

Abstract
Despite a growing number of successful therapies, heart failure remains the most common cause of death and disability worldwide. Thus, new and novel therapeutic strategies are urgently needed. Mitochondria of cardiomyocytes generate ATP that is needed to power cardiac contraction. Mitochondrial-derived ATP activate myosin ATPase at the sarcomere and the sarcoplasmic reticular (SR) ATPase Ca2+ pump, both which intersect with Ca2+ during contraction. Failure to maintain the relationship between mitochondria and SR can lead to cardiomyocyte dysfunction and heart failure. Here, we discuss recent discoveries that reveal Ca2+ transport via the voltage dependent anion channel (VDAC) into the mitochondria can favorably impact cardiac contraction and prevent cardiac arrhythmias. In a broader view, discussion of the opening of a new era for HF therapeutics that will address the sarcomere, SR and mitochondria as a functional unit.
Keywords: Voltage dependent anion channels, Calcium, Contractility, Mitochondria, Heart failure, Excitation Contraction Coupling, Mitochondrial Ca2+ uptake transporter
Graphical Abstract
1. Introduction
With each heartbeat, blood is ejected from the left ventricle and delivered to the body. The amount of blood ejected changes based on the metabolic demands of the heart. As such, intensity of cardiac contraction, or contractility, must be regulated over a range of workloads such as during sleep or intense exercise and requires adjustments in oxygen consumption [1] [2]. A balance of the demand for myocardial oxygen consumption is met by the energy produced (i.e. ATP) by mitochondrial metabolism in order to ensure adequate contractility[3]. When oxygen consumption exceeds supply, contractility is impaired and cardiac function declines[4]. In addition to this energy balance for ATP, contractility also requires the transient flux of cytosolic Ca2+ on a beat-to-beat basis in order to activate key Ca2+ dependent steps of actin-myosin crossbridge shortening[5]. Indeed, the height of the Ca2+ transient and ATP availability are key determinants of the force of contraction of individual cardiomyocytes which involves communication between the sarcomere, sarcoplasmic reticulum (SR) and mitochondria[6, 7].
Ca2+ transients in cardiomyocytes are triggered by the opening of the L-type Ca2+ channels in the T-tubule membrane and then the emptying of the ryanodine receptor (RYR2) Ca2+ stores by Ca2+ induced Ca2+ release[8, 9]. Dispersal of Ca2+ throughout the cytosol enables Ca2+ to interact with troponin C to relieve inhibition of the actomyosin complex and shorten the cardiomyocyte in a mechanism involving ATP hydrolysis by myosin. ATP hydrolysis is also required for Ca2+ re-sequestration in the sarcoplasmic reticulum (SR) by the Ca2+ ATPase, SERCA2a. Thus, ATP and Ca2+ must both be available to ensure proper systolic and diastolic function of the heart[10]. The ATP levels must remain constant in the cardiomyocyte and require matching of the ATP production and degradation. In this case, ATP must be regenerated and ADP and Pi removed from the sarcomere[11]. Much like Ca2+, sufficient supply of ATP must be available to the myosin-ATPase to power contraction[12]. Importantly, ADP, the reaction product of ATP hydrolysis, must be removed from the sarcomere or cardiomyocytes will fail to relax resulting in damage[11, 13]. On the other hand, cardiomyocytes rapidly move Ca2+ into and out of the cytosol in order to prevent cell damage from Ca2+overload[14]. Often this involves sequestration of Ca2+ in the SR or mitochondria and also involves extrusion of Ca2+ across the T-tubule membrane. Failure to remove Ca2+ from the cytosol during diastole prevents the relaxation of the cardiomyocyte and limits contractility. It is clear that cardiomyocytes must regulate the production and storage of both ATP and Ca2+ in order to maintain function and this involves collaboration between mitochondria and the SR.
The heart must function continuously to ensure survival and operates with limited energy reserve. In fact, reserves are sufficient to provide ATP for one minute of cardiac contractions until cardiac standstill ensues. To meet this challenge, an efficient metabolic system has evolved in the heart to produce and consume only the exact amount of ATP required to maintain homeostasis. Too little ATP resulting from energy depletion of the heart would limit contractility and trigger arrhythmias resulting in heart failure. Too great a demand for ATP can result in excessive redox damage that impairs cardiomyocytes and even triggers apoptosis inducing heart failure. ATP concentration remains constant over a range of workloads. To achieve balance, cardiomyocytes utilize different metabolism pathways. The phosphocreatine shuttle is an energy transfer system in cardiomyocytes that provides communication about ATP-ADP-Pi status between the cytosol and mitochondria whereby ATP is regenerated and delivered to the ATPases at the sarcomere (myosin) and the SR (SERCA2a). Muscle specific isoforms of creatine kinase enzymes localize to the sarcomere or the SR whereas mitochondrial isoforms can be found in the mitochondrial free space. ATP/ADP are then channeled from the cytosolic locations into the mitochondria where it is phosphorylated to ATP [15, 16]. In this way the phosphocreatine shuttle connects sites of consumption with ATP re-supply sites in the mitochondria. In addition, ADP is rapidly removed from the sarcomere and SR in order limit product-inhibition of the myosin-ATPase or SERCA2a-ATPase. The phosphocreatine transfer system ensures the rapid flux of energy equivalents to accommodate excitation contraction coupling (ECC).
Mitochondria adjust oxidative metabolism in order to maintain bioenergetics and ATP homeostasis for cardiac contraction[17]. Key metabolic enzymes sense the changes in mitochondrial Ca2+ that accompany Ca2+ transients and alter activity for those dehydrogenases involved in the TCA cycle[18, 19]. Fuel substrates derived from many carbon sources including oxidation of glucose, ketones, fatty acids and amino acids are available to the cardiomyocytes. During heart failure, substrate metabolism is limited and metabolic inflexibility develops when failing cardiomyocytes utilize glucose metabolism whereas healthy cardiomyocytes prefer fatty acid oxidation [20, 21]. Ca2+ binds to the solute carrier family members (SLC2512 and SLC25A13) via EF-binding domains that reside in the intermembranous space[22]. Ca2+ bound to SLC25A12 activates the transport of cytosolic glutamate across the IMM in exchange for mitochondrial aspartate. These Ca2+-dependent transporters are part of the cytosolic-mitochondrial aspartate/malate shuttle and supply reducing equivalents that are available to the electron transport chain (ETC) in a Ca2+ dependent manner[23]. Matrix Ca2+ can bind and activate the pyruvate dehydrogenase complex (PDH), the NAD-linked isocitrate dehydrogenase (ICDH) and the 2-alpha ketoglutaraldyde (KDH). Pyruvate is the end product of glycolysis and is converted to acetyl-CoA by the pyruvate dehydrogenase enzyme complex[24]. Importantly, the PDH-phosphatase (PTP1a) requires Ca2+ binding, and an increase in [Ca2+]M can accelerate acetyl-coA production which provides two carbons to the enzymes of the TCA-cycle located in the inner mitochondrial membrane (IMM)[25]. Ca2+ may also regulate the ATP-synthase via post-translational mechanism or through other indirect mechanism. What is clear from these studies is that mitochondrial Ca2+ influences energy production at several levels in the mitochondria and acts in a feed forward mechanism enabling energy demand to match efficiently with ATP supply[26].
2. Mitochondrial Calcium Signaling
Mitochondria occupy 30–40% of the cell volume of cardiomyocytes, have critical roles in the generation of ATP, protect cells from redox damage, and confer metabolic flexibility by which the cell ensures energetic adaptability[27]. How mitochondria coordinate these cellular functions is likely to require changes to the level of mitochondrial Ca2+ levels. Ca2+ signaling has emerged over the last several decades as a critical factor in cell homeostasis where it can interact with Ca2+ sensitive enzymes localized in the mitochondrial IMM and matrix in order to modulate oxidative metabolism. As such, mitochondria require an elaborate signaling system to control mitochondrial Ca2+ levels involving its uptake, efflux and buffering. Mitochondrial Ca2+ levels range from 100 nM in resting cells to 1μM following cellular stimulation and even levels around 1M under pathologic conditions[28]. Ca2+ influx to mitochondria can also influence cell Ca2+ signaling that protects the cardiomyocyte and provides optimal energy production. Mitochondria can functionally influence [Ca2+]c as ion channel modulators, as firewalls to separate vulnerable regions of the cell from very high [Ca2+]c levels, and in the formation of nanotunnels to promote intermitochondrial connections (Figure 1)[29–31]. Common to each of these interactions is the close proximity of the mitochondria to the cytosolic Ca2+ source that highlights the importance of the relationship of mitochondria with the architecture of the cardiomyocyte. For example, does the spatial relationship between the mitochondria and the source of cytosolic Ca2+ flux matter? The mitochondria have a low affinity mechanism for Ca2+ uptake and therefore require very high Ca2+ levels to operate these transport mechanisms. Often this requires mitochondria to have close contact with RYR2 or the inositol trisphosphate receptor (IP3R). In comparison to global Ca2+ signals, these local Ca2+ release events can deliver intense amounts of Ca2+ and rapidly fill mitochondria. In contrast, mitochondria in close apposition to the Ca2+ pumps (SERCA2a) are not exposed to very high levels of Ca2+ as Ca2+ is diverted into the SR. It is clear these spatial and temporal parameters can lead to heterogenous Ca2+ signals among different mitochondria.
