

Abstract
Lung and airway epithelial cells generated in vitro from human pluripotent stem cells (hPSC) have applications in regenerative medicine, modeling of lung disease, drug screening and studies of human lung development. This protocol is an extension to our previously published protocol on the directed differentiation of lung and airway epithelium from hPSC that modifies the technique and offers additional applications. Here we describe a strategy for directed differentiation of hPSC into mature lung and airway epithelial cells obtained through maturation of NKX2.1+ hPSC-derived lung progenitors in a 3-dimensional (3D) matrix of collagen I in the absence of glycogen synthase kinase 3 (GSK3) inhibition. This protocol is conducted in defined media conditions, has a duration of 50 to 80 days, does not require reporter lines, and results in cultures containing mature alveolar type II and I cells as well as airway basal, ciliated, club and neuroendocrine cells. We also present a flow cytometry strategy to assess maturation in the cultures. Several of these populations, including mature NGFR+ basal cells, can be prospectively isolated by cell sorting and expanded for further investigation.
Keywords: directed-differentiation, human pluripotent stem cells, lung, airway, 3D maturation
Introduction
Directed differentiation of human pluripotent stem cells (hPSC) into terminally mature tissues or cell types holds major promise for the development of regenerative medicine strategies, and provides tractable systems for studies into human development and cellular physiology, human relevant disease models and drug screening assays1.
The lung consists of a branching airway system that terminates in alveoli where gas exchange takes place, and contains multiple cell types, including basal, ciliated, secretory, goblet and neuroendocrine (NE) cells in the airways, and alveolar type I (ATI) and surfactant-producing, cuboidal alveolar type II (ATII) cells in the alveoli. In the recent years, major efforts have resulted in considerable progress in the generation of lung epithelium from hPSC2,3,12–15,4–11. Although there are differences between protocols2,3,12–15,4–11, at their core all try to recapitulate in vitro the development that occurs in vivo through carefully dosed and timed activation and/or inhibition of key signaling pathways16,17. Thus to generate lung, definitive endoderm (DE), the germ layer from which the lung epithelium arises18, is first specified, followed by its patterning to anterior foregut endoderm (AFE) and induction of lung progenitor specification with subsequent lineage specification and maturation16,17.
The protocol presented here is an extension to our previous protocol19 (doi:10.1038/nprot.2015.023 ) for the in vitro generation of lung and airway progenitor cells from hPSC in 2-dimensional (2D) cultures. The current extension shows how 3D culture promotes improved maturation of ATI and ATII cells as well as airway basal (BCs), ciliated, club and NE cells and is applicable to any hPSC line capable of generating endoderm. Importantly, BCs, the stem cells of the airway, can be generated, isolated and expanded from these cultures.
The initial stages of the protocol, the specification and expansion of NKX2.1+ lung progenitors, remain mostly unchanged. The major change is maturation of these lung progenitors in a 3D matrix of collagen I. We have used this protocol to yield the most mature multi-lineage alveolar and airway cells reported so far13. More specifically, this protocol is currently the only method available to generate BCs that express NGFR, a marker that only appears after birth5,20, and morphologically recognizable ATI cells in vitro13.
Comparison between this protocol and other methods available
We were the first to describe the generation of NKX2.1+ lung progenitors in 2D adherent cultures with efficiencies as high as >90% of total cells in culture5. As the in vitro generation of NKX2.1+ lung progenitors with high efficiency is challenging, several groups have developed protocols that require the use of strategies to purify or enrich these prior to inducing maturation. One such strategy is the use of reporter lines to isolate lung progenitors. In two reports from the same lab McCauley et al.10 specified for lung progenitors, and isolated NKX2.1GFP+ cells at day 15 for further culture in Matrigel 3D media to yield proximal airway spheroids devoid of distal potential, while Jacob et al.11 resorted to a NKX2.1GFPSFTPCtdTomato reporter cell line to isolate NKX2.1GFP+SFTPCtdTomato+ cells for further culture in Matrigel to generate spheroids containing ATII cells. While these protocols are advantageous when differentiation efficiency is modest, they do not allow for investigations such as lineage tracing studies, that require presence of broad variety of lung and airway cells, and are not universally applicable.
Another strategy is the identification of surface markers by which NKX2.1+ lung progenitors can be isolated from the cultures. Gotoh et al.7 have identified carboxypeptidase M (CPM) as a specific marker of NKX2.1+ progenitors which was then used by Konishi et al.8 to select for NKX2.1+ cells, followed by maturation in Matrigel co-culture with fetal human lung fibroblasts to yield spheroids containing ciliated and NE cells, and by Yamamoto et al.12 to generate ATII containing spheroids in Matrigel. Yamamoto et al.12 also relied on reporter lines to perform serial isolation of SFTPCGFP+ cells to form new spheroids in Matrigel as a strategy to incrementally enrich the spheroids for ATII cells. Hawkins et al. used differential expression of CD47 and CD26 to show that CD47hiCD26lo cells are enriched in NKX2.1+ lung progenitors21.
Our own group has published an alternative strategy that leads to the generation of branching organoids9. At variance with the current protocol, this strategy involves generating ‘lung bud organoids’ from AFE in suspension culture, which, after plating in Matrigel, generate branching organoids. The advantage of this latter protocol is that not only pulmonary epithelium but also some pulmonary mesenchyme is generated, allowing to model of at least some forms of pulmonary fibrosis22 and that the organoid morphology and genome-wide expression signature is most similar to that of fetal lung a the second trimester of human gestation9. Disadvantages however are that maturation beyond the second trimester of human gestation was, thus far, not achievable in this model and that, while proximal cells are present, the organoids predominantly contain developing distal cells, a large fraction of which were ATII cells. Another group also reported generation of human lung organoids14,23. However, while these small structures contained cells expressing markers of lung14 and airway and have some airway potential after subcutaneous xenografting in mice23, they did not show branching structures and were, similarly to our reported organoids, staged as early fetal lung15.
The protocol described here allows further maturation of lung cells in 3D, and produces most known lung and airway cells in proportions that can be manipulated by activating or inhibiting specific pathways13, and many of which can be prospectively isolated from the cultures for further study. This protocol has the advantage of not relying on reporter lines to isolate lung progenitors or desired differentiated lineages and thus can be universally applicable to any hPSC line. We also do not rely on commercially available endoderm kits, using instead an embryoid body approach, from which we obtain higher endoderm yield and, consequentially higher lung specification. Our model is also permissive for the specification of all distal and airway lineages. In addition, while in most studies the maturity of the generated cells goes unreported or is equivalent to 2nd trimester of human gestation13, the cells generated by our protocol exhibit transcriptomic profiles, ultrastructure and an array of lineage-specific markers that are also comparable to those found in postnatal lungs13. Several mature cell types can be isolated using surface markers. Most notably, by using this protocol we were the first to generate NGFR+ BCs13, the stem cells of the postnatal airway20, in vitro. Having generated these cells we were able to isolate and further expand them using techniques established by others 24.
Applications of the protocol
The technology we describe here has numerous potential applications. The wide variety of pulmonary cell types present in the cultures that have reached an advanced stage of maturation allows study of viral tropism and pathogenesis of respiratory viruses25,26. Viral respiratory infection is a major worldwide public health problem and for many respiratory viruses, no reliable models are available except for primates. Modeling monogenic genetic diseases such surfactant deficiency syndromes could be envisaged using this model27–29. In addition, timed-expression of oncogenes may allow modeling of lung cancer and determination of the cell-of-origin, pathogenesis and therapeutic strategies30–32.
In addition, this protocol is useful to study human lung development. As proximodistal specification of the lung is poorly understood, this model lends itself to hypothesis-driven studies or unbiased siRNA and CRISPR screens to find such mechanisms. For example, we described previously how Notch inhibition promotes proximal at the expense of distal development in this model13. The lineage relations among various epithelial cells in the human lung are furthermore not clear1. Since this protocol is permissive to the generation of all lung epithelial lineages simultaneously and with a high degree of maturity from a population of fully specified but not yet committed lung progenitors, this in vitro model may allow to establish lineage relations among human alveolar and airway cells and identify the differentiation branch points, using single cell RNA sequencing, retroviral integration site analysis or even lineage tracing experiments, as progress has been made in the development of lineage tracing technologies for hPSC lines33. Understanding these relations would not only advance our knowledge about human lung development but could prove essential to achieve more efficient generation and maturation of each lineage in vitro. Ultimately this technology may have applications in regenerative medicine of the lung.
This approach can furthermore be used to study the normal biology and physiology of specific cell populations, such as BCs and ATII cells that can be prospectively isolated and expanded from the cultures. Importantly, in contrast to other approaches (see above), genetically encoded reporters are not required, making this protocol much more universally applicable.
Limitations
One limitation of this protocol is that, while the cells bearing ATI markers generated in the cultures show changes in morphology and combined marker expression indicative of ATI specification, expression of AGER in these cells was not detected13, suggesting still incomplete maturation. There are currently no protocols capable of generating mature ATI cells.
A second limitation is that although 3D structures are generated, these are not true organoids as they do not recapitulate the structure of a developing lung. This is in contrast to our previously published lung organoids that also contain some mesoderm with a genome-wide expression pattern consistent with pulmonary mesoderm9. Mesodermal cells were not detectable in the current model.
Similar to the previous version of this protocol19, there is variation across different hPSC lines in the efficiency of lung progenitor generation.
This protocol generates a mixture of proximal and distal cells. This is a potential limitation if particular cell types are required in a study. We can, however, prospectively isolate cell types that have been generated by others using reporters10–12 by sorting for NGFR (BCs) and HT2–280 (ATII cells). Furthermore, for some applications cellular heterogeneity is a strength. As we showed in our original report13, the balance between proximal and distal specification can be modulated by manipulating Notch signaling. This is of practical use to enrich for or deplete of specific cell types but also shows that this model can be used to study mechanisms underlying proximodistal specification. An example of another application to which the presence of a high diversity of lung and airway cells is desirable is the study of respiratory viruses and viral tropism. For example by infecting the cells generated using this protocol with parainfluenza virus, we demonstrate predominant infection of proximal cells (see Anticipated Results).
