


Abstract
Protein synthesis in eukaryotic cell is spatially and structurally compartmentalized that ensures high efficiency of this process. One of the distinctive features of higher eukaryotes is the existence of stable multi-protein complexes of aminoacyl-tRNA synthetases and translation elongation factors. Here, we report a quaternary organization of the human guanine-nucleotide exchange factor (GEF) complex, eEF1B, comprising α, β and γ subunits that specifically associate into a heterotrimeric form eEF1B(αβγ)3. As both the eEF1Bα and eEF1Bβ proteins have structurally conserved GEF domains, their total number within the complex is equal to six. Such, so far, unique structural assembly of the guanine-nucleotide exchange factors within a stable complex may be considered as a ‘GEF hub’ that ensures efficient maintenance of the translationally active GTP-bound conformation of eEF1A in higher eukaryotes.
INTRODUCTION
Polypeptide synthesis on the ribosome requires aminoacylated tRNAs (aa-tRNAs) and a number of protein factors (1). To provide the substrate for polypeptide synthesis, translation elongation factor 1A (eEF1A) in a GTP-dependent manner binds aa-tRNA and delivers it to the ribosomal A-site. If the correct codon-anticodon interaction occurs, the ribosome induces GTP cleavage on eEF1A and promotes the release of GDP-bound eEF1A from the A-site (1). In higher eukaryotes, the translation elongation factor complex, eEF1B, mediates the GDP/GTP exchange on eEF1A, thus, restoring its active conformation. This complex consists of the eEF1Bα, eEF1Bβ and eEF1Bγ subunits (2). Herein we use the nomenclature for translation elongation factors proposed by Merrick and Nyborg (3). In the UniProtKB database, eEF1Bα is described as Elongation factor 1-beta (EF1B_human, accession number P24534), eEF1Bβ—as Elongation factor 1-delta (EF1D_human, accession number P29692), and eEF1Bγ—as Elongation factor 1-gamma (EF1G_human, accession number P26641). Both eEF1Bα and eEF1Bβ have guanine-nucleotide exchange (GEF) activity, whereas eEF1Bγ is thought to be a structural component of the complex (2). To accomplish the guanine-nucleotide exchange reaction, eEF1B assembles with eEF1A into a ‘heavy’ complex known as eEF1H (4). Besides, eEE1B forms a stable complex with valyl-tRNA synthetase (VRS-eEF1B) (5,6). The presence of this enzyme in the GEF complex facilitates the direct transfer (channeling) of valyl-tRNA from the enzyme to eEF1A*GTP (7).
Although the subunits composing eEE1B are known, their number and how they combine within this complex remains unclear. Up to date, several models of eEE1B structural organization have been proposed, however, there are significant inconsistencies among them (8). According to the simplest model, eEF1Bα and eEF1Bβ bind to the same eEF1Bγ subunit via their N-terminal domains to form the eEF1Bαβγ complex (9). Another model assumes that eEF1Bγ creates a dimeric core, and eEF1Bα and eEF1Bβ bind to the separate eEF1Bγ subunits to form the eEF1Bαβγ2 complex (10). The structural role for the catalytic eEF1Bα and eEF1Bβ subunits was also suggested. The protomer eEF1Bαβγ complex was proposed to dimerize (6) or even trimerize (11) in a larger entity via the leucine-zipper motif of the eEF1Bβ subunit. In turn, the protomer eEF1Bαβγ2 complex was proposed to dimerize via the eEF1Bα subunit (10). Thus, neither the reconstitution experiments nor analysis of the natively purified complexes resulted in the unambiguous determination of the eEF1B quaternary organization (6,10–13). It seems that due to high aggregation propensity of this complex, its structural characterization appeared to be a difficult task.
In this study, we decipher a quaternary architecture of the human eEF1B complex containing α, β and γ subunits. We show that eEF1Bβ self-associates in a stable trimer and its leucine-zipper motif is responsible for trimerization. eEF1Bγ carries distinct binding sites for the eEF1Bα and eEF1Bβ subunits and interacts with them in equimolar stoichiometry. Hence, eEF1Bα, eEF1Bβ and eEF1Bγ specifically associate into a heterotrimeric complex, eEF1B(αβγ)3, which encompasses six highly conserved GEF domains. We suggest that such multi-GEF assembly may ensure the efficient restoration of the GTP-bound eEF1A conformation for the translating ribosomes in higher eukaryotes.
MATERIALS AND METHODS
Protein expression and purification
The recombinant plasmid expressing N-terminally His-tagged full-length human eEF1Bα was prepared as follows: eEF1Bα ORF was excised from the pGEX6P-1/eEF1Bα construct (14) and cloned into the pET28α(+) vector (Novagene, Madison, WI, USA). The recombinant protein was purified to homogeneity by a two-step chromatographic procedure: affinity chromatography on a Ni-NTA column (Qiagen, Valencia, CA, USA) and anion-exchange chromatography on a HighTrapQ column (GE Healthcare, Buchinghamshire, UK) using linear NaCl gradient from 250 to 450 mM. The expression and purification procedures for eEF1Bα(19–225) (14); for full-length human eEF1Bγ and its truncated form eEF1Bγ(229–437) (15); for full-length eEF1Bβ and its truncated forms eEF1Bβ(43–281) and GST-eEF1Bβ(78–118) (16) were previously published.
Analytical gel filtration of proteins and protein complexes
The interaction between different proteins partners was studied by size-exclusion chromatography on a Superose 6 HR 10/30 column (24 ml, GE Healthcare) as previously described in (14,16). For the formation of binary eEF1Bαγ and eEF1Bβγ, and ternary eEF1Bαβγ complexes, the respective purified full-length proteins were mixed in buffer containing 25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 10% glycerol, 5 mM 2-mercaptoethanol in the final volume 0.12 ml and incubated for 5 min at 37°C. Then the protein mixture was centrifuged at 16 000 g for 15 min (RT) and loaded onto a Superose 6 HR 10/30 column. The final concentration of subunits in the binary complexes incubation mixtures was 10 μM and in the ternary complex incubation mixture was 8 μM. The interaction of full-length eEF1Bγ with the N-terminally truncated forms of eEF1Bα and eEF1Bβ, as well as the interaction of full-length eEF1Bα and eEF1Bβ with the C-terminal domain of eEF1Bγ were examined in the same way as described for the full-length proteins.
Analytical ultracentrifugation
Analytical ultracentrifugation (AUC) experiment was performed using a ProteomeLab XL-I analytical ultracentrifuge (Beckman-Coulter, Indianapolis, USA), equipped with An-60 Ti analytical rotor, using absorbance optics at 280 nm as described previously (14). Briefly, in the sedimentation velocity experiments, protein sample (400 μl) and buffer reference (410 μl) solutions were loaded onto 12 mm double-sector Epon charcoal-filled centerpieces (Beckman-Coulter). For each experiment, the rotor speed and temperature are indicated in the figure legends. The sedimentation velocity multiple scans at various time-points were fitted to a continuous size distribution model using Sedfit (17). All size distributions were solved and regularized at a confidence level of 0.95 by maximum entropy, using the best-fit mean anhydrous frictional ratio (f/f0). We calculated also a hydrodynamic parameter Smax/S that allows evaluating the shape of the proteins and the protein complexes. In this ratio, Smax is the maximum possible sedimentation coefficient for a protein of the given mass, corresponding to a sphere of the minimum diameter to contain this mass without water, and S is the sedimentation coefficient S20,w for the individual protein or protein complex estimated by size-distribution analysis. Smax was calculated using the formula Smax = 0.00361(Mr)2/3, where Mr is the molecular mass of the protein or protein complex in Daltons. Smax/S is in the range from 1.5 to 1.9 for moderately elongated proteins and from two to three for highly elongated proteins (18).
The sedimentation equilibrium experiments were done as described previously (14). Briefly, the protein samples (0.1 ml) were loaded into the sample channels, and a buffer solution (0.11 ml) was loaded into reference channels of six-channel Epon charcoal-filled centerpieces (Beckman–Coulter). For each experiment, the rotor speed and temperature are indicated in the figure legends. The sedimentation equilibrium absorbance data were collected every four hours. The scan obtained at a single rotor speed or the scans obtained at different rotor speeds (multispeed equilibrium data) were then fitted to a non-interacting discrete species model assuming a single species by using SEDPHAT (19) with Equation (1):
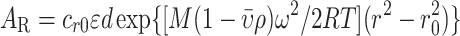 |
(1) |
in which r denotes the distance from the center of rotation; r0 is the arbitrary reference radius; ω is the angular velocity; T is the absolute temperature of the rotor; R is the gas constant; 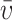 is the partial specific volume; ρ is the solvent density; ϵ is the extinction coefficient; d is the optical path length, and cr0 is the concentration at the reference radius. For a multispeed global data analysis at each channel, a single base-line parameter was included as a floating parameter common to all rotor speeds. The time-invariant and radial-invariant noise was also fitted for better fitting quality.
is the partial specific volume; ρ is the solvent density; ϵ is the extinction coefficient; d is the optical path length, and cr0 is the concentration at the reference radius. For a multispeed global data analysis at each channel, a single base-line parameter was included as a floating parameter common to all rotor speeds. The time-invariant and radial-invariant noise was also fitted for better fitting quality.
If the experimental data could not be fitted to the single species model, the monomer–dimer equilibrium model in SEDPHAT corresponding to Equation (2) was applied:
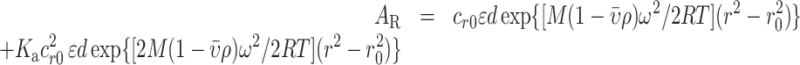 |
(2) |
in which Ka denotes the association constant of the dimer.
The buffer solution used for the sedimentation velocity and equilibrium experiments contained 25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 5% glycerol (v/v) and 1 mM dithiothreitol. The solvent density of 1.0216 g/cm3 and viscosity of 0.01962 Poise at 2.3°C, and 1.02164 g/cm3 and 0.01857 Poise at 4°C were calculated using Sednterp software (https://www.spinanalytical.com/auc-software.php). Before analytical ultracentrifugation, the individual eEF1Bα, eEF1Bβ, eEF1Bγ proteins and eEF1Bαγ, eEF1Bβγ, eEF1Bαβγ complexes were additionally purified on a Superose 6 HR 10/30 column (GE Healthcare) equilibrated in the buffer solution specified above.
