

Abstract
Background
This study sought to identify candidate biomarkers associated with gastric cancer (GC) prognosis based on an integrated bioinformatics analysis.
Methods
First, the GSE54129 and GSE79973 data sets were downloaded from the Gene Expression Omnibus (GEO) database. The differentially expressed genes (DEGs) identified between the 2 data sets were screened using the limma software package in R, and the intersection DEGs were obtained by a Venn analysis. Subsequently, gene clustering and a functional analysis were performed to explore the roles of the DEGs. The protein-protein interaction (PPI) network of the genes in clusters was constructed using the Search Tool for the Retrieval of Interacting Genes/Proteins. A survival analysis evaluated the associations between the candidate genes and the overall survival of GC patients. A drug-gene interaction analysis and an external data set analysis were conducted using The Cancer Genome Atlas-Stomach Adenocarcinoma (TCGA-STAD) data set to validate the prognostic genes.
Results
We extracted 421 intersection DEGs from the 2 GEO data sets. There were 5 gene clusters, and the functional analysis revealed that they were mainly associated with the extracellular matrix-receptor interaction pathway. The PPI interaction analysis identified the top 36 hub genes. The survival analysis revealed that 7 upregulated genes [i.e., platelet-derived growth factor receptor beta (PDGFRB), angiopoietin 2 (ANGPT2), vascular endothelial growth factor C (VEGFC), collagen type IV alpha 2 chain (COL4A2), collagen type IV alpha 1 chain (COL4A1), thrombospondin 1 (THBS1), and fibronectin 1 (FN1)] were associated with the survival prognosis of GC patients. The 20 drug-gene interaction pairs among the 4 genes and 18 drugs were obtained. Finally, TCGA-STAD data set was used to validate the expression levels of COL4A1, PDGFRB, and FN1.
Conclusions
We found that 7 upregulated genes (i.e., PDGFRB, ANGPT2, VEGFC, COL4A2, COL4A1, THBS1, and FN1) were promising markers of prognosis in GC patients.
Keywords: Gastric cancer (GC), differentially expressed gene, protein-protein interaction analysis, survival analysis
Introduction
Gastric cancer (GC) is the 4th most common cancer worldwide, and is characterized by increasing incidence and mortality rates, especially in China (1). Standard treatments, such as surgical resection, chemotherapy, and radiotherapy, have greatly increased the survival outcomes of GC patients. However, the global 5-year overall survival (OS) rate remains <15%, which is mainly due to the late diagnosis and lack of early detection of GC (2,3). A high recurrence rate and tendency to metastasize also lead to poor clinical outcomes (4,5).
Previous studies based on multivariate regression analyses have identified some prognostic biomarkers in GC. For example, Jin et al. (6) found that increased spondin-2 in GC tissues was significantly related to lymph node metastasis and advanced tumor, node, metastasis stages, and that the high expression of spondin-2 leads to a poor prognosis in GC patients. This regression analytic strategy may enable the identification of important prognostic targets for cancer management. Du et al. (7) recently identified adenoma polyposis coli (APC) as a new prognostic factor for patients with T4 GC based on APC expression and a methylation profile analysis.
Over the decades, gene chips have been proven to be a reliable technology. They can rapidly screen for differentially expressed genes (DEGs) and generate large amounts of genomic information for public databases. Some prognostic biomarkers have been identified and applied in clinical treatment (8,9); different from the above studies, our study not only identified novel biomarkers related to GC, but also we have predicted the new drugs for patients with GC based on the GSE54129 and GSE79973 data sets.
We downloaded the GSE54129 and GSE79973 data sets from the Gene Expression Omnibus (GEO) database. The DEGs between the 2 data sets were distinguished using R’s limma package and Venn diagram software. We further explored the function of these DEGs, including the Gene Ontology function and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, using the Database for Annotation, Visualization, and Integrated Discovery (DAVID). Next, we constructed a protein-protein interaction (PPI) network to analyze the DEGs further. Kaplan-Meier (KM) curves revealed an association between the DEGs and OS in GC patients. Further, a gene-drug interaction analysis was conducted to explore the association between drugs and the DEGs. Finally, we selected a data set from The Cancer Genome Atlas (TCGA) to validate these key prognostic genes. Our results will promote the discovery of novel prognostic markers for GC patients.
