in our previous work [24]. Gene set enrichment analysis (GSEA) was conducted to identify the PTPRO‐associated biological pathways.
2.2. Human CRC samples
CRC tissue samples were collected from patients diagnosed at Fudan University Shanghai Cancer Center (FUSCC) (Shanghai, China) between January 2008 and June 2014. The patients with (1) pathologically confirmed colorectal adenocarcinoma, (2) complete clinicopathological information, (3) no concurrent other malignancies, and (4) no neoadjuvant chemoradiation therapy were included, while those with multiple primary CRC or hereditary CRC were excluded. These tissues were used for evaluating mRNA and protein expression levels of related genes and tissue microarray preparation. According to the stipulations of the Institutional Review Board of FUSCC, informed consent was obtained from all patients for sample and data use in research.
2.3. Transcriptome profiling
Primary CRC and liver metastasis tissues were collected at FUSCC, which were subjected to RNA isolation by TRIzol reagent (15596026, Invitrogen, Carlsbad, CA, USA) according to the protocol provided. After qualification testing, RNA sequencing libraries were established, which were used to analyze the massive ends of cDNA. In brief, biotinylated poly (dT) primers (5′d PO4 [(T)12‐18] 3′) were applied to purify the poly‐adenylated RNA from the total RNA isolated by the TRIzol reagent. Afterwards, it was reversely transcribed to cDNA, which then was fragmented with a size ranging from 150 to 600 bp. Streptavidin magnetic beads were adopted to capture the biotinylated ends, which were subsequently added to the modified adapters. Polymerase chain reaction (annealing: 55°C‐65°C, 5 min, 4°C, 2 min; polymerization: 10°C‐50°C, 10‐90 min; deactivation: 85°C, 10 min) was conducted to amplify the aforementioned sequencing libraries, purification was done using SPRI beads, and then sequencing was performed (HiSeq2000, Illumina, San Deigo, CA, USA). After normalization, those with false discovery rate (FDR) < 0.05 and |log2fc| > 1.5 would be deemed as differentially expressed genes.
2.4. CRC cell lines
The human CRC cell lines DLD1, HCT116, SW480, SW620, LoVo, RKO and human normal colon epithelial cell line NCM460 were purchased from the National Cancer Institute (Bethesda, MD, USA). Human 293T cells were obtained from the American Type Culture Collection (Rockville, MD, USA). CRC cell lines and 293T cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) (SH30243.01, HyClone, Logan, UT, USA) coupled with 10% fetal bovine serum (FBS; 10099141, Thermo Fisher Scientific, Waltham, MA, USA), while NCM460 cells were cultured in RPMI‐1640 (11875093, Gibco, Waltham, MA, USA) with 10% FBS. All cell lines used in this study were verified by short tandem repeat genotyping.
2.5. Establishment of stable cell lines and transfection
PTPRO shRNAs were purchased from WZ Biosciences (SH899102, Jinan, Shandong, China). pCDH‐CMV‐PTPRO was mixed with the packaging plasmids psPAX2 and PMD2.G. With Lipofectamine 3000 reagent (L3000008, Thermo Fisher Scientific) and the protocol provided by the manufacturer, the mixture was then added to 293T cells. After culture for 48 h, viral particles were harvested from the medium, then added into SW480 and LoVo cells to establish stable cells with negative control and PTPRO knockdown (SW480‐PTPRO‐NC/KD, LoVo‐PTPRO‐NC/KD). Puromycin (P8230, Solarbio, Shanghai, China) was used to screen cells with successful PTPRO knockdown. Likewise, stable cells with empty vector and PTPRO overexpression (HCT116‐Vector/PTPRO, SW620‐Vector/PTPRO) were established through the same procedure. What's more, Lipofectamine RNAi Max (13778075, Thermo Fisher Scientific) was utilized for RNAi transfection according to the guidelines provided.
2.6. Application of inhibitors
For the application of the FA synthetase inhibitor orlistat (O4139‐25MG, Merck, Darmstadt, Germany) in vitro, it was prepared by referencing the procedure of Browne et al. [25], and 50 μmol/L orlistat was adopted to treat CRC cells for 48 h when necessary. As for in vivo experiments, it was prepared as described by Kridel et al. [26], and it was intraperitoneally injected into nude mice with a concentration of 240 mg/kg. The phosphoInositide‐3 kinase (PI3K) inhibitor LY294002 (20 μmol/L) (HY‐10108, MedChemExpress, Monmouth Junction, NJ, USA) was used to treat CRC cells for 24 h as per a report from Chen et al. [27]. Based on a previous study [28], 40 μmol/L mitogen‐activated protein kinase (MAPK) inhibitor (E)‐osmundacetone (HY‐N1966, MedChemExpres) was applied for 24 h for in vitro experiments.
2.7. Cell viability and migration assays
Cell Counting Kit‐8 (CK04, DOJINDO, Kumamoto, Japan) was used to evaluate cell viability. Transwell assays were performed by seeding 4 × 104‐8 × 104 cells into the upper chamber (CLS3464, Corning Costar, Corning, NY, USA) with no FBS supplementation while the lower chamber was added 500 μL DMEM with 10% FBS. After 36‐72 h of culture, migrated cells were fixed with 4% paraformaldehyde (G1101, Servicebio, Wuhan, Hubei, China), stained with Crystal Violet Staining Solution (C0121, Beyotime, Shanghai, China), and counted under a microscope.
2.8. Establishment of mouse models
Female 6‐ to 8‐week‐old BALB/c nude mice were purchased from Shanghai SLAC Laboratory Animal Co., Ltd (Shanghai, China). Mouse‐related studies were conducted in specific pathogen‐free facilities, and all the procedures were approved by the Institutional Animal Care and Use Committee of Fudan University (Shanghai, China), which complies with the regulations from the US National Institutes of Health for the care and use of laboratory animals.
To establish the mouse models of CRC liver metastasis, 1 × 106 LoVo‐NC, LoVo‐PTPRO‐KD, SW480‐NC, and SW480‐PTPRO‐KD cells were injected into the spleens of the nude mice. At 8‐10 weeks after injection, the mice were euthanized (pentobarbital overdose), and liver specimens were collected and photographed to count metastatic lesions.
Azoxymethane (AOM) (A5486, Sigma‐Aldrich, St. Louis, MO, USA) and dextran sulfate sodium (DSS) (0216011080, MP Biomedicals, Santa Ana, CA, USA) were applied to treat PTPRO‐knockout (PTPRO–/–) and PTPRO wild‐type (WT) 6‐ to 8‐week‐old male C57BL/6J mice to establish CRC tumorigenesis model. AOM (12 mg/kg body weight) was intraperitoneally injected into mice, after which drinking water with 2% DSS was fed for 5 successive days, and afterwards, regular drinking water was provided. This cycle would be repeated once. Then tumor number and load (sum of tumor diameters) were measured.
LoVo‐NC and LoVo‐PTPRO‐KD cells (5 × 106) were injected subcutaneously into the right and left hind flanks, respectively, of the aforementioned BALB/c nude mice. Likewise, HCT116‐vector and HCT116‐PTPRO cells (5 × 106) were injected subcutaneously into the hind flanks of nude mice. Tumors were gauged for length every 3 or 4 days by utilizing a caliper. The formula Volume = 1/2 × length × width2 was adopted to calculate the size of tumors. Lastly, the mice were euthanized (pentobarbital overdose) when reaching humane endpoint, then the tumors were resected from these nude mice for imaging and weighing. Subsequently, these tumor xenografts were stored in liquid nitrogen until the metabolomic assays were performed.
2.9. Metabolomic profiling
A combination of gas chromatography‐time‐of‐flight mass spectrometry (GC‐TOFMS, LECO Corp., St Joseph, MI, USA) and ultra‐performance liquid chromatography‐tandem mass spectrometry (UPLC‐MS/MS, Waters Corp., Milford, MA, USA) was used to quantify small‐molecule metabolites in the tumor xenografts. The metabolites were identified by comparison with an internal library built using standard reference chemicals.
2.10. Quantification of triacylglycerol and neutral lipids
For the quantitative estimation of triglycerides in cells, a Triglyceride Assay Kit (ab65336, Abcam, Cambridge, UK) was used in accordance with the manufacturer's protocols. The lipophilic fluorescence dye BODIPY 493/503 (D3922, Invitrogen) was applied to stain the neutral lipid droplets, and flow cytometry (MoFlo XDP, Beckman Coulter, Pasadena, CA, USA) was conducted to quantify the neutral lipid content.
2.10.1. FAO quantification
The FAO rate in CRC cells with and without PTPRO knockdown and overexpression was assessed using a fatty acid oxidation assay kit (ab118183, Abcam) in accordance with the manufacturer's instructions.
2.11. Establishment of organoids
Organoids derived from CRC samples were established based on an establishedprocedure. Briefly, fresh cancer tissues were cut into pieces and washed with cold PBS supplemented with 10% penicillin/streptomycin (S110JV, BasalMedia, Shanghai, China) for at least three times. The tissues were incubated with digestion buffer containing Advanced DMEM/F12 medium (12634010, Gibco) with 2.5% FBS, 1% penicillin/streptomycin (15140122, Gibco), 75 U/mL collagenase type IV (C4‐BIOC, Sigma‐Aldrich),125 g/mL dispase type II (17105041, Gibco) for 30 min at 37°C. The tissues were washed another 3 times with PBS, and the crypt fraction and tumor pellets were collected. Then, the crypts and tumor pellets were suspended in Basement Membrane Extract (BME; Cultrex RGF Basement Membrane Extract, Type 2; R&D Systems, Minneapolis, MN, USA) and dispensed into 24‐well culture plates (40 μL BME/well). The BME was then solidified by a 20‐min incubation at 37°C in a 5% CO2 cell culture incubator and overlaid with 500 μL of complete human organoid media.
