

Abstract
A number of new 2-methyl- and 2-arylvinyl-3-nitropyridines were synthesized and their reactions with thiols were studied. It was found that 3-NO2 tends to be selectively substituted under the action of sulfur nucleophiles in the presence of another nucleofuge in position 5. Correlations between the substitution pattern and regioselectivity as well as photophysical properties were established. Some synthesized compounds possessed a large Stokes shift.
Keywords: nitro group, nitropyridines, bis-(het)aryl ethenes, nucleophilic substitution, thiols, UV–Vis spectroscopy
1. Introduction
Pyridine is an important heterocyclic motif and a part of various natural products. The pyridine ring system is incorporated into alkaloids, medicines (for example, omeprazole, lorlatinib, ivosidenib and many others), fungicides, herbicides and insecticides. Application of the pyridine derivatives as biologically active precursors and coordination complexes was reviewed recently [1]. Nitropyridines are of particular interest due to their biological significance [2,3,4,5]. In addition, some nitropyridines are considered to be promising energetic compounds [6,7,8,9,10] and efficient organic optical materials [11]. The introduction of the nitro group into the pyridine ring facilitates its functionalization in different ways. Recently, we investigated reactions of 3-R-5-nitropyridines with various types of nucleophiles [12]. It was found that in the case of anionic S-, N- and O-nucleophiles, the substitution of the non-activated nitro group occurred while carbon nucleophiles underwent dearomatization of the pyridine ring with the formation of 1,2- or 1,4-addition products. As a result, a number of novel or hardly accessible pyridines and their dihydro derivatives were synthesized [12].
In this work we report on the synthesis, reactivity and photophysical properties of 2-methyl- and 2-(2-arylvinyl)-3-nitropyridines, as shown in Figure 1. 2-Alkenylpyridines are widely employed as precursors to pharmaceuticals (vorapaxar, axitinib, nifurpirinol) and other biologically active compounds [13]. In addition, 2-(2-arylvinyl)pyridines were proven to be the fluorescent molecules, with the fluorescence quantum yield showing a large dependence on the acidity of media [14].
Figure 1.

2-Methyl- and 2-(2-arylvinyl)-3-nitropyridines synthesized and studied in this work.
2. Results and Discussion
2-Methyl-3-nitropyridines 2a–c were synthesized from the corresponding commercially available 2-chloro-3-nitropyridines 1a–c by the reaction with diethyl malonate, followed by acidic hydrolysis/decarboxylation, as shown in Scheme 1. The oxidation of compounds 2b,c with the hydrogen peroxide–urea complex gave N-oxides 3b,c in moderate yields.
Scheme 1.

Synthesis of 2-methyl-3-nitropyridines.
We examined 2-methylpyridines 2 and 3 in reactions with aldehydes under piperidine catalysis. Our attempts to isolate condensation products of compounds 2b,c failed, whereas 2-methyl-3,5-dinitropyridine 2a and N-oxides 3b,c gave diarylethenes 4a–g in high yields, as shown in Scheme 2. It should be noted that compound 2a reacts several times faster than pyridine N-oxides 3b,c, indicating that para-NO2 is a more potent activating group for this reaction than the N-oxide moiety neighboring the 2-methyl group. The functional group tolerance, along with the relatively mild conditions and availability of aromatic aldehydes, makes this method a valid alternative for Pd-catalyzed coupling reactions [15,16,17,18]. Deoxygenation of pyridine N-oxides 4c,e–g with PCl3 allowed us to obtain four additional 2-ethenylpyridines 4h–k, which were inaccessible via direct condensation of 2-methylpyridines 2b,c with aldehydes, as shown in Scheme 2.
Scheme 2.

Synthesis of 2-(2-arylvinyl)-3-nitropyridines.
In all cases, only trans-diarylethenes are formed, which was confirmed by NMR spectroscopy: coupling constants of 15–16 Hz were observed for proton signals of the double bonds. In addition, X-ray analysis for compounds 4a,i was performed, undoubtedly proving our assumption, as shown in Figure 2.
Figure 2.

X-ray crystal structures of compounds 4a (left) and 4i (right) with thermal ellipsoids at 50% probability level.
The possibility of the substitution of the non-activated nitro group in pyridines was studied recently by our group [12]. In 3-nitro-5-Cl(Br)-pyridines, 3-NO2 was found to be more nucleofugal than halogen in position 5. The reactions of 2-methyl-3-nitropyridines 2 and 3 with thiols are summarized in Scheme 3 and Table 1. Upon heating the reactants in DMF in the presence of K2CO3, the selective formation of 3-R2S-products 5 was observed in all cases; however, the reaction of 2a with BnSH gave 5a with a trace amount of the isomer 6a.
Scheme 3.

Reactions of 2-methyl-3-nitropyridines with thiols.
Table 1.
Reactions of 2-methyl-3-nitropyridines with thiols.
| Substrate | R1 | n | R2 | Product, Isolated Yield (%) |
|---|---|---|---|---|
| 2a | NO2 | 0 | PhCH2 | 5a, 70 |
| 3b | Br | 1 | PhCH2 | 5b, 96 |
| 3b | Br | 1 | 4-Cl-C6H4 | 5c, 95 |
| 2b | Br | 0 | PhCH2 | 5d, 65 |
| 2c | CF3 | 0 | PhCH2 | 5e, 60 |
| 3c | CF3 | 1 | PhCH2 | 5f, 52 |
Interestingly, diarylethenes 4 react with thiols under the same mild conditions, but with lower selectivity. 1H NMR spectra of the crude products generally contain an additional set of signals corresponding to the 5-R2S-isomer, whereas the ratio of 5/6 varied from 2:1 to 20:1 depending on the substrate and thiol. Moreover, compounds 6g,h were isolated and fully characterized, but in all other cases we were unable to isolate isomers 6, as shown in Scheme 4 and Table 2.
Scheme 4.

Reactions of 2-(arylvinyl)-3-nitropyridines with thiols.
Table 2.
Reactions of 2-(arylvinyl)-3-nitropyridines with thiols.
| Substrate | R1 | Ar | n | R2 | Ratio 5/6 a | Product, Isolated Yield (%) |
|---|---|---|---|---|---|---|
| 4a | NO2 | 4-Cl-C6H4 | 0 | PhCH2 | 3:1 |
5g, 56 6g, 18 |
| 4a | NO2 | 4-Cl-C6H4 | 0 | i-C4H9 | 2:1 |
5h, 62 6h, 31 |
| 4a | NO2 | 4-Cl-C6H4 | 0 | 2-furylmethyl | >20:1 | 5i, 56 |
| 4a | NO2 | 4-Cl-C6H4 | 0 | 4-Cl-C6H4 | >20:1 | 5j, 67 |
| 4b | NO2 | 4-Me2N-C6H4 | 0 | 4-Cl-C6H4 | >20:1 | 5k, 83 |
| 4b | NO2 | 4-Me2N-C6H4 | 0 | i-C4H9 | 8:1 | 5l, 84 |
| 4b | NO2 | 4-Me2N-C6H4 | 0 | PhCH2 | 10:1 | 5m, 88 |
| 4d | NO2 | 1-(4-fluorophenyl)pyrazol-4-yl | 0 | PhCH2 | 10:1 | 5n, 89 |
| 4d | NO2 | 1-(4-fluorophenyl)pyrazol-4-yl | 0 | 4-Cl-C6H4 | >20:1 | 5o, 93 |
| 4j | Br | 4-Cl-C6H4 | 0 | PhCH2 | >20:1 | 5p, 60 |
| 4f | Br | 4-Me2N-C6H4 | 1 | PhCH2 | >20:1 | 5q, 67 |
a Determined from 1H NMR spectrum of crude product.
