


Abstract
Algorithms have been widely used to predict G-quadruplexes (G4s)-prone sequences. However, an experimental validation of these predictions is generally required. We previously reported a high-throughput technique to evidence G4 formation in vitro called FRET-MC. This method, while convenient and reproducible, has one known weakness: its inability to pin point G4 motifs of low thermal stability. As such quadruplexes may still be biologically relevant if formed at physiological temperature, we wanted to develop an independent assay to overcome this limitation. To this aim, we introduced an isothermal version of the competition assay, called iso-FRET, based on a duplex-quadruplex competition and a well-characterized bis-quinolinium G4 ligand, PhenDC3. G4-forming competitors act as decoys for PhenDC3, lowering its ability to stabilize the G4-forming motif reporter oligonucleotide conjugated to a fluorescence quencher (37Q). The decrease in available G4 ligand concentration restores the ability of 37Q to hybridize to its FAM-labeled short complementary C-rich strand (F22), leading to a decrease in fluorescence signal. In contrast, when no G4-forming competitor is present, PhenDC3 remains available to stabilize the 37Q quadruplex, preventing the formation of the F22 + 37Q complex. Iso-FRET was first applied to a reference panel of 70 sequences, and then used to investigate 23 different viral sequences.
INTRODUCTION
Different from the classical double-helix, G-quadruplexes (G4) constitute a family of specific DNA and RNA secondary structures. G4s result from the stacking of two or more G-quartets, i.e. planar layers of four guanines held together by Hoogsteen hydrogen bonding (1,2). G-quadruplexes have been studied from different perspectives and have been found in a variety of genomes, including pathogens (e.g. Nipah (3) and Ebola (4) viruses), bacteria and Archaea (5), as well as many eukaryotes, where G4 motifs are often found in promoters (6) and close to the origins of replication for mammals (7). G-quadruplexes play important roles in biological processes, including genome stability (8,9), regulation of gene expression (10–12), specific chromatin remodeling and replication (13,14), and RNA metabolism (1). How to find potential G4 motifs and then characterize their structures are basic questions concerning G-rich sequences in genomes. In 2016, we introduced a novel prediction method, G4Hunter, to discover potential G-rich sequences located within genes or genomes, also able to provide a rough estimation about the possibility of the target sequence to form a G4 (15). G4Hunter has been used to find G-rich sequences in a variety of species, including human (16), Plasmodium (17), Dictyostelium (18) and viral (19,20) genomes, and is now available as a web application (21).
Compared to model G4 structures, natural G4 sequences are often more complex and irregular. Genomic G4 motifs vary in length and may form a number (or variety) of non-canonical topologies. The first bioinformatic approaches performed in 2005 estimated that there were >300 000 G4-prone sequences in the human genome (22,23). With the development of sequencing technologies, 736 689 G4 structures have been identified in vitro (24). Even if the real number of G4-motifs actually formed may be lower than the one so far identified, most biophysical approaches are unable to deal with so many candidate sequences. For example, high resolution structural methods such as nuclear magnetic resonance (NMR) (25) cannot easily handle hundreds or thousands of samples. For this reason, rapid high-throughput assays able to deal with hundreds of motifs are needed.
Previously, we developed a thermal competition assay, the so-called FRET-MC assay, to characterize if an unknown sequence forms a quadruplex (26). The two ends of a short single-stranded DNA oligonucleotide mimicking ≈4 copies of the human telomeric motif, Tel21, were labeled with fluorescein (FAM) and TAMRA. This FAM-Tel21-TAMRA sequence, hereafter abbreviated to F21T, was used as a fluorescent probe sequence in the FRET-MC assay, which is based on the competitive binding of a selective G4 ligand between F21T and an unknown competitor. In principle, any G4-forming sequence would act as decoy for a specific G4 ligand, meaning that less compound would be available to stabilize F21T. This assay is highly reliable, with one weakness: it fails to identify G4-motifs with a low thermal stability as they would be single-stranded at the temperature where F21T unfolds.
For this reason, we wished to develop an isothermal version of this competition assay, which would overcome the issues caused by differences in thermal stabilities. To this aim, we designed a duplex-quadruplex competition assay derived from the system developed by Lacroix et al. (27), with a pair of probe strands consisting of a quencher (37Q, a strand from a telomeric sequence hTERC) and a partially complementary strand labeled with FAM and named F22 (27). In a manner similar to FRET-MC, a well-characterized and highly specific G4 ligand such as PhenDC3 (28) was used. The sequence of interest (competitor X) would compete with 37Q for PhenDC3 binding if, and only if, it adopts a quadruplex structure. The difference with FRET-MC is that the isothermal system here relies on a duplex-quadruplex competition: hybridization between 37Q and F22 would occur only if PhenDC3 is not present, or unavailable due to binding with the sequence to be tested. All steps are processed at a constant temperature, allowing true high-throughput. We validated this iso-FRET assay with a training set of DNA and RNA sequences containing positive (quadruplexes of different stabilities and topologies) and negative (single-strands and duplexes) controls. Kinetic considerations, advantages and disadvantages of this method are discussed.
MATERIALS AND METHODS
Samples
Non-labeled oligonucleotides were purchased RP cartridge purified from Eurogentec (Seraing, Belgium) as dried samples. Fluorescently labeled oligonucleotides (F22, F22m, 37Q, 37Qm and Cy5-37merR) were purchased from IBA (Göttingen, Germany). All sequences are provided in Supplementary Tables S1–S3. Except for salmon sperm DNA for which concentration is expressed in nucleotides, all other DNA/RNA concentrations were expressed as strand concentrations. DNA samples were stored at –20°C and RNA samples were kept at –80°C. Stock solution of PhenDC3 was prepared in DMSO at 2 mM concentration and stored at –20°C.
Determination of the equilibrium constant (Kd) between a G-quadruplex and PhenDC3
The Kd was measured as described by Le et al. (29). The Kd measurement was performed in 96-well plates with a Tecan Infinite M1000 Pro plate reader (France). A cyanine fluorophore (Cy5) was attached to the 5′ end and the fluorescently labeled strand was named as Cy5-37merR. 5 μl of 100 nM Cy5-37merR and 5 μl of PhenDC3 were added to give final concentrations in each well of 10 nM for Cy5-37merR and 0/2.5/5/7.5/10/25/50/75/100/250/500/750/1,000/2,500/5,000/7,500/10,000 nM for PhenDC3, in 20K buffer (20 mM KCl, 80 mM LiCl, 10 mM lithium cacodylate, pH 7.2) with 0.4% (v/v) DMSO. The final volume was 50 μl. Plates were kept at room temperature (RT, around 25°C) for 2 h before measurements. Cy5 was excited at 633 nm and the emission wavelength was set at 647 nm, excitation and emission bandwidths were set to 5 nm, with an integration time of 20 μs. Each experimental condition was tested at least in triplicate. Fluorescence quenching was used to normalize the measurements in terms of % bound. The Kd was processed with a single-site binding model (GraphPad Prism V 8.4.2) for curve fitting.
Kinetics
Kinetics experiments were performed in 96-well plates with a Tecan Infinite M1000 Pro plate reader (France).
For 37Q folding: (i) 200 nM 37Q was kept in 25 μl of 10 mM lithium cacodylate buffer (pH 7.2) containing potassium at different concentrations; or (ii) 200 nM 37Q in 25 μl of corresponding buffers contained 1 μM salmon sperm DNA and 1 μM PhenDC3 (0.4% v/v DMSO). Absorbance was recorded at 295 nm, interval time was set at 7.5 s, with 200 kinetics cycles and a settle time of 0 ms.
For hybridization: 250 nM 37Q (or 37Qm) was kept in 20 μl of 10 mM lithium cacodylate buffer (pH 7.2) containing potassium at different concentrations for 5 min, then 5 μl of 100 nM F22 (or F22m) were added to the corresponding buffer. The fluorophore (FAM) attached to F22 (or F22m) was excited at 492 nm and the emission wavelength was set at 520 nm, excitation and emission bandwidths were set to 10 nm, with an integration time of 20 μs. Each experimental condition was tested at least in triplicate.