2.12. RNA isolation and quantitative real‐time polymerase chain reaction (qRT‐PCR)
TRIzol reagent (15596026, Invitrogen) was used to isolate total RNA from CRC tissues and cell lines, and PrimeScript RT reagent (RR036A, TaKaRa, Kusatsu, Japan) was used to obtain cDNA. The expression levels of target genes and β‐actin were determined by qRT‐PCR on the ABI 7900HT Real‐Time PCR System (Applied Biosystems, Frederick, MD, USA).
2.13. Western blotting
In brief, NCM460 and CRC cells with and without the intervention of PTPRO expression were lysed by RIPA buffer (89900, Thermo Fisher Scientific) and quantified by BCA Protein Assay Kit (23225, Thermo Fisher Scientific).Then protein samples were added to the polyacrylamide gel for electrophoresis. After the completion of electrophoresis, the protein would be transferred onto polyvinylidene difluoride membranes and then subjected to immunoblotting using primary antibodies for target proteins, and the membranes would be incubated overnight at 4°C. The next day, second antibodies (7074P2 and 7076P2, Cell Signaling, Danvers, MA, USA) were added for 1‐hour incubation. Finally, the BeyoECL Plus buffer (P0018S, Beyotime) was used to visualize the results on ImageQuant LAS 4000 (General Electric, Fairfield, CT, USA) The primary antibodies used were as follows: anti‐β‐actin (4970, Cell Signaling), anti‐PTPRO (12161‐1‐AP, Proteintech, Chicago, IL, USA), anti‐ ac‐CoA carboxylase 1 (ACC1) (21923‐1‐AP, Proteintech), anti‐ATP Citrate lyase (ACLY) (15421‐1‐AP, Proteintech), anti‐FASN (10624‐2‐AP, Proteintech), anti‐sterol regulatory element‐binding protein 1 (SREBP1) (114088‐1‐AP, Proteintech), anti‐SREBP2 (28212‐1‐AP, Proteintech), anti‐ACOX1 (10957‐1‐AP, Proteintech), anti‐carnitine palmitoyltransferase 1A (CPT1A) (15184‐1‐AP, Proteintech), anti‐peroxisome proliferator‐activated receptor α (PPARα) (15540‐1‐AP, Proteintech), anti‐PPARγ (16643‐1‐AP, Proteintech), anti‐AKT serine/threonine kinase (AKT) (10176‐2‐AP, Proteintech), anti‐p‐AKT (66444‐1‐Ig, Proteintech), anti‐mammalian target of rapamycin (mTOR) (20657‐1‐AP, Proteintech), anti‐p‐mTOR (67778‐1‐Ig, Proteintech), anti‐p38 (14064‐1‐AP, Proteintech.), anti‐p‐p38 (28796‐1‐AP, Proteintech), anti‐extracellular signal‐regulated kinase (ERK) (16443‐1‐AP, Proteintech), and anti‐p‐ERK (28733‐1‐AP, Proteintech).
2.14. Immunohistochemistry (IHC) staining
IHC staining was conducted as described in our previous article [29]. CRC tissue sections with IHC staining were evaluated and scored independently by two pathologists who were blinded to the clinicopathological information of the patients. The scores of 0 (negative), 1 (weak), 2 (medium), and 3 (strong) were used for staining intensity quantification, while the scores of 0 (< 5%), 1 (5%–25%), 2 (26%–50%), 3 (51%–75%) and 4 (> 75%), which were based on the percentage of the positive staining areas in relation to the whole cancerous area, were used to evaluate the extent of staining. Then, the immunoreactivity score was calculated by multiplying scores of staining intensity and percentage of positivity. Cases with a final staining score of ≤ 4 were deemed as low expression, and those with score of > 4 were reckoned as high expression.
2.15. Statistical analysis
R software (R version 3.2.5, https://www.r‐project.org/) was used for statistical analysis. Data are depicted as the mean ± standard deviation. The Wilcoxon rank‐sum test or Student's t‐test was applied to compare the discrepancies between two groups. Overall survial (OS) and disease‐free survival (DFS) were depicted by the Kaplan‐Meier method, and the statistical differences were compared with the log‐rank test. OS was defined as the time from treatment initiation to death or last follow‐up, while DFS was the time from treatment initiation to recurrence/metastasis or last follow‐up. The relationship between PTPRO expression and other genes was evaluated by the Spearman rank correlation test. P < 0.05 was deemed as statistically significant.
3. RESULTS
3.1. PTPRO was downregulated in liver metastatic sites of CRC, and low PTPRO expression was associated with poor prognosis
To detect the molecules that mediate the progression of CRC, transcriptome profiling was performed between three non‐metastatic CRC (nmCRC) tissues and three paired metastatic CRC (mCRC) and liver metastatic tissues (Supplementary Table S1). The results showed that compared with nmCRC tissues, PTPRO expression was strikingly downregulated in mCRC tissues and liver metastatic sites (Figure 1A–1B). Further qRT‐PCR conducted in 11 matched normal colorectal epithelial tissues, mCRC tissues, and liver metastasis tissues from patients with CRC liver metastasis confirmed the notably lower expression of PTPRO in CRC and liver metastatic tissues compared with that in normal tissues (Figure 1C).
FIGURE 1.
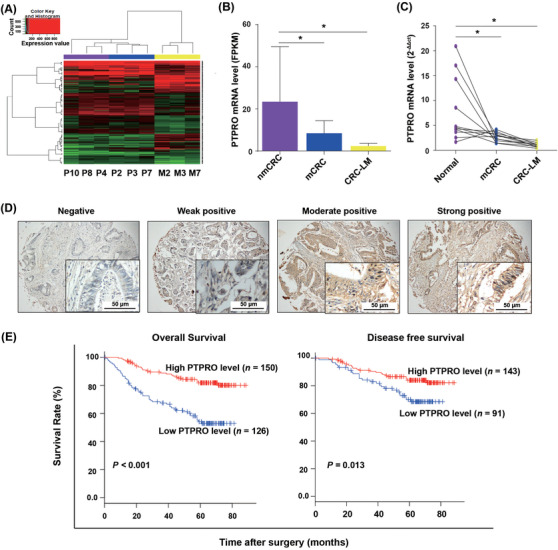
PTPRO was downregulated in CRC and liver metastasis, and its low expression was linked with poor prognosis. (A) RNA sequencing and cluster analysis of mRNAs with differential expression in three nmCRC tissues (P4, P8, P10), and three paired tissues of mCRC (P2, P3, P7) and CRC‐LM (M2, M3, M7). The x axis of color key represents the expression value, and the y axis respesents the corresponding density information. (B) FPKM value of PTPRO expression in the three groups. (C) qRT‐PCR validation of PTPRO expression in 11 matched normal colon epithelial, mCRC, and CRC‐LM tissues. (D) Representative images of immunohistochemistry staining of PTPRO protein with different intensities. (E) The relationship between PTPRO expression and overall survival or disease‐free survival by Kaplan‐Meier method. *P < 0.05. PTPRO: protein tyrosine phosphatase receptor type O; CRC: colorectal cancer; nmCRC: non‐metastatic CRC; mCRC: metastatic CRC; CRC‐LM: CRC liver metastasis
Given the expression difference between metastatic and primary sites, we hypothesized that PTPRO might be a potential prognostic factor in CRC. Therefore, an IHC assay using samples from 276 patients with CRC (42 with and 234 without metastasis) was performed (Figure 1D) to divide them into low and high PTPRO expression groups. The patients with low expression of PTPRO showed significantly shorter OS and DFS (Figure 1E).
3.2. PTPRO inhibited CRC cell growth and liver metastasis
Basic mRNA and protein expression levels of PTPRO were tested in normal colon epithelial NCM460 cells and six CRC cell lines. Notably, high expression of PTPRO was detected in NCM460 cells, and strikingly lower expression of PTPRO was detected in CRC cells (HCT116, SW620, and DLD1) with aggressive features (Figure 2A). To assess the roles of PTPRO in the growth and metastatic potential of CRC cells, we knocked down PTPRO in SW480 and LoVo cells (Figure 2B) and enhanced its expression in HCT116 and SW620 cells (Figure 2C). Attenuated PTPRO expression significantly enhanced cell migration ability in vitro (Figure 2D) and promoted cell viability (Supplementary Figure S1A). In contrast, the ectopic PTPRO expression dramatically attenuated cell migration ability (Figure 2E) and suppressed cell viability (Supplementary Figure S1B). Furthermore, in vivo studies revealed that LoVo cells with PTPRO silenced exhibited accelerated subcutaneous tumor growth (Figure 2F–2H) and enforced PTPRO expression dramatically inhibited the tumor‐forming ability of HCT116 cells (Supplementary Figure S1C‐E). In addition, the liver metastasis model showed that cells with PTPRO silenced formed significantly more metastatic lesions than the control cells (Figure 2I–2J).
