


Abstract
Mitochondrial tRNAs are indispensable for the intra-mitochondrial translation of genes related to respiratory subunits, and mutations in mitochondrial tRNA genes have been identified in various disease patients. However, the molecular mechanism underlying pathogenesis remains unclear due to the lack of animal models. Here, we established a mouse model, designated ‘mito-mice tRNALeu(UUR)2748’, that carries a pathogenic A2748G mutation in the tRNALeu(UUR) gene of mitochondrial DNA (mtDNA). The A2748G mutation is orthologous to the human A3302G mutation found in patients with mitochondrial diseases and diabetes. A2748G mtDNA was maternally inherited, equally distributed among tissues in individual mice, and its abundance did not change with age. At the molecular level, A2748G mutation is associated with aberrant processing of precursor mRNA containing tRNALeu(UUR) and mt-ND1, leading to a marked decrease in the steady-levels of ND1 protein and Complex I activity in tissues. Mito-mice tRNALeu(UUR)2748 with ≥50% A2748G mtDNA exhibited age-dependent metabolic defects including hyperglycemia, insulin insensitivity, and hepatic steatosis, resembling symptoms of patients carrying the A3302G mutation. This work demonstrates a valuable mouse model with an inheritable pathological A2748G mutation in mt-tRNALeu(UUR) that shows metabolic syndrome-like phenotypes at high heteroplasmy level. Furthermore, our findings provide molecular basis for understanding A3302G mutation-mediated mitochondrial disorders.
Graphical Abstract
Graphical Abstract.
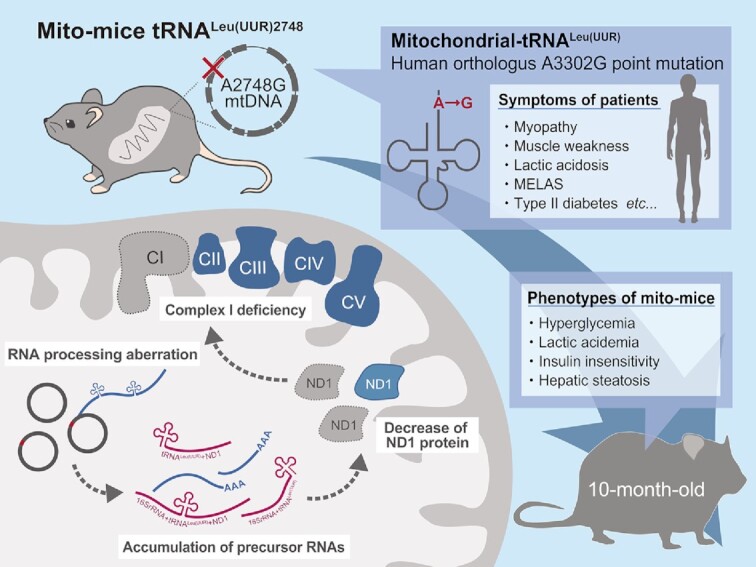
Novel mito-mice carrying the A2748G point mutation in mitochondrial tRNALeu(UUR) show age-dependent metabolic disorders and hepatic steatosis as the A2748G mutation induces mitochondrial dysfunction via RNA processing aberration.
INTRODUCTION
Mitochondrial DNA (mtDNA) encodes a set of protein-coding mRNA related to the respiratory complex subunits as well as non-coding genes including ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs). Deletion or point mutations in mtDNA can cause a decrease in the translation of respiratory complex subunits, resulting in defects in the mitochondrial metabolism, ultimately leading to the development of mitochondrial diseases as well as other disorders including diabetes, neurodegenerative diseases, infertility, and cancer (1,2). To date, over 700 mitochondrial disease-related mutations have been identified in both coding and non-coding genes of mtDNA. Notably, among all mtDNA mutations, ∼40% of the mutations have been found in mt-tRNA genes, with the largest number of pathological mutations being identified in mt-tRNALeu(UUR) gene. For example, the A3243G mutation of mt-tRNALeu(UUR) gene (an A-to-G base substitution at nucleotide position 3243 on the mtDNA) is one of the most frequent mutations found in patients with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome (3). In addition to A3242G, at least 35 pathological mutations have been reported in the mt-tRNALeu(UUR) gene.
Despite the known roles of mtDNA mutations in pathogenesis of mitochondrial diseases, the underlying molecular mechanism remains unclear. One of the largest obstacles is the lack of animal models because the conventional homologous recombination strategy developed for nuclear DNA is not applicable to mtDNA. To date, only a few lines of mutant mice carrying pathogenic mtDNA mutations have been reported (4–9). Kauppila et al. has developed a clonal expansion strategy using mtDNA mutator mice (10) and generated a mouse line carrying the C5024T mutation in mt-tRNAAla (9). Recently, protein-based genome editing methods, such as TALEN and base-editing technologies, have been developed, which enable cleavage (11–14) or site-specific editing (15–17) of mtDNA. mitoTALEN technology has been applied to mutant mice that have the C5024T mutation in mt-tRNAAla to cleave the mutant mtDNA (18). However, these methods are highly sequence-dependent, and establishing mouse models having mitochondrial disease-related mutations in their mtDNA remains a difficult endeavor. Notably, there is no mouse model carrying mutations in tRNALeu(UUR) despite the clinical significance of this mutation. Considering the growing number of pathological mutations identified in patients, there is a pressing need to establish animal models with mtDNA mutations.
Our group has used trans-mitochondria techniques, by which we fused the enucleated cells carrying mutant mDNA with mouse female karyotype embryonic stem (ES) cells in which mitochondria were pharmacologically eliminated. Subsequent transplantation of the chimeric ES cells into fertilized mouse oocytes successfully yielded the ‘mito-mice’ that carry mutant mtDNA and exhibit mitochondrial disease-like phenotypes (5,7,8). As mentioned above, among the pathogenic mtDNA gene mutations identified previously from patients with mitochondrial disease, point mutations in mt-tRNALeu(UUR) are known to be frequent. Therefore, in our previous study, we focused on point mutations in mt-tRNALeu(UUR) and performed random mutagenesis in mouse fibrosarcoma cells (B82 cells) to expand the repertoire of mutant mtDNA-bearing cells and successfully obtained a cell line carrying the A2748G mutation at a rate as high as ≥95% heteroplasmy (19). Importantly, mouse mt-tRNALeu(UUR) A2748G mutation is orthologous to human pathological A3302G mutation identified in mitochondrial disease patients (20–26; also See Supplemental Table S1). Patients with the A3302G mutation exhibit a marked reduction in Complex I activity in various tissues and exhibit muscle weakness. In addition, some patients developed non-insulin-dependent (type II) diabetes mellitus and polycystic ovary syndrome (26).
In the present study, we successfully generated a unique mouse model, ‘Mito-mice tRNALeu(UUR)2748’, by transferring A2748G mtDNA containing mitochondria to ES cells that were pharmacologically depleted of mitochondria and investigated the molecular nature of the observed pathological phenotypes to determine the cause of mitochondrial dysfunction in various tissues.
MATERIALS AND METHODS
Cell lines and cell culture
Mouse fibrosarcoma B82 cells were cultured in RPMI 1640 (Nissui Seiyaku) supplemented with 10% FBS (Sigma-Aldrich), uridine (50 μg/ml), pyruvate (0.1 mg/ml), and an antibiotic-antimycotic mixed stock solution (Nacalai Tesque) in an incubator at 37°C with 5% CO2. Mouse ES cells TT2-F and an XO subline established from XY TT2 cells (27), were used to generate mito-mice. ES cells and ES cybrids were cultivated on mitomycin C-inactivated feeder cells derived from fetal fibroblasts in KnockOut D-MEM (Invitrogen) supplemented with 15% KnockOut Serum Replacement (Invitrogen), non-essential amino acids (10 μg/ml; Wako), leukemia inhibitory factor (1000 U/ml, Invitrogen), 100 μM 2-mercaptoethanol (Sigma-Aldrich), 1 μM PD0325901 (Wako), 3 μM CHIR99021 (Wako), uridine (50 μg/ml), and pyruvate (0.1 mg/ml).
Measurement of lactate level in the cell medium
The level of lactate in the cell supernatant was measured using F-kit l-Lactic Acid (Roche). NAD+ and l-lactate dehydrogenase were mixed in the culture supernatant, and the amount of NADH at an absorption wavelength of 340 nm was measured with a Centro LB 960 (Belsalt Japan) based on the reaction of l-lactate and NAD+ catalyzed by l-lactate dehydrogenase into pyruvate and NADH. The level of lactate produced per cell was calculated by normalizing by the number of cells.
Isolation of ES cybrids carrying A2748G mtDNA
The host ES cells in ES medium were pretreated with rhodamine 6G (R6G; 0.75 μg/ml in 3% ethanol) for 48 h to eliminate endogenous mitochondria and mtDNA (28). The mtDNA donor B82mt2748 cells were pretreated with cytochalasin B (10 g/ml) for 10 min and centrifuged at 13 000 × g for 30 min at 37°C for enucleation.