Isothermal FRET competition assay
Iso-FRET was performed in 96-well plates with a Tecan Infinite M1000 Pro plate reader (France). For competitor samples: To 5 μl of 25 μM competitor oligonucleotide mixed with 5 μl of 1 μM 37Q in corresponding buffer, 10 μl of 2.5 μM PhenDC3 were added and left to stand for 5 min. Then 5 μl of 100 nM F22 were added. Final concentrations in each well were: 5 μM competitor, 200 nM 37Q, 1 μM PhenDC3 and 20 nM F22 containing 0.4% (v/v) DMSO. Control samples contained 20 nM F22 in the presence or absence of 200 nM 37Q in 25 μl of 20K buffer. Plates were kept at RT or in an incubator at 37°C before measurements. Fluorescence measurement settings were the same as described the above.
Data analysis
F value
We first defined the Fvalue parameter to evaluate the extent to which a competitor affects F22 + 37Q hybridization in the presence of PhenDC3, which is related to the fluorescence intensities (FI) of F22 alone [FI F22], F22 in the presence of 37Q [FI (F22 + 37Q duplex)] and F22 in the presence of 37Q, PhenDC3 and X, abbreviated as [FI competitors]:
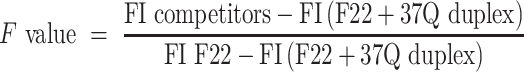 |
We then defined a threshold to distinguish between G4s and non-G4s competitors
F values were separated into the G4 group and the non-G4 group depending on the competitor structure. χ² test was employed to check if these two groups displayed a normal distribution, and non-normal distributions were transformed into normal by Johnson transformation. [μ ± 2σ] was used to calculate boundaries of Fvalues to distinguish G4s from non-G4s group (95% prediction interval) based on the three-sigma rule of thumb.
CF factor
The global alignment analysis (30) based on Needleman-Wunsch algorithm (31) was used to search reverse complementary base pairs between X and F22. EDNAFULL (NUC4.4; https://ftp.ncbi.nlm.nih.gov/blast/matrices/NUC.4.4) was adopted as scoring matrix, gap penalty was at 10.0 and extension penalty was set as 0.5. CF factor was defined to quantify the complementarity between X and F22:
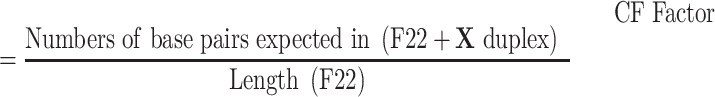 |
Other biophysical methods
All sequences were kept in corresponding buffers and denatured at 95°C for 5 min, and then cooled down to room temperature before use.
Isothermal differential absorbance spectrum (IDS) corresponds to the difference between the absorbance spectra obtained in the absence or in the presence of 100 mM KCl. Samples were tested at 3 μM strand concentration in 1 mL of 10 mM lithium cacodylate pH 7.2 buffer. Absorbance spectra were recorded on a Cary 300 spectrophotometer (Agilent Technologies, France) at 25°C. Scan range was set as 500–200 nm with scan rate: 600 nm/min, baseline was corrected automatically.
Thermal differential absorbance spectrum (TDS) corresponds the difference between the absorbance spectra obtained at high (95°C) and low (25°C) temperature of the sample, tested at 3 μM strand concentration, pre-folded in 1 mL of 100 mM KCl, 10 mM lithium cacodylate pH 7.2 buffer. Absorbance spectra were recorded at 25 and 95°C, respectively; other settings were as the same as IDS.
FRET-melting competition assay (FRET-MC): 15 μM competitor sequences and 5 μM F21T were pre-folded in 10K buffer (10 mM KCl, 90 mM LiCl, 10 mM lithium cacodylate, pH 7.2). Each well contained 25 μl solution comprising 0.2 μM F21T, 3 μM competitor and 0.4% (v/v) DMSO, in the absence or presence of 0.4 μM PhenDC3. FRET-MC experiments were performed in 96-well plates using a HT7900 RT-PCR instrument (Applied BioSystem), the FAM channel was used to collect the fluorescence signal. qPCR process was set as: 25°C 5 min; increasing temperature 0.5°C per minute, recording fluorescence, 140 cycles; then keeping plates at 25°C after measurements. ΔTm was calculated as the difference between Tm of F21T with or without PhenDC3; where Tm was identified as the temperature related to 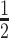 fluorescence. The S Factor provides a normalized value (26) of the stabilization (ΔTm) remaining in the presence of X. S is close to 0 for G4-forming sample, and remains close to 1 when X is not forming a quadruplex.
fluorescence. The S Factor provides a normalized value (26) of the stabilization (ΔTm) remaining in the presence of X. S is close to 0 for G4-forming sample, and remains close to 1 when X is not forming a quadruplex.
Circular dichroism (CD) spectra were recorded on a J-1500 spectropolarimeter (Jasco, France). Pre-folded samples were tested at 3 μM strand concentration in 1 ml of 100 mM KCl, 10 mM lithium cacodylate pH 7.2 buffer. The spectra were measured over the wavelength range of 200–340 nm at 25°C with a scan rate of 100 nm/min with and automatic baseline correction.
RESULTS
Principle of the assay
Several methods have been developed to characterize G4 structures in vitro: some of them are based on the spectral properties of G-quadruplexes (32,33), others on specific fluorescence light-up dyes such as NMM (34), Thioflavin T (35), and DASPMI (36). Both types of techniques consider only the structure of sequences of interest. We introduced the concept of competition in the FRET-MC assay (26), in which the sequence of interest X is in competition with a labeled probe. X, being added in large excess, can outcompete the labeled G4 probe for PhenDC3 binding if, and only if, X adopts a thermally stable quadruplex structure. In any other situation, X is unable to act as decoy for the specific G4 ligand, which remains bound to F21T and stabilizes it, as shown by an increase in Tm value. FRET-MC is therefore a thermal denaturation assay, and the thermal stability of the G4 structure adopted by X plays a decisive role: G4 competitors with a low Tm are unfolded before F21T starts to melt, meaning that they are seen as single-strands, leading to false negatives.
In contrast, in the isothermal competition assay developed here, the system uses two mono-labeled rather than one double-labeled fluorescence oligonucleotide, and the competition process is tested at a constant temperature (room temperature in most experiments described below, but the assay can be easily transposed to 37°C) to avoid issues related to thermal stability: what matters here is whether X is predominantly folded into a quadruplex or not at ≈ 25°C, not if its Tm is 40, 60 or 90°C.
The two mono-labeled partially complementary RNA strands, 37Q and F22, were initially used to design an isothermal assay to pick novel G4 ligands (27). In brief, potent G4 ligands stabilize the intramolecular quadruplex formed by 37Q, preventing it from hybridizing to F22. As a consequence, F22 remains single-stranded and its fluorescence is high. In contrast, when a compound has little or no affinity for the 37Q quadruplex, formation of the F22 + 37Q duplex is possible, leading to fluorescence quenching. This assay is transposable into 96-well format and allows the screening of many ligands or conditions.
In here, rather than testing a variety of compounds, we introduce F22 and 37Q to evidence G4 structures in vitro. As shown in Figure 1, the G4-characterization assay can be divided into three main steps:
Figure 1.

Principle of the iso-FRET competition assay. The modified strands (37Q, F22) are shown in color. In the first scenario (top) (i), the competitor (grey) forms a quadruplex and traps PhenDC3 (orange oval), allowing 37Q (red) to quench F22 (green) by forming a duplex. In the second scenario (bottom) (ii), the competitor does not form a quadruplex, PhenDC3 remains bound to 37Q, which cannot hybridize to F22 which emits a strong fluorescence signal.
An excess of the sequence of interest X (unknown competitor) is added to 37Q (Figure 1, left);
A well-known high affinity G4 ligand, PhenDC3, is added to the competitor-37Q mixture. PhenDC3 is therefore ‘given the choice’ between X and 37Q (Figure 1, center);
-
F22 is then added, and the fluorescence signal can be recorded. Fluorescence intensity indicates whether the sequence of interest adopts or not a G4 structure (Figure 1, right).
(Figure 1, upper right part) X forms a quadruplex which can trap PhenDC3. As X is in excess (as compared to both PhenDC3 and 37Q), far less G4 ligand will be available to bind and stabilize the 37Q quadruplex, allowing duplex formation with F22, and ultimately fluorescence quenching due to fluorescein – quencher proximity.
(Figure 1, lower right part) X does not form a quadruplex and does not act as a decoy for PhenDC3, which remains bound to the G4 structure formed by 37Q: formation of the F22 + 37Q duplex remains disfavored and the fluorescence emission of F22 remains high.
To validate this assay, we selected a variety of DNA and RNA sequences for which we know which structure(s) they adopt (26). This collection of 70 sequences includes a variety of quadruplex-forming motifs with various topologies, as well as single- and double-stranded DNAs and RNAs (sequences shown in Supplementary Table S1 (26)).