Figure 1:
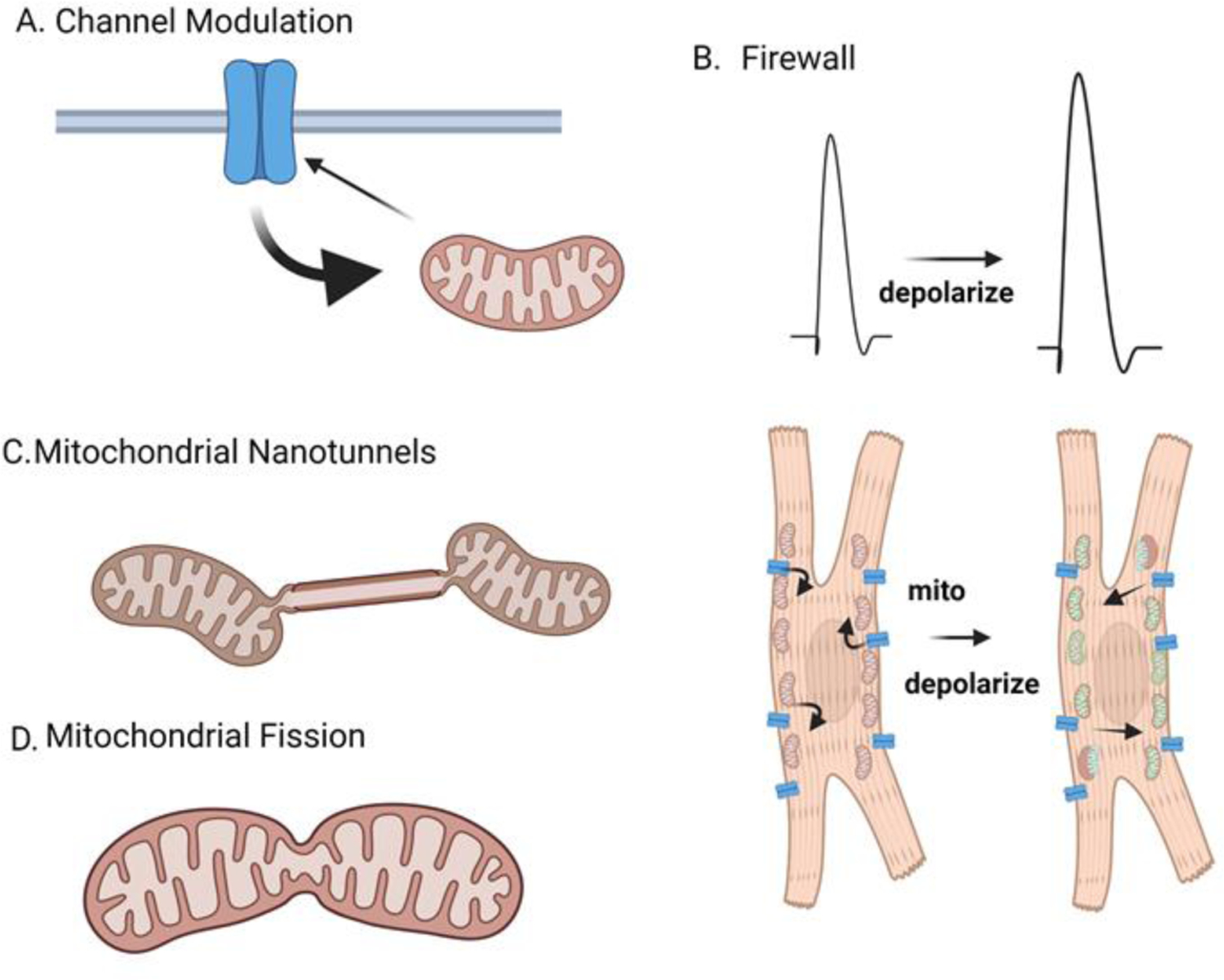
Mitochondria can influence Ca2+ transients of cardiomyocytes. The cartoon depicts different mechanisms by which mitochondria respond to altered cytosolic Ca2+levels. A) Mitochondria take up Ca2+ from store operated Ca2+ channels and prevent Ca2+ dependent inactivation of the channel. B) Mitochondria can serve as a firewall to prevent global increases in Ca2+. In atrial cardiomyocytes, Ca2+ entry across the sarcolemma (via cav1.2) is loaded into mitochondria that are present underneath the surface membrane. The amplitude of the Ca2+ transients becomes larger when cells are pretreated with agent that depolarize the mitochondrial membrane. C) Nanotunnels are specialized membrane extensions that connect two mitochondria. This phenomenon develops in cardiomyocytes with leaky Ca2+ release channels (RYR2). D) Elevated Ca2+ levels in the cytosol can trigger morphologic change to mitochondria called fission.
Like the rest of the cell, individual mitochondria require a separate repertoire of ion channels, transporters and exchangers that direct Ca2+uptake into the matrix (Figure 2). Ca2+ collects in two spaces of the mitochondria that include the intermembranous space and the interior matrix. Ca2+ must therefore pass through two separate membranes to reach the mitochondrial matrix where it is heavily buffered by phosphate. First, cytosolic Ca2+ crosses the outer mitochondrial membrane (OMM) via the voltage dependent anion channel (VDAC) into the intramitochondrial space[32]. Ca2+ then is transported across the inner mitochondrial membrane (IMM) into the mitochondrial matrix via the mitochondrial Ca2+ uptake (MCU) transporter. A second mechanism to alter mitochondrial Ca2+ levels is the mitochondrial permeability transition pore (mPTP). Under unusual cellular conditions that involve matrix Ca2+ overload (e.g. ischemia), the ATP synthase can act in reverse to hydrolyze ATP and form a mega-Ca2+ channel that shares features with the mPTP. The mPTP can then uncouple oxidative phosphorylation when the IMM potential dissipates, resulting in increased permeability to Ca2+. Ca2+ extrusion mechanisms for removal of Ca2+ across the IMM and mitochondria matrix include the sodium Ca2+ exchanger (NCLx) and H+/Ca2+ antiporter assumed to be the Leucine Zipper and EF handing containing transmembrane protein 1 (LETM1)[33–35]. Mitochondrial Ca2+ levels are set by a number of factors including the actions of these transporters, mitochondria size and fusion-fission capacity, membrane potential and through interactions with other organelles including ER, lysosomes, surface membrane and nucleus[29, 36, 37]. Mitochondrial Ca2+ signals differ not only between cell types but also vary among different mitochondria within the same cell[38, 39]. In atrial cardiomyocytes, mitochondria sequester Ca2+ released during excitation contraction coupling (ECC). As shown by the Bootman lab, cytosolic Ca2+ signals originate from the periphery of atrial cell, unlike ventricular cardiomyocytes, and do not spread toward the interior of the cell. Rather mitochondria sequester Ca2+ released from the RYR2 stores. In fact, inhibiting mitochondrial Ca2+ signaling led to globalized Ca2+ transients in atrial cells following pacing[30]. These results highlight the importance of mitochondria in the shaping and control of cytosolic Ca2+ signals. Given the clinical importance of atrial fibrillation worldwide, targeting mitochondrial Ca2+ signaling in atrial cell might represent an important novel strategy.
Figure 2:
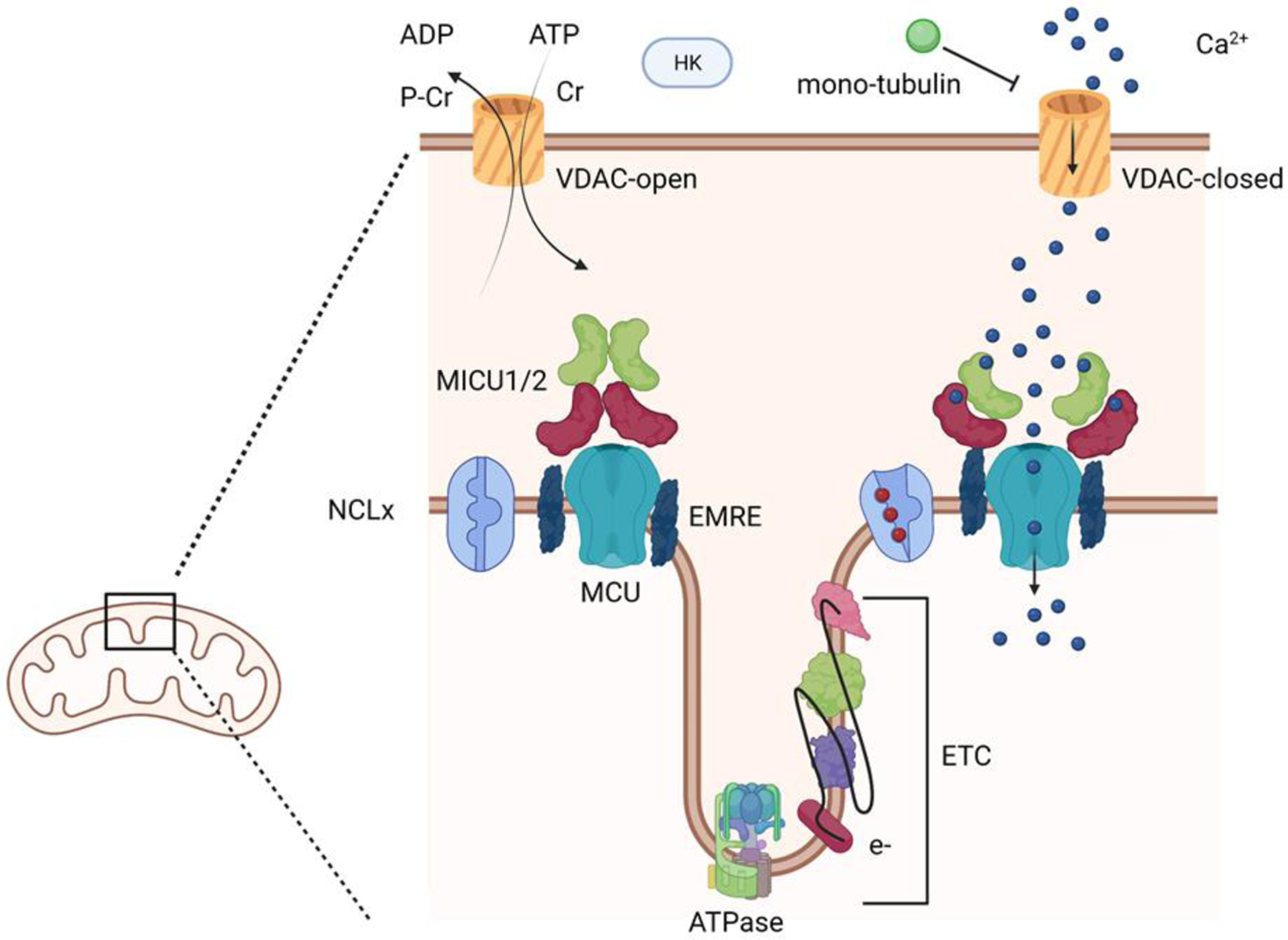
Mitochondrial Ca2+ signaling toolkit. The schematic demonstrates the outer and inner mitochondrial membranes. VDAC is present in the OMM where it exists in open and closed conductance states. Ca2+ flows through the closed VDAC channels. At the IMM, mitochondrial uptake transporter (MCU) exits in a complex with MICU1/2 and EMRE. MICU1/2 act as Ca2+ sensor in the mitochondrial outer space. EF hands bind Ca2+ and stimulate conformation changes that activate MCU-dependent Ca2+ transport.
Mitochondria are dispersed throughout the cardiomyocyte at various locations including the perinuclear region (PNM), subsarcolemmal domains (SLM) and intramyofibrillar (IFM) regions[40]. These populations of mitochondria exhibit differences in Ca2+ handing, redox balance, motility and morphology. These differences may reflect the spatial relationship of these mitochondria to its neighboring source of ATP consumption. The clearest example of this is that IFM, compared to PNM, have more robust Ca2+ handling, are more resistant to redox challenge and are static without much migration. The IFM mitochondria are closest to the sarcomere and have the greatest ATP demands. This has raised the hypothesis that subpopulations of mitochondria must adapt to their immediate environment. Notably, IFM reside in the rigid architecture of the myofibrils and are subject to rhythmic contractions. The restricted environment of the sarcomeric lattice and the mechanical signals ensure IFM mitochondria do not migrate along microtubules and do not undergo extensive changes in shape. In contrast, PNM are more mobile and seems to have different functional roles through interactions with the nucleus and lysosomes. One explanation for the difference in Ca2+ handling for these different mitochondria is that Ca2+ handling proteins are located in specific domains of the mitochondria themselves and therefore create a heterogynous mitochondrial Ca2+ response. For example, it has also been shown that differential localization of mitochondrial Ca2+ uptake transporter (MCU) near the junctional SR ensures exposure to high Ca2+ levels to activate MCU whereas NCLx is expressed away from the junctional SR[41]. The mechanism underlying these different populations is presently unknown but may involve interaction with different Ca2+ signals (e.g. RYR2 or IP3R), connections with the cytoskeleton (desmin, tubulin or a-synuclein) or other factors[42, 43]. Most studies of mitochondrial function have been performed over the years using isolated mitochondrial preparations where spatial considerations are lost as are connections to the cytoskeleton. Given that different phenotypes have been assigned to these distinct populations, it will be important to understand how these regions of mitochondria behave in isolated mitochondrial preparation.
