

Abstract

Functionalized alicyclic amines are important building blocks for the synthesis of bioactive natural compounds and drugs. Existing methods of functionalization are typically limited to the synthesis of protected amines or the use of highly basic organometallic reagents that can compromise functional group tolerance. Here, we report a novel approach that enables the transformation of O-benzoyl hydroxylamines into α-functionalized cyclic secondary amines by means of a copper-catalyzed regio-, stereo-, and chemoselective coupling with allenes and bis(pinacolato)diboron. A key feature of the present transformation is the use of a catalytic Lewis base additive which inhibits the competing C–N bond forming reaction between the catalytically generated boron-substituted allylcopper intermediate with the O-benzoyl hydroxylamine, thus enabling in situ transformation of the latter into an alicyclic imine which undergoes selective C–C bond formation with the allylcopper species.
Introduction
Saturated N-heterocycles represent one of the most important classes of compounds in drug discovery.1 As such, synthetic procedures to diversely functionalize the alicyclic amine framework are in high demand. Among the different methodologies that have been developed for this purpose, a particularly attractive strategy to access substituted saturated azaheterocycles is the α-C–H bond functionalization of cyclic amines.2 Despite great advances in the field, current procedures typically rely on the use of a directing group on the nitrogen atom, limiting their utility to the synthesis of tertiary or protected secondary alicyclic amines. A commonly used approach includes the α-lithiation of N-Boc-protected azaheterocycles, followed by transmetalation to an organozinc and subsequent palladium-catalyzed Negishi coupling.3 Other strategies based on protecting groups have relied on transition metal-catalyzed direct C–H arylation reactions,4 photoredox catalysis,5 intramolecular hydride transfer,6 and C–H insertions via metal carbenoids7 (Figure 1A).
Figure 1.

Methods for α-C–H functionalization of alicyclic amines.
An alternative strategy to generate α-substituted saturated nitrogen heterocycles is the generation of a cyclic imine and its engagement with a nucleophile. However, alicyclic imines are unstable, and they tend to undergo trimerization, resulting in nonreactive compounds.8 Recently, Seidel and co-workers reported a method for the in situ generation of alicyclic imines based on the reduction of a sacrificial ketone hydride acceptor by the corresponding N-lithiated amine.9 The cyclic imine can then be trapped by an organolithium compound9a or a Grignard reagent using Lewis acid activation,9b thus resulting in a net α-C–H functionalization (Figure 1B). Although this method represents an interesting entry to α-functionalized secondary alicyclic amines, cryogenic temperatures are required, and the stoichiometric use of organometallic reagents can impose extra steps associated with the preparation and purification of these sensitive compounds. Moreover, in some cases, their high basicity can compromise its compatibility with certain functional groups.
An attractive way to produce α-functionalized alicyclic amines would be the catalytic transformation of an unsaturated hydrocarbon into an organometallic intermediate, which can then react with an alicyclic imine. Following this idea, we focused on the development of a catalytic transformation based on the in situ generation of boron-substituted allyl-copper species by addition of Cu-Bpin to an allene.10,11 Given the inherent instability of saturated cyclic imines, a catalytic carboboration process involving in situ formation of this electrophilic coupling partner would be highly desirable. Inspired by the work of You and co-workers, where they showed that O-benzoyl cyclic hydroxylamines can evolve into the corresponding cyclic imines under basic conditions,12 we envisioned a borylative coupling of allenes with B2pin2 and cyclic O-benzoyl hydroxylamines where the catalytic allylcopper intermediate should selectively react with the cyclic imine generated from the O-benzoyl hydroxylamine (Figure 1C). Three-component carboboration of allenes typically involve several challenges associated with the regio- and stereoselective generation of the catalytic Bpin-substituted allyl copper intermediate and the control over the stereo- and site selectivity of its electrophilic trapping. Furthermore, the proposed transformation imposes an important additional selectivity issue since it requires a catalytic system capable of generating an allyl copper intermediate reactive enough to promote an efficient C–C bond formation with the imine while precluding the direct C–N coupling with the O-benzoyl hydroxylamine precursor. Transient organocopper reagents have indeed been reported to react with O-benzoyl hydroxylamines,13 and that reactivity must be shut down. Several requirements must be met to achieve this successfully. The rate of the reaction between the allylcopper intermediate and the cyclic O-benzoyl hydroxylamine must be slower than the rate of imine formation. Moreover, the imine trapping by the allylcopper intermediate, i.e., the C–C bond forming step, must be fast enough to avoid imine trimerization. We here report the successful implementation of this idea and thus the development of a catalytic process that allows for the selective synthesis of boron-substituted α-allylated cyclic secondary amines. We have found that the use of a catalytic Lewis base additive in combination with a Cu/bisphosphine catalyst is crucial to inhibit the C–N coupling and thus to enable an efficient imine trapping.
Results and Discussion
Preliminary Studies and Optimization of the Reaction Conditions
We began our studies by surveying the reaction between phenylallene 1, B2pin2 and morpholino benzoate 2 (Table 1).14 Initial experiments using several copper catalysts already showed the challenging nature of this multicomponent reaction. The use of monodentate phosphine ligands provided almost exclusively the C–N coupling product 4 in low yields together with regioisomeric mixtures of allene protoboration15 products 5 and 6 (entries 1–2). With NHC–Cu complexes derived from sterically bulky aryl-substituted heterocyclic ligands, chemoselectivity improved, favoring the C–C bond formation, although α-functionalized morpholine 3 was obtained in very low yields (entries 3 and 4). We then turned to evaluate bidentate phosphines and found that large bite-angle phosphines favored the formation of product 3 (entries 5–9), although with yields and selectivity still far from satisfactory.
Table 1. Optimization Studies.

| entrya | ligand | additive | 3 (%)b | 4 (%)b | 5 (%)b | 6 (%)b |
|---|---|---|---|---|---|---|
| 1 | PPh3 | <5 | 30 | 30 | 25 | |
| 2 | PCy3 | 15 | 42 | 23 | 14 | |
| 3 | IMes | 24 | 36 | 19 | <5 | |
| 4 | IPr | 22 | <5 | 5 | <5 | |
| 5 | Xantphos | 30 | <5 | 30 | <5 | |
| 6 | DPEphos | 50 | 24 | 25 | <5 | |
| 7 | dppf | 35 | 14 | 12 | <5 | |
| 8 | dppe | 11 | <5 | <5 | <5 | |
| 9 | dppp | 40 | 18 | 20 | <5 | |
| 10 | dcpe | 25 | 16 | 12 | <5 | |
| 11 | dcpe*c | 60 | <5 | <5 | 15 | |
| 12 | dcpe | P(O)Ph3 | 72 | <5 | <5 | 8 |
| 13 | dcped | 35 | <5 | <5 | <5 | |
| 14 | <5 | 30 | 22 | <5 | ||
| 15 | P(O)Ph3 | <5 | 8 | 6 | <5 | |
| 16e | dcpe | P(O)Ph3 | 60 | <5 | <5 | 10 |
| 17f | dcpe | P(O)Ph3 | 23 | 28 | 30 | 15 |
Reactions run on a 0.3 mmol scale. Diastereomeric ratio of 3 >95:5 in all cases.
Determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as the internal standard.
dcpe* = dcpe:dcpe(O):dcpe(O)2 in a 3:1:3 ratio.
12 mol %.
NaOtBu used instead of LiOtBu.
NaOMe used instead of LiOtBu.