Partial specific volume (cm3/g) and extinction coefficient (M–1cm–1) for full-length eEF1Bβ were calculated using SEDNTERP software to be 0.72259 and 22 590, respectively; those for GST-eEF1Bβ(7–118) were 0.73865 and 43 110; those for eEF1Bβ(117–281) were 0.72446 and 14 110; those for eEF1Bγ were 72 288 and 87 230; those for the eEF1Bαγ complex (1:1) were 0.72225 and 117 300; those for the eEF1Bβγ complex (1:1) were 0.72274 and 109 820; those for the eEF1Bαβγ complex (1:1:1) were 0.72330 and 139 855. All sedimentation velocity and equilibrium graphs were prepared in GUSSI program (version 1.0.8d, Chad Brautigam, UT Southwestern).
Hydrogen–deuterium exchange coupled to mass spectrometry (HDX-MS)
The HDX-MS experiments were carried out as previously described in (20). Briefly, freshly prepared individual eEF1Bα, eEF1Bβ and eEF1Bγ proteins were additionally purified on a Superose 6 HR 10/30 column (GE Healthcare), dialyzed (25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 55% glycerol and 5 mM 2-mercaptoethanol) and kept at –20°C. The respective protein complexes were prepared as follows: 10 μM of each subunit were mixed and incubated for 5 min at 37°C, then concentrated on the AmiconUltra-4 (50 kDa, Merck) membrane to the volume of 200 μl and injected onto a Superose 6 HR 10/30 column. The most concentrated fractions of each complex were combined and dialyzed against the same buffer indicated above. The initial concentrations of samples used for the HDX-MS experiments: eEF1Bα – 56.6 μM, eEF1Bβ – 36.4 μM, eEF1Bγ – 46 μM, eEF1Bαγ – 39.2 μM, eEF1Bβγ – 35 μM, eEF1Bαβγ – 53.6 μM.
A 5 μl aliquot of the individual protein or the protein complex stock solution was combined with 45 μl of D2O (99.8% Cambridge Isotope Laboratories) reaction buffer containing 25 mM Tris–HCl pH 7.5, 150 mM NaCl, 150 mM KSCN, and 5 mM 2-mercaptoethanol and incubated at 20°C for 10 s, 1 min, 5 min, 25 min or 2.5 h before quenching by addition 10 μL of 2 M glycine pH 2.5 in D2O. The samples were immediately frozen in the liquid nitrogen and stored at –80°C until use. Out-time point controls were performed by incubation of the protein in D2O buffer for 24 h to obtain maximum exchange for each peptide and then, quenched with 10 μl of 2 M glycine. The deuteration level was calculated and denoted as 100% exchange. Mass spectrometry measurements and data analysis were done as described in (21). The experiments were repeated three times, the results represent the mean of all replicates. The peptides were identified using ProteinLynx Global Server software (PLGS, Waters) and further filtered in the DynamX 3.0 program (Waters) with the following acceptance criteria: minimum intensity threshold of 3000, minimum products per amino acids of 0.3, minimum score of 7.5 and theoretical value for parent ions below 10 ppm. The values reflecting experimental mass of each peptide in all possible states, replicates, time points and charge states were exported from the DynamX 3.0 and further data analysis was carried out using in house written script (21).
To depict the kinetic of exchange for each peptide, we built a plot with an experimentally measured level of H/D exchange (in %) at 10 s, 1, 5, 25 and 150 min. The obtained curve dissects the kinetic plot into two parts. The area above the kinetic curve was integrated overall incubation time and divided by the whole area of the kinetic plot. The obtained value we call ‘aggregated protection’ of the peptide, which may be within the range from 0 (no protection) to 1 (full protection). We distinguish three categories of peptides with respect to their aggregated protection value: <0.05—the absence of aggregated protection; >0.05 and <0.15—weak aggregated protection; >0.15—high aggregated protection. The peptides with no and weak aggregated protection belong to dynamically structured regions while the peptides with high aggregated protection—to rigidly structured regions. ‘Differential aggregated protection’ graph shows the difference between the values of aggregated protection measured for the same peptide in different states, namely free and bound to a partner. A positive value of the differential aggregated protection indicates that the peptide becomes more protected in the complex with a partner, while a negative value means a decrease of protection.
Native gel electrophoresis of protein complexes
The eEF1Bαγ complex was prepared as follows: 5 μM eEF1Bα was mixed with increasing (2–7 μM) concentrations of eEF1Bγ and vice versa 5 μM eEF1Bγ was mixed with increasing (2–7 μM) concentrations of eEF1Bα in buffer containing 25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 10% glycerol, 5 mM 2-mercaptoethanol in a final volume 20 μl. The eEF1Bβγ complex was prepared in the same way.
The complexes between eEF1Bβ and eEF1A2 were prepared as follows: 10 μM eEF1A2 was mixed with increasing (0.36–21 μM) concentrations of eEF1Bβ and 150 μM GDP in buffer containing 25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 10% glycerol, 5 mM 2-mercaptoethanol in a final volume 25 μl.
The eEF1Bαβγ complex was prepared by mixing three individual subunits at 6 μM concentrations in buffer containing 25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 10% glycerol, 5 mM 2-mercaptoethanol and incubated for 5 min at 37°C. Titration of the eEF1Bαβγ complex by eEF1A2 was performed as follows: 3 μM eEF1Bαβγ complex was mixed with increasing (3–30 μM) concentrations of eEF1A2 and 150 μM GDP in buffer indicated above in a final volume 20 μl.
The protein mixtures were incubated for 5 min at 37°C and loaded onto a 1.5% agarose gel (for eEF1B and eEF1Bβγ complexes) and 1% agarose gel (for the eEF1A2-eEF1Bβ and eEF1A2-eEF1Bαβγ complexes) containing 89 mM Tris-borate, pH 8.3. The gel was run at 100 V/31–34 mA for 2–3 h at room temperature, then stained and photographed as described previously (14).
Dynamic light scattering
To measure a hydrodynamic radius of eEF1Bα we used the Dynamic Light Scattering technique (DLS) and the Adaptive Correlation approach described in (22). DLS experiments were performed using a Zetasizer Nano ZS (Malvern Panalytical Ltd, UK) at 25°C with a scattering angle of 173° in air. All samples (1 ml) were measured in a 1 cm glass cuvette. Briefly, three measurements for each eEF1Bα concentration were done. Each measurement included 30 sub-measurements with duration time of 1, 2 and 3 s. The steady-state sub-measurements were analyzed using cumulants analysis. The correlation functions of the steady-state sub-measurements were averaged to report the hydrodynamic radius (Zave) and polydispersity index (PdI) values. Zave values were obtained for four different eEF1Bα concentrations. RH0 – a hydrodynamic radius of eEF1Bα at the infinite dilution was calculated from the plot of Zave versus eEF1Bα concentration by extrapolation to zero concentration.
Prior to the DLS measurements, eEF1Bα was subjected to gel filtration on a Sephacryl S200 column (GE Healthcare) equilibrated in buffer, containing 25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 5% glycerol (v/v) and 1 mM dithiothreitol. Fractions with the highest eEF1Bα concentration were centrifuged at 16 000 g for 2.5 hours at 10°C. Buffer was prepared using ultrapure deionized water, filtered (pore size 0.2 μm) and degassed.
Homology structure modeling and molecular docking
The unstructured regions in proteins we predicted by a MetaDisorderMD2 meta-server (23). The 3D structure models of eEF1Bα, eEF1Bβ (monomer) and eEF1Bγ were generated by Modeller (version 9.14) (24). The unstructured regions in the proteins were modeled using the loops reconstruction option in this program (25). Further high-resolution protein structure refinement for the best predicted 3D model was done by ModRefiner (26). Additionally, all structures were verified using the MolProbity web server (http://molprobity.biochem.duke.edu) (27) and refined by YASARA Energy Minimization Server (28). Visualization and analysis of the protein were done using UCSF Chimera (29).
To model full-length eEF1Bα we used the available structures of its isolated GEF domain (PDB ID: 1B64), its N-terminal domain complexed with the GST-like domain of eEF1Bγ (PDB ID: 5DQS) and highly homologous C-terminal region of eEF1Bβ (PDB ID: 2N51) as templates. From the ensemble of eEF1Bα conformations created by Modeller, we selected a model with the hydrodynamic radius that matches the experimentally measured RH0 value.
eEF1Bβ monomer was modeled using the available structure of its C-terminal region (PDB ID: 2N51) and reported model of its N-terminal domain possessing a dynamic α-helical organization (20). Five structural ensembles with 40 structures in each were generated by Modeller. The best five structures were selected from each ensemble using the Discrete Optimized Protein Energy (DOPE) and Modeller Objective Function (MOF) scores (30) (Supplementary, Table S1). The model 1, which has the best DOPE score (Supplementary Table S1), was chosen for further computing of the eEF1Bβ trimer. Symmetric docking of the eEF1Bβ monomers was done by the SymmDock webserver (31) taking into account that the LZ-motif is responsible for trimerization. The best model of eEF1Bβ trimer was chosen according to the geometric shape complementarity score and then refined by the YASARA Energy Minimization Server (the energy value – 500 712 kJ/mol and energy score – 0.21).
Homology modeling of eEF1Bγ was performed using a structure of its C-terminal domain (PDB ID: 1PBU), structures of its GST-like N-terminal domain complexed with the N-terminal domain of eEF1Bα (PDB ID: 5DQS) and with the short N-terminal peptide of eEF1Bβ (PDB ID: 5JPO) as templates. From the ensemble of eEF1Bγ conformations created by Modeller, we selected one model with the extended conformation taking into account a moderately elongated shape of eEF1Bγ in solution.
In silico docking between the eEF1Bβ and eEF1Bγ proteins was performed using PatchDock web server (32). The N-terminal domain (residues 1–77) of eEF1Bβ was set as ligand and the N-terminal domain (residues 1–210) of eEF1Bγ was set as receptor. The coordinates for eEF1Bβ(1–77) and eEF1Bγ(1–210) were from the respective atomistic models mentioned above. Docking results were individually inspected and compared with HDX-MS data for the eEF1Bβγ complex. The best high scoring model was used for the refinement and re-scoring step with FireDock (33). Analysis and visualization of the molecular interfaces between proteins were performed by Cocomaps web server (34,35). The docking procedure between the full-length eEF1Bβ and eEF1Bγ proteins was done in the same way.