Sub-populations of mitochondria differ in mitochondrial Ca2+ uptake capacity, buffering capacity and Ca2+ release. In particular, IFM take up the greatest amount of Ca2+ compared to the SLM and PNM. Lu et al used the genetically encoded Ca2+ indicator (mitycam) to measure [Ca2+]M in isolated and intact cardiomyocytes[40]. Detailed measurements of Ca2+ enabled comparisons of the different fractions that led to the conclusion that mitochondria located at the Z-line exhibited markedly elevated amplitude of [Ca2+]M that rise more rapidly than Ca2+ uptake for mitochondria located at the M-line. These different populations of mitochondria may account for the spatial and temporal Ca2+ signals in cardiomyocytes. Cardiomyocytes must adapt their energetic need to events at the sarcomere and the membrane and local control of mitochondrial Ca2+ signals can confer adaptability. Similarly, mitochondria are subject to fission-fusion events and migrate via microtubules throughout the cell. Another key parameter for mitochondrial Ca2+ uptake is the source of the cytosolic Ca2+ flux. Global changes in Ca2+ transients offer much less Ca2+ than microdomains of the mitochondria that involve close proximity of OMM to ER membranes or lysosomal membranes[40, 44]. How mitochondrial Ca2+ contributes to these events is increasingly been recognized and will likely lead to a greater understanding of the physiologic role of mitochondria Ca2+ signaling confers on the cardiomyocyte.
The interdependence of mitochondrial and cytosolic Ca2+ signaling has been recognized with increasing frequency and recent studies have established the pathogenic implications as well as potential therapeutic possibilities[11, 42, 45]. Disordered Ca2+ release from the SR of cardiomyocytes is a well-established pathogenic mechanism that contributes to cardiomyocyte death for patients subjected to acute cardiac injury such as ischemia or in patients with genetic mutations in key Ca2+ handling proteins. Leaky RYR2 channels, faulty SERCA2a pumping and inefficient buffering by calsequestrin are all associated with arrhythmia and depressed contractility[46–48]. Here inhomogeneous Ca2+ gradients, spontaneous sparks, elevated diastolic Ca2+ levels can give rise to dysfunctional Ca2+ transients[49]. For example, mutations in the RYR2 Ca2+ release channel cause catecholaminergic polymorphic ventricular tachycardia (CPVT), a rare genetic cause of sudden cardiac death. Dysregulated Ca2+ release can be sequestered into mitochondria at high levels.
Mitochondrial Ca2+ overload compromises energetic efficiency by impairing oxidative phosphorylation resulting in reduced contractility, greater production of reactive oxygen species (ROS) and even trigger cardiomyocyte apoptosis. The increased ROS produced by faulty RYR2 Ca2+ release oxidize the RYR2 channel and amplify greater Ca2+ leak. Cardiomyocytes from mice bearing the RYR2 mutant exhibited Ca2+ signaling and mitochondrial defects, including the presence of nanotunnels that develop between adjacent mitochondria[29]. These nanotunnels facilitate communication between mitochondria and protect myocardial energy reserves. Excessive Ca2+ leak causes impaired ATP production by mechanism that involve inhibition of complex I or TCA cycle enzymes pyruvate dehydrogenase (PDH) and α-ketoglurate dehydrogenase (αKGDH) and the adenine nucleotide translocase (ATN). The emerging view of mitochondria Ca2+ signaling is that key relationships between the mitochondria and surrounding organelles modulate ATP production to promote optimal energetic efficiency.
3. Cardiac VDAC channels
The outer mitochondrial membrane (OMM) is a key barrier for mitochondria and provides insulation from events occurring in the cytosol. As a consequence, OMM permeability which is mediated by the VDACs is the primary means of communication between mitochondria and cytosol. VDAC1–3 comprise a family of channels that assemble into tetramers with a beta-barrel structure[50, 51]. Functionally, VDAC porins open and close in order to exchange nucleotides, metabolites and ions across the OMM, but recent work has revisited the role of VDACs in mitochondrial Ca2+ handling[52, 53]. The gating mechanism for VDACs, as established from lipid bilayer studies, show they exist in high and low conductance closed states. In the high conductance state, VDACs are permeable to metabolites and nucleotides and less permeable to Ca2+. In contrast, the closed state of VDAC are less permeable to metabolites and yet exhibit much greater permeability to Ca2+[32]. Importantly, voltage changes across the bilayer govern the transitions of VDAC from the high conductance states at neutral voltages to the closed states at more negative membrane potentials. Because the OMM potential does not ordinarily vary in resting cells, it has been difficult to extrapolate the relevance of the required membrane change to gating properties of VDACs. Recent evidence has accrued to indicate VDAC gating is influenced by lipid content of the bilayer, post translation modification of VDAC (ubiquitination, phosphorylation, and acetylation), Ca2+ binding or even interaction with protein partners (e.g. mono-tubulin, Bcl-2 or other factors)[54–57]. These different post-translational modifications to VDACs support the idea that VDACs are subject to regulation by different signaling pathways that are located on both sides of the OMM. It is therefore likely that VDACs must interpret signals emanating from different cellular context. Consequently, VDACs have now emerged as a key therapeutic target for a range of illnesses from cardiac arrhythmias to cancers. Notably, VDACs can impact Ca2+ signaling, oxidative metabolism and generation of reactive oxygen species[32]. The different VDAC isoforms are highly homologous and are expressed ubiquitously in most tissues and cells types, with the exception of VDAC3 channels which are most often found in the testis. While VDAC1 and VDAC3 appear to have overlapping function based on mitochondrial studies from MEFs lacking either or both isoforms, it appears VDAC2 has non-overlapping functions since VDAC2 KO mice are embryonically lethal[58]. These studies also show that VDAC2 has a key role in BCl-2 apoptotic signaling. It remains unknown whether cardiogenesis was impacted by the deletion of VDAC2 as has been described for adenine nucleotide translocator (ANT) KO mice[59, 60].
The role of VDAC2 in cardiac rhythmicity emerged from a landmark study by Shimizu et al[61]. These investigators used the zebrafish mutant called temblor as a model of cardiac arrythmia. Tremblor cardiomyocytes lack Na/Ca2+ exchanger (NCX1) and display dysfunctional Ca2+ transients. A suppressor screen for compounds that could restore proper cardiac rhythmicity identified Efsevin as a hit. Unexpectantly, biochemical studies showed that the drug bound to VDAC2 and promoted Ca2+ uptake into the mitochondria. Additionally, VDAC2 overexpression in the tremblor cardiomyocytes was also able to restore cardiac rhythmicity. These studies establish the idea that VDAC2 mediated Ca2+ uptake could correct abnormal Ca2+ events occurring in the cytosol. While mitochondrial Ca2+ microdomains have been described for VDACs and RYR2 or IP3R, no such associations have been demonstrated for NCX1. Enhancing mitochondria Ca2+ uptake can rescue cardiomyocytes lacking NCX1 and implies that NCX1 may interact with mitochondria in a microdomain which is separate from that described for ECC. Spontaneous Ca2+ release events in embryonic cardiomyocytes trigger NCX1 and triggers contraction as part of the Ca2+ clock mechanism in order to create regenerative Ca2+ transients rather than ECC that focuses mostly on the cav1.2-RYR2 mechanism. In the case of Ca2+ clock, NCX1 depolarizes cells to the threshold potential for cav1.2 that can trigger an action potential and contraction. It has also been proposed that Ca2+ clock mechanism re-emerging in the failing heart to cause arrhythmias and is likely the mechanism underlying the Tremblor mutant. The notion that increasing Ca2+ uptake into the mitochondria under conditions of faulty NCX1 function and elevated diastolic Ca2+ implies that mitochondria may function as a popoff valve for elevated diastolic Ca2+ levels. However, it is equally likely that greater VDAC2-Ca2+ uptake leads to greater ATP production that can fuel SERCA2 pumping and myosin-ATPase activity to match demand. Other possibilities include the altered association of VDAC2 with CPT1a on the OMM that would alter acetyl-carnitine and fatty acid oxidation. Similarly, VDAC2 has an important role in the removal of ADP from the contractile apparatus. Increased sarcomeric ADP levels is known to inhibit the myosin-ATPase activity as is described in failing cardiomyocytes. Thus, agents that might increase VDAC2 permeability can increase ADP mitochondrial transport and thereby promote myofibril relaxation and contractility.
How mitochondria interact with surrounding organelles, such as the S/ER, lysosomes and plasma membrane, provides insight into how mitochondria can shape cytosolic Ca2+ signaling as well as cellular processes such as apoptosis and ROS generation[62]. Several different protein partners exist for different VDACs and these interactions can regulate OMM permeability and therefore influence mitochondrial Ca2+ levels, energy transfer and apoptosis. VDAC partners with IP3R and RYR2 located in the S/ER membrane to form Ca2+ microdomains. Here, these channels govern local Ca2+ release, a fraction of which can be transferred to the mitochondria. Increased matrix Ca2+ can bind PTP1a and activate pyruvate dehydrogenase. VDACs have also been shown to interact with enzymes linked to glycolytic metabolism. Hexokinases (HK) are part of the glycolytic cascade and localize to the OMM. HKI and HKII can phosphorylate glucose to form G-6P which commits glucose to mitochondrial oxidation. HKI/II both interact with VDAC1 and can reduce VDAC conductance[63]. VDAC has been shown to interact with the OMM cholesterol transport protein TSPO[64]. TSPO can inhibit VDAC mediated Ca2+ transport and has been implicated in mitophagy[65]. Along these same lines, other studies established that carnitine palmitoyltransferase (CPT1a) is an OMM protein that mediates the transfer an acyl groups from long chain fatty acid to carnitine. Acylcarnitine can that transport to the matrix for fatty acid oxidation. Reports that CPT1a interacts with VDAC are intriguing and suggest a cross talk between fatty acid oxidation and OMM permeability[66]. It is clear from these studies that VDAC can interact with several metabolic pathways in a bidirectional manner to link events in the cytosol with mitochondrial function whether involving fuel metabolism, ROS signaling, ATP production or apoptosis.