An intriguing observation was that the use of different batches of bis(dicyclohexylphosphino)ethane (dcpe) resulted in drastically different results (entries 10 and 11). Analysis of these two batches revealed that the use of pure dcpe led to a mixture of C–C coupling product 3 and C–N coupling product 4 in a 1.5:1 ratio (entry 10). However, the use of a partially oxidized sample consisting of a 3:1:3 mixture of dcpe, dcpe monoxide (dcpe(O)), and dcpe bis(oxide) (dcpe(O)2) provided 3 with total chemoselectivity as a single diastereomer in 60% yield (entry 11). Motivated by this finding, we surveyed the cooperative effect of several Lewis bases with the Cu/dcpe catalyst (see the Supporting Information, Tables S2 and S3).16 Among the Lewis bases tested, the use of catalytic amounts of P(O)Ph3 provided the best results. The optimized CuCl/dcpe/P(O)Ph3 catalyst allowed for almost full control with respect to side reactions and provided the α-functionalized product 3 as a single isomer in 72% yield with total diastereocontrol (entry 12). Excess of dcpe ligand also led to the selective formation of 3, although in a diminished yield (entry 13). Importantly, in the absence of a phosphorous ligand, selectivity was switched toward C–N coupling product 4, which was obtained in 30% yield (entry 14). The same selectivity was observed when a catalytic amount of P(O)Ph3 was used, although 4 was obtained in an almost negligible yield (entry 15). The choice of base was also important to achieve high levels of selectivity. Indeed, while the use of NaOtBu led to a similar result, the use of NaOMe instead or LiOtBu caused a significant drop in the chemoselectivity, providing a mixture of C–C and C–N coupling products (entries 16–17).
Substrate Scope and Structural Modification of Products
Having established optimized conditions, we set out to explore the scope of this three-component borylative α-C–H allylation (Figure 2A). In general, the reaction proved to be remarkably effective for a wide range of allenes and cyclic amines, providing the corresponding α-functionalized alicyclic amines with excellent levels of regio- and stereocontrol. Common functional groups such as esters, silyl ethers, alkenes, halides, and carbamates were well tolerated under the reaction conditions. In some cases, alkenyl boronates were unstable to silica gel and an additional oxidation step was carried out for a more facile purification of the resulting ketone. Allenes bearing aryl (3, 7–9), alkenyl (10), and aliphatic (11–14) groups proved to be efficient substrates for this three-component coupling. Remarkably, full diastereoselectivity was achieved in nearly all cases, and it was only eroded when allenes bearing linear alkyl substituents (12, 13) were used. Notably, an allene bearing a conjugated alkenyl group, which adds extra points of reactivity, could be used without formation of side products arising from competitive Cu-Bpin olefin insertion or vinylogous SE2″ trapping of the organocopper intermediate. Although this allene showed diminished reactivity, the reaction still afforded product 10 with complete regio- and diastereoselectivity. 1,1-Disubstitution in the allene caused a slight decrease on the regioselectivity (r.r. = 76:24), although it allowed to obtain pure α-allylated amine 14 bearing contiguous quaternary and tertiary stereocentres. Gratifyingly, this method could be extended to other alicyclic amine frameworks such as piperidine (15–20), piperazine (21), pyrrolidine (22), and azepane (23), obtaining in all cases the product in good yield with perfect regio- and diastereoselectivity. Of note is the reaction with 4-substituted piperidine derivatives, which afforded α-functionalized products 16–19 with total stereocontrol over the three newly created stereocenters. Interestingly, the use of an acrylate-substituted piperidine benzoate allowed for the synthesis of endocyclic olefin 20 through an in situ elimination process (see the Supporting Information for details). Relative configuration of products was assigned based on the X-ray crystallographic analysis of products 7 and 16 (Figure 2B). Interestingly, it was also observed that these products exist as azaboraspiro compounds where the boron center features a sp3-hybridization resulting from a dative nitrogen–boron coordination. 11B NMR analysis of products 3 and 7–23 (11B: δ = 10–16 ppm) also suggests the presence of this interaction in solution.
Figure 2.
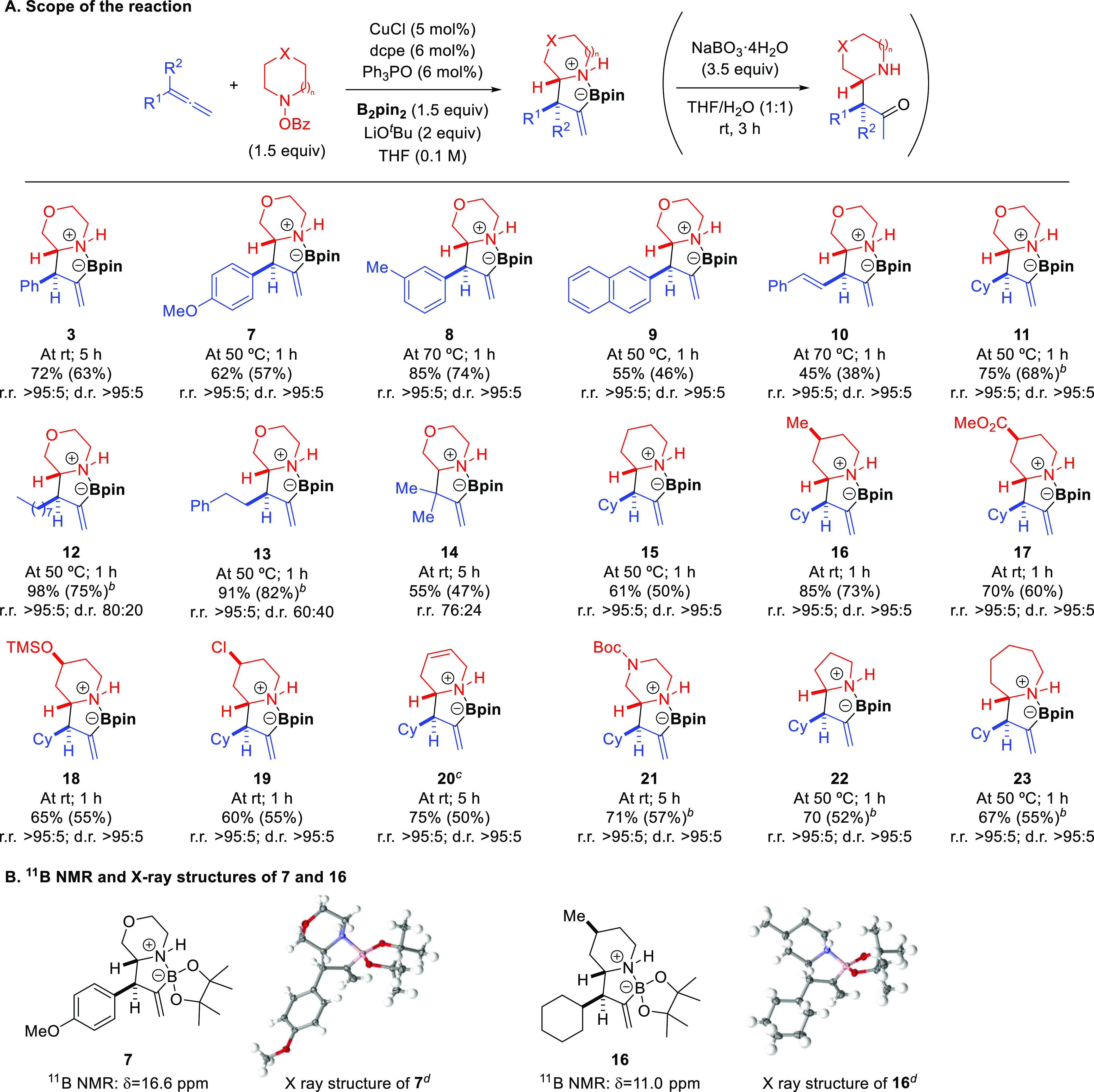
(A) Scope of the copper-catalyzed borylative α-allylation of O-benzoyl cyclic hydroxylamines. aReactions run on a 0.3 mmol scale. Regioisomeric ratios (r.r.; C–C vs C–N coupling) and diastereomeric ratios (d.r.) were determined by 1H NMR analysis. Yields of isolated products are shown in brackets. bThose values refer to the oxidized product. cSynthesized from 4-(acryloyloxy)piperidin-1-yl benzoate. (B) dEllipsoids shown at 50% probability.