Experimental design
The first half of the protocol described here is very similar to the previous version19. This protocol starts with the induction of DE by exposing hPSC to high concentrations of Activin A for approximately 72h (steps 1–10, Fig. 1) in suspension culture where embryoid bodies (EBs) are generated. We found that we can bypass the previously reported mouse embryonic fibroblasts (MEF) depletion step and the primitive streak induction phase, which shortens the DE induction stage by two days. AFE is specified in accordance with our previous protocol19, more specifically by dual inhibition of bone morphogenetic protein (BMP) and transforming growth factor beta (TGFβ) signaling for 24h followed by dual inhibition of WNT and TGFβ for another 24h (steps 11–26, Fig. 1). The subsequent generation of lung progenitors from AFE requires 9 days of activation of WNT signaling (simulated in vitro by the small molecular GSK3 inhibitor, CHIR9902134), fibroblast growth factor (FGF), bone morphogenic protein (BMP) and retinoic acid (RA) signaling (steps 27–31, Fig. 2). Lung progenitors are then expanded for an additional 10 days in the presence of CHIR99021 and FGF (steps 32–39, Fig. 2).
Figure 1. Differentiation of hPSC to AFE.

A. Schematic representation of the differentiation protocol until day 6. B-E. Bright-field views after culture of RUES2 ESC to confluence (B), to DE (C) and to AFE (D,E) (representative of n>10 independent experiments). F. Representative example of the flow cytometric profile after staining day 4 RUES2 ESC and sviPSC DE cells for CXCR4 and cKIT (representative of n>10 independent experiments). DE, definitive endoderm; AFE, anterior foregut endoderm.
Figure 2. Differentiation of hPSC to lung progenitors.

A. Schematic representation of the differentiation protocol until day 25. B,C. Bright-field views after culture of RUES2 ESC to lung progenitors at day 15 (B) and day 25 (C) (representative of n>10 independent experiments). D. Representative example of the expression of NKX2.1, SOX2 and SOX9 in RUES2 ESC at day 25 of the differentiation protocol (representative of n>10 independent experiments, scale bar: 100μm). E,F. Representative examples of the expression of NKX2.1, MUC1 and PDPN in RUES2 ESC at days 15 (E) and 25 (F) of the differentiation protocol (representative of n=4 independent experiments, scale bar: 50μm). DE, definitive endoderm; AFE, anterior foregut endoderm; LP, lung progenitor.
The second half of the protocol describes the long-term differentiation and maturation of the lung progenitors. In this updated protocol we have taken the lung progenitors to mature in a 3D matrix of collagen I and in the absence of GSK3 inhibition while agonizing FGF signaling as well as pathways known to induce alveolar maturation35 (steps 40–55, Fig. 3–9). After 25 days in collagen I and maturation media defined airway and alveolar epithelial populations can be found (Fig. 4,5). Extending cultures up to 80 days of culture allows for further maturation of BCs (Fig. 6), without compromising other epithelial lineages (Fig. 7).
Figure 3. Practical aspects of the culture protocol.

A. Representative example of what to expect when disrupting the cell monolayer into cell clumps for day 15 and day 25 replating (steps 35 and 50). The clumps will pellet to the bottom of the conical tube and the media will be cloudy from single cells/very small cell aggregates in suspension. B. Representative example of the bright-field appearance of appropriately sized cell clumps immediately after replating into collagen I gel (steps 49–52) (scale bar 500μM). C. Representative example of the bright-field appearance of small sized cell clumps that exhibit lower survival rate upon replating immediately after replating into collagen I gel (steps 49–52) (scale bar 500μM). D-F. Potential appearances of the collagen I layer over the course of the differentiation. (D) Intact collagen layer at day 50 of the differentiation protocol; (E) Collagen layer partially detached and retracted from the well wall; (F) Contracted collagen I gel (D-F scale bar 2.5mm).
Figure 9. Applications of the differentiation protocol.

A. Schematic representation of HPIV3 infection of day 50 cells differentiated according to our protocol. B. Bright-field view and GFP expression in a representative culture well of day 58 RUES2 ESC differentiated according to the protocol shown in A and infected with GFP expressing-HPIV3 at day 50 (n=3 independent experiments, scale bar: 5mm). C. Higher magnification bright-field views and GFP expression in the cultures 8-days post infection with GFP expressing-HPIV3 (representative of n=3 independent experiments). D. Immunofluorescence staining for viral particle HN and mature airway and distal lung markers 8 days post-infection with HPIV3 (representative of n=3 independent experiments, inset scale bar: 30μm). E. Schematic representation of lentiviral transduction of lung progenitors before embedding in collagen I gels and further differentiation and maturation according to our protocol. F. Representative bright-field views and ZsGreen expression in lung progenitors 44h after transduction with a ZsGreen expressing-lentivirus, according to established protocols (representative of n=3 independent experiments). G. Representative bright-field views and ZsGreen expression of transduced lung progenitors further cultured in collagen I gels according to the protocol shown in A, at the indicated differentiation days (representative of n=3 independent experiments). DE, definitive endoderm; AFE, anterior foregut endoderm; LP, lung progenitor.
Figure 4. Differentiation of hESC-derived lung progenitors to airway and distal cells at day 50 of the differentiation protocol.

A. Schematic representation of the differentiation protocol until day 50. B. Bright-field view after culture of RUES2 ESC to day 50 of the differentiation protocol (representative of n>10 independent experiments, scale bar: 500μm). C-J. Representative examples of the expression of mature markers of distal and airway epithelial cells after culturing RUES2 ESC to day 50 according to the protocol shown in A (representative of n>10 independent experiments, inset scale bar: 20μm). DE, definitive endoderm; AFE, anterior foregut endoderm; LP, lung progenitor.
Figure 5. Differentiation of hiPSC-derived lung progenitors to airway and distal cells at day 50 of the differentiation protocol.

A. Schematic representation of the differentiation protocol until day 50. B. Bright-field view after culture of sviPSC to day 50 of the differentiation protocol (representative of n>10 independent experiments, scale bar: 500μm). C-J. Representative examples of the expression of mature markers of distal and airway epithelial cells after culturing sviPSC to day 50 according to the protocol shown in A (representative of n>5 independent experiments, inset scale bar: 20μm). DE, definitive endoderm; AFE, anterior foregut endoderm; LP, lung progenitor.
Figure 6. Extension of the maturation period yields more mature basal cells.

A. Schematic representation of the differentiation protocol until day 80. B. Representative examples of the expression of mature markers of basal cells, NGFR and KRT14, after culturing RUES2 ESC to day 80 according to the protocol shown in A (representative of n=6 independent experiments, inset scale bar: 20μM). DE, definitive endoderm; AFE, anterior foregut endoderm; LP, lung progenitor.
Figure 7. Expression of mature lung and airway markers in cultures carried to day 80.

A. Schematic representation of the differentiation protocol until day 80. B. Representative examples of the expression of mature markers of distal and airway epithelial cells after culturing RUES2 ESC to day 80 according to the protocol shown in A (representative of n=6 independent experiments). DE, definitive endoderm; AFE, anterior foregut endoderm; LP, lung progenitor.
The cultures can be analyzed at any step of the differentiation using several approaches (Box 1). Here we document differentiation by immunofluorescence (IF) (Fig. 2,4–7) and flow cytometry (Fig 1, 8). Antibodies used are listed in Table 1. Cultures can be analyzed as well by quantitative reverse transcription polymerase chain reaction (qRT-PCR) (Fig 8D) and we provide primer sequences in Suppl. Table 1. Additional methods for analyzing the cultures can be found in our original paper13, and include electron microscopy, RNA sequencing, single cell RNA sequencing, western blot, light sheet microscopy and high-speed live imaging.
Box 1. Options for further analysis of cells.
Immunofluorescence
Remove the media, lift the collagen I gels with the help of a tweezer and transfer to a cryomold.
-
Embed in OCT and snap freeze on top of dry ice for later sectioning.
PAUSEPOINT. Samples in OCT can be stored up to 1 year at −80°C.
Section and stain according to established protocols (see Suppl. Methods for details and Table 1 for suggested antibodies to phenotypically characterize the cultures).
RNA digestion for qRT-PCR:
Remove the media, lift the collagen I gels with the help of a tweezer and transfer each gel to an Eppendorf tube.
Digest the collagen I gels with 150U/mL collagenase type I in IMDM for 45–60 mins at 37°C. We use 750uL-1mL collagenase solution per Eppendorf tube/sample and perform the digestion inside an incubator.
-
Pellet the cells at 300g for 5min at RT, discard the supernatant and lyse with RLT buffer.
PAUSEPOINT Samples in RLT buffer can be stored up to 1 year at −80°C.
Perform qRT-PCR using your preferred protocol. Suggested amplification conditions can be found in Suppl. Methods and primers for lung and airway-specific mRNAs can be found in Suppl. Table 1.
Flow cytometry analysis:
Remove the media, lift the collagen I gels with the help of a tweezer and transfer each gel to an Eppendorf tube.
Digest the collagen I gels with 150U/mL collagenase type I in IMDM for 45–60 mins at 37°C. We use 750uL-1mL collagenase solution per Eppendorf tube/sample and perform the digestion inside an incubator.
Centrifuge at 300g for 5min at RT, discard the supernatant and further dissociate the cells with 500μl of pre-warmed 0.05% (wt/vol) trypsin-EDTA to single cells, which usually takes 10–15 mins.
Once cultures are fully dissociated, stop the reaction by adding 1mL of wash media on top of the trypsin. Centrifuge at 300g for 5min at RT. Discard the supernatant.