RESULTS
Structural organization of eEF1Bα
eEF1Bα is a monomeric non-globular protein with a moderately elongated shape (14). Two conserved regions can be delineated in its primary structure: the non-catalytic N-terminal domain (residues 1–62) and the catalytic C-terminal region (residues 97–225). Both parts are connected by a non-conserved linker (Figure 1A). The structure of the C-terminally located GEF domain (residues 135–225) of human eEF1Bα was solved by NMR (36). The presence of the structurally independent α-helical central acidic region (CAR) upstream the GEF domain in human eEF1Bα was suggested based on the structure of a long eEF1Bβ C-terminal fragment, which amino acid sequence is highly homologous to eEF1Bα (37). Besides, the crystallographic data on the eEF1Bα N-terminal domain (residues 1–90) complexed with the GST-like domain of eEF1Bγ are present in the PDB database (PDB ID: 5DQS). However, the complete three-dimensional structure of full-length eEF1Bα has never been reported.
Figure 1.
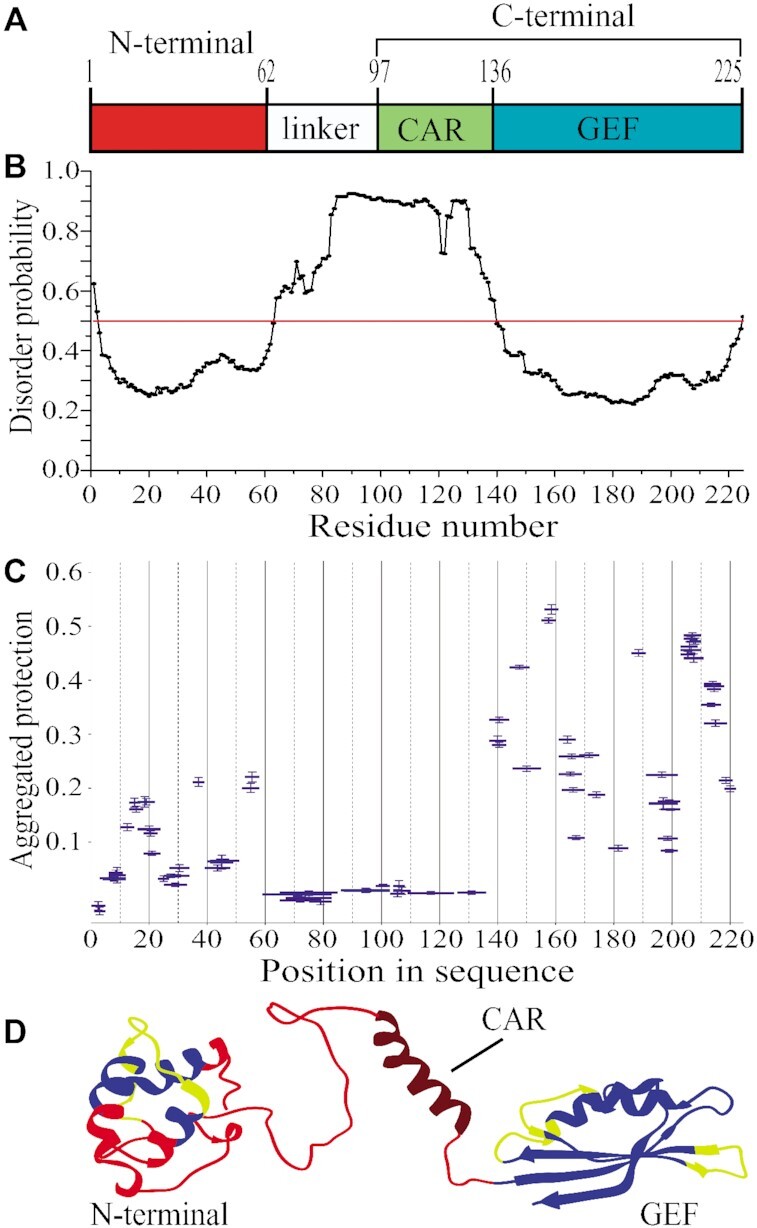
Structural organization of full-length eEF1Bα. (A) Schematic representation of the eEF1Bα domain structure. Abbreviations: CAR – the central acidic region, GEF – the guanine-nucleotide exchange factor domain. (B) Prediction of the disordered regions in eEF1Bα. All residues whose disorder probability is over 0.5 (red line) are considered as disordered. (C) The aggregated protection plot of the eEF1Bα peptides. The aggregated protection values for peptides (mean ± SD, n = 3 measurements) are plotted versus their position in the protein sequence. (D) The 3D model of eEF1Bα colored according to the HDX-MS data. Red color indicates unprotected and unstructured regions (<0.05), dark red – the CAR domain that displays no protection, but is predicted to have α-helical organization, yellow – weakly protected dynamic segments (0.05–0.15), blue – highly protected rigidly structured regions (>0.15).
MetaDisorderMD2 meta-server predicts high disorder probability for fragment 64–139 that includes the linker region and the CAR domain (Figure 1A and B). To confirm this prediction, we characterized the structural dynamics of eEF1Bα by the method of hydrogen-deuterium exchange coupled to mass spectrometry (HDX-MS) (38). The unstructured and highly dynamic regions exchange with D2O very rapidly (39). In the aggregated protection plot of eEF1Bα, the peptides covering the linker region and the CAR domain show near zero protection against H/D exchange (Figure 1C). The majority of peptides from the GEF domain display high protection except few weakly protected segments. In turn, unprotected, weakly and highly protected peptides are present in the N-terminal domain (Figure 1C). Thus, the linker region and the CAR domain are highly dynamic that is consistent with the predicted disorder probability profile for this protein (Figure 1B and C).
We created a 3D model of eEF1Bα (Figure 1D) that matches the experimentally measured hydrodynamic radius, RH0 = 3.35 ± 0.24 nm, for this protein (Supplementary Figure S1) and agrees well with the HDX-MS data. The peptides comprising the core of the N-terminal three-helix bundle are protected against H/D exchange (Figure 1D, colored in blue), while the loop regions with the adjacent parts of α-helices show weak or no protection suggesting their dynamic conformation (Figure 1D, colored in yellow and red, respectively). The linker region is disordered that is consistent with the absence of protection against H/D exchange (Figure 1C and D, colored in red). Notably, no protection was detected for the peptides composing the CAR domain – an isolated α-helical element in eEF1Bα (Figure 1C and D). This α-helix is located between structurally dynamic linkers and may undergo local fluctuations that result in H-bonds breaking and exposure of the amide hydrogens to attack by deuterium (40). The conventional HDX method used in this study most probably is not sensitive enough to detect weakly structured (weak hydrogen bonding) and/or rapidly fluctuating secondary elements (41). We colored the unprotected α-helical CAR domain in dark red in order to distinguish it from the unstructured regions (Figure 1D). The GEF-domain represents a compact and tightly packed two-layer α/β sandwich in which most of peptides have substantial protection (Figure 1D, colored in blue) excluding few loop regions, which probably are conformationally flexible (Figure 1D, colored in yellow).
Hence, we established that eEF1Bα consists of two rigidly structured domains connected by a long dynamic linker region.
Structural organization of eEF1Bβ
It has been shown that eEF1Bβ forms oligomers in solution (12,16). To elucidate the exact number of monomers in the oligomeric eEF1Bβ structure, we performed analytical ultracentrifugation experiments. eEF1Bβ sedimented as one major species with a molecular mass of 92 ± 4 kDa calculated for the best-fit frictional ratio f/f0 = 1.97 ± 0.07 (Supplementary Figure S2A). The hydrodynamic parameter Smax/S was estimated to be 2.02 ± 0.04 that is characteristic of highly elongated proteins (18). Analysis of the sedimentation equilibrium data (Supplementary Figure S2B) gave the molecular mass value of 97.6 ± 2.4 kDa that corresponds to the theoretical mass of the eEF1Bβ trimer (95.7 kDa). Therefore, we conclude that recombinant eEF1Bβ self-associates in a stable trimer with a highly elongated shape.
Four conserved regions can be delineated in the primary structure of eEF1Bβ (Figure 2A): the N-terminal domain, leucine-zipper (LZ) motif, CAR domain, and GEF domain. The isolated N-terminal domain is a monomer with a dynamic α-helical organization (20). The structure of the C-terminal fragment (residues 153–281) including CAR and GEF domains was solved by NMR (37). This fragment is also monomeric in solution. Thus, it leaves a trimer-forming role to the middle part of the eEF1Bβ molecule that comprises the LZ motif and the linker region (Figure 2A). Previously, we have reported that the chimeric GST-eEF1Bβ(78–118) protein, which consists of the LZ-motif and GST, forms oligomers in vitro (16). Here, using sedimentation velocity and equilibrium approaches we established that GST-eEF1Bβ(78–118) forms trimers and hexamers in solution (Supplementary Figure S3) confirming the intrinsic trimerization capacity of LZ-motif. Additionally, the possible contribution of the linker region into eEF1Bβ self-association was tested by using its truncated form, eEF1Bβ(117–281), that comprises the linker region, CAR and GEF domains, but not LZ-motif (Figure 2A). Sedimentation equilibrium analysis proved that eEF1Bβ(117–281) is a monomeric protein (Supplementary Figure S3C). Hence, we conclude that the linker region does not mediate eEF1Bβ self-association. Altogether, the obtained results clearly indicate that the LZ-motif is responsible for eEF1Bβ trimerization.
Figure 2.

Structural organization of full-length eEF1Bβ. (A) Domain organization of eEF1Bβ. Abbreviations: LZ – the leucine-zipper motif, CAR – the central acidic region, GEF – the guanine-nucleotide exchange factor domain. (B) Structural model of the LZ-motif built by CCBuilder 2.0. Three α-helices twist around each other to form a bundle with following parameters: radius – 5.6 Å, interface angle – 19º, pitch – 61.8 Å, number of residues per turn – 3.62. The model has the lowest (–425.6 kJ/mol) BUDE score (the interaction energy between the helices). All leucine residues involved onto assembly of the α-helical coiled-coils are shown by colored sticks. The amino acid sequence of the LZ-motif is shown on the top of the figure. (C) Prediction of the disordered regions in eEF1Bβ. All residues whose disorder probability is over 0.5 (red line) are considered as disordered. (D) The aggregated protection plot of eEF1Bβ peptides. The aggregated protection values for peptides (mean ± SD, n = 3 measurements) are plotted versus they position in the protein sequence. (E) The 3D-model of eEF1Bβ colored according to the HDX-MS data. Red color indicates unprotected and unstructured regions (<0.05), dark red – the CAR domain and the N-terminal α-helixes that display no protection, but are predicted to have α-helical organization, yellow – weakly protected dynamic segments (0.05-0.15), blue – highly protected rigidly structured regions (>0.15), black – the regions with missing peptides.