Structures of compounds 5 and 6 were confirmed by NMR, HRMS, X-ray and elemental analysis. 1H-1H NOESY spectra of compounds 5m and 5q revealed interactions of the spatially close protons of the double bond and benzyl substituent, as shown in Figure 3. The structures of 5h,l were determined by the X-ray diffraction single-crystal method, as shown in Figure 4.
Figure 3.
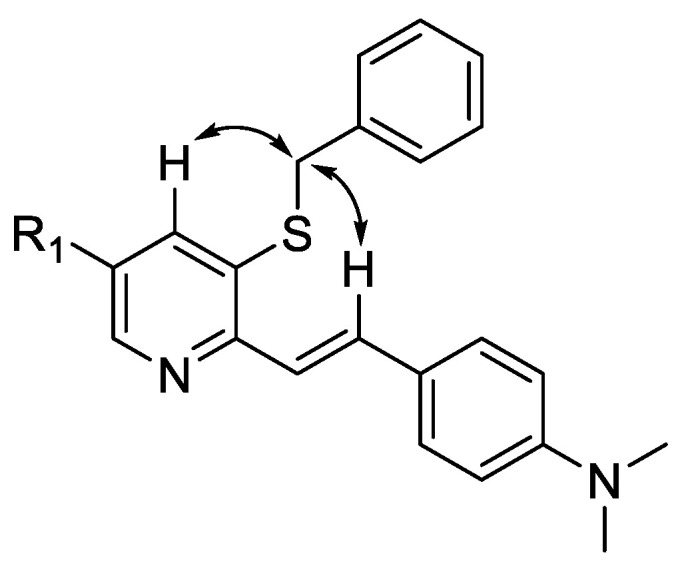
Cross-peaks observed in 1H-1H NOESY spectrum of compounds 5m (R1 = NO2) and 5q (R1 = Br).
Figure 4.

X-ray crystal structures of compounds 5h (left) and 5l (right) with thermal ellipsoids at 50% probability level.
The above-mentioned results allow us to conclude that electron-releasing substituents in the aryl group and the bulky thiolate anion favor substitution at position 3: the best selectivity was observed for reactions of 4b with α-toluene thiol, whereas 4a with isobutyl mercaptan gave the lowest selectivity (2:1). Reactions with 4-chlorothiophenol afforded exclusively a 3-substituted product.
Compounds with multiple conjugated double bonds, such as diarylethenes 4–6, can be expected to have strong absorbance in the UV and visible region; therefore, the photophysical properties of some representative compounds with various substitution patterns were studied. Indeed, it was found that all recorded UV–Vis spectra in the MeCN solution have a strong and distinctive absorption band in the 326-509 nm region accompanied by one or more weaker and non-informative bands around 260-300 nm (Figure 5, Table 3). Notable exceptions are compounds 4a,i with only one dominant absorption maximum and compound 4e with a stronger shortwave band, which can be attributed to the N-oxide moiety.
Figure 5.

UV–Vis spectra of selected synthesized compounds in MeCN (2 × 10−5 M).
Table 3.
Photophysical properties of some synthesized compounds.
| Compound | λmax nm | ε1 dm3 mol−1 cm−1 | λmax nm | ε2 dm3 mol−1 cm−1 |
|---|---|---|---|---|
| 4a | - | - | 368 | 46,400 |
| 4b | 302 | 13,500 | 509 | 42,800 |
| 4c | 263 | 16,200 | 397 | 34,900 |
| 4e | 267 | 28,700 | 332 | 24,700 |
| 4i | - | - | 326 | 39,000 |
| 4k | 266 | 14,600 | 429 | 32,800 |
| 5g | 296 | 16,000 | 384 | 20,300 |
| 5m | 305 | 13,600 | 476 | 31,400 |
| 5n | 266 | 18,100 | 403 | 25,900 |
| 5p | 302 | 25,200 | 344 | 19,000 |
| 6g | 267 | 15,000 | 337 | 31,400 |
In the case of dinitro compounds 4a–c, the electron-releasing Me2N group in the phenyl ring leads to a considerable red shift of the absorption maximum (by 141 nm) with respect to the electron-withdrawing chlorine atom (compounds 4a and 4b, Figure 6). Compound 4b, with a strong electron-releasing 4-dimethylaminophenyl group, absorbs light in the visible region, whereas compounds 4a and 4c have their absorption maxima at the border between the visible and UV regions. This can be explained by the difference in the degree of charge transfer along the conjugation chain between the strongly withdrawing nitropyridine ring and the second electron-donating ring through the double bond.
Figure 6.

UV–Vis spectra of compounds 4a–c in MeCN (2 × 10−5 M).
On the other hand, the replacement of 5-NO2 with the CF3 group in a pyridine cycle caused a 42 nm blue shift for compounds 4a/4i and an 80 nm shift for 4-dimethylaminophenyl derivatives 4b/4k, as well as a small decrease in molar absorptivity (Figure 7). It can be concluded that substituents at the double bond as well as in position 5 can be independently altered to predictably fine-tune absorption spectra of these compounds.
Figure 7.

UV–Vis spectra of compounds 4a,b,i,k in MeCN (2 × 10−5 M).
The study of isomeric substitution products of 3- and 5-NO2 in compound 4a revealed an important dependence, as shown in Figure 8. Substitution of the nitro group at position 5 gave compound 6g, whose absorption spectrum generally resembles that of the parent compound and follows the same pattern described above for the 5-NO2/5-CF3 pair. On the other hand, substitution of the nitro group at position 3 gave compound 5g, which is qualitatively different from both compounds. In this case, the absorption maximum shifts slightly towards the visible region, and the absorption spectrum itself acquires a more complex structure. From this we can conclude that the combination of 2-alkenyl and 3-alkylthio substituents leads to the appearance of a characteristic electronic structure. A similar pattern was observed for the substitution of 3-NO2 in compound 4c, but not for compound 4b, which can be explained by the predominance of a strong charge transfer over the finer electronic structure. It should be noted that the alkylthio substituent does not significantly affect the photophysical properties of the obtained compounds.
Figure 8.

UV–Vis spectra of compounds 4a, 5g, 6g in MeCN (2 × 10−5 M).
Compounds 5g,n showed fluorescence upon excitation by light with the wavelength equal to the λmax in the visible region, as shown in Figure 9 and Figure 10. Compound 5g has an emission maximum at 538 nm and a Stokes shift of 154 nm, whereas for compound 5n, these values are 571 nm and 168 nm, respectively. The large values of Stokes shifts (150–170 nm) almost completely eliminate the overlap between the absorption and emission regions. In addition, the properties of these fluorescent molecules can be tuned by changing the substituent at the double bond.