Resuspend the cells in flow cytometry buffer, and stain according to established protocols (see Suppl. Methods for details, Table 1 for a list of antibodies frequently used in our lab for flow cytometry and Suppl. Fig. 1 for gating strategy). Specific cell populations can be sorted based on marker expression (see Fig. 8). Sorted populations, such as NGFR+ BCs, can be further expanded on 3T3-J2 feeders24 (Suppl. Fig. 2A), cultured according to established protocols in air-liquid interphase conditions13 (Suppl. Fig. 2B) and cultured in Matrigel as 3D cultures36 (Suppl. Fig. 2C).
Figure 8. Assessment of the culture by flow cytometry.

A. Schematic representation of the differentiation protocol until day 80. B. Representative examples of the flow cytometric profile after staining of day 4 DE, day 15 and day 25 RUES2 ESC differentiated according to A for EPCAM, PDPN and MUC1 (representative of n>10 independent experiments). C. Representative example of the flow cytometric profile after staining of day 50 RUES2 ESC differentiated according to A for EPCAM, MUC1, PDPN, ITGB4, ITGA6, HT2–280 and NGFR (representative of n>10 independent experiments). D. Expression of mRNA for differentiation markers in the indicated populations sorted from day 50 RUES2 ESC differentiated according to A (mean± s.d., n=3 independent experiments, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 one-way ANOVA with Dunnett’s multiple comparison test). E. Representative example of the flow cytometric profile after staining of day 80 RUES2 ESC differentiated according to A for EPCAM, MUC1, PDPN, ITGB4, ITGA6, HT2–280 and NGFR (representative of n=6 independent experiments). F. Representative example of the flow cytometric profile after staining of day 50 RUES2 ESC differentiated in the continued presence of CHIR99021 (CHIR+) for EPCAM, MUC1, PDPN, ITGB4, ITGA6, HT2–280 and NGFR (representative of n>10 independent experiments). Flow cytometry data were acquired on a BD Fortessa analyzer and analyzed on Flowjo v10.2 software. DE, definitive endoderm; AFE, anterior foregut endoderm; LP, lung progenitor.
Table 1:
Antibodies for culture caracterization
| Antibodies used for immunofluorescent staining | ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Name | Host species | Conjugate | Clone number | Manufacturer and Catalog number | RRID | URL | Dilution factor | |
| acTUB | rabbit | unconjugated | D20G3 | Cell Signaling Technology Cat# 5335 | RRID:AB_10544694 | https://scicrunch.org/resolver/RRID:AB_10544694 | 1:400 | |
| AQP5 | goat | unconjugated | G-19 | Santa Cruz Biotechnology Cat# sc-9890 | RRID:AB_2059877 | https://scicrunch.org/resolver/RRID:AB_2059877 | 1:100 | |
| CAV1 | rabbit | unconjugated | D46G3 | Cell Signaling Technology Cat# 3267 | RRID:AB_2275453 | https://scicrunch.org/resolver/RRID:AB_2275453 | 1:400 | |
| COL4 | mouse | unconjugated | COL-94 | Abcam Cat# ab6311 | RRID:AB_305414 | https://scicrunch.org/resolver/RRID:AB_305414 | 1:200 | |
| HN | rat | unconjugated | Moscona Lab - Center for Host Pathogen Interaction, Department of Pediatrics, CUIMC, Cat# VT-10F7-B11 | RRID:AB_2847948 | https://antibodyregistry.org/search.php?q=AB_2847948 | 1:1000 | ||
| HN | rat | unconjugated | Moscona Lab - Center for Host Pathogen Interaction, Department of Pediatrics, CUIMC, Cat# VT-3G9-B9 | RRID:AB_2847947 | https://antibodyregistry.org/search.php?q=AB_2847947 | 1:1000 | ||
| HOPX | rabbit | unconjugated | FL-73 | Santa Cruz Biotechnology Cat# sc-30216 | RRID:AB_2120833 | https://scicrunch.org/resolver/RRID:AB_2120833 | 1:200 | |
| HT1–56 | mouse | unconjugated | Terrace biotech, Cat# TB-29AHT1–56 | RRID:AB_2847898 | https://antibodyregistry.org/search.php?q=AB_2847898 | 1:150 | ||
| HT2–280 | mouse | unconjugated | Terrace biotech, Cat# TB-27AHT2–280 | RRID:AB_2832931 | https://antibodyregistry.org/search.php?q=AB_2832931 | 1:150 | ||
| KRT5 | rabbit | unconjugated | EP1601Y | Thermo Fisher Scientific Cat# MA5–14473 | RRID:AB_10979451 | https://scicrunch.org/resolver/RRID:AB_10979451 | 1:200 | |
| KRT14 | rabbit | unconjugated | LL002 | Abcam Cat# ab7800 | RRID:AB_306091 | https://scicrunch.org/resolver/RRID:AB_306091 | 1:100 | |
| MUC1 | armenian hamster | unconjugated | MH1 (CT2) | Thermo Fisher Scientific Cat# MA5–11202 | RRID:AB_11000874 | https://scicrunch.org/resolver/RRID:AB_11000874 | 1:100 | |
| MUC1 | mouse | APC | 16A | BioLegend Cat# 355608 | RRID:AB_2565312 | https://scicrunch.org/resolver/RRID:AB_2565312 | 1:100 | |
| MUC5B | rabbit | unconjugated | H-300 | Santa Cruz Biotechnology Cat# sc-20119 | RRID:AB_2282256 | https://scicrunch.org/resolver/RRID:AB_2282256 | 1:100 | |
| NGFR | mouse | FITC | ME20.4 | BioLegend Cat# 345104 | RRID:AB_2282828 | https://scicrunch.org/resolver/RRID:AB_2282828 | 1:100 | |
| NKX2.1 (TTF1) | mouse | unconjugated | 8G7G3/1 | Thermo Fisher Scientific Cat# MA5–13961 | RRID:AB_10984070 | https://scicrunch.org/resolver/RRID:AB_10984070 | 1:100 | |
| NKX2.1 (TTF1) | rabbit | unconjugated | polyclonal | Seven Hills Bioreagents, Cat# WRAB-1231 | RRID:AB_2832953 | https://antibodyregistry.org/search.php?q=AB_2832953 | 1:1000 | |
| P63 | mouse | unconjugated | 4A4 | Abcam Cat# ab3239 | RRID:AB_303633 | https://scicrunch.org/resolver/RRID:AB_303633 | 1:100 | |
| PDPN | rat | PE | NC-08 | BioLegend Cat# 337004 | RRID:AB_1595457 | https://scicrunch.org/resolver/RRID:AB_1595457 | 1:100 | |
| SCGB1A1 (CC10) | rat | unconjugated | 394324 | R and D Systems Cat# MAB4218 | RRID:AB_2183286 | https://scicrunch.org/resolver/RRID:AB_2183286 | 1:200 | |
| SFTPB | rabbit | unconjugated | polyclonal | Seven Hills Bioreagents Cat# WRAB-SPB?? | RRID:AB_451724 | https://scicrunch.org/resolver/RRID:AB_451724 | 1:1000 | |
| SOX2 | goat | unconjugated | Y-17 | Santa Cruz Biotechnology Cat# sc-17320 | RRID:AB_2286684 | https://scicrunch.org/resolver/RRID:AB_2286684 | 1:100 | |
| SOX9 | rabbit | unconjugated | polyclonal | Millipore Cat# AB5535 | RRID:AB_2239761 | https://scicrunch.org/resolver/RRID:AB_2239761 | 1:300 | |
| Antibodies used for FACS | ||||||||
|
| ||||||||
| Name | Host species | Conjugate | Clone number | Manufacturer and Catalog number | RRID | URL | Dilution factor | |
| cKIT | mouse | PE | 104D2 | BioLegend Cat# 983304 | RRID:AB_2650654 | https://scicrunch.org/resolver/RRID:AB_2650654 | 1μL per 200.000 cells | |
| CXCR4 | mouse | APC | 12G5 | BioLegend Cat# 306510 | RRID:AB_314616 | https://scicrunch.org/resolver/RRID:AB_314616 | 1μL per 200.000 cells | |
| EpCAM | mouse | PerCP | 1B7 | Thermo Fisher Scientific Cat# 46–9326-42 | RRID:AB_1834413 | https://scicrunch.org/resolver/RRID:AB_1834413 | 1μL per 200.000 cells | |
| ITGA6 | rat | AF-647 | GoH3 | BioLegend Cat# 313610 | RRID:AB_493637 | https://scicrunch.org/resolver/RRID:AB_493637 | 1μL per 200.000 cells | |
| ITGB4 | mouse | PE | 58XB4 | BioLegend Cat# 327808 | RRID:AB_2129146 | https://scicrunch.org/resolver/RRID:AB_2129146 | 1μL per 200.000 cells | |
| MUC1 | mouse | PE-Cy7 | 16A | BioLegend Cat# 355606 | RRID:AB_2564195 | https://scicrunch.org/resolver/RRID:AB_2564195 | 1μL per 200.000 cells | |
| MUC1 | mouse | APC | 16A | BioLegend Cat# 355608, | RRID:AB_2565312 | https://scicrunch.org/resolver/RRID:AB_2565312 | 1μL per 200.000 cells | |
| PDPN | rat | AF-488 | NC-08 | BioLegend Cat# 337006, | RRID:AB_2161951 | https://scicrunch.org/resolver/RRID:AB_2161951 | 1μL per 200.000 cells | |
| PDPN | rat | PE | NC-08 | BioLegend Cat# 337004, | RRID:AB_1595457 | https://scicrunch.org/resolver/RRID:AB_1595457 | 1μL per 200.000 cells | |
| NGFR | mouse | APC | ME20.4 | BioLegend Cat# 345108, | RRID:AB_10645515 | https://scicrunch.org/resolver/RRID:AB_10645515 | 1μL per 200.000 cells | |
| HT2–280 | mouse | unconjugated | Terrace biotech, Cat# TB-27AHT2–280 | RRID:AB_2832931 | https://antibodyregistry.org/search.php?q=AB_2832931 | 1μL per 200.000 cells | ||
| anti-mouse IgM | goat | AF-647 | polyclonal | Thermo Fisher Scientific Cat# A-21042 | RRID:AB_2535711 | https://scicrunch.org/resolver/RRID:AB_2535711 | 1μL per 200.000 cells | |
| 2.00E+05 | ||||||||
Materials
Biological Materials
hPSC: This protocol was developed and optimized for differentiation of RUES2 human embryonic stem cells (ESC) (Rockefeller University Embryonic Stem Cell Line 2, NIH approval number NIHhESC-09–0013, registration number 0013; RRID:CVCL_B810; https://scicrunch.org/resolver/RRID:CVCL_B810, passage 17–28) and has also been shown to work with two human induced pluripotent stem cell (hiPSC) lines generated using Sendai Virus (sv) and modified mRNA from human dermal fibroblasts (purchased from Mount Sinai Stem Cell Core facility, passage 17–26)13,19. We anticipate that this protocol could also be applied to other hESC and hiPSC lines, although some optimization might be required. See Troubleshooting.
