

Abstract
KRAS mutation is a major driver of pancreatic carcinogenesis and will likely be a therapeutic target. Due to lack of sensitive assays for clinical samples of pancreatic cancer with low cellularity, KRAS mutations and their prognostic association have not been fully examined in large populations. In a multi‐institutional cohort of 1162 pancreatic cancer patients with formalin‐fixed paraffin‐embedded tumor samples, we undertook droplet digital PCR (ddPCR) for KRAS codons 12/13/61. We examined detection rates of KRAS mutations by clinicopathological parameters and survival associations of KRAS mutation status. Multivariable hazard ratios (HRs) and 95% confidence intervals (CIs) for disease‐free survival (DFS) and overall survival (OS) were computed using the Cox regression model with adjustment for potential confounders. KRAS mutations were detected in 1139 (98%) patients. The detection rate did not differ by age of tissue blocks, tumor cellularity, or receipt of neoadjuvant chemotherapy. KRAS mutations were not associated with DFS or OS (multivariable HR comparing KRAS‐mutant to KRAS‐wild‐type tumors, 1.04 [95% CI, 0.62–1.75] and 1.05 [95% CI, 0.60–1.84], respectively). Among KRAS‐mutant tumors, KRAS variant allele frequency (VAF) was inversely associated with DFS and OS with HRs per 20% VAF increase of 1.27 (95% CI, 1.13–1.42; p trend <0.001) and 1.31 (95% CI, 1.16–1.48; p trend <0.001), respectively. In summary, ddPCR detected KRAS mutations in clinical specimens of pancreatic cancer with high sensitivity irrespective of parameters potentially affecting mutation detections. KRAS VAF, but not mutation positivity, was associated with survival of pancreatic cancer patients.
Keywords: cohort study, oncogene, pancreatectomy, pancreatic neoplasm, sequence analysis
Droplet digital PCR provides highly sensitive mutation calling in clinical tumor samples irrespective of quality and quantity of tumor DNA. KRAS variant allele frequency, but not mutation positivity, can be a prognostic biomarker in pancreatic cancer.

Abbreviations
- CI
confidence interval
- ddPCR
droplet digital polymerase chain reaction
- DFS
disease‐free survival
- FFPE
formalin‐fixed paraffin‐embedded
- HR
hazard ratio
- OS
overall survival
- PARP
poly(ADP‐ribose) polymerase
- VAF
variant allele frequency
1. INTRODUCTION
Somatic gain‐of‐function mutations in the KRAS oncogene have been major drivers in pancreatic carcinogenesis 1 , 2 and have been attributed to resistance to anti‐epidermal growth factor receptor therapy in colorectal cancer. 3 Given recent advances in effective molecular‐targeted agents and the resultant global trend toward precision oncology, 4 , 5 molecular profiling of tumors is of increasing importance in clinical decision making. Pancreatic cancer exhibits abundant desmoplastic stroma, resulting in low tumor cellularity, 6 , 7 , 8 which has inhibited robust molecular annotation based on clinical tissue samples. Genomic analyses of FFPE tissue samples have been particularly challenging due to DNA degradation during the processes of fixation, long‐term storage, and preanalytical preparation. 9 , 10 Consequently, research on KRAS mutations using clinical samples of pancreatic cancer has been limited by measurement errors due to low mutation detectability of conventional technologies. Therefore, clinical outcomes according to KRAS mutation status have not been examined extensively in unselected populations of pancreatic cancer patients.
Droplet digital PCR has emerged as a promising diagnostic technique that allows sensitive and quantitative characterization of genetic aberrations, including point mutations and copy number alterations. 11 , 12 Given the higher sensitivity of variant calling compared to real‐time PCR and next‐generation sequencing, ddPCR has been applied for noninvasive specimens containing low abundance of tumor DNA, such as blood and urine (so‐called liquid biopsy). 13 , 14 , 15 The ddPCR assays have the potential of absolute quantification of VAF as well as technical advantages including short turnaround time, low assay costs, and low amount of DNA required. 12 However, the utility of ddPCR has not been fully investigated for molecular profiling based on clinical FFPE samples of pancreatic cancer. In addition, the prognostic association of KRAS VAF has not been examined in pancreatic cancer. Given the carcinogenic effects of activating KRAS mutations and downstream signaling pathways, 11 , 16 , 17 , 18 we hypothesized that higher levels of KRAS VAF might be associated with shorter survival times among patients with pancreatic cancer.
Therefore, we leveraged multiplex ddPCR for common KRAS mutations in a large multicenter cohort of consecutive patients with resected pancreatic cancer. We examined the overall feasibility of ddPCR for KRAS mutations and the mutation detectability according to clinicopathological parameters that might affect mutation detections. We also examined KRAS mutation load in relation to survival outcomes of patients with resected pancreatic cancer.
2. MATERIALS AND METHODS
2.1. Study cohort
We identified consecutive patients who underwent surgical resection of pancreatic carcinoma with curative intent at The Cancer Institute Hospital of Japanese Foundation for Cancer Research, The University of Tokyo Hospital, or Keio University Hospital (all in Tokyo, Japan) between 2005 and 2017. Among those patients, we included 1162 cases with pancreatic cancer (adenocarcinoma or adenosquamous carcinoma) where tissue specimens were available for ddPCR for KRAS mutations. We excluded patients with mixed tumors (e.g., mixed ductal‐neuroendocrine carcinoma) or ductal adenocarcinoma variants including carcinoma derived from intraductal papillary mucinous neoplasm, undifferentiated carcinoma, and colloid carcinoma. For analyses of OS, we excluded patients with concomitant advanced cancer of other origin and patients with 30‐day or in‐hospital mortality. For analyses of DFS, we further excluded patients with a resected metastatic lesion, R2 resection margin, or no available cross‐sectional imaging following the index surgery.
Informed consent was obtained from all participants on an opt‐out basis given the retrospective nature of the current study. This study was designed and carried out according to the guidelines in the Helsinki Declaration. The study was approved by the ethics committees at The Cancer Institute Hospital of Japanese Foundation for Cancer Research, The University of Tokyo, and Keio University School of Medicine, and was registered with the UMIN registry (registration number UMIN000044027).
2.2. Data collection
Utilizing a standardized database constructed using Microsoft Access software, study physicians reviewed medical charts and collected clinical data, including demographics, tumor characteristics, and treatment outcomes. Study pathologists (M.Tak., M.Tan., and Y.M.), blinded to clinical data, reviewed H&E‐stained tissue sections of FFPE tissue blocks and recorded histopathological features of pancreatic carcinomas. According to the guidelines of the Japan Pancreas Society, 19 we classified stroma type (medullary [scant stroma], intermediate, or scirrhous [abundant stroma]), resection margin status (R0, no residual tumor cells on the dissection or cut surface; R1, microscopic residual tumor; or R2, macroscopic residual tumor), and the degree of lymphatic, venous, or neural invasion (absent, mild, moderate, or marked). Percentages of tumor cells and inflammatory cells (e.g., lymphocytes, neutrophils, eosinophils, and plasma cells) were estimated microscopically within cancerous areas on guide H&E slides used for DNA extraction. Cancer stage was defined according to the eighth edition of the TNM staging system proposed by the UICC. 20 For adenocarcinoma cases, tumor differentiation was graded as well, moderate, or poor according to the WHO classification. 21
2.3. Droplet digital PCR for KRAS mutations
All tissue samples were obtained from surgical specimens of the primary pancreatic carcinomas. During the study period, the surgical specimens were fixed with 20% formalin neutral buffer solution at The Cancer Institute Hospital of the Japanese Foundation for Cancer Research and The University of Tokyo Hospital. The specimens were fixed with 10% nonbuffered formalin until May 2017 and with 10% formalin neutral buffer solution thereafter at Keio University Hospital. Genomic DNA was extracted from 10 μm‐thick sections of archival FFPE tissue blocks of pancreatic cancer using the GeneRead DNA FFPE Kit (Qiagen). The extraction protocol includes treatment with uracil‐DNA glycosylate that potentially reduces false‐positive signals in ddPCR derived from formalin fixation. 22 The study pathologists (M.Tak., M.Tan., and Y.M.) marked tumor areas in guide H&E‐stained slides. Using the guide H&E slides, DNA was extracted through macrodissection of tumor areas. The extracted DNA was quantified using the NanoDrop One spectrophotometer (Thermo Fisher Scientific) and has been stored at −20°C in well‐monitored freezers.