FIGURE 2.
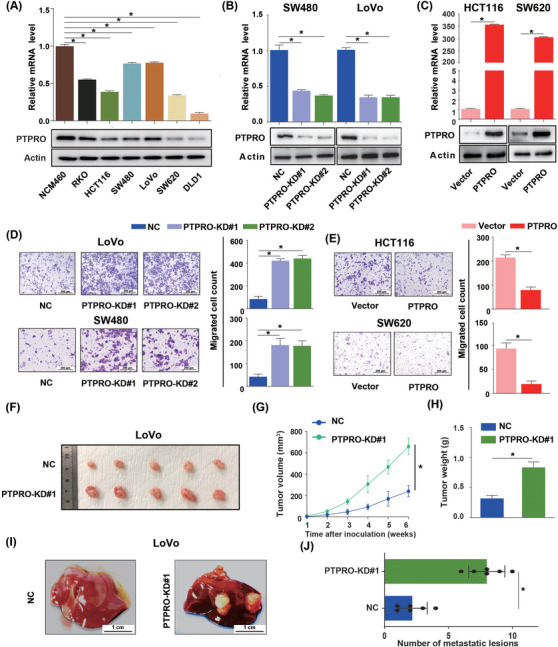
PTPRO represses CRC cell growth and liver metastasis. (A) PTPRO expression levels in six CRC cell lines and NCM460 determined by Western blotting and qRT‐PCR. (B) Western blotting and qRT‐PCR verify the shRNA‐mediated deletion of PTPRO in SW480 and LoVo cells. (C) Western blotting and qRT‐PCR verify the PTPRO expression in HCT116 and SW620 cells with transfection of the control PCDH retrovirus or the PCDH‐PTPRO retrovirus. (D) Migration assay of SW480 and LoVo cells with PTPRO‐NC or PTPRO‐KD. (E) Migration assay of HCT116 and SW620 cells with PTPRO‐Vector or overexpression. (F‐H) Subcutaneous xenograft tumor images and tumor growth of LoVo cells with PTPRO‐NC and PTPRO‐KD in nude mice. (I‐J) The gross images (I) and number (J) of liver metastasis induced by LoVo cells with PTPRO‐NC and PTPRO‐KD in nude mice. *P < 0.05. PTPRO: protein tyrosine phosphatase receptor type O; CRC: colorectal cancer; qRT‐PCR: quantitative real‐time polymerase chain reaction; NC, negative control; KD: knockdown
3.2.1. Abrogation of PTPRO expression promoted CRC tumorigenesis
To explore whether PTPRO could repress tumorigenesis, we employed PTPRO‐WT and PTPRO–/– mice for further experiments (Figure 3A). Both PTPRO‐WT and PTPRO–/‐ mice were treated with AOM and DSS. PTPRO–/– mice showed an earlier appearance of fecal blood and diarrhea than PTPRO‐WT mice (Figure 3B). The results showed that PTPRO–/– mice had more and larger tumors formed in the colorectum compared with PTPRO‐WT mice (Figure 3C–3E). Furthermore, PTPRO‐WT mice showed longer OS after treatment than PTPRO–/– mice (Figure 3F, P < 0.05).
FIGURE 3.
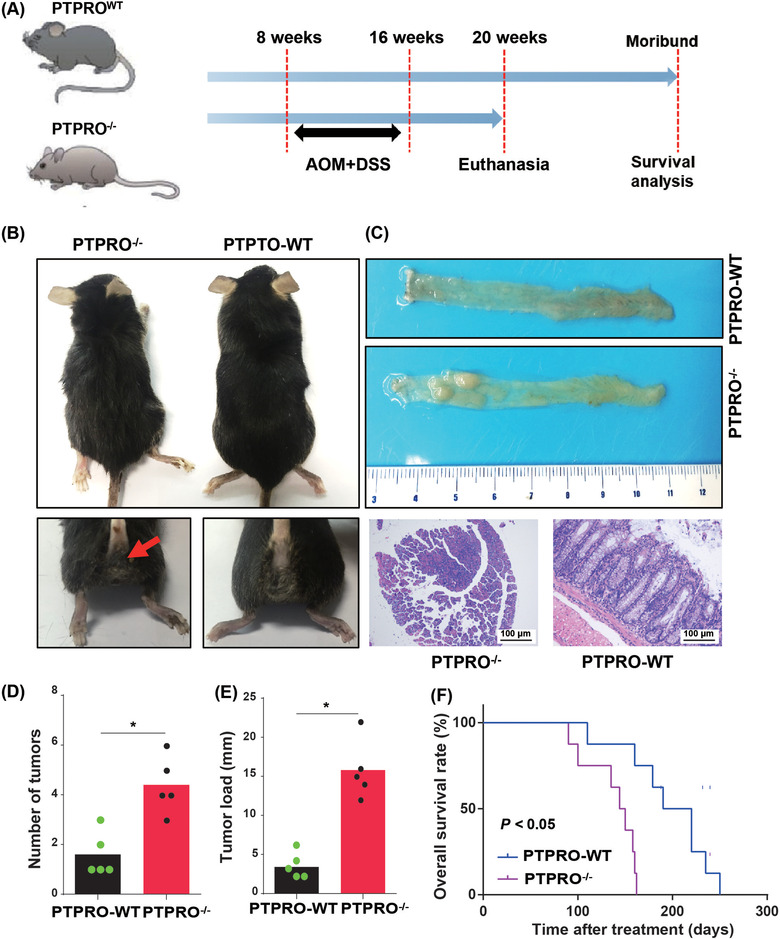
Abrogation of PTPRO expression promoted CRC tumorigenesis. (A) PTPRO–/‐ and PTPRO‐WT mice were treated by AOM and DSS to induce CRC tumorigenesis. (B) PTPRO–/– increased the bloody diarrhea rates during the AOM and DSS treatment. (C) Representative colon images of the PTPRO–/‐ and PTPRO‐WT mice. (D‐E) Comparison of the number of tumors and tumor load between PTPRO–/‐ or PTPRO‐WT mice. (F) Kaplan‐Meier analysis between PTPRO–/‐ and PTPRO‐WT mice. *P < 0.05. PTPRO: protein tyrosine phosphatase receptor type O; CRC: colorectal cancer; AOM: azoxymethane; DSS: dextran sulfate sodium; WT: wild type
3.2.2. Lipid accumulation mediated by PTPRO silencing was essential for CRC cell growth and liver metastasis
To explore the potential signaling pathways regulated by PTPRO, GSEA was conducted in GSE39582, one of the largest microarray datasets of CRC in the GEO database, which revealed that the FA metabolism signaling pathway was markedly enriched in patients with low PTPRO expression (Figure 4A). Furthermore, the metabolite analysis of the subcutaneous xenografts using GC‐TOF‐MS revealed that knockdown of PTPRO increased the content of free fatty acids dramatically, such as oleic acid, palmitoleic acid, and stearic acid, while PTPRO overexpression had the opposite effects (Figure 4B), indicating that PTPRO might play crucial roles in rewiring lipid metabolism in CRC. To verify the effects of PTPRO on FA metabolism in vitro, intracellular triglyceride (TAG) content and FAO level analyses were conducted, which showed that PTPRO knockdown strikingly increased the intracellular TAG content while decreasing the FAO level (Figure 4C; Supplementary Figure S2A). In contrast, PTPRO overexpression significantly restrained the intracellular TAG content and increased the FAO level (Figure 4C; Supplementary Figure S2A). What's more, the lipophilic dye BODIPY 493/503 was adopted for cellular staining, which showed that PTPRO attenuation increased the intracellular levels of neutral lipids in CRC cells (Figure 4D; Supplementary Figure S2B), whereas PTPRO overexpression decreased the levels of neutral lipids in CRC cells (Figure 4D; Supplementary Figure S2C). Based on the aforementioned results, we could infer that PTPRO might be pivotal for the reprogramming of FA metabolism in CRC cells.
FIGURE 4.