Irradiated mouse embryonic fibroblasts (MEF, GlobalStem, cat. no. GSC-6201G, RRID:CVCL_RB05; https://scicrunch.org/resolver/RRID:CVCL_RB05)
Reagents
Media and supplements
Iscove’s Modification of DMEM (IMDM) (Corning, cat. no. 10016CV)
DMEM and Ham’s F12 50/50 mix (Corning, cat. no. 10092CV)
Ham’s F12 Medium, 1X (Corning, cat. no. 10080CV)
-
Knockout Serum replacement (Life Technologies, cat. no.10828028)
Critical: This reagent exhibits lot-to-lot variation. Lot testing for DE differentiation is recommended.
-
B27 supplement (50X), serum free (Life Technologies, cat. no. 17504044)
Critical: This reagent exhibits lot-to-lot variation. Lot testing for DE differentiation is recommended.
-
N2 supplement (100X) (Life Technologies, cat. no. 17502048)
Critical: This reagent exhibits lot-to-lot variation. Lot testing for DE differentiation is recommended.
Penicillin-Streptomycin (10,000U/mL) (Life Technologies, cat. no. 15140122)
Primocin (Invivogen, cat. no. ANT-PM-2)
GlutaMAX supplement (Life Technologies, 35050061)
MEM non-essential amino acids solution (100X) (life technologies, cat. no. 11140076)
Bovine Serum Albumin (BSA) Fraction V, 7.5% (wt/vol) solution (Life Technologies, cat. no. 15260)
1-Thioglycerol (MTG) (Sigma-Aldrich, cat. no. M6145)
-
2-Mercaptoethanol (Sigma-Aldrich, cat. no. M3148)
Caution: This reagent is toxic.
L-Ascorbic Acid (Sigma-Aldrich, cat. no. A4544)
Growth factors and small molecules
Fibroblast Growth Factor (FGF) 2 (FGFb, R&D systems, cat. no. 233-FB-01M)
FGF10 (R&D systems, cat. no. 345-FG-250)
FGF7 (R&D systems, cat. no. 251-KG)
-
Activin A (R&D systems, cat. no. 333-AC)
Critical: This reagent exhibits lot-to-lot variation. Lot testing for DE differentiation is recommended.
Bone Morphogenic Protein 4 (BMP4) (R&D systems, cat. no. 314-BP)
Y-27632 dihydrochloride (ROCK inhibitor) (R&D systems, 1254/50)
CHIR99021 (R&D systems, cat. no. 4426/50)
All-trans-retinoic acid (ATRA) (Sigma-Aldrich, cat. no. R2625)
Dexamethasone (Sigma-Aldrich, cat. no. D4902–1G)
8-bromo-cyclic adenosine monophosphate (cAMP) (Tocris, cat. no. 1140)
3-Isobutyl-1-methylxanthine (IBMX) (Tocris, cat. no. 2845)
Dorsomorphin dihydrochloride (Tocris, cat. no. 3093)
IWP2 (Tocris, cat. no. 3533)
SB431542 (Tocris, cat. no.1614)
Enzymes
0.05% (wt/vol) Trypsin-EDTA (Life Technologies, cat. no. 25300120)
TrypLE Express Enzyme (1X) (Life Technologies, cat. no. 12605010)
Accutase-EDTA (Innovative Cell Technologies, cat. no. 12605010)
Collagenase type I (Life Technologies, cat. no. 17100017)
Extracellular Matrices
Gelatin from bovine skin, type B (Sigma-Aldrich, cat. no G9391)
Growth factor-reduced Matrigel basement membrane matrix (Corning, cat. no. 354230)
-
Cultrex Rat collagen I, 5mg/mL solution (R&D systems, cat. no. 3440–100-01)
Critical: This reagent might exhibit lot-to-lot variation. pH test prior to embedding the cells is recommended
Fibronectin (R&D systems, cat. no. 1918-FN)
Other Reagents
Antibodies. See Table 1 for a complete list of those used to produce the example results presented here.
Fetal Bovine Serum (FBS) (Atlanta Biologicals, cat. no. S11150H)
Cell Culture Phosphate Buffered Saline 10X (PBS 10X) (Corning, cat. no. 46013CM)
Dulbecco’s Phosphate-Buffered Salt Solution 1X (PBS) (Corning, cat. no. 21031CV)
-
Sodium hydroxide standard solution (NaOH) (Fluka Analytical, cat. no. 71463–1L)
Caution: This reagent is corrosive.
-
Dimethyl Sulfoxide (DMSO) (Fisher Scientific, cat. no. BP231–100)
Caution: This reagent is toxic.
0.5M EDTA, pH 8.0 (Fisher Scientific, cat. no. 15575–038)
Cell Culture Grade Water (Corning, cat. no. 25055CM)
Hank’s Balanced Salt Solutions (HBSS) (Corning, cat. no. 21023CV)
Tissue-Tek O.C.T. compound (Sakura Finetek, cat. no. 4583)
Tissue-Tek Intermediate Cryomold (Sakura Finetek, cat. no. 4566)
RNeasy (Qiagen, cat. no. 74004)
-
RLT buffer (Qiagen, cat. no. 1053393)
Caution: This reagent is toxic and corrosive.
-
Dry Ice
Caution: This reagent may cause burns and frostbite. Vapor may displace oxygen and cause suffocation.
Equipment
Ultra-low-attachment plate, 6-well (Corning, cat. no. 3471)
Tissue culture–treated plate, 6-well (Corning, cat. no. 353046)
Tissue culture–treated plate, 24-well (Corning, cat. no. 353047)
Purifier Logic+ Class II, type A2 biosafety cabinet (Labconco)
CO2 incubator at 37C, 95% humidity, 95% air and 5% CO2 (Fisher Scientific, HERAcell vios 160i)
CO2 incubator at 37C, 95% humidity, 5% O2, 90% N2 and 5%CO2 (Fisher Scientific, HERAcell vios 160i)
Eppendorf Centrifuge 5424R
Eppendorf Centrifuge 5810R, 15amp version
Falcon 50mL conical centrifuge tube (Corning, cat. no. 352098)
Falcon 15mL conical centrifuge tube (Corning, cat. no. 352097)
Pre-sterilized Posi-Click 1.7mL microcentrifuge tubes (Denville, cat. no. C2172)
Falcon Round-Bottom Polystyrene (Corning, cat. no. 352008)
500 mL Easy Grip Polystyrene Storage Bottles (Corning, cat. no. 430282)
1L Easy Grip Polystyrene Storage Bottles (Corning, cat. no. 430518)
500mL Bottle Top Filter Vacuum Filter, 0.22μm pore 33.2cm2 PES Membrane (Corning, cat. no. 431118)
Steriflip-GP Sterile Centrifuge Tube Top Filter Unit, 0.22 μm, Polyethersulfone, gamma irradiated (EMD Millipore, cat. no. SCGP00525)
Flow cytometry analyzer (BD Fortessa™, BD Bioscience, San Jose, CA)
Flowjo v10.2 software (BD Bioscience, San Jose, CA, RRID:SCR_008520; https://scicrunch.org/resolver/RRID:SCR_008520)
Motorized Leica DMi8 (Leica-Microsystems)
Leica DMi1 (Leica-Microsystems)
ImageJ image processing software (U. S. National Institutes of Health, Bethesda, MD, RRID:SCR_003070; https://scicrunch.org/resolver/RRID:SCR_003070)
Cell passaging tool (Life Technologies, cat. no. 23181010)
Cell scraper (Sarstedt, cat. no. 83.1830)
P1000 Barrier pipette tips (Denville Scientific, cat. no. P1126)
P200 Barrier pipette tips (Denville Scientific, cat. no. P1122)
P20 Barrier pipette tips (Denville Scientific, cat. no. 1121)
P10 Barrier pipette tips (Denville Scientific, cat. no. P1096-FR)
Serological pipette, 5 ml, individually wrapped (Fisher Scientific, cat. no. 13–678-11D)
Serological pipette, 10 ml, individually wrapped (Fisher Scientific, cat. no. 13–678-11E)
Serological pipette, 25 ml, individually wrapped (Fisher Scientific, cat. no. 13–678-11)
Serological pipette, 50 ml, individually wrapped (Fisher Scientific, cat. no. 13–678-11F)
pH test strips (Sigma-Aldrich, cat. no. P4786–100EA)
Anti-human CD326 (EPCAM) MicroBeads (Miltenyi Biotec, Cat# 130–061-101, RRID:AB_2832928, https://scicrunch.org/resolver/RRID:AB_2832928)
Reagent Setup
Growth factor and small molecule reconstitution
FGF2: reconstitute to a concentration of 10μg/mL in sterile PBS with 0.1% (wt/vol) BSA. Aliquot and store at −20°C. Keep thawed aliquots at 4°C and use within 1 month.