The resultant cytoplasts were fused with R6G-pretreated ES cells by polyethylene glycol and the fusion mixture cultivated in a selective medium with HAT (hypoxanthine-aminopterin-thymidine) and without uridine and pyruvate. Because of the absence of thymidine kinase activity, mouse cells carrying the nuclear genome from B82 cybrids could not survive in the presence of HAT. Seven days after fusion, growing colonies were picked for further evaluation.
Generation of chimeric mice and mito-mice tRNALeu(UUR)2748
Female ICR mice (Charles River Laboratories Japan Inc) were subcutaneously injected with PMSG (5 IU/0.1 ml; Serotropin; ASKA Animal Health Co., Ltd.). After 48 h, hCG (5 IU/0.1 ml; Gonatropin; ASKA Animal Health Co., Ltd.) was intraperitoneally administered to induce hyper-ovulation, and they were mated with male ICR mice. The day after mating, the embryos were collected at the two-cell stage and incubated in KSOM medium (Merck). The zona pellucidae were removed by treatment with acidified Tyrode's buffer (Sigma-Aldrich). Each treated embryo was incubated overnight with 15–20 ES clones carrying A2748G mtDNA in the wells of a 35-mm culture dish. The next day, blastocyst stage embryos were transferred into the uterus of the pseudo parents. Founder (F0) chimeric females were mated with C57BL/6J (B6; CLEA Japan Inc.) males to produce the F1 generation, and F1 female mice carrying A2748G mtDNA (female mito-mice tRNALeu(UUR)2748) were backcrossed with B6 male mice. The F1 and F2 generation male mice were used for the pathological analyses. Aged-matched B6 male mice were used as controls for the experiments. The mice were housed in specific pathogen-free facilities at the University of Tsukuba on a 14-h light/10-h dark cycle under controlled temperature (22–25°C) and fed with a standard diet and water.
Genotyping of A2748G mutant mtDNA
Detection of the A2748G mutation was achieved by restriction fragment length polymorphism analysis. Mouse mtDNA primer sets that bind to bases 2548 and 2838 were used for the analysis (all primer sequences are listed in Supplemental Table S5). PCR amplifications were performed using TakaRa Ex Taq DNA Polymerase Hot-Start Version (TaKaRa Bio) with the following cycling conditions: 30 cycles of 94°C for 30 s; 53°C for 30 s; and 72°C for 30 s. The above reactions yielded a 291-bp PCR fragment. The PCR fragments were then treated with the restriction enzyme XspI (TaKaRa Bio) at the mutation site to obtain a 93-bp fragment and an 81-bp fragment. The fragments were then separated by electrophoreses through 3.0% agarose gels containing ethidium bromide (0.1 mg/ml). Chemiluminescence of fragments was measured with a bioimaging analyzer, EZ-Capture ST (ATTO), and quantification of the percentage of A2748G mtDNA was performed by the CS Analyzer 3.0 (ATTO).
Sequencing of whole mtDNA
Total DNA extracted from the liver was used for the amplification of mtDNA fragments. The PCR reactions amplified 2.5–3.0 kbp mtDNA fragments using eight primer pairs (primer sequences are listed in Supplemental Table S5) designed to generate overlapping fragments of mtDNA. All PCR amplifications were performed using TakaRa Ex Taq DNA Polymerase Hot-Start Version (TaKaRa Bio) with the following cycling conditions: 30 cycles of 94°C for 30 s; 53°C for 30 s; and 72°C for 60 s. The PCR fragments were then separated on 1.0% agarose gels and extracted using the QIAquick Gel Extraction Kit (Qiagen). Purified PCR fragments were cut into approximately 300-bp fragments, after which 100 000 reads per sample were sequenced using Illumina MiSeq (Illumina). We calculated a Phred-scaled P-value score of Fisher's Exact test to assess the strand bias in candidate mutations detected by next generation sequencing. Genetic variants with a Phred-scaled P-value score under 60 are considered true mutations, and variants with a Phred-scaled P-value score larger than 60 are considered false-positive calls. Detected mutations were checked for homology with human pathogenic mtDNA mutations based on pathogenic mtDNA mutations listed in MITOMAP (http://mitomap.org).
Histological analyses
For lipid deposition analysis, cryosections of the liver were stained with Oil Red O and hematoxylin. Histochemical analyses for complexes II and IV activities were performed as previously described (29) using cryosections of heart and renal tissues and coverslips with ES cells. Images were obtained using a digital color camera DFC310 FX (Leica).
For electron microscopy, tissue samples were fixed with 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) at 4°C, overnight, followed by post-fixation with 2% osmium tetroxide in 0.1 M phosphate buffer for 2 h, and dehydration. Dehydrated samples were infiltrated with propylene oxide (PO) and placed into a 7:3 mixture of PO and resin (Nisshin EM) for 1 h, after which PO was volatilized overnight. The samples were transferred to fresh 100% resin and polymerized for 48 h. Ultrathin sections were prepared using an ultra-microtome (Leica), after which they were mounted on copper grids and stained with 2% uranyl acetate, followed by secondary staining with a lead stain solution (Sigma-Aldrich). The grids were then imaged using transmission electron microscopy (JEM-1400Plus; JEOL Ltd.). The area of the mitochondria was calculated using ImageJ software.
Blue native–PAGE
The mitochondrial fractions were isolated from tissue homogenates by centrifugation (900 × g for 5 min), after which the supernatants were collected and centrifuged again at 20 000 × g for 10 min. Isolated mitochondrial fractions were solubilized with solubilization buffer [1.5 M aminocaproic acid and 50 mM bis–Tris (pH 7.0) containing 1.5% n-dodecyl-β-d-maltoside] and centrifuged at 30 000 × g for 30 min. The resulting supernatants (15 μg/lane) were electrophoresed using native–PAGE 3–12% bis–Tris protein gels (Thermo Fisher Scientific) at a constant voltage (100 V). For evaluating in-gel activities, gels were incubated in Complex I activity substrate [2 mM Tris-HCl, 0.1 mg/ml NADH, and 2.5 mg/ml nitro blue tetrazolium chloride (pH 7.4)] and Complex IV activity substrate [50 mM phosphate buffer, 0.5 mg/ml diaminobenzidine, and 1 mg/ml cytochrome c (pH 7.4)]. The activities of Complexes I and IV were quantified using ImageJ.
Measurement of blood glucose and lactate
Mouse tail veins were punctured with 25 G needles, and blood glucose levels were determined using Glucocard PlusCare (ARKRAY), and lactate levels were determined using Lactate Pro 2 (ARKRAY). For the oral glucose tolerance test, after starvation overnight, glucose (1.5 g/kg body weight) was orally administered to mice, and blood glucose levels were measured after 15, 30, 60, 90 and 120 min. For the insulin tolerance test, after starvation for 1 h, insulin (0.7 U/kg body weight; Humulin R; Eli Lily Japan) was intraperitoneally administered to mice, and blood glucose levels were measured after 15, 30, 60, 90 and 120 min. For each experiment, the area under the curve was calculated.
Blood analysis
Blood samples were collected from mice by cardiocentesis. HbA1c levels were measured in whole blood using CinQ HbA1c (ARKRAY). After centrifugation of the blood at 2000 rpm for 20 min at 4°C, the plasma fraction was collected. Plasma samples were used to determine the levels of blood urea nitrogen (UN-S SEIKEN kit; Denka Co., Ltd.), triglycerides (L-type TG M test; Wako), alanine aminotransferase (L-type ALT J2 test; Wako), aspartate aminotransferase (L-type AST J2 test; Wako), total cholesterol (L-type CHO M test; Wako), high-density lipoprotein cholesterol (Cholestest N HDL; SEKISUI Medical Co., Ltd.), low-density lipoprotein cholesterol (Cholestest LDL; SEKISUI Medical Co., Ltd.), creatinine (L-type CRE M test; Wako), non-esterified fatty acid (NEFA-HR; Wako), total ketone bodies (Auto Wako T-KB; Wako) using the Hitachi 7180 automatic analyzer (Hitachi, Tokyo, Japan). Blood insulin levels were analyzed using a mouse insulin ELISA kit (Morinaga Institute of Biological Science).
Northern blotting
Total RNA was extracted from mouse livers using ISOGEN (Nippon Gene). For analyzing mt-tRNA expression levels, total RNA was separated by electrophoresis through a 7 M urea 10% acrylamide gel. For aminoacyl-tRNA analysis, total RNA was extracted and electrophoresed on a 7 M urea 8% acrylamide gel under acidic conditions (pH 5.0) and then transferred to Immobilon-Ny + membranes (Merck). For expression level analysis of precursor RNAs, total RNA was separated by electrophoresis on a formaldehyde–2% agarose gel along with DynaMarker RNA High (BioDynamics Laboratory Inc.), and then transferred to Immobilon-Ny + membranes (Merck). The membranes were hybridized with DIG-modified probes that bind to mt-tRNALeu(UUR), mt-tRNAIle, 5.8S rRNA, ND1, 16S rRNA and 18S rRNA (the probe sequences are listed in Supplemental Table S5). The membranes were then blocked using a DIG wash and Block buffer kit (Roche) and incubated with anti-Digoxigenin-AP (1:10 000; #11093274910; Roche). Signals were detected using the ImageQuant LAS4000 (GE Healthcare) using CDP-star (Sigma-Aldrich).