Typical LZ-motif consists of a periodic repetition of a leucine residue at every seventh position known as a heptad repeat and forms a continuous α-helix, which mediates dimerization and in some cases oligomerization of proteins (42,43). In the eEF1Bβ primary structure, a heptad repeat contains six leucine residues that occupy every seventh position and create a hydrophobic stripe along the helix (Figure 2B, upper part). This heptad repeat self-associates in the trimeric coiled-coil conformation (Figure 2B) according to CCBuilder software (44).
The LZ-motif, the GEF domain and a part of the N-terminal domain were predicted by MetaDisorderMD2 meta-server to be ordered, whereas the long linker region between the LZ-motif and the GEF domain, and two short regions within the N-terminal domain—disordered (Figure 2C). This prediction was further supported by the HDX-MS analysis of eEF1Bβ (Figure 2D). All peptides that constitute the LZ-motif display substantial protection against H/D exchange indicating a rigidly structured region. The most of the GEF domain peptides are highly protected except few weakly protected and unprotected segments (Figure 2D). The N-terminal domain, linker region and CAR domain display near zero protection indicating highly dynamic structures (Figure 2D). It has been shown that the CAR domain is an independent and structurally dynamic element of eEF1Bβ (37). The absence of protection for the N-terminal domain is also in agreement with the previous result obtained on the isolated eEF1Bβ(1–77) confirming its rapidly fluctuating tertiary structure (20). As mentioned above, the sensitivity of the conventional HDX method may be not sufficient to detect weakly structured and/or rapidly fluctuating secondary elements. Using homology modeling, we built an atomistic model of full-sized eEF1Bβ (Figure 2E) that correlate with the HDX-MS (Figure 2D) and analytical ultracentrifugation data (Supplementary Figure S2A). In Figure 2E, the elements of the CAR and N-terminal domain, which are highly dynamic according to HDX-MS but predicted to possess α-helical organization, are colored in dark red in order to differentiate them from the factual unstructured regions (colored in red). The tightly packed LZ-motif and GEF-domain are colored in blue except for weakly protected and unprotected loop regions of the latter, which most probably are conformationally flexible (Figure 2E, colored in yellow and red, respectively).
Thus, we conclude that eEF1Bβ is an elongated trimeric molecule, in which the monomers are kept together by the α-helical coil-coiled bundle. The C-terminal fragment of each monomer comprising highly dynamic linker and CAR, and rigidly structured GEF domain extends from one side of this bundle. Three α-helical N-terminal fragments are located at the opposite side.
Structural organization of eEF1Bγ
Purified recombinant eEF1Bγ has strong self-aggregation propensities. Its apparent molecular mass varies from 100 to 1000 kDa according to analytical size-exclusion chromatography (12,15,45). Here, we used the analytical ultracentrifugation analysis to describe the oligomeric state of full-length eEF1Bγ in more detail. The sedimentation velocity experiment revealed minor and major eEF1Bγ sedimentating species of 50 and 91 kDa, calculated for the best-fit frictional ratio f/f0 = 1.65, respectively (Supplementary Figure S4A). These values are close to those of monomer (52.6 kDa) and dimer (105.2 kDa). The calculated hydrodynamic parameter Smax/S of 1.74 for the monomer and 1.84 for the dimer indicates a moderately elongated shape of both species (18). The sedimentation equilibrium experiment showed that depending on protein concentration eEF1Bγ may form a mixture of dimeric and tetrameric forms, stable dimers, and a mixture of monomeric and dimeric forms (Supplementary Figure S4B). Different values of the apparent dissociation constant obtained at different protein concentrations and different centrifugation velocities indicate that self-association of eEF1Bγ is irreversible (Supplementary Figure S4B).
eEF1Bγ consists of two conserved domains connected by a lysine-rich linker (46) (Figure 3A). Importantly, MetaDisorderMD2 meta-server predicts with the highest probability for this inter-domain linker region (residues 215–280) to be disordered (Figure 3B). The HDX-MS analysis also showed the absence of protection against H/D exchange for this region (Figure 3C) confirming its high structural dynamics. In contrast, most peptides from the N- and C-terminal domains are substantially protected with few exceptions (Figure 3C).
Figure 3.
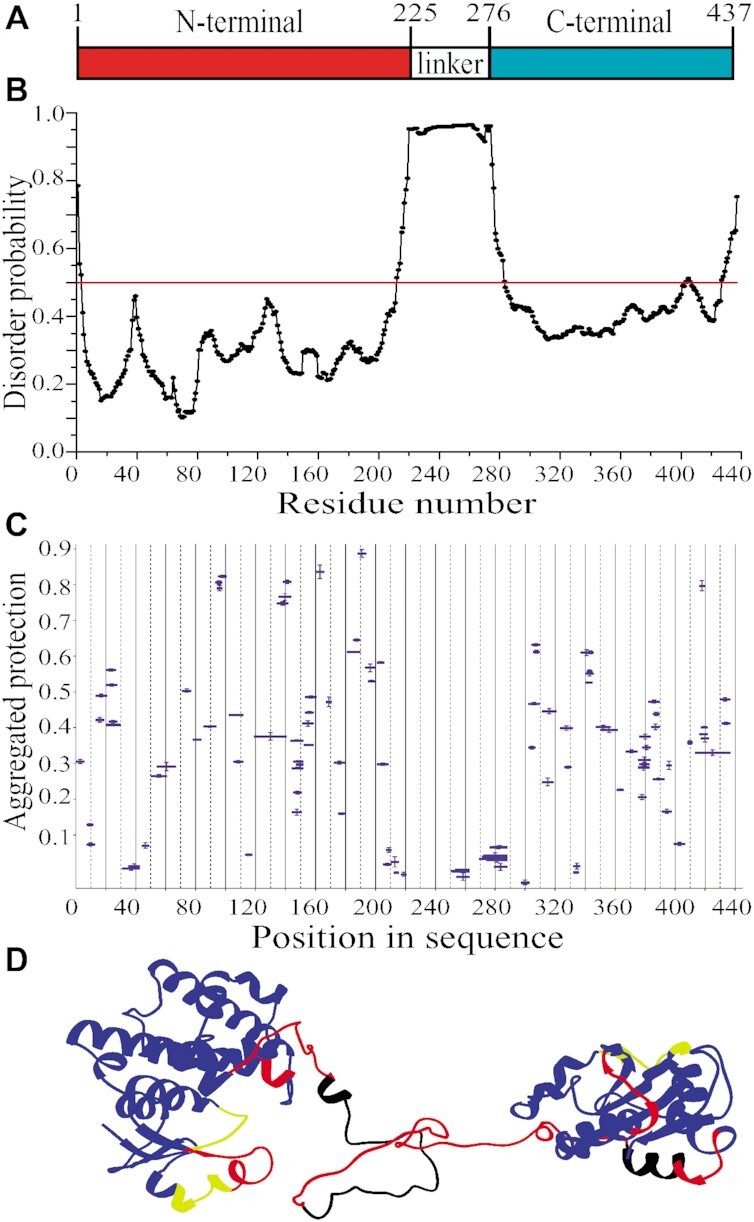
Structural organization of full-length eEF1Bγ. (A) Domain organization of eEF1Bγ. (B) Prediction of the disordered region in eEF1Bγ. All residues whose disorder probability is over 0.5 (red line) are considered as disordered. (C) The aggregated protection plot of the eEF1Bγ peptides. The aggregated protection values for peptides (mean ± SD, n = 3 measurements) are plotted versus their position in the protein sequence. (D) A 3D model of eEF1Bγ colored according to the HDX-MS data. Red color indicates unprotected and unstructured regions (<0.05), yellow – weakly protected dynamic segments (0.05–0.15), blue – highly protected rigidly structured regions (>0.15), black – the regions with missing peptides.
Until now, the 3D structure of full-length eEF1Bγ has not been reported. However, the structure of its isolated C-terminal domain (PDB ID: 1PBU) has been published (46) and the structures of its GST-like N-terminal domain in the complex with both the N-terminal domain of eEF1Bα (PDB ID: 5DQS) and short N-terminal peptide of eEF1Bβ (PDB ID: 5JPO) have been deposited in the PDB database. Using homology modeling we built an atomistic model of full-sized eEF1Bγ (Figure 3D) that is consistent with the HDX-MS data. Indeed, the majority of peptides from both folded domains display significant protection against H/D exchange (Figure 3C and D, colored in blue). Only few loop segments in both domains display weak or no protection probably due to their local conformational fluctuations (Figure 3D, colored in yellow and red, respectively). The long inter-domain linker region is disordered that also correlates with the absence of protection (Figure 3D, colored in red). Thus, we conclude that eEF1Bγ is a non-globular protein with a moderately elongated shape, which consists of two rigidly structured domains connected by a long highly dynamic linker region.
Reconstitution of the eEF1B complex
Several techniques were employed to estimate the stoichiometry of the subunits in the binary eEF1Bαγ and eEF1Bβγ, and ternary eEF1Bαβγ complexes. First, the native gel electrophoresis showed the formation of both the eEF1Bαγ and eEF1Bβγ complexes at equimolar concentrations of subunits (Supplementary Figure S5). Then, the molecular masses of the complexes were estimated by analytical size-exclusion chromatography, sedimentation velocity and equilibrium analysis (Figure 4, Supplementary Figures S6 and 7).
Figure 4.