The ddPCR procedures were undertaken using the QX200 system (Bio‐Rad Laboratories) at a single centralized center (The University of Tokyo). All reactions were prepared using a multiplex screening kit for seven mutations in KRAS codons 12 and 13 (G12A, G12C, G12D, G12R, G12S, G12V, and G13D; ddPCR KRAS Screening Multiplex Kit; Bio‐Rad Laboratories). The total volume of 20 μl ddPCR reaction mix was prepared with 100 ng DNA, 1 μl of 20× multiplex assay mix, 10 μl of 2× ddPCR Supermix for Probes (no dUTP), and water in a variable volume. The reaction mix and 70 μl Droplet Generation Oil for Probes (Bio‐Rad Laboratories) were loaded into the corresponding wells in the DG8 cartridge. The cartridge was placed into a QX200 Droplet Generator (Bio‐Rad Laboratories), which partitioned each PCR mix into approximately 20000 droplets. Emulsified mixes were transferred to a 96‐well plate, and the plate was heat sealed in PX1 PCR Plate Sealer (Bio‐Rad Laboratories). Polymerase chain reaction was carried out using T100 Thermal Cycler (Bio‐Rad Laboratories) with the following thermal cycling conditions: 95°C for 10 min, 40 cycles of 94°C for 30 s and 55°C for 1 min, 98°C for 10 min, and 4°C for holding. Each run included positive and negative controls (DNA from CFPAC‐1 cells and HPNE cells, respectively). The plate was then loaded to a QX200 Droplet Reader (Bio‐Rad Laboratories), and the droplets from each well were analyzed. The data were processed and analyzed using the QuantaSoft software (version 1.7.4, Bio‐Rad Laboratories; Figure S1). To define positive and negative calls, we utilized the R package twoddpcr and undertook k‐means clustering based on the Mahalanobis distance. 23 Using 50 randomly selected cases at each institution, we defined site‐specific criteria for positive and negative calls. The results were in accordance with the visual inspection. The VAF of KRAS was calculated as the ratio of the number of KRAS‐mutant droplets to that of droplets including KRAS‐mutant signal and/or KRAS‐WT signal. Tumors were classified as KRAS‐mutant when the VAF was 1% or higher; otherwise, as KRAS‐WT. 24 When DNA from HPNE cells was analyzed as negative control (one well per run, total n = 21), the mean fractional abundance of KRAS was 0.44% (SD, 0.37%). For cases negative for KRAS codons 12/13, we additionally undertook ddPCR using a multiplex screening kit for five mutations in KRAS codon 61 (Q61K, Q61L, Q61R, Q61H c.183A > T, and Q61H c.183A > C; ddPCR KRAS Q61 Screening Kit; Bio‐Rad Laboratories) in the same analytical pipeline. Given the limited number of cases at each institution, we pooled all cases negative for KRAS codons 12/13 and defined criteria for positive and negative calls for codon 61. KRAS mutations were detected successfully in pancreatic carcinomas with quite low tumor cellularity (Figure S2). Representative microscopic images of pancreatic cancer according to strata of KRAS VAF are shown in Figure S3.
2.4. Statistical analysis
In our primary analyses, we pooled data from the three institutional cohorts. To compare clinical and pathological characteristics between KRAS categories, we used the χ2‐test or Fisher’s exact test, as appropriate, for categorical variables, and Student’s t‐test or ANOVA, as appropriate, for continuous variables. To compare KRAS mutation rates between nonordinal and ordinal subgroups, we used the χ2‐test and the Cochran–Armitage trend test, respectively.
In survival analyses, we examined associations between KRAS mutation status and DFS and OS among patients with pancreatic cancer. Disease‐free survival was defined as time from the index surgery to the first recurrence of the cancer or death, whichever came first. When any of these end‐points was not observed, the patients were censored at the time‐point of the last cross‐sectional imaging study. Overall survival was defined as time from the index surgery to death of any cause, where patients who were alive at the last follow‐up were censored. In analyses of pancreatic cancer‐specific survival, deaths from causes other than pancreatic cancer were censored. Cumulative survival probabilities were estimated using the Kaplan–Meier product‐limit method and were compared using the log–rank test. A linear trend in survival probabilities across ordinal categories of KRAS VAF was assessed using the log–rank test for trend. The Cox proportional hazards regression models stratified by institutional cohort were used to calculate HRs and 95% CIs for DFS and OS by KRAS mutation status. Tests for trend were carried out by entering KRAS VAF as a continuous variable in the Cox regression models and evaluating the Wald test. To adjust for potential confounding factors, the multivariable Cox regression model initially included the following variables: age at the time of surgery (continuous), sex (female vs. male), year of diagnosis (continuous), tumor location (head vs. body/tail of the pancreas), histological type (well/moderately differentiated vs. poorly differentiated vs. adenosquamous), tumor stroma type (nonscirrhous vs. scirrhous), lymphatic invasion (absent/mild vs. moderate/marked), venous invasion (absent/mild vs. moderate/marked), neural invasion (absent/mild vs. moderate/marked), cancer stage (I vs. II vs. III/IV), resection margin status (R0 vs. R1/2), receipt of neoadjuvant chemotherapy (yes vs. no), and receipt of adjuvant chemotherapy (yes vs. no). Backward elimination with a threshold p value of 0.05 was carried out to select variables for the final models. Complete data on the covariates were available for all cases. The assumption of proportional hazards was generally satisfied by assessing a time‐dependent covariate, which was the cross‐product of KRAS mutation status and DFS or OS (p > 0.09). We observed no statistically significant heterogeneity in the survival associations of KRAS mutation status between the institutional cohorts using Cochran’s Q statistic for the random‐effects model (p heterogeneity > 0.05) 25 , 26 and thus, pooled the institutional cohorts for survival analyses. We fitted a restricted cubic spline curve with four knots to examine a possible nonlinear association between KRAS VAF and pancreatic cancer survival. 27 We assessed the nonlinearity using the likelihood ratio test that compared the model with only the linear term to the model with the linear and the cubic spline terms. A statistical interaction was assessed using the Wald test on the cross‐product of KRAS VAF and a variable of interest (stroma status or cellularity of tumor or inflammatory cells) in the Cox regression model. We calculated HRs in strata of stroma status or cellularity of tumor or inflammatory cells based on a single regression model with a reparameterization of the interaction term. 28
All statistical analyses were carried out using SAS software (version 9.4; SAS Institute). To account for multiple comparisons, we used the two‐sided α level of 0.005 for statistical significance according to experts’ recommendations. 29
3. RESULTS
We included 1162 patients with resected pancreatic cancer from the three institutions (Figure 1). Tables 1 and S1 summarize clinical and pathological characteristics of the patients with pancreatic cancer, overall and by institution, respectively. During the median follow‐up time of 56.0 months (interquartile range, 42.8–83.7 months) for all censored patients, 825 patients (71% of the total study population) were deceased. Median quantity of the extracted DNA per case was 5646 ng (interquartile range, 2664–9744 ng).
