

Abstract
Post-translational modifications (PTMs) of histones play a key role in DNA-based processes and contribute to cell differentiation and gene function by adding an extra layer of regulation. Variations in histone sequences within each family of histones expands the chromatin repertoire and provide further mechanisms for regulation and signaling. While variants are known to be present in certain genomic loci and carry out important functions, much remains unknown about variant-specific PTMs and their role in regulating chromatin. This ambiguity is in part due to the limited technologies and appropriate reagents to identify and quantitate variant-specific PTMs. Nonetheless, histone variants are an integral portion of the chromatin system and the understanding of their modifications and resolving how PTMs function differently on specific variants is paramount to the advancement of the field. Here we review the current knowledge on post-translational modifications specific to histone variants, with an emphasis on well-characterized PTMs of known function. While not every possible PTM is addressed, we present key variant-specific PTMs and what is known about their function and mechanisms in convenient reference tables.
Keywords: variants, post-translational modifications, histones, DNA, replication-independent
1. Introduction
The principal structural repeating unit of chromatin is the nucleosome, composed of an octameric histone protein complex consisting of two copies of core histone families H2A, H2B, H3, and H4, joined occasionally by the linker histone family, H1. Nucleosomes also play a critical role in the recruitment of downstream activities often through acting as a signaling hub for the writing, erasing, and reading of histone post-translational modifications and histone variant occupancy. The genes of mammalian and human histone families, our primary focus here, exist as multiple copies (~10-20 per family) that are found in both clusters and in isolation. Many of these genes diverge in sequence and encode slightly or moderately different amino acid sequences referred to as “variants”. These variants impart effects on many DNA-templated processes and are deposited throughout the cell cycle often by variant-specific histone chaperones. The number of genes in each family differs between species; however, the theme of multiple variants per family is generally conserved and many variants have remarkably close homologues even across kingdoms.
Replacement of canonical histones by variants affects nucleosome structure, accessibility of the histone tail, and introduces a variety of novel sequence motifs [1]. Multiple variants have been well-described for histones H2A, H3, and H1. H2B is less studied and there is little evolutionary divergence in H4[2]. Some variants have been discovered in all eukaryotes, while others are specific to higher eukaryotic lineages [3]. The monikers “canonical” and “variant” are most generally used to distinguish the common histones from those of more specialized function; however, this categorization is also informed by differences in physiology. Canonical histone genes are conspicuously intron-less. Their mRNA is also not polyadenylated as almost all eukaryotic mRNAs but regulated by an unusual and seemingly more primitive stem-loop structure. Canonical histones are also closely associated with replication. Variant histones have few to none of these traits. They are expressed in a cell-cycle independent manner with standard RNA processing and often have established specialized functions. However, these defining characteristics are not absolute. For example, some canonical histone mRNA has been observed as polyadenylated and expressed outside of S-phase and some variant histone genes are intron-less. The processing of histone variant RNA by both stem-loop and Poly A tails has been shown to be necessary for essential processes such as appropriate amounts of H2A.X throughout the cell cycle[4]. These variants also influence the post-translational modifications (PTMs) enriched or absent on histone tails.
In this review, we address post-translational modifications associated with specific histone sequences with greater granularity than usual and this requires further terms and definitions. Here we use the term “canonical variants” to individually address the many distinct protein sequences that are “canonical”. There is no single canonical histone sequence for most histone families. Thus, “canonical histone” or terms like “canonical histoneH2A” ambiguously refers to multiple unique genes and multiple distinct protein sequences. These “canonical variants” are indeed prone to differential expression, modification, and function.
Histone variants can exhibit unique PTMs or notably exclude PTMs common on other related sequences and this can profoundly affect their function. Some histone variants have unique amino acid sequences that harbor PTMs that are crucial to their variant-specific function. Other histone sequences lack otherwise well-conserved sites of modification, inferring a unique role by exclusion. Many well-studied histone PTM sites that are conserved across multiple variants have been observed to exhibit similar occupancy stoichiometry [5]; however, multiple examples of well-conserved sites being moderately to dramatically non-stoichiometrically occupied across different sequences have been observed [5,6]. Variants can also display hierarchical modification in which the presence of a particular residue is imperative for the modifications of other residues to occur (Figure 1). Unambiguous observation of variant-specific PTMs can be technically challenging. Most methods observe variants and PTMs as independent attributes without connecting the PTM to a sequence. Furthermore, it is well known that histone PTMs primarily exist and function in concert with other PTMs on the same molecule or proteoform[7]. Our sparse knowledge of histone variant-specific proteoforms indicates important mechanisms and functions yet to be revealed. Thus, understanding how histone variant-specific PTMs regulate DNA will lend greater understanding to chromatin biology.
Figure 1: Histone variants can be non-conserved, semi-conserved, exhibit unique or extended sequences, and be modified in a hierarchical manner.
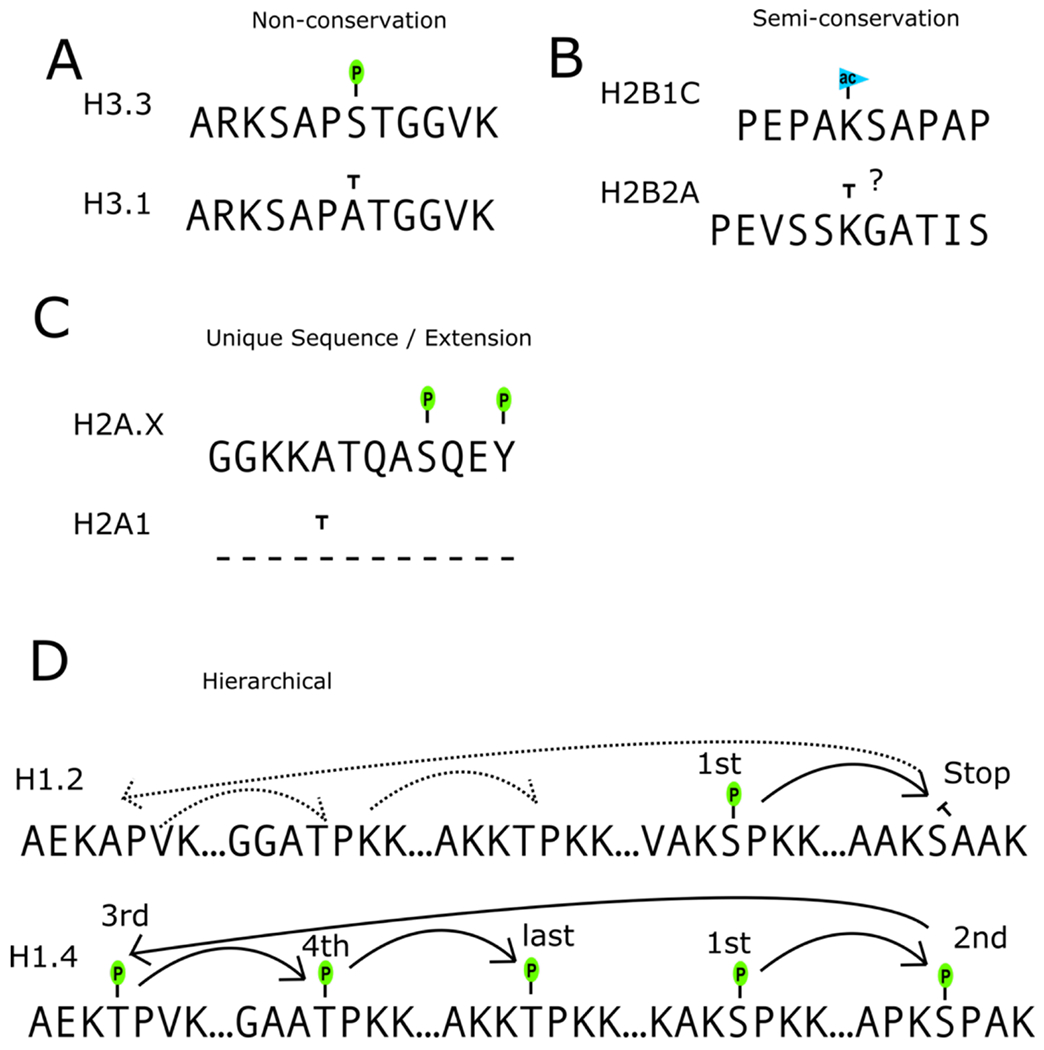
A. Non-conservation of sites of modification between variants can lead to variant PTM specificity. B. Sites of modifications can be semi-conserved existing on multiple variants but varying in neighboring residues which alters modification potential, either completely or alter abundance. C. Some variants have unique sequences compared to their canonical or variant counterpart sequences or exhibit extended sequences that are unique. D. As seen in H1.2 and H1.4, variants can be modified in a hierarchical manner. Additionally, the lack of conserved sites between variants can result in disruption to the modification hierarchy. For example, the proline after S187 is not conserved in H1.2, thereby breaking the CDK motif and precluding phosphorylation at this site. Because this is a necessary step in the primary hierarchy, the latter phosphorylation events do not occur despite good conservation between these sequences. Hierarchies can be driven by multiple factors including PTM mediated conformational changes or the reading or prior PTMs in the hierarchy.
1.1-. Histone Properties: N-terminal Acetylation
Histone N-terminal acetylation (N-α-Acetylation) is an abundant modification of unknown function [8]. This modification is co-translational and constitutive on many histones but completely absent in certain variants. N-terminal acetylation is thought to be irreversible and is not observed to occur in degrees like most post-translational modifications; it is either completely present or absent on the N-terminus of the histone it modifies. In the H2A family, only some members are N-terminally acetylated and this appears to be linked with differential function. N-terminal acetylation, while not extensively studied, has been shown to mediate essential functions in many cases [9]. The N-terminus of histones are arguably the most important part of the protein. Because this information is not readily available, we summarize confirmed histone N-terminal acetylation in the various histone families below.
1.2 -. Histone Variants- defined and described
Except for histone H4, which has at least one variant [14], multiple variants have been reported for all core histone families and the linker histone family, H1[15]. Variant sequences represent one, few or many amino acid differences that are usually expressed at low levels and inserted into specific nucleosomes by histone chaperones. The opposite of a histone variant is usually described as a ‘canonical’ histone and is defined by being synthesized in S phase. However, the term ‘canonical’ histone often refers to several different amino acid sequences either collectively or ambiguously. Given that these ‘canonical’ sequences vary slightly from each other, they are also sometimes referred to as canonical variants, sequence variants, or simply variants (broadly defined). To complicate matters further, some histone genes can be both expressed as stem loop S-phase specific mRNA and polyadenylated cell cycle independent mRNA and are thus both ‘canonical’ and ‘variant’[16, 17]. In some contexts, rather than being minor species, variant histones can be vastly more abundant than canonical histones [18]. The fraction polyadenylated also appears to be regulated by external cues [17]. Functional differences of variants are also evident in the chemical modifications and downstream impacts on variant-containing DNA.
