

Abstract
Background
Oculocutaneous albinism (OCA) is an autosomal recessive disease with hypopigmentation in skin, hair, and eyes, causing by the complete absence or reduction of melanin in melanocytes. Many types of OCA were observed based on the mutation in different causing genes relating to albinism. OCA can occur in non‐syndromic and syndromic forms, where syndromic OCA coexists with additional systemic consequences beyond hypopigmentation and visual‐associated symptoms.
Methods
We performed whole exome sequencing in seven affected individuals (P1‐P7) for mutation identification, and then, Sanger sequencing was used for verifications.
Results
Among them, five patients (P1‐P5) have mutations on TYR gene including c.346C > T, c.929insC, c.115 T > C, and c.559_560ins25. The mutation on OCA2 and HPS1 genes was found in patient 6 (P6, OCA2 c.2323G > A) and patient 7 (P7, HPS1 c.972delC), respectively. Confirmation in parents (except the family of the elderly patient, P5) showed that the mother and the father in each family carried one of the variants that were detected in patients. Additionally, the effective genetic counseling was applied in the third pregnancy of a family with two OCA children (P1 and P2).
Conclusion
To our best knowledge, this is the first case with a novel homozygous missense mutation (c.115 T > C, p.W39R) in the TYR gene. This study provides a broader spectrum of mutations linked to the oculocutaneous albinism, an additional scientific basis for diagnosis, and appropriate genetic counseling for risk couples.
Keywords: HPS1, OCA2, oculocutaneous albinism, TYR, vietnamese, WES
Oculocutaneous albinism (OCA) is an autosomal recessive disease with hypopigmentation in skin, hair and eyes, causing by the complete absence or reduction of melanin in melanocytes. We performed whole exome sequencing in seven affected individuals (P1‐P7) for mutation identification. Among them, five patients (P1‐P5) have mutations on TYR gene, one (P6) in OCA2 and one (P7) in HPS1. To our best knowledge, this is the first case with a homozygous missense mutation (c.115T>C, p.W39R) in the TYR gene. This study provides a broader spectrum of mutations linked to the oculocutaneous albinism, an additional scientific basis for diagnosis, and appropriate genetic counselling for risk couples.

1. INTRODUCTION
Albinism is a rare genetic disorder caused by the reduction or absence of polymeric pigment melanin that affects the skin, hair, and/or eyes. Defective melanin production from tyrosine through a complex pathway of metabolic reactions leads to hypopigmentation, severe visual deficits, and finally albinism. Most patients with albinism have white hair and very light‐colored skin. Skin and hair color can range from white to brown and eyes color can range from light blue to brown. Vision impairment is a major feature of all albinism types. Several vision problems can occur, including nystagmus, iris transillumination, macular hypoplasia, strabismus, reduced visual acuity, and depth perception. There are two main albinism categories, which are classified based on the affection of skin, hair, and eyes, or only the eyes in oculocutaneous albinism (OCA) and ocular albinism (OA), respectively. In OCA, the number and structure of melanin are not significantly altered in any degree of observed pigmentation, whereas the appearance of a large number of distinct pigment cells within melanosomes is characteristic in OA. The OCA is a genetically heterogeneous and autosomal recessive disorder characterized by the hypopigmentation of skin, hair, and eyes. To date, at least seven autosomal genes have been associated with seven non‐syndromic OCA (OCA1, OCA2, OCA3, OCA4, OCA6, OCA7, and OCA8), including TYR, OCA2, TYRP1, SLC45A2, SLC24A5, C10orf11, and DCT. 1 , 2 For OCA5, the causative locus was mapped on chromosome 4q24, this is the only OCA subtype that causing genes have not been determined. 3 Other genes included HPS1, AP3B1, HPS3, HPS4, HPS5, HPS6, DTNBP1, BLOC1S3, BLOC1S6, AP3D1, BLOC1S5, and LYST were also involved in causing two syndromic OCA: Hermansky–Pudlak syndrome (HPS1‐11) and Chediak–Higashi syndrome (CHS). 4 , 5 , 6 Syndromic OCA can be more severe and associated with additional symptoms than only alteration of pigmentation and vision. The other type of albinism is ocular albinism (OA) that affected only the eyes, caused by mutation in OA1/GPR143 gene on X chromosome. 7 Vision acuity and photophobia of patients with OA are reduced and strabismus or nystagmus is also observed. 1
In Vietnam, until now, the genetic data of albinism have remained unknown. Thereby, for the first time, genetic analysis was performed on seven Vietnamese albinism patients in our study.