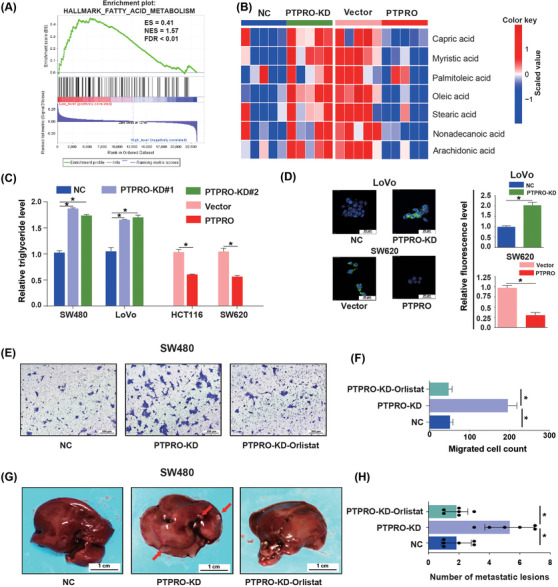
Lipid accumulation mediated by PTPRO silencing was essential for CRC cell growth and liver metastasis. (A) FA metabolism signaling pathway was significantly enriched in patients with low PTPRO expression (FDR < 0.01). (B) GC‐TOF‐MS showed the contents of FFA in PTPRO‐silenced or ‐enforced xenografts. (C) Relative triglyceride levels in PTPRO‐NC/KD and Vector/PTPRO cells. (D) The content of neutral lipids in LoVo (PTPRO‐NC/KD) and SW620 (Vector/PTPRO) cells. (E‐F) Migration assay analysis of orlistat in SW480 cells with PTPRO attenuation. (G‐H) The effect of orlistat on liver metastasis formation in nude mice bearing SW480 cells with PTPRO attenuation. *P < 0.05. Abbreviations: PTPRO: protein tyrosine phosphatase receptor type O; CRC: colorectal cancer;FA: fatty acid; FFA: free fatty acid; GC‐TOF‐MS: gas chromatography‐time‐of‐flight mass spectrometry; NC: negative control; KD: knockdown
To further evaluate whether PTPRO‐mediated lipid accumulation was essential for CRC growth and liver metastasis, we used orlistat, a FA synthase inhibitor, to decrease the lipid content in CRC cells with PTPRO silenced. The results showed that orlistat treatment completely abrogated the effects exerted by PTPRO attenuation on cell viability (Supplementary Figure S2D), cell migration (Figure 4E–4F), and liver metastasis formation (Figure 4G–4H), confirming the fundamental roles of FA accumulation in promoting CRC cell growth and liver metastasis.
3.2.3. PTPRO suppressed the expression of lipogenic enzyme by downregulating SREBP1
To explore whether PTPRO silence triggered lipid accumulation was due to its regulation on de novo FA synthesis, we examined the mRNA expression levels of key de novo FA synthesis enzymes, including FASN, ACC1, ACLY, and stearoyl‐CoA desaturase (SCD1), in CRC cells with different PTPRO expression levels. We found that ACC1 was notably upregulated in PTPRO‐silenced cells, while it was downregulated in PTPRO‐enforced cells (Supplementary Figure S3A‐B). Further Western blotting confirmed the ACC1 protein level changes in PTPRO‐silenced or ‐enforced cells (Figure 5A–5B; Supplementary Figure S3C). Previous researches have shown that SREBPs with central roles in FA synthesis regulation could transactivate FA synthesis‐related enzymes [30, 31]. Therefore, we detected the expression levels of two SREBPs, which are encoded by SREBF1 and SREBF2 genes, in CRC cells with PTPRO knockdown and overexpression, respectively. qRT‐PCR and Western blotting experiments revealed that only SREBP1 was upregulated in PTPRO‐silenced cells (Figure 5C; Supplementary Figure S4A‐B) and downregulated in PTPRO‐enforced cells (Figure 5D; Supplementary Figure S4B‐C). Further nuclear expression analysis confirmed the mature form of SREBP1 was notably upregulated in PTPRO‐silenced cells (Supplementary Figure S4D‐E). To confirm whether PTPRO inhibited the lipogenic enzyme ACC1 via SREBP1, we silenced the expression of SREBP1 in PTPRO‐knocked down cells and enforced SREBP1 expression in PTPRO‐overexpressing cells. Enhanced SREBP1 expression significantly abrogated PTPRO‐induced ACC1 downregulation and decreases in the intracellular TAG content and neutral lipid levels as reflected by the BODIPY staining results, with knockdown of SREBP1 showing the opposite effects (Figure 5E–5I). To further support these findings, we evaluated the correlations between the mRNA levels of PTPRO, SREBP1, and ACC1 in the GSE39582 cohort, which suggested that the expression of PTPRO was negatively correlated with SREBP1 and ACC1 (Figure 5J). This correlation was further validated by qRT‐PCR in 64 CRC patients from FUSCC (Figure 5K).
FIGURE 5.
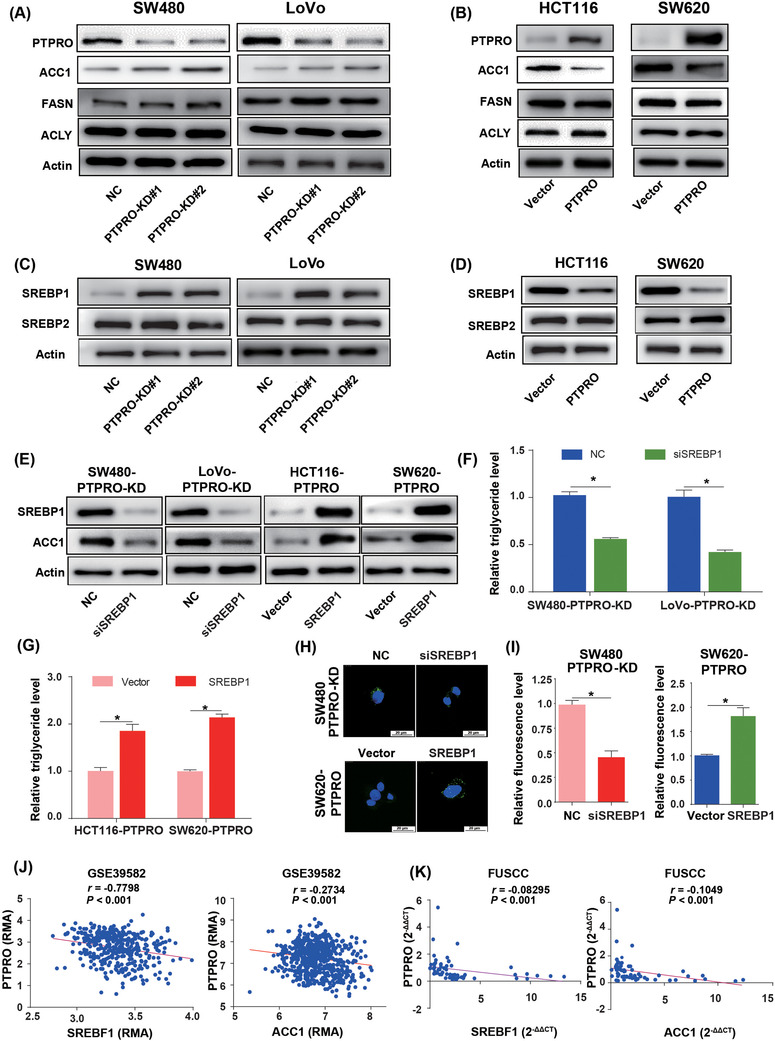
PTPRO suppressed the expression of ACC1 by down‐regulating SREBP1. (A) Western blotting demonstrates the effects of PTPRO silencing on the expression of lipogenic enzymes in CRC cells. (B) Western blotting demonstrates the effects of PTPRO overexpression on the expression of lipogenic enzymes in CRC cells. (C) Western blotting demonstrates the effects of PTPRO silencing on the expression of SREBP1 in CRC cells. (D) Western blotting demonstrates the effects of PTPRO overexpression on the expression of SREBP1 in CRC cells. (E) SREBP1 was critical for the expression of ACC1 regulated by PTPRO. (F) The silencing of SREBP1 decreased the triglyceride levels in PTPRO‐KD cells. (G) SREBP1 overexpression enhanced the triglyceride levels in PTPRO‐enforced cells. (H‐I) SREBP1 influenced the neutral lipid content in PTPRO‐attenuated or ‐enforced cells. (J) PTPRO expression was negatively correlated with SREBP1 and ACC1 expression levels in the GSE39582 cohort. (K) Validation of the negative correlation between PTPRO expression and SREBP1 and ACC1 in the FUSCC dataset. *P < 0.05. Abbreviations: PTPRO: protein tyrosine phosphatase receptor type O; CRC: colorectal cancer; ACC1: acetyl‐CoA carboxylase; SREBP1: sterol regulatory element‐binding protein 1; FASN: FA synthetase; ACLY: ATP Citrate lyase; NC: negative control; KD: knockdown; FUSCC: Fudan University Shanghai Cancer Center
To further test the roles of SREBP1 and ACC1 in guiding the regulating effects of PTPRO on tumorigenesis and metastasis, we silenced SREBP1 and ACC1 expression in PTPRO‐attenuated human organoids. It was found that SREBP1 or ACC1 knockdown abrogated the promoting effects of PTPRO silence on organoid sphere formation (Supplementary Figure S5A‐B), xenografts formation and growth (Supplementary Figure S5C‐E) and liver metastasis (Supplementary Figure S5F‐G), confirming that PTPRO represses tumorigenesis and metastasis by downregulating SREBP1 and ACC1.