Elucidating the functional roles and regulatory mechanisms of the variants of known importance remains of intense interest, yet technical limitations in reagents, assays, and variant-specific detection methods have stunted our advancement in these studies. Many histone variants (broadly defined) lack antibodies that distinguish them from closely related sequences. PTM-specific antibodies generally do not distinguish the variant on which the PTM is located, unless the PTM is at a variant-specific residue or sequence. Bottom-up proteomic approaches can overcome some specific challenges but is not completely effective in resolving all ambiguities [19]. Middle- and top-down proteomics resolve many of these remaining ambiguities [20]. In addition, these more challenging proteomics approaches can quantitate single molecule combinations of PTMs [21], or proteoforms[7]. However, genomic localization of variants and variant-specific PTMs remains incompletely addressed.
Imprecise language, the rapid evolution of the field, and unfounded inference pose additional challenges to understanding the literature of variant-specific PTMs. It is common for authors to ambiguously address variant identity and PTM localization or degree. This is often reflective of experimental limitations or historical artefacts. For example, H2A.Z can either refer to H2A.Z.1 or the combination of H2A.Z.1. and H2A.Z.2. The existence of splice variants, such as macroH2A.1.2, further complicates this issue [22]. Secondly, abbreviations like ‘me’ are often used to denote methylation in conjunction with the BRNO nomenclature without definition to degree [23]. The advancement of this field is dependent on clarity on these matters. We strive to untangle these ambiguities and define the specificity of knowledge on this subject. While not every possible PTM will be presented, we do highlight those with documented variant-specific functions.
2. Variants and their PTMs
Variant specificity of PTMs can occur in several different ways. The most obvious and easiest to study is when the modified residue is unique to one sequence variant. A prominent example of this is histone H3.3S31ph. The serine 31 residue is not present in other H3 sequences and thus can only be phosphorylated in this variant. Variations in sequence proximate to a modification site can modulate or even block modification despite the presence of the modified residue. More distal variations in sequence can also result in variant specificity, especially when distal modifications are prerequisite for or inhibitory of writing a PTM. Least directly, variants may be modified due to their localization in chromatin regions with greater enzymatic activity. The variant specificity of PTMs can also be reflected in subtle but quantitative differences in PTM abundances between variants. Somewhat surprisingly, the less direct mechanisms can be highly specific. Discussed below are the variants and PTMs of variants for each core histone and the linker histone family, H1, with a focus on the human and mammalian histones. We focus on clear examples of variant PTMs as well as prominent histone PTMs with variant specificity. These are organized into accessible tables for quick reference. Again, while extensive, we refrain from exhaustive documentation of all histone PTMs for the sake of clarity.
2.1-. H1 variants
Before being known as a dynamic component of chromatin, linker histones of the H1 family were seen as rigid structural components of chromatin that repressed transcription [24]. Knockout studies refuted this belief when the absence of H1 had no adverse effect on viability in tetrahymena revealing that H1 did not act as a general repressor of transcription but a regulator of certain genes [25]. Structurally, H1 remains crucial to the spacing and stability of the nucleosome particle with increased flexibility in both its N and C termini [26]. H1 is rich in lysine and post-translationally modified mostly by phosphorylation with the possibility of acetylation, ribosylation, ubiquitination, and methylation as well [27]. Somatic variants of H1 include H1.1 to H1.5 which are replication dependent while H1.0 and H1X are not. Typically, H1 incorporation is studied by antibody- based genome wide analysis methods; however, variant studies are limited by the availability of reagents [28]. Our understanding of the linker histone family is rapidly progressing as shown in recent work [28–30]. Functionally, the roles of H1 have been discussed in a variety of processes including heterochromatin formation, DNA repair and specific gene expression [31]. The PTMs associated with H1 are discussed below, organized by PTM type.
2.2 -. H1 PTMs
Phosphorylation
Phosphorylation remains the most well-characterized modification of H1 mainly occurring in the C terminal tail where kinases can readily mediate the modification; however, phosphorylation sites are spread throughout the sequence [32]. Note that H1 is most commonly indexed with the first amino acid as the residue two (S2), wherein other histones this would be indexed as residue one (S1). However, some studies do not follow this indexing rule. This should not be confused with cases where the analogous sequence and PTM is actually at a different index under the same indexing (e.g., H1.4S173ph is the analogous site to H1.2S172ph). H1 phosphorylations have been studied and linked to the cyclin dependent kinase family with phosphorylation by CDK1 notably observed during mitosis. Multiple variants of H1 and associated kinases have been studied for their role in the cell cycle [32].
Chen et al. found variant specificity of phosphorylation on different H1 variants even with local sequence conservation of modification sites [33]. H1 and its variants undergo sequential modifications and these may be interrupted by the lack of conserved sites between variants. Using top-down mass spectrometry, sequential H1.4 phosphorylation was observed during mitosis first at S173 followed by S187, T18, T146, and T154[33]. In H1.2, the lack of conserved sites early in this hierarchy resulted in the absence of progressive phosphorylation even for conserved sites. While the first phosphorylation event (S172) occurs in H1.2, S187 and T18 are not conserved and thus do not occur. Surprisingly, the later sites in this hierarchy, T146 and T154, are conserved but were not observed to be phosphorylated on H1.2. Histone H1.2 was phosphorylated at S2 independent of cell cycle and not observed on other variants. Similarly, S187ph, T18ph, T146ph, and T154ph were not found on H1.3 despite local sequence conservation at these sites. Notably, H1.3 lacks conservation at S172, thereby blocking the progressive phosphorylation. Thus, much of the cell cycle phosphorylation function of the H1 family appears to be variant-specific to H1.4, to a lesser extent H1.2, and H1.3 appears to not participate in most of these events. Several publications have also confirmed H1 phosphorylation to activate transcription by being present at promoter regions [34].
Methylation
Unlike phosphorylation, methylation of H1 occurs less frequently and represses transcription in variants H1.2-H1.5[35]. Methylation (degree undetermined) of lysine 26 (K26) in the N-terminal tail of H1.4 is the most abundant methylation found on a human linker histone [36]. This mark was traced to the methyltransferase EZH2 and G9a and removed by KDM4D [37]. H1.4 K26 is at a sequence unique to this variant, and not well conserved among the other H1 sequences. However, various methylation sites on H1 variants have been identified including H1.2 K187me2 and H1.4K26me2 which have different readers, HP1 binds to histone H1.4 and reads the modification but does not bind to H1.2 methylation supporting specific proteins are necessary for variant-specific methylation events [37]. Though many sequences where variable modification may occur are conserved between H1 variants, this is not sufficient to infer existence on a given variant or equivalent stoichiometry between variants.
Acetylation
H1 acetylation sites are thought to modulate DNA binding and displacement [8]. In H1.4, K34 acetylation mediated by the acetyltransferase GCN5 leads to the recruitment of TAF1, a subunit of the transcription factor TFIID. This reduces H1 affinity to chromatin and promotes active transcription [38]. Other acetylation events have been reported on H1, modulated by unknown enzymes. Removal of acetylation on H1 has also been reported to result in heterochromatin formation [39]. Unlike phosphorylation and acetylation, fewer studies have been conducted on H1 ubiquitination. Wong et. al and Andres et.al identify this PTM as associated with double stranded break response and repair [40, 41]. There is still much work to be done that can reveal the complete story of the variant specificity of H1 PTMs and their effect on the linker histone family.
Nonetheless, histone H1 and its PTMs are important to chromatin dynamics and many of these PTMs are surprisingly variant-specific. Many H1 modifications have been mapped by mass spectrometry however the functions remain unclear. We report the most noted sites in the table below using traditional indexing.
2.3-. H2A variants
Of the core histones, the largest number of variants are found in the H2A family with about 20 reported in mammals [51]. H2A variants have been found in almost all organisms. Of those studied, various roles have been reported for H2A variants including DNA repair, nucleosome assembly and are often implicated in the progression of cancers [52]. Only histone H2A variants that exhibit specific PTMs are addressed here. Bonisch et al. previously described other H2A variants in a 2012 review [53].
Histone variants H2A.X and H2A.Z were first described in 1980[54]. In sequence, H2A.X differs from the canonical H2As by approximately four amino acids in the core domain but has a unique extended C-terminus of approximately 22 amino acids where much of its variant-specific function is fulfilled by variant-specific PTMs. H2A.X is at the core of the DNA damage response which is regulated by H2A.X occupancy at the site of damage, H2A.X phosphorylation and additional PTMs [55, 56]. This recognized process connects H2A.X to the preservation of genomic integrity ensuring DNA damage is acknowledged and repair initiated. H2A.X is also involved in sex chromosome inactivation in male mammals leading to heterochromatic regions where chromatin is silenced. Upon silencing, unpaired regions of the X and Y chromosomes form what is known as the sex body [57]. The sex body is characterized by phosphorylated H2A.X, the exclusion of RNA polymerase and condensed gonosomes [25]. Notably, when H2A.X deficient spermatocytes were studied, spermatocytes fail to condense to form the sex body and severe defects in meiotic pairing occurred. In the absence of H2A.X, other sex body forming proteins failed to localize to sex chromosomes demonstrating the importance of H2A.X to chromosome silencing in males. Combinations of PTMs can also modulate biological outcomes and this is observed in H2A.X where both serine 139 and tyrosine 142 phosphorylation were implicated in the DNA damage response [55].
Evolutionarily, the H2A variant H2A.Z is conserved and encoded by two distinct genes H2AZ1 and H2AZ2 (previous commonly used gene names: H2AFZ and H2AFV, respectively) [3]. These genes encode proteins that differ by three amino acids, commonly referred to as H2AZ.1 and H2AZ.2, although the Uniprot recommended protein names are H2A.Z and H2A.V, respectively. This is not to be confused with the drosophila-specific histone variant H2A.V which is indeed a homologue of human H2A.V (H2AZ.2) but also H2A.Z.1 and H2A.X. Additionally, the H2AZ2 gene also produces a splice variant commonly denoted H2AZ.2.2 [56]. In the literature, the generic H2A.Z moniker can either refer to H2A.Z.1 alone or an ambiguous mixture of H2A.Z.1, H2A.Z.2, and possibly H2A.Z.2.2. Here we endeavor to use precise language to explicitly clarify variant sequences, although prior evidence and/or language is frequently not variant-specific. Within this review, we use H2A.Z to mean ambiguously H2A.Z.1 or H2A.Z.2 and otherwise specify the specific sequence(s).
Unlike H2A.X, H2A.Z.1 and H2A.Z.2 vary significantly in their sequence from canonical H2A, although their sequences largely resemble each other [58]. Lack of conservation between major H2A sequences decreases the possibility for similar modification patterns and suggests differential functions.