Practical considerations: sequential order of addition and choosing the concentrations of each component
The isothermal assay involves (at least) three different nucleic acid sequences (i.e. 37Q, F22 and X) and one G4 ligand (PhenDC3). This leaves six possible bimolecular interactions between two different partners; three of them are directly relevant for this study: (i) duplex formation between 37Q and F22, and binding between (ii) PhenDC3 and 37Q or (iii) PhenDC3 and X. Other possible interactions are not considered here, such as PhenDC3 binding to the F22 + 37Q duplex or to the C-rich F22 single strand: previous results have unambiguously confirmed the selectivity of PhenDC3, with little or no binding to single- and double-strands (26,28). On the other hand, possible interactions (e.g. partial Watson–Crick complementarity) between X and F22 or X and 37Q may generate artefacts (see below).
The key competing equilibria to be considered here are around the 37Q sequence; whether it binds to its F22 complementary sequence or to the PhenDC3 ligand, and the later event is modulated by PhenDC3 availability (whether X can act as decoy or not). To give a proper ‘choice’ to PhenDC3, we reasoned that adding the ligand to a well-mixed solution containing both 37Q and the competitor X (both were given enough time to properly fold) should favor a ‘fair’ competition for PhenDC3. Otherwise, if one of these two oligonucleotides is added after PhenDC3, re-equilibration time of the system should depend on the lifetime (koff) of the quadruplex–ligand complex, for which we have little information, especially when considering a putative G4 formed by X.
F22 will be the last component to be added. This was previously established (27), as it is nearly impossible to reverse F22 + 37Q complex formation, as the lifetime of this duplex is extremely long at room temperature. PhenDC3 is a G4 ligand, not a duplex-destabilizing agent: one should have to first unfold the F22 + 37Q duplex to allow PhenDC3 binding to 37Q (27). The sequential addition of each component is represented in Figure 1.
We chose not to start with the addition of PhenDC3 since non-specific binding of this compound with the surface of microplate wells may be problematic. We indeed observed that artefacts may result in 96-well plates even when treated to prevent hydrophobic and ionic interactions (data not shown). This effect is abrogated by adding 1 μM salmon sperm DNA as a non-specific competitor.
As mentioned above, there are several non-covalent interactions involved in the isothermal assay, starting with the interaction of PhenDC3 with either X or 37Q. The principle of this experiment is that the fraction occupancy of 37Q by PhenDC3 should be significantly reduced by X addition if X adopts a quadruplex fold. The concentration of each component has to be carefully considered. As we have no a priori assumption of the exact Kd of PhenDC3 for the X quadruplex, we reasoned that X should be in excess as compared to 37Q and PhenDC3 in order to act as an efficient competitor. Assuming that kon and koff of the ligand to both structures are relatively high, what is relevant for these equilibria are the equilibrium constants (Kd). In the simpler situation where X does not adopt a G4 fold, PhenDC3 affinity should be low or negligible, and one can ignore this competing equilibrium. In that case, duplex formation should be strongly inhibited, and this should happen provided that most 37Q quadruplexes are bound to PhenDC3. This can be obtained by making sure that (i) PhenDC3 is in molar excess as compared to 37Q and (ii) PhenDC3 concentration is significantly higher than the Kd for 37Q (64 nM, as determined in Supplementary Figure S1).
To summarize, the concentrations of each component should be ranked as follows:
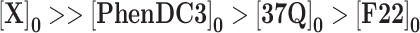 |
To study how X affects results, we defined 4 groups of competitors based on FRET-MC results (26), which contained strong G4s (Pu24T, cmyc, 25TAG), moderately stable G4s (KRAS-22RT, SP-PGQ-1, UpsB-Q3), poor G4s (SP-PGQ-3, TBA, BmU16) and non G4s (single- or double-strands such as ds26, dT26 or Hairpin1) at different competitor concentrations in 20K buffer. The novel isothermal competition assay was used to test them. Since absolute fluorescence values are always relative (expressed in arbitrary units), they were normalized with the F value (26) based on the F22 fluorescence before or after F22 + 37Q hybridization; Fvalue reflected the amount of PhenDC3 bound to 37Q.
As shown in Supplementary Figure S2, stable quadruplexes trap a significant fraction of PhenDC3, even at 1:1 ligand to competitor equivalents, and increasing [X]0 had little effect. For duplexes and single strands, no trapping was observed at any concentration. For other quadruplexes such as KRAS-22RT, SP-PGQ-1, SP-PGQ-3, or TBA, a concentration effect was observed: raising X concentration led to further reduction in fluorescence intensity, until X concentration reached 5 μM: at this stage, Fvalues were low enough and we did not investigate higher ones. Based on these observations, we chose a concentration of 5 μM for X, and the molar ratio between X, PhenDC3, 37Q and F22 was therefore fixed at 250:50:10:1.
Kinetic considerations
It is commonly believed that intramolecular G4 folding is relatively slow compared to hairpin duplex formation (37,38). Intermolecular duplex formation in the nanomolar concentration range takes a few minutes (39), while intramolecular G4 folding kinetics are impacted by a series of factors, including sequence effects (40), metal cations (41), the presence of intermediates (‘dead ends’) (42) and the presence of G4 ligands acting as molecular chaperones, such as PIPER (a perylene derivative) (43) and 360A (44), which have been demonstrated to accelerate G4 folding. PhenDC3 is even able to promote G4 folding within minutes in the absence of potassium (45).
To follow G4 folding under different conditions, we chose to keep the ionic strength constant, with a total concentration of mono-cations of 100 mM. G4 folding may be followed by recording absorbance at 295 nm (46). 37Q folding into G4 was followed at different K+ concentrations, adjusting ionic strength with Li+. As expected, folding of 37Q in the absence of potassium (0K) proceeded extremely slowly. In the presence of K+, 37Q achieved near-complete folding in ≈ 5 minutes (Figure 2). Some G4 ligands such as PhenDC3 have been shown to promote G4 folding and increase kon, acting as G4 chaperones (44): this is the case here, where addition of PhenDC3 shortens the G4 folding time to less than 100 s (Supplementary Figure S3).
Figure 2.
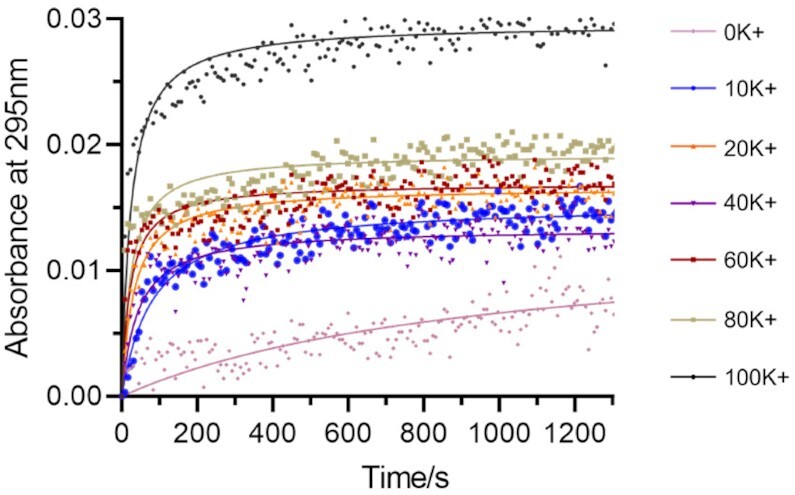
Kinetics of 37Q G4 folding (200 nM strand concentration) at different potassium concentrations. G4 folding was processed at RT.
To study F22 + 37Q duplex formation, we first considered the two oligonucleotides alone, with no competitor or PhenDC3 to simplify the model. As shown in Figure 3A, fluorescence quenching was only partial (25%) even 5 h after 37Q addition in a 100 mM K+ buffer, while quenching reached 74% in 0K buffer. This difference can be explained by the selective stabilization of G4 structures by potassium, which partially hinders or delays F22 + 37Q hybridization, even in the absence of a G4 ligand, as previously reported (47). As expected for an intermolecular duplex, kinetics depended on strand concentration: lowering 37Q concentration delayed F22 + 37Q duplex formation (Supplementary Figure S4).
Figure 3.