Size-exclusion chromatography and sedimentation velocity analysis of the eEF1Bαγ, eEF1Bβγ and eEF1Bαβγ complexes. (Plots A, C and E) 100 μl of 10 μM indicated binary and ternary complexes or individual subunits were injected into a Superose 6 HR column. Inset: SDS-PAGE of the binary and ternary complexes. The central fractions of each protein complex peak were combined, concentrated and loaded (5 μg) onto 12% polyacrylamide gels. (Plots B, D and F) Left: absorbance scans of the sedimentation velocity data (symbols show only every third data point of every third scan for clarity) and best-fit boundary model from the c(s) analysis (solid lines). Residuals are indicated. Right: continuous size distribution analysis, c(s), plotted as a function of sedimentation coefficient. Inset: 2D grayscale ‘bitmap’ residual plot shows a high quality of fit. The initial concentration of the complexes was: eEF1Bαγ – 0.3 mg/ml (3.8 μM), eEF1Bβγ – 0.15 mg/ml (1.9 μM), eEF1Bαβγ – 0.36 mg/ml (3 μM).
The eEF1Bαγ complex (1:1 subunit ratio) demonstrated the apparent molecular mass of about 400 kDa in the gel filtration experiment (Figure 4A) that is five times higher than its theoretical value (79.9 kDa). Sedimentation velocity analysis of the same complex showed the presence of two protein species sedimenting with Sw = 1.973 S (S(20,w) = 3.998 S), and Sw = 2.716 S (S(20,w) = 5.503 S) that correspond to the molecular masses of 80 and 130 kDa, respectively, calculated with the best-fit friction ratio f/f0 = 1.68. These values are close to those of the heteromeric eEF1Bαγ and heterodimeric eEF1B(αγ)2 forms of this complex (Figure 4B, Supplementary Figure S6A). The sedimentation equilibrium analysis revealed the presence of the stable eEF1B(αγ)2 complex and eEF1Bαγ-eEF1B(αγ)2 mixture (Supplementary Figure S6B).
The eEF1Bβγ complex (1:1 subunit ratio) showed the apparent molecular mass of about 1000 kDa in analytical gel filtration (Figure 4C) that is almost twelve times higher than its theoretical value (84.4 kDa). Sedimentation velocity analysis of the same complex revealed the presence of two major protein species sedimenting at Sw = 4.30 S (S(20,w) = 8.729 S), and Sw = 7.66 S (S(20,w) = 15.551 S) that correspond to the molecular masses of 250 and 590 kDa, respectively (Figure 4D, Supplementary Figure S6C), calculated with the best-fit friction ratio f/f0 = 1.64. The molecular mass of the first species is close to the value of a heterotrimer eEF1B(βγ)3, whereas the second species represents supposedly a heterohexamer eEF1B(βγ)6. Sedimentation equilibrium analysis of eEF1Bβγ confirmed the presence of the heterotrimer-heterohexamer mixture in solution (Supplementary Figure S6D). However, the presence of some admixtures of self-associated single subunits and/or species with other eEF1Bβγ stoichiometry cannot be excluded.
The ternary eEF1Bαβγ complex (1:1:1 subunit ratio) migrated as a single peak of more than 1 MDa during analytical gel filtration on a Superose 6HR (Figure 4E). Sedimentation velocity analysis of the same complex showed the presence of three protein species sedimenting at Sw = 3.588 S (S(20,w) = 6.965 S), Sw = 5.021 S (S(20,w) = 9.746 S), and Sw = 6.297 S (S(20,w) = 12.224 S) that correspond to the molecular masses of 200, 340 and 480 kDa, respectively (Figure 4F, Supplementary Figure S7A), calculated with the best-fit friction ratio f/f0 = 1.85. Considering that the theoretical molecular mass of the heteromeric eEF1Bαβγ complex (1:1:1 ratio) is 111.8 kDa, the molecular mass of the major species (340 kDa) corresponds to a heterotrimer eEF1B(αβγ)3. The first and third sedimenting species remain undefined. Sedimentation equilibrium analysis of eEF1Bαβγ showed the presence of the heterotrimer-heterohexamer mixture in solution (Supplementary Figure S7B). Hence, the α, β and γ subunits of eEF1B preferably associate in a stable heterotrimeric complex as revealed by analytical ultracentrifugation. This is consistent with the observed 1:1 stoichiometric complex formation between the α and γ as well as β and γ subunits (Supplementary Figure S5). However, the higher order oligomers are also present in the in vitro preparation of this complex. Like in the case of eEF1B(βγ)3, further oligomerization of eEF1B(αβγ)3 most probably is mediated by the eEF1Bγ subunit. We cannot exclude that some species with other eEF1Bαβγ stoichiometry may be present among higher order oligomers while the slowest sedimenting protein fraction may also contain self-associated single subunits. Of note, all complexes possess the moderately elongated shapes (18) (Smax/S = 1.68 for eEF1Bαγ, Smax/S = 1.73 for eEF1Bβγ, and Smax/S = 1.79 for eEF1Bαβγ) that also contributes to the overestimation of their molecular masses by the analytical gel filtration technique (Figure 4A-E).
Macromolecular architecture of the eEF1B complex
In order to map the interaction sites on the subunits involved in the eEF1B complex, we applied two approaches: the site-directed mutagenesis and HDX-MS. Deletion of the first 19 amino acids in eEF1Bα and the first 43 amino acids in eEF1Bβ completely abolished their interaction with full-length eEF1Bγ (Supplementary Figure S8A and B). As expected, eEF1Bγ(228–437), comprising the linker region and the C-terminal domain, interacts with neither full-length eEF1Bα nor eEF1Bβ (Supplementary Figure S8C and D), thus confirming the exclusive role of the eEF1Bγ N-terminal domain in eEF1B complex formation (9).
Next, the regions of the eEF1Bα, eEF1Bβ and eEF1Bγ subunits that contribute to the protein-protein binding sites was determined by HDX-MS. Usually, the interacting proteins create the binding interfaces with increased structural rigidity and/or decreased solvent accessibility that results in reducing of H/D exchange. To map the eEF1Bγ binding site on eEF1Bα and eEF1Bβ, we compared the peptide protection patterns of the isolated eEF1Bα and eEF1Bβ proteins and their complex with eEF1Bγ (Figure 5A and B). The ‘differential aggregated protection’ plots display the peptides that changed their protection. The majority of peptides composing the N-terminal domain, except the first five amino acids, increase their protection against H/D exchange, while the other part of eEF1Bα remains unaffected (Figure 5C). This suggests that the entire N-terminal domain of eEF1Bα undergoes a global decrease in dynamics upon binding to eEF1Bγ (Figure 5E). In contrast, only the peptides encompassing a narrow region, residues 11–29, of eEF1Bβ increase their protection within eEF1Bβγ complex (Figure 5D and F). According to the biochemical data, eEF1Bγ binds the eEF1Bα and eEF1Bβ simultaneously (Figure 4E). Upon binding to eEF1Bα, the eEF1Bγ peptides covering the regions 144–161 and 170-190 became more protected (Figure 6A and C) that indicates their involvement in the formation of the eEF1Bα binding interface (Figure 6E). No significant difference of protection was observed for the C-terminal domain and the linker region (Figure 6C). Binding of eEF1Bβ to eEF1Bγ results in a dramatic increase of protection for the majority of peptides covering the N-terminal domain of the latter, indicating that upon interaction with eEF1Bβ the whole N-terminal domain of eEF1Bγ experiences the global increase of structural rigidity excluding the short region involved in the interaction with eEF1Bα (Figure 6B, D and F). The C-terminal domain and the linker region of eEF1Bγ remain unaffected (Figure 6D).
Figure 5.

Mapping the protein-protein interactive surfaces on the eEF1Bα and eEF1Bβ subunits by HDX-MS. The aggregated protection of peptides derived from eEF1Bα (A) and eEF1Bβ (B) in a free state (blue color) and involved into the eEF1Bαγ and eEF1Bβγ complexes (red color) is depicted. The differential aggregated protection plots show the eEF1Bα (C) and eEF1Bβ (D) peptides that change their protection (brown color) against H/D exchange in the complex with eEF1Bγ (the difference is statistically significant for at least three or more incubation time-points). The peptides indicated in blue in the plots (C) and (D) do not change their protection. The regions of eEF1Bα (E) and eEF1Bβ (F) that change their aggregated protection values upon interaction with eEF1Bγ are painted in gray.
Figure 6.

Mapping the protein-protein interactive surfaces on the eEF1Bγ subunits by HDX-MS. The aggregated protection of peptides derived from eEF1Bγ (blue color) and involved into the eEF1Bαγ (A) and eEF1Bβγ (B) complexes (red color) are depicted. The differential aggregated protection plots (C) and (D) show the eEF1Bγ peptides that change their protection (brown color) in the complexes with eEF1Bα and eEF1Bβ, respectively (the difference is statistically significant for at least three or more incubation time-points). The peptides indicated in blue in the plots (C) and (D) do not change their protection. The regions of eEF1Bγ that change their aggregated protection values upon interaction with eEF1Bα (E) and eEF1Bβ (F) are painted in gray.
We also compared the HDX protection patterns of the binary eEF1Bαγ and eEF1Bβγ, and ternary eEF1Bαβγ complexes. The eEF1Bα and eEF1Bβ protection patterns in the eEF1Bαβγ complex do not significantly differ from those observed in the eEF1Bαγ and eEF1Bβγ complexes, respectively, as jugged by the differential aggregation protection plots of the ternary and binary complexes (Supplementary Figure S9A and B). However, a region comprising the residues 137–154 in the GEF domain of eEF1Bα display higher protection in the ternary complex as compared to the binary one (Supplementary Figure S9A). This fragment corresponding to the first β-strand of the GEF domain is not involved into direct interaction between eEF1Bα and eEF1Bγ. The observed decrease of deuterium uptake in this case may be attributed to the local conformational change in this region of eEF1Bα. In the case of eEF1Bβ, two regions slightly altered their protection in the ternary complex as compared to the binary one (Supplementary Figure S9B). A region comprising the residues 80–120 that corresponds to the LZ-motif increases its protection indicating a greater stabilization of the α-helical coil-coiled bundle in the ternary complex. Unlike eEF1Bα, a small region of eEF1Bβ (residues 192–200) that belongs to the first β-strand of the GEF domain displays lower protection in the eEF1Bαβγ complex compared to eEF1Bβγ, probably due to local conformational fluctuations.