The primary role of H2A.Z (ambiguously H2A.Z.1 and/or H2A.Z.2) remains controversial being reported as both a transcriptional activator and repressor but is believed to be mostly involved in gene expression and transcription with little understanding of how it regulates these events [59]. The location of variant histones within the genome impacts downstream functions like transcription and gene regulation which some PTMs directly influence. H2A.Z acetylation is enriched at promoters involved in transcription in yeast, mammals, and plants while ubiquitination is associated with repressed regions of chromatin and polycomb targets [60]. H2A.Z is important in plant biology where it plays essential regulatory roles, including modulating growth based on ambient temperature [61]. When several forms of mononucleosomes were reconstituted and studied for stability, authors found those with H2A.Z containing particles exhibit increased compact confirmation indicative of increased nucleosome stability [51].
Macro H2A
Unique to the H2A family are the macro variants, (mH2A1.1, mH2A1.2 and mH2A2) which are vertebrate-specific and characterized by the presence of an evolutionarily conserved 25 kDa globular region called the macro domain where interaction with histone-modifying enzymes often occur [62]. Two spliced forms of mH2A1 exist, mH2A1.1, mH2A1.2 differing in their expression patterns during development [63]. mH2A1.2 has been reported to have multifaceted functions in gene transcription and also shown to localize to the H3K27me3 marked promoter region of repressed genes [64]. Only moderate sequence variation exists within the canonical H2As with some H2A PTMs being identified as specific to the canonical sequences but our goal here is to present those PTMs that are specific to the noted variant sequences.
2.4. H2A PTMs
Phosphorylation
Phosphorylated serine 139 of H2A.X is referred to as gamma-H2A.X (ɣH2A.X), a commonly used hallmark signal of DNA double stranded break that accumulates at sites of DNA damage [57]. ATM is well-established as the kinase that mediates S139ph of H2A.X[65]. In mammals, γH2A.X is read by MDC1 forming a complex with phosphorylated H2A.X[66]. Using proteomic approaches, Kleiner et. al identified additional readers of γH2A.X such as 53BP1 a DNA repair mediator that recognizes γH2A.X through its BRCT domains [67]. Once present, ɣH2A.X is necessary to retain and ensure efficient repair by recruiting remodeling factors at the site of damage [68]. Apart from S139ph, the C terminal domain of H2A.X can also be phosphorylated at Y142 by the WSTF remodeling factor kinase [69]. Cook et al. show that upon DNA damage, dephosphorylation of Y142 on H2A.X avoids apoptosis. Using phosphosynthetic peptides, they demonstrated binding of pro-apoptotic factors like JNK1 to S139ph/Y142ph doubly phosphorylated peptides revealing Y142ph is abundant and related to DNA damage [70].
As for H2A.Z, it has been overexpressed in malignancies like melanoma, breast, prostate, and bladder where in some cases it is involved in cell proliferation [71]. However, these studies make few or little acknowledgement of which isoform was measured. In another study, extracted lesions of melanoma patients revealed H2A.Z.1 and H2A.Z.2 increased expression correlated to significantly lower survival [72].
Acetylation
H2A.Z.2 has also been shown to be acetylated. Increased acetylation levels have been observed in cancers expressing H2A.Z.2 occupied chromatin via a BRD2-E2F1 axis [68]. A 2016 study reported cancer cells also exhibit increased marks of methylation at K101 when H2A.Z.1 was present. The dimethyl mark was written by methyltransferase SMYD3 and linked to promoting proliferation by activating cell cycle regulators like cyclin A1 [73]. Thus, H2A variants and their PTM landscape are critical factors in diseases, especially malignancies.
Macro PTMs
The PTM landscape of macro H2As have not been widely characterized apart from hosting a special PTM: ADP-ribosylation (ADPR) [49]. Scarcity in additional studies is mostly due to limitations in reagents, their larger size and lower abundance. However, mH2A1.2 was reported to have several PTMs when analyzed by mass spectrometry. Ubiquitination of K115 was confirmed on mH2A1.2 by Chu et al and has been reported by other groups [49]. Additional PTMs for macro H2A identified in this study include K17me, K122me, and T128ph. Despite their identification, the exact functions of these PTMs remain unclear.
Canonical histone H2A variants and any PTM specificity within the canonical sequences are poorly studied. The limited reports show at least some canonical variants being disproportionately acetylated at K5 (H2A2A and H2A1H) and phosphorylated at S1 (H2A1 and H2A2A) at least within some contexts [20]. The role of each of these canonical sequences in transducing these signals will require further study for validation of differential function.
2.5-. H2B Variants
H2B variants are less studied and divided into two classes. H2B sequences are most often reported as S-phase dependent, yet polyadenylation has also been reported [17]. H2B variants have been documented to play roles in chromatin compaction during gametogenesis; however, the mechanistic role of H2B remains unclear [82]. One difficulty in studying H2B variants is the high sequence divergence and the lack of conserved sites between variant and canonical H2B [83]. In humans, phosphorylation on S14 and S36 of H2B is present but excluded on variants H2BFM and H2BFWT where these sites are not conserved. Even amongst conserved sequences of H2B, there are multiple sites where modifiable residues exist but modifications are excluded, suggesting functional differences amongst sequences. Why so many variants of H2B exist remains unclear. Additionally, for H2B variant-PTM specificity is mostly presented as the lack of a PTM on one or two variants rather than a unique presence on one or two variants as observed in other histone families. However, multiple histone H2B sequence variants are present in all higher eukaryotes. This is in stark contrast to histone H4 that is almost always a single sequence per organism despite encoding multiple independently evolving genes. In organisms like plants, H2B variants have been functionally described in detail, suggesting variant-specific functions unique to plants, separate from human and mammalian histone H2B [84]. Humans have at least 20 different H2B amino acid sequences. H2B proteins are largely involved in germinal cell production and identified in gonadic tissue, specific to sperm/testis are variants H2B1A (aka htSH2B) and H2BFWT shown to affect nucleosome stability [85–87]. H2B variants are implicated in various processes important to many studied pathologies today but detection of PTMs within this histone family have remained scarce when compared with H3 and H2A.
2.6-. H2B PTMs
Ubiquitination, phosphorylation, and acetylation
Many have speculated the possible PTMs of H2B with few confirmed and functionally validated studies. Ubiquitination is a less commonly discussed but large modification confirmed in H2B variants [88]. H2B is often ubiquitinated (H2bub) at K120 and this modification is found on most variants except the variants H2BFWT and H2BFM where this site is particularly not conserved [85]. If there is a variant-specific function of H2BFWT and H2BFM it is likely connected to being refractory to this important modification. H2Bub has been shown to physically disrupt chromatin strands, fostering a more open chromatin structure accessible to transcription factors and DNA repair proteins. Other roles for this modification include histone cross-talk, influencing methylation events on histone H3, including H3K4[89]. H2B acetylation at K5 is a contributor to active transcription in cells [90]. Confirmed by multiple studies is the phosphorylation of H2B at S14 by the kinase MST1 involved in apoptosis, that is also excluded on variants H2BFWT and H2BFM [91, 92]. AMPK mediates phosphorylation of H2B at S36 and was found on actively transcribed genes [93]. Still, the lack of accurate discrimination amongst H2B sequences in most work limits our current understanding of H2B variant-specific PTMs. Many of the narratives proposed in the literature for H2B modifications likely have some variant specificity and should be tested further in future work.
2.7-. H3 Variants
H3 variants have been found in most eukaryotic organisms, including lower eukaryotes such as yeast [98]. One H3 variant CENP-A, expressed specifically in the centromere, has been linked to cell survival due to its critical role in centromere function during mitosis [99]. There are also tissue-specific H3 variants, such as H3.1t. The somatic H3 variants H3.1, H3.2 and H3.3 play crucial roles in transcription and chromatin silencing [100]. In mammals, H3.2 has been enriched in marks associated with gene silencing, while H3.3 is reported as enriched at transcriptionally active regions [100]. Differing by a single amino acid, H3.1 and H3.2 have historically been functionally classified as indistinct. However, the dramatic serine (H3.2) to cysteine (H3.1) difference at position 96 suggests unique function and modification potential, such as accepting S-Glutathionylation, S-palmitoylation, or intermolecular disulfide (figure 2). However, no such roles for H3.1C96 have been rigorously validated. We have since learned that these variants differ in both their expression and PTM patterns suggesting these variants may have individual biological functions [95]. Still, many investigations do not confidently distinguish between all H3 sequences. This is true especially when resolving H3.1 and H3.2; however, H3.3 is more readily distinguished, including by commercially available antibodies, and is more clearly distinct. Although the differences in sequence in the H3 family can be subtle they often translate into distinct function.
Figure 2: Frequently modified and non-conserved sites of Histone H3 variants.
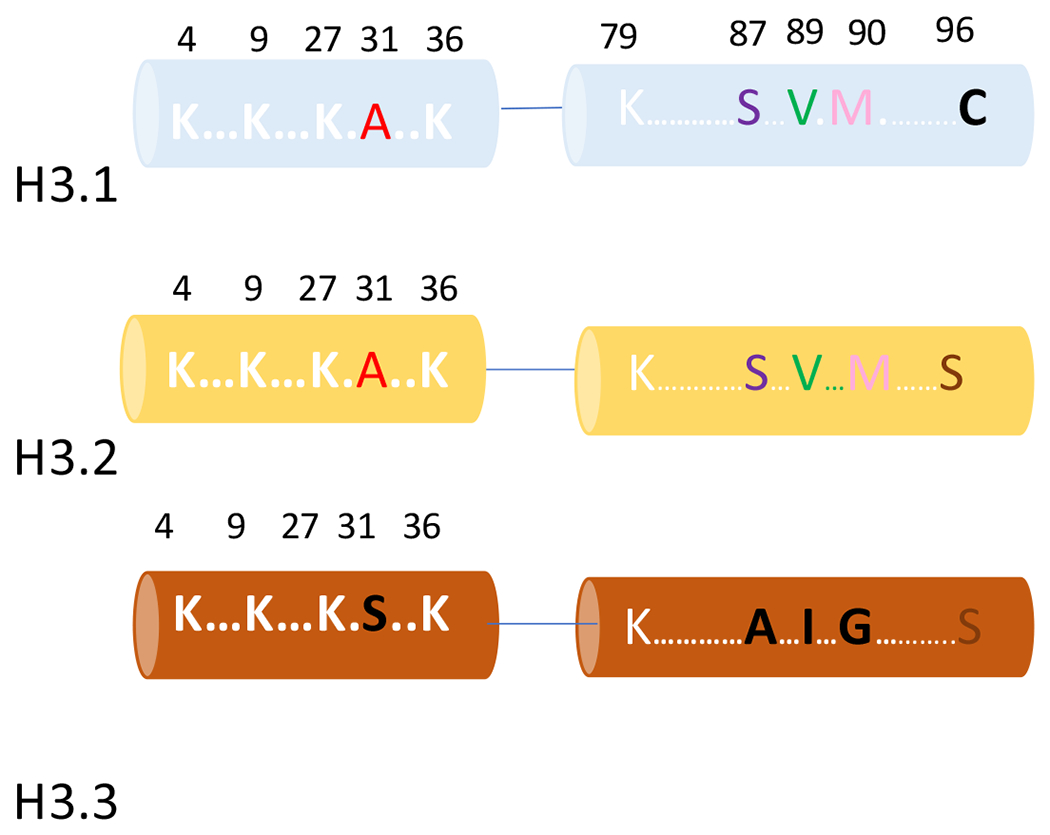
H3 variant sequences contain many similarities but there are also points of difference that are prone to modifications. We discuss Serine 31 in H3.3, a common site of H3 phosphorylation unique to this variant. Histone H3.1 and H3.2 share a serine at position 87 and a valine at position 89 which are alanine and Isoleucine in H3.3 respectively. Position 90 is conserved between H3.1 and H3.2 but is changed to a Glycine on H3.3. H3.1 and H3.2 differ by a single amino acid, H3.1 having a cysteine and H3.2 a serine at position 96.