Time-dependent fluorescence of the F22 + 37Q system at different potassium concentrations. Concentration of F22 and F22m were 20 nM, 37Q and 37Qm were 200 nM. 37Q and 37Qm were kept in corresponding buffer for 5 min before adding F22 or F22m, respectively. Hybridizations were processed at RT. Panel A and B related to the same normalized fluorescence scale.
To evaluate the impact of 37Q quadruplex formation on duplex formation, we studied in parallel a control system (F22m + 37Qm), in which the F22 + 37Q pair has been mutated. F22m and 37Qm are a couple of partially complementary RNA strands also able to form a duplex involving the same number of mismatches as F22 + 37Q. However, 37Qm is unable to form a quadruplex and has no affinity for G4 ligands (27). As expected for duplex formation when no competing quadruplex is present, hybridization between F22m and 37Qm is fast, even in the nanomolar strand concentration range (39) (Figure 3B). Therefore, the relatively long (hours) equilibrium time required for proper F22 + 37Q hybridization results from quadruplex formation by 37Q, which delays, but does not prevent duplex formation.
Effects of interaction time and potassium concentration
Potassium concentration not only impacts F22 + 37Q hybridization (Figure 3A), but also the folding of G4 competitors, and the competition between 37Q and X for PhenDC3 binding. We initially defined the boundary between G4 X and non-G4 X roughly based on an arbitrary threshold: F ≥ 0.5 related to non-G4s X, while F < 0.5 meant G4-forming X. Twelve different competitors including different G4s and non-G4s were taken as examples to analyze the influence of potassium. They were divided in three groups based on this rough boundary: (i) sequences in group A (Supplementary Figure S5A) showed high affinities to PhenDC3 under all ionic conditions and at all time points (G4 competitors, low Fvalues); (ii) in contrast to group A, PhenDC3 remained bound to 37Q in the presence of sequences belonged to group B (Supplementary Figure S5B, non-G4 competitors, high Fvalues); (iii) sequences in group C (Figure 4) are reported to fold into G4s, while their Fvalues depend on specific ionic conditions and time points (poor competitors, with intermediate F values).
Figure 4.
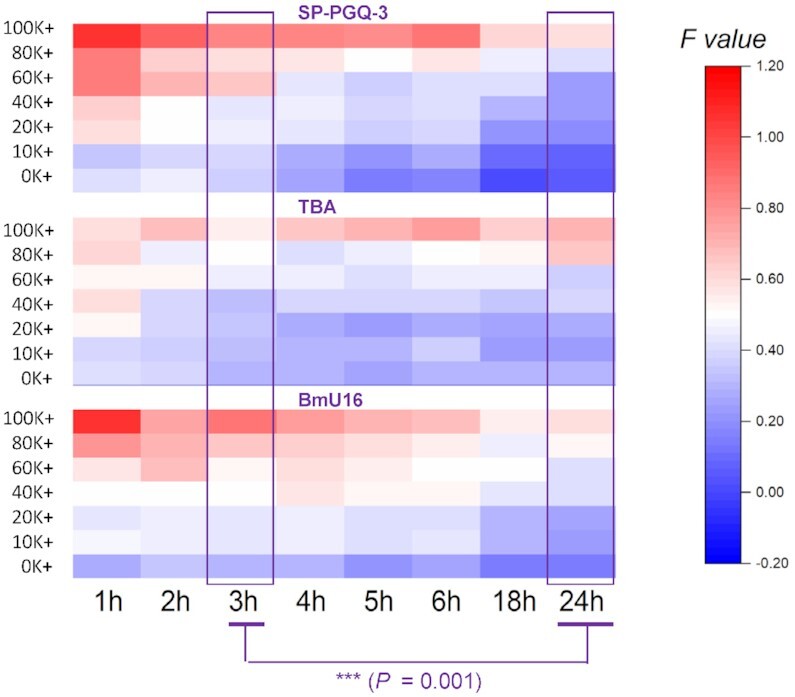
Effects of incubation time and ionic conditions on fluorescence intensity for group C competitors (G4-forming oligos with moderate affinity for PhenDC3). Each well contained 5 μM competitor, 200 nM 37Q, 1 μM PhenDC3 and 20 nM F22; control wells (F22 and F22-37Q) included 20 nM F22 in the presence or absence of 200 nM 37Q. All samples were tested in triplicate at various potassium concentrations (0–100 mM) at RT. The difference between 3 h and 24 h was performed by Wilcox sign test. All sub-panels related to the same F value scale.
For poor competitors, F values went gradually up with increasing potassium concentrations: when [K+] exceeded 20 mM, F values were above 0.5, and this phenomenon was more apparent at short incubation times. We wanted to test whether Fvalues would allow to discriminate between G4 X and non-G4 X. Although the differences between five ‘good’ G4 X and non-G4 X were highly significant in both 0K and 20K buffer, this difference between average F values (ΔMedian) was actually higher in 20K (0.82) than in 0K (0.64), meaning that the discrimination between the two groups (G4 vs non-G4) is better in the presence of 20 mM potassium (20K).
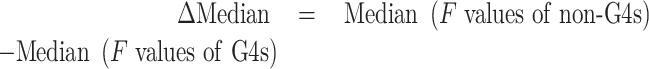 |
According to the kinetics results based on the simplified model (Figure 3A), we selected an equilibrium time of 3 h or longer as a result of the slow F22 + 37Q hybridization step. In the following experiments, we started to optimize the interaction time. An extended incubation had nearly no effect on the results obtained with groups A in all buffers (low Fvalues), as well as for non-G4 competitors in group B, which kept high Fvalues at all times. In contrast, SP-PGQ-3, TBA, and BmU16 in group C (Figure 4) exhibited a time-dependent behavior, with F values decreasing over time (P = 0.001 between 3 h and 24 h). This result illustrates the fact that even mediocre G4 competitors (G-quadruplex oligonucleotides with moderate affinities for PhenDC3 such as the thrombin binding aptamer) can be properly classified as G4-forming provided that appropriate precautions are taken. For this reason, we selected 24 h as the default interaction time in 20K buffer.
Validation of the isothermal competition assay
We employed a validated training set (26) containing a series of identified DNA and RNA G4 sequences with different topologies, as well as single- and double-stranded controls. Examples of positive controls are cmyc and Pu24T, which both form very stable quadruplexes, while ds26 (duplex) and dT26 (single-strand) were chosen as negative controls. Each competitor oligonucleotide X was tested at a single concentration, and we determined the fluorescence intensity for each condition.
We divided the training sequences into 3 categories according to Fvalues: (i) F < 0.33: G4 competitor; (ii) 0.33 ≤ F < 0.54: unknown; (iii) F ≥ 0.54: non-G4 competitor. With these intervals, we found no outlier (false positive or negative), and all known samples behaved as predicted (Figure 5).
Figure 5.
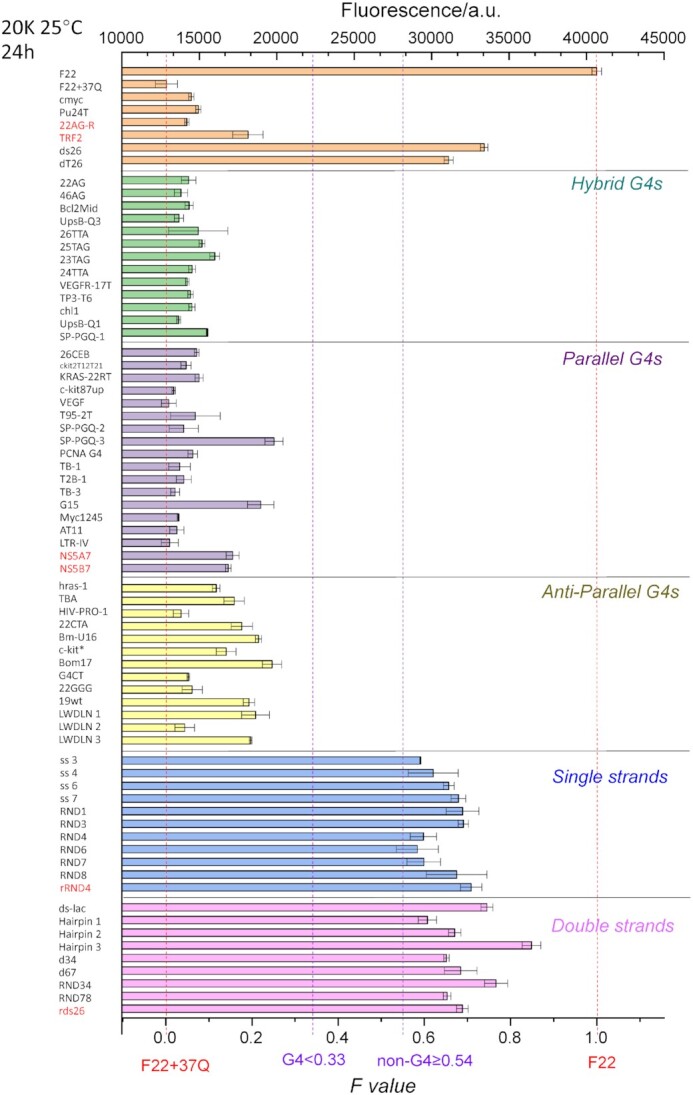
Fluorescence quenching in the presence of various competitors. 20 nM F22 is incubated for 24 h in the presence of 200 nM 37Q and 1 μM PhenDC3, alone or in the presence of 5 μM of a variety of X competitors (mostly DNA with 6 RNA samples shown in red), listed on the left. The Fvalues (bottom X-axis) provides a normalized value. The four different vertical dotted lines correspond to (i) the level of fluorescence in the absence of a competitor (F = 0); (ii) the first threshold value at 0.33 chosen for positive samples (G4-forming sequences all exhibit Fvalues between 0 and 0.33); (iii) the second threshold value at 0.54: negative controls/non G4-forming sequences all exhibit Fvalues between 0.54 and 1; (iv) the level of fluorescence of F22 alone, with no 37Q added (F = 1). Samples were measured in 20K buffer at RT.