VDACs have also been shown to interact with cytoskeletal elements that have important functional impact on VDAC permeability[67]. Tubulin polymerizes to assemble microtubules that are used by mitochondria to migrate throughout the cell. Tubulins can also influence fusion and fission events that are important to mitochondrial quality control[55, 68, 69]. Importantly, monomeric tubulin can interact with VDAC where it can block the channel function by plugging the pore. Another cytoskeletal element desmin was identified as a VDAC partner where it may regulate Ca2+ transfer to the mitochondrial matrix[70]. Desmin is expressed in all muscle types and contributes to the structural support of these contractile cells. DES mutant mice develop dilated cardiomyopathy and heart failure that models human disease. Interestingly, dysmorphic mitochondria and defects in respiration are a part of the phenotype of DES mutant mice that can be rescued by expressing the heat shock protein α-crystalin beta (CRYAB). Alpha-synnuclein (αSyn) is a Parkinson’s disease gene that is a cytoskeletal element that can also interact with VDAC and mediate Ca2+ transport[71, 72]. Collectively, these studies demonstrate that VDAC interacts with key cytosolic proteins that can regulate OMM permeability and therefore have impact of Ca2+ signaling, ATP production, ROS production and apoptosis.
4. VDAC2 and Dilated Cardiomyopathy
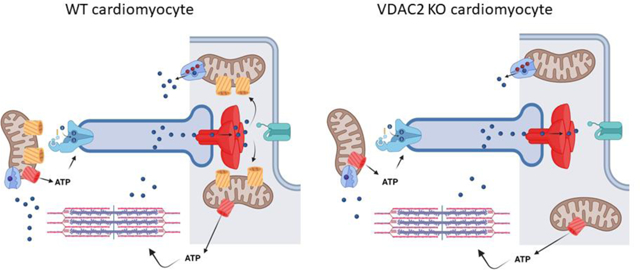
Given the recent evidence that VDAC2 has a key role in cardiac rhythmicity, focus on the function of this VDAC in the heart has been intensely pursued. Germline VDAC2 deletion from mice has hampered studies on the role of VDAC2 in the adult heart because of the embryonically lethality that occurred at early stages such as embryonic day 10. Whether this embryonic lethality results from defects cardiac formation or absence of rhythmically beating cells was not addressed in these early studies. MEFs derived from the VDAC2 KO mice exhibited increased sensitivity to apoptosis and Bcl-2 signaling that involved greater release of cytochrome C. Informed by these early studies, Shankar et al. generated a cardiac restricted VDAC2 KO mouse mice[73]. These mice develop features consistent with a dilated cardiomyopathy (DCM) including reduced ejection fraction, increased cardiac fibrosis, and elevations of the serum biomarker brain natriuretic peptide (BNP). These mice exhibit reduced survival that is often seen in patients with DCM. Mechanistic studies show that cardiomyocytes from VDAC2 KO mice exhibit severely reduced mitochondrial Ca2+ transfer. The functional consequence of this reduced Ca2+ transfer is complex and impacts cytosolic events including contractility. Is matrix Ca2+ reduced in the VDAC2 KO mice? Is there an impairment in PDH activity or other TCA cycle enzymes that are Ca2+ dependent? Answers to these questions may explain why VDAC KO mice have a clear cardiac phenotype when compared with the MCU KO mice[74, 75]. While deletion of MCU subunits like EMRE and MICU1/2 result in decompensated cardiac function MCU-Ca2+ uptake is not necessary for adequate contractility[76–78].
Shankar and colleagues found that VDAC2-mediated Ca2+ uptake regulates cytosolic Ca2+ signaling including ECC and the diastolic oscillations of the Ca2+ clock. Cardiomyocytes from VDAC2 KO mice show Ca2+ transients with depressed amplitudes and prolonged tau compared to WT. Extensive electrical remodeling occurs in the VDAC2 KO cardiomyocytes that change properties of the action potential (AP) including a significantly prolonged AP50 duration. These results are rather surprising given prior reports showing that reduced mitochondria Ca2+ uptake actually had the opposite effect. Cardiomyocytes (HL-1 cells) treated with VDAC2 silencing lentiviruses revealed no change in cytosolic Ca2+ transients in response to action potentials or store depletion with caffeine, but the cardiomyocytes with VDAC2 depletion revealed increased the intensity, width and duration of Ca2+ sparks. Similar approaches to alter MCU-mediated Ca2+ uptake in cardiomyocytes increased contractility and reduced RYR2 leak[79]. It is likely that these differences reflect bidirectional Ca2+ signaling between the SR and mitochondria. Shankar and colleagues actually hypothesize that deletion of VDAC2 altered the connection between OMM and the SR membrane and therefore reduced a critical Ca2+ microdomain. Specifically, VDAC2 was shown to co-localize and co-precipitate with NCX1 and SERCA2a. VDAC is known to interact with RYR1 but the notion that VDAC localize together with SERCA2 and NCX1 implies a different mechanism for VDAC2. NCX1 is localized in the t-tubule membrane whereas SERCA2 lines the longitudinal SR which is quite a distance from the terminal cisternae and dyads. Certainly, these findings seem to resolve a controversy in the role mitochondria play in failing cardiomyocytes. Studies for years have shown differences in the connections between mitochondria and the SR in the failing cardiomyocyte. Reduced mitochondria Ca2+ transfer was thought to impose energetic starvation on the cardiomyocyte. In contrast, others have suggested that loss of the mito-SR connections are detrimental. In fact, actions to augment mito-Ca2+, such as overexpressing MCU, can mitigate the contractile defects imposed in heart failure[79]. Thus, many questions can be raised from these studies: is there a hierarchy for the VDAC2 interaction and these different microdomains? A greater understanding of how these different microdomains are established by VDAC2 can help understand how VDAC2 and Efsevin affect SR Ca2+ content and action potentials of cardiomyocytes. Mitochondria may enhance Ca2+ uptake under conditions of elevated diastolic Ca2+ and thereby function as a popoff valve for elevated diastolic Ca2+ levels. Here the interaction of VDAC2 and SERCA2 could mediate such a connection. However, it is equally likely that reduced VDAC2-Ca2+ uptake leads to impaired ATP production that must fuel SERCA2 pumping and myosin-ATPase activity. Similarly, VDAC2 has an important role in the removal of ADP from the contractile apparatus. Deletion of VDAC2 would prevent ADP transfer to the mitochondria and would inhibit the myosin-ATPase activity leading to impaired cardiac relaxation. An interesting observation made in this work is that VDAC2 depletion from the heart did not alter mitochondrial ROS production or respiration. This is rather unexpected but is line with the notion that VDAC2 specifically regulates mitochondrial Ca2+, whereas cardiac VDAC1 has been shown to increase ROS production and promote apoptosis[32]. It is well established that SR-Ca2+ release is delivered to the outer mitochondrial membranes where Ca2+ can then concentrate in the mitochondrial matrix in order to facilitate metabolism and energetics. In contrast, VDAC2 deletion from cardiomyocyte did alter profiles of gene expression for enzymes involved in glucose metabolism.
5. Mitochondria as a therapeutic target for heart failure
Heart failure is a disease syndrome with very high mortality resulting from progressive pump failure and sudden cardiac death from arrhythmias. Available treatments have focused on modifying actions of neurohormones such β-adrenergic receptor antagonists and inhibitors of renin-angiotensin-II cascade that are activated deleteriously in the HF patients or by use of implanted defibrillators for treat life threatening arrhythmias. Targeting mitochondrial metabolism in the failing heart has long been pursued as therapeutic strategy but has never proven to be clinically effective. Most often approaches are designed to enhanced mitochondrial biogenesis, enhance electron transport chain induced ATP generation, target substrate utilization and/or prevent oxidative damage. These approaches invariably alter the bioenergetic balance in the mitochondria without regard for the SR. As a result, cardiomyocytes with increased mitochondrial mass and greater ATP generation will invariably have greater myocardial oxygen consumption which contributes to greater mortality in HF. Review of the clinic trails.gov found 42 trials linked to heart failure and mitochondria. Table 1 identifies many of these drugs and nutrients being tested. Given the growing number of small molecules that appear to modify mitochondrial Ca2+, like the gliflozins and Efsevin, it is likely that a new era for treating HF has arrived and will seek to target the mitochondria to enhance cardiac performance. Iron replacement therapy in HF patients has emerged as an important target. While the O2 carrying capacity of hemoglobin is definitely important in HF patient with anemia, the expectation is that iron replacement also enhances ETC as cytochromes require iron. Along this same line, protocols are testing nutrients to improve mitochondrial function in HF. For example, beetroot juice and epicatechin (dark chocolate) are being tested as enhancers of mitochondrial function in HF. MitoQ is an antioxidant that consists of ubiquinol attached to a lipophilic cation. MitoQ has been tested in a small group of aging subjects with vascular disease and shown to improve aortic stiffness and as well as cholesterol metabolism. MitoQ is being tested in humans with HF to see in the reduced oxidative stress and improve mitochondria function and heart failure (Table 1).
Table 1:
Clinical Trials registered in ClinicTrials.gov targeting mitochondria function in patient with heart failure.
| Intervention | Target | Citations |
|---|---|---|
| Mito Q | Mitochondrial Antioxidant | [95] |
| Epicatechin | ETC | [96] |
| Ferric Carboxymaltose | ETC- Cytochome-Fe binding | [97] |
| 14N Sodium Nitrate | NO-CGAMP signaling | [98] |
| Nicotinamide ribose | SIRT/NAD+ signaling | [99, 100] |
| 3-hydroxybutyrate | Ketone oxidation | [101, 102] |
| Clonidine | a-adrenergic blocker | [103] |
| Empagliflozin | Sodium Glucose Transporter 2 inhibitor | [104] |
| Beet Root Juice | Mitochondrial efficiency; NO availability | [105] |
| KNO3 + L-carnitine | Enhances oxidative phos | [106] |
| Dapagliflozin | Sodium Glucose Transporter 2 inhibitor | [107, 108] |
| Sodium Valproate | FAO | [109, 110] |
6. VDAC activators
Efsevin was recently identified as potential antiarrhythmic agent as it suppressed ventricular dysrhythmias in a Zebrafish deficient in NCX1[61, 80]. Efsevin is a dihydropyrrole carboxylic ester compound that has a short half-life. Lipid bilayer experiments with VDAC2 show that Efsevin directly binds VDAC2 to reduce open probability and shifts to a closed state. This shift leads to greater cation permeability and exclusion of anion and metabolites. Recent studies have demonstrated that Efsevin binding site on VDAC2 occurs between the inner channel wall and the N-terminal helix associated with pore which is important for the Ca2+ transfer from the SR into the mitochondria[81]. Efsevin is unstable with a short half-life which is limiting its clinical applications but recent studies have shown that Efsevin can reduce arrhythmia in cardiomyocytes from CPVT mice.
While it is established that Efsevin can restore rhythmic Ca2+ oscillation in cardiomyocytes, it is unclear how it can alter contractility[61, 80]. To address this issue the Shankar et al. utilized an ex vivo assay for force production from cardiac slices. WT and VDAC2 KO mice were subjected to pressure overload by transaortic constriction (TAC) as a model of heart failure. Efsevin was superfused over cardiac slices and contractile force was measured. Acute drug delivery in this model of cardiac hypertrophy demonstrated that Efsevin augmented contractility. These preliminary studies broaden the mechanism by which Efsevin works but will await future chronic and in vivo studies to provide insight to the preclinical application of this drug. It will be important to test a more bioavailable analogue of Efsevin in other models of heart failure. Additional agents that activate VDAC2 that were also identified in a suppressor screen that include ezetimibe and disulfiram, both of which are FDA approved drugs[82]. Ezetimide limits cholesterol absorption from the gastrointestinal track but at present no connection to mitochondria has been established. Disulfiram is an inhibitor of the mitochondrial enzyme aldehyde dehydrogenase 2 (ALDH2) that is known to detoxify reactive aldehydes from cardiomyocytes. These agents require urgent testing in arrhythmia in vivo models and, if successful, transition to human trials.