2. MATERIAL AND METHOD
2.1. Subject
A total of seven affected individuals with OCA, including six (P1, P2, P3, P4, P6, and P7) and their parents (Figure 2–5) from four unrelated nonconsanguineous families, and a single man (P5) were recruited from Hanoi Medical University Hospital, Hanoi, Vietnam. All patients presented with typical clinical features of albinism, including various degrees of eyes, hair, and skin hypopigmentation. Written informed consents were obtained from all patients and family members before sample collection. This study was approved by the Institute of Genome Research Institutional Review Board, Vietnam Academy of Science and Technology.
For patients and family members, 2 ml of whole blood was collected, preserved in EDTA‐containing tubes, and stored at −20°C. Genomic DNA extraction was performed by using Exgene™ Blood SV (GeneAll Biotechnology), following the manufacturer's guidelines. Qubit™ dsDNA HS Assay Kit (Thermo Fisher Scientific) was used for DNA quantification.
2.2. Method
2.2.1. Exome analysis
The DNA library was constructed by Sure Select V6‐Post (Agilent Technologies) following the manufacturer's guidelines. The sequencing was performed by using an Illumina NovaSeq 6000 platform (Illumina) with paired reads of 150 bp. The reads were mapped to the hg19/GRCh37 human reference genome by the BWA.v0.7.12 tool, and Picard was used to mark the duplicates. Genome Analysis Toolkit (GATK) and Samtools were used to detect single nucleotide variants (SNVs) and short insertions/deletions (Indels). To exclude false positive, all variants with depth read lower than 20× were removed. Short Indels in the repeat regions and within the ten bp range from the start and end of the read were also excluded. After that, the remaining variants were filtered from the public databases comprising 1000G and gnomAD. All variants with minor allele frequency (MAF) under 0.01% were selected for further evaluation.
The variants were annotated with the ANNOVAR program. The in silico analysis was performed by SIFT, Polyphen‐2, and Mutation Taster to anticipate the functional effect of missense and nonsense variants. The candidate variants were classified according to the Guideline and Standard of the American College of Medical Genetics and Genomics (ACMG).
2.2.2. Sanger validation
The candidate variants were validated by direct Sanger sequencing in patients as well as their parents. Primers for PCR and sequencing were provided by PHUSA Biochem Company (Can Tho, Vietnam). For PCR amplification, 10 ng of total genomic DNA was used as a template in 20 μl of reaction mixture containing 1X Neb Master mix (New England Biolabs, Ipswich), 0.8 μl of each primer (10 pmole), and 8.4 μl of deionized water. The thermocycling was 95°C for 2 min, followed by 40 cycles of 95°C for 30 s, 58°C for 30 s, 68°C for 20 s, and a final extension at 68°C for 5 min. The PCR products were purified using Multiscreen PCR 96 Filter Plate (Merck‐Millipore) and sequenced by ABI Prism BigDye Terminator Cycle Sequencing Kit Version 3.1 (Applied BioSystems) on ABI 3500 Genetic Analyzer (Applied BioSystems).
3. RESULT
3.1. Clinical features
In all the seven patients, hypopigmented eyes, hair and skin, and photophobia were observed. The other ophthalmological findings, including nystagmus, reduced visual acuity, strabismus, foveal hypoplasia, ecchymosis, and reduced stereopsis, were different in each patient (Table 1).
TABLE 1.
Patient characteristics
| Patient | Dermatological findings | Iris color | Photophobia | Visual acuity problem | Nystagmus | Strabismus | Foveal hypoplasia | Ecchymosis | Reduced stereopsis |
|---|---|---|---|---|---|---|---|---|---|
| 1 | White skin, fair hair | Blue | + | Myopia of 14 diop | + | − | + | · | + |
| 2 | White skin, fair hair | Blue | + | Myopia 1/10 (1,5‐2D) | + | − | + | · | + |
| 3 | Pinkish‐white skin, blond hair | Brown | + | Amblyopia | + | − | − | − | + |
| 4 | Pinkish‐white skin, blond hair | Brown | + | Amblyopia | + | + | − | + | + |
| 5 | Pinkish‐white skin, white hair | Brown | + | Myopia 6/10 (~0,5D) | − | − | − | · | + |
| 6 | Pinkish‐white skin, light brown hair | Brown | + | None | − | − | − | · | − |
| 7 | White skin, brown hair | Blue | + | Astigmatism | + | − | − | · | + |
(+) Positive; (−) Negative; (·) Not available.
In brothers of P1 (7 years old) and P2 (5 years old), the clinical manifestation included white hair and skin, and blue eyes (Figure 1A,B). The presentation of photophobia with discomfort and eye pain when exposed to light, nystagmus (increased when illness and stress), and choroidal metaplasia were detected. Myopia −14 diopters (D) and visual acuity of 1/10 were identified in P1 and P2, respectively, and the problem to perceive depth was also observed in both patients (Table 1). Additionally, the younger brother showed a history of repeated pneumonia (P2).