Regarding the temperature of the assay, while working at room temperature may be simple, homeotherms maintain body temperatures in the range of 36–42°C (48). For natural G-rich sequences found in animal genomes, it may therefore be biologically relevant to characterize them at physiological temperature. Taking Homo sapiens as an example, we determined if this assay would provide comparable results at 37°C. Using the same training set as previously, we found the same qualitative results: (i) F < 0.27: G4 competitor; (ii) 0.27 ≤ F < 0.52: unknown; (iii) F ≥ 0.52: non-G4 competitor. (Supplementary Figure S6).
Interestingly, compared to the training set at 25°C, the difference in Fvalues for the G4 and non-G4 groups was even more significant at 37°C, as a consequence of smaller standard deviations for each category. This illustrates the interest of working at physiological temperature. As a further bonus, a higher temperature accelerates F22 + 37Q hybridization, meaning that shorter incubation times may be selected.
Can poor G4 competitor be evidenced by the iso-FRET assay?
As mentioned above, the competitor X is added in large excess as compared to 37Q, facilitating the competition with PhenDC3. However, if X a forming a quadruplex with a very low affinity for PhenDC3 (as compared to 37Q), this competition may not be effective enough to give a positive result in the iso-FRET assay. We consider the assay to be ineffective when [37Q + PhenDC3] is equal or lower than [X + PhenDC3], meaning that the interaction between X and PhenDC3 is no longer dominant in the competitive binding.
Knowing the final concentrations in each well (typically 200 nM for 37Q, 5 μM for X, i.e. a 25× fold excess, and 1 μM for PhenDC3) and based on Mass action law, we can determine the equilibrium dissociation constant for the interaction between X and PhenDC3 (KdX) that would lead to [37Q + PhenDC3] = [X + PhenDC3], given that we determined the dissociation constant for the interaction between a close analog of 37Q and PhenDC3 (KdQ) to be 64 nM. A rapid calculation gives a value for KdX of ≈17 μM, a far worse (several orders of magnitude) affinity for a quadruplex that was previously determined, or measured here for PhenDC3. The affinity we determined here for Cy5-37merR is actually a bit weaker than what has been previously reported in the literature for other quadruplexes (49,50) under different experimental conditions, with Kd as low as 2 nM. The risk of a false negative in the iso-FRET assay (a G4-forming sequence that would not act as an effective competitor) is therefore extremely low.
A potential limitation: G-rich sequences complementary to F22
The iso-FRET assay involves two probe strands, F22 and 37Q. In this part we investigate what happens when the X sequence is complementary to any of the probe strands:
We can quickly discard the case in which X is Watson-Crick complementary to 37Q, as this would mean that X is C-rich; G4 formation would therefore be very unlikely. In any case, adding this C-rich strand would decrease the ability of the 37Q sequence to quench the fluorescence of F22 (due to hybridization to X in large excess), giving high Fvalues indicative of non-G4 formation by X.
The situation where X is complementary to F22 is more complex, especially given that the F22 + 37Q duplex contains three point-mismatches, and one bulge. Given that the X oligonucleotide is in molar excess as compared to 37Q, it is clear that, if the F22 + X duplex is thermally more stable than F22 + 37Q, the assay will lead to an artefactual result: a FAM fluorescence signal is expected for F22 + X as X is not conjugated to a quencher. In other words, provided that X is able to form a stable hybrid with F22, it will appear as non G4-forming in this test no matter what is its real G4-forming propensity. This prediction was experimentally verified for a variety of G-rich probes with various levels of complementarity to F22. We defined the CF factor as a simple way to quantify this complementarity for each X and F22, and compare it to F22 + 37Q duplex.
As shown in Supplementary Table S1, 37Q held the highest CF Factor among all sequences in the training set, 0.77; CF for G4 X were in the range of 0.18 to 0.68, which means F22 + X duplex was weaker than 37Q + F22. To investigate how F22 + X duplex influence G4 X, we designed six G4 X (Supplementary Table S2) with a high tendency to form quadruplexes (G4Hunter scores > 1.5), as well as a good complementarity to F22 (CF ≥ 0.68). All CF sequences led to significant decrease in ΔTm of F21T in the FRET-MC assay (Supplementary Figure S7), indicating they are indeed forming G4 structures.
As shown in Figure 6, Fvalues provide an excellent assessment of G4 propensity (F < 0.35; green horizontal line) for all sequences with CF factor ≤ 0.77. Negative controls (duplexes and single strands) all give an Fvalue above 0.53 (purple horizontal line) and the separation between G4 and non-G4 forming sequences is very clear. On the other hand, the assay fails to account for G4 formation with sequences having a very high level of complementarity to F22 (CF factors ≥ 0.86; green triangles on the right part of the figure). In other words, this assay is reliable as long as X is not highly complementary to F22, and this caveat can easily be anticipated when the sequence of X is known: one can first check F22 + X complementarity and discard sequences that would form too stable hybrids.
Figure 6.
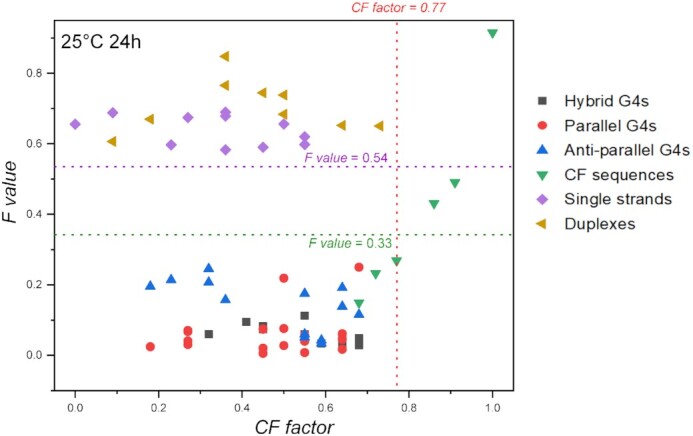
Fluorescence quenching in the presence of various DNA competitors. 20 nM F22 is incubated for 24 h in the presence of 200 nM 37Q and 1 μM PhenDC3, alone or in the presence of 5 μM of a variety of competitors. Samples were measured in 20K buffer at RT.
Why using fluorescent RNA probe strands rather than DNA?
Compared to DNA oligonucleotides, RNA strands are significantly more expensive and susceptible to degradation by RNases. These inherent disadvantages prompted us to test whether one could convert the RNA system designed by Lacroix et al. into a duplex-quadruplex competition assay involving oligodeoxynucleotides, dF22 and d37Q (27). As shown in Supplementary Figure S8, hybridization of dF22 to d37Q reached a plateau after two hours at room temperature, as found for F22 + 37Q hybridization. Unfortunately, quenching with the DNA oligonucleotides was not as pronounced as for the RNA system, which showed higher quenching efficiency than dF22 + d37Q under all ionic conditions at the beginning of the hybridization step (0 h). One possible reason for this difference is that the RNA to DNA substitution modifies the kinetics and thermodynamics of both the duplex and quadruplex species. For example, a RNA-RNA duplex with high G/C content has a higher kon than a similar DNA duplex at 25°C (51).