Western blotting
Total protein was extracted from livers using the EzRIPA Lysis Kit (ATTO). Proteins separated by using 8% or 12% SDS-PAGE gels were transferred to polyvinylidene difluoride (PVDF) membranes and blocked with PVDF blocking reagent for Can Get Signal (Toyobo) for 1 h. Membranes were then incubated with primary antibodies against MT-ND1 (1:1000; #ab181848; Abcam), MT-ND2 (1:1000; #19704-1-AP; Proteintech), MT-ND5 (1:1000; #PA5-36600; Thermo Fisher Scientific), NDUFB8 (1:1000; #ab110242; Abcam), NDUFS4 (1:1000; #ab139178; Abcam), SDHA (1:1000; #11998S; Cell Signaling Technology), MT-CO1 (1:1000; #ab14705; Abcam), COX4 (1:1000; #4850S; Cell Signaling Technology), ATP5A (1:1000; #ab14748; Abcam), VDAC (1:1000; #4866S; Cell Signaling Technology), TOM20 (1:1000; #sc-17764; Santa Cruz Biotechnology) or β-ACTIN (1:3000; #A1978; Sigma-Aldrich) overnight at 4°C [Can Get Signal immunoreaction enhancer solution 1 (Toyobo) was used for dilution]. The membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies against rabbit IgG (1:5000; #G-21234; Thermo Fisher Scientific) or mouse IgG (1:5000; #G-21040; Thermo Fisher Scientific) for 1 h at room temperature [Can Get Signal immunoreaction enhancer solution 2 (Toyobo) was used for dilution]. The membranes were then incubated with ECL substrate (GE Healthcare), and signals were detected using the Amersham ImageQuant 800 (Cytiva).
Statistics
Data are presented as mean ± SD. Statistical significance was calculated by the Student's t-test or Tukey–Kramer test using BellCurve for Excel (Social Survey Research Information Co., Ltd.). A P-value of <0.05 was considered to indicate statistically significant differences between samples.
Study approval
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Tsukuba (Approval No. 20-335).
RESULTS
Generation of mito-mice tRNALeu(UUR)2748
A2748 is localized to tRNALeu(UUR) gene of mouse mtDNA and is highly conserved in mammals (Figure 1A, B). Its human orthologue A3302 is mutated to G (A3302G) in some mitochondrial disease patients who exhibit multiple symptoms including myopathy and diabetes (Figure 1A, Supplementary Table S1). We have previously generated a mouse B82 cell line carrying a high proportion of A2748G mutation in its mtDNA (B82mt2748 cells) (19). Prior to the generation of the mutant mouse model, we analyzed the mitochondrial function of B82mt2748 cells. It is generally known that cells with reduced mitochondrial function often have increased lactate production due to upregulation of anaerobic glycolysis. Accordingly, we observed elevated lactate production in B82mt2748 cells compared to control cybrids that were transplanted with the mitochondrial genome of wild-type mice (B82mtB6 cells) (Supplementary Figure S1A). Upon investigating the protein levels of mitochondrial respiratory subunits, we found that mitochondrial DNA-encoded ND1 and nuclear DNA-encoded NDUFA9, both of which are subunits of Complex I, were selectively and markedly reduced in B82mt2748 cells (Supplementary Figure S1B). Consistent with this result, blue-native page and in-gel assay showed that amount and the activity of Complex I was strikingly decreased in B82mt2748 (Supplementary Figure S1C). These results demonstrate that the A2748G mutation of mitochondrial tRNALeu(UUR) confers pathogenicity to mitochondrial functions by affecting Complex I.
Figure 1.

Generation of trans-mitochondrial mice carrying A2748G mutation in mitochondrial tRNALeu(UUR). (A) Clover-leaf structure of mitochondrial tRNALeu(UUR) in human and mouse. The positions of the A2748G mutation in mouse tRNALeu(UUR) and orthologous mutations (the A3302G mutation) in human tRNALeu(UUR) are indicated in red text. (B) Comparison of the A3302G point mutation in tRNALeu(UUR) genes from other species. The GenBank accession numbers of the sequences used in the alignment are: Homo sapiens, NC_01290; Callithrix jacchus, NC_025586; Bos taurus, NC_006853; Sus scrofa, NC_000845; Canis lupus familiaris, NC_002008; Oryctolagus cuniculus, NC_001913; Rattus norvegicus, NC_001665; and Mus musculus, AY172335. (C) Diagram illustrating the strategy for generating trans-mitochondrial mice from mouse cybrids carrying A2748G mtDNA via cytoplasmic transplantation. (D) Cytochemical analysis of Complex IV activity in wild-type ES cells and ES cybrids with 94% A2748G mtDNA. Cells expressing Complex IV activity are stained in brown. Scale bar: 50 μm. (E) Generation of F0 chimeric mice and F1 offspring with A2748G mtDNA. Chimerism was judged by coat color. Details of offspring born from chimeric mice #4-1 to 4 are listed in Supplemental Table S3. (F) Estimation of the proportion of A2748G mtDNA in tail tissues from F1 offspring by XspI digestion of the PCR products. The A2748G mtDNA produced 117-bp, 93-bp and 81-bp fragments due to the gain of an XspI site through the A2748G substitution in the tRNALeu(UUR) gene, whereas WT mtDNA produced 174-bp and 117-bp fragments.
To generate mito-mice carrying the A2748G mutation, we treated XO mouse ES cells (TT2F) (27) with rhodamine 6G to remove their mitochondria (28) and fused these cells with enucleated B82mt2748 cells (Figure 1C). We established 92 ES cell clones after extensive selection and single cell cloning. A total of 51 clones carried B82mt2748 cell-derived A2748G mutant mtDNA with the mutant load ranging from 70% to 97% (Supplementary Table S2). Meanwhile, 41 ES cell clones were devoid of mutant mtDNA (Supplementary Table S2). Notably, the hybrid ES cells carrying 94% of A2748G mtDNA showed a marked decrease in cytochrome c oxidase (Complex IV) activity (Figure 1D), indicating defective mitochondrial respiration.
We then selected four ES cell clones that carried 84%, 94%, 96% and 97% A2748G mtDNA to establish F0 generation chimeric mice (Figure 1E). While the ES cell clone carrying 84% A2748G mutation rate was successful in generating F1 mutant mice, as discussed below, the ES cells with 97%, 96% and 94% mutation rates yielded no F1 offspring. Each of these three ES cell clones yielded only one chimeric F0 female mouse with chimerism of 0%, 10% and 50%, respectively (corresponding to #1–#3 in Figure 1E). Moreover, the highest A2748G mutation rate in tail mtDNA was less than 14%. Given that mammalian mtDNA is maternally inherited (30,31), we crossed the two female F0 chimeric mice with 10% and 50% chimerism, respectively, with male wild-type mice (C57BL/6J) to obtain F1 generation mice with systemic A2748G mtDNA. However, no F1 offspring carrying detectable A2748G mtDNA were born (Figure 1E). Therefore, we conclude that ES cells with a high load (>94%) of A2748G mtDNA are unsuitable for generating mutant mouse lines, possibly due to germline abnormalities resulting from severe mitochondrial respiratory defects.
Interestingly, the ES cell clone carrying an 84% A2748G mutation rate yielded the birth of four F0 female mice, which exhibited 100% chimerism (corresponding to #4 in Figure 1E). In this case, the A2748G mtDNA rate in the tail ranged from 71%∼81%. When the four chimeric F0 female mice were mated to male wild-type mice, 35 out of 37 offspring inherited mutant mtDNA from their mothers, with the mutant mtDNA rate ranging from 5% to 87% in the tail (Figure 1F, Supplementary Table S3). Importantly, we have analyzed the whole sequence of mtDNA extracted from liver tissues of F1 mutant mice and detected five mutations besides the A2748G mutation but did not find off-target mutations except previously known genetic polymorphisms (Supplementary Table S4, also see Discussion). Furthermore, the 10-week-old F1 female mutant mice gave birth to F2 female offspring that carried mutant mtDNA at the same rate of their respective mothers (Supplementary Figure S2A). We noticed that the age of female mice at pregnancy seemed to influence the mutation rate in their offspring (Supplementary Figure S2B–E), with the mutation rate of A2748G in the F2 offspring born from old mutant F1 mice being significantly lower than that in the F2 offspring born from young F1 mutant mice (Supplementary Figure S2D–E). Nevertheless, given the transmission of A2748G mtDNA over two successive generations, we conclude that the experiments have established a mutant mouse line carrying A2748G mtDNA, which we have designated mito-mice tRNALeu(UUR)2748.