3.3. SGLT2 inhibitors.
In the last several years, new medications that inhibit the sodium-glucose transporter type II (SGLT2) (e.g. empagliflozin, canaglifozin and dapagliflozin) were found to significantly reduce HF mortality and hospitalization and improve quality of life[83–85]. Interestingly, while these medications act by preventing reabsorption of glucose in the kidney, their benefits seem mostly related to improvement in heart failure outcomes, independent of the effect on blood glucose. How these agents confer cardioprotection has led to some speculation that these agents may influence mitochondrial function and thereby improve cardiac function[86]. Specifically, by altering total glucose load in the body and a reduction in the insulin/glucagon ratio create conditions that favor a shift in metabolism towards ketogenesis. Ketone production may well be protective in the heart as it is the ideal fuel source in heart failure[87]. As a result, mitochondrial efficiency improves and promotes greater contractility and less arrhythmias. Some gliflozins might also inhibit SGLT1 that is highly enriched in the heart. Molecular docking studies have indicated that Empagliflozin binds glut-4 as well as SGLT1 and NHE1[88]. In this model, limiting glucose uptake in cardiomyocytes activates AMPK and inhibits mTor signaling which would be cardioprotective. The beneficial effects of empagliflozin may be related to LV remodeling that is favorable and protective. Finally, studies have directly addressed the role of empagliflozin on mitochondrial Ca2+ content[89, 90]. Studies show that altered Na in the mitochondria reduced NCLX activity leading to higher mitochondrial Ca2+ content. As a class the gliflozins offer novel ways to impact mitochondrial function and Ca2+ signaling in cardiomyocytes and offer new therapies to treat patients with heart failure.
7. MCU activation
Ca2+ crosses the IMM through the actions of the MCU complex that include MICU1, MICU2, ERME. Matrix Ca2+ content plays a key role in oxidative phosphorylation, ROS production and cell death pathways. Loss of function studies in mice for MCU have been challenging as MCU KO mice in certain genetic background have no particular phenotype, whereas in the C57/B6 background this mutation is embryonically lethal. Moreover, loss of function and gain of function studies with MCU and its components of the complex demonstrate a relative role for MCU in different cardiac conditions. Enhancing Ca2+ transfer to the matrix may well be an approach to enhance cardiomyocyte survival during heart, ischemia and even arrhythmias[79]. Thus, the therapeutic targeting of the MCU complex is logical. High throughput screens for small molecules that act as directly on the MCU complex have been characterized[91, 92]. Using a clever approach, Arduino et al reconstituted human MCU complex in yeast mitochondria. They exploited the D-lactate shunt in yeast to bypass ETC and TCA cycling on the Ca2+ transfer. They identified the chemotherapeutic agent mitoanxtrone as an MCU inhibitor. Interestingly, MTX is known to cause cardiotoxicity in patients[93]. Another agent, Kaempferol, is a plant flavonoid with anti-oxidant properties. Kaempferol can directly activate MCU and enhance the Ca2+ transfer from the S/ER into the mitochondria. In cardiomyocyte, MCU complex activators might enhance mitochondrial Ca2+ uptake and offer favorable energetic conditions that can be cardioprotective.
8. Elamipretide for mitoprotection
Elamipretide (SS-31) is an aromatic cationic tetrapeptide that was identified as a modulator of damaged mitochondria. Specifically, SS-31 concentrates in the mitochondrial inner membrane where it interacts with cardiolipin to stabilize components of the cytochrome system as well as MCUs. In this way, Elamipretide could target damaged mitochondria to enhance bioenergetic efficiency while leaving healthy mitochondria intact. Elamipretide is currently being tested in a phase III trial for patients with the inherited mitochondrial disorder Barth’s syndrome. Many preclinical studies using animal models of heart failure show that SS-31 can repair mitochondrial dysfunction that can improve contractility. Elamipretide has been tested as a single dose infusion in patient with heart failure with reduced ejection fraction (HFrEF). This early phase trial established the safety of the single infusion of this peptide in HF patients. Mechanistic insight to the actions of Elamipretide in the human heart was obtained from studies using heart explanted at the time of cardiac transplantation. Here heart from patient with heart failure and control hearts (not used for transplant) represented the non-failing control were studied using a cardiac splice approach. Importantly Elamipretide did not alter mitochondrial respiration of the non-failing heart slices. In contrast, Elamipretide improved mitochondrial respiration that was supported by specific substrates including pyruvate and malate or glutamate whereas succinate respiration was not different in Elamipretide treated slices. Indeed, oxygen flux improved in HF-heart-section treated with Elamipretide that involved supercomplexes of the electron transport chain. Based on the success of these preclinical and safety studies, a phase II study with Elamipretide was designed to test a longer treatment course (i.e. 28 days) in a similar population of HFrEF patients who were already on guideline directed medical therapy. Results confirmed the safety of the drug but did not identify improvement in the primary end point of LV function as assessed by cardiac magnetic resonance imaging (cMRI). Elamipretide did not improve cardiac volumes or ejection fraction in these patients. There was a trend for improved quality of life assessments in these patients but this was not significant. Based on this study, despite many positive preclinical studies, the use of Elamipreptide in heart failure cannot be supported for the treatment HFrEF. It should be noted that treatment for 28 days might not be sufficient time to remodel mitochondria in cardiomyocytes and subsequently improve LV function, the primary end point of the trial. Repair of the mitochondria would then need to be translated to reverse remodeling of the heart which might be expected to take much longer as it does for other therapies including β-blockers and biventricular pacing. Thus, discarding Elamipreptide at this point might be premature. Finally, it is possible that Elamipreptide induced improvement in mitochondrial respiration and fitness might reduce arrhythmias, as occurs with Efsevin, but was not assessed in any trial to date.
9. Conclusion
Despite tremendous progress over the last several decades, treatments for heart failure have offered only modest benefit for reducing mortality and deferring hospitalization in patients with compromised cardiac function. Optimism has been raised by novel strategies designed to improve mitochondrial fitness in the failing cardiomyocyte[94]. Previous drug design strategies for mitochondria have aimed to improve oxidative metabolism, stimulate mitochondrial biogenesis and ATP availability which would alter the energy balance and potentially improve contractility. However, several obstacles led to failure for these approaches and include unwanted ROS generation, increased oxygen consumption and damage to healthy mitochondria. Recent evidence establishes VDAC in the OMM of cardiomyocyte mitochondria as an important target. In fact, unbiased screens, discussed here, have identified drugs that target opening of VDAC2 in the improvement of cardiac function and automaticity. Promoting the flux of Ca2+ from the cytosol (and SR) into the mitochondria seems to active metabolic enzymes needed to power ATP production. Depletion of VDAC2 in mice clearly reproduced many features of dilated cardiomyocyte and targeting VDAC2 activation reduces arrhythmias. This strategy will likely open new lines of thinking involving the connection between mitochondria and the energy deficits in the failing heart.
Figure 3:
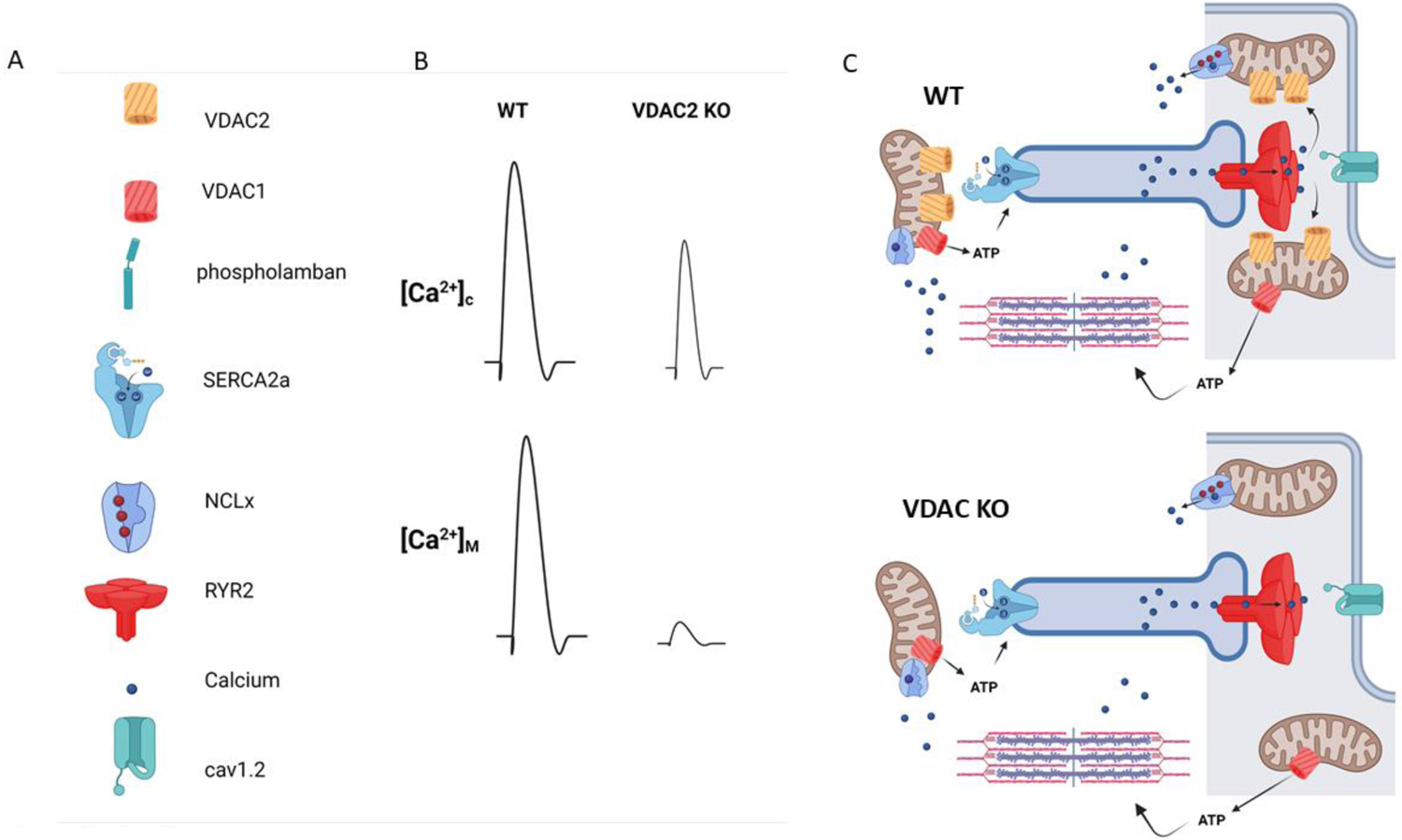
VDAC2 deletion in cardiomyocytes causes defects in ECC. A) legend for Ca2+ handling proteins in C. B) Cytosolic and Mitochondria Ca2+ responses in WT and VDAC2 KO cells. Deletion of VDAC2 leads to a reduction in Ca2+ release and reduced Ca2+ transfer into mitochondria. VDAC1 may have more a role in regulating redox signaling and apoptosis, whereas VDAC2 has a dominant role in Ca2+ signaling.