FGF10: reconstitute to a concentration of 10μg/mL in sterile PBS with 0.1% (wt/vol) BSA. Aliquot and store at −20°C. Keep thawed aliquots at 4°C and use within 1 month.
FGF7: reconstitute to a concentration of 10μg/mL in sterile PBS with 0.1% (wt/vol) BSA. Aliquot and store at −20°C. Keep thawed aliquots at 4°C and use within 1 month.
Activin A: reconstitute to a concentration of 100μg/mL in sterile PBS with 0.1% (wt/vol) BSA. Aliquot and store at −20°C. Keep thawed aliquots at 4°C and use within 1 month.
BMP4: reconstitute to a concentration of 10μg/mL in sterile PBS with 0.1% (wt/vol) BSA. Aliquot and store at −20°C. Keep thawed aliquots at 4°C and use within 1 month.
Y-27632 dihydrochloride (ROCK inhibitor (RI)): reconstitute to a concentration of 10mM in DMSO. Aliquot and store at −20°C. Keep thawed aliquots at 4°C and use within 1 month.
CHIR99021: reconstitute to a concentration of 3mM in DMSO. Aliquot and store at −20°C. Keep thawed aliquots at 4°C and use within 1 month.
All-trans-retinoic acid (ATRA): reconstitute to a concentration of 0.5mM in DMSO. Aliquot and store at −20°C. Keep thawed aliquots at 4°C, protected from light, and use within 1 week.
Dexamethasone: reconstitute dexamethasone in ethanol to a concentration of 1mg/mL and then add PBS to a final concentration of 20μg/mL. Aliquot and store at −20°C. Keep thawed aliquots at 4°C and use within 1 month.
cAMP: reconstitute to a concentration of 0.1M in water. Aliquot and store at −20°C. Keep thawed aliquots at 4°C and use within 1 month.
3-Isobutyl-1-methylxanthine (IBMX): reconstitute to a concentration of 0.1M in DMSO. Aliquot and store at −20°C. Keep thawed aliquots at 4°C and use within 1 month.
Dorsomorphin dihydrochloride: reconstitute to a concentration of 2mM in DMSO. Aliquot and store at −20°C. Keep thawed aliquots at 4°C and use within 1 month.
IWP2: reconstitute to a concentration of 1mM in DMSO. Aliquot and store at −20°C. Keep thawed aliquots at 4°C and use within 1 month.
SB-431542: reconstitute to a concentration of 10mM in DMSO. Aliquot and store at −20°C. Keep thawed aliquots at 4°C and use within 1 month.
Ascorbic acid: Prepare a 50 mg/ml ascorbic acid solution by dissolving 500 mg of L-ascorbic acid powder in 10 ml of water. Filter with a 0.22-μM Steriflip-GP filter. Aliquot and store at −20°C. Keep thawed aliquots at 4°C and use within 24h.
Culture media
hPSC Maintenance Media: combine 400mL DMEM and Ham’s F12 50/50 mix with 100mL knockout serum replacement, 3.5μL 2-mercaptoethanol, 20ng/mL FGF2 (1mL from stock), 5mL glutaMAX, 5mL MEM non-essential amino acids and 1mL primocin. Filter the media through a 0.22μm pore filter unit into a 500mL sterile bottle and store at 4°C, protected from light, and use within 1 month.
Serum-free differentiation (SFD) Media: in an 1L sterile bottle combine 750mL IMDM with 250mL Ham’s F12, 5mL N2 supplement, 10mL B27 supplement, 10mL glutaMAX, 10mL penicillin-streptomycin and 7.5mL BSA. Store at 4°C, protected from light, and use within 1 month.
Feeder Media: combine 425mL IMDM with 75mL ESC-quality FBS, 20μL MTG, 5mL glutaMAX and 5mL penicillin-streptomycin. Store at 4°C and use within 1–2 months.
Wash Media: Combine 500mL IMDM with 25mL ESC-quality FBS. Store at 4°C and use within 1–2 months.
Differentiation Media
Definitive endoderm induction media (days 1–4): Supplement SFD media with 50μg/mL ascorbic acid, 0.4μM MTG, 100ng/mL Activin A (1μL stock per mL media), 10μM Y-27632 (1μL stock per mL media), 0.5ng/mL BMP4 (0.05μL stock per mL media) and 2.5ng/mL FGF2 (0.25μL stock per mL media). Store at 4°C and use within 24h.
Anterior foregut endoderm induction media (days 4–5): Day 4 to 5 media is SFD media supplemented with 50μg/mL ascorbic acid (1μL stock per mL media), 0.4μM MTG, 10μM SB431542 (1μL stock per mL media) and 2μM dorsomorphin dihydrochloride (1μL stock per mL media). Store at 4°C and use within 24h.
Anterior foregut endoderm induction media (days 5–6): Day 5 to 6 media is SFD media supplemented with 50μg/mL ascorbic acid (1μL stock per mL media), 0.4μM MTG, 10μM SB431542 (1μL stock per mL media) and 1μM IWP2 (1μL stock per mL media) for media switch at day 5. Store at 4°C and use within 24h.
Lung progenitor specification media (days 6–15): Supplement SFD media with 50μg/mL ascorbic acid (1μL stock per mL media), 0.4μM MTG, 3μM CHIR99021 (1μL stock per mL media), 10ng/mL FGF10 (1μL stock per mL media), 10ng/mL FGF7 (1μL stock per mL media), 10ng/mL BMP4 (1μL stock per mL media) and 50nM ATRA (0.1μL stock per mL media). Store at 4°C and use within 24h.
Lung progenitor expansion media (days 15–25): Supplement SFD media with 50μg/mL ascorbic acid (1μL stock per mL media), 0.4μM MTG, 3μM CHIR99021 (1μL stock per mL media), 10ng/mL FGF10 (1μL stock per mL media) and 10ng/mL FGF7 (1μL stock per mL media). Store at 4°C and use within 24h.
Lung progenitor maturation media (days 25–50+): Supplement SFD media with 50μg/mL ascorbic acid (1μL stock per mL media), 0.4μM MTG, 10ng/mL FGF10 (1μL stock per mL media), 10ng/mL FGF7 (1μL stock per mL media), 50ng/mL dexamethasone (1μL stock per mL media), 0.1mM 8-bromo-cAMP (1μL stock per mL media) and 0.1mM IBMX (1μL stock per mL media). Store at 4°C and use within 24h.
Other Reagents
Rat tail Collagen I: On ice, mix the collagen I solution (90% of final volume) with cold PBS 10X (10% of final volume) and 23μL per mL of collagen I solution of cold 1N NaOH for an approximate collagen I gel concentration of 4.5mg/mL. If using other rat tail collagen I products follow manufacturer’s instructions to obtain a 4.5mg/mL collagen I gel. Make fresh just before use and keep it on ice.
Gelatin: prepare a 0.1% (wt/vol) gelatin solution in 1X PBS by dissolving 2g of bovine gelatin powder in 1L of tissue culture grade water and boil for 10 min. Add 200mL of cold 10X PBS and an additional 800mL of cold tissue culture grade water and filter the solution through a 0.22μm pore filter unit into 500mL or 1L sterile bottles. Keep at 4°C and use it within 1 month.
Collagenase type I: reconstitute to a 50mg/mL solution in HBSS. Aliquot and store at −20°C for long-term storage. Keep thawed aliquots for short-term usage at 4°C and use within 1–2 months.
Flow cytometry buffer: mix 300mL of 1X PBS with 8mL of 7.5% (wt/vol) BSA and 120μL of 0.5M EDTA. Keep at 4°C and use it within 1–2 months.
0.1% (wt/vol) gelatin-coated tissue culture plates: distribute 2mL/well of 0.1% (wt/vol) gelatin solution (see above) to 6-well tissue culture plates. Coat at room temperature for 15 min. Plates with a lid can be stored at 4°C, sealed with parafilm and used within 1 week. Aspirate the coating solution just before use.
0.33% (vol/vol) fibronectin-coated tissue culture plates: Dilute fibronectin in 1X PBS (1:300). Distribute 300μL/well of diluted fibronectin to 24-well tissue culture plates. Coat at room temperature for 2h. Store the plates with a lid at 4°C, sealed with parafilm, up to 1 week. Aspirate the coating solution just before use.
1% (vol/vol) growth factor reduced Matrigel-coated tissue culture plates: Thaw Matrigel on ice. Dilute Matrigel in IMDM (1:100). Distribute 300μL/well of diluted Matrigel to 24-well tissue culture plates. Coat at room temperature for 2h. Store the plates with a lid at 4°C, sealed with parafilm, up to 1 week. Aspirate the coating solution just before use.
Human pluripotent stem cell culture setup
CRITICAL Further details covering how to properly maintain an hPSC culture can be found in ‘ES Cell International Pte Ltd: Methodology Manual Human Embryonic Stem Cell Culture 2005’ and ‘Harvard Stem Cell Institute (HSCI) StemBook Protocols for pluripotent cells, URL: http://www.stembook.org/protocols/pluripotent-cells’.
Prepare 0.1% (wt/vol) gelatin-coated 6-well tissue culture plates.
-
Thaw a MEF vial in a 37°C water bath for 2 min.
CRITICAL We use vials of MEF from GlobalStem, cat. no. GSC-6201G. Each vial contains 2M cells, which we use to make two 6-well plates of feeders. If MEF from a different source are used the volumes stated might need to be adjusted.
Transfer the thawed MEF cells into a 15mL conical tube and add 14mL wash media. Spin the cells at 300g for 5min at room temperature (RT) (18–22 °C).
While the cells are spinning, aspirate the gelatin solution from the 6-well tissue culture plates prepared in step 1 and add 1 mL of feeder media to each well.