An important feature of the present methodology is that it allows the introduction of a functionalized allyl group at the amine ring, which can be diversely modified to access more complex structures in a straightforward manner. Remarkably, the nitrogen–boron coordination present in the α-allylated alicyclic amines served as the basis for a chemoselective structural modification. Reaction of 16 with different aryl iodides catalyzed by a Pd/SPhos complex afforded Heck coupling products 24–26 in good yields with total E-selectivity (Figure 3a). Importantly, the alkenylboronate ester group remained intact and no traces of Suzuki cross-coupling products were detected.17 This excellent chemoselectivity may be explained by the effect of the B–N coordination, which likely hampers transmetalation, a key step in the Suzuki cross-coupling, thus favoring the double bond insertion into the Pd(II)–Ar intermediate, which guides the reaction through the Heck coupling pathway. Interestingly, the B–N coordination could be broken by protonation of the nitrogen atom with hydrochloric acid, which afforded salt 27 (Figure 3b). Despite the strong B–N coordination, the boronic ester moiety could be readily transformed into the corresponding alkenyl trifluoroborate 28 and alkenyl iodide 29 by treatment with KHF2 and NIS, respectively (Figure 3c). C–C bond formation was also possible through an iodination/Suzuki coupling sequence as illustrated with the synthesis of products 30 and 31. Curiously, treatment of compound 17 with Cu(OAc)2 under Chan–Lam conditions18 did not result in the azetidine formation but produced N-acetylated ketone 32, which may result from a copper-promoted C–B acetoxylation followed by an intramolecular acetyl transfer (see the Supporting Information for details). The alkenyl boronate moiety was also used as a handle to install another boron functionality in the carbon chain by means of a site-selective copper-catalyzed protoboration.19 Thus, by using ICyCuCl as the catalyst, reaction of compound 17 with B2pin2 and NaOtBu in tetrahydrofuran (THF) resulted in the diastereoselective formation of vicinal diboronate 33.
Figure 3.

(A–C) Structural modification of products. (i) KHF2 (4.5 equiv), MeOH/H2O 2:1, rt, 3 h. (ii) NIS (1.5 equiv), THF, rt, 2 h. (iii) ArB(OH)2 (1.5 equiv), Pd(PPh3)4 (5 mol %), K2CO3 (3 equiv), 1,4-dioxane/H2O 6:1, 100 °C, 2 h. (iv) Cu(OAc)2 (1 equiv), Et3N (2 equiv), 4 Å MS, MeCN, 80 °C, 20 h. (v) ICyCuCl (10 mol %), NaOtBu (1.2 equiv), B2(pin)2 (1.5 equiv), THF, rt, 16 h. a Ellipsoids shown at 30% probability.
Mechanistic Investigations
The most intriguing observation done during our optimization studies was the high level of chemoselectivity (C–C vs C–N coupling) achieved in this transformation when a catalytic amount of a Lewis base (i.e., P(O)Ph3) was used. It is important to note that this effect was only observed for the dcpe and dppe ligands (see the Supporting Information, Table S4), thus pointing at a special behavior of the resulting copper catalysts. To gather insight about the origin of these high levels of chemoselectivity, density functional theory (DFT) calculations were performed using the coupling of allene 1, morpholino benzoate 2, and B2pin2 as the model reaction (see the Supporting Information for details).
Based on literature precedents,10,11 formation of allylcopper intermediate C would proceed through regio- and stereoselective insertion of 1 into Cu(dcpe)Bpin complex A. Indeed, calculations showed that this step features a low activation energy barrier of 9.5 kcal/mol (Figure 4). We then investigated the evolution of C either to C–N coupling product 4 or C–C coupling product 3 (Figure 4). For the formation of C–N coupling product 4, we initially considered the oxidative addition of the N–O bond of 2 into copper complex C followed by reductive elimination (Figure 4, gray pathway). Note that the dcpe ligand readily dissociates one phosphine unit upon Cu–N coordination to generate intermediate D. Benzoate-assisted SN2-type oxidative addition20 into D features a high energy barrier (ΔG‡ = 28.0 kcal/mol) and results in intermediate E, which would undergo facile reductive elimination (ΔG‡ = 3.9 kcal/mol). An alternative pathway for the formation of C–N coupling product 4 involving the formation of the branched isomeric allylcopper species by metallotropic rearrangement15b of C and subsequent SE2′ substitution with morpholino benzoate 2 (see the Supporting Information, Figure S18) was found to be an even more kinetically demanding process (ΔG‡ = 29.4 kcal/mol). We then explored the reaction between allylcopper intermediate C and the cyclic imine generated from 2 (Figure 4, red pathway). The most favorable route for this transformation involves a six-membered transition state (TSG-H) in which copper coordinates the imine nitrogen and which features an energy of only 16.3 kcal/mol.21 Other possible pathways such as half-chair-like transition structure involving coordination of the imine nitrogen atom to boron were found to be much higher in energy (see the Supporting Information, Figure S22). The fact that the pathway for imine trapping is much more favorable than the oxidative addition/reductive elimination pathway raises the question of why a mixture of C–C coupling product 3 and C–N coupling product 4 is obtained in the absence of a Lewis base (Table 1, entry 10). By spectroscopic studies, we observed that the alicyclic imine is formed gradually during the reaction (see the Supporting Information for details). Furthermore, DFT calculations showed that this transformation features an energy barrier of 16.6 kcal/mol (see the Supporting Information, Figure S23). Thus, the initial concentration of imine might be low to produce product 3 at the beginning of the reaction, thus allowing competing reactions to take place. However, the energetic barrier for the N–O bond oxidative addition in the Cu/dcpe system is too high to make this pathway competitive at the working temperature.22
Figure 4.
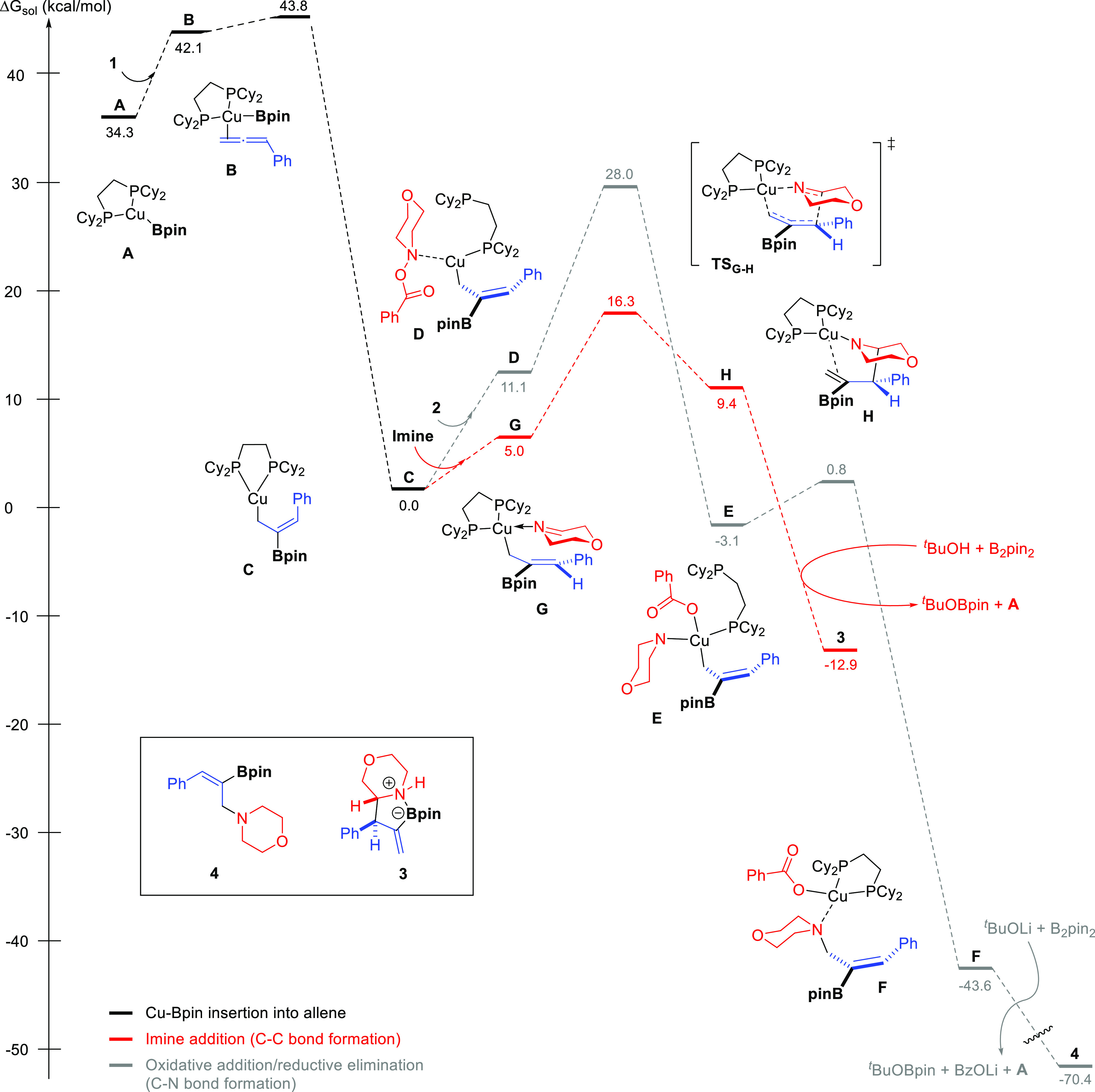
Free energy profile for the most favored pathways leading to the formation of C–C coupling product 3 and C–N coupling product 4 involving Cu/dcpe intermediates. Computational studies were performed at the B3LYP-D3/6-311++G(d,p)-SDDTHF(SMD)//B3LYP-D3/6-31G(d)-SDD level of theory. Energies are relative to complex C combined with those of the relevant substrates.