FIGURE 1.
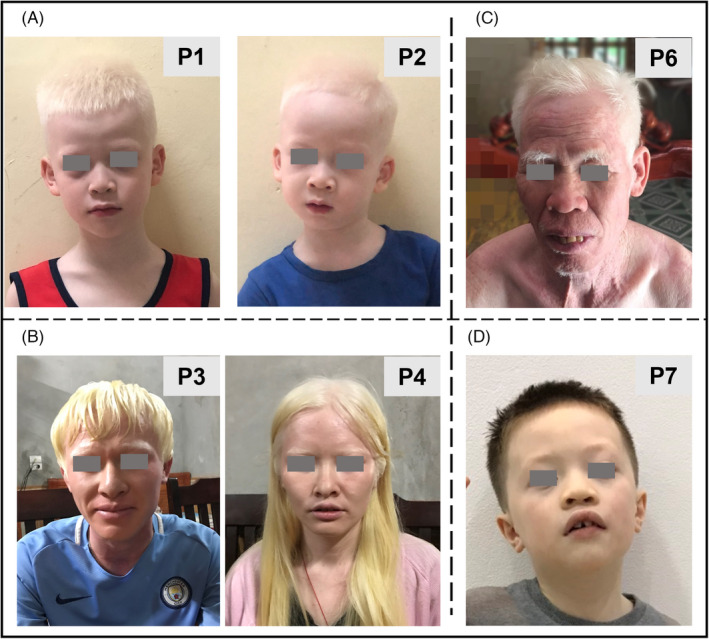
Clinical features of six albinism cases in Vietnam (patient 6 was not present) with hypopigmentation in skin, hair (white skin and hair in P1, P2, and P5; pinkish‐white skin and light yellow hair in P3 and P4; white skin and brown hair in P7), and iris color arrange from blue (P1, P2, and P7) to brown (P3, P4, and P5)
Patients 3 (P3, 27 years old) and 4 (P4, 23 years old) were the first and the second child in a family of three children. The fair skin, light blonde hair, brown eyes (Figure 1C,D), photophobia, nystagmus, amblyopia, and decreased perception of depth were observed in both patients. The younger sister (P4) presented with strabismus in her right eye (Table 1 and Figure 1D), a history of glomerulonephritis, scaly dermatitis, and subcutaneous hemorrhage.
Patient 5 (P5) was a 63‐year‐old man with white skin and hair, blue eyes, and photophobia (Figure 1E). The patient had been visual acuity of 6/10 in both eyes and reduced stereopsis. Manifestations of nystagmus and foveal hypoplasia were not observed in this patient (Table 1).
Patient 6 (P6) was a 26‐year‐old woman, the second child in the family. Clinical features of OCA consist of pinkish‐white skin, hair and eyebrows, light brown eyelashes, brown eyes, and manifestations of photophobia (patient's images were not provided). The patient had normal physical, mental, and motor development, with no manifestations of nystagmus, foveal hypoplasia, and refractive errors (Table 1).
Patient 7 (P7) was a 5‐year‐old boy, who presented with white skin, reddish yellow to brown hair, blue eyes (Figure 1F), increased sensitivity to light (photophobia), astigmatism, and must wear glasses. Other manifestations, including nystagmus and reduced depth perception, were observed in the patient (Table 1).
3.2. Genetic analysis
WES data analysis revealed six candidate pathogenic variants with very rare or unknown allele frequency, including four variants in the TYR gene (NM_000372.3, #MIM:606993) (P1, P2, P3, P4, and P5), two variants in OCA2 (NM_000275.3, #MIM:611409) (P6), and HPS1 (NM_000195, #MIM:604982) (P7) genes, respectively (Table 2).
TABLE 2.