This study sought to screen candidate biomarkers associated with GC prognosis based on an integrated bioinformatics analysis. We present the following article in accordance with the REMARK reporting checklist (available at https://jgo.amegroups.com/article/view/10.21037/jgo-22-651/rc).
Methods
Data source and pre-processing
The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013). We obtained the GSE54129 data set (comprising 111 GC and 21 normal control samples) and GSE79973 data set (comprising 10 GC and 10 normal control samples) from the GEO (http://www.ncbi.nlm.nih.gov/geo/) database using the keywords “gastric cancer, Homo sapiens and tissue” based on the GPL570 (HG-U133_Plus_2) Affymetrix Human Genome U133 Plus 2.0 Array. TCGA-Stomach Adenocarcinoma (TCGA-STAD) data set, comprising 415 tumor samples and 35 adjacent non-tumor samples, was obtained from TCGA database (https://cancergenome.nih.gov/). There was relevant clinical information in 408 samples. Next, the R affy package (version 1.58.1; http://bioconductor.org/help/search/index.html?q=affy/) was used to normalize the data by, for example, format conversion or the correction of missing data (10). The MicroArray Suite algorithm and the quantiles method were used for the data standardization. Subsequently, the probes were mapped onto the corresponding genes. The mean value of multiple probes was taken as the final expression value of the gene when the probes were mapped to the same gene.
Identification of DEGs
We used the R limma package (version 3.10.3; http://www.bioconductor.org/packages/2.9/bioc/html/limma.html) to screen for DEGs between the tumor and control samples (11). The P value was calculated and adjusted using the Benjamini-Hochberg method (12). The thresholds of an adjusted P value <0.05 and a |log2 fold change (FC)| >1 were set as the screening criteria for DEG identification. Additionally, a Venn analysis was conducted to extract the intersection DEGs between the GSE54129 and GSE79973 data sets. These DEGs were regarded as the candidate targets and used in the following analysis.
Clustering and functional analyses
A clustering analysis identified the gene clusters with similar functions. The ConsensusClusterPlus algorithm (version 1.44.0; http://bioconductor.org/packages/release/bioc/html/ConsensusClusterPlus.html) was used to identify gene clusters based on the expression values of the intersection DEGs between the GSE54129 and GSE79973 data sets (13). The cumulative distribution function (CDF) was used to determine the optimal number of clusters (14). Subsequently, a KEGG pathway enrichment analysis of the genes in the extracted clusters was performed using the online analytic tool DAVID (15,16). Gene counts ≥2 and a P value <0.05 indicated a statistically significant difference.
PPI analysis
We evaluated the relationships between the protein products and genes in the KEGG enriched pathways based on the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (17). The PPI score was 0.4, and the species was Homo sapiens. Cytoscape software (version 3.2.0; https://cytoscape.org/) was used to construct the PPI network (18). Further, the topology relation of the PPI network was analyzed by Cytoscape, and the node scores were obtained. Gene nodes with a degree ≥5 were regarded as the hub genes in the PPI network. The KEGG analysis of these hub genes was carried out based on the DAVID (19).
Analysis of the candidate genes using KM curves
All the genes in the significant clusters were used for the survival analysis. First, we collected the gene expression matrix data and corresponding clinical information from TCGA-STAD data set. All the candidate genes were classified into either the high- or low-expression group using the R survival package (version 2.42-6; https://cran.r-project.org/web/packages/survival/index.html). Finally, the survival curve was constructed using the KM method. A correlation coefficient P value <0.05 was set as the significance threshold for the survival analysis of the prognosis-related genes. Additionally, the overlapping genes between the hub genes in the PPI network and the prognosis-associated genes were further extracted and served as the hub genes for the survival prognosis of GC patients.
Prediction of drug-gene interaction
Drug development has benefited from the Drug-Gene Interaction database (DGIdb), which is widely used to identify the drugs that target genes. The potential relationships between drugs and prognosis-related hub genes were predicted by the DGIdb using the following preset filter parameters: Food and Drug Administration (FDA) approved and antineoplastic. Next, the drug-gene interaction network was constructed using Cytoscape software.