The N-terminal domain of eEF1Bγ interacts simultaneously with both eEF1Bα and eEF1Bβ. The HDX protection patterns of eEF1Bγ in the eEF1Bαβγ complex strongly resemble those obtained for the eEF1Bαγ and eEF1Bβγ complexes, respectively, (Supplementary Figure S9C-F) confirming an independent binding mode of eEF1Bα and eEF1Bβ to eEF1Bγ (47).
Importantly, the HDX-MS data obtained for eEF1Bαγ are in agreement with the crystal structure of their N-terminal domains complex (PDB ID: 5DQS). However, there is no crystal structure for eEF1Bβγ with 1:1 stoichiometry. Using the molecular docking algorithm (32,48) we successfully modeled such complex, in which the first two α-helices of eEF1Bβ insert into a cleft inside the eEF1Bγ N-terminal domain (Supplementary Figure S10A). A superposition of the crystal structure of the eEF1Bαγ N-terminal domains (PDB ID: 5DQS) with the atomistic model of the eEF1Bβγ N-terminal domains using the N-terminal domain of eEF1Bγ as a common part resulted in the ternary complex model that agrees well with the HDX-MS data (Supplementary Figure S10B). The same docking procedure applied for the full-length subunits resulted in the reconstruction of the eEF1B(αβγ)3 complex (Figure 7, Supplementary video). The modeled complex has an extended overall shape and contains six structurally conserved GEF domains.
Figure 7.
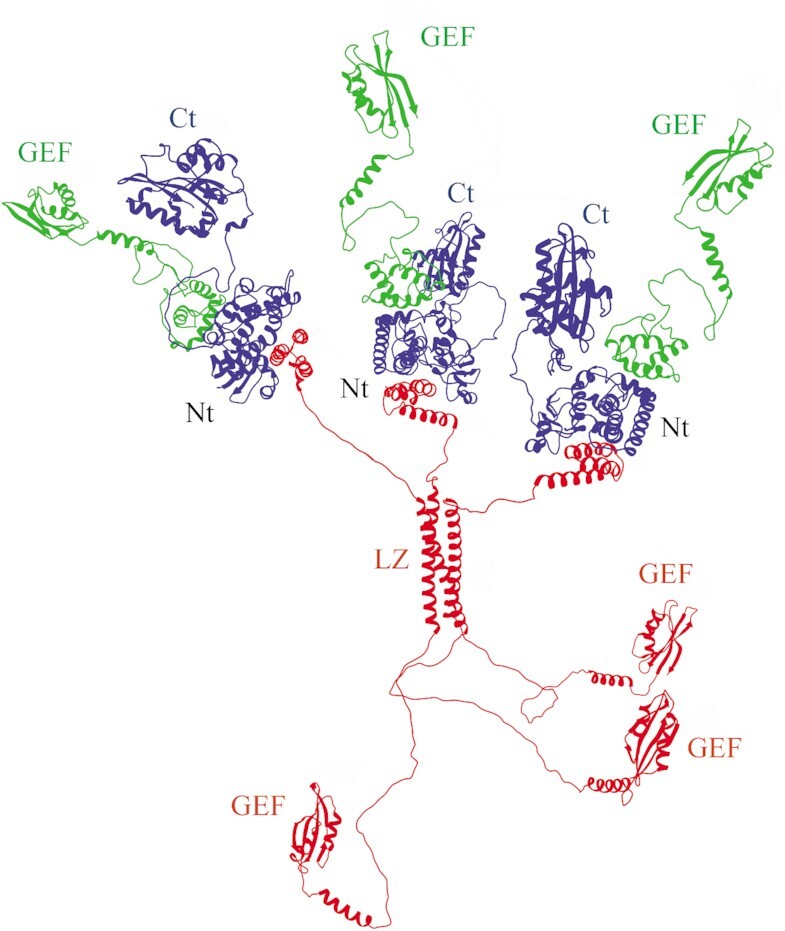
The quaternary organization of the eEF1B(αβγ)3 complex. eEF1Bα is in green, eEF1Bβ – in red, and eEF1Bγ – in blue. Abbreviations: GEF – the GEF domain of eEF1Bα and eEF1Bβ, Nt – the complex of eEF1Bα, eEF1Bβ and eEF1Bγ N-terminal domains, Ct – the C-terminal domain of eEF1Bγ, LZ – the LZ-motif of eEF1Bβ.
We have previously reported that eEF1A forms a complex in equimolar stoichiometry with eEF1Bα through its GEF domain (14). In the case of eEF1Bβ, one molecule of this trimeric protein is expected to bind minimum one and maximum three molecules of eEF1A. To verify this assumption, we incubated eEF1A2 with increasing concentrations of eEF1Bβ and resolved the samples by native gel electrophoresis. Indeed, three discrete eEF1Bβ-eEF1A2 complexes with different electrophoretic mobility were detected in the gel (Figure 8A) demonstrating the ability of all eEF1Bβ GEF domains to concurrently bind eEF1A. Consequently, there are as many as six GEF domains within the eEF1B(αβγ)3 complex located at the C-terminal extremities of the α and β subunits (Figure 7). The titration experiment with the whole eEF1B complex revealed that 3 μM eEF1B(αβγ)3 was saturated by 18 μM eEF1A2 that corresponds to a ratio 1:6 (Figure 8B). Thus, one molecule of the eEF1B complex is indeed capable of binding up to six molecules of eEF1A2.
Figure 8.
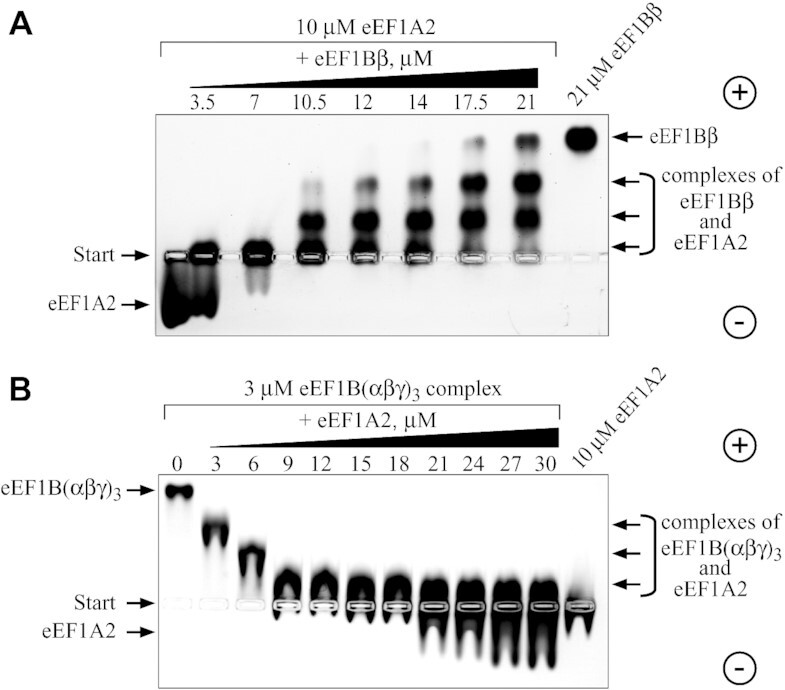
Interaction of eEF1A2 with eEF1Bβ and the eEF1B(αβγ)3 complex. eEF1A2 was incubated with increasing amounts of eEF1Bβ (A) and the eEF1B(αβγ)3 complex was incubated with increasing amounts of eEF1A2 (B) The protein mixtures were resolved by 1% native agarose gel electrophoresis and the proteins were visualized by Coomassie Brilliant Blue staining.
DISCUSSION
In this work, we presented the structural models of the eEF1Bα, eEF1Bβ and eEF1Bγ subunits and reconstruction of the whole eEF1B complex. Two proteins play a structural role within eEF1B: eEF1Bβ that is a stable homotrimer (Figure 2E and Supplementary Figure S2) and eEF1Bγ that binds eEF1Bβ and eEF1Bα (Figure 4E).
With the aid of site-directed mutagenesis and HDX-MS analysis, we outlined the protein interactive surfaces on eEF1Bα, eEF1Bβ and eEF1Bγ (Figures 5 and 6 and Supplementary Figure S8). As mentioned above, the HDX-MS data obtained for the eEF1Bαγ complex correlate well with the crystal structure of the eEF1Bαγ N-terminal domain complex (PDB ID: 5DQS). In this structure, eEF1Bα domain contacts eEF1Bγ via the loop region (D21-V29) and third α-helix (C50-I59). Indeed, the peptides composing these two regions significantly increase their protection against H/D exchange upon binding to eEF1Bγ (Figure 5C). Besides, an increase of protection is also observed for the peptides from the first (S8-Y18) and second (N32-S42) α-helices and the loop region (S43-A47) that do not interact with eEF1Bγ directly but may contribute to the appropriate conformation of the binding surface (Figure 5E). This holds true at least for the first α-helix (S8-Y18) of eEF1Bα since its deletion prevents the interaction with eEF1Bγ (Supplementary Figure S8A). According to the 5DQS structure, loop K147-E155 and α-helix N186-N196 of eEF1Bγ directly interact with eEF1Bα in the complex. The HDX-MS data reveal the same regions of eEF1Bγ that become significantly more protected upon interaction with eEF1Bα (Figure 6C and E).
According to the crystal structure of the eEF1Bβ N-terminal fragment (residues 1-32) complexed with the eEF1Bγ N-terminal domain (PDB ID: 5JPO), two α-helices of the eEF1Bβ fragment in a straight conformation form a complex with the eEF1Bγ N-terminal domain tetramer. The first α-helix is squeezed between two N-terminal domains and the second one is bound to the cleft of the third domain. Importantly, an increase of protection against H/D exchange was observed for the same region of eEF1Bβ in the complex with eEF1Bγ (Figure 5D). It worth noting that our data indicate the equimolar stoichiometry of eEF1Bβ and eEF1Bγ in the complex (Figure 4C and Supplementary Figure S5) that contradicts to the 5JPO crystal structure. This may be due to a short size of the eEF1Bβ fragment used for crystallization and a tendency of the eEF1Bγ N-terminal domain to form oligomers at a high protein concentration (Supplementary Figure S4B).