Figure 9.

Normalized absorption and emission spectra of compounds 5g and 5n in MeCN.
Figure 10.

Fluorescence of compounds 5g and 5n in solid state and in MeCN solution under 365 nm UV lamp.
3. Conclusions
In conclusion, a number of the previously unknown 2-methyl- and 2-arylvinyl-3-nitropyridines were synthesized. Their reactions with S-nucleophiles proceeded under mild conditions and were found to be regioselective with a predominance of 3-NO2 substitution; the influence of the substituents in positions 2 and 5 on the observed selectivity was revealed. The synthesized compounds showed promising tunable photophysical properties, such as large Stokes shifts. The reported synthetic approach can be considered as a convenient tool for rapid access to pyridine-based building blocks of various potential applications.
4. Materials and Methods
4.1. General Information
All chemicals were of commercial grade and used directly without purification. Melting points were measured on a Stuart SMP20 apparatus (Stuart (Bibby Scientific), UK). 1H and 13C NMR spectra were recorded on a Bruker AM-300 (at 300.13 and 75.13 MHz, respectively, Bruker Biospin, Germany) or Bruker Avance DRX 500 (at 500 and 125 MHz, respectively, Bruker Biospin, Germany) in DMSO-d6 or CDCl3. J values are given in Hz. HRMS spectra were recorded on a Bruker micrOTOF II mass spectrometer using ESI. UV–Vis absorption spectra were recorded in MeCN (2 × 10−5 M) in standard 10 × 10 × 45 mm quartz cuvettes on a Cary 60 UV–Vis spectrophotometer (Agilent Technologies, Santa Clara, CA, USA). Fluorescence spectra were recorded in MeCN (2 × 10−6 M) in standard 10 × 10 × 45 mm quartz cuvettes on a Cary Eclipse fluorescence spectrophotometer (Agilent Technologies). All reactions were monitored by TLC analysis using ALUGRAM SIL G/UV254 plates, which were visualized with UV light. Compounds 1a–c were purchased from commercial suppliers. In some cases we were unable to record the 13C NMR spectra of products due to insufficient solubility in common organic solvents (compounds 4d, 5,i,j,o and 6j).
4.2. General Procedure for the Synthesis of 2-Methyl-3-nitropyridines 2a–c
To a stirred suspension of NaH (60% in mineral oil, 0.80 g, 20 mmol) in anhydrous THF (30 mL), diethyl malonate (1.52 mL, 10 mmol) was added dropwise. The suspension was stirred for 15 min until hydrogen evolution ceased and a solution of the corresponding 2-chloropyridine 1 (10 mmol) in THF (20 mL) was added. The reaction mixture was stirred at r.t. (room temperature) for 6 h, poured in water (200 mL) and acidified with conc. HCl to pH 3. This was then extracted with CHCl3, evaporated and 50% H2SO4 (30 mL) was added to the residue. The mixture was stirred for 6 h at 120 °C, cooled, neutralized with Na2CO3 to pH 8 and extracted with CHCl3. The organic phase was dried over Na2SO4, evaporated and the residue was purified via column chromatography (SiO2/CHCl3).
2-Methyl-3,5-dinitropyridin (2a) [19], brown oil; yield 62%; 1H NMR (300 MHz, CDCl3): δ 9.55 (d, 1H, J = 2.1 Hz), 9.06 (d, 1H, J = 2.1 Hz), 3.03 (s, 3H).
5-Bromo-2-methyl-3-nitropyridin (2b) [20], yellowish oil; yield 90%; 1H NMR (300 MHz, CDCl3): δ 8.80 (d, 1H, J = 2.1 Hz), 8.44 (d, 1H, J = 2.1 Hz), 2.84 (s, 3H).
2-Methyl-3-nitro-5-trifluoromethylpyridine (2c), yellowish oil; yield 81%; 1H NMR (300 MHz, CDCl3): δ 9.00 (s, 1H), 8.54 (s, 1H), 2.97 (s, 3H).
4.3. General Procedure for the Oxidation of 2-Methyl-3-nitropyridines 2b,c
To a solution of the corresponding 2-methyl-3-nitropyridine 2 (10 mmol) in CH2Cl2 (30 mL), a freshly prepared complex urea/H2O2 (1.88 g, 20 mmol) was added. The resulting suspension was cooled to 0 °C and trifluoroacetic anhydride (5 mL, 36 mol) was added dropwise. The reaction mixture was stirred for 30 min at 0 °C and 4 h at r.t. A saturated aqueous solution of Na2S2O3 (50 mL) was added and the organic phase was separated. An aqueous layer was additionally extracted with CH2Cl2 and combined organic solutions were washed with the saturated solution of NaHCO3, dried over Na2SO4 and evaporated. The residue was recrystallized from aqueous EtOH.
5-Bromo-2-methyl-3-nitropyridine N-oxide (3b), pale-yellow solid; yield 47%; mp 104–105 °C; 1H NMR (300 MHz, CDCl3): δ 8.60 (s, 1H), 7.86 (s, 1H), 2.67 (s, 3H, CH3). 13C NMR (125 MHz, CDCl3): δ 148.5, 145.5, 143.5, 122.9, 116.4, 13.8. HRMS (ESI, m/z): calcd for C6H5BrN2O3 [M + H]+: 232.9556; found: 232.9563.
2-Methyl-3-nitro-5-trifluoromethylpyridine N-oxide (3c), pale-yellow solid; yield 65%; mp 82–83 °C; 1H NMR (300 MHz, CDCl3): δ 8.69 (s, 1H), 7.89 (s, 1H), 2.75 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 149.9, 148.8, 139.5, 127.1 (q, 2JCF = 36 Hz), 121.1 (q, 1JCF = 274 Hz), 116.1, 14.3. HRMS (ESI, m/z): calcd for C7H5F3N2O3 [M + H]+: 223.0325; found: 223.0331.
4.4. General Procedure for the Synthesis of Compounds 4a–g
To a solution of the corresponding 2-methylpyridine 2 or 3 (5 mmol) in toluene (30 mL), aromatic aldehyde (5 mmol) and 50 μL of piperidine was added. The reaction mixture was stirred under reflux with a Dean–Stark adapter until water separation was completed. The solvent was evaporated and the residue was triturated with 20 mL of cold EtOH. The precipitate was filtered off and air-dried.
(E)-2-[(4-Chlorophenyl)vinyl]-3,5-dinitropyridine (4a), yellow solid; yield 78%; mp 204–205 °C; 1H NMR (300 MHz, CDCl3): δ 9.58 (s, 1H), 9.05 (s, 1H), 8.30 (d, 1H, J = 15.3 Hz), 7.83 (d, 1H, J = 15.3 Hz), 7.64 (d, 2H, J = 8.4 Hz), 7.45 (d, 2H, J = 8.4 Hz). 13C NMR (150 MHz, DMSO-d6): δ 152.0, 147.5, 143.2, 141.9, 140.3, 135.0, 134.0, 130.2, 129.2, 129.1, 121.6. HRMS (ESI, m/z): calcd for C13H8ClN3O4 [M + H]+: 306.0276; found: 306.0283.