FIGURE 1.
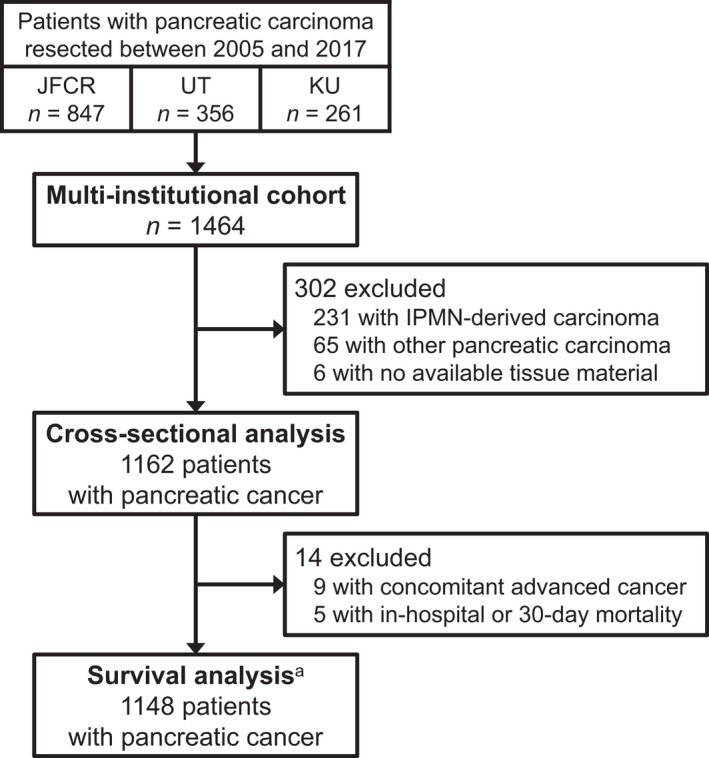
Flow diagram of selection of patients with resected pancreatic cancer in a multi‐institutional cohort. aFor analyses of disease‐free survival, we further excluded 53 patients with a resected metastatic lesion, R2 resection margin, or no available cross‐sectional imaging following the index surgery. IPMN, intraductal papillary mucinous neoplasm; JFCR, Japanese Foundation for Cancer Research; KU, Keio University; UT, The University of Tokyo
TABLE 1.
Clinical and pathological characteristics of pancreatic cancer cases, overall or by KRAS mutation status
| Characteristic a | All cases | KRAS mutation | p value | KRAS‐mutant tumors (n = 1139) | p value | |||
|---|---|---|---|---|---|---|---|---|
| KRAS VAF | ||||||||
| Wild type | Mutant | 1%–9% | 10%–19% | ≥20% | ||||
| (n = 1162) | (n = 23) | (n = 1139) | (n = 276) | (n = 503) | (n = 360) | |||
| Mean age ± SD (years) | 67.2 ± 9.7 | 66.0 ± 13.7 | 67.2 ± 9.6 | 0.55 | 67.1 ± 8.9 | 67.3 ± 9.4 | 67.0 ± 10.4 | 0.91 |
| Sex | 0.52 | 0.41 | ||||||
| Female | 480 (41) | 8 (35) | 472 (41) | 108 (39) | 205 (41) | 159 (44) | ||
| Male | 682 (59) | 15 (65) | 667 (59) | 168 (61) | 298 (59) | 201 (56) | ||
| Year of diagnosis | 0.23 | 0.037 | ||||||
| 2005–2010 | 337 (29) | 3 (13) | 334 (29) | 78 (28) | 160 (32) | 96 (27) | ||
| 2011–2014 | 393 (34) | 9 (39) | 384 (34) | 81 (29) | 162 (32) | 141 (39) | ||
| 2015–2017 | 432 (37) | 11 (48) | 421 (37) | 117 (43) | 181 (36) | 123 (34) | ||
| Tumor location | 0.31 | 0.43 | ||||||
| Head of the pancreas | 741 (64) | 17 (74) | 724 (64) | 184 (67) | 312 (62) | 228 (63) | ||
| Body to tail of the pancreas | 421 (36) | 6 (26) | 415 (36) | 92 (33) | 191 (38) | 132 (37) | ||
| Histological type | 0.001 | <0.001 | ||||||
| Adenocarcinoma | 1132 (97) | 20 (87) | 1112 (98) | 276 (100) | 494 (98) | 342 (95) | ||
| Adenosquamous carcinoma | 30 (2.6) | 3 (13) | 27 (2.4) | 0 | 9 (1.8) | 18 (5.0) | ||
| Tumor differentiation b | 0.21 | 0.20 | ||||||
| Well to moderate | 664 (59) | 9 (45) | 655 (59) | 169 (61) | 298 (60) | 188 (55) | ||
| Poor | 468 (41) | 11 (55) | 457 (41) | 107 (39) | 196 (40) | 154 (45) | ||
| Stroma type | 0.70 | 0.010 | ||||||
| Non‐scirrhous | 764 (66) | 16 (70) | 748 (66) | 166 (60) | 325 (65) | 257 (71) | ||
| Scirrhous | 398 (34) | 7 (30) | 391 (34) | 110 (40) | 178 (35) | 103 (29) | ||
| Lymphatic invasion | 0.58 | 0.013 | ||||||
| Absent/mild | 745 (64) | 16 (70) | 729 (64) | 189 (68) | 331 (66) | 209 (58) | ||
| Moderate/marked | 417 (36) | 7 (30) | 410 (36) | 87 (32) | 172 (34) | 151 (42) | ||
| Venous invasion | 0.38 | <0.001 | ||||||
| Absent/mild | 403 (35) | 6 (26) | 397 (35) | 128 (46) | 175 (35) | 94 (26) | ||
| Moderate/marked | 759 (65) | 17 (74) | 742 (65) | 148 (54) | 328 (65) | 266 (74) | ||
| Neural invasion | 0.20 | <0.001 | ||||||
| Absent/mild | 410 (35) | 11 (48) | 399 (35) | 123 (45) | 162 (32) | 114 (32) | ||
| Moderate/marked | 752 (65) | 12 (52) | 740 (65) | 153 (55) | 341 (68) | 246 (68) | ||
| Mean tumor size ± SD (cm) | 3.4 ± 1.6 | 3.6 ± 1.5 | 3.4 ± 1.6 | 0.64 | 3.2 ± 1.6 | 3.4 ± 1.5 | 3.7 ± 1.6 | <0.001 |
| Tumor cellularity | 0.20 | <0.001 | ||||||
| <30% | 505 (43) | 13 (57) | 492 (43) | 248 (90) | 210 (42) | 34 (9.4) | ||
| ≥30% | 657 (57) | 10 (43) | 647 (57) | 28 (10) | 293 (58) | 326 (91) | ||
| Cellularity of inflammatory cells | 0.41 | <0.001 | ||||||
| <25% | 111 (9.6) | 2 (8.7) | 109 (9.6) | 13 (4.7) | 35 (7.0) | 61 (17) | ||
| 25–49% | 738 (63) | 12 (52) | 726 (64) | 123 (45) | 338 (67) | 265 (74) | ||
| ≥50% | 313 (27) | 9 (39) | 304 (27) | 140 (51) | 130 (26) | 34 (9.4) | ||
| UICC cancer stage | 0.47 | 0.25 | ||||||
| I | 301 (26) | 8 (35) | 293 (26) | 80 (29) | 130 (26) | 83 (23) | ||
| II | 506 (44) | 11 (48) | 495 (43) | 117 (42) | 223 (44) | 155 (43) | ||
| III | 305 (26) | 4 (17) | 301 (26) | 68 (25) | 134 (27) | 99 (28) | ||
| IV | 50 (4.3) | 0 | 50 (4.4) | 11 (4.0) | 16 (3.2) | 23 (6.4) | ||
| Resection margin status | 0.80 | 0.78 | ||||||
| R0 | 899 (77) | 19 (83) | 880 (77) | 209 (76) | 397 (79) | 274 (76) | ||
| R1 | 257 (22) | 4 (17) | 253 (22) | 66 (24) | 103 (20) | 84 (23) | ||
| R2 | 6 (0.5) | 0 | 6 (0.5) | 1 (0.4) | 3 (0.6) | 2 (0.6) | ||
| Neoadjuvant therapy | 0.51 | <0.001 | ||||||
| None | 1013 (87) | 19 (83) | 994 (87) | 219 (79) | 442 (88) | 333 (93) | ||
| Chemotherapy c | 149 (13) | 4 (17) | 145 (13) | 57 (21) | 61 (12) | 27 (7.5) | ||
| Adjuvant therapy | 0.28 | 0.73 | ||||||
| None | 248 (21) | 7 (30) | 241 (21) | 61 (22) | 101 (20) | 79 (22) | ||
| Chemotherapy c | 914 (79) | 16 (70) | 898 (79) | 215 (78) | 402 (80) | 281 (78) | ||
Note: Data are shown as n (%) unless otherwise indicated.