Highlights.
VDAC2 deletion from cardiomyocytes cause dilated cardiomyopathy and increased mortality.
Cytosolic Ca2+ transients are reduced as a cause of impaired contractility.
VDAC2 inteacts with RYR2, SERCA2a and NCX1 in cardiomyocytes.
VDAC2-enhanced Ca2+ uptake in CM prevents heart failure.
Enhanced Ca2+ uptake in mitochondria is a novel strategy to treat heart failure.
Acknowledgements
This project was supported by Award Number R01DZK109911 (PBR) from the National Institute for Diabetes, Digestion and Kidney, and R01AG045551 (PBR).
Abbreviations
- Ca2+
Calcium
- CM
cardiomyocyte
- [Ca2+]i
Cytoplasmic calcium concentration
- GoF
Gain of function
- HF
Heart Failure
- LoF
Loss of function
- RYR2
Ryanodine Receptor type 2
- S/ER
Sarco(endo)plasmic reticulum
- SOCE
Store-operated calcium entry
- TG
Thapsigargin
- VSM
Vascular smooth muscle
- Cav1.2
Voltage-dependent calcium channels
- ATP
Adenine Trisphosphate
- ETC
Electron Transport Chain
- SGLT2
sodium-glucose transporter type II
- ECC
Excitation Contraction Coupling
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The author has declared that no conflict of interest exists.
References
- [1].Braunwald E, Ross J Jr., Sonnenblick EH, Mechanisms of contraction of the normal and failing heart, N Engl J Med, 277 (1967) 1012–1022 concl. [DOI] [PubMed] [Google Scholar]
- [2].Katz AM, Regulation of cardiac muscle contractility, J Gen Physiol, 50 (1967) Suppl:185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Francis GS, Bartos JA, Adatya S, Inotropes, J Am Coll Cardiol, 63 (2014) 2069–2078. [DOI] [PubMed] [Google Scholar]
- [4].Zhou B, Tian R, Mitochondrial dysfunction in pathophysiology of heart failure, J Clin Invest, 128 (2018) 3716–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Reichel H, Bleichert A, Excitation-contraction coupling in heart muscle, Nature, 183 (1959) 826–827. [DOI] [PubMed] [Google Scholar]
- [6].Dibb KM, Eisner DA, Trafford AW, Regulation of systolic [Ca2+]i and cellular Ca2+ flux balance in rat ventricular myocytes by SR Ca2+, L-type Ca2+ current and diastolic [Ca2+]i, J Physiol, 585 (2007) 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dibb KM, Graham HK, Venetucci LA, Eisner DA, Trafford AW, Analysis of cellular calcium fluxes in cardiac muscle to understand calcium homeostasis in the heart, Cell Calcium, 42 (2007) 503–512. [DOI] [PubMed] [Google Scholar]
- [8].Bers DM, Cardiac excitation-contraction coupling, Nature, 415 (2002) 198–205. [DOI] [PubMed] [Google Scholar]
- [9].Eisner DA, Caldwell JL, Kistamas K, Trafford AW, Calcium and Excitation-Contraction Coupling in the Heart, Circ Res, 121 (2017) 181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Briston SJ, Dibb KM, Solaro RJ, Eisner DA, Trafford AW, Balanced changes in Ca buffering by SERCA and troponin contribute to Ca handling during beta-adrenergic stimulation in cardiac myocytes, Cardiovasc Res, 104 (2014) 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Boyman L, Karbowski M, Lederer WJ, Regulation of Mitochondrial ATP Production: Ca(2+) Signaling and Quality Control, Trends Mol Med, 26 (2020) 21–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bottomley PA, Panjrath GS, Lai S, Hirsch GA, Wu K, Najjar SS, Steinberg A, Gerstenblith G, Weiss RG, Metabolic rates of ATP transfer through creatine kinase (CK Flux) predict clinical heart failure events and death, Sci Transl Med, 5 (2013) 215re213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Balaban RS, Domestication of the cardiac mitochondrion for energy conversion, J Mol Cell Cardiol, 46 (2009) 832–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bers DM, Calcium fluxes involved in control of cardiac myocyte contraction, Circ Res, 87 (2000) 275–281. [DOI] [PubMed] [Google Scholar]
- [15].Piquereau J, Veksler V, Novotova M, Ventura-Clapier R, Energetic Interactions Between Subcellular Organelles in Striated Muscles, Front Cell Dev Biol, 8 (2020) 581045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Weiss RG, Gerstenblith G, Bottomley PA, ATP flux through creatine kinase in the normal, stressed, and failing human heart, Proc Natl Acad Sci U S A, 102 (2005) 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Boyman L, Chikando AC, Williams GS, Khairallah RJ, Kettlewell S, Ward CW, Smith GL, Kao JP, Lederer WJ, Calcium movement in cardiac mitochondria, Biophys J, 107 (2014) 1289–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Heineman FW, Balaban RS, Control of mitochondrial respiration in the heart in vivo, Annu Rev Physiol, 52 (1990) 523–542. [DOI] [PubMed] [Google Scholar]
- [19].Glancy B, Balaban RS, Role of mitochondrial Ca2+ in the regulation of cellular energetics, Biochemistry, 51 (2012) 2959–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lopaschuk GD, Kelly DP, Signalling in cardiac metabolism, Cardiovasc Res, 79 (2008) 205–207. [DOI] [PubMed] [Google Scholar]
- [21].Hak JB, Van Beek JH, Eijgelshoven MH, Westerhof N, Mitochondrial dehydrogenase activity affects adaptation of cardiac oxygen consumption to demand, Am J Physiol, 264 (1993) H448–453. [DOI] [PubMed] [Google Scholar]
- [22].del Arco A, Satrustegui J, Molecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain, J Biol Chem, 273 (1998) 23327–23334. [DOI] [PubMed] [Google Scholar]
- [23].Contreras L, Gomez-Puertas P, Iijima M, Kobayashi K, Saheki T, Satrustegui J, Ca2+ Activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: role in the heart malate-aspartate NADH shuttle, J Biol Chem, 282 (2007) 7098–7106. [DOI] [PubMed] [Google Scholar]
- [24].Szibor M, Gizatullina Z, Gainutdinov T, Endres T, Debska-Vielhaber G, Kunz M, Karavasili N, Hallmann K, Schreiber F, Bamberger A, Schwarzer M, Doenst T, Heinze HJ, Lessmann V, Vielhaber S, Kunz WS, Gellerich FN, Cytosolic, but not matrix, calcium is essential for adjustment of mitochondrial pyruvate supply, J Biol Chem, 295 (2020) 4383–4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Boja ES, Phillips D, French SA, Harris RA, Balaban RS, Quantitative mitochondrial phosphoproteomics using iTRAQ on an LTQ-Orbitrap with high energy collision dissociation, Journal of proteome research, 8 (2009) 4665–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Balaban RS, The role of Ca(2+) signaling in the coordination of mitochondrial ATP production with cardiac work, Biochim Biophys Acta, 1787 (2009) 1334–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tian R, Colucci WS, Arany Z, Bachschmid MM, Ballinger SW, Boudina S, Bruce JE, Busija DW, Dikalov S, Dorn GW II, Galis ZS, Gottlieb RA, Kelly DP, Kitsis RN, Kohr MJ, Levy D, Lewandowski ED, McClung JM, Mochly-Rosen D, O’Brien KD, O’Rourke B, Park JY, Ping P, Sack MN, Sheu SS, Shi Y, Shiva S, Wallace DC, Weiss RG, Vernon HJ, Wong R, Schwartz Longacre L, Unlocking the Secrets of Mitochondria in the Cardiovascular System: Path to a Cure in Heart Failure-A Report from the 2018 National Heart, Lung, and Blood Institute Workshop, Circulation, 140 (2019) 1205–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Garbincius JF, Elrod JW, Mitochondrial calcium exchange in physiology and disease, Physiol Rev, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lavorato M, Iyer VR, Dewight W, Cupo RR, Debattisti V, Gomez L, De la Fuente S, Zhao YT, Valdivia HH, Hajnoczky G, Franzini-Armstrong C, Increased mitochondrial nanotunneling activity, induced by calcium imbalance, affects intermitochondrial matrix exchanges, Proc Natl Acad Sci U S A, 114 (2017) E849–E858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mackenzie L, Roderick HL, Berridge MJ, Conway SJ, Bootman MD, The spatial pattern of atrial cardiomyocyte calcium signalling modulates contraction, J Cell Sci, 117 (2004) 6327–6337. [DOI] [PubMed] [Google Scholar]
- [31].Bakowski D, Parekh AB, Regulation of store-operated calcium channels by the intermediary metabolite pyruvic acid, Curr Biol, 17 (2007) 1076–1081. [DOI] [PubMed] [Google Scholar]
- [32].Sander P, Gudermann T, Schredelseker J, A Calcium Guard in the Outer Membrane: Is VDAC a Regulated Gatekeeper of Mitochondrial Calcium Uptake?, Int J Mol Sci, 22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jiang D, Zhao L, Clish CB, Clapham DE, Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome, Proc Natl Acad Sci U S A, 110 (2013) E2249–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Jiang D, Zhao L, Clapham DE, Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter, Science, 326 (2009) 144–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bassani JW, Bassani RA, Bers DM, Ca2+ cycling between sarcoplasmic reticulum and mitochondria in rabbit cardiac myocytes, J Physiol, 460 (1993) 603–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Viola HM, Hool LC, How does calcium regulate mitochondrial energetics in the heart? - new insights, Heart Lung Circ, 23 (2014) 602–609. [DOI] [PubMed] [Google Scholar]
- [37].Giorgi C, Marchi S, Pinton P, Publisher Correction: The machineries, regulation and cellular functions of mitochondrial calcium, Nat Rev Mol Cell Biol, 19 (2018) 746. [DOI] [PubMed] [Google Scholar]
- [38].Wescott AP, Kao JPY, Lederer WJ, Boyman L, Voltage-energized Calcium-sensitive ATP Production by Mitochondria, Nat Metab, 1 (2019) 975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Williams GS, Boyman L, Chikando AC, Khairallah RJ, Lederer WJ, Mitochondrial calcium uptake, Proc Natl Acad Sci U S A, 110 (2013) 10479–10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lu X, Thai PN, Lu S, Pu J, Bers DM, Intrafibrillar and perinuclear mitochondrial heterogeneity in adult cardiac myocytes, J Mol Cell Cardiol, 136 (2019) 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].De La Fuente S, Lambert JP, Nichtova Z, Fernandez Sanz C, Elrod JW, Sheu SS, Csordas G, Spatial Separation of Mitochondrial Calcium Uptake and Extrusion for Energy-Efficient Mitochondrial Calcium Signaling in the Heart, Cell Rep, 24 (2018) 3099–3107 e3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Min CK, Yeom DR, Lee KE, Kwon HK, Kang M, Kim YS, Park ZY, Jeon H, Kim DH, Coupling of ryanodine receptor 2 and voltage-dependent anion channel 2 is essential for Ca(2)+ transfer from the sarcoplasmic reticulum to the mitochondria in the heart, Biochem J, 447 (2012) 371–379. [DOI] [PubMed] [Google Scholar]