Dysregulation of precursor mRNA processing in mito-mice tRNALeu(UUR)2748
In mice, A2748 (equivalent to the human A3302) is located at position 71 near the 3′-end of the acceptor stem of tRNALeu(UUR), which base pairs with the uridine at position 2 of 5′-end (Figure 2A). An A-to-G substitution might influence the structure of the amino acid-acceptor stem, thereby affecting the stability or aminoacylation of tRNALeu(UUR) (Figure 2A). We examined the steady-state level and aminoacylation level of mitochondrial tRNALeu(UUR) in total RNA purified from liver tissues of wild-type mice, mito-mice tRNALeu(UUR)2748 having, respectively, a low and high mutation rate. Northern blot analysis revealed that neither the steady-state of mature mitochondrial tRNALeu(UUR) nor the aminoacylation level was altered in mito-mice tRNALeu(UUR)2748 regardless of mutation rate (Figure 2B-E, Supplementary Figure S3A).
Figure 2.

Steady-state level, aminoacylation, and processing of tRNALeu(UUR) in mito-mice tRNALeu(UUR)2748. (A) Schematic illustration of the secondary structural of tRNALeu(UUR) with and without the A2748G mutation. Note that the A2748-to-G mutation in the acceptor stem will impair Watson–Crick base-pairing. (B) Northern blotting of mature tRNA levels in total RNA isolated from livers of 10-month-old mice. mt-tRNALeu(UUR) levels were normalized to mt-tRNAIle and nuclear DNA-encoded 5.8S rRNA. Quantification of tRNALeu(UUR) levels (C) showed no statistically significant difference between the three groups. Data are presented as the mean ± SD. n = 3 for each group. (D) Representative aminoacylated (upper band) and nonaminoacylated (lower band) tRNALeu(UUR) levels in total RNA from livers of 10-month-old mice were examined by northern blotting under acidic conditions. (E) Quantification of aminoacylated tRNALeu(UUR) levels based on Supplementary Figure S3A. Levels of aminoacylated tRNALeu(UUR) were normalized by the sum of aminoacylated and nonaminoacylated tRNA. Data are presented as the mean ± SD. n = 4 for each group. (F) Schematic diagram of the primary and secondary structure of precursor pre-mRNA that contains 16S rRNA, tRNALeu(UUR), and ND1. RNase P and RNase Z are responsible for cleaving 16S rRNA-tRNALeu(UUR), and tRNALeu(UUR)-ND1, respectively. (G) Representative northern blotting of precursor transcripts in total RNA obtained from the livers of 10-month-old mice. Hybridization of three different probes designed to recognize tRNALeu(UUR), ND1 and 16S rRNA revealed expression of the 2.6 kb precursor transcript RNA19, the precursor 16S rRNA-tRNALeu(UUR) (1.6 kb), and the precursor tRNALeu(UUR)-ND1 mRNA (1.0 kb). (H) Quantification of precursor transcript RNA19 levels detected with each probe based on Supplementary Figure S3B. Precursor RNA19 levels were normalized by the 18S rRNA transcripts. Data are presented as the mean ± SD; *P < 0.05, **P < 0.01 by Tukey–Kramer test. n = 4 for each group. (I) Quantification of precursor transcript levels of tRNALeu(UUR)-ND1 (blue arrowheads in Supplementary Figure S3B) and 16S rRNA-tRNALeu(UUR) (red arrowheads in Supplementary Figure S3B) detected with tRNALeu(UUR) probe based on the low exposure time membrane in Supplementary Figure S3B. Each precursor transcript levels were normalized by the 18S rRNA transcripts. *P < 0.05, **P < 0.01 by Tukey–Kramer test. Data are presented as the mean ± SD. n = 4 for each group.
Mitochondrial genes are initially transcribed as a long polycistronic transcript, followed by RNase-mediated processing and maturation (Figure 2F). Point mutations at A2748 might dysregulate the structure of the primary transcript, leading to abnormal processing. Indeed, previous studies have shown that mutations in tRNALeu(UUR), including A3302G, are associated with an abnormal accumulation of a precursor transcript named RNA19 (Figure 2F), comprising 16S rRNA, tRNALeu(UUR), and ND1 genes (32,33). To investigate whether the mouse A2748G mutation affects RNA processing, we performed northern blotting of RNA extracted from liver tissues of 10-month-old mice and detected RNA19 using probes targeting tRNALeu(UUR) or ND1 mRNA or 16S rRNA (Figure 2G and Supplementary Figure S3B). All three probes detected a ∼2.6 kb transcript, which clearly indicates that the long transcript is RNA19. Most importantly, RNA19 was significantly accumulated in the high % mito-mice tRNALeu(UUR)2748 group compared to wild-type or low % mito-mice groups (Figure 2G, H and Supplementary Figure S3B). In addition to RNA19, a ∼1.6 kb band corresponding to the 16S rRNA-tRNALeu(UUR) fusion transcript was detected by probes targeting tRNALeu(UUR) or 16S rRNA, and a ∼1.0 kb band corresponding to the tRNALeu(UUR)-ND1 fusion transcript was detected by probes targeting tRNALeu(UUR) or ND1 (Figure 2F, G and Supplementary Figure S3B). Notably, both the 16S rRNA-tRNALeu(UUR) fusion transcript and tRNALeu(UUR)-ND1 fusion transcript were significantly accumulated in the high % mito-mice tRNALeu(UUR)2748 group (Figure 2I). Taken together, these results strongly suggest that the A2748G mutation has a deleterious effect on the processing of the tRNALeu(UUR)2748-containing transcript, which results in the accumulation of immature transcripts, thereby leading to the impairment of intra-mitochondrial translation.
Decrease in mtDNA-encoded proteins and respiratory complex activities in mito-mice tRNALeu(UUR)2748
The aberrant processing of the 16S rRNA-tRNALeu(UUR)-ND1 transcript in mito-mice tRNALeu(UUR)2748 prompted us to examine the levels of respiratory subunit proteins in the mutant mice. We examined mtDNA-derived proteins in the liver tissues of the 10-month-old mice from the high and low % groups as well as wild-type controls. Consistent with the abnormal processing of transcripts encoding ND1, we observed that the protein level of ND1 was markedly decreased in the liver tissues of the high % group compared to other groups (Figure 3A). Interestingly, Complex I subunits ND2 and ND5 as well as Complex IV subunit CO1 did not differ between wild-type and mito-mice tRNALeu(UUR)2748 regardless of mutation rate (Figure 3A). The decrease of ND1 in the high % group was similar to the result observed in B82mt2748 cells, the origin of mito-mice tRNALeu(UUR)2748 (Supplementary Figure S1B).
Figure 3.

Mitochondrial complex activities and morphology in mito-mice tRNALeu(UUR)2748. (A) Representative images of the levels of indicated subunits of Complexes I, II, IV, and V in livers of 10-month-old mice examined by western blotting. ND1, ND2, ND5 and CO1 are mtDNA-derived proteins, while others are nuclear DNA (nDNA)-derived. TOM20, VDAC and β-ACTIN were used as loading controls. (B) Native–PAGE gel images show the amount of each mitochondrial respiratory complexes (top) and the activity of Complexes I and IV (bottom) in kidney, liver, and brain tissues of 10-month-old mice in the gel. Quantification of Complex I activity (C) and Complex IV activity (D) in the kidney, liver, and brain (Each group, n = 3). Data are presented as the mean ± SD; *P < 0.05, **P < 0.01 by Tukey–Kramer test. (E) Histochemical analysis of Complexes II and IV activities in heart and kidney from 10-month-old mice. Complex IV-positive (COX+) cells are stained in brown, and Complex IV-negative/Complex II-positive (COX-SDH+) cells are stained in blue. Note that kidney and heart of high % group mice contain COX-SDH + cells. Glomeruli (‘G’) of renal cells were not sufficiently stained owing to a scarcity of mitochondria. Scale bar = 100 μm. (F) Electron microscopy images of mitochondria in soleus muscle and liver from 10-month-old mice. The ‘L’ indicates lipid droplets. Scale bars = 1 μm. (G) The mitochondrial areas in soleus muscle and liver were analyzed by histogram and violin plot (each group has at least 200 mitochondria). **P < 0.01, ***P < 0.001, ****P < 0.0001 by Kruskal–Wallis test.
We also investigated the nuclear DNA-derived respiratory complex subunit proteins. NDUFS4 and NDUFB8, the Complex I subunits, showed a substantial decrease in liver tissues of the high % group when compared to other groups (Figure 3A). However, SDHA, COX4, ATP 5A, which encode the nuclear DNA-derived Complexes II, IV, V subunit proteins, respectively, did not differ among all groups (Figure 3A). Thus, among all Complexes examined in this study, components of Complex I were most susceptible to the A2748G mutation when the mutant mtDNA accumulated. Notably, ND1 showed a striking decrease in the high % group, which was possibly caused by the aberrant processing of the tRNALeu(UUR)-ND1-containing transcript.
We next examined the levels of individual respiratory complex and complex activity in kidney, liver, and brain from 10-month-old wild-type and mutant mice by blue-native PAGE in combination with In-Gel activity assay. Compared to other groups, the high % group showed significantly decreased levels of intact Complex I in all examined tissues. (Figure 3B and Supplementary Figure S4). Accordingly, the high % group showed a significant decrease in Complex I activity in kidney, liver, and brain, with liver Complex I activity exhibiting the largest reduction (86.3 ± 2.6%, 43.3 ± 11.7%, 50.1 ± 3.3% in high % group compared to WT in the kidney, liver, and brain, respectively, **P < 0.01, *P < 0.05, Figure 3B, C).