(E)-4-(2-(3,5-Dinitropyridin-2-yl)vinyl)-N,N-dimethylaniline (4b), dark-violet solid; yield 91%; mp 260 °C (dec.); 1H NMR (300 MHz, CDCl3): δ 9.41 (d, 1H, J = 2.1 Hz), 8.93 (d, 1H, J = 2.1 Hz), 8.28 (d, 1H, J = 15.0 Hz), 7.64 (d, 1H, J = 15.0 Hz), 7.57 (d, 2H, J = 8.8 Hz), 6.77 (d, 2H, J = 8.8 Hz), 3.06 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 154.8, 152.4, 147.3, 146.4, 139.5, 131.1, 128.9, 123.2, 114.4, 111.9, 40.1. HRMS (ESI, m/z): calcd for C15H14N4O4 [M + H]+: 315.1088; found: 315.1085.
(E)-5-Bromo-2-(4-dimethylaminophenyl)vinyl-3-nitropyridine N-oxide (4c), red-brown solid; yield 73%; mp 217 °C (dec.); 1H NMR (300 MHz, CDCl3): δ 8.55 (d, 1H, J = 15.9 Hz), 8.49 (s, 1H), 7.64 (s, 1H), 7.49 (d, 2H, J = 8.7 Hz), 7.00 (d, 1H, J = 15.6 Hz), 6.69 (d, 2H, J = 8.7 Hz), 3.05 (s, 6H). 13C NMR (75 MHz, CDCl3): δ 151.8, 147.2, 143.9, 142.8, 141.2, 129.9, 123.8, 123.0, 113.0, 111.9, 107.1, 40.1. HRMS (ESI, m/z): calcd for C15H14BrNO3 [M + H]: 364.0291; found: 364.0292.
(E)-2-(2-(1-(4-Fluorophenyl)-1H-pyrazol-4-yl)vinyl)-3,5-dinitropyridine (4d), brown solid; yield 62%; mp 255 °C; 1H NMR (300 MHz, CDCl3): δ 9.53 (d, 1H, J = 2.1 Hz), 9.03 (d, 1H, J = 2.1 Hz), 8.30 (d,1H, J = 15.3 Hz), 8.19 (s, 1H), 8.06 (s, 1H), 7.70 (m, 3H), 7.20 (m, 2H). HRMS (ESI, m/z): calcd for C16H10FN5O4 [M + H]+: 356.0790; found: 356.0789.
(E)-2-(4-Chlorophenyl)vinyl-3-nitro-5-trifluoromethylpyridine N-oxide (4e), yellow solid; yield 81%; mp 161–162 °C; 1H NMR (300 MHz, CDCl3): δ 8.65 (d,1H, J = 15.9 Hz), 8.64 (s, 1H), 7.73 (s, 1H), 7.52 (d, 2H, J = 8.4 Hz), 7.39 (d, 2H, J = 8.4 Hz), 7.17 (d, 1H, J = 15.9 Hz). 13C NMR (75 MHz, CDCl3): δ 147.9, 143.6, 142.3, 140.4, 136.7, 134.1, 129.4, 129.3, 125.5 (q, 2JCF = 36 Hz), 121.1 (q, 1JCF = 273 Hz), 116.0 (d, 3JCF = 3.2 Hz), 112.8. HRMS (ESI, m/z): calcd for C14H8ClF3N2O3 [M + H]+: 345.0248; found: 345.0239.
(E)-5-Bromo-2-(4-chlorophenyl)vinyl-3-nitropyridine N-oxide (4f), yellow solid; yield 69%; mp 173–174 °C; 1H NMR (300 MHz, CDCl3): δ 8.45 (s, 1H), 8.30 (d, 1H, J = 16.1 Hz), 7.60 (s, 1H), 7.41 (d, 2H, J = 8.4 Hz), 7.29 (d, 2H, J = 8.4 Hz), 7.02 (d, 1H, J = 16.1 Hz). 13C NMR (125 MHz, CDCl3): δ 147.8, 144.1, 140.3, 140.2, 136.1, 134.4, 129.2, 129.1, 122.7, 115.7, 113.1. HRMS (ESI, m/z): calcd for C13H8BrClN2O3 [M + H]+: 354.9480; found: 354.9472.
(E)-2-(4-Dimethylaminophenyl)vinyl-3-nitro-5-trifluoromethylpyridine N-oxide (4g), red-brown solid; yield 71%; mp 198-199 °C; 1H NMR (300 MHz, CDCl3): δ 8.85 (d, 1H, J = 15.6 Hz), 8.58 (s, 1H), 7.68 (s, 1H), 7.52 (d, 2H, J = 8.4 Hz), 7.10 (d, 1H, J = 15.6 Hz), 6.69 (d, 2H, J = 8.4 Hz), 3.06 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 152.2, 146.8, 145.1, 144.4, 140.1, 130.5, 123.4, 122.7 (q, 2JCF = 36 Hz), 121.4 (q, 1JCF = 273 Hz), 116.3 (q, 3JCF = 3.6 Hz), 111.9, 106.6, 40.0. HRMS (ESI, m/z): calcd for C16H14F3N3O3 [M + H]+: 354.1060; found: 354.1060.
4.5. General Procedure for the Synthesis of Compounds 4h–k
To a solution of the corresponding pyridine N-oxide (0.5 mmol), CH2Cl2 (10 mL) was added PCl3 (0.13 mL) and the mixture was stirred under reflux for 2 h. After cooling, the reaction was washed with a saturated NaHCO3 solution, dried over Na2SO4 and evaporated.
(E)-4-(2-(5-Bromo-3-nitropyridin-2-yl)vinyl)-N,N-dimethylaniline (4h), dark-brown solid; yield 89%; mp 202–203 °C; 1H NMR (300 MHz, CDCl3): δ 8.75 (s, 1H), 8.34 (s, 1H), 8.05 (d, 1H, J = 15.3 Hz), 7.55 (d, 1H, J = 15.6), 7.53 (d, 2H, J = 8.4), 6.71 (d, 2H, J = 8.4 Hz), 3.04 (s, 6H). 13C NMR (75 MHz, CDCl3): δ 153.8, 151.3, 148.8, 145.5, 142.8, 140.5, 135.0, 129.8, 115.5, 115.5, 112.3, 40.4, 29.7. HRMS (ESI, m/z): calcd for C15H14BrN3O2 [M + H]+: 348.0342; found: 348.0342.
(E)-2-(4-Chlorophenyl)vinyl-3-nitro-5-trifluoromethylpyridine (4i), yellow solid; yield 89%; mp 138 °C; 1H NMR (300 MHz, CDCl3): δ 9.02 (s, 1H), 8.50 (s, 1H), 8.16 (d, 1H, J = 15.6 Hz), 8.77 (d, 1H, J = 15.6 Hz), 7.59 (d, 2H, J = 8.4 Hz), 7.41 (d, 2H, J = 8.4 Hz). 13C NMR (75 MHz, CDCl3): δ 152.0, 149.2 (q, 3JCF = 3.6 Hz), 143.0, 140.8, 136.1, 133.9, 130.7 (q, 3JCF = 3.6 Hz), 129.4, 129.3, 124.9 (q, 2JCF = 35 Hz), 120.6. HRMS (ESI, m/z): calcd for C14H8ClF3N2O2 [M + H]+: 329.0299; found: 329.0311.