2.8-. H3 PTMs
Phosphorylation
H3 is rich in lysine residues and is extensively modified on its N-terminal tail. Replication coupled H3.1 and replication-independent H3.3 are arguably some of the better studied variants of the H3 family [101]. H3.3 phosphorylation at S31 is a mitotic signal not conserved and thus not observed in H3.1 and H3.2[100]. Apart from its role in mitosis, other roles for H3.3 phosphorylation including activation of transcription, p300 activity, and enhancer acetylation has been studied and described [102, 103]. H3.3 has also been reported as enriched for di-acetylation at K9 and K14, a mark of active transcription [102].
Methylation and acetylation
The extraordinarily high lysine content and other commonly modified amino acids make H3 prone to many PTMs, with acetylation and lysine methylation as the most abundant. These abundant modifications often coexist on the same molecule in complex combinations [104, 105]. Differences in sequence of H3 variants also affect the PTMs which can be present on each variant. For example, in organisms like Arabidopsis, H3.1 is preferentially methylated at K27 over H3.3 due to steric hinderance present on H3.3 and this modification is important for the maintenance of H3K27 methylation after DNA replication. In contrast in the Arabidopsis sperm-specific H3 variant H3.10, K27 cannot be methylated preventing H3K27 methylation in spermatogenesis [103]. An important line of evidence for the significance of variant-specific PTMs is the presence of at least one endogenous variant-specific PTM reader of H3.3K36me3 in ZMYND11[106].
H3 acetylation is nearly always associated with some aspect of activation of transcription while H3 methylation has a complex relationship with transcription. Many H3 lysine methylation events are associated with repressed transcriptionally silent chromatin; however, some are linked to active transcription and other chromatin states. K4 methylation is notably linked to active transcription [107]. K79 methylation promotes chromatin accessibility [108, 109]. The interpretation of H3 methylation is also nuanced, such as K36 methylation that is laid down by active transcription but plays a largely repressive role once present [110]. These nuances can also be modulated by the context of other PTMs present in cis or nearby. Di and tri-methylation of lysine 27, a mark of gene silencing, is greatly enriched in H3.2 fractions in many contexts, including HEK293 cells [98, 100]. Much of what is known about the variant specificity of H3 PTMs exhibits quantitative differences in abundance rather than uniqueness [101–103, 107, 108]. While H3 remains at the core of many histone studies and its PTMs are intensively studied, these PTMs are mostly not accurately assigned to specific variants nor studied for variant-specific function. A more accurate accounting of the variant specificity is needed to fully understand the more nuanced mechanisms of regulation at play. Functionally characterized H3 variant-specific PTMs are found below.
2.9-. H4 Variants
Histone H4 remains the most conserved sequence decorated mostly with abundant sites of acetylation on its lysine residues at K5, K8, K12 and K16 and methylation on lysine 20 [113]. Many proteomic studies have focused on histone H4[12, 114, 115]. To date, only one variant has been confirmed and reported in this family, with only about 85% sequence similarity to canonical H4 sequence [12, 14]. Why this histone is exceptionally conserved is not clearly understood; however, mutations are rarely observed due to embryonic lethality [116].
3. Overcoming Current Limitations
We have described and documented the evidence of histone variant-specific PTMs and what is known or can be inferred about their variant-specific function; however, the vast majority of the chromatin literature provides limited insights for several key reasons. Ambiguous language and incomplete description of precise sequence often confound a ready understanding of this subject. The methods used are often equally ambiguous because the technology does not distinguish variant-specific PTMs. However, there are solutions available or emerging that can address these issues and reveal a more complete understanding of chromatin biology.
A lack of clearly defined sequences and variant identity perpetuates ambiguity; however, this is readily addressed. We simply need to elevate the expectation of specificity and clarity in the language used and provide ready access to detail. For example, when using a sequence to represent the canonical variants of a family, it is important to explicitly state the sequence name and that it is representative of a collection of canonical sequences. A system of protein accession numbers as well as recommended names have been developed for such purposes with brevity. It is similarly trivial to list out multiple sequences when the experiment does not distinguish between sequences.
Most importantly, there is a scarcity of technologies that address variant-specific PTMs conveniently and effectively; however, there are both available and emerging technologies to address this. Resolving histone variant-specific biology is essential to building the framework of knowledge to understand and ultimately manipulate chromatin. Yet, few methods exist that can help us detect, quantify, or isolate variants.
Antibody-based detection of histones has long been the standard of chromatin studies. Some variant-specific PTMs that are more divergent, such as H2A.XS139ph, are easily distinguished by superb antibodies. More generally, these approaches have limited capacity to quantify PTMs, localize these PTMs to specific variants, and are mostly incapable of detecting combinations of PTMs. Nonetheless, carefully designed experiments have successfully revealed variant-specific PTM functions, although less directly and conveniently than desired. Sequential ChIP approaches have potential but are especially prone to false detection without proper controls due to the near unlimited capacity to amplify signal and limited (even if excellent) specificity of reagents. Epitope tagged variants have been successfully used to study histone variants and their PTMs [117, 118]; however, this introduces a perturbation that may affect results. This limitation is more pronounced when addressing smaller differences in sequence. It is possible in some situations to develop anti-bodies that cover sufficient sequence to recognize both a PTM and a variant-specific sequence. A related approach that may offer outstanding insights would be to use the endogenous readers, such as ZMYND11[106]to enrich and or detect variant-specific PTMs.
Mass spectrometry-based proteomics offers an excellent capacity to address questions regarding the variant specificity of histone PTMs. Mass spectrometry (MS) allows for the qualitative and quantitative analysis of histones including variant-specific PTMs and co-occurring PTMs. Bottom-up proteomic methods rely on protein digestion and the analysis of peptides, while top-down proteomics analyzes the intact protein. Top-down proteomics preserves the most variant specificity and PTM combination information but is challenging. In between, middle down proteomics uses limited digestion to maintain more variant specificity and PTM combination information, while enabling analysis of densely modified proteins such as histone H3. With MS, we can elucidate the possible complex combinations of PTMs and their variant-specific PTM patterns. Hundreds of individual PTMs have been mapped with MS analysis but these approaches remain technically challenging [107,108]. Proteomic approaches do not excel at genomic or spatial localization, although this is possible. ChIP-mass spec approaches have the potential to report both genomic localization and inform or validate the variant specificity of the PTMs. This overcomes some of the limitations of sequential ChIP by using a rigorous method of identity and quantity validation. In general, MS-based validation in combination with other enrichment approaches has significant promise moving forward. Finally, the field would benefit from use or development of approaches that provide absolute instead of relative abundance of modifications. Proteomic approaches should be more widely used in chromatin research to validate results, provide variant specificity, and reveal combinations of PTMs acting in cis. While not the prevailing approach in chromatin biology, collaborations with chromatin-focused proteins offer new perspectives for ongoing projects around the world.
There are also emerging technologies to allow more variant-specific PTM studies. Recombinant nucleosomes with specific variants and PTMs show exceptional promise for the in vitro testing of variant-specific mechanisms [119, 120]. Mass spectrometric analysis of intact nucleosomes with detection of variants has also been demonstrated [121]. These approaches and similar future innovations will inevitably allow greater specificity and enable more variant-specific PTM studies. As the building block of the physiological state of the genome, chromatin affords us a potential plethora of knowledge. Yet there exists a significant incomplete understanding of chromatin, including histone variant-specific PTMs. As a field we can bridge this knowledge gap by innovating and continuing to pursue publications that present a level of detail necessary to better understand histone variants and their PTMs.
Conclusion
Histone variants remain of intense interest due to their critical role in many cellular processes. However, due to the lack of reagents and methods that effectively localize PTMs to specific variants conveniently and accurately, there is still much to learn. Variant-specific PTM studies have a significant promise to reveal chromatin function more completely. Variant containing nucleosomes are key players in the regulation of the genome. The PTMs present on these variants are obvious mediators of these variant-specific functions. As the technology and understanding in this area advances, chromatin biology will be increasingly focused on variant-specific PTMs. Our hope is that advances in approach will alleviate the current ambiguities that exist and allow us to identify variant-specific mechanisms of chromatin regulation confidently and rapidly. While evidence justifies ongoing variant-specific PTM effort, there remains a limited body of work addressing variant-specific PTMs. Although this paper aims to present confirmed functions of variant-specific PTMs we still do not understand many of the functions PTMs possess. Our goal has been to present the major variant-specific modifications of histone in a single publication to document the current knowledge on complexities of histone variants and their PTMs.
Table 1.
Evidence of N-terminal Acetylation (N-α-Acetylation) in histone families
Table 2 -.