In contrast to Complex I, the levels of intact Complex II, III, IV, and V in liver, kidney, and brain were comparable between the wild-type and high % groups (Figure 3B and Supplementary Figure S4). Furthermore, the activity of Complex IV did not differ between the wild-type and high % groups (Figure 3B and D). Besides the biochemical examination, we performed a sequential COX-SDH staining, which has been used to visualize Complex IV activity and Complex II activity at the histological level for pathological examination of biopsy samples (34). SDH activity is only detectable with low levels of Complex IV activity and strong SDH-positive staining indicates a loss of Complex IV activity. We observed sparse but clear SDH staining in kidney and heart tissues from high % mito-mice tRNALeu(UUR)2748 but not from wild-type and low % groups (Figure 3E). These results suggest that Complex IV activity was affected in a limited population of cells in high % mutant mice. It should be noted that among all Complexes, Complex I is the most affected respiratory complex in patients carrying the A3302G mutation (20,23–25). Thus, the mito-mice tRNALeu(UUR)2748 appears to recapitulate the molecular phenotype of human patients.
Dysregulation of mitochondrial morphology in mito-mice tRNALeu(UUR)2748
Since defective mitochondrial proteostasis could induce morphological abnormality of mitochondria, a hallmark of mitochondrial dysfunction, we examined mitochondrial morphology in various tissues of 10-month-old wild-type and mutant mice using transmission electron microscopy. We observed loss of cristae structure in the liver and soleus muscle of high % mice but not low % or wild-type groups (Figure 3F). During the morphological examination, we noticed an accumulation of small mitochondria in tissues of the high % mice. Statistical analysis revealed that the mitochondria in the liver and soleus muscle of high % mice were significantly reduced in area compared to those of wild-type mice (soleus, wild-type: 0.655 μm2, low %: 0.571 μm2, high %: 0.470 μm2, ****P < 0.0001 versus wild-type; liver, wild-type: 0.276 μm2, low %: 0.270 μm2, high %: 0.224 μm2, **P < 0.01 vs wild-type, Figure 3F-G). We also observed an increase in lipid droplets that directly contacted mitochondria in the soleus muscle fiber of high % group compared with the other two groups (Figure 3F). Notably, the abnormal cristae and increase of lipid droplets were also present in muscle fibers of patients with the A3302G mutation (20,22,24). In addition, a comparison between the high % and low % groups revealed that the mitochondria in the brain of the high % group were significantly smaller than those of the low % group (low %: 0.150 μm2, high %: 0.135 μm2, ***P < 0.001, Supplementary Figure S5). However, the structure of cristae in the brain and kidney appeared to be preserved regardless of mutational load (Supplementary Figure S5).
Metabolic disorder of mito-mice tRNALeu(UUR)2748
Patients with mitochondrial disease frequently exhibit metabolic disorders that include high blood lactate acidosis and the severity of the symptoms can be accelerated by aging (2). Given the broad mitochondrial dysfunction in high % mutant mice, we investigated the impact of A2748G mutation on metabolic profiles in mice of different ages. We divided 4-week-old mice into two groups for metabolic analysis at 3 and 10 months of age. Each group comprised three subgroups including wild-type, low %, and high % mice. Mito-mice tRNALeu(UUR)2748 were classified into low % (tail mutation rate; <50%) and high % (tail mutation rate; ≥50%) groups based on the tail mutation rate at 4 weeks of age.
To exclude the possibility that the mutation rate can be variable during aging, we examined the mutation load in the tail of individual 4-week-old mutant mice and traced the mutation rate at 3 and 10 months of age. We confirmed that the mutation rate in the tail did not change during aging (Figure 4A). In addition, liver biopsies were taken at 10 weeks and 10 months of age in the same individual mito-mice tRNALeu(UUR)2748 to assess the age-related change of A2748G mtDNA. The results showed that the mutation rate in the liver did not change with age (Figure 4B). Notably, the mutation rate in the tail was essentially identical to the mutation rate in the major tissues at both 3-month-old and 10-month-old (Figure 4C). Moreover, in all tissues, there was no difference in the average mutation rate between the 3-month-old and 10-month-old groups (Supplementary Figure S6). These findings indicate that the A2748G mutant mtDNA is equally distributed throughout the whole body in this mouse model and that the mutation rate does not change until 10 months of age.
Figure 4.

Metabolic phenotypes of mito-mice tRNALeu(UUR)2748. (A) The proportion of A2748G mtDNA in the tail of the same mutant mouse did not change over time (n = 16 each for comparison of 4-week-old and 3-month-old; n = 17 each for comparison of 4-week-old and 10-month-old mice). (B) For individual mutant mice, liver biopsy was taken at 10-week-old and 10-month-old, followed by PCR to examine the mutation rate. No age-dependent variation was observed in the liver biopsies. (C) The mutation rate of A2748G mtDNA in tail was correlated with that in the indicated tissues from both 3-month-old (open circle) and 10-month-old (black circle) mice. The black dotted line and the blue dotted line represent linear regressions for the data of 10-month-old and 3-month-old mice, respectively. (D) Body weight of mito-mice tRNALeu(UUR)2748 and wild-type mice at 3 and 10 months of age (n = 8 for each group). **P < 0.01 by Tukey–Kramer test. (E) Blood lactate levels in mito-mice tRNALeu(UUR)2748 and wild-type mice at 3 and 10 months of age (WT, n = 8; low %, n = 11; high %, n = 8). *P < 0.05 by Tukey–Kramer test. (F) Blood glucose levels in mito-mice tRNALeu(UUR)2748 and wild-type mice at 3 and 10 months of age (WT, n = 8; low %, n = 11; high %, n = 7). *P < 0.05, **P < 0.01 by Tukey–Kramer test. (G, H) Oral glucose tolerance test and area under the curve (AUC) of 3-month-old mice (G) (WT, n = 8; low %, n = 8; high %, n = 7) or 10-month-old mice (H) (WT, n = 6; low %, n = 10; high %, n = 7). *High % group versus wild-type; †high % versus low % groups. *P < 0.05, **P < 0.01 by Tukey–Kramer test. (I, J) Insulin tolerance test and area under the curve (AUC) of 3-month-old mice (I) (WT, n = 8; low %, n = 8; high %, n = 7) or 10-month-old mice (J) (WT, n = 6; low %, n = 10; high %, n = 7). *High % group versus wild-type; †high % versus low % groups. *P < 0.05, **P < 0.01 by Tukey–Kramer test. (K) Plasma insulin levels in mito-mice tRNALeu(UUR)2748 and wild-type mice at 3 and 10 months at steady state (3-month-old: WT, n = 5; low %, n = 5; high %, n = 6; 10-month-old: WT, n = 8; low %, n = 8; high %, n = 6). *P < 0.05, **P < 0.01 by Tukey–Kramer test. All data are presented as the mean ± SD.
Our phenotype analysis at 3 and 10 months of age revealed that the mito-mice tRNALeu(UUR)2748 exhibited age-dependent development of a metabolic disorder. The high % group of the mutant mice exhibited a higher body weight (Figure 4D), blood lactate level (Figure 4E), and blood glucose level (Figure 4F) compared to 10-month-old but not 3-month-old wild-type mice. The elevated blood lactate level in high % group, which is a hallmark of mitochondrial dysfunction, was consistent with the mitochondrial abnormality observed in the high % group in the 10-month-old mice (Figure 3).
Given the apparent hyperglycemia of the high % mutant mice, we investigated the glucose metabolism in detail. We performed glucose tolerance test at 3- and 10-months of age. In the 3-month-old mice, the change of blood glucose levels after glucose administration did not differ between wild-type and mutant mice (Figure 4G). However, in the 10-month-old mice, the high % group exhibited a clear glucose intolerance characterized by a significantly high blood glucose level 60, 90, and 120 min after glucose challenge compared to other groups (Figure 4H). Moreover, an insulin tolerance test revealed that the blood glucose lowering effect of insulin was significantly blunted in the high % group compared to other groups at 10 months (Figure 4J) but not 3 months of age (Figure 4I). Finally, we examined blood insulin level at 10-month-old mice and found a significant elevation of blood insulin level in the high % group (Figure 4K). Together, these results suggest that mito-mice tRNALeu(UUR)2748 develop a metabolic syndrome-like phenotype with insulin resistance in a mutation rate- and age-dependent manner.