Gene mutations in seven Vietnamese OCA
| Patient | Gene | Variant change | Zygosity | Type of variant | Region | ID (dbSNP/HGMD) | genomAD | ClinVar/ACMG classification† | OCA subtype |
|---|---|---|---|---|---|---|---|---|---|
| 1 and 2 | TYR | c.346C > T (p.R116*) | het | stopgain | exon 1 | rs61753256 | 0.00002829 | P | OCA1A |
| c.929insC (p.R311fs*7) | het | FS ins | exon 2 | rs281865527 | 0.000044 | P | |||
| 3 and 4 | TYR | c.115 T > C (p.W39R) | homo‡ | nonsyn | exon 1 | CM100987 | · |
LP† (PM2, PM3, PP1‐PP4) |
OCA1B |
| 5 | TYR | c.559_560ins25 (p.G190fs*12) | homo | FS ins | exon 1 | · | · |
P† (PVS1, PM4, PP1, PP3, PP4) |
OCA1A |
| 6 | OCA2 | c.2323G > A (p.G775S) | homo | nonsyn | exon 22 | rs774822330 | 0.000008028 |
LP† (PM2, PM3,PP1‐PP4) |
OCA2 |
| 7 | HPS1 | c.972delC (p.M325fs*6) | homo | FS del | exon 11 | rs281865082 | 0.0001251 | P | HPS1 |
Abbreviations: FS del, frameshift deletion; FS ins, frameshift insertion; het, heterozygous; hom, homozygous; HPS, Hermansky–Pudlak syndrome; LP, Likely pathogenic; nonsyn, non‐synonymous; OCA, oculocutaneous albinism; P, pathogenic; PM, pathogenic moderate; PP, pathogenic supporting; PVS, pathogenic very strong.
†In current study; ‡Previously unknown zygosity; (·) Not available.
A TYR compound heterozygous mutation (c.346C > T and c.929insC) that resulted in two premature termination codons (PTCs) (p.R116* and p.R311fs*7) was observed in two brothers (P1 and P2). Both mutations were reported in dbSNP (rs61753256 and rs281865527) and were known as pathogenicity in ClinVar (Table 2). Sanger sequencing confirmed that the affected individuals inherited the c.3416C > T from the father and the c.929insC from the mother (Figure 2A). In addition, genetic testing detected c.929insC heterozygous mutation from the amniotic fluid sample of the third pregnancy of the mother (Figure 2A). Two homozygous TYR variations were found in the patients P3 and his younger sister P4 (c.115 T > C, p.W39R; Figure 2B) and P5 (c.559_560ins25, p.G190fs*12; Figure 2C). In which, the c.115 T > C has been reported in the human genome mutation database (HGMD: CM100987) and was not found in dbSNP as ClinVar. This variant resulted in a substitution of conserved amino acid tryptophan by arginine (p.W39R) and was predicted as probably damaging/deleterious by SIFT, Polyphen2, and disease causing in MutationTaster. The c.559_560ins25 was not identified in any online databases (Table 2), but three carriers with OCA were reported in previous publications (PMID: 25577957, 31,196,117 and 31,077,556). Verification by directed sequencing showed that the heterozygous c.115 T > C variant was observed in the parent of P3 and P4 and did not inherit to the third child (Figure 2B). In brief, OCA1 subtype diagnosis was considered in five affected individuals with the finding of TYR mutations.
FIGURE 2.
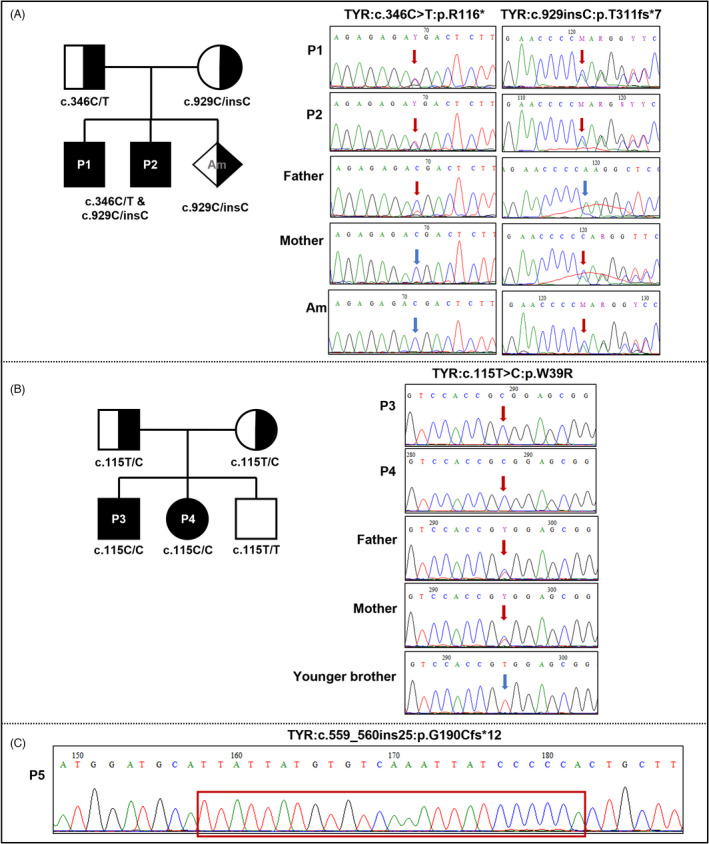
Pedigree charts of two families (A and B) and electropherograms of five affected individuals and their families (except the family of P5) with TYR mutations. Full/half black represents patient/carrier individuals. Mutated/normal nucleotides (A and B) and insertion sequence (C) were marked with red/blue arrows and red box, respectively.