Data validation
To further validate the candidate genes associated with GC prognosis, we performed a gene differential expression analysis for TCGA-STAD data set. The screening cutoffs for the DEGs were set as an adjusted P value <0.05 and a |log2FC| >1.0. Finally, a Venn analysis was conducted for the DEGs and the intersection DEGs based on the GEO data sets.
Results
Identification of DEGs
A total of 1,815 (830 upregulated and 985 downregulated) DEGs were screened between the GC and normal tissues in GSE54129. There were 704 (356 upregulated and 348 downregulated) DEGs in the GSE79973 data set. Additionally, the hierarchical clustering analyses revealed that the DEGs in the GSE54129 (see Figure 1A) and GSE79973 (see Figure 1B) data sets could significantly discriminate between the GC and normal samples. We further extracted 421 intersection DEGs between these 2 data sets by a Venn analysis (see Figure 1C).
Figure 1.

Gene differential expression analysis. (A) Heatmap of the DEGs in the GSE54129 data set. (B) Heatmap of the DEGs in the GSE79973 data set. (C) The Venn diagram shows the intersection DEGs between the GSE54129 and GSE79973 data sets. The orange bars indicated the GC samples, and the green bars indicated the controls. GC, gastric cancer; DEGs, differentially expressed genes.
Intersection DEG clustering and functional analyses
We carried out a clustering analysis of the intersection DEGs to identify the gene sets with a similar function. The Consensus Cluster Plus algorithm was used for the gene clustering analysis, and the CDF was calculated to determine the optimal cluster number. From the CDF curve and CDF delta area curve, we found that k=5 represented the most stable clustering outcome (Figure 2A,2B). Thus, these intersection DEGs were grouped into 5 clusters (see Figure 2C). Our results also showed that there were 178 genes in cluster 1, 63 in cluster 2, 174 in cluster 3, 5 in cluster 4, and 1 in cluster 5. Subsequently, we conducted functional analyses of the genes in the 5 clusters. We found that the genes in clusters 1 and 3 were all significantly enriched in 9 KEGG pathways (see Figure 2D). For example, the genes in cluster 1 mainly participated in the xenobiotics metabolism by cytochrome P450 and chemical carcinogenesis pathways. The genes in cluster 3 were primarily associated with the extracellular matrix (ECM)-receptor interaction and focal adhesion pathways. The genes in cluster 2 were strongly related to the gastric acid secretion and retinol metabolism pathways. The genes in clusters 4 and 5 were not enriched in any pathway.
Figure 2.

Gene clustering and functional analyses. (A) The consensus CDF curve; (B) the CDF delta area curve, which represents the area difference between the area of CDF under ki and horizontal axis and CDF under ki + 1 and horizontal axis; (C) the consensus cluster under k=5; (D) The KEGG pathway enrichment analysis of genes in clusters 1–3. k represents the number of gene clusters. CDF, cumulative distribution function, KEGG, Kyoto Encyclopedia of Genes and Genomes.
PPI network construction and functional analysis
The PPI network based on the intersection DEGs was constructed. It contained 58 nodes and 211 interaction pairs. Notably, there were 22 genes in cluster 1, 7 in cluster 2, and 29 in cluster 3 (see Figure 3A). Further, there were 36 hub genes with a degree ≥5 (see Table 1). Additionally, we performed a KEGG enrichment analysis for these hub genes. The results suggested that they were predominantly enriched in 18 KEGG pathways, such as the ECM-receptor interaction and focal adhesion pathways (see Figure 3B).
Figure 3.

PPI and functional analyses. (A) The PPI network. The circular nodes represent the upregulated genes, and the square nodes represent the downregulated genes. The blue nodes show the genes in cluster 1, the green nodes show the genes in cluster 2, and the pink nodes show the genes in cluster 3. The size of each node represents the degree value. (B) The KEGG pathway enrichment analysis of the hub genes in the PPI network. The hub genes were those with a degree ≥5. PPI, protein-protein interaction, KEGG, Kyoto Encyclopedia of Genes and Genomes.