We nevertheless tested whether this DNA system would be applicable to the analysis of various X sequences. As for the experiments described above, competitors from the three groups (A = G4; B = non-G4 and C = moderate G4-forming competitors) were tested at the same concentrations as for the RNA version. On the positive side, stable G4 such as cmyc and 25TAG (group A) were correctly identified as G4-competitors in this DNA-based assay, as they gave low F values in all settings while non-G4 forming sequences such as ds26 (group B) gave high Fvalues (Supplementary Figure S9A-B), as expected in both cases. Unfortunately, Group C (modest G4 competitors) gave more erratic results with the DNA system: BmU16 showed low Fvalues while both SP-PGQ-3 and TBA gave relatively high Fvalues, sometimes even a bit higher than ds26 (Supplementary Figure S9C).
In other words, the DNA system worked, but not as well as the RNA one. Multiple factors may explain this difference, such as changes in duplex stability (52–54) which will also affect the stability of the F22 + X or dF22 + X duplex when X is partially complementary to F22 or dF22. As a consequence, we chose to work with the F22 + 37Q RNA system to characterize unknown sequences in vitro, despite the potential problems created by RNA oligonucleotides. The actual cost per point remains low, even if RNA synthesis is expensive, as the reaction volume and concentrations are low, and could be further reduced by using 384-well plates. 20 nM strand concentration for F22 in 10 μl corresponds to 0.1 picomoles, or less than 1/10 000 of a 200 nanomole synthesis. In reality, the higher cost of RNA synthesis would start to make a difference if tens of thousands or millions of candidate sequences were to be tested. In addition, stability over time of these two labeled RNA was excellent, and stock solutions could be kept for months, if not years, with no loss of activity.
Experimental validation on viral DNA and RNA G-rich sequences
To validate the new isothermal assay, we employed twenty-three G4-prone sequences including DNAs and RNAs from pathogenic viruses (Supplementary Table S3). All of the sequences were previously predicted or demonstrated to form G4 structures. We performed classical biophysical assays to characterize their G4-forming potential before testing the isothermal assay. With the exception of HPV-16 (DNA), we found IDS and TDS spectra compatible with G4 formation for all sequences, with a negative peak around 295 nm (Supplementary Figure S10A), in agreement with FRET-MC results (Supplementary Figure S10B). CD spectra were also recorded for the DNA samples (Supplementary Figure S10C) and the proposed topologies are summarized in Supplementary Table S3 (the interpretation of CD spectra of RNA samples is more tricky, as A-form RNA duplexes give a positive peak close to the one expected for RNA parallel G4s (55)).
We analyzed these motifs with the isothermal assay at 37°C. As shown in Figure 7, isothermal assay results of all DNA competitor sequences were consistent with classical characterizations (Supplementary Figure S10): they all formed G4 structures, except HPV-16. FRET-MC of HPV-16 also gave a high S Factor value (>0.6), again suggesting that no G4 is formed (26) (Supplementary Figure S10). Among RNA oligomers, Nipah-NV2 gave a Fvalue in the ‘undetermined’ zone (0.27 ≤ F< 0.52), while all other methods indicated that this RNA is actually forming a G4 structure. Its relatively high Fvalue could be the result of its relatively high complementarity to F22 (CF = 0.68; the boundary chosen for DNA should probably be lowered for RNA given the higher stability of the corresponding RNA–RNA duplex).
Figure 7.
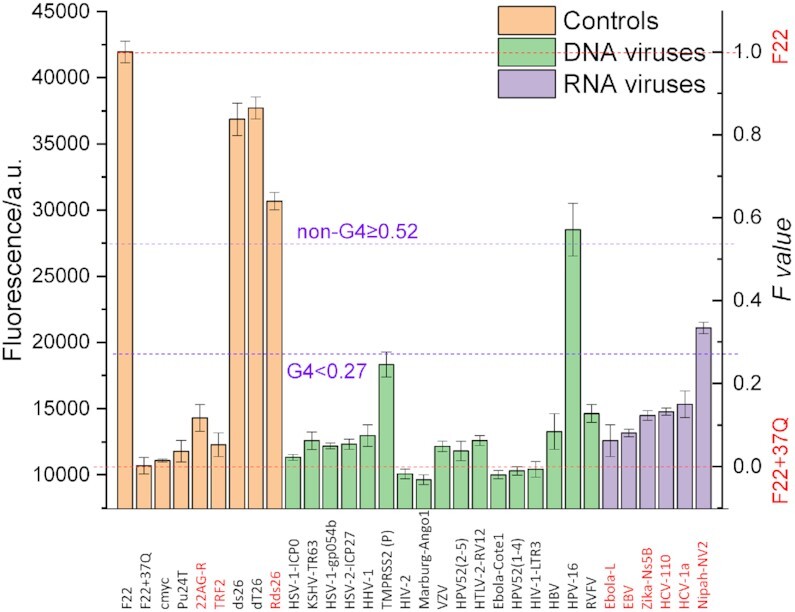
Fluorescence quenching in the presence of viral competitor sequences. 20 nM F22 is incubated for 24 h in the presence of 200 nM 37Q and 1 μM PhenDC3, alone or in the presence of 5 μM of DNA (green bars) and RNA (purple bars) virus competitors, the complementarity of each sequence to F22 is indicated by the CF factor value, shown above each bar. Measurements were performed in 20K buffer at 37°C. The names of RNA sequences are highlighted in red. Sequences are provided in Supplementary Table S3.
Altogether, these results illustrate that the iso-FRET assay may also be applied to RNA sequences, and that the conclusion reached by this method corroborates the analysis by other assays. One extra precaution should be taken with RNA samples though, where results can be biased by the possible complementarity between F22 and RNA X. For instance, the higher stability of the resulting RNA–RNA duplex may interfere with the isothermal assay. For this reason, we suggest to avoid testing sequences with a high complementarity (CF value). For example, the Fvalue for Nipah-NV2 is relatively high (Figure 7), although G4 formation by this sequence was validated by other means. This ambiguous result (intermediate Fvalue) may result from a high CF value (CF = 0.68, Supplementary Table S3), a relatively low affinity to PhenDC3 and/or low stability of the corresponding quadruplex.
DISCUSSION: ADVANTAGES AND LIMITATIONS OF THE ISOTHERMAL ASSAY
In this manuscript, we introduced a new method to demonstrate G4 formation in vitro. This isothermal assay is amenable to very high throughput. We summarize the advantages and shortcomings of this assay in Table 1, and compare its properties to the recently-developed FRET-MC assay in the next two paragraphs.
Table 1.
Comparison of FRET-MC and isothermal competition assay to characterize G4-forming sequences. Attractive features are shown in bold characters, while potential disadvantages are underlined
| Parameter | FRET-MC | Iso-competition |
|---|---|---|
| Signal | Fluorescence (T m ) | Fluorescence (quenching) |
| Data analysis | T m determination | Simple normalization |
| Temperature | Variable: 25–95°C | Isothermal (adjustable 20–37°C) |
| System | At equilibrium | Not at equilibrium (under kinetic control) |
| Number of partners | 3 F21T, PhenDC3, X | 4 37Q, F22, PhenDC3, X |
| Throughput | Hundreds/day | Thousands/day |
| Analyzed samples | DNA & RNA | DNA & RNAa |
| Ionic strength | Near-physiological | Near-physiological |
| Volume | 25 μl | 25 μl |
| Competitor concentration | μM range | μM range |
| Main Limitations/Artefacts | Thermally unstable G4 | Complementarity to F22 |
aAlthough not tested, both assays should be transposable to other nucleic acids modifications (e.g. PNA; 2’OMe), keeping in mind that, for iso-competition, complementarity to F22 may be a problem if very stable duplexes are expected to form between X and this RNA.
Common features between FRET-MC and iso-FRET
Both assays share a number of features: they are fluorescence-based and can be read on an inexpensive plate reader. As a consequence, many samples can be tested in parallel, and multiple positive and negative controls can be included to increase the robustness of the assay. Even if most spectroscopic instruments (e.g. a spectropolarimeter) can be adapted using liquid handlers to improve throughput, this would correspond to a serial analysis. In contrast, FRET-MC and iso-FRET allow the testing of 96/384 samples in a parallel manner with a real time PCR machine or a fluorescence plate reader.