Major variant-specific PTMs of histone H1, “?” indicates uncertainty of modification in the respective variant.
| H1 PTM | Site conserved | Variant Specificity | Writer | Reader | Eraser | Function | Dependency | Citation |
|---|---|---|---|---|---|---|---|---|
| S2ph | H1.2, H1.3, H1.4, H1.5 | H1.2, H1.3, H1.4, H1.5 | Aurora B Kinase, PKA, GSK3 | Cell cycle/Mitosis | Zheng,[42] Talasz[43] Alexandrow[44] |
|||
| T18ph | H1.3, H1.4 | H1.3, H1.4 | Aurora B Kinase, PKA, GSK3 | Mitosis | Andres[40] | |||
| T146ph | H1.2, H1.3, H1.4 | H1.2, H1.4 | Aurora B Kinase, PKA, GSK3 | Chen[33] | ||||
| S17ph | H1.5 | H1.5 | Activates Transcription | Izzo[45] | ||||
| T154ph | H1.2, H1.3, H1.4, H1.5 | H1.2, H1.4, H1.5 | Aurora B Kinase, PKA, GSK3 | Izzo Chen [33, 45] |
||||
| S172/173ph | H1.2, H1.3, H1.4 | S173 is high in H1.2, S172 is high in H1.4 | Aurora B Kinase, PKA, GSK3 | Activates Transcription | Chen Zheng[33, 42] |
|||
| S187ph | H1.3, H1.4, H1.5 | H1.3, H1.4, H1.5 | Aurora B Kinase, PKA, GSK3 | Izzo [45] |
||||
| S35ph | H1.2, H1.3, H1.4 | High in H1.4 | PKA | Happel[29] | ||||
| S2ac | H1.2, H1.3, H1.4, H1.5 | GNC5 | PHD/BRDs | HDAC | Activates Transcription | Andres[40] | ||
| K26ac | H1.2, H1.3, H1.4, H1.5 | H1.4 | SirT1 | Activates Transcription | Vaquero[39] | |||
| K34ac | H1.2, H1.3, H1.4, H1.5 | ? in H1.5, high in H1.4 | GCN5 | Activates Transcription | Kamieniarz Izzo[38, 45] |
|||
| K26me | H1.2, H1.3, H1.4, H1.5 | High in H1.4 | G9a/EZH2 | KDM4 | Represses Transcription | Blocked by S27ph | Trojer Walport Daujat [46–48] |
|
| K84ac | H1.1, H1.2, H1.3, H1.4, H1.5 | H1.4 | PCAF | HDAC1 | DNA Damage | Chu[49] | ||
| K52me | H1.2, H1.3, H1.4, H1.5 | ? in H1.5 | Represses Transcription | Allfrey[50] | ||||
| K46ub | H1.2, H1.3, H1.4, H1.5 | ? in H1.5 | RNF8 | Double stranded break repair | Wong[41] | |||
| K63ub | H1.2, H1.3, H1.4, H1.5 | H1.2 | RNF8-UBC3 | Double stranded break repair | Andres[40] |
Table 3-.
Major variant-specific PTMs of histone H2A
| H2A PTM | Site conserved | Variant Specificity | Writer | Reader | Eraser | Function | Dependency | Citation |
|---|---|---|---|---|---|---|---|---|
| S1ph | All except: H2AB2, H2A.Z, H2AJ, H2AV, H2AB1 | H2A, H2A. X, H2A.bbd, mH2A1, H2A1, H2A2A | MSK1 | PKC | Dang, Tropberger[20, 74] | |||
| K5ac | All except: H2AB1, H2AB2 | Higher in H2A2A and H2A1H, but not exclusive | Dang[20] | |||||
| K13ac | All except: H2AB2, H2AV, H2AB1, H2AJ | H2A.Z | Tip60, HAT1, CBP/p300 | BPTF | Activates Transcription | Corujo, Kimura[75, 76] | ||
| K15ac | All except: H2AB1, H2AV, H2A.Z, H2AB2, H2A1 | Tip60, HAT1, CBP/p300 | Brd2 | Activates Transcription | Corujo, Kimura[75, 76] | |||
| K7me | All except: H2AB1, H2AB2 | H2A.Z | SETD6 | Cellular differentiation | Binda,[77] | |||
| K17me | mH2A1.2 | mH2A1.2 | Activates or represses transcription | Variant associated with H3K27me | Chu,[49] | |||
| T128ph | mH2A1.1, mH2A1.2 | mH2A1.1 | Heterochromatin exclusion | Chu, Corujo [49, 75] |
||||
| S139ph | H2A.X, mH2A1 | H2A.X | ATM, ATR, DNA-PK, JNK | MDC1 | DNA Damage/repair | on T142ph | Brown[78] | |
| Y142ph | H2A.X | H2A.X | WSTF | EYA | Double strand break repair/Apoptosis | on S139ph | Hatimy, Xiao [55, 69] |
|
| K115ub | mH2A1, H2A.Z | mH2A1.2 | RING1B PRC1/USP10 |
USP3, USP11, USP16, USP44, BRCC36, Dub3 | Variant associated with H3K27me | Chu, Hernandez [49, 79] |
||
| K119ub | All except H2AB1, H2AB2 | H2A.X | UBC13 | Involved in Double stranded break recognition | Ikura,[80] | |||
| K121ub | All | H2A.Z | BRCA1 | USP10 | Transcriptional silencing | Corujo Monteiro [75, 81] |
Table 4-.
Major variant-specific PTMs of histone H2B
| H2B PTM | Site conserved | Variant Specificity | Writer | Reader | Eraser | Function | Dependency | Citation |
|---|---|---|---|---|---|---|---|---|
| K5ac | Except H2BFWT, H2BFM | All except H2BFWT, H2BFM | Promotes gene transcription | Chitsazian[90] | ||||
| K12ac | All except H2B1A, H2BWT, H2BFM | Except H2B1A, H2BWT, H2BFM | Activates Transcription | Magic, Bonenfant[94, 95] | ||||
| K15ac | All except H2B1A, H2BWT, H2BFM | Except H2B1A, H2BWT, H2BFM | Activates Transcription | Magic [94] | ||||
| K20ac | All except H2B1M, H2B1A, H2BWT | Except H2B1M, H2B1A, H2BWT. Well conserved in H2BFS |
Activates Transcription | Magic, Bonenfant[94, 95] | ||||
| S14ph | Except H2B1A, H2BFWT, H2BFM | All except H2B1A, H2BFWT, H2BFM | MST1 | Apoptosis | Zhao, Cheung[91, 92] | |||
| S36ph | Except H2BFWT, H2BFM | All Except H2BFWT, H2BFM | AMPK | Activates Transcription | Bungard[93] | |||
| K120ub | except H2BFWT, H2BFM | All Except H2BFWT, H2BFM | RNF20/40 | Activates Transcription | Mark can be inhibited by the presence of H2A.Z | Wojcik, Wright [96, 97] |
Table 5-.
Major variant-specific PTMs of histone H3
| H3 PTM | Site conserved | Variant Specificity | Writer | Reader | Eraser | Function | Dependency | Citation |
|---|---|---|---|---|---|---|---|---|
| K9ac | H3.1, H3.2, H3.3 | Enriched : H3.3 | GCN5/G9a | JHDM2 | Activates Transcription | Loyola, Kutateladze[104, 105] | ||
| K14ac | H3.1, H3.2, H3.3 | Enriched : H3.3 | TIP60 | ANCCA, PB-2 | JHDM2 | Activates Transcription | Hake, Kutateladze[98, 105] | |
| K18ac | H3.1, H3.2, H3.3 | Enriched : H3.3 | CBP/P300 | BRDT | JHDM2 | Activates Transcription | Hake, Kutateladze [98, 105] | |
| K23ac | H3.1, H3.2, H3.3 | Enriched : H3.3 | SAS3 | TRIM24 | JHDM2 | Activates Transcription | Hake, Kutateladze[98, 105] | |
| K27ac | H3.1, H3.2, H3.3 | Enriched : H3.3 | BRDT | Kutateladze[105] | ||||
| K4me | H3.1, H3.2, H3.3 | Low H3.1 | MLL1 | LSD1 | Activates Transcription | Kutateladze[105] | ||
| K9me | H3.1, H3.2, H3.3 | High in H3.1 | SUV39H2 | Tudor domain | JHDM2 | Represses Transcription | Loyola[104] | |
| K27me | H3.1, H3.2, H3.3 | Enriched : H3.2/H3.3 | EZH2 | Tudor/chromodomain | JARID2 | Represses Transcription | MacroH2A | Hake, Kutateladze [98, 105] |
| K36me | H3.1, H3.2, H3.3 | Enriched : H3.3 | SET2 | Tudor domain ZMYND11 (H3.3-specific) | JARID2 | Activates Transcription | Kutateladze[105] Wen[106] |
|
| S28ph | H3.1, H3.2, H3.3 | MSK1 | 14-3-3 | Kutateladze, Sawicka[105, 111] | ||||
| S31ph | H3.1, H3.2, H3.3 | Exclusively: H3.3 | 14-3-3 | Geneactivation | Hake, Kutateladze[105, 112] | |||
| K79me | Enriched : H3.1/3 | DOT1L | TP53 BP1 | H2B Ubiquitination | Sullivan, Wood[99, 109] |
Acknowledgements
This work was supported in part by NIH grants P01AG066606, R01GM139295, and R01CA193235. The authors thank members of the Young lab and Michael-Christopher Keogh for helpful discussions. The authors have no competing interests to declare.