Table 1. Hub genes with a degree ≥5 in the PPI network.
| Nodes | Regulation | Cluster | Degree |
|---|---|---|---|
| COL3A1 | Up | Cluster 3 | 18 |
| COL1A2 | Up | Cluster 3 | 17 |
| COL1A1 | Up | Cluster 3 | 17 |
| COL18A1 | Up | Cluster 3 | 17 |
| COL4A1 | Up | Cluster 3 | 15 |
| COL5A1 | Up | Cluster 3 | 15 |
| COL5A2 | Up | Cluster 3 | 14 |
| THBS1 | Up | Cluster 3 | 14 |
| FN1 | Up | Cluster 3 | 14 |
| ALDH1A1 | Down | Cluster 2 | 13 |
| COL4A2 | Up | Cluster 3 | 13 |
| COL6A3 | Up | Cluster 3 | 13 |
| COL11A1 | Up | Cluster 3 | 13 |
| COL10A1 | Up | Cluster 3 | 12 |
| PTGS2 | Up | Cluster 3 | 12 |
| AKR1C3 | Down | Cluster 1 | 11 |
| COL12A1 | Up | Cluster 3 | 11 |
| THBS2 | Up | Cluster 3 | 11 |
| IL8 | Up | Cluster 3 | 10 |
| ALDH3A1 | Down | Cluster 1 | 9 |
| UGT2B15 | Down | Cluster 1 | 9 |
| CYP2C9 | Down | Cluster 1 | 9 |
| COMP | Up | Cluster 3 | 9 |
| AKR1C1 | Down | Cluster 1 | 7 |
| CYP3A5 | Down | Cluster 1 | 7 |
| CYP2C19 | Down | Cluster 1 | 7 |
| VEGFC | Up | Cluster 3 | 7 |
| SPP1 | Up | Cluster 3 | 7 |
| ADH1A | Down | Cluster 1 | 6 |
| AKR1C2 | Down | Cluster 1 | 6 |
| ADH7 | Down | Cluster 2 | 6 |
| PDGFRB | Up | Cluster 3 | 6 |
| ANGPT2 | Up | Cluster 3 | 6 |
| DHRS9 | Down | Cluster 1 | 5 |
| CBR1 | Down | Cluster 1 | 5 |
| ATP4A | Down | Cluster 2 | 5 |
PPI, protein-protein interaction.
Analyses of candidate genes using KM curves
To further study the association between the hub genes and the OS of GC patients, survival analyses for all the genes in the 5 clusters were carried out. The results suggested that 66 genes were significantly correlated with the clinical outcomes of GC patients. Subsequently, the 7 overlapping genes between the prognosis-related genes and hub genes in the PPI network were extracted. They included angiopoietin 2 (ANGPT2) (see Figure 4A), collagen type IV alpha 1 chain (COL4A1) (see Figure 4B), collagen type IV alpha 2 chain (COL4A2) (see Figure 4C), fibronectin 1 (FN1) (see Figure 4D), platelet-derived growth factor receptor beta (PDGFRB) (see Figure 4E), thrombospondin 1 (THBS1) (see Figure 4F), and vascular endothelial growth factor C (VEGFC) (see Figure 4G). These genes were all upregulated, and their high expression levels were associated with a poor prognosis.
Figure 4.

The Kaplan-Meier survival curves for (A) ANGPT2; (B) COL4A1; (C) COL4A2; (D) FN1; (E) PDGFRB; (F) THBS1; and (G) VEGFC.
Prediction of drug-gene interactions
The correlations between the 7 prognosis-associated genes and drugs were predicted using the DGIdb database. There were 20 drug-gene interaction pairs, including 4 upregulated genes (i.e., PDGFRB, ANGPT2, VEGFC, and THBS1) and 18 drugs (see Figure 5).
Figure 5.

Drug-gene interactions based on the DGIdb database. The pink circles represent the upregulated genes in cluster 3, and the yellow squares represent the drugs.
Data validation
A differential expression analysis was conducted on TCGA-STAD data set to verify our GEO data set findings. In total, 1,178 DEGs were identified between the GC and normal samples, including 737 upregulated genes and 441 downregulated genes. Next, a Venn analysis was conducted with the intersection DEGs from the GEO data set and the DEGs from TCGA-STAD data set (see Figure 6). We identified 131 overlapping genes, including 3 prognosis-related genes (i.e., COL4A1, PDGFRB, and FN1).