- [43].Dia M, Gomez L, Thibault H, Tessier N, Leon C, Chouabe C, Ducreux S, Gallo-Bona N, Tubbs E, Bendridi N, Chanon S, Leray A, Belmudes L, Coute Y, Kurdi M, Ovize M, Rieusset J, Paillard M, Reduced reticulum-mitochondria Ca(2+) transfer is an early and reversible trigger of mitochondrial dysfunctions in diabetic cardiomyopathy, Basic Res Cardiol, 115 (2020) 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lu X, Ginsburg KS, Kettlewell S, Bossuyt J, Smith GL, Bers DM, Measuring local gradients of intramitochondrial [Ca(2+)] in cardiac myocytes during sarcoplasmic reticulum Ca(2+) release, Circ Res, 112 (2013) 424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hamilton S, Terentyeva R, Clements RT, Belevych AE, Terentyev D, Sarcoplasmic reticulum-mitochondria communication; implications for cardiac arrhythmia, J Mol Cell Cardiol, 156 (2021) 105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Santulli G, Xie W, Reiken SR, Marks AR, Mitochondrial calcium overload is a key determinant in heart failure, Proc Natl Acad Sci U S A, 112 (2015) 11389–11394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhao YT, Valdivia CR, Gurrola GB, Powers PP, Willis BC, Moss RL, Jalife J, Valdivia HH, Arrhythmogenesis in a catecholaminergic polymorphic ventricular tachycardia mutation that depresses ryanodine receptor function, Proc Natl Acad Sci U S A, 112 (2015) E1669–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Glukhov AV, Kalyanasundaram A, Lou Q, Hage LT, Hansen BJ, Belevych AE, Mohler PJ, Knollmann BC, Periasamy M, Gyorke S, Fedorov VV, Calsequestrin 2 deletion causes sinoatrial node dysfunction and atrial arrhythmias associated with altered sarcoplasmic reticulum calcium cycling and degenerative fibrosis within the mouse atrial pacemaker complex, Eur Heart J, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hamilton S, Terentyeva R, Martin B, Perger F, Li J, Stepanov A, Bonilla IM, Knollmann BC, Radwanski PB, Gyorke S, Belevych AE, Terentyev D, Increased RyR2 activity is exacerbated by calcium leak-induced mitochondrial ROS, Basic Res Cardiol, 115 (2020) 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Blachly-Dyson E, Baldini A, Litt M, McCabe ER, Forte M, Human genes encoding the voltage-dependent anion channel (VDAC) of the outer mitochondrial membrane: mapping and identification of two new isoforms, Genomics, 20 (1994) 62–67. [DOI] [PubMed] [Google Scholar]
- [51].Sampson MJ, Lovell RS, Craigen WJ, The murine voltage-dependent anion channel gene family. Conserved structure and function, J Biol Chem, 272 (1997) 18966–18973. [DOI] [PubMed] [Google Scholar]
- [52].Gincel D, Zaid H, Shoshan-Barmatz V, Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function, Biochem J, 358 (2001) 147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tan W, Colombini M, VDAC closure increases calcium ion flux, Biochim Biophys Acta, 1768 (2007) 2510–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tong Z, Xie Y, He M, Ma W, Zhou Y, Lai S, Meng Y, Liao Z, VDAC1 deacetylation is involved in the protective effects of resveratrol against mitochondria-mediated apoptosis in cardiomyocytes subjected to anoxia/reoxygenation injury, Biomed Pharmacother, 95 (2017) 77–83. [DOI] [PubMed] [Google Scholar]
- [55].Martel C, Wang Z, Brenner C, VDAC phosphorylation, a lipid sensor influencing the cell fate, Mitochondrion, 19 Pt A (2014) 69–77. [DOI] [PubMed] [Google Scholar]
- [56].Das S, Wong R, Rajapakse N, Murphy E, Steenbergen C, Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation, Circ Res, 103 (2008) 983–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sun Y, Vashisht AA, Tchieu J, Wohlschlegel JA, Dreier L, Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy, J Biol Chem, 287 (2012) 40652–40660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ, VDAC2 inhibits BAK activation and mitochondrial apoptosis, Science, 301 (2003) 513–517. [DOI] [PubMed] [Google Scholar]
- [59].Narula N, Zaragoza MV, Sengupta PP, Li P, Haider N, Verjans J, Waymire K, Vannan M, Wallace DC, Adenine nucleotide translocase 1 deficiency results in dilated cardiomyopathy with defects in myocardial mechanics, histopathological alterations, and activation of apoptosis, JACC Cardiovasc Imaging, 4 (2011) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kokoszka JE, Waymire KG, Flierl A, Sweeney KM, Angelin A, MacGregor GR, Wallace DC, Deficiency in the mouse mitochondrial adenine nucleotide translocator isoform 2 gene is associated with cardiac noncompaction, Biochim Biophys Acta, 1857 (2016) 1203–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Shimizu H, Schredelseker J, Huang J, Lu K, Naghdi S, Lu F, Franklin S, Fiji HD, Wang K, Zhu H, Tian C, Lin B, Nakano H, Ehrlich A, Nakai J, Stieg AZ, Gimzewski JK, Nakano A, Goldhaber JI, Vondriska TM, Hajnoczky G, Kwon O, Chen JN, Mitochondrial Ca(2+) uptake by the voltage-dependent anion channel 2 regulates cardiac rhythmicity, Elife, 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Roy SS, Madesh M, Davies E, Antonsson B, Danial N, Hajnoczky G, Bad targets the permeability transition pore independent of Bax or Bak to switch between Ca2+-dependent cell survival and death, Mol Cell, 33 (2009) 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kerner J, Lee K, Hoppel CL, Post-translational modifications of mitochondrial outer membrane proteins, Free Radic Res, 45 (2011) 16–28. [DOI] [PubMed] [Google Scholar]
- [64].Ilic A, Todorovic D, Mutavdzin S, Boricic N, Bozic Nedeljkovic B, Stankovic S, Simic T, Stevanovic P, Celic V, Djuric D, Translocator Protein Modulation by 4’-Chlorodiazepam and NO Synthase Inhibition Affect Cardiac Oxidative Stress, Cardiometabolic and Inflammatory Markers in Isoprenaline-Induced Rat Myocardial Infarction, Int J Mol Sci, 22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gatliff J, Campanella M, TSPO is a REDOX regulator of cell mitophagy, Biochem Soc Trans, 43 (2015) 543–552. [DOI] [PubMed] [Google Scholar]
- [66].Lee K, Kerner J, Hoppel CL, Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex, J Biol Chem, 286 (2011) 25655–25662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Guzun R, Gonzalez-Granillo M, Karu-Varikmaa M, Grichine A, Usson Y, Kaambre T, Guerrero-Roesch K, Kuznetsov A, Schlattner U, Saks V, Regulation of respiration in muscle cells in vivo by VDAC through interaction with the cytoskeleton and MtCK within Mitochondrial Interactosome, Biochim Biophys Acta, 1818 (2012) 1545–1554. [DOI] [PubMed] [Google Scholar]
- [68].Maldonado EN, Sheldon KL, DeHart DN, Patnaik J, Manevich Y, Townsend DM, Bezrukov SM, Rostovtseva TK, Lemasters JJ, Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: regulation by free tubulin and erastin, J Biol Chem, 288 (2013) 11920–11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Appaix F, Kuznetsov AV, Usson Y, Kay L, Andrienko T, Olivares J, Kaambre T, Sikk P, Margreiter R, Saks V, Possible role of cytoskeleton in intracellular arrangement and regulation of mitochondria, Exp Physiol, 88 (2003) 175–190. [DOI] [PubMed] [Google Scholar]
- [70].Diokmetzidou A, Soumaka E, Kloukina I, Tsikitis M, Makridakis M, Varela A, Davos CH, Georgopoulos S, Anesti V, Vlahou A, Capetanaki Y, Desmin and alphaB-crystallin interplay in the maintenance of mitochondrial homeostasis and cardiomyocyte survival, J Cell Sci, 129 (2016) 3705–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Khan A, Kuriachan G, Mahalakshmi R, Cellular Interactome of Mitochondrial Voltage-Dependent Anion Channels: Oligomerization and Channel (Mis)Regulation, ACS Chem Neurosci, 12 (2021) 3497–3515. [DOI] [PubMed] [Google Scholar]
- [72].Rosencrans WM, Rajendran M, Bezrukov SM, Rostovtseva TK, VDAC regulation of mitochondrial calcium flux: From channel biophysics to disease, Cell Calcium, 94 (2021) 102356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Shankar TS, Ramadurai DKA, Steinhorst K, Sommakia S, Badolia R, Thodou Krokidi A, Calder D, Navankasattusas S, Sander P, Kwon OS, Aravamudhan A, Ling J, Dendorfer A, Xie C, Kwon O, Cheng EHY, Whitehead KJ, Gudermann T, Richardson RS, Sachse FB, Schredelseker J, Spitzer KW, Chaudhuri D, Drakos SG, Cardiac-specific deletion of voltage dependent anion channel 2 leads to dilated cardiomyopathy by altering calcium homeostasis, Nat Commun, 12 (2021) 4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T, The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter, Nat Cell Biol, 15 (2013) 1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Muller M, Miller R, Kolesar JE, Molgo J, Kaufman B, Hajnoczky G, Foskett JK, Madesh M, MCUR1 is an essential component of mitochondrial Ca(2+) uptake that regulates cellular metabolism, Nat Cell Biol, 17 (2015) 953. [DOI] [PubMed] [Google Scholar]
- [76].Liu JC, Syder NC, Ghorashi NS, Willingham TB, Parks RJ, Sun J, Fergusson MM, Liu J, Holmstrom KM, Menazza S, Springer DA, Liu C, Glancy B, Finkel T, Murphy E, EMRE is essential for mitochondrial calcium uniporter activity in a mouse model, JCI Insight, 5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs AM, Kriek M, Phadke R, Johnson CA, Roberts NY, Bonthron DT, Pysden KA, Whyte T, Munteanu I, Foley AR, Wheway G, Szymanska K, Natarajan S, Abdelhamed ZA, Morgan JE, Roper H, Santen GW, Niks EH, van der Pol WL, Lindhout D, Raffaello A, De Stefani D, den Dunnen JT, Sun Y, Ginjaar I, Sewry CA, Hurles M, Rizzuto R, Consortium UK, Duchen MR, Muntoni F, Sheridan E, Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling, Nature genetics, 46 (2014) 188–193. [DOI] [PubMed] [Google Scholar]