References
- [1].Wu RS, Bonner WM. Separation of basal histone synthesis from S-phase histone synthesis in dividing cells. Cell. 1981;27(2):321–330. doi: 10.1016/0092-8674(81)90415-3 [DOI] [PubMed] [Google Scholar]
- [2].Henikoff S, Smith MM. Histone Variants and Epigenetics. Cold Spring Harbor Perspectives in Biology. 2015;7(1):a019364. doi: 10.1101/cshperspect.a019364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Talbert PB, Ahmad K, Almouzni G, Ausió J, Berger F, Bhalla PL, Bonner WM, Cande WZ, Chadwick BP, Chan SWL, et al. A unified phylogeny-based nomenclature for histone variants. Epigenetics & Chromatin. 2012;5(1):7. doi: 10.1186/1756-8935-5-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Griesbach E, Schlackow M, Marzluff WF, Proudfoot NJ. Dual RNA 3′-end processing of H2A.X messenger RNA maintains DNA damage repair throughout the cell cycle. Nature Communications. 2021;12(1):359. doi: 10.1038/s41467-020-20520-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gunjan A, Alexander BT, Sittman DB, Brown DT. Effects of H1 histone variant overexpression on chromatin structure. The Journal of Biological Chemistry. 1999;274(53):37950–37956. doi: 10.1074/jbc.274.53.37950 [DOI] [PubMed] [Google Scholar]
- [6].Lu C, Coradin M, Janssen KA, Sidoli S, Garcia BA. Combinatorial Histone H3 Modifications Are Dynamically Altered in Distinct Cell Cycle Phases. Journal of the American Society for Mass Spectrometry. 2021;32(6):1300–1311. doi: 10.1021/jasms.0c00451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].The Consortium for Top Down Proteomics, Smith LM, Kelleher NL. Proteoform: a single term describing protein complexity. Nature Methods. 2013;10(3):186–187. doi: 10.1038/nmeth.2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wiśniewski JR, Zougman A, Krüger S, Mann M. Mass Spectrometric Mapping of Linker Histone H1 Variants Reveals Multiple Acetylations, Methylations, and Phosphorylation as Well as Differences between Cell Culture and Tissue. Molecular & Cellular Proteomics. 2007;6(1):72–87. doi: 10.1074/mcp.M600255-MCP200 [DOI] [PubMed] [Google Scholar]
- [9].Ree R, Varland S, Arnesen T. Spotlight on protein N-terminal acetylation. Experimental & Molecular Medicine. 2018;50(7):1–13. doi: 10.1038/s12276-018-0116-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hole K, Van Damme P, Dalva M, Aksnes H, Glomnes N, Varhaug JE, Lillehaug JR, Gevaert K, Arnesen T. The Human N-Alpha-Acetyltransferase 40 (hNaa40p/hNatD) Is Conserved from Yeast and N-Terminally Acetylates Histones H2A and H4 Imhof A, editor. PLoS ONE. 2011;6(9):e24713. doi: 10.1371/journal.pone.0024713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dang X, Singh A, Spetman BD, Nolan KD, Isaacs JS, Dennis JH, Dalton S, Marshall AG, Young NL. Label-Free Relative Quantitation of Isobaric and Isomeric Human Histone H2A and H2B Variants by Fourier Transform Ion Cyclotron Resonance Top-Down MS/MS. Journal of Proteome Research. 2016;15(9):3196–3203. doi: 10.1021/acs.jproteome.6b00414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dang X, Scotcher J, Wu S, Chu RK, Tolić N, Ntai I, Thomas PM, Fellers RT, Early BP, Zheng Y, et al. The first pilot project of the consortium for top-down proteomics: a status report. Proteomics. 2014;14(10):1130–1140. doi: 10.1002/pmic.201300438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Demetriadou C, Koufaris C, Kirmizis A. Histone N-alpha terminal modifications: genome regulation at the tip of the tail. Epigenetics & Chromatin. 2020;13(1):29. doi: 10.1186/s13072-020-00352-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Long M, Sun X, Shi W, Yanru A, Leung STC, Ding D, Cheema MS, MacPherson N, Nelson CJ, Ausio J, et al. A novel histone H4 variant H4G regulates rDNA transcription in breast cancer. Nucleic Acids Research. 2019;47(16):8399–8409. doi: 10.1093/nar/gkz547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Martire S, Banaszynski LA. The roles of histone variants in fine-tuning chromatin organization and function. Nature Reviews Molecular Cell Biology. 2020;21(9):522–541. doi: 10.1038/s41580-020-0262-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Arnaudo AM, Molden RC, Garcia BA. Revealing histone variant induced changes via quantitative proteomics. Critical Reviews in Biochemistry and Molecular Biology. 2011;46(4):284–294. doi: 10.3109/10409238.2011.577052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kari V, Karpiuk O, Tieg B, Kriegs M, Dikomey E, Krebber H, Begus-Nahrmann Y, Johnsen SA. A Subset of Histone H2B Genes Produces Polyadenylated mRNAs under a Variety of Cellular Conditions Tian B, editor. PLoS ONE. 2013;8(5):e63745. doi: 10.1371/journal.pone.0063745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tvardovskiy A, Schwämmle V, Kempf SJ, Rogowska-Wrzesinska A, Jensen ON. Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nucleic Acids Research. 2017;45(16):9272–9289. doi: 10.1093/nar/gkx696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].El Kennani S, Crespo M, Govin J, Pflieger D. Proteomic Analysis of Histone Variants and Their PTMs: Strategies and Pitfalls. Proteomes. 2018;6(3):29. doi: 10.3390/proteomes6030029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dang X, Singh A, Spetman BD, Nolan KD, Isaacs JS, Dennis JH, Dalton S, Marshall AG, Young NL. Label-Free Relative Quantitation of Isobaric and Isomeric Human Histone H2A and H2B Variants by Fourier Transform Ion Cyclotron Resonance Top-Down MS/MS. Journal of Proteome Research. 2016;15(9):3196–3203. doi: 10.1021/acs.jproteome.6b00414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang T, Holt MV, Young NL. The histone H4 proteoform dynamics in response to SUV4-20 inhibition reveals single molecule mechanisms of inhibitor resistance. Epigenetics & Chromatin. 2018;11(1):29. doi: 10.1186/s13072-018-0198-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Schmidt BH, Osheroff N, Berger JM. Structure of a topoisomerase II—DNA—nucleotide complex reveals a new control mechanism for ATPase activity. Nature Structural & Molecular Biology. 2012;19(11):1147–1154. doi: 10.1038/nsmb.2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Turner BM. Reading signals on the nucleosome with a new nomenclature for modified histones. Nature Structural & Molecular Biology. 2005;12(2):110–112. doi: 10.1038/nsmb0205-110 [DOI] [PubMed] [Google Scholar]
- [24].Schlissel MS, Brown DD. The transcriptional regulation of Xenopus 5S RNA genes in chromatin: The roles of active stable transcription complexes and histone H1. Cell. 1984;37(3):903–913. doi: 10.1016/0092-8674(84)90425-2 [DOI] [PubMed] [Google Scholar]
- [25].Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Müller WG, McNally JG, Bazett-Jones DP, Nussenzweig A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. Journal of Cell Biology. 2006;172(6):823–834. doi: 10.1083/jcb.200510015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hergeth SP, Schneider R. The H1 linker histones: multifunctional proteins beyond the nucleosomal core particle. EMBO reports. 2015;16(11):1439–1453. doi: 10.15252/embr.201540749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Terme J-M, Millán-Ariño L, Mayor R, Luque N, Izquierdo-Bouldstridge A, Bustillos A, Sampaio C, Canes J, Font I, Sima N, et al. Dynamics and dispensability of variant-specific histone H1 Lys-26/Ser-27 and Thr-165 post-translational modifications. FEBS Letters. 2014;588(14):2353–2362. doi: 10.1016/j.febslet.2014.05.035 [DOI] [PubMed] [Google Scholar]
- [28].Harshman SW, Young NL, Parthun MR, Freitas MA. H1 histones: current perspectives and challenges. Nucleic Acids Research. 2013;41(21):9593–9609. doi: 10.1093/nar/gkt700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Happel N, Doenecke D. Histone H1 and its isoforms: Contribution to chromatin structure and function. Gene. 2009;431(1–2):1–12. doi: 10.1016/j.gene.2008.11.003 [DOI] [PubMed] [Google Scholar]
- [30].McBryant SJ, Lu X, Hansen JC. Multifunctionality of the linker histones: an emerging role for protein-protein interactions. Cell Research. 2010;20(5):519–528. doi: 10.1038/cr.2010.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lu X, Wontakal SN, Kavi H, Kim BJ, Guzzardo PM, Emelyanov AV, Xu N, Hannon GJ, Zavadil J, Fyodorov DV, et al. DrosophilaH1 Regulates the Genetic Activity of Heterochromatin by Recruitment of Su(var)3-9. Science. 2013;340(6128):78–81. doi: 10.1126/science.1234654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hergeth SP, Dundr M, Tropberger P, Zee BM, Garcia BA, Daujat S, Schneider R. Isoform-specific phosphorylation of human linker histone H1.4 in mitosis by the kinase Aurora B. Journal of Cell Science. 2011;124(10):1623–1628. doi: 10.1242/jcs.084947 [DOI] [PubMed] [Google Scholar]
- [33].Chen Y, Hoover ME, Dang X, Shomo AA, Guan X, Marshall AG, Freitas MA, Young NL. Quantitative Mass Spectrometry Reveals that Intact Histone H1 Phosphorylations are Variant Specific and Exhibit Single Molecule Hierarchical Dependence. Molecular & Cellular Proteomics. 2016;15(3):818–833. doi: 10.1074/mcp.M114.046441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Vicent GP, Nacht AS, Font-Mateu J, Castellano G, Gaveglia L, Ballare C, Beato M. Four enzymes cooperate to displace histone H1 during the first minute of hormonal gene activation. Genes & Development. 2011;25(8):845–862. doi: 10.1101/gad.621811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tweedie-Cullen RY, Reck JM, Mansuy IM. Comprehensive Mapping of Post-Translational Modifications on Synaptic, Nuclear, and Histone Proteins in the Adult Mouse Brain. Journal of Proteome Research. 2009;8(11):4966–4982. doi: 10.1021/pr9003739 [DOI] [PubMed] [Google Scholar]
- [36].Lu A, Zougman A, Pudełko M, Bȩbenek M, Ziółkowski P, Mann M, Wiśniewski JR. Mapping of Lysine Monomethylation of Linker Histones in Human Breast and Its Cancer. Journal of Proteome Research. 2009;8(9):4207–4215. doi: 10.1021/pr9000652 [DOI] [PubMed] [Google Scholar]
- [37].Weiss T, Hergeth S, Zeissler U, Izzo A, Tropberger P, Zee BM, Dundr M, Garcia BA, Daujat S, Schneider R. Histone H1 variant-specific lysine methylation by G9a/KMT1C and Glp1/KMT1D. Epigenetics & Chromatin. 2010;3(1):7. doi: 10.1186/1756-8935-3-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kamieniarz K, Izzo A, Dundr M, Tropberger P, Ozretic L, Kirfel J, Scheer E, Tropel P, Wisniewski JR, Tora L, et al. A dual role of linker histone H1.4 Lys 34 acetylation in transcriptional activation. Genes & Development. 2012;26(8):797–802. doi: 10.1101/gad.182014.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 Interacts with Histone H1 and Promotes Formation of Facultative Heterochromatin. Molecular Cell. 2004;16(1):93–105. doi: 10.1016/j.molcel.2004.08.031 [DOI] [PubMed] [Google Scholar]