Abbreviation: VAF, variant allele frequency.
Percentage indicates the proportion of cases with a specific clinical or pathological characteristic in all cases or in each stratum of KRAS mutation status. Total percentages may not equal 100% due to rounding.
Tumor differentiation was assessed only for adenocarcinomas.
These categories include chemoradiotherapy.
Utilizing the ddPCR assay, we detected KRAS mutations in 1139 (98%) out of all 1162 patients (KRAS VAF distribution presented in Figure S4). KRAS mutations were detected in codons 12/13 in 1104 cases and in codon 61 in 35 cases out of the 58 cases negative for codons 12/13. KRAS‐WT cases had tumor cellularity of at least 5% (median, 20%; range, 5%–60%), suggesting a low possibility of false negatives in ddPCR due to a limited amount of tumor DNA. KRAS mutations were detected with comparable rates across the institutional cohorts and strata of clinicopathological characteristics that potentially affected the mutation detection (Figure 2). Of note, KRAS mutation rate did not differ by year of diagnosis (corresponding to the age of FFPE blocks). Clinical and pathological characteristics of pancreatic cancer cases by KRAS mutation status are summarized in Table 1. KRAS‐WT tumors were more likely to represent adenosquamous histology. Among KRAS‐mutant tumors, high levels of KRAS VAF were associated with adenosquamous histology, venous and neural invasions, large tumor size, high tumor cellularity, low cellularity of inflammatory cells, and no receipt of neoadjuvant chemotherapy.
FIGURE 2.
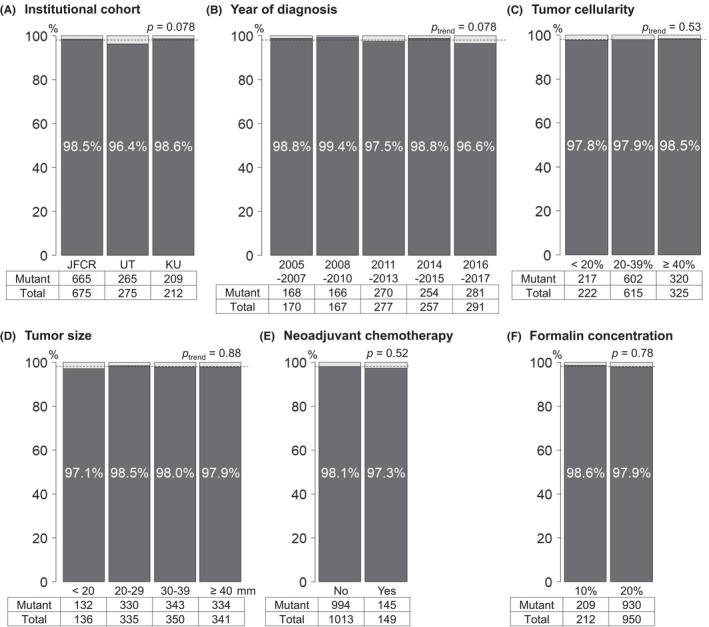
KRAS mutation rates by clinical and pathological parameters in a multi‐institutional cohort of patients with resected pancreatic cancer. (A) Institutional cohort. (B) Year of diagnosis (corresponding to the age of tissue blocks). (C) Tumor cellularity. (D) Tumor size. (E) Receipt of neoadjuvant chemotherapy. (F) Formalin concentration. Dotted lines indicate the overall KRAS mutation rate in the total study group. JFCR, Japanese Foundation for Cancer Research; KU, Keio University; UT, The University of Tokyo
We examined associations of KRAS mutation status with survival outcomes among pancreatic cancer (Tables 2 and S2). KRAS mutations were not associated with DFS (multivariable HR comparing KRAS‐mutant to KRAS‐wild‐type tumors, 1.04; 95% CI, 0.62–1.75; p = 0.87) or OS (multivariable HR, 1.05; 95% CI, 0.60–1.84; p = 0.86). In contrast, KRAS VAF was associated with DFS and OS among patients with KRAS‐mutant pancreatic cancer (p trend <0.001). Compared to patients with KRAS VAF of 1%–9%, patients with VAF of 10%–19% and ≥20% had multivariable HRs for DFS of 1.22 (95% CI, 1.02–1.45) and 1.60 (95% CI, 1.32–1.93), respectively; and multivariable HRs for OS of 1.19 (95% CI, 0.99–1.44) and 1.52 (95% CI, 1.25–1.85), respectively. Among KRAS‐mutant tumors, 20% VAF increase was associated with HRs of 1.27 (95% CI, 1.13–1.42) for DFS and 1.31 (95% CI, 1.16–1.48) for OS. Kaplan–Meier analyses yielded consistent results (Figure 3). For KRAS‐mutant and WT cases, median DFS times were 14.1 (95% CI, 13.1–15.6) and 12.3 (95% CI, 8.1‐NA) months, respectively; median OS times were 31.0 (95% CI, 28.8–33.1) and 34.3 (95% CI, 18.7‐NA) months, respectively. For patients with KRAS VAF of 1%–9%, 10%–19%, and ≥20%, median DFS times were 18.6 (95% CI, 15.1–23.6), 15.2 (95% CI, 13.1–17.2), and 11.5 (95% CI, 10.4–12.8) months, respectively; median OS times were 42.0 (95% CI, 35.8–49.7), 31.7 (95% CI, 27.9–36.6), and 25.5 (95% CI, 21.4–28.8) months, respectively. We fitted a restricted cubic spline curve for KRAS VAF in relation to DFS or OS, which suggested largely linear associations of KRAS VAF with HRs among KRAS‐mutant tumors (Figure S5). In analyses of pancreatic cancer‐specific survival, we observed a similar prognostic association of KRAS mutation status. The higher risk of pancreatic cancer‐specific mortality was noted for higher KRAS VAF (HR per 20% increase, 1.33; 95% CI, 1.17–1.51) among KRAS‐mutant tumors, but not for KRAS mutation positivity (HR, 0.98; 95% CI, 0.56–1.71). Among patients with KRAS‐mutant tumors, higher levels of KRAS VAF were associated with higher likelihood of liver metastasis at the time of the first recurrence (Table 3).
TABLE 2.