The other non‐syndromic OCA subtype with a molecular diagnosis of OCA2 (P6) was identified to be homozygous for OCA2 c.2323G > A (dbSNP: rs774822330), leading to a substitution of glycine to serine at codon 775 in polypeptide (p.G775S) (Table 2). This missense variant was predicted to be damaging effect on protein function by SIFT, Polyphen2, and disease causing by MutationTaster. Sequencing analysis indicated that both father and mother were heterozygous carriers for c.2323G > A (Figure 3).
FIGURE 3.
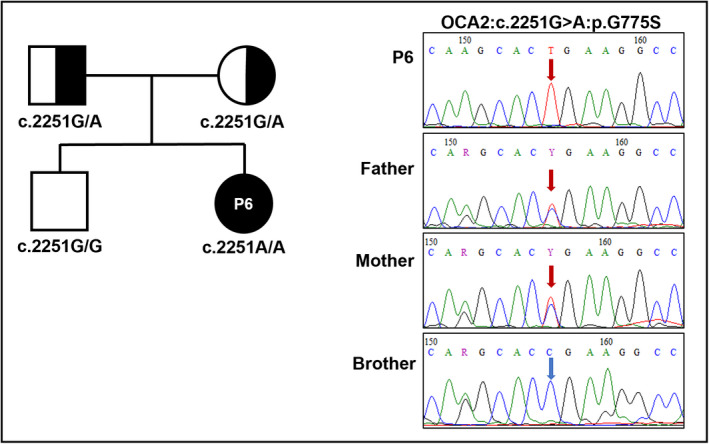
Pedigree chart and electropherograms of P6 family with OCA2 mutation. Full/half black represents patient/carrier individuals. Mutated/normal nucleotides were marked with red/blue arrows
Only patient 7 (P7) was classified as the subtype of syndromic albinism due to the fact that the c.972delC homozygote of the HPS1 gene was found to cause Hermansky–Pudlak syndrome. The c.972delC was pathogenic mutation and was reported in dbSNP as ClinVar (Table 2). The familial segregation was confirmed for c.972delC with the heterozygous trait in both parents by Sanger sequencing (Figure 4).
FIGURE 4.
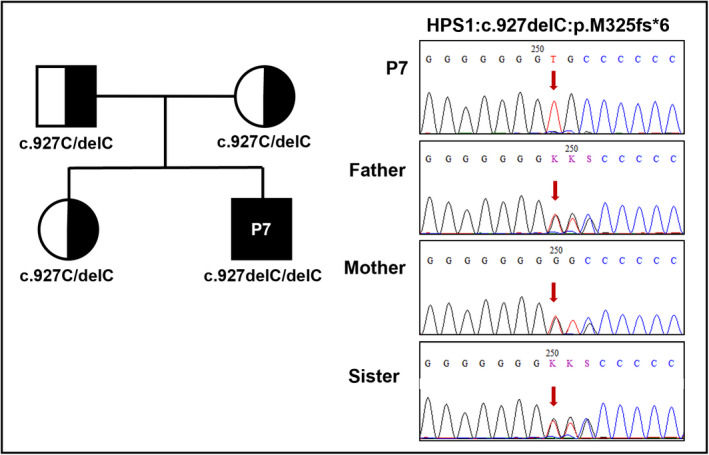
Pedigree chart and electropherograms of P7 family with HPS1 mutation. Full/half black represents patient/carrier individuals. Mutated nucleotides were marked with red arrows
4. DISCUSSION
In this study, the molecular diagnosis for seven affected individuals with albinism was provided, including identified mutations in TYR, OCA2, and HPS1 genes associated with OCA subtype 1 (P1, P2, P3, P4, and P5), subtype 2 (P6), and Hermansky–Pudlak syndrome (HPS) (P7), respectively. OCA1 and OCA2 were the common subtypes of non‐syndromic OCA, while HPS was the rare syndromic OCA.
The TYR mutations causing to OCA1 included compound heterozygote (c.346C > T and c.929insC), two homozygous of c.115 T > C (p.W39R), and c.559ins25 (p.G190Cfs*12), which were observed in two siblings (P1, P2, and P3, P4) and the elderly patient (P5), respectively. There were two subtypes of OCA1 (OCA1A and OCA1B), of which OCA1A was produced by null mutations that resulted in absence or inactive tyrosinase. In OCA1B, the activity of tyrosinase was reduced compared with normal form, caused by leaky mutations, allowing some accumulation of melanin pigment over time. 8 , 9 Therefore, genetic testing would be considered as the important investigation for the accurate diagnosis of OCA1 subtypes.