- [40].Andrés M, García-Gomis D, Ponte I, Suau P, Roque A. Histone H1 Post-Translational Modifications: Update and Future Perspectives. International Journal of Molecular Sciences. 2020;21(16):5941. doi: 10.3390/ijms21165941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wong TK, Marushige K. Modification of histone binding in calf thymus chromatin and in the chromatin-protamine complex by acetic anhydride. Biochemistry. 1976;15(10):2041–2046. doi: 10.1021/bi00655a003 [DOI] [PubMed] [Google Scholar]
- [42].Zheng Y, John S, Pesavento JJ, Schultz-Norton JR, Schiltz RL, Baek S, Nardulli AM, Hager GL, Kelleher NL, Mizzen CA. Histone H1 phosphorylation is associated with transcription by RNA polymerases I and II. Journal of Cell Biology. 2010;189(3):407–415. doi: 10.1083/jcb.201001148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Talasz H, Sarg B, Lindner HH. Site-specifically phosphorylated forms of H1.5 and H1.2 localized at distinct regions of the nucleus are related to different processes during the cell cycle. Chromosoma. 2009;118(6):693–709. doi: 10.1007/s00412-009-0228-2 [DOI] [PubMed] [Google Scholar]
- [44].Alexandrow MG, Hamlin JL. Chromatin decondensation in S-phase involves recruitment of Cdk2 by Cdc45 and histone H1 phosphorylation. Journal of Cell Biology. 2005;168(6):875–886. doi: 10.1083/jcb.200409055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Izzo A, Schneider R. The role of linker histone H1 modifications in the regulation of gene expression and chromatin dynamics. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 2016;1859(3):486–495. doi: 10.1016/j.bbagrm.2015.09.003 [DOI] [PubMed] [Google Scholar]
- [46].Trojer P, Zhang J, Yonezawa M, Schmidt A, Zheng H, Jenuwein T, Reinberg D. Dynamic Histone H1 Isotype 4 Methylation and Demethylation by Histone Lysine Methyltransferase G9a/KMT1C and the Jumonji Domain-containing JMJD2/KDM4 Proteins. Journal of Biological Chemistry. 2009;284(13):8395–8405. doi: 10.1074/jbc.M807818200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Walport LJ, Hopkinson RJ, Chowdhury R, Zhang Y, Bonnici J, Schiller R, Kawamura A, Schofield CJ. Mechanistic and structural studies of KDM-catalysed demethylation of histone 1 isotype 4 at lysine 26. FEBS Letters. 2018;592(19):3264–3273. doi: 10.1002/1873-3468.13231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Daujat S, Zeissler U, Waldmann T, Happel N, Schneider R. HP1 Binds Specifically to Lys26-methylated Histone H1.4, whereas Simultaneous Ser27 Phosphorylation Blocks HP1 Binding. Journal of Biological Chemistry. 2005;280(45):38090–38095. doi: 10.1074/jbc.C500229200 [DOI] [PubMed] [Google Scholar]
- [49].Chu F, Nusinow DA, Chalkley RJ, Plath K, Panning B, Burlingame AL. Mapping Post-translational Modifications of the Histone Variant MacroH2A1 Using Tandem Mass Spectrometry. Molecular & Cellular Proteomics. 2006;5(1):194–203. doi: 10.1074/mcp.M500285-MCP200 [DOI] [PubMed] [Google Scholar]
- [50].Allfrey VG, Faulkner R, Mirsky AE. ACETYLATION AND METHYLATION OF HISTONES AND THEIR POSSIBLE ROLE IN THE REGULATION OF RNA SYNTHESIS. Proceedings of the National Academy of Sciences. 1964;51(5):786–794. doi: 10.1073/pnas.51.5.786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ishibashi T, Dryhurst D, Rose KL, Shabanowitz J, Hunt DF, Ausió J. Acetylation of Vertebrate H2A.Z and Its Effect on the Structure of the Nucleosome. Biochemistry. 2009;48(22):5007–5017. doi: 10.1021/bi900196c [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Giaimo BD, Ferrante F, Herchenröther A, Hake SB, Borggrefe T. The histone variant H2A.Z in gene regulation. Epigenetics & Chromatin. 2019;12(1):37. doi: 10.1186/s13072-019-0274-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bonisch C, Hake SB. Histone H2A variants in nucleosomes and chromatin: more or less stable? Nucleic Acids Research. 2012;40(21):10719–10741. doi: 10.1093/nar/gks865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].West MHP, Bonner WM. Histone 2A, a heteromorphous family of eight protein species. Biochemistry. 1980;19(14):3238–3245. doi: 10.1021/bi00555a022 [DOI] [PubMed] [Google Scholar]
- [55].Hatimy AA, Browne MJG, Flaus A, Sweet SMM. Histone H2AX Y142 phosphorylation is a low abundance modification. International Journal of Mass Spectrometry. 2015;391:139–145. doi: 10.1016/j.ijms.2015.07.028 [DOI] [Google Scholar]
- [56].van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends in Cell Biology. 2009;19(5):207–217. doi: 10.1016/j.tcb.2009.03.001 [DOI] [PubMed] [Google Scholar]
- [57].Bao Y Chromatin response to DNA double-strand break damage. Epigenomics. 2011;3(3):307–321. doi: 10.2217/epi.11.14 [DOI] [PubMed] [Google Scholar]
- [58].Luger K, Suto RK, Clarkson MJ, Tremethick DJ. [No title found]. Nature Structural Biology. 2000;7(12):1121–1124. doi: 10.1038/81971 [DOI] [PubMed] [Google Scholar]
- [59].Mylonas C, Lee C, Auld AL, Cisse II, Boyer LA. A dual role for H2A.Z.1 in modulating the dynamics of RNA polymerase II initiation and elongation. Nature Structural & Molecular Biology. 2021;28(5):435–442. doi: 10.1038/s41594-021-00589-3 [DOI] [PubMed] [Google Scholar]
- [60].Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431(7010):873–878. doi: 10.1038/nature02985 [DOI] [PubMed] [Google Scholar]
- [61].Kumar SV, Wigge PA. H2A.Z-Containing Nucleosomes Mediate the Thermosensory Response in Arabidopsis. Cell. 2010;140(1):136–147. doi: 10.1016/j.cell.2009.11.006 [DOI] [PubMed] [Google Scholar]
- [62].Pehrson JR, Fried VA. MacroH2A, a Core Histone Containing a Large Nonhistone Region. Science. 1992;257(5075):1398–1400. doi: 10.1126/science.1529340 [DOI] [PubMed] [Google Scholar]
- [63].Pehrson JR, Costanzi C, Dharia C. Developmental and tissue expression patterns of histone macroH2A1 subtypes. Journal of Cellular Biochemistry. 1997;65(1):107–113. doi: [DOI] [PubMed] [Google Scholar]
- [64].Dell’Orso S, Wang AH, Shih H-Y, Saso K, Berghella L, Gutierrez-Cruz G, Ladurner AG, O’Shea JJ, Sartorelli V, Zare H. The Histone Variant MacroH2A1.2 Is Necessary for the Activation of Muscle Enhancers and Recruitment of the Transcription Factor Pbx1. Cell Reports. 2016;14(5):1156–1168. doi: 10.1016/j.celrep.2015.12.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. The Journal of Biological Chemistry. 2001;276(45):42462–42467. doi: 10.1074/jbc.C100466200 [DOI] [PubMed] [Google Scholar]
- [66].Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123(7):1213–1226. doi: 10.1016/j.cell.2005.09.038 [DOI] [PubMed] [Google Scholar]
- [67].Kleiner RE, Verma P, Molloy KR, Chait BT, Kapoor TM. Chemical proteomics reveals a γH2AX-53BP1 interaction in the DNA damage response. Nature Chemical Biology. 2015;11(10):807–814. doi: 10.1038/nchembio.1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Pinto DMS, Flaus A. Structure and Function of Histone H2AX. In: Nasheuer H-P, editor. Genome Stability and Human Diseases. Vol. 50. Dordrecht: Springer Netherlands; 2010. p. 55–78. (Subcellular Biochemistry). http://link.springer.com/10.1007/978-90-481-3471-7_4. doi: 10.1007/978-90-481-3471-7_4 [DOI] [Google Scholar]
- [69].Xiao A, Li H, Shechter D, Ahn SH, Fabrizio LA, Erdjument-Bromage H, Ishibe-Murakami S, Wang B, Tempst P, Hofmann K, et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature. 2009;457(7225):57–62. doi: 10.1038/nature07668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009;458(7238):591–596. doi: 10.1038/nature07849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Vardabasso C, Gaspar-Maia A, Hasson D, Pünzeler S, Valle-Garcia D, Straub T, Keilhauer EC, Strub T, Dong J, Panda T, et al. Histone Variant H2A.Z.2 Mediates Proliferation and Drug Sensitivity of Malignant Melanoma. Molecular Cell. 2015;59(1):75–88. doi: 10.1016/j.molcel.2015.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].The Histone Variant H2A.Z.2 Regulates Proliferation in Melanoma. Cancer Discovery. 2015;5(8):795.1–795. doi: 10.1158/2159-8290.CD-RW2015-112 [DOI] [Google Scholar]
- [73].Tsai C-H, Chen Y-J, Yu C-J, Tzeng S-R, Wu I-C, Kuo W-H, Lin M-C, Chan N-L, Wu K-J, Teng S-C. SMYD3-Mediated H2A.Z.1 Methylation Promotes Cell Cycle and Cancer Proliferation. Cancer Research. 2016;76(20):6043–6053. doi: 10.1158/0008-5472.CAN-16-0500 [DOI] [PubMed] [Google Scholar]
- [74].Tropberger P, Pott S, Keller C, Kamieniarz-Gdula K, Caron M, Richter F, Li G, Mittler G, Liu ET, Bühler M, et al. Regulation of Transcription through Acetylation of H3K122 on the Lateral Surface of the Histone Octamer. Cell. 2013;152(4):859–872. doi: 10.1016/j.cell.2013.01.032 [DOI] [PubMed] [Google Scholar]
- [75].Corujo D, Buschbeck M. Post-Translational Modifications of H2A Histone Variants and Their Role in Cancer. Cancers. 2018;10(3):59. doi: 10.3390/cancers10030059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kimura A, Horikoshi M. Tip60 acetylates six lysines of a specific class in core histonesin vitro: Lysine site specificity of Tip60. Genes to Cells. 1998;3(12):789–800. doi: 10.1046/j.1365-2443.1998.00229.x [DOI] [PubMed] [Google Scholar]