At this point, we envisaged that the formation of other Cu species may be responsible for the formation of the C–N coupling product. Since the absence of a phosphine ligand led to the selective formation of C–N coupling product 4 (Table 1, entry 14), we carried out calculations on the reaction involving phosphine-free copper species (Figure 5). Assuming the formation of Cu(Bpin)(OtBu)Li complex I,23 generated from reaction of CuOtBu and B2Pin2 in the presence of excess of LiOtBu, and subsequent allene insertion, we calculated the possible pathways for the resulting allylcopper complex K. Inclusion of explicit solvent molecules was done given their importance for calculation of Li complexes.24 The pathway involving a disolvated complex was found to be the most favored one (see the Supporting Information, Figure S19) and thus was used for comparison with the Cu(dcpe) system. In this case, after coordination of 2 to K to form complex L, a lower activation energy barrier of 12.9 kcal/mol was found for the N–O bond oxidative addition, thus representing a feasible pathway for the formation of 4 at the reaction temperature (Figure 5, green pathway). In sharp contrast, the activation energy barrier for the formation of C–C coupling product 3 from imine complex O (Figure 5, orange pathway) was significantly higher for this system (ΔG‡ = 24.6 kcal/mol).
Figure 5.
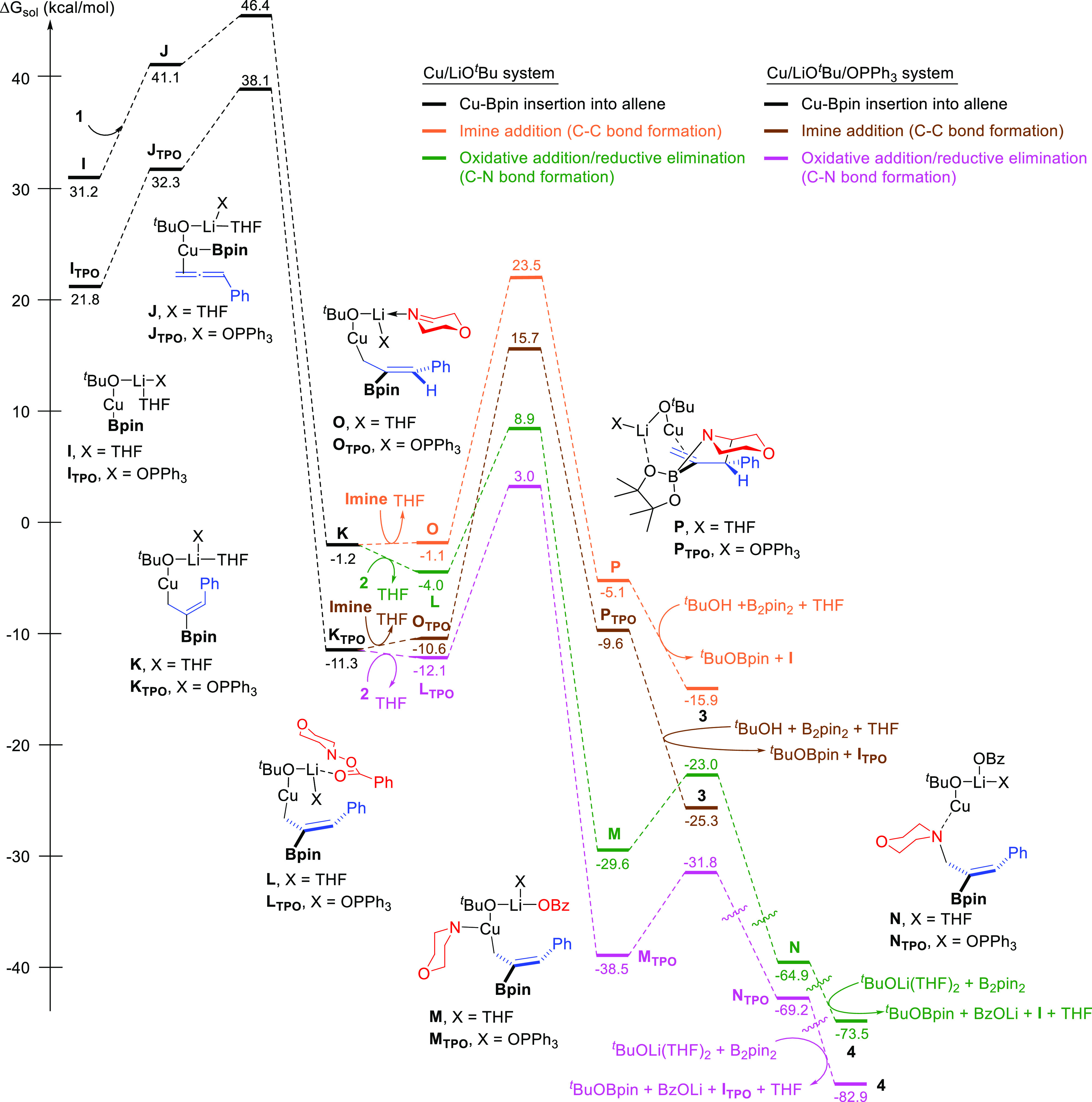
Free energy profile for the pathways leading to the formation of C–C coupling product 3 and C–N coupling product 4 involving dcpe-free copper systems. Computational studies were performed at the B3LYP-D3/6-311++G(d,p)-SDDTHF(SMD)//B3LYP-D3/6-31G(d)-SDD level of theory. Energies are relative to complex C combined with those of the relevant substrates.