KRAS mutation status and survival among patients with pancreatic cancer
| Disease‐free survival | Overall survival | |||||||
|---|---|---|---|---|---|---|---|---|
| No. of cases | No. of events | Univariable HR (95% CI) | Multivariable HR a (95% CI) | No. of cases | No. of events | Univariable HR (95% CI) | Multivariable HR a (95% CI) | |
| KRAS mutation | ||||||||
| Wild type | 23 | 15 | 1.00 (referent) | 1.00 (referent) | 23 | 13 | 1.00 (referent) | 1.00 (referent) |
| Mutant | 1072 | 850 | 1.23 (0.74–2.05) | 1.04 (0.62–1.75) | 1125 | 799 | 1.26 (0.73–2.19) | 1.05 (0.60–1.84) |
| p value | 0.43 | 0.87 | 0.40 | 0.86 | ||||
| KRAS VAF | ||||||||
| 1%–9% | 258 | 189 | 1.00 (referent) | 1.00 (referent) | 269 | 172 | 1.00 (referent) | 1.00 (referent) |
| 10%–19% | 479 | 374 | 1.22 (1.02–1.45) | 1.22 (1.02–1.45) | 498 | 348 | 1.27 (1.05–1.52) | 1.19 (0.99–1.44) |
| ≥20% | 335 | 287 | 1.63 (1.35–1.96) | 1.60 (1.32–1.93) | 358 | 279 | 1.70 (1.40–2.05) | 1.52 (1.25–1.85) |
| p trend b | <0.001 | <0.001 | <0.001 | <0.001 | ||||
Abbreviations: CI, confidence interval; HR, hazard ratio; VAF, variant allele frequency.
The multivariable Cox regression model initially included age, sex, year of diagnosis, tumor location, histological type, stroma type, lymphatic invasion, venous invasion, neural invasion, cancer stage, resection margin status, receipt of neoadjuvant chemotherapy, and receipt of adjuvant chemotherapy. Backward elimination with a threshold p of 0.05 was conducted to select variables for the final models. The variables that remained in the final models are described in Table S2.
p trend was calculated by entering KRAS VAF (continuous) in the Cox regression model.
FIGURE 3.
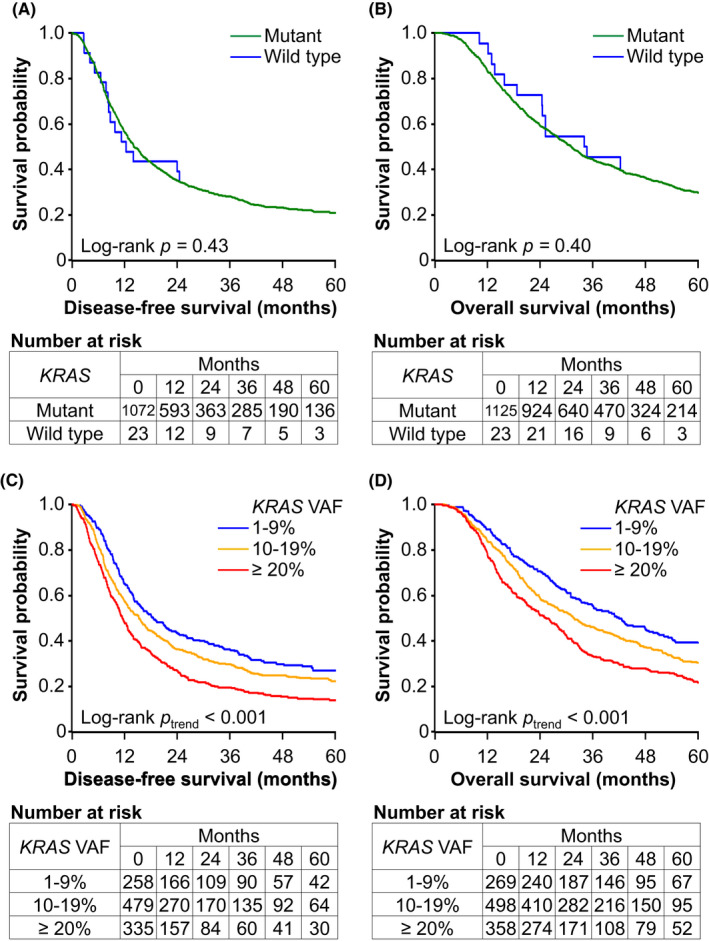
Kaplan–Meier survival curves of patients with pancreatic cancer according to KRAS mutation status. (A) Disease‐free survival by KRAS mutations. (B) Overall survival by KRAS mutations. (C) Disease‐free survival by variant allele frequency (VAF) of KRAS among KRAS‐mutant tumors. (D) Overall survival by KRAS VAF among KRAS‐mutant tumors
TABLE 3.
Recurrence patterns of resected KRAS‐mutant pancreatic cancer by KRAS variant allele frequency (VAF)
| Site of recurrence a | All cases | KRAS VAF | p trend b | ||
|---|---|---|---|---|---|
| 1%–9% | 10%–19% | ≥20% | |||
| (n = 1139) | (n = 276) | (n = 503) | (n = 360) | ||
| Liver | 299 (26) | 45 (16) | 112 (22) | 142 (39) | <0.001 |
| Local | 266 (23) | 70 (25) | 122 (24) | 74 (21) | 0.14 |
| Peritoneum | 160 (14) | 39 (14) | 75 (15) | 46 (13) | 0.58 |
| Lung | 153 (13) | 45 (16) | 63 (13) | 45 (13) | 0.19 |
| Lymph node | 144 (13) | 34 (12) | 58 (12) | 52 (14) | 0.38 |
| Remnant pancreas | 56 (4.9) | 8 (2.9) | 30 (6.0) | 18 (5.0) | 0.28 |
| Others | 36 (3.2) | 10 (3.6) | 15 (3.0) | 11 (3.1) | 0.71 |
Note: Data are shown as n (%).
Sites of recurrence were assessed on the cross‐sectional imaging study delineating the first recurrence. Multiple sites might be assigned for one case.
p trend was calculated by the Cochran–Armitage trend test.
We undertook secondary subgroup analyses to examine factors that potentially affected KRAS VAF and its survival associations. In survival analyses stratified by tumor cellularity, the survival associations of KRAS VAF were attenuated, but a similar trend toward high HRs for DFS and OS associated with high levels of KRAS VAF was observed (Table S3). Tumors with higher levels of KRAS VAF were associated with lower infiltrates of inflammatory cells (Table 1), but the inverse associations of KRAS VAF with survival times were consistently observed across strata of cellularity of inflammatory cells (Table S4). In addition, we did not observe any statistical interaction between KRAS VAF and stromal fibrosis in relation to survival times (Table S5). Given the potential effect of neoadjuvant chemotherapy on tumor molecular features, we undertook a subgroup analysis limited to patients without neoadjuvant chemotherapy, which yielded similar results (Table S6).
4. DISCUSSION
In a large multi‐institutional cohort of consecutive patients with resected pancreatic cancer, we utilized multiplex ddPCR for archival FFPE tumor samples and detected KRAS mutations with high sensitivity. The high analytical sensitivity was not susceptible to clinicopathological factors that might have an impact on the mutation detection. Of note, we successfully detected KRAS mutations in tumor samples preserved for up to 15 years with a comparable detection rate. Our survival analyses have shown that high KRAS mutation load, but not KRAS mutation positivity, is associated with worse survival outcomes of patients with resected pancreatic cancer. Our study supports the utility of ddPCR for genomic characterization in personalized management of patients with pancreatic cancer and the prognostic role of KRAS mutation load.