As both eEF1Bα and eEF1Bβ have similar guanine nucleotide exchange activity in vitro (14,16) a reason of the existence of two different nucleotide exchanging proteins in one complex as well as their functional equivalence remain unknown. Binding eEF1Bγ enhances the catalytic activity of eEF1Bα (6,49) due to elimination of the self-inhibitory action of the eEF1Bα N-terminal domain (14) while no effect of eEF1Bγ on the eEF1Bβ activity was observed (6,16). The functional activity of the eEF1Bαβγ complex has been also compared with eEF1Bα and eEF1Bβ by polyphenylalanine synthesis in vitro, eEF1Bαβγ was found to be several times more active than eEF1Bα or eEF1Bβ alone (12). It suggests that association of these nucleotide exchanging subunits within the ternary complex allows them to execute their activity more efficiently as compared to the individual proteins.
eEF1Bα and eEF1Bβ may perform a different role within the eEF1B complex containing valyl-tRNA synthetase. It has been shown that the N-terminal domain of VRS interacts with the eEF1Bβ subunit, however the enzyme interaction site on eEF1Bβ has not been mapped (6). Taking into account that eEF1Bβ is a homotrimer (Figure 3), one may expect the binding of up to three molecules of valyl-tRNA synthetase to eEF1Bβ. Although, two parallel reactions take place in the VRS-eEF1B complex, namely the GDP/GTP exchange on eEF1A that catalyzes the GEF subunit and valylation of tRNA on VRS, a ternary complex valyl-tRNA*eEF1A*GTP is finally formed as a common product of both reactions (7). This ternary complex is a result of the ‘hand to hand’ transfer from the enzyme to eEF1A and such transfer can be realized when eEF1A bound to the GEF domain is located in a close vicinity to the valyl-tRNA synthetase catalytic site. Thus, if three molecules of VRS are attached to the eEF1B complex, they may act in concert with three of six available GEF domains. The remaining GEF domains most probably function independently to provide eEF1A*GTP for other aa-tRNAs. Regarding the functional significance of VRS-eEF1B complex, an important question arises why the only valyl-tRNA synthetase is exclusively associated with eEF1B? One of the possible explanations is that valyl-tRNA has a lowest affinity to eEF1A among other aa-tRNAs. In the bacterial system, the lowest valyl-tRNA affinity to EF-Tu is compensated by its higher amount in cell as compared to the other aminoacylated tRNAs (50,51). In turn, in higher eukaryotes poor affinity of valyl-tRNA to the elongation factor 1A may be compensated by its ‘hand-to-hand’ transfer within the VRS-eEF1B complex. In such a way valyl-tRNA may avoid competition with other aa-tRNAs that have stronger affinity for eEF1A.
Importantly, at least two cases of a disease-causing loss of function variants in the human gene EEF1B2 that encodes eEF1Bα have been reported to date (52,53). In both cases, the pathogen variants in EEF1B2 led to moderate intellectual disability in patients. Of note, the complete loss of function of the eEF1Bα protein could not be compensated by the presence of eEF1Bβ. To explain the pathologic neurological consequences in the case of EEF1B2 mutation, it has been hypothesized that neurons are more susceptible to perturbation of the translation than other types of cells (54). Taking into account that a neuron-specific isoform eEF1A2 is more dependent on GDP/GTP exchange than eEF1A1 (14,55), one may suggest that proper functioning of the eEF1B complex could be critical for efficient translation in neurons.
It has been reported that the subunits involved into eEF1B can interact with several protein partners in human cancer cells (56). However, only translationally controlled tumor protein (TCTP) was shown to bind the CAR domain of eEF1Bβ (57) that resulted in inhibition of its guanine-nucleotide exchange activity (58). Further studies are required to establish the interaction sites of other identified protein partners of eEF1B in order to understand structural and functional consequences of these interactions.
Apparently, the multi-GEF eEF1B complex appeared lately in evolution, as the leucine-zipper-containing eEF1Bβ sequence is present in all metazoans from cnidarians to mammals while it is not found in the available sequences from fungi and plants (2). Its appearance could become favorable because voluminous metazoan cell requires some sort of compartmentalization or increase in local concentration of the metabolic components in particular places of the cytoplasm. Consequently, one obvious explanation of the existence of the multi-GEF complex might be the necessity to maintain high efficiency of eEF1A conversion into active GTP-bound conformation in the translational compartment. Besides, a part of the eEF1B complex associated with VRS functions as an exclusive valyl-tRNA*eEF1A*GTP supplier to the translating ribosome.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (59) partner repository with the dataset identifier PXD031783.
Supplementary Material
ACKNOWLEDGEMENTS
Authors are grateful to Dr A. N. Zaderko for the help with DLS measurements, Drs O.V. Novosylna, and D.S. Gurianov for the help with protein expressions and purifications. The technical assistance of N. Kolodka and D. Futorny is gratefully acknowledged.
Contributor Information
Tetiana V Bondarchuk, Institute of Molecular Biology and Genetics, NAS of Ukraine, 150 Zabolotnogo St., 03143 Kyiv, Ukraine.
Vyacheslav F Shalak, Institute of Molecular Biology and Genetics, NAS of Ukraine, 150 Zabolotnogo St., 03143 Kyiv, Ukraine.
Dmytro M Lozhko, Institute of Molecular Biology and Genetics, NAS of Ukraine, 150 Zabolotnogo St., 03143 Kyiv, Ukraine.
Agnieszka Fatalska, Institute of Biochemistry and Biophysics, PAN, Pawinskiego 5a, 02-109 Warsaw, Poland; Department of Genetics, University of Cambridge, Cambridge CB2 3EH, UK.
Roman H Szczepanowski, International Institute of Molecular and Cell Biology, Trojdena 4, 02-109 Warsaw, Poland.
Vladyslava Liudkovska, Institute of Molecular Biology and Genetics, NAS of Ukraine, 150 Zabolotnogo St., 03143 Kyiv, Ukraine; International Institute of Molecular and Cell Biology, Trojdena 4, 02-109 Warsaw, Poland.
Oleksandr Yu Tsuvariev, Institute of High Technologies, Taras Shevchenko National University of Kyiv, Akademik Glushkov Ave. 4-g, 03022 Kyiv, Ukraine.
Michal Dadlez, Institute of Biochemistry and Biophysics, PAN, Pawinskiego 5a, 02-109 Warsaw, Poland.
Anna V El'skaya, Institute of Molecular Biology and Genetics, NAS of Ukraine, 150 Zabolotnogo St., 03143 Kyiv, Ukraine.
Boris S Negrutskii, Institute of Molecular Biology and Genetics, NAS of Ukraine, 150 Zabolotnogo St., 03143 Kyiv, Ukraine.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Research Foundation of Ukraine [2020.02/0028 ‘Study on the structural features of human translation elongation complex eEF1B’]; A.F., R.H.S., M.D. acknowledge support by the National Science Centre: MAESTRO [UMO-2014/14/A/NZ1/00306]; Centre of Preclinical Research and Technology [POIG.02.02.00-14-024/08-00]; National Multidisciplinary Laboratory of Functional Nanomaterials [POIGT.02.02.00-00-025/09-00]; Foundation of Polish Science: TEAM-Tech Core Facility grant [TEAM TECH CORE FACILITY/2016-2/2]; T.V.B. was a recipient of short-term FEBS fellowship ‘Collaborative and Experimental Scholarship for Central & Eastern Europe’. Funding for open access charge: We have no source of funding for the publication charges.
Conflict of interest statement. None declared.
REFERENCES
- 1. Dever T.E., Green R.. The elongation, termination, and recycling phases of translation in eukaryotes. CSH Perspect. Biol. 2012; 4:a013706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Le Sourd F., Boulben S., Le Bouffant R., Cormier P., Morales J., Belle R., Mulner-Lorillon O. eEF1B: At the dawn of the 21st century. Biochim. Biophys. Acta. 2006; 1759:13–31. [DOI] [PubMed] [Google Scholar]
- 3. Clark B.F.C., Grunberg-Manago M., Gupta N.K., Hershey J.W.B., Hinnebusch A.G., Jackson R.J., Maitra U., Mathews M.B., Merrick W.C., Rhoads R.E., Sonenberg N., Spremulli L.L., Trachsel H., Voorma H.O.. Prokaryotic and eukaryotic translation factors: International Union of Biochemistry and Molecular Biology (IUBMB). Biochimie. 1996; 78:1119–1122.9150893 [Google Scholar]
- 4. Carvalho J.F., Carvalho M.D., Merrick W.C.. Purification of various forms of elongation factor 1 from rabbit reticulocytes. Arch. Biochem. Biophys. 1984; 234:591–602. [DOI] [PubMed] [Google Scholar]
- 5. Motorin Y.A., Wolfson A.D., Lohr D., Orlovsky A.F., Gladilin K.L.. Purification and properties of a high-molecular-mass complex between Val-tRNA synthetase and the heavy form of elongation factor 1 from mammalian cells. Eur. J. Biochem./FEBS. 1991; 201:325–331. [DOI] [PubMed] [Google Scholar]
- 6. Bec G., Kerjan P., Waller J.P.. Reconstitution in vitro of the valyl-tRNA synthetase-elongation factor (EF) 1 beta gamma delta complex. Essential roles of the NH2-terminal extension of valyl-tRNA synthetase and of the EF-1 delta subunit in complex formation. J. Biol. Chem. 1994; 269:2086–2092. [PubMed] [Google Scholar]
- 7. Negrutskii B.S., Shalak V.F., Kerjan P., El'skaya A.V., Mirande M.. Functional interaction of mammalian valyl-tRNA synthetase with elongation factor EF-1alpha in the complex with EF-1H. The J. Biol. Chem. 1999; 274:4545–4550. [DOI] [PubMed] [Google Scholar]
- 8. Sasikumar A.N., Perez W.B., Kinzy T.G.. The many roles of the eukaryotic elongation factor 1 complex. Wiley Interdiscipl. Rev. RNA. 2012; 3:543–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Janssen G.M., van Damme H.T., Kriek J., Amons R., Moller W.. The subunit structure of elongation factor 1 from Artemia. Why two alpha-chains in this complex?. The J. Biol. Chem. 1994; 269:31410–31417. [PubMed] [Google Scholar]
- 10. Sheu G.T., Traugh J.A.. A structural model for elongation factor 1 (EF-1) and phosphorylation by protein kinase CKII. Mol. Cell. Biochem. 1999; 191:181–186. [PubMed] [Google Scholar]
- 11. Minella O., Mulner-Lorillon O., Bec G., Cormier P., Belle R.. Multiple phosphorylation sites and quaternary organization of guanine-nucleotide exchange complex of elongation factor-1 (EF-1betagammadelta/ValRS) control the various functions of EF-1alpha. Biosci. Rep. 1998; 18:119–127. [DOI] [PubMed] [Google Scholar]