In line with these DFT calculations, 31P NMR spectroscopic studies revealed that the dcpe ligand dissociates from the metal center during the formation of the copper tert-butoxide complex. When CuCl (1.0 equiv), dcpe (1.1 equiv), and LiOtBu (2.0 equiv) were mixed (Figure 6A, spectrum I), we observed the formation of Cu(dcpe)-OtBu (Q), derived aggregates R,23 and a free dcpe ligand that most probably involves formation of (CuOtBu)n,25 which in the presence of excess LiOtBu would form phosphine-free copper bis(tert-butoxide) species S. Addition of B2pin2 led to the disappearance of the LCu-OtBu signals, and a new peak assigned to Cu(dcpe)-Bpin (A) was formed (Figure 6A, spectrum II). Subsequent addition of phenylallene afforded the allyl-Cu(dcpe) complex C with ∼50% conv. (Figure 6A, spectrum III). In both cases, the presence of a free dcpe ligand suggests that there is no ligand reassociation and points at the presence of nonligated Cu-Bpin complex I and allyl-Cu complex K, respectively.
Figure 6.
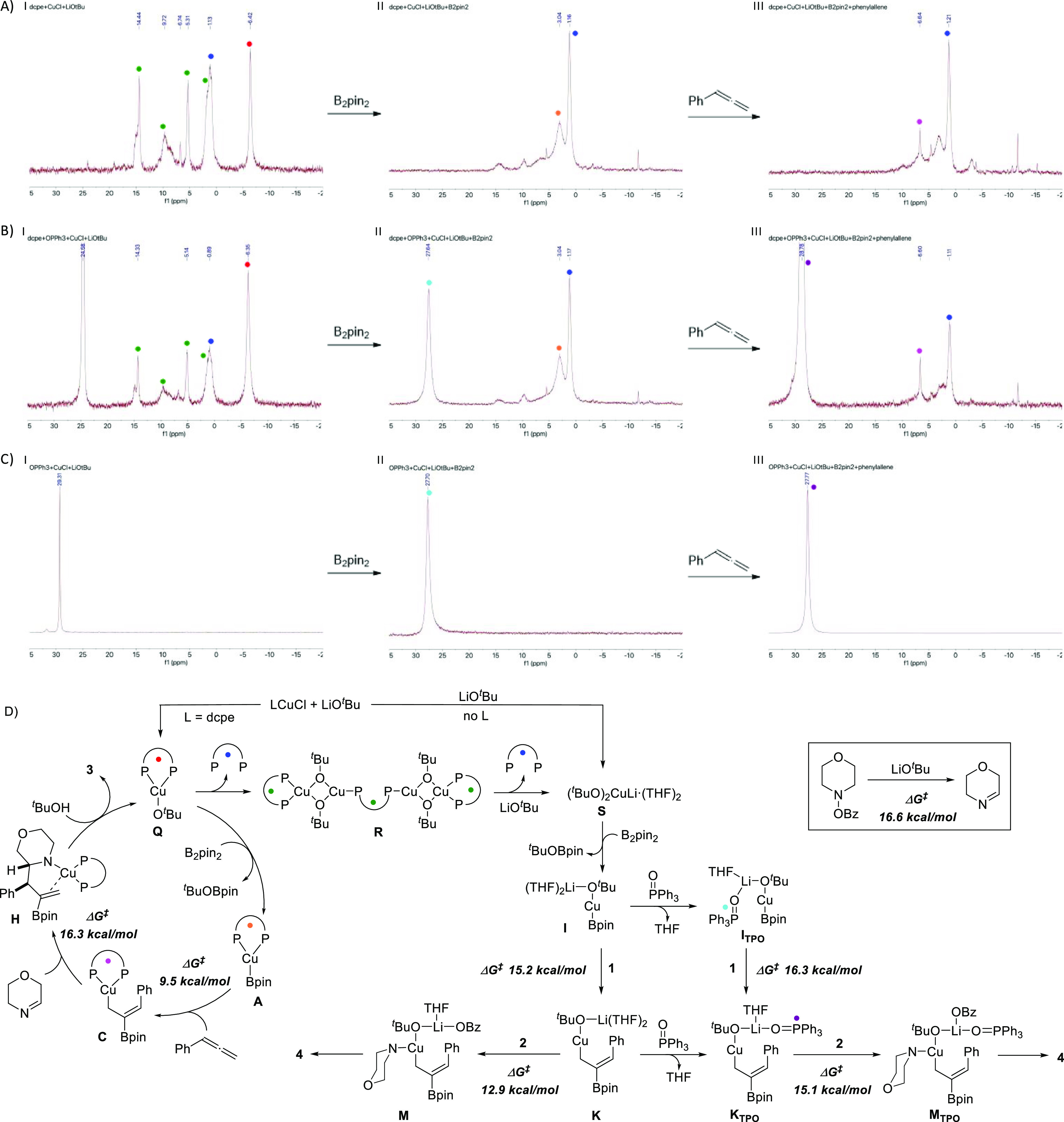
Spectroscopic 31P NMR study of (A) CuCl/dcpe/LiOtBu system, (B) CuCl/dcpe/P(O)Ph3/LiOtBu system, and (C) CuCl/P(O)Ph3/LiOtBu system. (D) Proposed mechanism.
In Cu(I) complexes, alkoxide ligands mainly act as σ-donors, thus providing the oxygen atom enough Lewis basicity to facilitate aggregation by alkoxo bridging in a process that can lead to ligand dissociation.23,25 Accordingly, the large size of the dcpe ligand and its hemilability may favor the formation of oligomeric species and ligand dissociation at the Cu-OtBu stage. Taken together, these experiments suggest that dcpe ligand dissociation is problematic and leads to phosphine-free Cu species, which may be responsible for the formation of side-product 4 especially at the beginning of the reaction when the imine concentration is low. In this sense, the lower chemoselectivity observed when sodium methoxide is used as the base (Table 1, entry 17) may be due to the higher propensity of the resulting copper alkoxide complexes to undergo aggregation.23
So, what is the role of the catalytic Lewis base additive and why does it increase chemoselectivity? 31P NMR of a mixture of CuCl (1.0 equiv), dcpe (1.1 equiv) P(O)Ph3 (1.0 equiv), and LiOtBu (2.0 equiv) also revealed the formation of Cu aggregates R and ligand dissociation, although in a minor extent (Figure 6B, spectrum I).26 Under these conditions, formation of Cu(dcpe)-OtBu Q was mainly observed, while broadening of the P(O)Ph3 may suggest coordination to other Cu species (Figure 6B, spectrum I). Addition of B2pin2 yielded Cu(dcpe)-Bpin A, a free dcpe ligand, and new coordinated P(O)Ph3 species ITPO (Figure 6B, spectrum II). Interestingly, this same species ITPO (27.7 ppm) was formed when CuCl (1.0 equiv), P(O)Ph3 (1.1 equiv), LiOtBu (2.0 equiv), and B2pin2 (1.2 equiv) were mixed in the absence of dcpe (Figure 6C, spectrum II). This, together with the fact that the same dcpe intermediates are observed in both experiments (Figure 6A,B), may suggest that no heteroleptic Cu-Bpin complex is formed in the presence of P(O)Ph3. A similar behavior was observed upon allene addition that yielded allyl-Cu(dcpe) complex C and bound P(O)Ph3 species, which appear together with a free dcpe ligand (Figure 6B, spectrum III). These experiments suggest that P(O)Ph3 has a dual role by reducing aggregation (and thus ligand dissociation) and, in a higher extent, by interacting with the Cu intermediates generated from the phosphine-free CuOtBu to produce a likely less active system KTPO for C–N bond formation. Indeed, DFT calculations systematically showed higher activation energy barriers for all the pathways related to the Cu/LiOtBu/POPh3 system (Figure 5, pink and brown pathways) when compared to those of the phosphine-free Cu system (Figure 5, green and orange pathways). The fact that only a trace amount of product 4 was obtained when P(O)Ph3 was used as the sole ligand (Table 1, entry 15) also supports such a deactivating interaction.