Pancreatic cancer develops through a stepwise accumulation of genetic and epigenetic alterations, including those for KRAS, CDKN2A (p16), SMAD4, and TP53. 1 , 2 An activating point mutation of the KRAS gene serves as a critical driver of this carcinogenic process, which not only dysregulates various cellular processes, including proliferation and survival, but also impairs antitumor immune response. 11 , 16 , 30 In parallel with the global trend of precision oncology, molecular profiling of tumors is of increasing importance. In the I‐PREDICT trial, targeting of a larger proportion of molecular alterations was associated with better survival outcomes of patients with refractory malignancy. 31 In pancreatic cancer, the PARP1 and PARP2 inhibitor, olaparib, has shown great promise in treating patients with a germline BRCA1 or BRCA2 mutation. 32 KRAS has long been considered undruggable, but tumors harboring KRAS G12C mutation might respond to highly specific molecular‐targeted agents (e.g., sotorasib and adagrasib). 33 , 34 Despite the relatively low frequency of KRAS G12C mutation reported in pancreatic cancer (<6%), 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 further research on tumor characteristics according to specific patterns of KRAS mutations is warranted. Taken together, there is an increasing need for rapid and sensitive screening of specific mutations in clinical samples of pancreatic cancer.
To overcome the hurdle to clinical sequencing of pancreatic cancer characterized by desmoplastic cellular stroma and low tumor cellularity, we utilized multiplex ddPCR for archival FFPE samples and successfully detected KRAS mutations in up to 98% of cases. Compared to prior studies, summarized in Table 4, 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 the use of ddPCR provided more sensitive detection of KRAS mutations in pancreatic cancer. Studies reporting the KRAS mutation rate of more than 90% were all based on next‐generation sequencing, 44 , 45 , 46 , 47 , 49 which required high levels of DNA quality and quantity during the quality control process. 47 , 50 , 51 In contrast, the current study included consecutive patients with resected pancreatic cancer as long as FFPE blocks of the primary pancreatic tumors were available and successfully detected KRAS mutations with high sensitivity irrespective of age of the tissue blocks, tumor cellularity, and other clinical and pathological factors affecting the mutation detection. Given that ddPCR can be readily applied for other genetic loci, our data support the potential of ddPCR in genetically characterizing various tumors in the current oncology practice. During our ddPCR procedures, we used 100 ng DNA per case to ensure the sensitivity of mutation calling and the robustness of measurements of KRAS VAF. However, given the reported usefulness of ddPCR in biospecimens containing low amounts of tumor DNA, 13 , 14 , 15 a reduced amount of DNA may be effective for ddPCR for KRAS mutations.
TABLE 4.
Summary of studies investigating KRAS mutations in clinical tissue samples of pancreatic cancer
| Sequencing | Specimen | No. (%) of patients | Ref. a | ||||
|---|---|---|---|---|---|---|---|
| Assay | Target KRAS codons | Preservation | Tumor extension | Examined lesion | KRAS mutant | Total | |
| Sanger | 12/13/61 | FFPE | All | Primary or metastatic | 71 (52) | 136 | 35 |
| Sanger | 12/13 | FFPE | Resectable | Primary | 105 (68) | 153 | 36 |
| Sanger | 12/13/61 | FFPE | Resectable | Primary | 136 (80) | 170 | 37 |
| Sanger | 12/13/61 | FFPE or fresh tissue | Locally advanced + metastatic | Primary | 214 (88) | 242 | 38 |
| Sanger | 12/13 | FFPE | All | Primary | 92 (79) | 117 | 39 |
| Pyrosequencing | 12/13 | FFPE | Resectable | Primary | 109 (87) | 126 | 48 |
| Pyrosequencing | 12/13 | FFPE | Locally advanced + metastatic | Primary or metastatic | 121 (70) | 173 | 40 |
| TaqMan allelic discrimination | 12/13 | Fresh frozen | Locally advanced + metastatic | Primary | 147 (67) | 219 | 41 |
| PCR (SSCP) | 12/13/61 | Fresh frozen | Resectable | Primary | 134 (78) | 171 | 42 |
| PCR (RFLP) | 12/13 | FFPE | Resectable | Primary | 126 (54) | 234 | 43 |
| NGS | All | Fresh frozen | Resectable | Primary | 96 (96) | 100 | 44 |
| NGS | All | Fresh frozen | Resectable | Primary | 100 (92) | 109 | 45 |
| NGS | All | FFPE | Resectable | Primary | 262 (93) | 283 | 49 |
| NGS | All | FFPE | Resectable | Primary | 328 (92) | 356 | 46 |
| NGS | All | Fresh frozen | Resectable | Primary | 420 (92) | 456 | 47 |
| ddPCR | 12/13/61 | FFPE | Resectable | Primary | 1139 (98) | 1162 | Current study |
Abbreviations: ddPCR, droplet digital PCR; FFPE, formalin‐fixed paraffin‐embedded; NGS, next‐generation sequencing; Ref., reference; RFLP, restriction fragment length polymorphism; SSCD, single‐strand conformation polymorphism.
We included studies examining KRAS mutations for ≥100 patients.
In the current study, the absolute quantification of variant alleles based on ddPCR allowed us to demonstrate the inverse association of KRAS VAF with postoperative survival times of patients with pancreatic cancer. Our data support the linear increase in the mortality hazard according to the VAF increase among KRAS‐mutant tumors. This result is consistent with the mechanistic evidence indicating the contribution of an increased dosage of mutant KRAS gene to rapid progression and metastasis of pancreatic neoplasms. 17 , 18 It should be noted that the VAF is a multifactorial index reflecting intratumoral KRAS mutation load and tumor cellularity; therefore, the low mortality hazard associated with low levels of KRAS VAF might be attributable at least in part to intense lymphocytic infiltrates resulting in low tumor purity. 52 , 53 , 54 However, the inverse associations of KRAS VAF with survival times were similarly observed across strata of cellularity levels of tumor and inflammatory cells as well as strata of stromal fibrosis status. In turn, our data suggest that tumors with high KRAS VAF might be more likely to represent the adenosquamous phenotype, 55 , 56 which has been associated with unfavorable survival outcomes of pancreatic cancer. 57 , 58 In addition, our data indicate that tumors with higher levels of KRAS VAF might be more likely to metastasize to the liver, suggesting the high metastatic potential associated with high KRAS mutation load. 49 However, we found no statistically significant association of KRAS positivity with survival outcomes, which was somehow inconsistent with prior reports. 11 The null association might be due to differences in study cohorts, chance findings, or unmeasured confounding factors. Nonetheless, there is a possibility that KRAS VAF‐low tumors, which were associated with long survival, were misclassified as KRAS‐WT tumors due to the less sensitive assays used in the prior studies, potentially overestimating survival times of patients with KRAS‐WT tumors. A large validation study is warranted to examine characteristics of pancreatic cancer according to KRAS mutation positivity utilizing sensitive assays such as ddPCR. In aggregate, our data highlight the importance of considering VAF in addition to mutation positivity when molecularly characterizing pancreatic cancer.
The current study has notable strengths, including the large sample size derived from three independent institutional cohorts. The large sample size allowed us to carry out various subgroup analyses and support the applicability of highly sensitive ddPCR for mutation detection in archival FFPE tissue samples of pancreatic cancer. Of note, we did not exclude cases with resected pancreatic cancer in terms of tumor characteristics but included the cases as long as FFPE blocks of the tumors were available. The multicenter study design was another strength. We did not observe statistically significant heterogeneity between the institutional cohorts in our main findings, supporting the generalizability of our data.
We acknowledge limitations of our study. The use of multiplex panels inhibited examinations of specific patterns of KRAS mutations occurring in codons 12/13 or codon 61. Different mutations in a single gene could represent distinct biological effects on tumor development and progression, and thereby have different clinical implications. 59 , 60 For survival analyses, there might be confounding factors that were unaccounted for; nonetheless, we adjusted for a variety of clinical and pathological characteristics, and the adjustment did not alter the results substantially.