Both methods are relatively fast and inexpensive; low volumes (typically 25 μl) and the concentration needed for the sample to be tested (μM range) imply that minimal amount of the sample are required. In addition, the sequence does not need a high level of purity: the presence of minor contaminants (e.g. shorter sequences) will not perturb the assay. Consequently, expensive or time-consuming purification protocols for the X oligonucleotides are not needed. In addition, both tests should be applicable to DNA and RNA samples (as well as chemically modified nucleic acids), long sequences, and mixtures or crude oligos.
Main differences between FRET-MC and iso-FRET
Even if both assays share a number of advantages, there are significant differences between the two. First of all, iso-FRET involves one more component, as the double-labeled F21T oligo in FRET-MC is replaced by a pair of mono-labeled sequences. This makes the system slightly more complex, and the sequences to be tested should not directly interact with any of these oligonucleotides.
Iso-FRET has a potentially higher throughput, for two main reasons: (i) it is isothermal and (ii) data analysis is extremely simple, objective and direct (normalizing fluorescence intensity): there is no need to determine a Tm value and calculate a ΔTm.
More importantly, iso-FRET can identify low-stability G4s. This can be an advantage for G4-poor genomes (some viruses such as SARS-CoV-2 contain a low density of G4-prone motifs). On the negative side, the iso-competition system is not at thermodynamic equilibrium – the result depends on the incubation time; this means that proper precautions should be taken to insure reproducibility. In addition, given that an interaction between X and F22 would perturb this assay, the sequence of X must be known; in contrast, FRET-MC can be performed blindly to characterize G-rich sequences. Finally, the fluorescence threshold in iso-FRET, chosen to distinguish between G4 and non-G4, requires a prior calibration with known controls.
CONCLUSION
The high throughput isothermal FRET assay enabled us to characterize structures of unknown G-rich sequences in vitro: if the unknown sequence X folds in a G4 structure, it induces fluorescence quenching by binding PhenDC3. In contrast, if X remains single-stranded or forms a different structure, the fluorescence signal remains high. This reliable so-called iso-FRET assay has been validated by several known oligonucleotide sequences forming G4 structures with different topologies, including parallel, anti-parallel, and hybrid structures, as well as double- and single-stranded DNAs and RNAs. Compared to the previous FRET-MC assay, this isothermal assay allows to process samples at 37°C, which could help indicate whether a particular sequence forms a G4 under physiological conditions. Importantly, iso-FRET eliminates the false negative results generated by low thermally stable G4s identified by FRET-MC. Conversely, limited by the slow hybridization rate (and slow competition process for weak G4s), iso-FRET is not a system working at thermodynamics equilibrium. In addition, the use of two mono-labelled fluorescent oligonucleotides, impedes the application of this method to G-rich sequences that show high complementary to F22 (one of probe strands). We finally applied iso-FRET to G4-prone motifs in virus genomes, and its results were confirmed by classical spectroscopic methods. The proposed isothermal competition assay constitutes a new biophysical method that can be added to the G4 toolbox required to characterize G4 structures in vitro.
DATA AVAILABILITY
All data is available in the supplementary information section.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Laurent Lacroix and Anne Cucchiarini for helpful discussions.
Contributor Information
Yu Luo, Laboratoire d’Optique et Biosciences, Ecole Polytechnique, CNRS, Inserm, Institut Polytechnique de Paris, 91128 Palaiseau, France; CNRS UMR9187, INSERM U1196, Université Paris-Saclay, F-91405 Orsay, France.
Daniela Verga, CNRS UMR9187, INSERM U1196, Université Paris-Saclay, F-91405 Orsay, France; CNRS UMR9187, INSERM U1196, Institut Curie, PSL Research University, F-91405 Orsay, France.
Jean-Louis Mergny, Laboratoire d’Optique et Biosciences, Ecole Polytechnique, CNRS, Inserm, Institut Polytechnique de Paris, 91128 Palaiseau, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
ANR G4Access [ANR-20-CE12-0023] and ICARE [ANR-21-CE44 to J.L.M.]; Chinese Scholarship Council [201906340018 to Y.L.]. Funding for open access charge: Inserm.
Conflict of interest statement. None declared.
REFERENCES
- 1. Agarwala P., Pandey S., Maiti S.. The tale of RNA G-quadruplex. Org. Biomol. Chem. 2015; 13:5570–5585. [DOI] [PubMed] [Google Scholar]
- 2. Sen D., Gilbert W.. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature. 1988; 334:364–366. [DOI] [PubMed] [Google Scholar]
- 3. Majee P., Mishra SK., Pandya N., Shankar U., Pasadi S., Muniyappa K., Nayak D., Kumar A.. Identification and characterization of two conserved G-quadruplex forming motifs in the nipah virus genome and their interaction with G-quadruplex specific ligands. Sci. Rep. 2020; 10:1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang S.R., Zhang Q.Y., Wang J.Q., Ge X.Y., Song Y.Y., Wang Y.F., Li X.D., Fu B.S., Xu G.H., Shu B.et al.. Chemical targeting of a G-quadruplex RNA in the ebola virus l gene. Cell Chem Biol. 2016; 23:1113–1122. [DOI] [PubMed] [Google Scholar]
- 5. Brazda V., Luo Y., Bartas M., Kaura P., Porubiakova O., Stastny J., Pecinka P., Verga D., Da Cunha V., Takahashi T.S.et al.. G-Quadruplexes in the archaea domain. Biomolecules. 2020; 10:1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Qin Y., Hurley L.H.. Structures, folding patterns, and functions of intramolecular DNA G-quadruplexes found in eukaryotic promoter regions. Biochimie. 2008; 90:1149–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Prorok P., Artufel M., Aze A., Coulombe P., Peiffer I., Lacroix L., Guédin A., Mergny J.L., Damaschke J., Schepers A.et al.. Involvement of G-quadruplex regions in mammalian replication origin activity. Nat. Commun. 2019; 10:3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Paeschke K., Bochman M.L., Garcia P.D., Cejka P., Friedman K.L., Kowalczykowski S.C., Zakian V.A.. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature. 2013; 497:458–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Paeschke K., Capra J.A., Zakian V.A.. DNA replication through G-quadruplex motifs is promoted by the saccharomyces cerevisiae pif1 DNA helicase. Cell. 2011; 145:678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Balasubramanian S., Hurley L.H., Neidle S.. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy. Nat. Rev. Drug Discov. 2011; 10:261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lago S., Nadai M., Cernilogar F.M., Kazerani M., Dominiguez Moreno H., Schotta G., Richter S.N.. Promoter G-quadruplexes and transcription factors cooperate to shape the cell type-specific transcriptome. Nat. Commun. 2021; 12:3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hansel-Hertsch R., Beraldi D., Lensing S.V., Marsico G., Zyner K., Parry A., Di Antonio M., Pike J., Kimura H., Narita M.et al.. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016; 48:1267–1272. [DOI] [PubMed] [Google Scholar]
- 13. Law M.J., Lower K.M., Voon H.P., Hughes J.R., Garrick D., Viprakasit V., Mitson M., De Gobbi M., Marra M., Morris A.et al.. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell. 2010; 143:367–378. [DOI] [PubMed] [Google Scholar]
- 14. Valton A.L., Hassan-Zadeh V., Lema I., Boggetto N., Alberti P., Saintome C., Riou J.F., Prioleau M.N.. G4 motifs affect origin positioning and efficiency in two vertebrate replicators. EMBO J. 2014; 33:732–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bedrat A., Lacroix L., Mergny J.L.. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 2016; 44:1746–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Toshniwal P., Nguyen M., Guédin A., Viola H., Ho D., Kim Y., Bhatt U., Bond C.S., Hool L., Hurley L.H.et al.. TGF-beta-induced fibrotic stress increases G-quadruplex formation in human fibroblasts. FEBS Lett. 2019; 593:3149–3161. [DOI] [PubMed] [Google Scholar]