3.2.4. PTPRO promoted the expression of FAO enzyme by upregulating PPARα
Decreased FAO could reduce the consumption of lipids, thus contributing to lipid accumulation. Hence, to determine whether PTPRO also participated in FAO regulation, we detected the expression of FAO‐related key enzymes, including CPT1A, CPT1B, ACOX1, and ACOX2. qRT‐PCR and Western blotting experiments showed that the expression of ACOX1 was pronouncedly decreased in cells with PTPRO knockdown and increased in those with PTPRO overexpression (Figures 6A–6B; Supplementary Figure S6A‐B), implying that PTPRO might modulate the expression of ACOX1 transcriptionally. Previous studies reported that PPARs could play important roles in lipid metabolism and are critical regulators in FAO [32, 33]. We hypothesized that PTPRO may regulate ACOX1 expression via PPARs. In order to verify this hypothesis, the expression of PPARα, PPARβ/δ, and PPARγ was evaluated by qRT‐PCR and Western blotting, and it was found that PPARα was downregulated in PTPRO‐silenced cells (Figure 6C; Supplementary Figure S6C) and upregulated in PTPRO‐enforced cells (Figure 6D; Supplementary Figure SD). To further confirm that PTPRO regulated FAO by targeting PPARα, we enhanced PPARα expression in PTPRO‐silenced cells and knocked down PPARα expression in PTPRO‐overexpressing cells. Silencing PPARα significantly abrogated PTPRO‐induced ACOX1 upregulation and decreases in the levels of intracellular TAG content and neutral lipid, with PPARα overexpression showing the opposite effects (Figure 6E–6I). Further correlation analysis suggested that the expression of PTPRO was positively correlated with ACOX1 and PPARα in the GSE39582 (Figure 6J) and FUSCC cohorts (Figure 6K).
FIGURE 6.
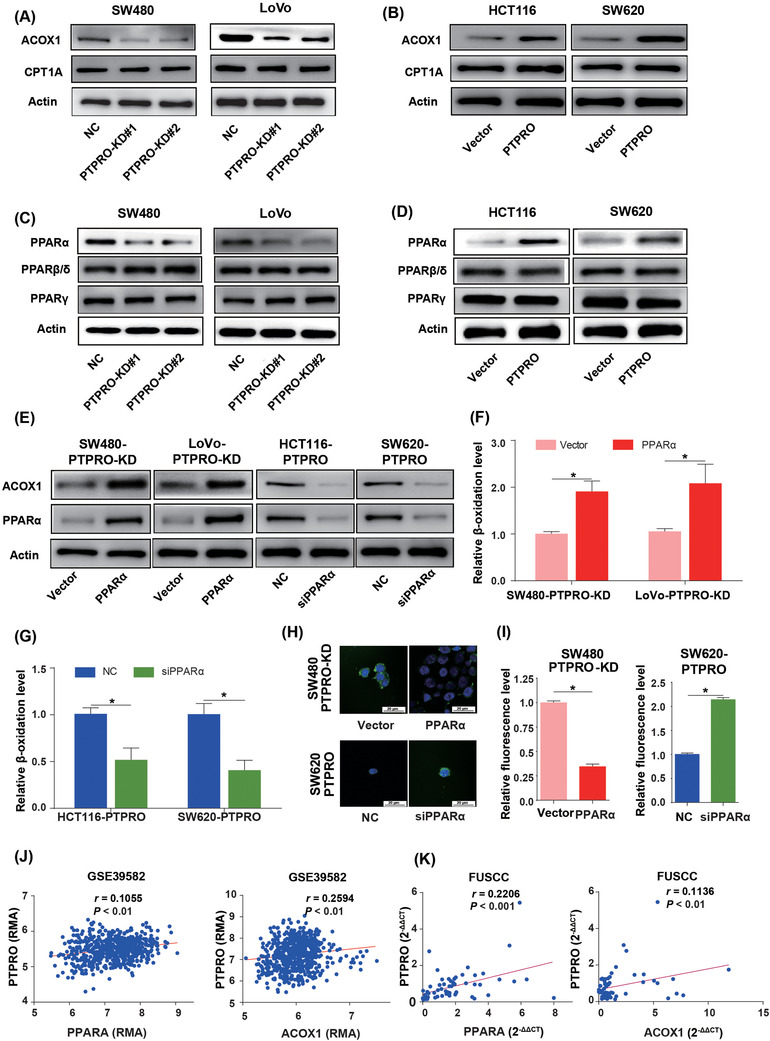
PTPRO enhanced the expression of ACOX1 by upregulating PPARα. (A) Western blotting demonstrates the effects of PTPRO silencing on the expression of critical enzymes for FAO. (B) Western blotting demonstrates the effects of PTPRO overexpression on the expression of FAO‐related enzymes. (C) Western blotting demonstrates the effects of PTPRO silencing on the expression of PPARα. (D) Western blotting demonstrates the effects e of PTPRO overexpression on the expression of PPARα. (E) PPARα was critical for the expression of ACOX1 regulated by PTPRO. (F) PPARα overexpression increased the FAO rate in PTPRO‐KD cells. (G) The silencing of PPARα decreased the FAO rate in PTPRO‐enforced cells. (H‐I) PPARα influenced the content of neutral lipids in PTPRO‐attenuated or ‐enforced cells. (J) PTPRO expression was positively correlated with the expression levels of PPARα and ACOX1 in the GSE39582 cohort. (K) Validation of the positive correlation between PTPRO expression and PPARα and ACOX1 in the FUSCC dataset. *P < 0.05. Abbreviations: PTPRO: protein tyrosine phosphatase receptor type O; FAO: fatty acid oxidation; ACOX1: Peroxisomal acyl‐coenzyme oxidase 1; PPARα: peroxisome proliferator‐activated receptor alpha; CPT1A: carnitine palmitoyltransferase 1A; NC: negative control; KD: knockdown; FUSCC: Fudan University Shanghai Cancer Center
3.2.5. PTPRO reprogramed FA metabolism by modulating the AKT/mTOR/SREBP1 and MAPK/PPARα signaling axes
We demonstrated that PTPRO could regulate FA and FAO through modulating the SREBP1/ACC1 and PPARα/ACOX1 pathways. Nevertheless, no studies have yet reported the mechanisms of PTPRO regulating SREBP1 and PPARα. Previous studies revealed that activation of the AKT/mTOR pathway could elevate the expression of SREBP1 [34, 35, 36], while PTPRO deficiency could activate AKT [23]. Therefore, we postulated that PTPRO might dictate the expression of SREBP1 and ACC1 via AKT/mTOR modulation. Meanwhile, it has been revealed that ERK and p38 mitogen‐activated protein kinase (MAPK) could rewire FA metabolism by suppressing the expression of PPARα [37, 38]. Based on the finding that PTPRO could repress the MAPK signaling axis [39], we assumed that PTPRO might be able to modulate PPARα and ACOX1 via MAPK regulation. Considering that PTPRO mainly functions through dephosphorylation, we examined the expression levels of AKT, mTOR, p38, ERK and their phosphorylation levels by Western blotting. The phosphorylation levels of AKT and mTOR were dramatically elevated in cells with PTPRO knocked down (Figures 7A). In addition, we found that both p38 and ERK were also notably phosphorylated in PTPRO‐attenuated cells (Figures 7B). As expected, AKT, mTOR, p38, and ERK were all dephosphorylated in cells overexpressing PTPRO (Figure 7C–7D). To determine whether PTPRO reprogrammed the FA metabolism through activation of the AKT/mTOR and MAPK signaling axes, we used the PI3K inhibitor LY294002 and and MAPK inhibitor (E)‐osmundacetone to inhibit the AKT/mTOR and MAPK pathways, respectively, in CRC cells with PTPRO silenced. LY294002 markedly diminished the expression of SREBP1 and ACC1 (Figure 7E) and reduced the content of intracellular TAG (Figure 7F) mediated by AKT/mTOR signaling activation. (E)‐osmundacetone enhanced the expression of PPARα and ACOX1 (Figure 7G) and promoted FAO (Figure 7H). BODIPY staining of neutral lipids further confirmed that PTPRO regulated the FA content by modulating the AKT/mTOR and MAPK pathways (Figure 7I). Moreover, compared with PTPRO‐WT mice, the AKT/mTOR and MAPK pathways were significantly activated in PTPRO–/‐ mice (Figure 7J). As expected, the SREBP1/ACC1 axis was upregulated in PTPRO–/‐ mice, while the PPARα/ACOX1 axis was downregulated (Figure 7J), indicating that PTPRO‐mediated AKT/mTOR/SREBP1/ACC1 and MAPK/PPARα/ACOX1 signaling axes might be able to promote CRC initiation by reprogramming FA metabolism.
FIGURE 7.