(E)-5-Bromo-2-(4-chlorostyryl)-3-nitropyridine (4j), yellow solid; yield 94%; mp 184–185 °C; 1H NMR (300 MHz, CDCl3): δ 8.83 (d, 1H, J = 1.8 Hz), 8.40 (d, 1H, J = 1.8 Hz), 8.02 (d, 1H, J = 15.6 Hz), 7.68 (d, 1H, J = 15.6 Hz), 7.56 (d, 2H, J = 8.4 Hz), 7.38 (d, 2H, J = 8.4 Hz). 13C NMR (75 MHz, CDCl3): δ 154.0, 147.4, 143.7, 138.4, 135.5, 135.1, 134.2, 129.2, 129.1, 124.9, 117.6. HRMS (ESI, m/z): calcd for C13H8BrClN2O2 [M + H]+: 338.9530; found: 338.9533.
(E)-N,N-Dimethyl-4-(2-(3-nitro-5-trifluoromethylpyridin-2-yl)vinyl)aniline (4k), brown solid; yield 82%; mp 150–151 °C; 1H NMR (300 MHz, CDCl3): δ 8.40 (s, 1H), 8.36 (s, 1H), 8.12 (d, 1H, J = 15.3 Hz), 7.56 (d, 1H, J = 15.3 Hz), 7.49 (d, 2H, J = 8.7 Hz), 6.66 (d, 2H, J = 8.7 Hz), 2.99 (s, 6H). 13C NMR (75 MHz, CDCl3): δ 153.4, 151.9, 149.1 (q, 3JCF = 4 Hz), 143.3, 141.8, 130.8 (q, 3JCF = 4 Hz), 130.3, 122.8 (q, 1JCF = 271 Hz), 123.6, 122.9 (q, 2JCF = 34 Hz), 115.0, 112.1, 40.2. HRMS (ESI, m/z): calcd for C16H14F3N3O2 [M + H]+: 338.1111; found: 338.1120.
4.6. General Procedure for the Synthesis of Compounds 5 and 6
To a solution of compound 2, 3 or 4 (1 mmol) in anhydrous DMF (5 mL), thiol (1 mmol) and K2CO3 (0.138 g, 1 mmol) were added. The reaction mixture was stirred for 1-2 h at 60 °C, poured into H2O (50 mL), acidified with conc. HCl to pH 3 and extracted with CHCl3 (3 × 20 mL). The organic phase was dried over Na2SO4, evaporated and the residue was purified via column chromatography (SiO2/CHCl3) or recrystallized from EtOH.
3-(Benzylsulfanyl)-2-methyl-5-nitropyridine (5a), beige solid; yield 70%; mp 79–80 °C; 1H NMR (300 MHz, CDCl3): δ 9.06 (d, 1H, J = 2.1 Hz), 7.35 (m, 5H), 4.24 (s, 2H, CH2), 2.65 (s, 3H, CH3). 13C NMR (125 MHz, CDCl3): δ 162.7, 142.8, 140.1, 134.9, 134.8, 128.9, 128.8, 128.0, 127.8, 37.1, 23.4. HRMS (ESI, m/z): calcd for C13H12N2O2S [M + H]+: 261.0692; found: 261.0699.
3-(Benzylsulfanyl)-5-bromo-2-methylpyridine N-oxide (5b), grey needles; yield 96%; mp 106–107 °C; 1H NMR (300 MHz, CDCl3): δ 8.25 (s, 1H), 7.26-7.32 (m, 5H), 7.24 (s, 1H), 4.14 (s, 2H, SCH2), 2.49 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3): δ 147.3, 137.8, 136.8, 134.9, 128.9, 128.1, 127.7, 116.3, 38.5, 14.5. HRMS (ESI, m/z): calcd for C13H12BrNOS [M + H]+: 309.9896; found: 309.9896.
5-Bromo-3-(4-chlorophenylsulfanyl)-2-methylpyridine N-oxide (5c), brown solid; yield 95%; mp 131–132 °C; 1H NMR (300 MHz, CDCl3): δ 8.27 (s, 1H), 7.44 (d, 2H, J = 8.4 Hz), 7.38 (d, 2H, J = 8.4 Hz), 6.89 (s, 1H), 2.56 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 146.7, 138.1, 137.3, 136.0, 134.8, 130.4, 129.0, 127.7, 116.6, 14.6. HRMS (ESI, m/z): calcd for C12H9BrClNOS [M + H]+: 329.9350; found: 329.9349.
3-(Benzylsulfanyl)-5-bromo-2-methylpyridine (5d), pale-yellow crystals; yield 65%; mp 55 °C; 1H NMR (300 MHz, CDCl3): δ 8.33 (s, 1H), 7.58 (s, 1H), 7.31 (m, 5H), 4.09 (s, 2H, SCH2), 2.48 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3): δ 155.5, 146.5, 137.3, 135.6, 134.4, 128.9, 128.8, 127.8, 117.9, 37.8, 22.5. HRMS (ESI, m/z): calcd for C13H12BrNS [M + H]+: 293.9947; found: 293.9956.
3-(Benzylsulfanyl)-2-methyl-5-(trifluoromethyl)pyridine (5e), yellow oil; yield 60%; 1H NMR (300 MHz, CDCl3): δ 8.53 (s, 1H), 7.64 (s, 1H), 7.31 (s, 5H), 4.14 (s, 2H, SCH2), 2.61 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3): δ 161.0, 142.3 (q, 3JCF = 4 Hz), 135.5, 133.4, 131.7 (q, 3JCF = 3.5 Hz), 128.9, 128.8, 127.8, 124.7 (q, 2JCF = 34 Hz), 123.5 (q, 1JCF = 273 Hz), 37.5, 23.2. HRMS (ESI, m/z): calcd for C14H12F3NS [M + H]+: 284.0715; found: 284.0719.
3-(Benzylsulfanyl)-2-methyl-5-(trifluoromethyl)pyridine N-oxide (5f), pale-yellow crystals; yield 52%; mp 85–86 °C; 1H NMR (300 MHz, CDCl3): δ 8.33 (s, 1H), 7.29 (m, 5H), 7.22 (s, 1H), 4.15 (s, 2H, SCH2), 2.57 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3): δ 151.8, 137.3, 134.7, 134.1, 128.9, 128.9, 128.1, 126.6 (q, 2JCF = 35 Hz), 121.9 (q, 1JCF = 273 Hz), 120.1, 38.4, 15.0. HRMS (ESI, m/z): calcd for C14H12F3NOS [M + H]+: 300.0664; found: 300.0662.