In five OCA1 cases of our study, three patients (P1, P2, and P5) were classified as OCA1A, carrying PTC mutations (p.R116*; p.R311fs*7; and p.G190Cfs*12) in both alleles, leading to a complete loss of tyrosine function, thereby not producing melanin in the melanocytes. These mutations were known, in which the c.346C > T (p.R116*) had been reported previously in Chinese and Caucasian patients 10 , 11 , 12 and the c.929insC mutation (p.R311fs*7) was quite common ones in the Chinese 12 , 13 , 14 , 15 and some other East Asian countries such as Japan and Korea with OCA1. 16 , 17 , 18 Particular, the c.559_560ins25 (p.G190Cfs*12) was a relatively rare mutation. To the best of our knowledge, P5 was the fourth case carrying this mutation and was the first finding case outside the Chinese. 11 , 15 , 19 Moreover, genetic counseling was performed in the third pregnancy of the mother's P1 and P2, and the baby showed no signs of OCA after birth. This confirms that prenatal genetic screening in risk couples was effective and possible for this disorder.
The P3 and P4 were classified as OCA1B, both carried a homozygous mutation c.115 T > C (p.W39R), leading to decreased tyrosine activity and subsequently reduce melanin creation. This mutation was just reported in a Chinese male patient in a heterozygous state, who was initially diagnosed with OCA2 based on clinical features. However, upon genetic analysis, he was classified as OCA1B with compound heterozygous for c.115 T > C (p.W39R) and c.1265G > A (p.R422Q) of the TYR gene. 14 Thus, molecular screening has played an important role in the accurate diagnosis of the albinism subtype. In our study, the TYR c.115 T > C (p.W39R) was the first observed in a homozygous state, finding in two patients of P3 and P4. In addition, not only the substitution at nucleotide 115 (c.115 T > C, p.W39R) but two others at position 116 (c.116G > A, p.W39*) and 117 (c.117G > T, p.W39C) were also reported to functionally affect codon 39 in the polypeptide, all implicated in OCA1B. 14 , 20 For these reasons, the TYR c.115 T > C has been classified as “likely pathogenic” following the ACMG criteria (Table 2).
An OCA2 case detected in this study was patient P6, carrying the OCA2 homozygous variant c.2323G > A (p.G775S). This variant was previously found with homozygous in a Vietnamese patient, but the authors assumed that this substitution had no harmful consequence on protein. Preising et al. explained that, when conducting in silico analysis using SIFT and Polyphen‐2, the results were contradictory, so c.2323G > A (p.G775S) was not considered to be the causative agent of the disease. Instead of the c.2323G > A (p.G775S), the homozygous c.1113 T > C (p.G371=) was reported as the causative mutation in the study of Preising et al., according to novel splice donor site prediction by Splice Sequence Finder Server. 21 However, until now, c.1113 T > C (p.G371=) has been reported as a benign variant on the ClinVar database (VCV000193573.7). On the other hand, in current work, in silico prediction by SIFT and Polyphen‐2, both showed the c.2323G > A (p.G775S) to be potentially damaging the encoded protein. In addition, at codon position 775 exist, two other alterations previously found in OCA2 patients including c.2323G > C (p.G775R) 22 and c.2324G > A (p. G775D). 23 The glycin at c.775 was highly conserved and located within the transmembrane 11 region in total of 12 domains on P polypeptide, encoded by the OCA2 gene. Therefore, the substitutions of G775 may possibly inhibit the folding of P‐protein and lead to harmful consequences. Based on these suggestions, we predict that c.2323G > A (p.G775S) could be considered a “likely pathogenic” variant according to ACMG classification (Table 2) and should be functionally demonstrated in further studies.
Patient 7 was the only syndromic OCA case associated with Hermansky–Pudlak syndrome (HPS) caused by homozygous frameshift HPS1 c.972delC mutation, making to the appearance of PTC on the polypeptide chain (p.M325fs*6), which was previously reported in causing of failure to the formation or resulting in a protein loss of function after translation. 24 The HPS1 loss of function was established as a known mechanism of disease in autosomal recessive HPS. 1 In fact, nucleotide C at position 972 was identified as a mutation “hotspot” of the HPS1 gene. 10 The p.M325fs*6 was the co‐product of the insertion (c.972insC) or deletion (c.972delC) at position 972 on the nucleotide sequence, which has been known to be the most common mutation in the European HPS patients. 25 Therein, c.972delC was observed in some Puerto Rican, 26 Mexican, 27 Chinese 28 and African American 29 patients. The c.972insC was found mainly in Puerto Rican 25 and some Japanese patients. 30 In HPS, the classic clinical features included OCA, a prolonged bleeding due to storage pool‐deficient platelets, and development of granulomatous, pulmonary fibrosis, or neutropenia in some cases. 1 However, apart from the main OCA findings, the other features specialty of HPS was not identified in P3. For this reason, molecular tests could be needed for the specific classification of albinism subtypes.