Hepatic abnormalities in mito-mice tRNALeu(UUR)2748
The apparent insulin insensitivity suggests that the liver function in the high % group might be impaired at 10-month-old. Therefore, we investigated markers of liver dysfunction using plasma from 10-month-old mice. As expected, the high % group exhibited a significant elevation of blood alanine aminotransferase level (ALT) at 10-month-old, an indicative of liver dysfunction. Consistent with this result, blood urea nitrogen (BUN), which is the major product of protein metabolism, was decreased in the high % group (Figure 5A, B). Notably, we observed a marked increase in blood triglyceride (TG) level in the high % group, suggesting altered lipid metabolism in the liver (Figure 5C). Other major serum parameters did not differ among the three groups (Supplementary Figure S7). In accordance with the high TG level, livers from the high % group looked pale and enlarged compared to those of the wild-type and low % group (Figure 5D). Indeed, the wet weight of liver tissues of the high % group was significantly higher than that of other groups (Figure 5E). Furthermore, Oil-Red staining and electron microscopic examination revealed a marked accumulation of lipid droplet in the liver of the high % group mice (Figure 5F). Together, the findings indicate that a high mutational load of A2748G mtDNA in mito-mice tRNALeu(UUR)2748 leads to liver dysfunction and hepatic steatosis.
Figure 5.

Liver dysfunction in mito-mice tRNALeu(UUR)2748. (A–C) Plasma levels of alanine aminotransferase (A), blood urea nitrogen (B), and triglyceride (C) as hallmarks of liver dysfunction (WT, n = 8; low %, n = 10; high %, n = 7). *P < 0.05 by Tukey–Kramer test. (D) Representative images show livers of 10-month-old mice. Scale bars = 1 cm. (E) Comparison of liver mass (3-month-old: WT, n = 6; low %, n = 8; high %, n = 7; 10-month-old: WT, n = 9; low %, n = 9; high %, n = 8). *P < 0.05, **P < 0.01 by Tukey–Kramer test. (F) Oil Red O staining and representative electron microscopy images of liver sections from 10-month-old mice. Scale bars = 100 μm (top) and 20 μm (bottom). All data are presented as the mean ± SD.
DISCUSSION
In this study, we generated mito-mice tRNALeu(UUR)2748 that carry A2748G mtDNA orthologous to the mitochondrial disease-related A3302G mutation. To our knowledge, this is the first report describing the generation of mouse line that carries mtDNA with a pathological point mutation in the tRNALeu(UUR) gene, the hotspot for mitochondrial diseases-related mutations. Mito-mice tRNALeu(UUR)2748 are heteroplasmic mice that carry both wild-type mtDNA and A2748G mtDNA and could transmit the mutant mtDNA to their offspring. Intriguingly, mito-mice with a high mutation rate exhibited defective mitochondrial respiration and morphology, leading to the development of metabolic disorders characterized by lactic acidemia, hyperglycemia, insulin insensitivity, and hepatic steatosis. Furthermore, the development of metabolic disorder was late on-set despite the presence of mutant mtDNA from the beginning of life. The mitochondrial dysfunction and the age-dependent metabolic phenotypes have been reported in A3302G-related mitochondrial disease patients (22). Thus, our mouse model recapitulates the pathogenesis of A3302G-mediated mitochondrial disease and provide strong evidence that the A2748G mutation tRNALeu(UUR)2748 in mouse is responsible for the mitochondrial dysfunction.
The molecular and biochemical analysis of mito-mice tRNALeu(UUR)2748 suggests that abnormal RNA processing plays a causal role in the development of mitochondrial dysfunction. The A2748G mutation was associated with abnormal mRNA processing, as evidenced by aberrant accumulation of precursor transcripts, including RNA19 (16S rRNA-tRNALeu(UUR)-ND1 fusion transcript), 16S rRNA-tRNALeu(UUR) fusion transcript, and tRNALeu(UUR)-ND1 fusion transcript. Importantly, the accumulation of these precursors was clearly linked to the decrease in the protein level of ND1 in the mito-mice tRNALeu(UUR)2748 that have a high mutation rate. It should be noted that the A2748G mutation did not affect the steady-state level of tRNALeu(UUR) or tRNA aminoacylation even in the liver tissues of the high % group, despite the accumulation of precursor transcripts. In line with our arguments, a previous study using cybrids carrying the human orthologous tRNALeu(UUR) A3302G mutation reported that the stability of A3302G tRNA was comparable to that of the wild type tRNA (33). On the other hand, Katharina et al. reported that the steady-state level of tRNALeu(UUR) was moderately decreased in A3302G mutant cells (33). This discrepancy might be caused by the distinct turnover rate in different biological materials (i.e. cell lines versus mouse tissues). Collectively, the mouse A2748G (or human A3302G) mutation might affect the steady-state level of mt-tRNALeu(UUR) in a context-dependent manner. Further studies are needed to elucidate this molecular mechanism.
Our study revealed that the A2748G mutation differentially affects the protein abundance of Complex I subunits in mito-mice tRNALeu(UUR)2748. While ND1, NDUFS4 and NDUFB8 subunits of Complex I showed a substantial decrease in the high % group, ND5 and ND2 did not differ among the groups. Because ND1, ND2, and ND5 are all derived from mtDNA, it is reasonable to conclude that the selective decrease of ND1 protein is not attributed to the defective decoding of Leu codon by the mutant tRNALeu(UUR). It is likely that the unprocessed tRNALeu(UUR) at the 5′ terminus of ND1 mRNA will interfere with the translation of ND1, thereby causing the selective decrease of ND1 protein. According to Guerrero-Castillo et al., ND1 is indispensable for the assembly of Complex I because of its bridging of the Q module and Pp module (35). Accordingly, ND1-deficiency will induce disassembly of Complex I, rendering other subunits susceptible to proteolysis. Interestingly, a recent study has thoroughly investigated the turnover rate of mitochondrial proteins and found that the half-life of mitochondrial DNA-encoded subunits are longer than that of nuclear DNA-encoded subunits (NDUFA9: 21 h, NDUFS4: 24 h, NDUFB8: 59.9 h versus ND1: 253.2 h, ND2: Infinite, ND5: 121.4 h) (36). Therefore, the nuclear DNA-encoded subunits (NDUFA9, NDUFS4, NDUFB8) will be quickly degraded once disassembled, while ND2 and ND5 are resistant to proteolysis and will reside in the mitochondria for a longer duration. In contrast to the stability, the marked decrease of ND1 in mito-mice tRNALeu(UUR)2748 strongly suggests that the defective translation of ND1 transcripts is responsible for the Complex I deficiency and the subsequent mitochondrial dysfunction. Future studies are needed to elucidate the molecular mechanism by which the aberrant ND1 transcript induces the selective translational defect.
The mito-mice tRNALeu(UUR)2748 was generated by fusing enucleated ES cells with mitochondria-containing cytoplasm of B82mt2748 cells, in which A2748 of mtDNA was mutated to G using ENU as a mutagen. Because ENU can randomly induce DNA mutation, mito-mice tRNALeu(UUR)2748 may also bear other off-target mutation in mtDNA besides A2748G. We verified the off-target mutations by analyzing the full sequence of mtDNA extracted from liver tissues of mito-mice tRNALeu(UUR)2748 and identified five nucleotides showing sequence conflict with the canonical C57BL/6J mtDNA reference sequence: C4794T in ND2, T9461C in ND3, T12048C in ND5, G9348A in CO3, 9821 insA in tRNAArg (Supplementary Table S4). Among those, T9461C in ND3, G9348A in CO3, and 9821 insA in tRNAArg have been reported as genetic mtDNA polymorphisms caused by mouse strain differences (Supplementary Table S4) (37); C4794T in ND2 and T12048C in ND5 have been previously reported as genetic mtDNA polymorphisms in the mouse LA9 cell line (Supplementary Table S4) (38). Because mito-mice tRNALeu(UUR)2748 have a mixed genetic background including C57BL/6, CBA/J, and C3H/An and these mutations have been previously reported, it is most likely that these mutations are derived from the mixed genetic background and not from ENU-mediated mutagenesis.
It should be noted that previous studies have reported that C4794T in ND2 and T12048C in ND5 can potentially lower Complex I activity (39). Although we do not exclude the possibility that C4794T in ND2 and T12048C in ND5 partially contribute to the decrease of Complex I activity in mito-mice tRNALeu(UUR)2748, the aberrant processing of tRNALeu(UUR)-ND1 transcripts as well as the selective decrease of ND1 protein in the mutant mice cannot be explained by C4794T in ND2 and T12048C in ND5. In addition, the steady-state protein levels of ND2 and ND5 did not change in mito-mice tRNALeu(UUR)2748 regardless of mutation rate (Figure 3A). This data strongly suggests that the Complex I deficiency in tissues of mito-mice tRNALeu(UUR)2748 is not directly caused by C4794T in ND2 and T12048C in ND5 and primarily caused by the ND1 deficiency resulting from the A2748G mutation in tRNALeu(UUR). Taken together, the whole mtDNA sequencing results suggest that the mito-mice tRNALeu(UUR)2748 do not possess off-target mutations, and that A2748G mutation is the primary cause of mitochondrial dysfunction.