Further demonstration of the effect of the catalytic Lewis base additive on the reaction outcome was obtained by microkinetic analysis27 of the relative final distribution of products at 298 K according to the calculated Gibbs energy profiles (Figures 4 and 5), using the conditions shown in Table 1, entries 10 and 12. In the absence of P(O)Ph3, and assuming a 1:1 ratio between Cu(dcpe)Bpin (A) and Cu(OtBu)(Bpin)Li (I) complexes,28 microkinetic simulation shows that formation of C–N coupling product 4 is competitive at initial times when the imine concentration is still low. As a result, products 3:4 are formed in a 1.5:1 ratio (Figure 7a). In the presence of P(O)Ph3 (Figure 7b), the lower efficiency of complex ITPO results in almost the exclusive formation of C–C coupling product 3 (3:4, 7.5:1 ratio).
Figure 7.
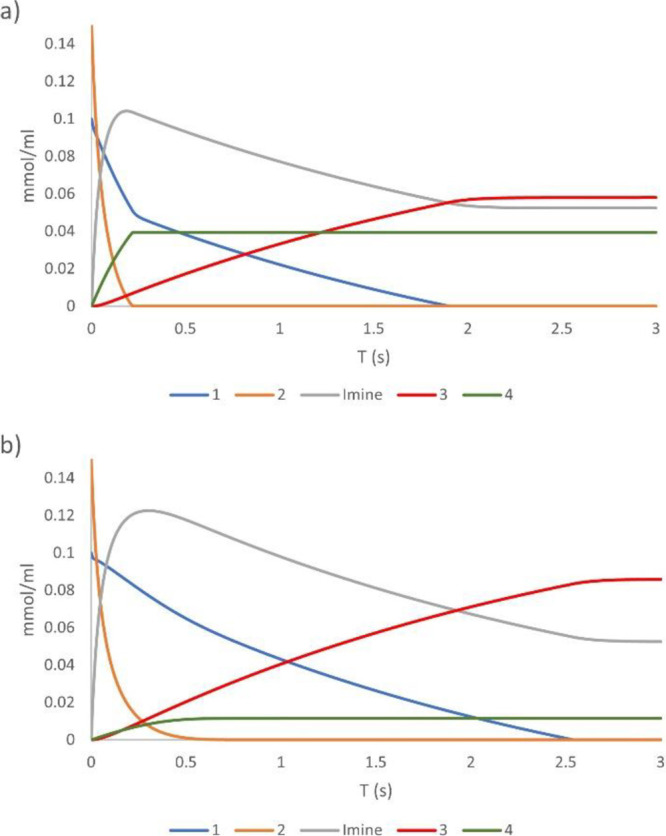
Microkinetic simulations according to the calculated Gibbs energy profiles under (a) P(O)Ph3-free conditions (Table 1, entry 10) and (b) in the presence of P(O)Ph3 (Table 1, entry 12).
The above findings show that the homoleptic Cu/dcpe system is responsible for the C–C bond formation (Figure 6D). Ligand dissociation leads to the generation of new phosphine-free copper species with different reactivities, which results in a decrease of chemoselectivity. The presence of P(O)Ph3 does not interfere in the Cu/dcpe catalytic cycle but coordinates to the phosphine-free Cu species likely by metal ion chelation29 leading to less-active intermediates.30 Thus, the Lewis base additive inhibits the catalytic activity toward C–N coupling but does not participate in the C–C bond forming event.
Conclusions
In summary, we have developed a copper-catalyzed borylative α-C–H allylation of cyclic O-benzoyl hydroxylamines. The method provides functionalized cyclic secondary amines with very high levels of chemo-, regio-, and stereoselectivity. Interesting structural features of these products are the presence of a synthetically versatile boron-containing allyl group and an azaboraspiro structure established by a dative nitrogen–boron coordination. This novel transformation occurs via the trapping of a catalytic allylcopper intermediate with a cyclic imine, which is in situ generated from the O-benzoyl hydroxylamine. Key to selectively achieve this transformation is the use of a catalytic Lewis base additive in combination with a Cu/dcpe catalyst. A combined spectroscopic and computational study reveals that the involved bis-phosphine Cu-alkoxide species are prone to ligand loss, resulting in phosphine-free Cu intermediates that lead to a lower chemoselectivity. The Lewis base additive deactivates these species by metal ion chelation, thus precluding C–N bond formation and allowing the C–C bond forming product to be obtained with excellent selectivity. We expect that these new findings will serve as the basis for the design of new transformations, especially those that involve the use of hemilabile ligands.
Acknowledgments
Financial support from the Spanish Ministry of Science and Innovation (PID2020-118237RB-I00, PID2020-118048GB-I00, and the ORFEO-CINQA network RED2018-102387-T), Xunta de Galicia (ED431C 2018/04; Centro singular de investigación de Galicia accreditation 2019-2022, ED431G 2019/03), and the European Union (European Regional Development Fund, ERDF) is gratefully acknowledged. B.P.-S., A.V.-R., and E.R.-C. thank Xunta de Galicia for predoctoral fellowships. We also acknowledge the use of RIAIDT-USC analytical facilities and CESGA (Xunta de Galicia) for computational time.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c07969.
List of starting materials and ligands, optimization studies, experimental procedures and compound characterization data, imine formation experiments, details of 31P NMR study of copper intermediates, microkinetic simulations and computational details, Cartesian coordinates, imaginary frequencies, and absolute energies in hartrees for all optimized geometries (PDF)
Author Contributions
† B.P.-S. and A.V.-R. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Vitaku E.; Smith D. T.; Njardarson J. T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- a Campos K. R. Direct sp3 C–H bond activation adjacent to nitrogen in heterocycles. Chem. Soc. Rev. 2007, 36, 1069–1084. 10.1039/B607547A. [DOI] [PubMed] [Google Scholar]; b Mitchell E. A.; Peschiulli A.; Lefevre N.; Meerpoel L.; Maes B. U. W. Direct α-functionalization of saturated cyclic amines. Chem. – Eur. J. 2012, 18, 10092–10142. 10.1002/chem.201201539. [DOI] [PubMed] [Google Scholar]
- a Campos K. R.; Klapars A.; Waldman J. H.; Dormer P. G.; Chen C.-Y. Enantioselective, palladium-catalyzed α-arylation of N-Boc-pyrrolidine. J. Am. Chem. Soc. 2006, 128, 3538–3539. 10.1021/ja0605265. [DOI] [PubMed] [Google Scholar]; b Stead D.; Carbone G.; O’Brien P.; Campos K. R.; Coldham I.; Sanderson A. Asymmetric deprotonation of N-Boc piperidine: react IR monitoring and mechanistic aspects. J. Am. Chem. Soc. 2010, 132, 7260–7261. 10.1021/ja102043e. [DOI] [PubMed] [Google Scholar]; c Seel S.; Thaler T.; Takatsu K.; Zhang C.; Zipse H.; Straub B. F.; Mayer P.; Knochel P. Highly Diastereoselective Arylations of Substituted Piperidines. J. Am. Chem. Soc. 2011, 133, 4774–4777. 10.1021/ja201008e. [DOI] [PubMed] [Google Scholar]; d Lin W.; Zhang K.-F.; Baudoin O. Regiodivergent enantioselective C–H functionalization of Boc-1,3-oxazinanes for the synthesis of β2- and β3-amino acids. Nat. Catal. 2019, 2, 882–888. 10.1038/s41929-019-0336-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pastine S. J.; Gribkov D. V.; Sames D. sp3 C–H bond Arylation Directed by Amidine Protecting Group: α-Arylation of Pyrrolidines and Piperidines. J. Am. Chem. Soc. 2006, 128, 14220–14221. 10.1021/ja064481j. [DOI] [PubMed] [Google Scholar]; b Jain P.; Verma P.; Xia G.; Yu J.-Q. Enantioselective Amine α-Functionalizations via Palladium-Catalysed C–H Arylation of Thioamides. Nat. Chem. 2017, 9, 140–144. 10.1038/nchem.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Greßies S.; Klauck F. J. R.; Kim J. H.; Daniliuc C. G.; Glorius F. Ligand-Enabled Enantioselective Csp3–H Activation of Tetrahydroquinolines and Saturated Aza-Heterocyles by RhI. Angew. Chem., Int. Ed. 2018, 57, 9950–9954. 10.1002/anie.201805680. [DOI] [PubMed] [Google Scholar]