- 12. Sheu G.T., Traugh J.A.. Recombinant subunits of mammalian elongation factor 1 expressed in Escherichia coli. Subunit interactions, elongation activity, and phosphorylation by protein kinase CKII. J. Biol. Chem. 1997; 272:33290–33297. [DOI] [PubMed] [Google Scholar]
- 13. Janssen G.M., Moller W.. Elongation factor 1 beta gamma from Artemia. Purification and properties of its subunits. Eur. J. Biochem./FEBS. 1988; 171:119–129. [DOI] [PubMed] [Google Scholar]
- 14. Trosiuk T.V., Shalak V.F., Szczepanowski R.H., Negrutskii B.S., El'skaya A.V.. A non-catalytic N-terminal domain negatively influences the nucleotide exchange activity of translation elongation factor 1Balpha. FEBS J. 2016; 283:484–497. [DOI] [PubMed] [Google Scholar]
- 15. Trosiuk T.V., Liudkovska V.V., Shalak V.F., Negrutskii B.S., El'skaya A.V.. Structural dissection of human translation elongation factor 1Bγ (eEF1Bγ): expression of full-length protein and its truncated forms. Biopolym. Cell. 2014; 30:10. [Google Scholar]
- 16. Bondarchuk T.V., Shalak V.F., Negrutskii B.S., El'skaya A.V.. Leucine-zipper motif is responsible for self-association of translation elongation factor 1Bβ. Biopolym. Cell. 2016; 32:11. [Google Scholar]
- 17. Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophysical. J. 2000; 78:1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Erickson H.P. Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol. Proced. Online. 2009; 11:32–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vistica J., Dam J., Balbo A., Yikilmaz E., Mariuzza R.A., Rouault T.A., Schuck P.. Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal. Biochem. 2004; 326:234–256. [DOI] [PubMed] [Google Scholar]
- 20. Bondarchuk T.V., Lozhko D.M., Shalak V.F., Fatalska A., Szczepanowski R.H., Dadlez M., Negrutskii B.S., El'skaya A.V.. The protein-binding N-terminal domain of human translation elongation factor 1Bbeta possesses a dynamic alpha-helical structural organization. Int. J. Biol. Macromol. 2019; 126:899–907. [DOI] [PubMed] [Google Scholar]
- 21. Fatalska A., Dzhindzhev N.S., Dadlez M., Glover D.M.. Interaction interface in the C-terminal parts of centriole proteins Sas6 and Ana2. Open Biol. 2020; 10:200221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Malm A.V., Corbett J.C.W.. Improved Dynamic Light Scattering using an adaptive and statistically driven time resolved treatment of correlation data. Sci. Rep. 2019; 9:13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kozlowski L.P., Bujnicki J.M.. MetaDisorder: a meta-server for the prediction of intrinsic disorder in proteins. BMC Bioinformatics. 2012; 13:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Webb B., Sali A.. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinformatics. 2014; 47:5.6.1–5.6.37. [DOI] [PubMed] [Google Scholar]
- 25. Fiser A., Do R.K., Sali A.. Modeling of loops in protein structures. Protein Sci. 2000; 9:1753–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu D., Zhang Y.. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011; 101:2525–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen V.B., Arendall W.B. 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C.. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krieger E., Joo K., Lee J., Raman S., Thompson J., Tyka M., Baker D., Karplus K.. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins. 2009; 77:114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 30. Shen M.Y., Sali A.. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006; 15:2507–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schneidman-Duhovny D., Inbar Y., Nussinov R., Wolfson H.J.. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005; 33:W363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Duhovny D.N.R., Wolfson H.J.. International Workshop on Algorithms in Bioinformatics. 2002; 2452:Lecture Notes in Computer Science; 185–200. [Google Scholar]
- 33. Mashiach E., Schneidman-Duhovny D., Andrusier N., Nussinov R., Wolfson H.J.. FireDock: a web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008; 36:W229–W232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vangone A., Spinelli R., Scarano V., Cavallo L., Oliva R.. COCOMAPS: a web application to analyze and visualize contacts at the interface of biomolecular complexes. Bioinformatics. 2011; 27:2915–2916. [DOI] [PubMed] [Google Scholar]
- 35. Zhu X., Mitchell J.C.. KFC2: a knowledge-based hot spot prediction method based on interface solvation, atomic density, and plasticity features. Proteins. 2011; 79:2671–2683. [DOI] [PubMed] [Google Scholar]
- 36. Perez J.M., Siegal G., Kriek J., Hard K., Dijk J., Canters G.W., Moller W.. The solution structure of the guanine nucleotide exchange domain of human elongation factor 1beta reveals a striking resemblance to that of EF-Ts from Escherichia coli. Structure. 1999; 7:217–226. [DOI] [PubMed] [Google Scholar]
- 37. Wu H., Wang C., Gong W., Wang J., Xuan J., Perrett S., Feng Y.. The C-terminal region of human eukaryotic elongation factor 1Bdelta. J. Biomol. NMR. 2016; 64:181–187. [DOI] [PubMed] [Google Scholar]
- 38. Engen J.R. Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal. Chem. 2009; 81:7870–7875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Balasubramaniam D., Komives E.A.. Hydrogen-exchange mass spectrometry for the study of intrinsic disorder in proteins. Biochim. Biophys. Acta. 2013; 1834:1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Skinner J.J., Lim W.K., Bedard S., Black B.E., Englander S.W.. Protein dynamics viewed by hydrogen exchange. Protein Sci. 2012; 21:996–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhu S., Shala A., Bezginov A., Sljoka A., Audette G., Wilson D.J.. Hyperphosphorylation of intrinsically disordered tau protein induces an amyloidogenic shift in its conformational ensemble. PloS One. 2015; 10:e0120416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Landschulz W.H., Johnson P.F., McKnight S.L.. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science. 1988; 240:1759–1764. [DOI] [PubMed] [Google Scholar]
- 43. Lupas A.N., Bassler J.. Coiled coils - a model system for the 21st century. Trends Biochem. Sci. 2017; 42:130–140. [DOI] [PubMed] [Google Scholar]
- 44. Wood C.W., Woolfson D.N.. CCBuilder 2.0: powerful and accessible coiled-coil modeling. Protein Sci. 2018; 27:103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Achilonu I., Siganunu T.P., Dirr H.W.. Purification and characterisation of recombinant human eukaryotic elongation factor 1 gamma. Protein Express. Purif. 2014; 99:70–77. [DOI] [PubMed] [Google Scholar]
- 46. Vanwetswinkel S., Kriek J., Andersen G.R., Guntert P., Dijk J., Canters G.W., Siegal G.. Solution structure of the 162 residue C-terminal domain of human elongation factor 1Bgamma. The J. Biol. Chem. 2003; 278:43443–43451. [DOI] [PubMed] [Google Scholar]
- 47. Mansilla F., Friis I., Jadidi M., Nielsen K.M., Clark B.F., Knudsen C.R.. Mapping the human translation elongation factor eEF1H complex using the yeast two-hybrid system. The Biochem. J. 2002; 365:669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guigo R., Gusfield D.. Second International Workshop, WABI 2002, Rome, Italy, September 17-21, 2002, Proceedings. 2002; 2452:Springer-Verlag Berlin Heidelberg; 554. [Google Scholar]
- 49. Janssen G.M., Moller W.. Kinetic studies on the role of elongation factors 1 beta and 1 gamma in protein synthesis. J. Biol. Chem. 1988; 263:1773–1778. [PubMed] [Google Scholar]
- 50. Louie A., Ribeiro N.S., Reid B.R., Jurnak F.. Relative affinities of all Escherichia coli aminoacyl-tRNAs for elongation factor Tu-GTP. J. Biol. Chem. 1984; 259:5010–5016. [PubMed] [Google Scholar]
- 51. Jakubowski H. Negative correlation between the abundance of Escherichia coli aminoacyl-tRNA families and their affinities for elongation factor Tu-GTP. J. Theor. Biol. 1988; 133:363–370. [DOI] [PubMed] [Google Scholar]
- 52. Najmabadi H., Hu H., Garshasbi M., Zemojtel T., Abedini S.S., Chen W., Hosseini M., Behjati F., Haas S., Jamali P.et al.. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011; 478:57–63. [DOI] [PubMed] [Google Scholar]
- 53. Larcher L., Buratti J., Heron-Longe B., Benzacken B., Pipiras E., Keren B., Delahaye-Duriez A.. New evidence that biallelic loss of function in EEF1B2 gene leads to intellectual disability. Clin. Genet. 2020; 97:639–643. [DOI] [PubMed] [Google Scholar]
- 54. McLachlan F., Sires A.M., Abbott C.M.. The role of translation elongation factor eEF1 subunits in neurodevelopmental disorders. Hum. Mutat. 2019; 40:131–141. [DOI] [PubMed] [Google Scholar]
- 55. Kahns S., Lund A., Kristensen P., Knudsen C.R., Clark B.F., Cavallius J., Merrick W.C.. The elongation factor 1 A-2 isoform from rabbit: cloning of the cDNA and characterization of the protein. Nucleic Acids Res. 1998; 26:1884–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Negrutskii B. Non-translational connections of eEF1B in the cytoplasm and nucleus of cancer cells. Front. Mol. Biosci. 2020; 7:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wu H., Gong W., Yao X., Wang J., Perrett S., Feng Y.. Evolutionarily conserved binding of translationally controlled tumor protein to eukaryotic elongation factor 1B. J. Biol. Chem. 2015; 290:8694–8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cans C., Passer B.J., Shalak V., Nancy-Portebois V., Crible V., Amzallag N., Allanic D., Tufino R., Argentini M., Moras D.et al.. Translationally controlled tumor protein acts as a guanine nucleotide dissociation inhibitor on the translation elongation factor eEF1A. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:13892–13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Perez-Riverol Y., Bai J., Bandla C., Garcia-Seisdedos D., Hewapathirana S., Kamatchinathan S., Kundu D.J., Prakash A., Frericks-Zipper A., Eisenacher M.et al.. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022; 50:D543–D552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (59) partner repository with the dataset identifier PXD031783.