In conclusion, the current study supports the feasibility of ddPCR for assessment of KRAS mutations in clinical tumor samples of pancreatic cancer. Given its high sensitivity and cost‐effectiveness, ddPCR can serve as a first‐line analytical assay for mutation detection using FFPE tumor samples in the era of precision oncology. Utilizing high‐quality data on KRAS mutations in unselected populations of patients with resected pancreatic cancer, our study has shown that high levels of KRAS mutation load could contribute to aggressive tumor behavior and, thereby, have a prognostic value beyond KRAS mutation positivity, independent of clinical and pathological characteristics.
DISCLOSURE
K.K., K.Tak., and M.S. are Associate Editors of Cancer Science. The other authors have no conflict of interest.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI grants (JP21K15368 to T.S., JP20K07414 to Y.M., JP19K08362 to T.H., and JP21K15393 to M.Tak.), the Practical Research for Innovative Cancer Control Program from AMED (JP21ck0106557 to Y. Nakai), and by grants from Takeda Science Foundation (to T.H.) and Daiwa Securities Health Foundation (to T.T.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We would like to thank the following collaborators for their valuable support in tissue processing and/or data collection: Satoko Baba and Motoyoshi Iwakoshi, Department of Pathology, The Cancer Institute Hospital of Japanese Foundation for Cancer Research, Tokyo, Japan; Kikuko Kaji, Department of Hepato‐Biliary‐Pancreatic Medicine, The Cancer Institute Hospital of Japanese Foundation for Cancer Research, Tokyo, Japan; Sachiyo Nagumo and Kei Sakuma, Department of Pathology, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan; Noriko Koga, Hepato‐Biliary‐Pancreatic Surgery Division, Department of Surgery, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan; Kensuke Hara, Department of Pathology, Keio University School of Medicine, Tokyo, Japan; and the staff of the Fourth Laboratory of Department of Pathology in Keio University School of Medicine, Tokyo, Japan.
Suzuki T, Masugi Y, Inoue Y, et al. KRAS variant allele frequency, but not mutation positivity, associates with survival of patients with pancreatic cancer. Cancer Sci. 2022;113:3097‐3109. doi: 10.1111/cas.15398
Tatsunori Suzuki, Yohei Masugi, and Yosuke Inoue contributed equally as co‐first authors.
Kiyoshi Hasegawa, Minoru Kitago, Yu Takahashi, and Mitsuhiro Fujishiro contributed equally as co‐last authors.
Use of standardized official symbols: We use HUGO (Human Genome Organization)‐approved official symbols for genes and gene products, including BRCA1, BRCA2, CDKN2A, EGFR, KRAS, PARP1, PARP2, SMAD4, and TP53, all of which are described at www.genenames.org. Gene names are italicized, and gene product names are non‐italicized.
Funding information
Daiwa Securities Health Foundation; Japan Society for the Promotion of Science, Grant/Award Number: JP19K08362, JP20K07414, JP21K15368, JP21K15393; Japan Agency for Medical Research and Development, Grant/Award Number: JP21ck0106557; Takeda Science Foundation.
REFERENCES
- 1. Hayashi A, Hong J, Iacobuzio‐Donahue CA. The pancreatic cancer genome revisited. Nat Rev Gastroenterol Hepatol. 2021;18:469‐481. [DOI] [PubMed] [Google Scholar]
- 2. Cancer Genome Atlas Research Network . Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32:185‐203.e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB. Colorectal cancer. Lancet. 2019;394:1467‐1480. [DOI] [PubMed] [Google Scholar]
- 4. Pishvaian MJ, Blais EM, Brody JR, et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the know your tumor registry trial. Lancet Oncol. 2020;21:508‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ogino S, Nowak JA, Hamada T, et al. Integrative analysis of exogenous, endogenous, tumour and immune factors for precision medicine. Gut. 2018;67:1168‐1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ogawa Y, Masugi Y, Abe T, et al. Three distinct stroma types in human pancreatic cancer identified by image analysis of fibroblast subpopulations and collagen. Clin Cancer Res. 2021;27:107‐119. [DOI] [PubMed] [Google Scholar]
- 7. Hosein AN, Brekken RA, Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol. 2020;17:487‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Puleo F, Nicolle R, Blum Y, et al. Stratification of pancreatic ductal adenocarcinomas based on tumor and microenvironment features. Gastroenterology. 2018;155:1999‐2013.e1993. [DOI] [PubMed] [Google Scholar]
- 9. Do H, Dobrovic A. Sequence artifacts in DNA from formalin‐fixed tissues: causes and strategies for minimization. Clin Chem. 2015;61:64‐71. [DOI] [PubMed] [Google Scholar]
- 10. Greytak SR, Engel KB, Bass BP, Moore HM. Accuracy of molecular data generated with FFPE biospecimens: lessons from the literature. Cancer Res. 2015;75:1541‐1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2020;17:153‐168. [DOI] [PubMed] [Google Scholar]
- 12. Olmedillas‐López S, García‐Arranz M, García‐Olmo D. Current and emerging applications of droplet digital PCR in oncology. Mol Diagn Ther. 2017;21:493‐510. [DOI] [PubMed] [Google Scholar]
- 13. Okada T, Mizukami Y, Ono Y, et al. Digital PCR‐based plasma cell‐free DNA mutation analysis for early‐stage pancreatic tumor diagnosis and surveillance. J Gastroenterol. 2020;55:1183‐1193. [DOI] [PubMed] [Google Scholar]
- 14. Holm M, Andersson E, Osterlund E, et al. Detection of KRAS mutations in liquid biopsies from metastatic colorectal cancer patients using droplet digital PCR, Idylla, and next generation sequencing. PLoS One. 2020;15:e0239819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takai E, Totoki Y, Nakamura H, et al. Clinical utility of circulating tumor DNA for molecular assessment in pancreatic cancer. Sci Rep. 2015;5:18425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moore AR, Rosenberg SC, McCormick F, Malek S. RAS‐targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. 2020;19:533‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mueller S, Engleitner T, Maresch R, et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature. 2018;554:62‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chan‐Seng‐Yue M, Kim JC, Wilson GW, et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet. 2020;52:231‐240. [DOI] [PubMed] [Google Scholar]
- 19. Japan Pancreas Society . Classification of Pancreatic Carcinoma, 4th English edition. Kanehara; 2017. [Google Scholar]
- 20. Brierley JD, Gospodarowicz MK, Wittekind C. TNM Classification of Malignant Tumours. 8th ed. Wiley‐Blackwell; 2017. [Google Scholar]
- 21. Klöppel G, Lingenthal G, von Bülow M, Kern HF. Histological and fine structural features of pancreatic ductal adenocarcinomas in relation to growth and prognosis: studies in xenografted tumours and clinico‐histopathological correlation in a series of 75 cases. Histopathology. 1985;9:841‐856. [DOI] [PubMed] [Google Scholar]
- 22. Do H, Dobrovic A. Dramatic reduction of sequence artefacts from DNA isolated from formalin‐fixed cancer biopsies by treatment with uracil‐ DNA glycosylase. Oncotarget. 2012;3:546‐558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chiu A, Ayub M, Dive C, Brady G, Miller CJ. Twoddpcr: an R/Bioconductor package and shiny app for droplet digital PCR analysis. Bioinformatics. 2017;33:2743‐2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alcaide M, Cheung M, Bushell K, et al. A novel multiplex droplet digital PCR assay to identify and quantify KRAS mutations in clinical specimens. J Mol Diagn. 2019;21:214‐227. [DOI] [PubMed] [Google Scholar]