- a McNally A.; Prier C. K.; MacMillan D. W. C. Discovery of an α-Amino C–H Arylation Reaction using The Strategy of Accelerated Serendipity. Science 2011, 334, 1114–1117. 10.1126/science.1213920. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Beatty J. W.; Stephenson C. R. J. Amine Functionalization via Oxidative Photoredox Catalysis: Methodology Development and Complex Molecule Synthesis. Acc. Chem. Res. 2015, 48, 1474–1484. 10.1021/acs.accounts.5b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shaw M. H.; Shurtleff V. W.; Terrett J. A.; Cuthbertson J. D.; MacMillan D. W. C. Native functionality in triple catalytic cross-coupling: sp3 C–H bonds as latent nucleophiles. Science 2016, 352, 1304–1308. 10.1126/science.aaf6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haibach M. C.; Seidel D. C–H bond functionalization through intramolecular hydride transfer. Angew. Chem., Int. Ed. 2014, 53, 5010–5036. 10.1002/anie.201306489. [DOI] [PubMed] [Google Scholar]
- Davies H. M. L.; Hansen T.; Hopper D. W.; Panaro S. A. Highly Regio-, Diastereo-, and Enantioselective C–H Insertions of Methyl Aryldiazoacetates into Cyclic N-Boc-Protected Amines. Asymmetric synthesis of Novel C2-Symmetric Amines and Threo-Methylphenidate. J. Am. Chem. Soc. 1999, 121, 6509–6510. 10.1021/ja9910715. [DOI] [Google Scholar]
- a Rouchaud A.; Braekman J.-C. Synthesis of New Analogues of the Tetraponerines. Eur. J. Org. Chem. 2009, 2009, 2666. 10.1002/ejoc.200900064. [DOI] [Google Scholar]; b Gu R.; Flidrova K.; Lehn J.-M. Dynamic Covalent Metathesis in the C=C/C=N Exchange between Knoevenagel Compounds and Imines. J. Am. Chem. Soc. 2018, 140, 5560–5568. 10.1021/jacs.8b01849. [DOI] [PubMed] [Google Scholar]
- a Chen W.; Ma L.; Paul A.; Seidel D. Direct α-C–H bond functionalization of unprotected cyclic amines. Nat. Chem. 2018, 10, 165–169. 10.1038/nchem.2871. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Paul A.; Seidel D. α-Functionalization of Cyclic Secondary Amines: Lewis Acid Promoted Addition of Organometallics to Transient Imines. J. Am. Chem. Soc. 2019, 141, 8778–8782. 10.1021/jacs.9b04325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent reviews, see:; a Fujihara T.; Tsuji Y. Cu-Catalyzed Borylative and Silylative Transformations of Allenes: Use of β-Functionalized Allyl Copper Intermediates in Organic Synthesis. Synthesis 2018, 50, 1737–1749. 10.1055/s-0036-1591777. [DOI] [Google Scholar]; b Whyte A.; Torelli A.; Mirabi B.; Zhang A.; Lautens M. Copper-Catalyzed Borylative Difunctionalization of π-Systems. ACS Catal. 2020, 10, 11578–11622. 10.1021/acscatal.0c02758. [DOI] [Google Scholar]; c Das K. K.; Manna S.; Panda S. Transition metal catalyzed asymmetric multicomponent reactions of unsaturated compounds using organoboron reagents. Chem. Commun. 2021, 57, 441–459. 10.1039/d0cc06460b. [DOI] [PubMed] [Google Scholar]
- For selected examples, see:; a Meng F.; Jang H.; Jung B.; Hoveyda A. H. Cu-Catalyzed Chemoselective Preparation of 2-(Pinacolato)boron-Substituted Allylcopper Complexes and their In Situ Site-, Diastereo-, and Enantioselective Additions to Aldehydes and Ketones. Angew. Chem. Int. Ed. 2013, 52, 5046–5051. 10.1002/anie.201301018. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Meng F.; McGrath K. P.; Hoveyda A. H. Multifunctional organoboron compounds for scalable natural product synthesis. Nature 2014, 513, 367–374. 10.1038/nature13735. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rae J.; Yeung K.; McDouall J. J. W.; Procter D. J. Copper-Catalyzed Borylative Cross-Coupling of Allenes and Imines: Selective Three-Component Assembly of Branched Homoallyl Amines. Angew. Chem., Int. Ed. 2016, 55, 1102–1107. 10.1002/anie.201508959. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jang H.; Romiti F.; Torker S.; Hoveyda A. H. Catalytic diastereo- and enantioselective additions of versatile allyl groups to N–H ketimines. Nat. Chem. 2017, 9, 1269–1275. 10.1038/nchem.2816. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Fujihara T.; Sawada A.; Yamaguchi T.; Tani Y.; Terao J.; Tsuji Y. Boraformylation and Silaformylation of Allenes. Angew. Chem., Int. Ed. 2017, 56, 1539–1543. 10.1002/anie.201611314. [DOI] [PubMed] [Google Scholar]; f Yeung K.; Talbot F. J. T.; Howell G. P.; Pulis A. P.; Procter D. J. Copper-Catalyzed Borylative Multicomponent Synthesis of Quaternary α-Amino Esters. ACS Catal. 2019, 9, 1655–1661. 10.1021/acscatal.8b04563. [DOI] [Google Scholar]; g Deng H.; Meng Z.; Wang S.; Zhang Z.; Zhang Y.; Shangguan Y.; Yang F.; Yuan D.; Guo H.; Zhang C. Enantioselective Copper-Catalyzed Three-Component Carboboronation of Allenes: Access to Functionalized Dibenzo[b,f][1,4]oxazepine Derivatives. Adv. Synth. Catal. 2019, 361, 3582–3587. 10.1002/adsc.201900503. [DOI] [Google Scholar]
- Li G.; Qian S.; Wang C.; You J. Palladium(II)-Catalyzed Dehydrogenative Cross-Coupling between Two Csp3-H Bonds: Unexpected C=C Bond Formation. Angew. Chem. Int. Ed. 2013, 52, 7837–7840. 10.1002/anie.201303099. [DOI] [PubMed] [Google Scholar]
- For selected examples, see:; a Matsuda N.; Hirano K.; Satoh T.; Miura M. Regioselective and Stereospecific Copper-Catalyzed Aminoboration of Styrenes with Bis(pinacolato)diboron and O-Benzoyl-N, N-dialkylhydroxylamines. J. Am. Chem. Soc. 2013, 135, 4934–4937. 10.1021/ja4007645. [DOI] [PubMed] [Google Scholar]; b Sakae R.; Hirano K.; Miura M. Ligand-Controlled Regiodivergent Cu-Catalyzed Aminoboration of Unactivated Terminal Alkenes. J. Am. Chem. Soc. 2015, 137, 6460–6463. 10.1021/jacs.5b02775. [DOI] [PubMed] [Google Scholar]; c Zhu S.; Niljianskul N.; Buchwald S. L. Enantio- and Regioselective CuH-Catalyzed Hydroamination of Alkenes. J. Am. Chem. Soc. 2013, 135, 15746–15749. 10.1021/ja4092819. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Shi S.-L.; Buchwald S. L. Copper-catalysed selective hydroamination reactions of alkynes. Nat. Chem. 2015, 7, 38–44. 10.1038/nchem.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Yang Y.; Shi S.-L.; Niu D.; Liu P.; Buchwald S. L. Catalytic asymmetric hydroamination of unactivated internal olefins to aliphatic amines. Science 2015, 349, 62–66. 10.1126/science.aab3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For full optimization studies see the Supporting Information.