Figure 6.
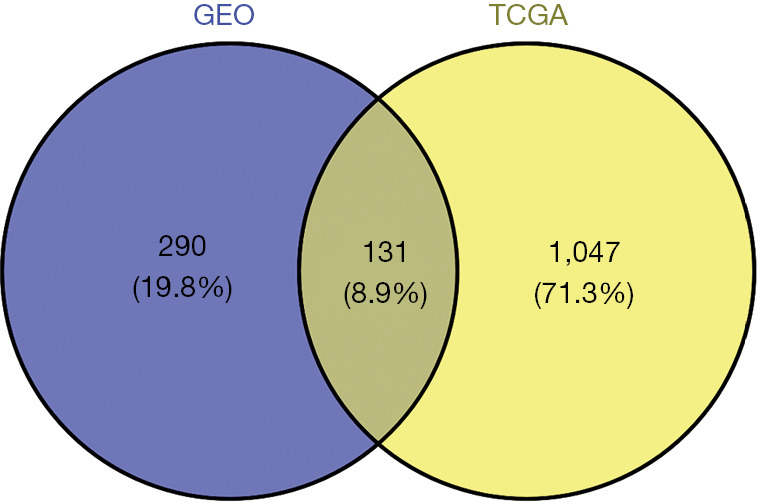
The Venn analysis. The overlapping genes were extracted between the intersection DEGs from the GEO data set and the DEGs from TCGA-STAD data set. DEGs, differentially expressed genes; GEO, Gene Expression Omnibus; TCGA-STAD, The Cancer Genome Atlas-stomach adenocarcinoma.
Discussion
In this study, the differential expression analysis identified 421 intersection DEGs between the GSE54129 and GSE79973 data sets. These were divided into 5 gene clusters, and the corresponding genes in these clusters mainly participated in the ECM-receptor interaction pathway. The survival analyses revealed that 7 upregulated genes (i.e., PDGFRB, ANGPT2, VEGFC, COL4A1, COL4A2, THBS1, and FN1) were strongly associated with the OS of GC patients, and these genes were also hub genes in the PPI network. There were close relationships between numerous drugs and 4 prognosis-related genes (i.e., PDGFRB, ANGPT2, VEGFC, and THBS1). Finally, the expression levels of COL4A1, PDGFRB, and FN1 were validated with TCGA data set.
Functional analyses have revealed that most DEGs are predominantly involved in the ECM-receptor interaction pathway (20,21). Notably, Liu et al. previously performed a graph-based clustering analysis and found that the ECM-receptor interaction pathway was correlated with the underlying molecular mechanisms of GC development (22). Several recent studies have implicated that many DEGs in GC tissues may regulate the progression of GC via this signaling pathway (23-25). However, the detailed mechanisms of the effects of this pathway on GC progression require clarification.
PDGFB is a member of the PDGF family and encodes a tyrosine kinase receptor. Our results indicated that this gene was upregulated in the GC samples, and its overexpression indicated an unfavorable prognosis. Further, the expression of this gene was verified by TCGA-STAD data set analysis. Early research reported that PDGFB is more highly expressed in GC tissues than normal tissues (26). Subsequently, a number of studies have suggested that PDGFB is involved in developing GC via different biological processes (27,28). Recently, Wang et al. pointed out that PDGFB is closely associated with neuropilin 1 (NRP1). Increased levels of NRP1 and PDGFRB are strongly related to the many malignant phenotypes in GC patients (29). Further, the risk of death is approximately 2-fold higher in GC patients with a higher expression of NRP1 and PDGFRB than others. This provides support for our finding that an enhanced PDGFRB level signals a poor prognosis for GC patients.