After MEF cells have spun, discard the supernatant and resuspend cells in 12 mL of feeder media. Add 1 mL of MEF to each well or plate cells at a density of 17,000–20,000 cells cm-2. Transfer the feeder cells into a 5%CO2 incubator and let attach overnight.
The following day, aspirate media and wash the wells with 2mL 1X PBS.
-
Thaw a vial of hPSC in a 37°C water bath for 2min, transfer to a 15mL conical tube and add 14mL of wash media.
CRITICAL We usually freeze 200,000–250,000 RUES ESC per vial, so that we thaw 1 vial of RUES2 cells directly into one well of a 6-well plate of feeders. The volumes provided need to be adjusted if thawing vials with higher number of cells.
Spin the cells down at 300g for 5min at RT, aspirate the supernatant, resuspend in 2mL of hPSC maintenance media and transfer to one well of a 6-well plate of feeders or plate at a density of 20,000–25,000 cells cm-2. If working with hiPSC add 10μM Y-27632 to hPSC maintenance media for the first 24h following plating. Transfer the cells into a 5%CO2 incubator.
-
Replace media every day and check growth of cultures. When culture reaches 70–75% confluency prepare new feeder plates (steps 1–5) (typically 3 to 4 days after plating). Passage cells (following steps) when cultures reach 90–95% confluence (typically 4 to 5 days after plating).
CRITICAL We usually use one well of confluent hPCS to passage cells. Below we provide volumes and cell numbers to passage 1 confluent well of RUES2 ESC into 3 wells of feeders (half plate) at the seeding density of 7,000–9,000 cells cm-2. Adapt steps 13 through 16 accordingly if using a different set up. We also recommend adding 2 mL/well of hPSC maintenance media 2h prior to passaging.
CRITICAL STEP A well-maintained hPSC culture without identifiable differentiated colonies is critical for proper DE and lung progenitor differentiation. For less well-maintained hPSC cultures removing prematurely differentiated cell colonies with the help of a P10–20 pipette tip attached to a vacuum system is highly recommended before passage and/or differentiation. Differentiated colonies can be identified and marked for removal with the help of a tissue-culture microscope prior to passaging or, alternatively, place the tissue-culture plate against a dark background and remove any colonies that stand out for being brighter, taller, presenting any discoloration or depression in the center of the colony.
Remove and discard the hPSC maintenance media and wash once with 2mL 1X PBS.
Aspirate and discard the PBS and then add 2mL pre-warmed Accutase-EDTA per well and place in the 5%CO2 incubator for 3min.
-
Aspirate and discard Accutase-EDTA and add 2mL fresh hPSC maintenance media to each well. Lift the cells with the help of a passaging tool, cell scraper or by gently pipetting media over the colonies with a P1000 pipette tip.
CRITICAL STEP When passaging, achieving a homogenous cell suspension of similar sized cell clumps is important for proper maintenance of the hPSC. In a non-homogeneous cell suspension bigger clumps give rise to bigger colonies that expand faster and have a higher probability of differentiating before the culture reaches confluence.
Count the number of cells obtained in step 12. Transfer the number of cells to be passaged into a 15mL conical tube and spin at 300g for 5 min at RT. For RUES ESC transfer 200,000–250,000 cells.
While hPSC are spinning, retrieve new MEF plates from the incubator. Aspirate and discard feeder media and wash each well with 2mL 1X PBS.
After hPSC have spun, discard the supernatant and resuspend cells in hPSC maintenance media. For RUES2 ESC resuspend in 6mL of hPSC maintenance media.
-
Aspirate PBS from MEF plates and distribute the hPCS. For RUES2 ESC distribute 2mL of cells per well of MEF (this will be equivalent to plate cells at 7,000–9,000 cells cm−2). If working with hiPSC lines plate at 10,000–15,000 cells cm−2 and add 10μM Y-27632 to hPSC maintenance media for the first 24h following plating
CRITICAL STEP Optimization of cell seeding density may be required for different cell lines.
Procedures
Definitive Endoderm induction TIMING
CRITICAL This stage of the protocol is summarized in Figure 1A. Start differentiation when the hPSC cultures reach a confluence of 90–95% (Fig. 1B). We usually use 2 confluent wells from a 6-well plate of RUES2 to make a full 6-well low-attachment plate of EBs. If you wish to downsize your experiment use 1 confluent well of hPSC to make 3 wells (half plate) of EBs. For other plate sizes, adapt volumes listed below accordingly and plate hPCS at a density of 120,000–150,000 cells cm-2.
Day 1
-
1
Prepare definitive endoderm induction media.
-
2
Retrieve the hPSC cultures from the incubator. Aspirate and discard the hPSC maintenance media. Wash each hPSC well with 2mL 1X PBS, aspirate the PBS and add 2mL of pre-warmed Accutase-EDTA per well. Return the plates to the incubator for 3min.
-
3
Aspirate and discard the Accutase-EDTA and add 2mL fresh hPSC maintenance media per well. Lift the cells with the help of a passaging tool, cell scraper or by gently pipetting media over the colonies with a P1000 pipette tip. Make sure the cell clumps are of similar size (ideally 2–3cells/clump).
-
4
Transfer the cell suspension from both wells to a 15mL conical tube.
CRITICAL STEP Count cells if you wish to plate at a specific density in step 6.
-
5
Spin cells at 300g for 5min at RT. Aspirate and discard the supernatant and resuspend the cell pellet in 12mL definitive endoderm induction media.
CRITICAL STEP If you wish to only make half plate of EBs, resuspend the cells in 6mL of definitive endoderm induction media. If working with other plate sizes or working with plating densities, resuspend in the appropriate volume of media.
-
6
Distribute the cell suspension in 6-well low attachment plates, 2mL per well (this will be equivalent to plating at 120,000–150,000 cells cm−2). Return the plate to a 5%CO2/5%O2 incubator.
Day 2.
CRITICAL At day 2 the cells will have formed aggregates in the form of small dense spheroids that we refer to EBs.
-
7
Prepare definitive endoderm induction media and add 2mL of freshly made media to each well of EBs. Return the plate to a 5%CO2/5%O2 incubator.
CRITICAL STEP This step can be performed by adding the freshly made media on top of the existing media so that the final volume of media per well at day 2 will be of 4mL, or, alternatively, you can perform a complete media change in the same way as described in step 9.
Day 3.
-
8
Prepare definitive endoderm induction media.
-
9
Transfer the EBs and media from all the wells to a 15 or 50mL conical tube with a 5mL serological pipette and then add 2mL of freshly prepared definitive endoderm induction media to each well. Spin the EBs at 130g for 1min at RT. Aspirate the supernatant and resuspend the cells in a sufficient volume of definitive endoderm induction media to distribute 1mL of EBs per well (final volume of day 3 media per well will be 3mL. Therefore, cells should be resuspended in 6mL medium for a full plate of EBs). Return the plates to a 5%CO2/5%O2 incubator.
-
10
Prepare 0.33% (vol/vol) fibronectin-coated 24-well tissue culture-treated plates. Store at 4°C for use the next day.
Anterior foregut patterning TIMING
CRITICAL This stage of the protocol is summarized in Fig. 1A. A representative image of day 4 EBs is shown in Fig. 1C. At this stage, some variability in EB size and density can be observed without compromise of DE yield. Cultures with greater cell expansion might require more frequent media changes or feeding with larger volumes than the volumes and timelines given here.
Day 4: DE dissociation
CRITICAL Optimal time for dissociation of EBs generated from RUES2 ESC is 72 to 74h after Activin A treatment when DE generation peaks. When working with other hPSC the DE kinetics should be determined and the dissociation time established within 6h after the DE yield maximizes. Details on how to establish DE kinetics for new cell lines can be found in Huang et al, 201519.
-
11
Fill a 50mL conical tube with 40mL of wash media.
-
12
Collect the EBs into a 15mL conical tube (maximum of a full 6 well plate of EBs per conical tube). Let the EBs sediment, which usually takes 1–2min, or spin at 130g at RT for 1min.
-
13
Aspirate and discard supernatant. Add 6mL of pre-warmed trypsin-EDTA. Flick the tube every 15s until the solution gets cloudy (this usually takes 2–3 minutes). Let the EBs sediment by gravity (as described in previous step, do not spin cells at this time) and transfer 2–3mL of the cloudy supernatant into the 50mL conical tube containing the wash media with a 5mL serological pipette.
-
14
Proceed to further dissociate the remaining EBs by gently flicking the 15ml conical tube. Once the EBs are fully dissociated (usually after about 2–3 min of flicking) transfer all of the remaining cell-trypsin suspension into the 50mL conical tube containing the wash media. This will neutralize the trypsin.
-
15
Before proceeding to step 16, collect 2mL from the 50mL conical tube containing the dissociated EBs into a 15mL conical tube. Set aside.
CRITICAL STEP These cells will be used to check the DE induction efficiency, described in steps 20–24.
-
16
Spin the cells in the 50ml conical tube at 300g for 5min at RT.
-
17
Prepare day 4 to 5 anterior foregut endoderm induction media.
-
18
Aspirate and discard the supernatant from the spun down 50ml conical tube and resuspend the cells in day 4 to 5 anterior foregut endoderm induction media.
CRITICAL STEP We usually dissociate a half plate (3 wells) of EBs, which we resuspend in 24mL of day 4 to 5 anterior foregut endoderm induction media, to distribute in two 24-well tissue culture treated plates, 500μL per well (this will be equivalent to plating at a density of 50,000–75,000 cells cm−2). If dissociating a different number of EB wells or using other plate sizes, adapt volumes accordingly.
-
19
Retrieve the 0.33% (vol/vol) fibronectin-coated plates from step 10, aspirate the fibronectin solution and plate the cells prepared in the previous step at a density of 50,000–75,000 cells cm−2, in a volume of 400–500μL of media per well of a 24-well plate. Return the plates to a 5%CO2/5%O2 incubator.