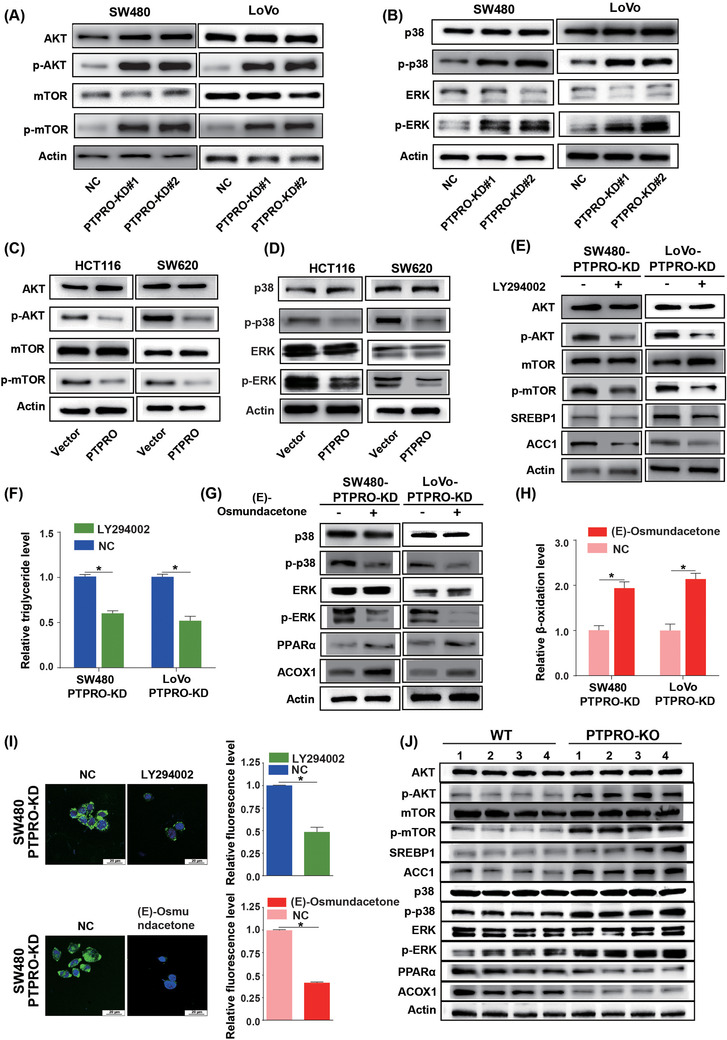
PTPRO reprogramed FA metabolism by regulating the Akt/mTOR/SREBP1 and P38/ERK/PPARα pathways. (A) Western blotting reveals the levels of AKT, p‐AKT, mTOR, and p‐mTOR in SW480 and LoVo cells with PTPRO‐NC/KD. (B) Western blotting reveals the levels of p38, p‐p38, ERK, and p‐ERK in SW480 and LoVo cells with PTPRO‐NC/KD. (C) Western blotting demonstrates the levels of AKT, p‐AKT, mTOR, and p‐mTOR in HCT116 and SW620 cells with Vector/PTPRO. (D) Western blotting reveals the levels of p38, p‐p38, ERK and p‐ERK in HCT116 and SW620 cells with Vector/PTPRO. (E) Western blotting demonstrates the expression changes of SREBP1 and ACC1 in PTPRO‐silenced cells treated with PI3K inhibitor LY294002. (F) PI3K inhibitor decreased the content of TAG in PTPRO‐silenced cells. (G) Western blotting reveals the expression changes of PPARα and ACOX1 in PTPRO‐silenced cells treated with MAPK inhibitor (E)‐osmundacetone. (H) MAPK inhibitor increased the rate of FAO in PTPRO‐silenced cells. (I) PI3K inhibitor or MAPK inhibitor attenuated the level of neutral lipids in PTPRO‐silenced cells. (J) The protein expression of total AKT, p‐AKT, mTOR, p‐mTOR, SREBP1, ACC1, p38, p‐p38, ERK, p‐ERK, PPARα, and ACOX1 in PTPRO‐KO and PTPRO‐WT mice tissues. *P < 0.05. Abbreviations: PTPRO: protein tyrosine phosphatase receptor type O; FA: fatty acid synthesis; FAO: fatty acid oxidation; TAG: tryglicerides; ACC1: acetyl‐CoA carboxylase; SREBP1: sterol regulatory element‐binding protein 1; NC: negative control; KD: knockdown; WT: wild type; KO: knockout; ACOX1: Peroxisomal acyl‐coenzyme oxidase 1; PPARα: peroxisome proliferator‐activated receptor alpha; MAPK: mitogen‐activated protein kinases; AKT: AKT serine/threonine kinase; p‐AKT: phospho‐AKT; ERK: extracellular signal‐regulated kinase; p‐ERK: phospho‐ERK; mTOR: mammalian target of rapamycin; p‐mTOR: phospho‐mTOR; p‐p38: phospho‐p38; PI3K: phosphoInositide‐3 kinase
In order to confirm the causal relationship between PTPRO and the AKT/mTOR/SREBP1‐MAPK/PPARα axis in tumor growth and metastasis, we treated PTPRO‐silenced cells with LY294002 and (E)‐osmundacetone. The results showed that LY294002 or (E)‐osmundacetone treatment almost completely eliminated the enforced cell proliferation (Supplementary Figure S7A), migration (Supplementary Figure S7B‐C) and liver metastasis (Supplementary Figure S7D‐E) induced by PTPRO silence. Overall, the regulation of FA metabolism by PTPRO is illustrated in Figure 8.
FIGURE 8.
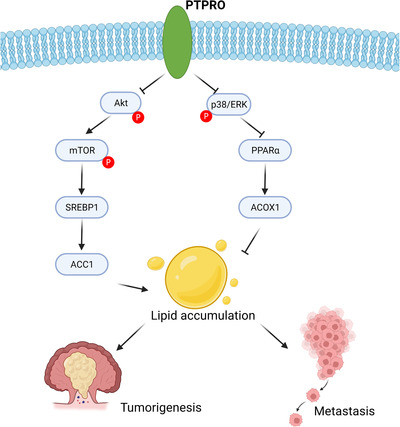
Schematic depiction of the regulation on FA metabolism by PTPRO in CRC cells. Abbreviations: PTPRO: protein tyrosine phosphatase receptor type O; CRC: colorectal cancer; FA: fatty acid; ACOX1: Peroxisomal acyl‐coenzyme oxidase 1; PPARα: peroxisome proliferator‐activated receptor alpha; AKT: AKT serine/threonine kinase; ERK: extracellular signal‐regulated kinase; mTOR: mammalian target of rapamycin; PPARα: peroxisome proliferator‐activated receptor alpha; ACC1: acetyl‐CoA carboxylase; SREBP1: sterol regulatory element‐binding protein 1
4. DISCUSSION
No previous studies have comprehensively explored the roles and mechanisms of PTPRO in CRC initiation and progression. In the present study, we revealed that PTPRO was downregulated in CRC and liver metastasis, and its low expression was linked with dismal OS and DFS, which indicated that PTPRO could be used as a prognosis predictor and that its low expression might predict a high risk of liver metastasis. Based on the findings that PTPRO might get involved in FA metabolism revealed by transcriptome profiling and GSEA analysis, subsequent in vitro and in vivo experiments were conducted, which demonstrated that dysregulation of PTPRO was indeed involved in the initiation and progression of CRC by regulating FA metabolism. Our findings revealed that the tumor suppressor PTPRO could repress de novo FA synthesis by decreasing the expression of ACC1 through modulating the AKT/mTOR/SREBP1 signaling axes and simultaneously promote FAO by upregulating the FAO‐related enzyme ACOX1 via the p38/ERK/PPARα signaling axes.
Researches have shown that a multitude of de novo FA synthesis‐related enzymes, such as FASN, ACLY, SCD, and ACSS2, were abnormally upregulated in several cancers, enhancing FA synthesis and contributing to the malignant phenotypes of cancer [40, 41, 42, 43, 44]. What's more, previous reports have confirmed that the expression of FA synthesis‐related key enzyme ACC1 was elevated in multiple cancer tissues and its high expression could accelerate cell proliferation strikingly [45, 46, 47]. Consistently, the present study showed that PTPRO silencing could enhance FA synthesis, thus promoting CRC cell growth and liver metastasis by upregulating ACC1 expression. SREBPs belong to the transcription factor family, which has been demonstrated to be a family of critical regulators of lipid homeostasis [6, 30, 31]. SREBP1 and SREBP2 are two isoforms found to express in mammalian cells. The expression of SREBP1 has been reported to be elevated in numerous cancers, including CRC, and its high expression has been demonstrated to expedite cancer progression [48, 49]. We found that PTPRO silencing‐induced SREBP1 expression could elevate the expression of ACC1. It was unexpected that only ACC1 was markedly upregulated in PTPRO‐knockdown cells, in which the expression level of SREBP1 was increased because lipogenic enzymes were commonly transcriptionally activated by SREBP1. However, it is also common that not all enzymes will be notably upregulated upon the activation of SREBP1. For instance, lncRNA ZFAS1 was found to upregulate FASN and SCD1 expression by stabilizing SREBP1 [50], whereas Li et al. [38] revealed that CD147 had no effect on SCD1 expression though nuclear SREBP1 expression was notably upregulated. Hence, the regulating effects of SREBP1 on lipogenic enzymes may be partially context‐dependent. In this study, ACC1 is the main target of SREBP1 in response to PTPRO silence, thus contributing to FA synthesis and lipid accumulation in CRC cells. In addition, it was established that the activation of the AKT/mTOR pathway could elevate the expression of SREBP1 [34, 36, 51]. More importantly, previous studies have indicated that PTPRO deficiency could activate the AKT pathway [23, 52]. Therefore, we postulated that PTPRO might exert its biological functions through modulating the AKT/mTOR pathway. In the present study, we demonstrated the AKT/mTOR activation through phosphorylation, which was mediated by PTPRO silencing, could induce the expression of SREBP1 and its downstream gene ACC1, facilitating FA synthesis and lipid accumulation.