- [77].Binda O, Sevilla A, LeRoy G, Lemischka IR, Garcia BA, Richard S. SETD6 monomethylates H2AZ on lysine 7 and is required for the maintenance of embryonic stem cell self-renewal. Epigenetics. 2013;8(2):177–183. doi: 10.4161/epi.23416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Brown JAL, Eykelenboom JK, Lowndes NF. Co-mutation of histone H2AX S139A with Y142A rescues Y142A-induced ionising radiation sensitivity. FEBS Open Bio. 2012;2(1):313–317. doi: 10.1016/j.fob.2012.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Hernandez-Munoz I, Lund AH, van der Stoop P, Boutsma E, Muijrers I, Verhoeven E, Nusinow DA, Panning B, Marahrens Y, van Lohuizen M. Stable X chromosome inactivation involves the PRC1 Polycomb complex and requires histone MACROH2A1 and the CULLIN3/SPOP ubiquitin E3 ligase. Proceedings of the National Academy of Sciences. 2005;102(21):7635–7640. doi: 10.1073/pnas.0408918102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ikura T, Tashiro S, Kakino A, Shima H, Jacob N, Amunugama R, Yoder K, Izumi S, Kuraoka I, Tanaka K, et al. DNA Damage-Dependent Acetylation and Ubiquitination of H2AX Enhances Chromatin Dynamics. Molecular and Cellular Biology. 2007;27(20):7028–7040. doi: 10.1128/MCB.00579-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Monteiro FL, Baptista T, Amado F, Vitorino R, Jerónimo C, Helguero LA. Expression and functionality of histone H2A variants in cancer. Oncotarget. 2014;5(11):3428–3443. doi: 10.18632/oncotarget.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Khadka J, Pesok A, Grafi G. Plant Histone HTB (H2B) Variants in Regulating Chromatin Structure and Function. Plants. 2020;9(11):1435. doi: 10.3390/plants9111435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Mariño-Ramírez L, Kann MG, Shoemaker BA, Landsman D. Histone structure and nucleosome stability. Expert Review of Proteomics. 2005;2(5):719–729. doi: 10.1586/14789450.2.5.719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Jiang D, Borg M, Lorković ZJ, Montgomery SA, Osakabe A, Yelagandula R, Axelsson E, Berger F. The evolution and functional divergence of the histone H2B family in plants Malik HS, editor. PLOS Genetics. 2020;16(7):e1008964. doi: 10.1371/journal.pgen.1008964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Boulard M, Gautier T, Mbele GO, Gerson V, Hamiche A, Angelov D, Bouvet P, Dimitrov S. The NH2 Tail of the Novel Histone Variant H2BFWT Exhibits Properties Distinct from Conventional H2B with Respect to the Assembly of Mitotic Chromosomes. Molecular and Cellular Biology. 2006;26(4):1518–1526. doi: 10.1128/MCB.26.4.1518-1526.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Li A, Maffey AH, Abbott WD, Conde e Silva N, Prunell A, Siino J, Churikov D, Zalensky AO, Ausió J. Characterization of Nucleosomes Consisting of the Human Testis/Sperm-Specific Histone H2B Variant (hTSH2B). Biochemistry. 2005;44(7):2529–2535. doi: 10.1021/bi048061n [DOI] [PubMed] [Google Scholar]
- [87].Zalensky AO, Siino JS, Gineitis AA, Zalenskaya IA, Tomilin NV, Yau P, Bradbury EM. Human Testis/Sperm-specific Histone H2B (hTSH2B). Journal of Biological Chemistry. 2002;277(45):43474–43480. doi: 10.1074/jbc.M206065200 [DOI] [PubMed] [Google Scholar]
- [88].DeVine T, Sears RC, Dai M-S. The ubiquitin-specific protease USP36 is a conserved histone H2B deubiquitinase. Biochemical and Biophysical Research Communications. 2018;495(3):2363–2368. doi: 10.1016/j.bbrc.2017.12.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Kwon M, Park K, Hyun K, Lee J-H, Zhou L, Cho Y-W, Ge K, Skalnik DG, Muir TW, Kim J. H2B ubiquitylation enhances H3K4 methylation activities of human KMT2 family complexes. Nucleic Acids Research. 2020;48(10):5442–5456. doi: 10.1093/nar/gkaa317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Chitsazian F, Sadeghi M, Elahi E. Confident gene activity prediction based on single histone modification H2BK5ac in human cell lines. BMC Bioinformatics. 2017;18(1):67. doi: 10.1186/s12859-016-1418-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Zhao Y, Garcia BA. Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harbor Perspectives in Biology. 2015;7(9):a025064. doi: 10.1101/cshperspect.a025064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Cheung WL, Ajiro K, Samejima K, Kloc M, Cheung P, Mizzen CA, Beeser A, Etkin LD, Chernoff J, Earnshaw WC, et al. Apoptotic Phosphorylation of Histone H2B Is Mediated by Mammalian Sterile Twenty Kinase. Cell. 2003;113(4):507–517. doi: 10.1016/S0092-8674(03)00355-6 [DOI] [PubMed] [Google Scholar]
- [93].Bungard D, Fuerth BJ, Zeng P-Y, Faubert B, Maas NL, Viollet B, Carling D, Thompson CB, Jones RG, Berger SL. Signaling Kinase AMPK Activates Stress-Promoted Transcription via Histone H2B Phosphorylation. Science. 2010;329(5996):1201–1205. doi: 10.1126/science.1191241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Magic Z, Supic G, Brankovic-Magic M. Towards targeted epigenetic therapy of cancer. Journal of B.U.ON.: official journal of the Balkan Union of Oncology. 2009;14 Suppl 1:S79–88. [PubMed] [Google Scholar]
- [95].Bonenfant D, Coulot M, Towbin H, Schindler P, van Oostrum J. Characterization of Histone H2A and H2B Variants and Their Post-translational Modifications by Mass Spectrometry. Molecular & Cellular Proteomics. 2006;5(3):541–552. doi: 10.1074/mcp.M500288-MCP200 [DOI] [PubMed] [Google Scholar]
- [96].Wojcik F, Dann GP, Beh LY, Debelouchina GT, Hofmann R, Muir TW. Functional crosstalk between histone H2B ubiquitylation and H2A modifications and variants. Nature Communications. 2018;9(1):1394. doi: 10.1038/s41467-018-03895-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Wright DE. Histone ubiquitylation and chromatin dynamics. Frontiers in Bioscience. 2012;17(1):1051. doi: 10.2741/3973 [DOI] [PubMed] [Google Scholar]
- [98].Hake SB, Allis CD. Histone H3 variants and their potential role in indexing mammalian genomes: The “H3 barcode hypothesis.” Proceedings of the National Academy of Sciences. 2006;103(17):6428–6435. doi: 10.1073/pnas.0600803103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Sullivan KF, Hechenberger M, Masri K. Human CENP-A contains a histone H3 related histone fold domain that is required for targeting to the centromere. Journal of Cell Biology. 1994;127(3):581–592. doi: 10.1083/jcb.127.3.581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Hake SB, Garcia BA, Duncan EM, Kauer M, Dellaire G, Shabanowitz J, Bazett-Jones DP, Allis CD, Hunt DF. Expression Patterns and Post-translational Modifications Associated with Mammalian Histone H3 Variants. Journal of Biological Chemistry. 2006;281(1):559–568. doi: 10.1074/jbc.M509266200 [DOI] [PubMed] [Google Scholar]
- [101].Wirbelauer C, Bell O, Schübeler D. Variant histone H3.3 is deposited at sites of nucleosomal displacement throughout transcribed genes while active histone modifications show a promoter-proximal bias. Genes & Development. 2005;19(15):1761–1766. doi: 10.1101/gad.347705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Jacob Y, Bergamin E, Donoghue MTA, Mongeon V, LeBlanc C, Voigt P, Underwood CJ, Brunzelle JS, Michaels SD, Reinberg D, et al. Selective methylation of histone H3 variant H3.1 regulates heterochromatin replication. Science (New York, N.Y.). 2014;343(6176):1249–1253. doi: 10.1126/science.1248357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Jiang D, Berger F. DNA replication-coupled histone modification maintains Polycomb gene silencing in plants. Science (New York, N.Y.). 2017;357(6356):1146–1149. doi: 10.1126/science.aan4965 [DOI] [PubMed] [Google Scholar]
- [104].Loyola A, Bonaldi T, Roche D, Imhof A, Almouzni G. PTMs on H3 Variants before Chromatin Assembly Potentiate Their Final Epigenetic State. Molecular Cell. 2006;24(2):309–316. doi: 10.1016/j.molcel.2006.08.019 [DOI] [PubMed] [Google Scholar]
- [105].Kutateladze TG. SnapShot: Histone Readers. Cell. 2011;146(5):842–842.e1. doi: 10.1016/j.cell.2011.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Wen H, Li Y, Xi Y, Jiang S, Stratton S, Peng D, Tanaka K, Ren Y, Xia Z, Wu J, et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature. 2014;508(7495):263–268. doi: 10.1038/nature13045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Liang G, Lin JCY, Wei V, Yoo C, Cheng JC, Nguyen CT, Weisenberger DJ, Egger G, Takai D, Gonzales FA, et al. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(19):7357–7362. doi: 10.1073/pnas.0401866101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Farooq Z, Banday S, Pandita TK, Altaf M. The many faces of histone H3K79 methylation. Mutation Research/Reviews in Mutation Research. 2016;768:46–52. doi: 10.1016/j.mrrev.2016.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Wood K, Tellier M, Murphy S. DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules. 2018;8(1):11. doi: 10.3390/biom8010011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nature Reviews Molecular Cell Biology. 2012;13(2):115–126. doi: 10.1038/nrm3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Sawicka A, Seiser C. Histone H3 phosphorylation – A versatile chromatin modification for different occasions. Biochimie. 2012;94(11):2193–2201. doi: 10.1016/j.biochi.2012.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Hake SB, Garcia BA, Kauer M, Baker SP, Shabanowitz J, Hunt DF, Allis CD. Serine 31 phosphorylation of histone variant H3.3 is specific to regions bordering centromeres in metaphase chromosomes. Proceedings of the National Academy of Sciences. 2005;102(18):6344–6349. doi: 10.1073/pnas.0502413102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].van Nuland R, Gozani O. Histone H4 Lysine 20 (H4K20) Methylation, Expanding the Signaling Potential of the Proteome One Methyl Moiety at a Time. Molecular & Cellular Proteomics. 2016;15(3):755–764. doi: 10.1074/mcp.R115.054742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Holt MV, Wang T, Young NL. High-Throughput Quantitative Top-Down Proteomics: Histone H4. Journal of the American Society for Mass Spectrometry. 2019;30(12):2548–2560. doi: 10.1007/s13361-019-02350-z [DOI] [PubMed] [Google Scholar]
- [115].Holt MV, Wang T, Young NL. One-Pot Quantitative Top- and Middle-Down Analysis of GluC-Digested Histone H4. Journal of the American Society for Mass Spectrometry. 2019;30(12):2514–2525. doi: 10.1007/s13361-019-02219-1 [DOI] [PubMed] [Google Scholar]
- [116].Oda H, Okamoto I, Murphy N, Chu J, Price SM, Shen MM, Torres-Padilla ME, Heard E, Reinberg D. Monomethylation of Histone H4-Lysine 20 Is Involved in Chromosome Structure and Stability and Is Essential for Mouse Development. Molecular and Cellular Biology. 2009;29(8):2278–2295. doi: 10.1128/MCB.01768-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Goldberg AD, Banaszynski LA, Noh K-M, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140(5):678–691. doi: 10.1016/j.cell.2010.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Duarte LF, Young ARJ, Wang Z, Wu H-A, Panda T, Kou Y, Kapoor A, Hasson D, Mills NR, Ma’ayan A, et al. Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nature Communications. 2014;5(1):5210. doi: 10.1038/ncomms6210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Lee JY, Greene EC. Assembly of recombinant nucleosomes on nanofabricated DNA curtains for single-molecule imaging. Methods in Molecular Biology (Clifton, N.J.). 2011;778:243–258. doi: 10.1007/978-1-61779-261-8_16 [DOI] [PubMed] [Google Scholar]
- [120].Morgan MAJ, Popova IK, Vaidya A, Burg JM, Marunde MR, Rendleman EJ, Dumar ZJ, Watson R, Meiners MJ, Howard SA, et al. A trivalent nucleosome interaction by PHIP/BRWD2 is disrupted in neurodevelopmental disorders and cancer. Genes & Development. 2021;35(23–24):1642–1656. doi: 10.1101/gad.348766.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Schachner LF, Jooß K, Morgan MA, Piunti A, Meiners MJ, Kafader JO, Lee AS, Iwanaszko M, Cheek MA, Burg JM, et al. Decoding the protein composition of whole nucleosomes with Nuc-MS. Nature Methods. 2021;18(3):303–308. doi: 10.1038/s41592-020-01052-9 [DOI] [PMC free article] [PubMed] [Google Scholar]