5. CONCLUSION
This is the first report on albinism‐causing genes in Vietnam, exploring the mutational spectrum relevant to this disorder. To the best of our knowledge, we also report a rare TYR mutation (c.115 T > C) with the novel zygosity being homozygous. Considering overlapped characteristics of OCA subtypes, molecular genetic analysis will substantially aid clinical diagnosis and genetic counseling of OCA.
AUTHOR CONTRIBUTIONS
Conceptualization: NDT, NVH; Funding acquisition: NDT; Data curation, Formal analysis, and Investigation: MTHT, VPN, NHH, TTBN, HTL, LTLA; Roles/Writing ‐ original draft: MTHT, NDT; Writing ‐ review & editing: MTHT, NHH, NDT, NVH.
CONFLICT OF INTEREST
All authors declare that they have no conflict of interest.
ACKNOWLEDGMENT
We are thankful to all patients for agreeing to participate in this study. This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 108.02‐2019.05.
Thuong MTH, Anh LTL, Nhung VP, et al. Genetic analyses of Vietnamese patients with oculocutaneous albinism. J Clin Lab Anal. 2022;36:e24625. doi: 10.1002/jcla.24625
DATA AVAILABILITY STATEMENT
The data used to support the findings of this study may be requested from the corresponding author.
REFERENCES
- 1. Kamaraj B, Purohit R. Mutational analysis of oculocutaneous albinism: a compact review. Biomed Res Int. 2014;2014:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Garrido G, Fernández A, Montoliu L. HPS11 and OCA8: two new types of albinism associated with mutations in BLOC1S5 and DCT genes. Pigment Cell Melanoma Res. 2021;34(1):10‐12. [DOI] [PubMed] [Google Scholar]
- 3. Kausar T, Bhatti M, Ali M, Shaikh R, Ahmed Z. OCA5, a novel locus for non‐syndromic oculocutaneous albinism, maps to chromosome 4q24. Clin Genet. 2013;84(1):91‐93. [DOI] [PubMed] [Google Scholar]
- 4. Li W, He M, Zhou H, Bourne JW, Liang P. Mutational data integration in gene‐oriented files of the Hermansky‐Pudlak syndrome database. Hum Mutat. 2006;27(5):402‐407. [DOI] [PubMed] [Google Scholar]
- 5. Wei AH, Li W. H ermansky–P udlak syndrome: pigmentary and non‐pigmentary defects and their pathogenesis. Pigment Cell Melanoma Res. 2013;26(2):176‐192. [DOI] [PubMed] [Google Scholar]
- 6. Pennamen P, Le L, Tingaud‐Sequeira A, et al. BLOC1S5 pathogenic variants cause a new type of Hermansky‐Pudlak syndrome. Genet Med. 2020;22(10):1613‐1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bassi MT, Schiaffino MV, Renieri A, et al. Cloning of the gene for ocular albinism type 1 from the distal short arm of the X chromosome. Nat Genet. 1995;10(1):13‐19. [DOI] [PubMed] [Google Scholar]
- 8. Grønskov K, Ek J, Brondum‐Nielsen K. Oculocutaneous albinism. Orphanet J Rare Dis. 2007;2(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Simeonov DR, Wang X, Wang C, et al. DNA variations in oculocutaneous albinism: an updated mutation list and current outstanding issues in molecular diagnostics. Hum Mutat. 2013;34(6):827‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oetting WS, Fryer JP, King RA. Mutations of the human tyrosinase gene associated with tyrosinase related oculocutaneous albinism (OCA1). Mutations in brief no. 204. Online. Hum Mutat. 1998;12(6):433‐434. [DOI] [PubMed] [Google Scholar]
- 11. Yang Q, Yi S, Li M, et al. Genetic analyses of oculocutaneous albinism types 1 and 2 with four novel mutations. BMC Med Genet. 2019;20(1):106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lin Y, Chen X, Yang Y, et al. Mutational analysis of TYR, OCA2, and SLC45A2 genes in Chinese families with oculocutaneous albinism. Mol Genet Genomic Med. 2019;7(7):e00687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Y, Guo X, Li W, Lian S. Four novel mutations of TYR gene in Chinese OCA1 patients. J Dermatol Sci. 2009;53(1):80‐81. [DOI] [PubMed] [Google Scholar]