- [78].Liu JC, Liu J, Holmstrom KM, Menazza S, Parks RJ, Fergusson MM, Yu ZX, Springer DA, Halsey C, Liu C, Murphy E, Finkel T, MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload, Cell Rep, 16 (2016) 1561–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Liu T, Yang N, Sidor A, O’Rourke B, MCU Overexpression Rescues Inotropy and Reverses Heart Failure by Reducing SR Ca(2+) Leak, Circ Res, 128 (2021) 1191–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Schweitzer MK, Wilting F, Sedej S, Dreizehnter L, Dupper NJ, Tian Q, Moretti A, My I, Kwon O, Priori SG, Laugwitz KL, Storch U, Lipp P, Breit A, Mederos YSM, Gudermann T, Schredelseker J, Suppression of Arrhythmia by Enhancing Mitochondrial Ca(2+) Uptake in Catecholaminergic Ventricular Tachycardia Models, JACC Basic Transl Sci, 2 (2017) 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Wilting F, Kopp R, Gurnev PA, Schedel A, Dupper NJ, Kwon O, Nicke A, Gudermann T, Schredelseker J, The antiarrhythmic compound efsevin directly modulates voltage-dependent anion channel 2 by binding to its inner wall and enhancing mitochondrial Ca(2+) uptake, Br J Pharmacol, 177 (2020) 2947–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Sander P, Feng M, Schweitzer MK, Wilting F, Gutenthaler SM, Arduino DM, Fischbach S, Dreizehnter L, Moretti A, Gudermann T, Perocchi F, Schredelseker J, Approved drugs ezetimibe and disulfiram enhance mitochondrial Ca(2+) uptake and suppress cardiac arrhythmogenesis, Br J Pharmacol, 178 (2021) 4518–4532. [DOI] [PubMed] [Google Scholar]
- [83].Luconi M, Raimondi L, Di Franco A, Mannucci E, Which is the main molecular target responsible for the cardiovascular benefits in the EMPA-REG OUTCOME trial? A journey through the kidney, the heart and other interesting places, Nutr Metab Cardiovasc Dis, 26 (2016) 1071–1078. [DOI] [PubMed] [Google Scholar]
- [84].Clegg LE, Heerspink HJL, Penland RC, Tang W, Boulton DW, Bachina S, Fox RD, Fenici P, Thuresson M, Mentz RJ, Hernandez AF, Holman RR, Reduction of Cardiovascular Risk and Improved Estimated Glomerular Filtration Rate by SGLT2 Inhibitors, Including Dapagliflozin, Is Consistent Across the Class: An Analysis of the Placebo Arm of EXSCEL, Diabetes Care, 42 (2019) 318–326. [DOI] [PubMed] [Google Scholar]
- [85].Lin X, Wang S, Huang J, Empagliflozin in Heart Failure, N Engl J Med, 384 (2021) 384–385. [DOI] [PubMed] [Google Scholar]
- [86].Pabel S, Hamdani N, Luedde M, Sossalla S, SGLT2 Inhibitors and Their Mode of Action in Heart Failure-Has the Mystery Been Unravelled?, Curr Heart Fail Rep, 18 (2021) 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Kruger M, Hoppel CL, Lewandowski ED, Crawford PA, Muoio DM, Kelly DP, The Failing Heart Relies on Ketone Bodies as a Fuel, Circulation, 133 (2016) 698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Li X, Lu Q, Qiu Y, do Carmo JM, Wang Z, da Silva AA, Mouton A, Omoto ACM, Hall ME, Li J, Hall JE, Direct Cardiac Actions of the Sodium Glucose Co-Transporter 2 Inhibitor Empagliflozin Improve Myocardial Oxidative Phosphorylation and Attenuate Pressure-Overload Heart Failure, J Am Heart Assoc, 10 (2021) e018298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Dietl A, Maack C, Targeting Mitochondrial Calcium Handling and Reactive Oxygen Species in Heart Failure, Curr Heart Fail Rep, 14 (2017) 338–349. [DOI] [PubMed] [Google Scholar]
- [90].Andreadou I, Efentakis P, Balafas E, Togliatto G, Davos CH, Varela A, Dimitriou CA, Nikolaou PE, Maratou E, Lambadiari V, Ikonomidis I, Kostomitsopoulos N, Brizzi MF, Dimitriadis G, Iliodromitis EK, Empagliflozin Limits Myocardial Infarction in Vivo and Cell Death in Vitro: Role of STAT3, Mitochondria, and Redox Aspects, Front Physiol, 8 (2017) 1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Arduino DM, Perocchi F, Pharmacological modulation of mitochondrial calcium homeostasis, J Physiol, 596 (2018) 2717–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Arduino DM, Wettmarshausen J, Vais H, Navas-Navarro P, Cheng Y, Leimpek A, Ma Z, Delrio-Lorenzo A, Giordano A, Garcia-Perez C, Medard G, Kuster B, Garcia-Sancho J, Mokranjac D, Foskett JK, Alonso MT, Perocchi F, Systematic Identification of MCU Modulators by Orthogonal Interspecies Chemical Screening, Mol Cell, 67 (2017) 711–723 e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Rossato LG, Costa VM, Dallegrave E, Arbo M, Silva R, Ferreira R, Amado F, Dinis-Oliveira RJ, Duarte JA, de Lourdes Bastos M, Palmeira C, Remiao F, Mitochondrial cumulative damage induced by mitoxantrone: late onset cardiac energetic impairment, Cardiovasc Toxicol, 14 (2014) 30–40. [DOI] [PubMed] [Google Scholar]
- [94].Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR, Cleland JG, Colucci WS, Butler J, Voors AA, Anker SD, Pitt B, Pieske B, Filippatos G, Greene SJ, Gheorghiade M, Expert consensus document: Mitochondrial function as a therapeutic target in heart failure, Nature reviews. Cardiology, 14 (2017) 238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Ribeiro Junior RF, Dabkowski ER, Shekar KC, KA OC, Hecker PA, Murphy MP, MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload, Free radical biology & medicine, 117 (2018) 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Ramirez-Sanchez I, Taub PR, Ciaraldi TP, Nogueira L, Coe T, Perkins G, Hogan M, Maisel AS, Henry RR, Ceballos G, Villarreal F, (−)-Epicatechin rich cocoa mediated modulation of oxidative stress regulators in skeletal muscle of heart failure and type 2 diabetes patients, Int J Cardiol, 168 (2013) 3982–3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Paterek A, Okninska M, Leszek P, Mackiewicz U, Jankowska EA, Ponikowski P, Maczewski M, Intravenous ferric carboxymaltose does not provide benefits in reperfused acute myocardial infarction in the rat with normal iron status, Biomed Pharmacother, 141 (2021) 111893. [DOI] [PubMed] [Google Scholar]
- [98].Satoh T, Wang L, Espinosa-Diez C, Wang B, Hahn SA, Noda K, Rochon ER, Dent MR, Levine AR, Baust JJ, Wyman S, Wu YL, Triantafyllou GA, Tang Y, Reynolds M, Shiva S, Hilaire CS, Gomez D, Goncharov DA, Goncharova EA, Chan SY, Straub AC, Lai YC, McTiernan CF, Gladwin MT, Metabolic Syndrome Mediates ROS-miR-193b-NFYA-Dependent Downregulation of Soluble Guanylate Cyclase and Contributes to Exercise-Induced Pulmonary Hypertension in Heart Failure With Preserved Ejection Fraction, Circulation, 144 (2021) 615–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Zingarelli B, Salzman AL, Szabo C, Protective effects of nicotinamide against nitric oxide-mediated delayed vascular failure in endotoxic shock: potential involvement of polyADP ribosyl synthetase, Shock, 5 (1996) 258–264. [DOI] [PubMed] [Google Scholar]
- [100].Mericskay M, Nicotinamide adenine dinucleotide homeostasis and signalling in heart disease: Pathophysiological implications and therapeutic potential, Arch Cardiovasc Dis, 109 (2016) 207–215. [DOI] [PubMed] [Google Scholar]
- [101].Horton JL, Davidson MT, Kurishima C, Vega RB, Powers JC, Matsuura TR, Petucci C, Lewandowski ED, Crawford PA, Muoio DM, Recchia FA, Kelly DP, The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense, JCI Insight, 4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Nielsen R, Moller N, Gormsen LC, Tolbod LP, Hansson NH, Sorensen J, Harms HJ, Frokiaer J, Eiskjaer H, Jespersen NR, Mellemkjaer S, Lassen TR, Pryds K, Botker HE, Wiggers H, Cardiovascular Effects of Treatment With the Ketone Body 3-Hydroxybutyrate in Chronic Heart Failure Patients, Circulation, 139 (2019) 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Middlekauff HR, Verity MA, Horwich TB, Fonarow GC, Hamilton MA, Shieh P, Intact skeletal muscle mitochondrial enzyme activity but diminished exercise capacity in advanced heart failure patients on optimal medical and device therapy, Clin Res Cardiol, 102 (2013) 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Mizuno M, Kuno A, Yano T, Miki T, Oshima H, Sato T, Nakata K, Kimura Y, Tanno M, Miura T, Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts, Physiological reports, 6 (2018) e13741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Woessner MN, Neil C, Saner NJ, Goodman CA, McIlvenna LC, Ortiz de Zevallos J, Garnham A, Levinger I, Allen JD, Effect of inorganic nitrate on exercise capacity, mitochondria respiration, and vascular function in heart failure with reduced ejection fraction, J Appl Physiol (1985), 128 (2020) 1355–1364. [DOI] [PubMed] [Google Scholar]
- [106].Chirinos JA, Zamani P, The Nitrate-Nitrite-NO Pathway and Its Implications for Heart Failure and Preserved Ejection Fraction, Curr Heart Fail Rep, 13 (2016) 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Lahnwong S, Palee S, Apaijai N, Sriwichaiin S, Kerdphoo S, Jaiwongkam T, Chattipakorn SC, Chattipakorn N, Acute dapagliflozin administration exerts cardioprotective effects in rats with cardiac ischemia/reperfusion injury, Cardiovasc Diabetol, 19 (2020) 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Moustafa DA, Imran Z, Ismail R, Rayan M, Gadeau AP, Eldassouki H, Abdulrahman N, Mraiche F, Evaluating the effects of sodium glucose co-transporter −2 inhibitors from a renin-angiotensin-aldosterone system perspective in patients infected with COVID-19: contextualizing findings from the dapagliflozin in respiratory failure in patients with COVID-19 study, Molecular biology reports, 49 (2022) 2321–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Evangeliou A, Vlassopoulos D, Carnitine metabolism and deficit--when supplementation is necessary?, Curr Pharm Biotechnol, 4 (2003) 211–219. [DOI] [PubMed] [Google Scholar]
- [110].Ferrari R, Merli E, Cicchitelli G, Mele D, Fucili A, Ceconi C, Therapeutic effects of L-carnitine and propionyl-L-carnitine on cardiovascular diseases: a review, Ann N Y Acad Sci, 1033 (2004) 79–91. [DOI] [PubMed] [Google Scholar]