One of the interesting findings of this study is that the loading proportion of the A2748G mtDNA inherited to progeny was negatively correlated with maternal age (Supplementary Figure S2), although the A2748G mtDNA was almost evenly distributed in 12 tissues (i.e. brain, heart, lung, stomach, intestine, liver, spleen, pancreas, kidney, testis, soleus, and quadriceps) with no apparent increase or decrease in the loading proportion in these tissues until at least 10 months of age (Figure 4C and Supplementary Figure S6). Such a phenomenon of mutant mtDNA being less inheritable in progeny with increased maternal age and generation has been observed in other mouse models with mutant mtDNA (6,40), suggesting that regardless of the type of mutant mtDNA, wild-type mtDNA may have a competitive advantage during oogenesis in mice. Further investigation is needed to reveal how mutant mtDNA is selected during oogenesis, since the content of A2748G mtDNA in oocytes may be controlled by mitochondrial quality control, or oocytes carrying A2748G mtDNA may prevent differentiation into ova depending on maternal age.
The mitochondrial tRNALeu(UUR) gene is the hotspot of mitochondrial diseases-related mutations (2,41,42). In fact, mutations in the mitochondrial tRNALeu(UUR) gene account for approximately 80% of the cases of mitochondrial diseases caused by pathological mutant mtDNA. The most frequent mutation in tRNALeu(UUR) is the A-to-G point mutation at the 3243 position, which is responsible for the development of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) symptoms (3). Compared to the A3243G mutation, mitochondrial disease related to A3302G is rare and manifest relatively mild peripheral symptoms such as myopathy and diabetes (20–24,26). However, recent studies have shown that the A3302G mutation can also affect central nervous system and causes severe symptoms. For example, the A3302G mutation has been identified in a MELAS patient (25). In addition, some A3302G patients show depression, hearing impairment, and migraine (23). Based on these clinical reports, exploring the phenotypes of the central nervous system will be important in future studies using the mito-mice tRNALeu(UUR)2748.
In conclusion, we established a mutant mouse model carrying a systemic A2748G mutation in mtDNA, that recapitulates the clinical symptoms of patients with mitochondrial disease who carry the orthologous A3302G mutation. Detailed examination of this mito-mice tRNALeu(UUR)2748 revealed that the A2748G mutation caused aberrant processing of tRNALeu(UUR)-ND1-containing precursor transcripts, which resulted in a marked reduction of ND1 and subsequent Complex I deficiency, ultimately leading to mitochondrial dysfunction and metabolic disorders.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Prof. Keiji Tanimoto at the University of Tsukuba for his helpful advice. We would like to thank Natalie D. DeWitt and Editage (www.editage.com) for English language editing.
Author contributions: H.T. designed the study, performed the experiments, analyzed the data, and wrote the paper. F.-Y.W. and K.I. helped with the design and coordination of the study and edited the paper. E.O., T.Y. assisted with the experiments and provided advice for the paper. H.T., S.M., A.S. and D.K. assisted with the experiments. J.-I.H. designed the study. K.N. designed the study, wrote the paper and supervised the project.
Contributor Information
Haruna Tani, Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Ibaraki 305-8572, Japan; Department of Modomics Biology and Medicine, Institute of Development, Aging and Cancer, Tohoku University, Aoba-ku, Sendai, Miyagi 980-8575, Japan.
Kaori Ishikawa, Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Ibaraki 305-8572, Japan; Faculty of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Ibaraki 305-8572, Japan.
Hiroaki Tamashiro, Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Ibaraki 305-8572, Japan.
Emi Ogasawara, Department of Biological Sciences, Graduate School of Science, Osaka University, Toyonaka, Osaka 560-0043, Japan.
Takehiro Yasukawa, Department of Clinical Chemistry and Laboratory Medicine, Graduate School of Medical Sciences, Kyushu University, Higashi-ku, Fukuoka, Fukuoka 812-8582, Japan; Department of Pathology and Oncology, Juntendo University School of Medicine, Bunkyo-ku, Tokyo 113-8421, Japan.
Shigeru Matsuda, Department of Modomics Biology and Medicine, Institute of Development, Aging and Cancer, Tohoku University, Aoba-ku, Sendai, Miyagi 980-8575, Japan; Department of Clinical Chemistry and Laboratory Medicine, Graduate School of Medical Sciences, Kyushu University, Higashi-ku, Fukuoka, Fukuoka 812-8582, Japan.
Akinori Shimizu, Department of Microbiology and Immunology, Faculty of Medicine, Fukuoka University, Jonan-ku, Fukuoka, Fukuoka 814-0180, Japan.
Dongchon Kang, Department of Clinical Chemistry and Laboratory Medicine, Graduate School of Medical Sciences, Kyushu University, Higashi-ku, Fukuoka, Fukuoka 812-8582, Japan; Kashiigaoka Rehabilitation Hospital, Higashi-ku, Fukuoka, Fukuoka 813-0002, Japan.
Jun-Ichi Hayashi, Life Science Center for Survival Dynamics, Tsukuba Advanced Research Alliance (TARA), University of Tsukuba, Tsukuba, Ibaraki 305-8572, Japan.
Fan-Yan Wei, Department of Modomics Biology and Medicine, Institute of Development, Aging and Cancer, Tohoku University, Aoba-ku, Sendai, Miyagi 980-8575, Japan.
Kazuto Nakada, Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Ibaraki 305-8572, Japan; Faculty of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Ibaraki 305-8572, Japan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
AMED-CREST [JP22gm1110006 to K.N.] from Japan Agency for Medical Research and Development; Japan Society for the Promotion of Science Fellows [18J20289, 21J00143 to H.T.]; Grants-in-Aid for Scientific Research B [16H04678 and 22H02536 to K.N., 19H03141 to J.-I.H., 21H02659 to F.-Y.W.]; Young Scientists B [16K18535 to K.I.]; Scientific Research C [18K06203 to K.I.]; Grant-in-Aid for Scientific Research on Innovative Areas [21H00225 to K.I.]; Grant-in-Aid for Transformative Research Areas A [21H05265 to F.-Y.W.] from the Japan Society for the Promotion of Science, JST FOREST Program [JPMJFR204M to K.I., JPMJFR205Y to F.-Y.W.]; JST ERATO Program [JPMJER2002 to F.-Y.W.] from Japan Science and Technology Agency; Cooperative Research Project Program of Joint Usage/Research Center at the Institute of Development, Aging, and Cancer, Tohoku University. Funding for open access charge: AMED-CREST grant [JP22gm1110006] from Japan Agency for Medical Research and Development
Conflict of interest statement. None declared.
REFERENCES
- 1. Larsson N.G., Clayton D.A.. Molecular genetic aspects of human mitochondrial disorders. Annu. Rev. Genet. 1995; 29:151–178. [DOI] [PubMed] [Google Scholar]
- 2. Wallace D.C. Mitochondrial diseases in man and mouse. Science. 1999; 283:1482–1488. [DOI] [PubMed] [Google Scholar]
- 3. Goto Y., Nonaka I., Horai S.. A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990; 348:651–653. [DOI] [PubMed] [Google Scholar]
- 4. Inoue K., Nakada K., Ogura A., Isobe K., Goto Y., Nonaka I., Hayashi J.I.. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat. Genet. 2000; 26:176–181. [DOI] [PubMed] [Google Scholar]
- 5. Kasahara A., Ishikawa K., Yamaoka M., Ito M., Watanabe N., Akimoto M., Sato A., Nakada K., Endo H., Suda Y.et al.. Generation of trans-mitochondrial mice carrying homoplasmic mtDNAs with a missense mutation in a structural gene using ES cells. Hum. Mol. Genet. 2006; 15:871–881. [DOI] [PubMed] [Google Scholar]
- 6. Fan W., Waymire K.G., Narula N., Li P., Rocher C., Coskun P.E., Vannan M.A., Narula J., Macgregor G.R., Wallace D.C.. A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science. 2008; 319:958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yokota M., Shitara H., Hashizume O., Ishikawa K., Nakada K., Ishii R., Taya C., Takenaga K., Yonekawa H., Hayashi J.-I.. Generation of trans-mitochondrial mito-mice by the introduction of a pathogenic G13997A mtDNA from highly metastatic lung carcinoma cells. FEBS Lett. 2010; 584:3943–3948. [DOI] [PubMed] [Google Scholar]
- 8. Shimizu A., Enoki S., Ishikawa K., Mito T., Obata K., Nagashima R., Yonekawa H., Nakada K., Hayashi J.I.. Transmitochondrial mice as models for primary prevention of diseases caused by mutation in the tRNA(Lys) gene. Proc. Natl. Acad. Sci. U.S.A. 2015; 111:3104–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kauppila J.H.K., Baines H.L., Bratic A., Simard M.L., Freyer C., Mourier A., Stamp C., Filograna R., Larsson N.G., Greaves L.C.et al.. A phenotype-driven approach to generate mouse models with pathogenic mtDNA mutations causing mitochondrial disease. Cell Rep. 2016; 16:2980–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Trifunovic A., Wredenberg A., Falkenberg M., Spelbrink J.N., Rovio A.T., Bruder C.E., Bohlooly-Y M., Gidlöf S., Oldfors A., Wibom R.. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004; 429:417–423. [DOI] [PubMed] [Google Scholar]