Day 4: Checking DE induction efficiency.
-
20
Spin down the cells set aside in step 15 at 300g for 5min at RT. Aspirate and discard the supernatant. Resuspend the cells in 100μL of flow cytometry buffer. Add 1μL of CXCR4 and 1μL of cKIT antibodies (see Table 1). Incubate at 4°C protected from light for 30–45min.
-
21
Wash the cells by adding 1mL flow buffer. Spin at 300g for 5min at RT. Aspirate and discard the supernatant.
-
22
Resuspend cells in 200μL flow buffer, filter through a 40μm mesh and transfer into a flow cytometry tube.
-
23
If you wish to exclude dead cells from the analysis, prepare a stock solution of 1μg/mL DAPI in flow buffer. Add 20uL of DAPI stock to each flow cytometry tube just prior to running samples in a flow cytometry analyzer.
-
24
Run samples in a flow cytometry analyzer to determine the efficiency of the DE induction. We recommend the following gating strategy: collect a total of 10.000 to 50.000 events per sample. Gate total cell population on FSC-A vs SCC-A. Viable cells (DAPI negative) can be gated on DAPI vs FSC-A plot. Perform doublet exclusion by gating on FSC-A vs FSC-W followed by gating on SCC-A vs SCC-W plots (Supp. Fig. 1). Finally determine the frequency of DE cells by gating on double positive CXCR4+cKIT+ cells or define quadrants based on unstained control (Fig. 1F).
CRITICAL STEP Subsequent steps ideally require ≥80% purity of CXCR4+c-KIT+ cells. It is thus recommended that hPSC lines are used that can generate sufficient proportions of CXCR4+c-KIT+ cells. RUES2 ESC consistently generate ≥95% DE (Fig. 1F) (see Troubleshooting for advice on how to improve DE yield).
Day 5
-
25
Prepare day 5 to 6 anterior foregut endoderm induction media. Check morphology of cells. A representative image of the bright field aspect of the culture at day 5 is shown in Fig. 1D.
-
26
~24h after plating the DE aspirate and discard the day 4 to 5 anterior foregut endoderm induction media and replace with day 5 to 6 anterior foregut endoderm induction media (500μL per well). Return the plates to a 5%CO2/5%O2 incubator.
Lung Progenitor induction TIMING
CRITICAL This stage of the protocol is summarized in Fig. 2A.
Day 6.
-
11
Prepare lung progenitor specification media. Check morphology of cells. A representative image of the bright field aspect of the culture at day 6 is shown in Fig. 1E.
-
12
~24h after adding day 5 to 6 anterior foregut endoderm induction media to the plates aspirate and discard the old media and replace it with lung progenitor specification media (500μL per well). Return the plates to a 5%CO2/5%O2 incubator.
Day 8 to 14.
-
13
Prepare lung progenitor specification media.
-
14
Aspirate and discard the old media and add freshly prepared lung progenitor specification media (500μL per well) every two days. Return the plates to a 5%CO2 incubator.
-
15
At day 14 prepare 1% (vol/vol) growth factor reduced Matrigel-coated 24-well tissue culture treated plates. Keep the plates at 4°C for next day use.
Lung progenitor expansion TIMING
CRITICAL This stage of the protocol is summarized in Fig. 2A.
Day 15.
CRITICAL As we have previously shown19, the differentiation efficiency of the cultures can be assessed at this stage (day 15). A purity of FOXA2+SOX2+NKX2.1+ of ~80% for RUES2 ESC and ~30–60% for hiPSC lines is anticipated. However, as differentiation efficiency increases up to day 25, assessing the differentiation at day 25 is rather more relevant.
-
11
Prepare lung progenitor expansion media. Check morphology of cells. A representative image of the bright field aspect of the culture at day 15 is shown in Fig. 2B.
-
12
Collect the culture media currently on the cells into a conical tube and retain. Add 300μL pre-warmed 0.05% (wt/vol) trypsin-EDTA to each well and return the plates to a 5%CO2 incubator for 1 to 1.5 mins. Retrieve the plates from the incubator, aspirate and discard the trypsin-EDTA and add the collected media (~500μL/well) back to the plates (this will neutralize any leftover trypsin. Alternatively, wash media can be used).
-
13
Mechanically detach the cell monolayer with a P1000 barrier pipette tip by scrapping the cells off the plate. Follow with gently pipetting the media up and down, further disrupting the cell monolayer into small cell clumps (1–2mm wide).
-
14
Collect the media and cells and transfer to a 50mL conical tube. Let the cell clumps pellet by force of gravity 1–2mins at room temperature and aspirate and discard the cloudy supernatant (Fig. 3A).
CRITICAL STEP Do not centrifuge at this step, it is essential to discard the single cells in suspension as these usually represent non-endodermal cells that grow in the culture between days 6 to 15. If performed as instructed, this step should remove >90% of non-endodermal cells that might be present in the culture at this stage without compromising the viability of the endodermal cell clumps (see Suppl. Table 2 for expected range of endodermal and non-endodermal cells present in the cultures at days 15 and 25 of the differentiation protocol).
-
15
Resuspend the cell clumps in lung progenitor expansion media (500μL per well).
-
16
Retrieve the 1% (vol/vol) growth factor reduced Matrigel-coated tissue culture plates from step 31, aspirate the Matrigel solution and replate the cells at 1:2 or 1:3 ratio (that is, 1 day 15 well to 2 or 3 wells in a Matrigel-coated plate). Return the plates to a 5%CO2 incubator.
CRITICAL STEP Performing these steps allows for better purity of NKX2.1+ lung progenitors at day 25. It also allows for post-day 15 lung specification, which is essential for those hPSC lines that do not achieve full lung specification by day 15, and promotes removal of non-endodermal cells present in the culture at day 155,19. The timed trypsinization preferentially digests the non-endodermal cells into single cells or small cell clusters that stay in the supernatant and are removed by aspiration19.
Day 17–24.
-
17
Prepare lung progenitor expansion media.
-
18
Aspirate and discard the old media and add freshly prepared lung progenitor expansion media (500μL per well) every two days. Return the plates to a 5%CO2 incubator.
Lung Progenitor Maturation TIMING
CRITICAL This stage of the protocol is summarized in Fig. 4A and 5A.
Day 25.
CRITICAL For improved multi-lineage maturation efficacy after subsequent collagen I embedding, it is preferable to work with cell lines and cultures that yield high lung progenitor generation efficiencies at day 25. When working with new hPSC lines it might be useful to spend some time optimizing steps 1 through 39 of this protocol to ensure optimal lung progenitor specification prior to carrying the cells through collagen I culture19.
-
11
Prepare lung progenitor maturation media. Check morphology of cells, a representative image of the bright field aspect of the culture at day 25 is shown in Fig. 2C.
-
12
Use one well of day 25 cells to determine the cell density for a given experiment. In the selected well, detach cells starting by aspirating and discarding the culture media. Then add 300–500μL 1X trypLE and return the plate to the incubator for 15mins.
-
13
Mechanically detach the monolayer with a P1000 barrier pipette tip by scrapping the cells off the plate. Transfer the trypLE and cells into an Eppendorf tube and then flick it a few times until trypLE gets cloudy and the cell clumps are fully dissociated.
-
14
Stop the reaction by adding 1mL wash media and spin cells at 300g for 5mins at RT.
-
15
Aspirate and discard the supernatant and resuspend cells in lung progenitor maturation media or flow buffer. Count the number of cells and determine how many wells you will need to dissociate (in step 47) to replate 100,000 cells cm−2 in collagen I.
CRITICAL STEP Performing steps 41–44 is important to assure that the same quantity of cells is reproducibly replated in collagen I in independent experiments, because the total number of cells obtained at day 25 varies slightly from experiment to experiment (1–1.5 million cells per well of a 24-well plate). Since this protocol works with cell clumps rather than single cells, and cell clumps vary in size, the way we found to be the most reproducible is to determine the cell density (or number of cells per well) for a specific experiment at day 25 and then calculate the amount of day 25 wells that needs to be dissociated taking into consideration the goal is to transfer to collagen I an average of 100.000 cells cm−2 (or 200.000 cells per well of a 24-well plate). Once the number of day 25 wells to be dissociated for a given experiment is determined, proceed to disrupt the monolayer into cell clumps and homogeneously distribute them within the collagen mix to the new plates.
-
16
Prepare the collagen I gel mix in a 15 or 50mL conical tube. Place the tubes on ice until needed.
-
17
Aspirate and retain in a conical tube the culture media from the number of wells determined in step 44.
-
18
Add 300uL/well pre-warmed 0.05% (wt/vol) trypsin-EDTA to the calculated number of wells in step 44 and return the plates to a 5%CO2 incubator for 1 to 1.5min.
-
19
Retrieve the plates, aspirate and discard the trypsin-EDTA and add the media retained in step 46 (~500uL/well) back to the wells. Alternatively, you can discard the media in step 46 and use wash media in this step.
-
20
Mechanically detach the cell monolayer with a P1000 barrier pipette tip by scrapping the cells off the plate. Follow by gently pipetting the medium up and down, further disrupting the cell monolayer until you obtain cell clumps with an average size of 1–2mm (representative images of appropriately sized cell clumps after replating in collagen I are shown in Fig. 3B).
-
21
Collect the medium and cell clumps with the help of a P1000 or a 5mL serological pipette and transfer into a 50mL conical tube. Let the cell clumps pellet by force of gravity, about 2 min, at room temperature and then aspirate the cloudy supernatant (Fig. 3A).
CRITICAL STEP Do not dissociate the cell monolayer into single cells or very small cell clumps (<0.5mm wide) as these exhibit low survival rates upon replating into Collagen I (Fig. 3C) (see Troubleshooting).