- 17. Gazanion E., Lacroix L., Alberti P., Gurung P., Wein S., Cheng M., Mergny J.L., Gomes A.R., Lopez-Rubio J.J.. Genome wide distribution of G-quadruplexes and their impact on gene expression in malaria parasites. PLoS Genet. 2020; 16:e1008917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saad M., Guédin A., Amor S., Bedrat A., Tourasse N.J., Fayyad-Kazan H., Pratviel G., Lacroix L., Mergny J.L.. Mapping and characterization of G-quadruplexes in the genome of the social amoeba dictyostelium discoideum. Nucleic Acids Res. 2019; 47:4363–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bohalova N., Cantara A., Bartas M., Kaura P., Stastny J., Pecinka P., Fojta M., Mergny J.L., Brazda V.. Analyses of viral genomes for G-quadruplex forming sequences reveal their correlation with the type of infection. Biochimie. 2021; 186:13–27. [DOI] [PubMed] [Google Scholar]
- 20. Brazda V., Porubiakova O., Cantara A., Bohalova N., Coufal J., Bartas M., Fojta M., Mergny J.L.. G-quadruplexes in H1N1 influenza genomes. BMC Genomics. 2021; 22:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brazda V., Kolomaznik J., Lysek J., Bartas M., Fojta M., Stastny J., Mergny J.L.. G4Hunter web application: a web server for G-quadruplex prediction. Bioinformatics. 2019; 35:3493–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huppert J.L., Balasubramanian S.. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005; 33:2908–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Todd A.K., Johnston M., Neidle S.. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005; 33:2901–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tu J., Duan M., Liu W., Lu N., Zhou Y., Sun X., Lu Z.. Direct genome-wide identification of G-quadruplex structures by whole-genome resequencing. Nat. Commun. 2021; 12:6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adrian M., Heddi B., Phan A.T.. NMR spectroscopy of G-quadruplexes. Methods. 2012; 57:11–24. [DOI] [PubMed] [Google Scholar]
- 26. Luo Y., Granzhan A., Verga D., Mergny J.-L.. FRET-MC: a fluorescence melting competition assay for studying G4 structures in vitro. Biopolymers. 2021; 112:e23415. [DOI] [PubMed] [Google Scholar]
- 27. Lacroix L., Seosse A., Mergny J.L.. Fluorescence-based duplex-quadruplex competition test to screen for telomerase RNA quadruplex ligands. Nucleic Acids Res. 2011; 39:e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Cian A., DeLemos E., Mergny J.-L., Teulade-Fichou M.-P., Monchaud D.. Highly efficient G-quadruplex recognition by bisquinolinium compounds. J. Am. Chem. Soc. 2007; 129:1856–1857. [DOI] [PubMed] [Google Scholar]
- 29. Le D.D., Di Antonio M., Chan L.K., Balasubramanian S.. G-quadruplex ligands exhibit differential G-tetrad selectivity. Chem. Commun. (Camb.). 2015; 51:8048–8050. [DOI] [PubMed] [Google Scholar]
- 30. Madeira F., Park Y.M., Lee J., Buso N., Gur T., Madhusoodanan N., Basutkar P., Tivey A.R.N., Potter S.C., Finn R.D.et al.. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019; 47:W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Needleman S.B., Wunsch C.D.. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970; 48:443–453. [DOI] [PubMed] [Google Scholar]
- 32. Del Villar-Guerra R., Trent J.O., Chaires J.B.. G-Quadruplex secondary structure obtained from circular dichroism spectroscopy. Angew. Chem. Int. Ed Engl. 2018; 57:7171–7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mergny J.L., Li J., Lacroix L., Amrane S., Chaires J.B.. Thermal difference spectra: a specific signature for nucleic acid structures. Nucleic Acids Res. 2005; 33:e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arthanari H., Basu S., Kawano T.L., Bolton P.H.. Fluorescent dyes specific for quadruplex DNA. Nucleic Acids Res. 1998; 26:3724–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Renaud de la Faverie A., Guédin A., Bedrat A., Yatsunyk L.A., Mergny J.L.. Thioflavin T as a fluorescence light-up probe for G4 formation. Nucleic Acids Res. 2014; 42:e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xie X., Zuffo M., Teulade-Fichou M.P., Granzhan A.. Identification of optimal fluorescent probes for G-quadruplex nucleic acids through systematic exploration of mono- and distyryl dye libraries. Beilstein J. Org. Chem. 2019; 15:1872–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lane A.N., Chaires J.B., Gray R.D., Trent J.O.. Stability and kinetics of G-quadruplex structures. Nucleic Acids Res. 2008; 36:5482–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gray R.D., Trent J.O., Arumugam S., Chaires J.B.. Folding landscape of a parallel G-quadruplex. J. Phys. Chem. Lett. 2019; 10:1146–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu S., Zhan J., Man B., Jiang S., Yue W., Gao S., Guo C., Liu H., Li Z., Wang J.et al.. Real-time reliable determination of binding kinetics of DNA hybridization using a multi-channel graphene biosensor. Nat. Commun. 2017; 8:14902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nguyen T.Q.N., Lim K.W., Phan A.T.. Folding kinetics of G-quadruplexes: duplex stem loops drive and accelerate G-quadruplex folding. J. Phys. Chem. B. 2020; 124:5122–5130. [DOI] [PubMed] [Google Scholar]
- 41. Bhattacharyya D., Mirihana Arachchilage G., Basu S.. Metal cations in G-quadruplex folding and stability. Front Chem. 2016; 4:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Harkness R.W., Hennecker C., Grun J.T., Blumler A., Heckel A., Schwalbe H., Mittermaier A.K.. Parallel reaction pathways accelerate folding of a guanine quadruplex. Nucleic Acids Res. 2021; 49:1247–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Han H., Cliff C.L., Hurley L.H.. Accelerated assembly of G-Quadruplex structures by a small molecule. Biochemistry. 1999; 38:6981–6986. [DOI] [PubMed] [Google Scholar]
- 44. De Cian A., Mergny J.L.. Quadruplex ligands may act as molecular chaperones for tetramolecular quadruplex formation. Nucleic Acids Res. 2007; 35:2483–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aznauryan M., Noer S.L., Pedersen C.W., Mergny J.-L., Teulade-Fichou M.-P., Birkedal V.. Ligand binding to dynamically populated G-quadruplex DNA. ChemBioChem. 2021; 22:1811–1817. [DOI] [PubMed] [Google Scholar]
- 46. Mergny J.-L., Phan A.-T., Lacroix L.. Following G-quartet formation by UV-spectroscopy. FEBS Lett. 1998; 435:74–78. [DOI] [PubMed] [Google Scholar]
- 47. Alberti P., Mergny J.-L.. DNA duplex–quadruplex exchange as the basis for a nanomolecular machine. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ivanov K.P. The development of the concepts of homeothermy and thermoregulation. J. Therm. Biol. 2006; 31:24–29. [Google Scholar]
- 49. Bonnat L., Bar L., Gennaro B., Bonnet H., Jarjayes O., Thomas F., Dejeu J., Defrancq E., Lavergne T.. Template-Mediated stabilization of a DNA G-Quadruplex formed in the HIV-1 promoter and comparative binding studies. Chemistry. 2017; 23:5602–5613. [DOI] [PubMed] [Google Scholar]
- 50. Bonnat L., Dautriche M., Saidi T., Revol-Cavalier J., Dejeu J., Defrancq E., Lavergne T.. Scaffold stabilization of a G-triplex and study of its interactions with G-quadruplex targeting ligands. Org. Biomol. Chem. 2019; 17:8726–8736. [DOI] [PubMed] [Google Scholar]
- 51. Rauzan B., McMichael E., Cave R., Sevcik L.R., Ostrosky K., Whitman E., Stegemann R., Sinclair A.L., Serra M.J., Deckert A.A.. Kinetics and thermodynamics of DNA, RNA, and hybrid duplex formation. Biochemistry. 2013; 52:765–772. [DOI] [PubMed] [Google Scholar]
- 52. Cheatham T.E., Kollman P.A.. Molecular dynamics simulations highlight the structural differences among DNA:DNA, RNA:RNA, and DNA:RNA hybrid duplexes. J. Am. Chem. Soc. 1997; 119:4805–4825. [Google Scholar]
- 53. Lesnik E.A., Freier S.M.. Relative thermodynamic stability of DNA, RNA, and DNA:RNA hybrid duplexes: relationship with base composition and structure. Biochemistry. 1995; 34:10807–10815. [DOI] [PubMed] [Google Scholar]
- 54. Gyi J.I., Lane A.N., Conn G.L., Brown T.. The orientation and dynamics of the C2′-OH and hydration of RNA and DNA·RNA hybrids. Nucleic Acids Res. 1998; 26:3104–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Weldon C., Eperon I.C., Dominguez C.. Do we know whether potential G-quadruplexes actually form in long functional RNA molecules. Biochem. Soc. Trans. 2016; 44:1761–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available in the supplementary information section.