Notably, we also found that PDGFRB was closely correlated with numerous drugs, including imatinib, sunitinib, regorafenib, sorafenib, pazopanib, axitinib, nilotinib, lenvatinib, imatinib mesylate, and dasatinib. A previous study showed that combining fluorouracil, leucovorin, and imatinib mesylate targeting PDGFRB was safe and effective for GC patients (30). Additionally, Qian et al. suggested that 9 drug molecules (i.e., imatinib, sunitinib, regorafenib, sorafenib, pazopanib, axitinib, dasatinib, nilotinib, and lenvatinib) were PDGFR tyrosine kinase inhibitors. They discussed the underlying roles of these drugs in signal transduction pathways based on pharmacogenetics (31). Overall, PDGFRB is not a potential prognostic gene for GC, but it may be a promising therapeutic target for GC treatment.
Our analysis validated the suggestion that another prognostic gene, COL4A1, was upregulated in GC patients. Currently, there is overwhelming evidence that this gene is associated with the possible mechanisms of GC (1,32). Further, Li et al. conducted a bioinformatics analysis and found that COL4A1 has an important prognostic value in the survival of GC patients (1). Consistent with our results, Li et al. found that COL4A1 is overexpressed in GC tissues compared to normal tissues, and a higher expression level of COL4A1 is associated with poorer overall survival for GC patients (33). Similarly, upregulated FN1 is a prognostic marker for GC. Many research groups have suggested that FN1 is highly expressed in GC tissues, and high levels are related to a poor prognosis for GC patients (34,35).
According to our bioinformatics analyses, the other 4 upregulated genes (i.e., ANGPT2, THBS1, COL4A2, and VEGFC) are prognostic markers for GC patients. Xu et al. previously argued that THBS1 and ANGPT2 are strong predictors of the survival of GC patients (35). A multivariate analysis by Eto et al. suggested that the negative expression of THBS1 is an independent prognostic indicator (36). Furthermore, the current targets for gastric cancer mainly include TP53, EGFR, HER-2, VEGF, VEGFR, MET, FGFR2, mTOR, etc. The protein encoded by VEGFC is a member of the platelet-derived growth factor/vascular endothelial growth factor (PDGF/VEGF) family. The encoded protein promotes angiogenesis and endothelial cell growth, and can also affect the permeability of blood vessels. We inferred that VEGFC might be a novel target for GC. Also, numerous studies have revealed that VEGFC acts as a promising marker in determining the prognosis of GC patients (37). Dai et al. stated that there were higher messenger ribonucleic acid and protein levels of VEGFC in GC samples and linked the expression of this gene with GC lymph node metastasis (38). There is extensive evidence implicating COL4A2 in the development of GC; however, few reports have investigated the effect of this gene on GC prognosis.
This work had some limitations. First, an integrated bioinformatics analysis based on a larger sample size needs to be conducted to validate our results. After that, functional experiments on the main targets need to be carried out to improve the meaning of this study. Second, the detailed regulatory mechanisms of the significant signaling pathways also need to be deciphered. Finally, the in vivo and in vitro experimental validation need to be performed to validate the results of our study, which might be performed in the future.
In conclusion, our study suggests that 7 key genes (PDGFRB, ANGPT2, VEGFC, COL4A2, COL4A1, THBS1, and FN1) can be used to predict the survival outcomes of GC patients, and these promising prognostic markers of GC may contribute to improving risk management and clinical outcomes of GC patients.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013).
Reporting Checklist: The authors have completed the REMARK reporting checklist. Available at https://jgo.amegroups.com/article/view/10.21037/jgo-22-651/rc
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at https://jgo.amegroups.com/article/view/10.21037/jgo-22-651/coif). The authors have no conflicts of interest to declare.