- 25. Cochran WG. The combination of estimates from different experiments. Biometrics. 1954;10:101‐129. [Google Scholar]
- 26. DerSimonian R, Laird N. Meta‐analysis in clinical trials. Control Clin Trials. 1986;7:177‐188. [DOI] [PubMed] [Google Scholar]
- 27. Durrleman S, Simon R. Flexible regression models with cubic splines. Stat Med. 1989;8:551‐561. [DOI] [PubMed] [Google Scholar]
- 28. Hamada T, Cao Y, Qian ZR, et al. Aspirin use and colorectal cancer survival according to tumor CD274 (programmed cell death 1 ligand 1) expression status. J Clin Oncol. 2017;35:1836‐1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Benjamin DJ, Berger JO, Johannesson M, et al. Redefine statistical significance. Nat Hum Behav. 2018;2:6‐10. [DOI] [PubMed] [Google Scholar]
- 30. Ischenko I, D'Amico S, Rao M, et al. KRAS drives immune evasion in a genetic model of pancreatic cancer. Nat Commun. 2021;12:1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sicklick JK, Kato S, Okamura R, et al. Molecular profiling of cancer patients enables personalized combination therapy: the I‐PREDICT study. Nat Med. 2019;25:744‐750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Golan T, Hammel P, Reni M, et al. Maintenance Olaparib for germline BRCA‐mutated metastatic pancreatic cancer. N Engl J Med. 2019;381:317‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roskoski R. Blockade of mutant RAS oncogenic signaling with a special emphasis on KRAS. Pharmacol Res. 2021;105806:105806. [DOI] [PubMed] [Google Scholar]
- 34. Hong DS, Fakih MG, Strickler JH, et al. KRAS(G12C) inhibition with Sotorasib in advanced solid tumors. N Engl J Med. 2020;383:1207‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim ST, Lim DH, Jang KT, et al. Impact of KRAS mutations on clinical outcomes in pancreatic cancer patients treated with first‐line gemcitabine‐based chemotherapy. Mol Cancer Ther. 2011;10:1993‐1999. [DOI] [PubMed] [Google Scholar]
- 36. Sinn BV, Striefler JK, Rudl MA, et al. KRAS mutations in codon 12 or 13 are associated with worse prognosis in pancreatic ductal adenocarcinoma. Pancreas. 2014;43:578‐583. [DOI] [PubMed] [Google Scholar]
- 37. Schultz NA, Roslind A, Christensen IJ, et al. Frequencies and prognostic role of KRAS and BRAF mutations in patients with localized pancreatic and ampullary adenocarcinomas. Pancreas. 2012;41:759‐766. [DOI] [PubMed] [Google Scholar]
- 38. Ogura T, Yamao K, Hara K, et al. Prognostic value of K‐ras mutation status and subtypes in endoscopic ultrasound‐guided fine‐needle aspiration specimens from patients with unresectable pancreatic cancer. J Gastroenterol. 2013;48:640‐646. [DOI] [PubMed] [Google Scholar]
- 39. da Cunha SG, Dhani N, Tu D, et al. Molecular predictors of outcome in a phase 3 study of gemcitabine and erlotinib therapy in patients with advanced pancreatic cancer: National Cancer Institute of Canada Clinical Trials Group Study PA3. Cancer. 2010;116:5599‐5607. [DOI] [PubMed] [Google Scholar]
- 40. Boeck S, Jung A, Laubender RP, et al. KRAS mutation status is not predictive for objective response to anti‐EGFR treatment with erlotinib in patients with advanced pancreatic cancer. J Gastroenterol. 2013;48:544‐548. [DOI] [PubMed] [Google Scholar]
- 41. Bournet B, Muscari F, Buscail C, et al. KRAS G12D mutation subtype is a prognostic factor for advanced pancreatic adenocarcinoma. Clin Transl Gastroenterol. 2016;7:e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rachakonda PS, Bauer AS, Xie H, et al. Somatic mutations in exocrine pancreatic tumors: association with patient survival. PLoS One. 2013;8:e60870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shin SH, Kim SC, Hong SM, et al. Genetic alterations of K‐ras, p53, c‐erbB‐2, and DPC4 in pancreatic ductal adenocarcinoma and their correlation with patient survival. Pancreas. 2013;42:216‐222. [DOI] [PubMed] [Google Scholar]
- 44. Hayashi H, Kohno T, Ueno H, et al. Utility of assessing the number of mutated KRAS, CDKN2A, TP53, and SMAD4 genes using a targeted deep sequencing assay as a prognostic biomarker for pancreatic cancer. Pancreas. 2017;46:335‐340. [DOI] [PubMed] [Google Scholar]
- 45. Witkiewicz AK, McMillan EA, Balaji U, et al. Whole‐exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Qian ZR, Rubinson DA, Nowak JA, et al. Association of Alterations in Main driver genes with outcomes of patients with resected pancreatic ductal adenocarcinoma. JAMA Oncol. 2018;4:e173420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47‐52. [DOI] [PubMed] [Google Scholar]
- 48. Zhou L, Baba Y, Kitano Y, et al. KRAS, BRAF, and PIK3CA mutations, and patient prognosis in 126 pancreatic cancers: pyrosequencing technology and literature review. Med Oncol. 2016;33:32. [DOI] [PubMed] [Google Scholar]
- 49. McIntyre CA, Lawrence SA, Richards AL, et al. Alterations in driver genes are predictive of survival in patients with resected pancreatic ductal adenocarcinoma. Cancer. 2020;126:3939‐3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen H, Luthra R, Goswami RS, Singh RR, Roy‐Chowdhuri S. Analysis of pre‐analytic factors affecting the success of clinical next‐generation sequencing of solid organ malignancies. Cancers (Basel). 2015;7:1699‐1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liudahl SM, Betts CB, Sivagnanam S, et al. Leukocyte heterogeneity in pancreatic ductal adenocarcinoma: phenotypic and spatial features associated with clinical outcome. Cancer Discov. 2021;11:2014‐2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Leinwand J, Miller G. Regulation and modulation of antitumor immunity in pancreatic cancer. Nat Immunol. 2020;21:1152‐1159. [DOI] [PubMed] [Google Scholar]
- 54. Masugi Y, Abe T, Ueno A, et al. Characterization of spatial distribution of tumor‐infiltrating CD8(+) T cells refines their prognostic utility for pancreatic cancer survival. Mod Pathol. 2019;32:1495‐1507. [DOI] [PubMed] [Google Scholar]
- 55. Lenkiewicz E, Malasi S, Hogenson TL, et al. Genomic and epigenomic landscaping defines new therapeutic targets for Adenosquamous carcinoma of the pancreas. Cancer Res. 2020;80:4324‐4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hayashi A, Fan J, Chen R, et al. A unifying paradigm for transcriptional heterogeneity and squamous features in pancreatic ductal adenocarcinoma. Nature Cancer. 2020;1:59‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Boecker J, Feyerabend B, Tiemann K, et al. Adenosquamous carcinoma of the pancreas comprise a heterogeneous group of tumors with the worst outcome: a clinicopathological analysis of 25 cases identified in 562 pancreatic carcinomas resected with curative intent. Pancreas. 2020;49:683‐691. [DOI] [PubMed] [Google Scholar]
- 58. Hester CA, Augustine MM, Choti MA, et al. Comparative outcomes of adenosquamous carcinoma of the pancreas: an analysis of the national cancer database. J Surg Oncol. 2018;118:21‐30. [DOI] [PubMed] [Google Scholar]
- 59. Zafra MP, Parsons MJ, Kim J, et al. An in vivo Kras allelic series reveals distinct phenotypes of common oncogenic variants. Cancer Discov. 2020;10:1654‐1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hobbs GA, Baker NM, Miermont AM, et al. Atypical KRAS(G12R) mutant is impaired in PI3K signaling and macropinocytosis in pancreatic cancer. Cancer Discov. 2020;10:104‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1