(E)-3-(Benzylsulfanyl)-2-((4-chlorophenyl)vinyl)-5-nitropyridine (5g), yellow crystals; yield 56%; mp 146–147 °C; 1H NMR (300 MHz, CDCl3): δ 9.17 (d, 1H, J = 2.1 Hz), 8.28 (d, 1H, J = 2.1 Hz), 7.92 (d, 1H, J = 15.6 Hz), 7.57 (d, 1H, J = 15.6 Hz), 7.53 (d, 2H, J = 8.7 Hz), 7.39 (d, 2H, J = 8.7 Hz), 7.30 (m, 5H), 4.18 (s, 2H, SCH2). 13C NMR (75 MHz, CDCl3): δ 159.0, 142.2, 142.1, 138.1, 135.5, 135.2, 134.5, 132.4, 132.3, 129.2, 129.2, 129.0, 128.9, 128.1, 122.8, 38.7. HRMS (ESI, m/z): calcd for C20H15ClN2O2S [M + H]+: 383.0616; found: 383.0603.
(E)-5-(Benzylsulfanyl)-2-((4-chlorophenyl)vinyl)-3-nitropyridine (6g), yellow crystals; yield 18%; mp 183–184 °C; 1H NMR (300 MHz, CDCl3): δ 8.63 (d, 1H, J = 1.8 Hz), 8.06 (d, 1H, J = 2.1 Hz), 7.97 (d, 1H, J = 15.6 Hz), 7.67 (d, 1H, J = 15.6 Hz), 7.56 (d, 2H, J = 8.7 Hz), 7.39 (d, 2H, J = 8.7 Hz), 7.33 (m, 5H), 4.22 (s, 2H, SCH2). HRMS (ESI, m/z): calcd for C20H15ClN2O2S [M + H]+: 383.0616; found: 383.0621.
2-[(E)-(4-Chlorophenyl)vinyl]-3-(isobutylsulfanyl)-5-nitropyridine (5h), yellow crystals; yield 62%; mp 139–140 °C; 1H NMR (300 MHz, CDCl3): δ 9.16 (d, 1H, J = 2.4 Hz), 8.31 (d, 1H, J = 2.1 Hz), 7.96 (d, 1H, J = 15.3 Hz), 7.63 (d, 1H, J = 15.3 Hz), 7.60 (d, 2H, J = 8.4 Hz), 7.41 (d, 2H, J = 8.4 Hz), 2.92 (d, 2H, J = 6.9 Hz), 1.99 (m, 1H), 1.14 (d, 6H, J = 6.6 Hz). 13C NMR (75 MHz, CDCl3): δ 158.0, 147.0, 142.4, 141.0, 137.9, 135.4, 134.5, 134.3, 129.9, 122.8, 42.3, 28.1, 22.2. HRMS (ESI, m/z): calcd for C17H17ClN2O2S [M + H]+: 349.0772; found: 349.0783.
2-[(E)-(4-Chlorophenyl)vinyl]-5-(isobutylsulfanyl)-3-nitropyridine (6h), yellow crystals; yield 31%; mp 106 °C; 1H NMR (300 MHz, CDCl3): δ 8.68 (d, 1H, J = 1.8 Hz), 8.09 (d, 1H, J = 1.8 Hz), 7.97 (d, 1H, J = 15.6 Hz), 7.68 (d, 1H, J = 15.6 Hz), 7.56 (d, 2H, J = 8.4 Hz), 7.38 (d, 2H, J = 8.4 Hz), 2.92 (d, 2H, J = 6.9 Hz), 1.96 (m, 1H), 1.10 (d, 6H, J = 6.6 Hz). 13C NMR (75 MHz, CDCl3): δ 152.0, 145.4, 143.9, 136.8, 135.1, 134.9, 134.6, 130.9, 129.1, 129.0, 121.5, 42.1, 28.3, 22.0. HRMS (ESI, m/z): calcd for C17H17ClN2O2S [M + H]+: 349.0772; found: 349.0764.
2-[(E)-2-(4-Chlorophenyl)vinyl]-3-[(2-furylmethyl)sulfanyl]-5-nitropyridine (5i), yellow crystals; yield 56%; mp 156 °C; 1H NMR (300 MHz, CDCl3): δ 9.24 (d, 1H, J = 1.8 Hz), 8.42 (d, 1H, J = 2.1 Hz), 7.95 (d, 1H, J = 15.6 Hz), 7.65 (d, 1H, J = 15.6 Hz), 7.58 (d, 2H, J = 8.4 Hz), 7.40 (d, 2H, J = 8.4 Hz), 7.33 (s, 1H), 6.26 (s, 1H), 6.14 (d, 1H, J = 3.0 Hz), 4.18 (s, 2H). HRMS (ESI, m/z): calcd for C18H13ClN2O2S [M + H]+: 373.0408; found: 373.0419.
3-[(4-Chlorophenyl)sulfanyl]-2-[(E)-2-(4-chlorophenyl)vinyl]-5-nitropyridine (5j), yellow solid; yield 67%; mp 172 °C; 1H NMR (300 MHz, DMSO-d6): δ 9.26 (s, 1H), 8.22 (s, 1H), 7.98 (d, 1H, J = 15.6 Hz), 7.74 (d, 2H, J = 8.4 Hz), 7.66 (d, 1H, J = 15.6 Hz), 7.52 (br.s, 4H), 7.49 (d, 2H, J = 8.4 Hz). HRMS (ESI, m/z): calcd for C19H12Cl2N2O2S [M + H]+: 403.0069; found: 403.0057.
(E)-4-(2-(3-((4-Chlorophenyl)sulfanyl)-5-nitropyridin-2-yl)vinyl)-N,N-dimethylaniline (5k), dark-red solid; yield 83%; mp 175 °C; 1H NMR (300 MHz, DMSO-d6): δ 9.21 (s, 1H), 8.20 (s, 1H), 7.96 (d, 1H, J = 15.6 Hz), 7.50 (m, 6H), 7.36 (d, 1H, J = 15.6 Hz), 6.74 (d, 2H, J = 8.4 Hz), 2.99 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 160.1, 151.6, 143.1, 141.3, 141.2, 134.9, 133.4, 133.2, 130.9, 130.1, 129.9, 123.7, 117.0, 111.9, 40.1. HRMS (ESI, m/z): calcd for C21H18ClN3O2S [M + H]+: 412.0881; found: 412.0870.
(E)-4-(2-(3-(Isobutylsulfanyl)-5-nitropyridin-2-yl)vinyl)-N,N-dimethylaniline (5l), dark-red solid; yield 84%; mp 165–166 °C; 1H NMR (300 MHz, CDCl3): δ 9.14 (d, 1H, J = 2.4 Hz), 8.27 (d, 1H, J = 2.1 Hz), 8.01 (d, 1H, J = 15.3 Hz), 7.58 (d, 2H, J = 8.7 Hz), 7.46 (d, 1H, J = 15.3 Hz), 6.74 (d, 2H, J = 8.7 Hz), 3.06 (s, 6H), 2.89 (d, 2H, J = 6.6 Hz), 1.96 (m, 1H), 1.13 (d, 6H, J = 6.6 Hz). 13C NMR (75 MHz, CDCl3): δ 159.5, 151.5, 141.4, 141.3, 140.2, 132.5, 130.0, 129.8, 124.1, 117.4, 112.0, 43.0, 40.2, 28.1, 22.2. HRMS (ESI, m/z): calcd for C19H23N3O2S [M + H]+: 358.1584; found: 358.1583.