(English Language Editor: L. Huleatt)
References
- 1.Li T, Gao X, Han L, et al. Identification of hub genes with prognostic values in gastric cancer by bioinformatics analysis. World J Surg Oncol 2018;16:114. 10.1186/s12957-018-1409-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chevallay M, Jung M, Morel P, Mönig S. Gastric cancer: management and multidisciplinary treatment. Rev Med Suisse 2018;14:2221-5. 10.53738/REVMED.2018.14.630.2221 [DOI] [PubMed] [Google Scholar]
- 3.Zhang JJ, Ze-Xuan-Zhu, Guang-Min-Xu, et al. Comprehensive Analysis of Differential Expression Profiles of Long Noncoding RNAs with Associated Co-expression and Competing Endogenous RNA Networks in the Hippocampus of Patients with Alzheimer's Disease. Curr Alzheimer Res 2021;18:884-99. 10.2174/1567205018666211202143449 [DOI] [PubMed] [Google Scholar]
- 4.Soeno T, Katoh H, Ishii S, et al. CD33+ Immature Myeloid Cells Critically Predict Recurrence in Advanced Gastric Cancer. J Surg Res 2020;245:552-63. 10.1016/j.jss.2019.07.095 [DOI] [PubMed] [Google Scholar]
- 5.Wang S, Chen X, Fu Y, et al. Relationship of ERCC5 genetic polymorphisms with metastasis and recurrence of gastric cancer. Rev Assoc Med Bras (1992) 2021;67:1538-43. 10.1590/1806-9282.20210209 [DOI] [PubMed] [Google Scholar]
- 6.Jin C, Lin JR, Ma L, et al. Elevated spondin-2 expression correlates with progression and prognosis in gastric cancer. Oncotarget 2017;8:10416-24. 10.18632/oncotarget.14423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du WB, Lin CH, Chen WB. High expression of APC is an unfavorable prognostic biomarker in T4 gastric cancer patients. World J Gastroenterol 2019;25:4452-67. 10.3748/wjg.v25.i31.4452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen S, Wei Y, Liu H, et al. Analysis of Collagen type X alpha 1 (COL10A1) expression and prognostic significance in gastric cancer based on bioinformatics. Bioengineered 2021;12:127-37. 10.1080/21655979.2020.1864912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pritzker KP. Predictive and prognostic cancer biomarkers revisited. Expert Rev Mol Diagn 2015;15:971-4. 10.1586/14737159.2015.1063421 [DOI] [PubMed] [Google Scholar]
- 10.Gautier L, Cope L, Bolstad BM, et al. affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004;20:307-15. 10.1093/bioinformatics/btg405 [DOI] [PubMed] [Google Scholar]
- 11.Colaprico A, Silva TC, Olsen C, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 2016;44:e71. 10.1093/nar/gkv1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glickman ME, Rao SR, Schultz MR. False discovery rate control is a recommended alternative to Bonferroni-type adjustments in health studies. J Clin Epidemiol 2014;67:850-7. 10.1016/j.jclinepi.2014.03.012 [DOI] [PubMed] [Google Scholar]
- 13.Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 2010;26:1572-3. 10.1093/bioinformatics/btq170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xue B, Oldfield CJ, Dunker AK, et al. CDF it all: consensus prediction of intrinsically disordered proteins based on various cumulative distribution functions. FEBS Lett 2009;583:1469-74. 10.1016/j.febslet.2009.03.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000;28:27-30. 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang da W , Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4:44-57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 17.Szklarczyk D, Franceschini A, Kuhn M, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 2011;39:D561-8. 10.1093/nar/gkq973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498-504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dennis G, Jr, Sherman BT, Hosack DA, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 2003;4:P3. 10.1186/gb-2003-4-5-p3 [DOI] [PubMed] [Google Scholar]
- 20.Zhu J, Luo C, Zhao J, et al. Expression of LOX Suggests Poor Prognosis in Gastric Cancer. Front Med (Lausanne) 2021;8:718986. 10.3389/fmed.2021.718986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu H, Wan H, Zhu M, et al. Discovery and Validation of an Epithelial-Mesenchymal Transition-Based Signature in Gastric Cancer by Genomics and Prognosis Analysis. Biomed Res Int 2021;2021:9026918. 10.1155/2021/9026918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu P, Wang X, Hu CH, et al. Bioinformatics analysis with graph-based clustering to detect gastric cancer-related pathways. Genet Mol Res 2012;11:3497-504. 10.4238/2012.September.26.5 [DOI] [PubMed] [Google Scholar]