- For examples on copper-catalyzed protoboration of allenes, see:; a Semba K.; Shinomiya M.; Fujihara T.; Terao J.; Tsuji Y. Highly Selective Copper-Catalyzed Hydroboration of Allenes and 1,3-Dienes. Chem. – Eur. J. 2013, 19, 7125–7132. 10.1002/chem.201300443. [DOI] [PubMed] [Google Scholar]; b Meng F.; Jung B.; Haeffner F.; Hoveyda A. H. NHC-Cu-Catalyzed Protoboration of Monosubstituted Allenes. Ligand-Controlled Site Selectivity, Application to Synthesis and Mechanism. Org. Lett. 2013, 15, 1414–1417. 10.1021/ol4004178. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yuan W.; Ma S. Ligand Controlled Highly Selective Copper-Catalyzed Borylcuprations of Allenes with Bis(pinacolato)diboron. Adv. Synth. Catal. 2012, 354, 1867–1872. 10.1002/adsc.201100929. [DOI] [Google Scholar]
- For examples of other metal-catalyzed transformations in which the use of a Lewis base additive plays a key role in the selectivity outcome, see:; a Evans D. A.; Fu G. C.; Anderson B. A. Mechanistic Study of the Rhodium(I)-Catalyzed Hydroboration Reaction. J. Am. Chem. Soc. 1992, 114, 6679–6685. 10.1021/ja00043a010. [DOI] [Google Scholar]; b Petrone D. A.; Yoon H.; Weinstabl H.; Lautens M. Additive Effects in the Palladium-Catalyzed Carboiodination of Chiral N-Allyl Carboxamides. Angew. Chem., Int. Ed. 2014, 53, 7908–7912. 10.1002/anie.201404007. [DOI] [PubMed] [Google Scholar]; c Yoon H.; Marchese A. D.; Lautens M. Carboiodination Catalyzed by Nickel. J. Am. Chem. Soc. 2018, 140, 10950–10954. 10.1021/jacs.8b06966. [DOI] [PubMed] [Google Scholar]
- Same chemoselectivity was observed for the coupling of aryl bromides and aryl triflates, although in these cases products were obtained in diminished yield. See Supporting Information for details.
- a Lam P. Y. S.; Clark C. G.; Saubern S.; Adams J.; Winters M. P.; Chan D. M. T.; Combs A. New aryl/heteroaryl C-N bond cross-coupling reactions via arylboronic acid/cupric acetate arylation. Tetrahedron Lett. 1998, 39, 2941–2944. 10.1016/S0040-4039(98)00504-8. [DOI] [Google Scholar]; b Vantourout J. C.; Law R. P.; Isidro-Llobet A.; Atkinson S. J.; Watson A. J. B. Chan–Evans–Lam Amination of Boronic Acid Pinacol (BPin) Esters: Overcoming the Aryl Amine Problem. J. Org. Chem. 2016, 81, 3942–3950. 10.1021/acs.joc.6b00466. [DOI] [PubMed] [Google Scholar]; c Chen J.-Q.; Li J.-H.; Dong Z.-B. A Review on the Latest Progress of Chan-Lam Coupling Reaction. Adv. Synth. Catal. 2020, 362, 3311–3331. 10.1002/adsc.202000495. [DOI] [Google Scholar]
- For copper-catalyzed protoboration of alkenyl boronates, see:; a Lee Y.; Jang H.; Hoveyda A. H. Vicinal Diboronates in High Enantiomeric Purity through Tandem Site-Selective NHC-Cu-Catalyzed Boron-Copper Additions to Terminal Alkynes. J. Am. Chem. Soc. 2009, 131, 18234–18235. 10.1021/ja9089928. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jung H.-Y.; Yun J. Copper-Catalyzed Double Borylation of Silylacetylenes: Highly Regio- and Stereoselective Synthesis of Syn-Vicinal Diboronates. Org. Lett. 2012, 14, 2606–2609. 10.1021/ol300909k. [DOI] [PubMed] [Google Scholar]; c Zhao W.; Montgomery J. Cascade Copper-Catalyzed 1,2,3-Trifunctionalization of Terminal Allenes. J. Am. Chem. Soc. 2016, 138, 9763–9766. 10.1021/jacs.6b05216. [DOI] [PubMed] [Google Scholar]
- Tobisch S. Copper-Catalysed Aminoboration of Vinylarenes with Hydroxylamine Esters—A Computational Mechanistic Study. Chem. – Eur. J. 2017, 23, 17800–17809. 10.1002/chem.201703803. [DOI] [PubMed] [Google Scholar]
- Protonation step from intermediate H was found to be almost barrierless. See Supporting Information, Figure S18.
- Ryu H.; Park J.; Kim H. K.; Park J. Y.; Kim S.-T.; Baik M.-H. Pitfalls in Computational Modeling of Chemical Reactions and How To Avoid Them. Organometallics 2018, 37, 3228–3239. 10.1021/acs.organomet.8b00456. [DOI] [Google Scholar]
- Lee J.; Radomkit S.; Torker S.; del Pozo J.; Hoveyda A. H. Mechanism-based enhancement of scope and enantioselectivity for reactions involving a copper-substituted stereogenic carbon centre. Nat. Chem. 2018, 10, 99–108. 10.1038/nchem.2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tallmadge E. H.; Jermaks J.; Collum D. B. Structure–Reactivity Relationships in Lithiated Evans Enolates: Influence of Aggregation and Solvation on the Stereochemistry and Mechanism of Aldol Additions. J. Am. Chem. Soc. 2016, 138, 345–355. 10.1021/jacs.5b10980. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Turlik A.; Ando K.; Mackey P.; Alcock E.; Light M.; McGlacken G. P.; Houk K. N. Mechanism and Origins of Stereoselectivity of the Aldol-Tishchenko Reaction of Sulfinimines. J. Org. Chem. 2021, 86, 4296–4303. 10.1021/acs.joc.0c02862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmen T. H.; Goeden G. V.; Huffman J. C.; Geerts R. L.; Caulton K. G. Alcohol Elimination Chemistry of (CuOtBu)4. Inorg. Chem. 1990, 29, 3680–3685. 10.1021/ic00344a012. [DOI] [Google Scholar]
- Excess of dcpe ligand completely precluded the formation of Cu aggregates. However, it did not lead to the formation of Cu(dcpe)-OtBu and a different Cu species was formed under these conditions. See the Supporting Information for details.
- Microkinetic analysis was performed using COPASI software:; a COPASI 4.36, Build 260. 2022, http://copasi.org/ (accessed 2022-05-24).; b Hoops S.; Sahle S.; Gauges R.; Lee C.; Pahle J.; Simus N.; Singhal M.; Xu L.; Mendes P.; Kummer U. COPASI—a COmplex PAthway Simulator. Bioinformatics 2006, 22, 3067–3074. 10.1093/bioinformatics/btl485. [DOI] [PubMed] [Google Scholar]
- The ratio between the different Cu species was estimated according to the NMR studies. See the Supporting Information for further details.
- For metal ion chelation in copper alkoxide complexes and its effect in reactivity, see:; Konovalov A. I.; Benet-Buchholz J.; Martin E.; Grushin V. V. The critical effect of the countercation in the direct cupration of fluoroform with [Cu(OR)2]−. Angew. Chem., Int. Ed. 2013, 52, 11637–11641. 10.1002/anie.201306272. [DOI] [PubMed] [Google Scholar]
- For an example in which in a system where two different copper catalysts are formed, and the action of the most active catalyst is delayed by means of a non-productive chemically fuelled side-cycle so that a second less active catalyst engages first, see; Zhang S.; del Pozo J.; Romiti F.; Mu Y.; Torker S.; Hoveyda A. H. Delayed catalyst function enables direct enantioselective conversion of nitriles to NH2-amines. Science 2019, 364, 45–51. 10.1126/science.aaw4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.