As a certain amount of lipids is required for cellular membrane synthesis and critical signal transduction in proliferating cancer cells, a decrease in the lipid content may restrict cell growth. FAO is one of the main processes that contribute to the consumption of FAs. A previous researchhas demonstrated that the inhibition of FAO caused by hypoxia was essential for cancer cell proliferation [53]. In addition, pathway enrichment analysis showed that the FAO pathway was suppressed in aggressive cancers in contrast with early‐stage cancers [54]. Thus, we hypothesized that PTPRO silencing‐mediated lipid accumulation might be triggered by its regulation on FAO too, based on which we revealed that the FAO‐related key enzyme ACOX1 was upregulated by PTPRO. Previous studies have shown that the FA accumulation caused by ACOX1 downregulation may promote carcinogenesis and tumor progression [55, 56]. Consistently, we demonstrated that ACOX1‐mediated FAO could exert a protective role in CRC initiation and growth. Researches have revealed that PPARs were essential transcriptional regulators for FAO, for which PPARα and PPARδ were shown to induce FAO, thus promoting tumorigenesis [32, 33]. Additionally, PPARα has been reported to be able to sustain the growth of cancer cells [57, 58]. We revealed that PTPRO positively regulated PPARα and its downstream gene ACOX1, thereby modulating FAO. What's more, prior studies have uncovered that MAPKs were important for the regulation of PPAR activity, and ERK and p38 MAPK are two major subclasses of the MAPK superfamily [37, 38]. It has also been suggested that PTPRO could repress the activity of the MAPK pathway in different cancer cells and biological processes [22, 23, 59, 60, 61]. Based on these researches, we demonstrated that PTPRO could dephosphorylate p38/ERK MAPK, promoting the expression of PPARα and its downstream gene ACOX1, thus enhancing FAO in CRC.
Therefore, with compelling evidence, we uncovered the functions of PTPRO in CRC and discovered that PTPRO silencing could enhance the expression of ACC1 and decrease the expression of ACOX1 in CRC cells via the activation of the AKT/mTOR and MAPK signaling axes. Thus, the lipid accumulation mediated by ACC1 and ACOX1 could further promote tumorigenesis and progression of CRC.
Though with intriguing findings, there were still some limitations in the present study: (1) without detecting the expression of PTPRO in other metastatic sites such as the lung and abdomen, we could not conclude whether low expression of PTPRO could predict metastasis to other sites except for the liver; (2) we have not explored why PTPRO is downregulated in CRC and liver metastasis, the mechanisms of which might provide more alternatives for developing therapeutic drugs for CRC; (3) the mechanism of PTPRO activating downstream pathways of PI3K/AKT and MAPK needs further study.
5. CONCLUSIONS
In summary, we investigated and uncovered the functions of PTPRO in CRC, revealing it to be a tumor suppressor and a prognosis predictor. Our findings demonstrated that the AKT/mTOR/SREBP1/ACC1 and MAPK/ PPARα/ACOX1 pathways are critical mechanisms contributing to CRC initiation and progression mediated by PTPRO silencing, which might facilitate future drug development for CRC patients.
DECLARATIONS
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
According to the stipulations of the Institutional Review Board of FUSCC, informed consent was obtained from pertinent patients for sample and data use in research. All mouse‐related experiments were carried out in accordance with the guidelines of the Institutional Animal Care and Use Committee of Fudan University.
AUTHOR CONTRIBUTIONS
Conceptualization: Weixing Dai, Wenqiang Xiang, Qingguo Li, and Guoxiang Cai
Sample collection: Weixing Dai, Lingyu Han, Zixu Yuan, Renjie Wang, Yanlei Ma, Yongzhi Yang
In vitro study: Weixing Dai and Wenqiang Xiang
Metabolomics assays: Ye Xu, Shaobo Mo, and Sanjun Cai
Mouse study: Lingyu Han and Wenqiang Xiang
Writing – Original Draft: Weixing Dai, Wenqiang Xiang, Qingguo Li, Guoxiang Cai
Writing – Review & Editing: All authors
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Upon reasonable request, data and materials supporting these findings in the present study will be available from the corresponding author.
Supporting information
Supporting Information
ACKNOWLEDGEMENTS
We would like to express our sincere appreciation for the platform and datasets that the team of GEO database provided, making scientific research boundaryless. This study was funded by the grant from the National Natural Science Foundation of China (No. 81871958 and No. 82103554), the Grant of Science and Technology Commission of Shanghai Municipality (No. 19140902100, No. 16401970502 and No. 17411951100).
Dai W, Xiang W, Han L, Yuan Z, Wang R, Ma Y, et al. PTPRO represses colorectal cancer tumorigenesis and progression by reprogramming fatty acid metabolism. Cancer Commun. 2022;42:848–867. 10.1002/cac2.12341
Contributor Information
Shaobo Mo, Email: shaobom@126.com.
Qingguo Li, Email: oncosurgeonli@sohu.com.
Guoxiang Cai, Email: gxcai@fudan.edu.cn.
REFERENCES
- 1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1), 7–33. [DOI] [PubMed] [Google Scholar]
- 2. Xia C, Dong X, Li H, Cao M, Sun D, He S, et al. Cancer statistics in China and United States, 2022: Profiles, trends, and determinants. Chin Med J (Engl). 2022;135(5), 584–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pavlova NN, Zhu J, Thompson CB. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022;34(3), 355–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Martinez‐Reyes I, Chandel NS. Cancer metabolism: Looking forward. Nat Rev Cancer. 2021;21(10), 669–80. [DOI] [PubMed] [Google Scholar]
- 5. Stine ZE, Schug ZT, Salvino JM, Dang CV. Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discov. 2022;21(2), 141–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun (Lond). 2018;38(1), 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cao Y. Adipocyte and lipid metabolism in cancer drug resistance. J Clin Invest. 2019;129(8), 3006–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12(1), 31–46. [DOI] [PubMed] [Google Scholar]
- 9. Fu Y, Zou T, Shen X, Nelson PJ, Li J, Wu C, et al. Lipid metabolism in cancer progression and therapeutic strategies. MedComm. 2021;2(1), 27–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bacci M, Lorito N, Smiriglia A, Morandi A. Fat and Furious: Lipid Metabolism in Antitumoral Therapy Response and Resistance. Trends Cancer. 2021;7(3), 198–213. [DOI] [PubMed] [Google Scholar]
- 11. Snaebjornsson MT, Janaki‐Raman S, Schulze A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020;31(1), 62–76. [DOI] [PubMed] [Google Scholar]
- 12. Munir R, Lisec J, Swinnen JV, Zaidi N. Lipid metabolism in cancer cells under metabolic stress. Br J Cancer. 2019;120(12), 1090–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu Q, Luo Q, Halim A, Song G. Targeting lipid metabolism of cancer cells: A promising therapeutic strategy for cancer. Cancer Lett. 2017;401, 39–45. [DOI] [PubMed] [Google Scholar]
- 14. Martinez‐Outschoorn UE, Peiris‐Pagés M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: A therapeutic perspective. Nat Rev Clin Oncol. 2017;14(1), 11–31. [DOI] [PubMed] [Google Scholar]
- 15. Ferraro GB, Ali A, Luengo A, Kodack DP, Deik A, Abbott KL, et al. Fatty Acid Synthesis Is Required for Breast Cancer Brain Metastasis. Nat Cancer. 2021;2(4), 414–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen XF, Tian MX, Sun RQ, Zhang ML, Zhou LS, Jin L, et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018;19(5), e45124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ostman A, Hellberg C, Böhmer FD. Protein‐tyrosine phosphatases and cancer. Nat Rev Cancer. 2006;6(4), 307–20. [DOI] [PubMed] [Google Scholar]
- 18. Tonks NK. Protein tyrosine phosphatases: From genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7(11), 833–46. [DOI] [PubMed] [Google Scholar]
- 19. Bollu LR, Mazumdar A, Savage MI, Brown PH. Molecular Pathways: Targeting Protein Tyrosine Phosphatases in Cancer. Clin Cancer Res. 2017;23(9), 2136–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sivaganesh V, Sivaganesh V, Scanlon C, Iskander A, Maher S, Le T, et al. Protein Tyrosine Phosphatases: Mechanisms in Cancer. Int J Mol Sci. 2021;22(23), 12865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ming F, Sun Q. Epigenetically silenced PTPRO functions as a prognostic marker and tumor suppressor in human lung squamous cell carcinoma. Mol Med Rep. 2017;16(1), 746–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dong H, Ma L, Gan J, Lin W, Chen C, Yao Z, et al. PTPRO represses ERBB2‐driven breast oncogenesis by dephosphorylation and endosomal internalization of ERBB2. Oncogene. 2017;36(3), 410–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang W, Hou J, Wang X, Jiang R, Yin Y, Ji J, et al. PTPRO‐mediated autophagy prevents hepatosteatosis and tumorigenesis. Oncotarget. 2015;6(11), 9420–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dai W, Li Y, Mo S, Feng Y, Zhang L, Xu Y, et al. A robust gene signature for the prediction of early relapse in stage I‐III colon cancer. Mol Oncol. 2018;12(4), 463–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Browne CD, Hindmarsh EJ, Smith JW. Inhibition of endothelial cell proliferation and angiogenesis by orlistat, a fatty acid synthase inhibitor. FASEB J. 2006;20(12), 2027–35. [DOI] [PubMed] [Google Scholar]