(E)-4-(2-(3-(Benzylsulfanyl)-5-nitropyridin-2-yl)vinyl)-N,N-dimethylaniline (5m), dark-red solid; yield 88%; mp 188 °C; 1H NMR (300 MHz, CDCl3): δ 9.13 (d, 1H, J = 2.1 Hz), 8.20 (d, 1H, J = 1.8 Hz), 7.98 (d, 1H, J = 15.3 Hz), 7.54 (d, 2H, J = 8.7 Hz), 7.43 (d, 1H, J = 15.3 Hz), 7.30 (m, 5H), 6.70 (d, 2H, J = 8.7 Hz), 4.16 (s, 2H), 3.05 (s, 6H). 13C NMR (75 MHz, CDCl3): δ 160.4, 151.5, 142.3, 141.0, 140.5, 135.5, 132.2, 130.6, 129.9, 129.0, 128.8, 127.9, 124.0, 117.4, 112.0, 40.2, 38.5. HRMS (ESI, m/z): calcd for C22H21N3O2S [M + H]+: 392.1427; found: 392.1417.
(E)-3-(Benzylsulfanyl)-2-(2-(1-(4-fluorophenyl)-1H-pyrazol-4-yl)vinyl)-5-nitropyridine (5n), orange solid; yield 89%; mp 145–146 °C; 1H NMR (300 MHz, CDCl3): δ 9.15 (d, 1H, J = 2.1 Hz), 8.26 (d, 1H, J = 2.1 Hz), 8.05 (s, 1H), 7.98 (s, 1H), 7.92 (d, 1H, J = 15.3 Hz), 7.67 (m, 2H), 7.40 (d, 1H, J = 15.3 Hz), 7.30 (m, 5H), 7.18 (m, 2H), 4.18 (s, 2H). 13C NMR (75 MHz, CDCl3): δ 161.4 (d, 1JCF = 247 Hz), 159.3, 142.1, 141.8, 140.3, 136.0 (d, 4JCF = 3 Hz), 135.3, 132.2, 131.5, 129.3, 129.0, 128.9, 128.0, 126.4, 121.9, 121.4, 121.1 (d, 3JCF = 8.5 Hz), 116.5 (d, 2JCF = 23 Hz), 38.6. HRMS (ESI, m/z): calcd for C23H17FN4O2S [M + H]+: 433.1129; found: 433.1123.
(E)-3-((4-Chlorophenyl)sulfanyl)-2-(2-(1-(4-fluorophenyl)-1H-pyrazol-4-yl)vinyl)-5-nitropyridine (5o), orange solid; yield 93%; mp 219 °C; 1H NMR (300 MHz, DMSO-d6): δ 9.24 (s, 1H), 8.99 (s, 1H), 8.22 (s, 1H), 8.18 (s, 1H), 7.80 (m, 4H), 7.55 (s, 4H), 7.40 (m, 3H). HRMS (ESI, m/z): calcd for C22H14ClFN4O2S [M + H]+: 453.0583; found: 453.0573.
(E)-3-(Benzylsulfanyl)-5-bromo-2-[(4-chlorophenyl)vinyl]pyridine (5p), beige solid, yield 60%; mp 142–143 °C; 1H NMR (300 MHz, CDCl3): δ 8.48 (s, 1H), 7.69–7.73 (m, 2H), 7.48–7.56 (m, 3H), 7.25-7.36 (m, 7H), 4.08 (s, 2H). 13C NMR (125 MHz, CDCl3): δ 153.2, 148.4, 141.1, 135.9, 135.1, 134.3, 133.6, 132.5, 128.9, 128.8, 128.7, 128.6, 127.7, 123.7, 118.3, 39.4. HRMS (ESI, m/z): calcd for C20H15BrClNS [M + H]+: 415.9869; found: 415.9870.
(E)-3-(Benylsulfanyl)-5-bromo-2-[(4-(dimethylamino)phenyl)vinyl]pyridine N-oxide (5q), dark-red solid; yield 67%; mp 177 °C; 1H NMR (300 MHz, CDCl3): δ 8.49 (d, 1H, J = 15.9 Hz), 8.23 (s, 1H), 7.51 (d, 2H, J = 8.7 Hz), 7.28 (m, 7H), 6.71 (d, 2H, J = 8.7 Hz), 4.14 (s, 2H), 3.03 (s, 6H, NCH3). 13C NMR (75 MHz, CDCl3): δ 151.2, 144.9, 140.5, 139.0, 135.9, 135.0, 129.2, 129.1, 128.8, 128.4, 127.9, 127.8, 124.8, 114.1, 112.1, 40.3, 39.0. HRMS (ESI, m/z): calcd for C22H21BrN2OS [M + H]+: 441.0631; found: 441.0635.
4.7. X-ray Crystallographic Data and Refinement Details
X-ray diffraction data for 5h were collected at 100 K on a Bruker Quest D8 diffractometer (Karlsruhe, Germany) equipped with a Photon III area detector, using graphite-monochromatized Mo Kα-radiation and a shutterless φ- and ω-scan technique. The intensity data were integrated by the SAINT program (version 8.04A) [21] and were semi-empirically corrected for absorption and decay using SADABS (2016/2) [22]. X-ray diffraction data for 4a, 4i and 5l were collected at 100 K on a four-circle Rigaku Synergy-S diffractometer (Wroclaw, Poland) equipped with a HyPix6000HE area detector, using monochromatized Cu Kα-radiation and a shutterless ω-scan technique. The intensity data were integrated and corrected for absorption and decay by the CrysAlisPro program [23]. All structures were solved by direct methods using SHELXT [24] and refined by the full-matrix least-squares method on F2 using SHELXL-2018 [25]. Positions of all atoms were found from the electron density difference map. Atoms were refined with individual anisotropic (non-hydrogen atoms) or isotropic (hydrogen atoms) displacement parameters. The Mercury program [26] was used for molecular graphics. Crystal data, data collection and structure refinement details are summarized in Supplementary Materials Table S1. The structures were deposited at the Cambridge Crystallographic Data Center with the reference CCDC numbers 2191010–2191013; they also contain the supplementary crystallographic data. These data can be obtained free of charge from the CCDC via http://www.ccdc.cam.ac.uk/data_request/cif, accessed on 30 August 2022.
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/molecules27175692/s1, X-ray data, 1H and 13C NMR spectra. References [19,20,21,22,23,24,25,26] are cited in the supplementary materials.