- 14. Wei A, Wang Y, Long Y, et al. A comprehensive analysis reveals mutational spectra and common alleles in Chinese patients with oculocutaneous albinism. J Invest Dermatol. 2010;130(3):716‐724. [DOI] [PubMed] [Google Scholar]
- 15. Zhong Z, Gu L, Zheng X, et al. Comprehensive analysis of spectral distribution of a large cohort of Chinese patients with non‐syndromic oculocutaneous albinism facilitates genetic diagnosis. Pigment Cell Melanoma Res. 2019;32(5):672‐686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goto M, Sato‐Matsumura KC, Sawamura D, Yokota K, Nakamura H, Shimizu H. Tyrosinase gene analysis in Japanese patients with oculocutaneous albinism. J Dermatol Sci. 2004;35(3):215‐220. [DOI] [PubMed] [Google Scholar]
- 17. Park SH, Chae H, Kim Y, Kim M. Molecular analysis of Korean patients with oculocutaneous albinism. Jpn J Ophthalmol. 2012;56(1):98‐103. [DOI] [PubMed] [Google Scholar]
- 18. Suzuki T, Tomita Y. Recent advances in genetic analyses of oculocutaneous albinism types 2 and 4. J Dermatol Sci. 2008;51(1):1‐9. [DOI] [PubMed] [Google Scholar]
- 19. Liu N, Kong XD, Shi HR, Wu QH, Jiang M. Tyrosinase gene mutations in the Chinese Han population with OCA1. Genet Res. 2014;96:e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. King RA, Pietsch J, Fryer JP, et al. Tyrosinase gene mutations in oculocutaneous albinism 1 (OCA1): definition of the phenotype. Hum Genet. 2003;113(6):502‐513. [DOI] [PubMed] [Google Scholar]
- 21. Preising MN, Forster H, Gonser M, Lorenz B. Screening of TYR, OCA2, GPR143, and MC1R in patients with congenital nystagmus, macular hypoplasia, and fundus hypopigmentation indicating albinism. Mol Vis. 2011;17:939‐948. [PMC free article] [PubMed] [Google Scholar]
- 22. Hongyi L, Haiyun W, Hui Z, et al. Prenatal diagnosis of oculocutaneous albinism type II and novel mutations in two Chinese families. Prenat Diagn. 2007;27(6):502‐506. [DOI] [PubMed] [Google Scholar]
- 23. Johanson HC, Chen W, Wicking C, Sturm RA. Inheritance of a novel mutated allele of the OCA2 gene associated with high incidence of oculocutaneous albinism in a Polynesian community. J Hum Genet. 2010;55(2):103‐111. [DOI] [PubMed] [Google Scholar]
- 24. Shotelersuk V, Hazelwood S, Larson D, et al. Three new mutations in a gene causing Hermansky‐Pudlak syndrome: clinical correlations. Mol Genet Metab. 1998;64(2):99‐107. [DOI] [PubMed] [Google Scholar]
- 25. Oh J, Ho L, Ala‐Mello S, et al. Mutation analysis of patients with Hermansky‐Pudlak syndrome: a frameshift hot spot in the HPS gene and apparent locus heterogeneity. Am J Hum Genet. 1998;62(3):593‐598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carmona‐Rivera C, Hess RA, O'Brien K, et al. Novel mutations in the HPS1 gene among puerto rican patients. Clin Genet. 2011;79(6):561‐567. [DOI] [PubMed] [Google Scholar]
- 27. Carmona‐Rivera C, Golas G, Hess RA, et al. Clinical, molecular, and cellular features of non‐Puerto Rican Hermansky‐Pudlak syndrome patients of Hispanic descent. J Invest Dermatol. 2011;131(12):2394‐2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wei A, Yuan Y, Bai D, et al. NGS‐based 100‐gene panel of hypopigmentation identifies mutations in Chinese Hermansky‐Pudlak syndrome patients. Pigment Cell Melanoma Res. 2016;29(6):702‐706. [DOI] [PubMed] [Google Scholar]
- 29. Merideth MA, Vincent LM, Sparks SE, et al. Hermansky‐Pudlak syndrome in two African‐American brothers. Am J Med Genet A. 2009;149A(5):987‐992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ito S, Suzuki T, Inagaki K, et al. High frequency of Hermansky‐Pudlak syndrome type 1 (HPS1) among Japanese albinism patients and functional analysis of HPS1 mutant protein. J Invest Dermatol. 2005;125(4):715‐720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study may be requested from the corresponding author.