- 11. Minczuk M., Papworth M.A., Kolasinska P., Murphy M.P., Klug A.. Sequence-specific modification of mitochondrial DNA using a chimeric zinc finger methylase. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:19689–19694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bacman R.S., Williams S.L., Pinto M., Peralta S., Moraes C.T.. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat. Med. 2013; 19:1111–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hashimoto M., Bacman S.R., Peralta S., Falk M.J., Chomyn A., Chan D.C., Williams S.L., Moraes C.T.. MitoTALEN: a general approach to reduce mutant mtDNA loads and restore oxidative phosphorylation function in mitochondrial diseases. Mol. Ther. 2015; 23:1592–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang Y., Wu H., Kang X., Liang Y., Lan T., Li T., Tan T., Peng J., Zhang Q., An G.et al.. Targeted elimination of mutant mitochondrial DNA in MELAS-iPSCs by mitoTALENs. Protein Cell. 2018; 9:283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mok B.Y., de Moraes M.H., Zeng J., Bosch D.E., Kotrys A.V., Raguram A., Hsu F., Radey M.C., Peterson S.B., Mootha V.K.et al.. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature. 2020; 583:631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee H., Lee S., Baek G., Kim A., Kang B.-C., Seo H., Kim J.-S.. Mitochondrial DNA editing in mice with DddA-TALE fusion deaminases. Nat. Commun. 2021; 12:1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cho S.I., Lee S., Mok Y.G., Lim K., Lee J., Lee L.M., Chung E., Kim J.S.. Targeted A-to-G base editing in human mitochondrial DNA with programmable deaminases. Cell. 2022; 185:1764–1776. [DOI] [PubMed] [Google Scholar]
- 18. Bacman S.R., Kauppila J.H.K., Pereira C.V., Nissanka N., Miranda M., Pinto M., Williams S.L., Larsson N.G., Stewart J.B., Moraes C.T.. MitoTALEN reduces mutant mtDNA load and restores tRNA(Ala) levels in a mouse model of heteroplasmic mtDNA mutation. Nat. Med. 2018; 24:1696–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shimizu A., Enoki S., Ishikawa K., Mito T., Obata K., Nagashima R., Yonekawa H., Nakada K., Hayashi J.. Mouse somatic mutation orthologous to MELAS A3302G mutation in the mitochondrial tRNA(Leu(UUR)) gene confers respiration defects. Biochem. Biophys. Res. Commun. 2015; 467:1097–1102. [DOI] [PubMed] [Google Scholar]
- 20. Watmough N.J., Bindoff L.A., Birch-Machin M.A., Jackson S., Bartlett K., Ragan C.I., Poulton J., Gardiner R.M., Sherratt H.S., Turnbull D.M.. Impaired mitochondrial beta-oxidation in a patient with an abnormality of the respiratory chain. Studies in skeletal muscle mitochondria. J. Clin. Invest. 1990; 85:177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shoffner J.M., Krawiecki N., Cabell M.F., Torroni A., Wallace D.C.. A novel tRNALeu (UUR) mutation in childhood mitochondrial myopathy. Am. J. Hum. Genet. 1993; 53:949. [Google Scholar]
- 22. van den Bosch B.J., de Coo I.F., Hendrickx A.T., Busch H.F., de Jong G., Scholte H.R., Smeets H.J.. Increased risk for cardiorespiratory failure associated with the A3302G mutation in the mitochondrial DNA encoded tRNALeu(UUR) gene. Neuromuscul. Disord. 2004; 14:683–688. [DOI] [PubMed] [Google Scholar]
- 23. Hutchison W.M., Thyagarajan D., Poulton J., Marchington D.R., Kirby D.M., Manji S.S., Dahl H.H.. Clinical and molecular features of encephalomyopathy due to the A3302G mutation in the mitochondrial tRNA(Leu(UUR)) gene. Arch. Neurol. 2005; 62:1920–1923. [DOI] [PubMed] [Google Scholar]
- 24. Ballhausen D., Guerry F., Hahn D., Schaller A., Nuoffer J.M., Bonafé L., Jeannet P.Y., Jacquemont S.. Mitochondrial tRNA(Leu(UUR)) mutation m.3302A>G presenting as childhood-onset severe myopathy: threshold determination through segregation study. J. Inherit. Metab. Dis. 2010; 33(Suppl. 3):S219–S226. [DOI] [PubMed] [Google Scholar]
- 25. Goto Y., Nonaka I., Horai S.. MELAS phenotype associated with m.3302A>G mutation in mitochondrial tRNA(Leu(UUR)) gene. Brain Dev. 1990; 36:180–182. [DOI] [PubMed] [Google Scholar]
- 26. Ding Y., Zhuo G., Zhang C.. The mitochondrial tRNALeu(UUR) A3302G mutation may be associated with insulin resistance in woman with polycystic ovary syndrome. Reprod Sci. 2016; 23:228–233. [DOI] [PubMed] [Google Scholar]
- 27. Yagi T., Tokunaga T., Furuta Y., Nada S., Yoshida M., Tsukada T., Saga Y., Takeda N., Ikawa Y., Aizawa S.. A novel ES cell line, TT2, with high germline-differentiating potency. Anal. Biochem. 1993; 214:70–76. [DOI] [PubMed] [Google Scholar]
- 28. Trounce I., Wallace D.C.. Production of transmitochondrial mouse cell lines by cybrid rescue of rhodamine-6G pre-treated L-cells. Somat. Cell Mol. Genet. 1996; 22:81–85. [DOI] [PubMed] [Google Scholar]
- 29. Vermulst M., Wanagat J., Kujoth G.C., Bielas J.H., Rabinovitch P.S., Prolla T.A., Loeb L.A.. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 2008; 40:392–394. [DOI] [PubMed] [Google Scholar]
- 30. Kaneda H., Hayashi J.I., Takahama S., Taya C., Lindahl K.F., Yonekawa H.. Elimination of paternal mitochondrial DNA in intraspecific crosses during early mouse embryogenesis. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:4542–4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shitara H., Hayashi J.I., Takahama S., Kaneda H., Yonekawa H.. Maternal inheritance of mouse mtDNA in interspecific hybrids: segregation of the leaked paternal mtDNA followed by the prevention of subsequent paternal leakage. Genetics. 1998; 148:851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bindoff L.A., Howell N., Poulton J., McCullough D.A., Morten K.J., Lightowlers R.N., Turnbull D.M., Weber K.. Abnormal RNA processing associated with a novel tRNA mutation in mitochondrial DNA. A potential disease mechanism. J. Biol. Chem. 1993; 268:19559–19564. [PubMed] [Google Scholar]
- 33. Maniura-Weber K., Helm M., Engemann K., Eckertz S., Möllers M., Schauen M., Hayrapetyan A., von Kleist-Retzow J.C., Lightowlers R.N., Bindoff L.A.et al.. Molecular dysfunction associated with the human mitochondrial 3302A>G mutation in the MTTL1 (mt-tRNALeu(UUR)) gene. Nucleic Acids Res. 2006; 34:6404–6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ross J.M. Visualization of mitochondrial respiratory function using cytochrome c oxidase/succinate dehydrogenase (COX/SDH) double-labeling histochemistry. J. Vis. Exp. 2011; 23:e3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guerrero-Castillo S., Baertling F., Kownatzki D., Wessels H.J., Arnold S., Brandt U., Nijtmans L.. The assembly pathway of mitochondrial respiratory chain complex I. Cell Metab. 2017; 25:128–139. [DOI] [PubMed] [Google Scholar]
- 36. Morgenstern M., Peikert P., Lubbert P., Suppanz I., Klemm C., Alka O., Steiert C., Naumenko N., Schendzielorz A., Melchionda L.et al.. Quantitative high-confidence human mitochondrial proteome and its dynamics in cellular context. Cell Metab. 2021; 33:2464–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moreno-Loshuertos R., Acín-Pérez R., Fernández-Silva P., Movilla N., Pérez-Martos A., Rodriguez de Cordoba S., Gallardo M.E., Enríquez J.A.. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat. Genet. 2006; 38:1261–1268. [DOI] [PubMed] [Google Scholar]
- 38. Bibb M.J., Van Etten R.A., Wright C.T., Walberg M.W., Clayton D.A.. Sequence and gene organization of mouse mitochondrial DNA. Cell. 1981; 26:167–180. [DOI] [PubMed] [Google Scholar]
- 39. Fan W., Lin C.S., Potluri P., Procaccio V., Wallace D.C.. mtDNA lineage analysis of mouse L-cell lines reveals the accumulation of multiple mtDNA mutants and intermolecular recombination. Genes Dev. 2012; 26:384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sato A., Nakada K., Shitara H., Kasahara A., Yonekawa H., Hayashi J.I.. Deletion-Mutant mtDNA increases in somatic tissues but decreases in female germ cells with age. Genetics. 2007; 177:2031–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maechler P., Wollheim C.B.. Mitochondrial function in normal and diabetic beta-cells. Nature. 2001; 414:807–812. [DOI] [PubMed] [Google Scholar]
- 42. Schon E.A., DiMauro S., Hirano M.. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat. Rev. Genet. 2012; 13:878–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.