Author Contributions
Conceptualization, V.V.N. and A.M.S.; methodology, V.V.N. and M.A.B.; investigation, V.V.N. and M.E.M.; resources, M.E.M.; writing—original draft preparation, V.V.N. and M.A.B.; writing—review and editing, A.M.S.; supervision, A.M.S. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
Funding Statement
This research received no external funding.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Khan E. Pyridine Derivatives as Biologically Active Precursors; Organics and Selected Coordination Complexes. ChemistrySelect. 2021;6:3041–3064. doi: 10.1002/slct.202100332. [DOI] [Google Scholar]
- 2.Yang S., Yan J., Wang L., Guo X., Li S., Wu G., Zuo R., Huang X., Wang H., Wang L., et al. Nitropyridinyl ethyleneimine compound, the pharmaceutical composition containing it, the preparation method and use thereof. 20140073796A1. U.S. Patent. 2015 January 27;
- 3.Jhons B.A., Spaltenstein A. HIV Integrase Inhibitors. WO 2007/09101 A2. U.S. Patent. 2007 January 18;
- 4.Romero D.L., Morge R.A., Biles C., Berrios-Pena N., May P.D., Palmer J.R., Johnson P.D., Smith H.W., Busso M. Discovery, Synthesis, and Bioactivity of Bis(heteroaryl)piperazines. 1. A Novel Class of Non-Nucleoside HIV-1 Reverse Transcriptase Inhibitors. J. Med. Chem. 1994;37:999–1014. doi: 10.1021/jm00033a018. [DOI] [PubMed] [Google Scholar]
- 5.Kuduk-Jaworska J., Puzsko A., Kubiak M., Pelczynska M. Synthesis, structural, phisico-chemical and biological properties of new palladium(II) complexes with 2,6-dimethyl-4-nitropyridine. J. Inorg. Biochem. 2004;98:1447–1456. doi: 10.1016/j.jinorgbio.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 6.Klapötke T.M., Stierstorfer J., Weyrauther M., Witkowski T.G. Synthesis and Investigation of 2,6-Bis(picrylamino)-3,5-dinitro-pyridine (PYX) and Its Salts. Chem.–A Eur. J. 2016;22:8619–8626. doi: 10.1002/chem.201600769. [DOI] [PubMed] [Google Scholar]
- 7.Agrawal J.P., Prasad U.S., Surve R.N. Synthesis of 1,3-bis(1,2,4-triazol-3-amino)-2,4,6-trinitrobenzene and its thermal and explosive behaviour. New J. Chem. 2000;24:583–585. doi: 10.1039/b003139i. [DOI] [Google Scholar]
- 8.Balachari D., Trudell M.L. Synthesis of new dipyridotetraazapentalenes. Tetrahedron Lett. 1997;38:8607–8610. doi: 10.1016/S0040-4039(97)10311-2. [DOI] [Google Scholar]
- 9.Balachari D., Stevens E.D., Trudell M.L., Beardall D., Wight C.A. Synthesis, Thermal Stability and Impact Stability of Novel Tetranitro-Dipyridotetraazapentalene Derivatives. Propell. Explos. Pyrotech. 2000;25:75–80. doi: 10.1002/(SICI)1521-4087(200004)25:2<75::AID-PREP75>3.0.CO;2-U. [DOI] [Google Scholar]
- 10.Kauer J.C., Carboni R.A. Aromatic azapentalenes. III. 1,3a,6,6a-Tetraazapentalenes. J. Am. Chem. Soc. 1967;89:2633–2637. doi: 10.1021/ja00987a022. [DOI] [Google Scholar]
- 11.Gryl M., Seidler T., Wojnarska J., Stadnicka K.M., Matulková I., Němec I., Němec P. Co-Crystals of 2-Amino-5-Nitropyridine Barbital with Extreme Birefringence and Large Second Harmonic Generation Effect. Chem.–A Eur. J. 2018;24:8727–8731. doi: 10.1002/chem.201802057. [DOI] [PubMed] [Google Scholar]
- 12.Bastrakov M.A., Nikol’Skiy V.V., Starosotnikov A.M., Fedyanin I.V., Shevelev S.A., Knyazev D.A. Reactions of 3-R-5-nitropyridines with nucleophiles: Nucleophilic substitution vs conjugate addition. Tetrahedron. 2019;75:130659. doi: 10.1016/j.tet.2019.130659. [DOI] [Google Scholar]
- 13.Frank R., Christoph T., Lesch B., Lee J. Substituted heterocyclic aza derivatives. 2013013817. WO Patent. 2013 January 31;
- 14.Van der Eycken E., Jidong Z., Kilonda A., Compernolle F., Toppet S., Hoornaert G., Van der Auweraer M., Jackers C., Verbouwe W., De Schryver F.C. Synthesis of (E)-5-(2-arylvinyl)-2-(hetero)arylpyridines, (E)-2-(2-arylvinyl)-5-methoxycarbonylpyridines and (E,E)-2,5-bis(2-arylvinyl)pyridines as polarity and pH probesElectronic supplementary information (ESI) J. Chem. Soc. Perkin Trans. 2002;2:928–937. doi: 10.1039/b200446c. [DOI] [Google Scholar]
- 15.Iyer S., Ramesh C. Aryl–Pd covalently bonded palladacycles, novel amino and oxime catalysts {di-μ-chlorobis(benzaldehydeoxime-6-C,N)dipalladium(II), di-μ-chlorobis(dimethylbenzylamine-6-C,N)dipalladium(II)} for the Heck reaction. Tetrahedron Lett. 2000;41:8981–8984. doi: 10.1016/S0040-4039(00)01594-X. [DOI] [Google Scholar]
- 16.Amatore C., Jutand A. Anionic Pd(0) and Pd(II) Intermediates in Palladium-Catalyzed Heck and Cross-Coupling Reactions. Accounts Chem. Res. 2000;33:314–321. doi: 10.1021/ar980063a. [DOI] [PubMed] [Google Scholar]
- 17.Lubisch W., Beckenbach E., Bopp S., Hofmann H.-P., Kartal A., Kästel C., Lindner T., Metz-Garrecht M., Reeb J., Regner F., et al. Benzoylalanine-Derived Ketoamides Carrying Vinylbenzyl Amino Residues: Discovery of Potent Water-Soluble Calpain Inhibitors with Oral Bioavailability. J. Med. Chem. 2003;46:2404–2412. doi: 10.1021/jm0210717. [DOI] [PubMed] [Google Scholar]
- 18.Lee D.-H., Taher A., Hossain S., Jin M.-J. An Efficient and General Method for the Heck and Buchwald–Hartwig Coupling Reactions of Aryl Chlorides. Org. Lett. 2011;13:5540–5543. doi: 10.1021/ol202177k. [DOI] [PubMed] [Google Scholar]
- 19.Liu M.-C., Lin T.-S., Sartorelli A.C. A One-Pot Synthesis of 3-Nitro-and 3,5-Dinitro-2-Picolines. Synth. Commun. 1990;20:2965–2970. doi: 10.1080/00397919008051513. [DOI] [Google Scholar]
- 20.Jones C.D., William R., Luke A., McCoull W. Aminopyrimidine Derivatives with TIE2 Inhibiting Activity. 20080194552 A1. U.S. Patent. 2008 August 14;
- 21.Bruker . APEX-III. Bruker AXS Inc.; Madison, WI, USA: 2019. [Google Scholar]
- 22.Krause L., Herbst-Irmer R., Sheldrick G.M., Stalke D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015;48:3–10. doi: 10.1107/S1600576714022985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.CrysAlisPro . Rigaku Oxford Diffraction. Rigaku Corporation; Oxford, UK: 2021. Version 1.171.41.106a. [Google Scholar]
- 24.Sheldrick G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015;71:3–8. doi: 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheldrick G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015;C71:3–8. doi: 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macrae C.F., Sovago I., Cottrell S.J., Galek P.T.A., McCabe P., Pidcock E., Platings M., Shields G.P., Stevens J.S., Towler M., et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020;53:226–235. doi: 10.1107/S1600576719014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.