- 23.Yan P, He Y, Xie K, et al. In silico analyses for potential key genes associated with gastric cancer. PeerJ 2018;6:e6092. 10.7717/peerj.6092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu ZH, Wang YM, Jiang YZ, et al. NID2 can serve as a potential prognosis prediction biomarker and promotes the invasion and migration of gastric cancer. Pathol Res Pract 2019;215:152553. 10.1016/j.prp.2019.152553 [DOI] [PubMed] [Google Scholar]
- 25.Bennett C, Paterson IM, Corbishley CM, et al. Expression of growth factor and epidermal growth factor receptor encoded transcripts in human gastric tissues. Cancer Res 1989;49:2104-11. [PubMed] [Google Scholar]
- 26.Wang JX, Zhou JF, Huang FK, et al. GLI2 induces PDGFRB expression and modulates cancer stem cell properties of gastric cancer. Eur Rev Med Pharmacol Sci 2017;21:3857-65. [PubMed] [Google Scholar]
- 27.Wang Y, Appiah-Kubi K, Lan T, et al. PKG II inhibits PDGF-BB triggered biological activities by phosphorylating PDGFRβ in gastric cancer cells. Cell Biol Int 2018;42:1358-69. 10.1002/cbin.11020 [DOI] [PubMed] [Google Scholar]
- 28.Liu B, Xiao X, Lin Z, et al. PDGFRB is a potential prognostic biomarker and correlated with immune infiltrates in gastric cancer. Cancer Biomark 2022;34:251-64. 10.3233/CBM-210335 [DOI] [PubMed] [Google Scholar]
- 29.Wang G, Shi B, Fu Y, et al. Hypomethylated gene NRP1 is co-expressed with PDGFRB and associated with poor overall survival in gastric cancer patients. Biomed Pharmacother 2019;111:1334-41. 10.1016/j.biopha.2019.01.023 [DOI] [PubMed] [Google Scholar]
- 30.Al-Batran SE, Atmaca A, Schleyer E, et al. Imatinib mesylate for targeting the platelet-derived growth factor β receptor in combination with fluorouracil and leucovorin in patients with refractory pancreatic, bile duct, colorectal, or gastric cancer—A dose-escalation Phase I trial. Cancer. 2007;109:1897-904. 10.1002/cncr.22622 [DOI] [PubMed] [Google Scholar]
- 31.Qian Y, Yu L, Zhang XH, et al. Genetic Polymorphism on the Pharmacokinetics and Pharmacodynamics of Platelet-derived Growth Factor Receptor (PDGFR) Kinase Inhibitors. Curr Drug Metab 2018;19:1168-81. 10.2174/1389200219666180629112943 [DOI] [PubMed] [Google Scholar]
- 32.Zhang QN, Zhu HL, Xia MT, et al. A panel of collagen genes are associated with prognosis of patients with gastric cancer and regulated by microRNA-29c-3p: an integrated bioinformatics analysis and experimental validation. Cancer Manag Res 2019;11:4757-72. 10.2147/CMAR.S198331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li F, Wang NN, Chang X, et al. Bioinformatics analysis suggests that COL4A1 may play an important role in gastric carcinoma recurrence. J Dig Dis 2019;20:391-400. 10.1111/1751-2980.12758 [DOI] [PubMed] [Google Scholar]
- 34.Li L, Zhu Z, Zhao Y, et al. FN1, SPARC, and SERPINE1 are highly expressed and significantly related to a poor prognosis of gastric adenocarcinoma revealed by microarray and bioinformatics. Sci Rep 2019;9:7827. 10.1038/s41598-019-43924-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu ZY, Chen JS, Shu YQ. Gene expression profile towards the prediction of patient survival of gastric cancer. Biomed Pharmacother 2010;64:133-9. 10.1016/j.biopha.2009.06.021 [DOI] [PubMed] [Google Scholar]
- 36.Eto S, Yoshikawa K, Shimada M, et al. The relationship of CD133, histone deacetylase 1 and thrombospondin-1 in gastric cancer. Anticancer Res 2015;35:2071-6. [PubMed] [Google Scholar]
- 37.Cao W, Fan R, Yang W, et al. VEGF-C expression is associated with the poor survival in gastric cancer tissue. Tumour Biol 2014;35:3377-83. 10.1007/s13277-013-1445-0 [DOI] [PubMed] [Google Scholar]
- 38.Dai Y, Jiang J, Wang Y, et al. The correlation and clinical implication of VEGF-C expression in microvascular density and lymph node metastasis of gastric carcinoma. Am J Transl Res 2016;8:5741-7. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as