- 26. Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004;64(6), 2070–5. [DOI] [PubMed] [Google Scholar]
- 27. Chen L, Jin XH, Luo J, Duan JL, Cai MY, Chen JW, et al. ITLN1 inhibits tumor neovascularization and myeloid derived suppressor cells accumulation in colorectal carcinoma. Oncogene. 2021;40(40), 5925–37. [DOI] [PubMed] [Google Scholar]
- 28. Kuriyama I, Nakajima Y, Nishida H, Konishi T, Takeuchi T, Sugawara F, et al. Inhibitory effects of low molecular weight polyphenolics from Inonotus obliquus on human DNA topoisomerase activity and cancer cell proliferation. Mol Med Rep. 2013;8(2), 535–42. [DOI] [PubMed] [Google Scholar]
- 29. Li Y, Liang L, Dai W, Cai G, Xu Y, Li X, et al. Prognostic impact of programed cell death‐1 (PD‐1) and PD‐ligand 1 (PD‐L1) expression in cancer cells and tumor infiltrating lymphocytes in colorectal cancer. Mol Cancer. 2016;15(1), 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Y, Mohibi S, Vasilatis DM, Chen M, Zhang J, Chen X. Ferredoxin reductase and p53 are necessary for lipid homeostasis and tumor suppression through the ABCA1‐SREBP pathway. Oncogene. 2022;41(12), 1718–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kanagasabai T, Li G, Shen TH, Gladoun N, Castillo‐Martin M, Celada SI, et al. MicroRNA‐21 deficiency suppresses prostate cancer progression through downregulation of the IRS1‐SREBP‐1 signaling pathway. Cancer Lett. 2022;525, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mana MD, Hussey AM, Tzouanas CN, Imada S, Barrera Millan Y, Bahceci D, et al. High‐fat diet‐activated fatty acid oxidation mediates intestinal stemness and tumorigenicity. Cell Rep. 2021;35(10), 109212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wickramasinghe NM, Sachs D, Shewale B, Gonzalez DM, Dhanan‐Krishnan P, Torre D, et al. PPARdelta activation induces metabolic and contractile maturation of human pluripotent stem cell‐derived cardiomyocytes. Cell Stem Cell. 2022;29(4), 559–76. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao S, Cheng L, Shi Y, Li J, Yun Q, Yang H. MIEF2 reprograms lipid metabolism to drive progression of ovarian cancer through ROS/AKT/mTOR signaling pathway. Cell Death Dis. 2021;12(1), 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yi J, Zhu J, Wu J, Thompson CB, Jiang X. Oncogenic activation of PI3K‐AKT‐mTOR signaling suppresses ferroptosis via SREBP‐mediated lipogenesis. Proc Natl Acad Sci USA. 2020;117(49), 31189–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu L, Yan H, Ruan M, Yang H, Wang L, Lei B, et al. An AKT/PRMT5/SREBP1 axis in lung adenocarcinoma regulates de novo lipogenesis and tumor growth. Cancer Sci. 2021;112(8), 3083–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mooli RGR, Rodriguez J, Takahashi S, Solanki S, Gonzalez FJ, Ramakrishnan SK, et al. Hypoxia via ERK Signaling Inhibits Hepatic PPARalpha to Promote Fatty Liver. Cell Mol Gastroenterol Hepatol. 2021;12(2), 585–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li J, Huang Q, Long X, Zhang J, Huang X, Aa J, et al. CD147 reprograms fatty acid metabolism in hepatocellular carcinoma cells through Akt/mTOR/SREBP1c and P38/PPARalpha pathways. J Hepatol. 2015;63(6), 1378–89. [DOI] [PubMed] [Google Scholar]
- 39. Chen L, Juszczynski P, Takeyama K, Aguiar RC, Shipp MA. Protein tyrosine phosphatase receptor‐type O truncated (PTPROt) regulates SYK phosphorylation, proximal B‐cell‐receptor signaling, and cellular proliferation. Blood. 2006;108(10), 3428–33. [DOI] [PubMed] [Google Scholar]
- 40. You X, Tian J, Zhang H, Guo Y, Yang J, Zhu C, et al. Loss of mitochondrial aconitase promotes colorectal cancer progression via SCD1‐mediated lipid remodeling. Mol Metab. 2021:Epub 2021 Mar 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang M, Chen X, Zhang J, Xiong E, Wang Q, Fang W, et al. ME2 Promotes Proneural‐Mesenchymal Transition and Lipogenesis in Glioblastoma. Front Oncol. 2021:eCollection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7(10), 763–77. [DOI] [PubMed] [Google Scholar]
- 43. Koobotse M, Holly J, Perks C. Elucidating the novel BRCA1 function as a non‐genomic metabolic restraint in ER‐positive breast cancer cell lines. Oncotarget. 2018;9(71), 33562–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Khwairakpam AD, Banik K, Girisa S, Shabnam B, Shakibaei M, Fan L, et al. The vital role of ATP citrate lyase in chronic diseases. J Mol Med (Berl). 2020;98(1), 71–95. [DOI] [PubMed] [Google Scholar]
- 45. Rios Garcia M, Steinbauer B, Srivastava K, Singhal M, Mattijssen F, Maida A, et al. Acetyl‐CoA Carboxylase 1‐Dependent Protein Acetylation Controls Breast Cancer Metastasis and Recurrence. Cell Metab. 2017;26(6), 842–55. e5. [DOI] [PubMed] [Google Scholar]
- 46. Li EQ, Zhao W, Zhang C, Qin LZ, Liu SJ, Feng ZQ, et al. Synthesis and anti‐cancer activity of ND‐646 and its derivatives as acetyl‐CoA carboxylase 1 inhibitors. Eur J Pharm Sci. 2019;137, 105010. [DOI] [PubMed] [Google Scholar]
- 47. Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, et al. Inhibition of acetyl‐CoA carboxylase suppresses fatty acid synthesis and tumor growth of non‐small‐cell lung cancer in preclinical models. Nat Med. 2016;22(10), 1108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gao Y, Nan X, Shi X, Mu X, Liu B, Zhu H, et al. SREBP1 promotes the invasion of colorectal cancer accompanied upregulation of MMP7 expression and NF‐κB pathway activation. BMC Cancer. 2019;19(1), 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sun Y, He W, Luo M, Zhou Y, Chang G, Ren W, et al. SREBP1 regulates tumorigenesis and prognosis of pancreatic cancer through targeting lipid metabolism. Tumour Biol. 2015;36(6), 4133–41. [DOI] [PubMed] [Google Scholar]
- 50. Wang H, Chen Y, Liu Y, Li Q, Luo J, Wang L, et al. The lncRNA ZFAS1 regulates lipogenesis in colorectal cancer by binding polyadenylate‐binding protein 2 to stabilize SREBP1 mRNA. Mol Ther Nucleic Acids. 2022;27, 363–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mashima T, Seimiya H, Tsuruo T. De novo fatty‐acid synthesis and related pathways as molecular targets for cancer therapy. Br J Cancer. 2009;100(9), 1369–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang X, Tan Z, Wang Y, Tang J, Jiang R, Hou J, et al. PTPRO‐associated hepatic stellate cell activation plays a critical role in liver fibrosis. Cell Physiol Biochem. 2015;35(3), 885–98. [DOI] [PubMed] [Google Scholar]
- 53. Huang D, Li T, Li X, Zhang L, Sun L, He X, et al. HIF‐1‐mediated suppression of acyl‐CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014;8(6), 1930–42. [DOI] [PubMed] [Google Scholar]
- 54. Louie SM, Roberts LS, Mulvihill MM, Luo K, Nomura DK. Cancer cells incorporate and remodel exogenous palmitate into structural and oncogenic signaling lipids. Biochim Biophys Acta. 2013;1831(10), 1566–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sun LN, Zhi Z, Chen LY, Zhou Q, Li XM, Gan WJ, et al. SIRT1 suppresses colorectal cancer metastasis by transcriptional repression of miR‐15b‐5p. Cancer Lett. 2017;409, 104–15. [DOI] [PubMed] [Google Scholar]
- 56. Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, et al. Hepatocyte‐specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113(12), 1774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dana N, Vaseghi G, Haghjooy Javanmard S. PPAR γ agonist, pioglitazone, suppresses melanoma cancer in mice by inhibiting TLR4 signaling. J Pharm Pharm Sci. 2019;22(1), 418–23. [DOI] [PubMed] [Google Scholar]
- 58. Borbath I, Leclercq I, Moulin P, Sempoux C, Horsmans Y. The PPARgamma agonist pioglitazone inhibits early neoplastic occurrence in the rat liver. Eur J Cancer. 2007;43(11), 1755–63. [DOI] [PubMed] [Google Scholar]
- 59. Hou J, Xu J, Jiang R, Wang Y, Chen C, Deng L, et al. Estrogen‐sensitive PTPRO expression represses hepatocellular carcinoma progression by control of STAT3. Hepatology. 2013;57(2), 678–88. [DOI] [PubMed] [Google Scholar]
- 60. Ramaswamy B, Majumder S, Roy S, Ghoshal K, Kutay H, Datta J, et al. Estrogen‐mediated suppression of the gene encoding protein tyrosine phosphatase PTPRO in human breast cancer: Mechanism and role in tamoxifen sensitivity. Mol Endocrinol. 2009;23(2), 176‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Asbagh LA, Vazquez I, Vecchione L, Budinska E, De Vriendt V, Baietti MF, et al. The tyrosine phosphatase PTPRO sensitizes colon cancer cells to anti‐EGFR therapy through activation of SRC‐mediated EGFR signaling. Oncotarget. 2014;5(20), 10070–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Upon reasonable request, data and materials supporting these findings in the present study will be available from the corresponding author.