-
22
Resuspend the cell clumps in the collagen I gel mix. Work fast and keep the collagen I-cell mix on ice as much as possible. Plate the collagen I-cell mix in a 24-well tissue culture treated plate, 300μL per well, with a 5mL serological pipette.
-
23
Place the plate in the incubator for 10 to 15mins or until the collagen I solidifies.
-
24
Add lung progenitor maturation media (500μL per well) on top of the collagen I gels. Return to a 5%CO2 incubator.
CRITICAL STEP We also recommend using some other wells of cells growing at day 25 for RNA extraction and IF staining of endodermal markers FOXA1 and FOXA25,19, the lung transcription factors NKX2.1 (Fig. 2D,F), SOX2 and SOX9 (Fig. 2D), and the surface mucin, MUC1 (Fig. 2E,F) for assessment of the efficiency of lung progenitor generation (see Table 1 and Suppl. Methods). For RUES2 the fraction of FOXA2+SOX2+NKX2.1+ cells is usually of 90–98% of the culture5,19. For other tested hPSC, such as sviPSC and mRNA generated iPSC, lung progenitor generation efficiency is 70 to 90% of the culture5,19. When working with new hPSC lines optimization of this protocol might be needed to achieve high yields of FOXA2+SOX2+NKX2.1+ lung progenitors. See Troubleshooting.
The efficiency of the differentiation at day 25 can also be assessed by flow cytometry for EPCAM, MUC1 and PDPN (see Table 1), which should be co-expressed by 70–80% of the cells (Fig. 8B, Suppl. Table 3).
Day 27 to desired analysis time point.
-
25
Prepare lung progenitor maturation media.
-
26
Aspirate and discard the old media from the cells and add freshly prepared lung progenitor maturation media (500μL per well) every two days.
CRITICAL STEP As maturation proceeds, collagen I gels (Fig. 3D) might detach from the wall of the wells (Fig. 3E) or contract (Fig. 3F). In these circumstances we recommend “repairing” the gel by aspirating all media, adding some newly prepared collagen I gel mix to fill the gap between the old gel layer and the walls of the well, allowing it to solidify and then adding fresh lung progenitor maturation media on top. The need to “repair” the collagen layer is variable from experiment to experiment and gets more frequent the longer the cells are maintained in culture, possibly due to active matrix cell remodeling.
-
27
Analyze cultures at desired timepoints (Box 1). Representative images of the bright field aspect of the culture at day 50 are shown in Fig. 3D, 4B and 5B. Per collagen I gel layer at day 50 you should get 10–20 “organoids”, that vary in size. Most markers for mature cells are present at day 50 (Fig. 4C–J, RUES2; Fig. 5C–J, sviPSC). To obtain basal cells expressing the adult marker NGFR, continue the culture until day 80 (Fig. 6,7).
CRITICAL We have uploaded example flow data sets from our cultures at different time points to the flow cytometry repository http://flowrepository.org. Data sets contain relevant FCS files and recommended gating strategy. Users are free to download data sets and practice gating and analysis themselves. Our data sets can be found under the IDs FR-FCM-Z2VH, FR-FCM-Z2VM, FR-FCM-Z2VN, FR-FCM-Z3Z6 and FR-FCM-Z3Z7.
Troubleshooting
Some common problems and troubleshooting advice for the early stages of this protocol (steps 1 to 39) have been thoroughly explored in the previous version of this protocol19 and remain relevant. These include problems related with low DE yield (steps 1 to 24), poor DE endoderm expansion at day 15 (steps 25 to 31) and good DE yield but low lung progenitor induction efficiency between day 15 and 25 (steps 32 to 39).
Additionally, if working with cell lines that cannot achieve efficient DE differentiation (step 24) even after troubleshooting, fluorescent-activated cell sorting (FACS) for CXCR4+c-KIT+ cells or magnetic-activated cell sorting (MACS) with EPCAM MicroBeads can be used to obtain a purer DE cell population. The sorting should be undertaken after step 14 and the protocol resumed at step 17. We found that MACS preserves cell viability better than FACS and recommend this.
Troubleshooting advice for common problems occurring during the lung progenitor maturation in the collagen I gels (steps 40–56) can be found in Table 2.
Table 2.
Troubleshooting
| Problem | Possible reason | Solution | Step |
|---|---|---|---|
|
| |||
| Cells detach from the culture plate prior to Collagen I transfer | High density of the culture (culture detaches as a monolayer) | If detachment occurs before day 15, reduce the DE plating density (step 19) or replate the cells a few days earlier (steps 23 and 37) | 30 and 39 |
| If detachment happens after day 15, increase the splitting ratio at step 37 | |||
| Loss or dysfunction of attachment substrate (cells detach as single cells or small cell clumps) | Replate the cells immediately into newly prepared 1% (vol/vol) growth factor reduced Matrigel-coated plates | ||
| If Matrigel solution has been thawed and kept at 4°C for more than 1 week, thaw a new vial or aliquot to coat plates or increase the concentration to 1.5–2% (vol/vol) | |||
|
| |||
| Low cell viability after collagen I embedding | Collagen I gel mix pH suboptimal (although rare, we have observed lot-to-lot pH variations in commercially available rat collagen I solutions) | When using a new lot, we recommend determining the pH of the prepared mix using a pH test strip prior to embedding the cells. If the pH is below or above 7, optimize the amount of NaOH to add to the collagen I mix | 55–56 |
| Size of the cell clumps transferred too small (bigger sized cell clumps exhibit better survival after embedding in collagen I as opposed to single cells or smaller cell clumps) | Transfer bigger sized cell clumps | ||
| Be gentler with the mechanical disruption of the monolayer at step 49 | |||
| Avoid over trypsinization at step 47, shorten the trypsinization time | |||
| Cell density in the collagen I too low | Increase cell replating density at day 25 (step 44) | ||
|
| |||
| Low cell viability later in the maturation stage | Insufficient feeding | Feed more often or increase the volume of media | 55–56 |
| Collagen I gels have contracted and cells are in direct contact with media | Repair the gel layer (see CRITICAL STEP in step 55) | ||
|
| |||
| Cells retain expression of NKX2.1 and express poor or incomplete set of lineage specific markers | Cell density in the collagen too high at the time of embedding | Plate at lower density in step 44 | 55–56 |
| Increase the duration of the maturation stage, delay the time point of analysis | |||
|
| |||
| Collagen I gels detach from the walls of the well, contract or thin out | This might happen due to the contracting forces of the cells growing inside the gel, matrix remodeling by the cells and/or long periods of culture | Repair the gel layer (see CRITICAL STEP in step 55) | 55–56 |
| Add a new layer of collagen I on top of the old one | |||
|
| |||
| Overgrowth of contaminating neuronal or non-endodermal lineages in the collagen I | Differentiation started from DE with low purity | Improve DE yield (see Huang et al. 201519) | 55–56 |
| Suboptimal trypsinization at day 15 | Increase the trypsinization period, wash the wells with 1x PBS prior to trypsinization, discard bottles of trypsin-EDTA that have been thawed for more than 1–2 months. | ||
| Too many non-endodermal cells were carried over when replating | Do not centrifuge at step 35. If necessary, wash the cells a few times with PBS until the supernatant gets clear | ||
| Overgrowth of non-endodermal cells from day 6 to day 15 of the differentiation protocol | Replate the cells twice into 1% (vol/vol) growth factor reduced Matrigel-coated plates. The first replate can be done a few days before day 15 and the second replate 1 to 2 days after the cells reach confluence. | ||
Timing
hPSC culture: 4–5 days
Definitive endoderm induction: 72–74h (steps 1–10)
Anterior foregut endoderm patterning: 48h (steps 11–26)
Lung progenitor induction: 9 days (steps 27–31)
Lung progenitor expansion: 10 days (steps 32–39)
Lung progenitor maturation: 25 to 55 days (steps 40–56)
Anticipated results
If steps 1 through 39 are performed as described above hPSC differentiation into lung progenitors should yield cultures highly enriched5,19 for the expression of the lung transcription factor NKX2.1 (Fig. 2D–F). The transcription factors SOX2 and SOX9, involved in proximodistal patterning of the lung, are co-expressed in NKX2.1+ cells at day 25 (Fig. 2D). Expression of the surface mucin, mucin 1 (MUC1), and podoplanin (PDPN) in the lung progenitors generated by the method described here can be detected by IF (Fig. 2E,F).
Culture of the lung progenitors generated at day 25 in collagen I gels and in lung progenitor maturation media yields 3D structures at day 50 where markers for the major proximal and distal epithelial cell types in the lung can be detected (Fig. 4C–J, RUES2; Fig. 5C–J, sviPSC). Areas similar to pseudo-stratified epithelium characteristic of the airways can be identified with PDPN expressing basal cells and polarized expression of MUC1 (Fig. 4C RUES2; Fig. 5C, sviPSC). BCs can be detected by staining for P63 and KRT5 (Fig. 4D RUES2; Fig. 5D, sviPSC. SCGB1A1-expressing club cells can be found interspersed with KRT5+ basal cells (Fig. 4E, RUES2; Fig. 5E, sviPSC) while ciliated (acTUB+) and goblet (MUC5B+) cells appear in clusters (Fig. 4F RUES2; Fig. 5F, sviPSC). Distal ATII cells expressing SFTPC, SFTPB, MUC1 and staining with the HT2–280 antibody37,38 (Fig. 4G,H, RUES2; Fig. 5G,H, sviPSC) can be easily detected in the culture and in close proximity with CAV1 expressing ATI cells (Fig. 4I,J, RUES2; Fig. 5I,J, sviPSC). Cells expressing markers characteristic of ATI cells such as HOPX and CAV1 tend to be found in areas of cell flattening and thinning of the cultures and exhibit morphologic characteristics of ATI in vivo cells with thin cytoplasmic extensions (Fig. 4J, RUES2; Fig. 5J, sviPSC. Some of these cells also stain with the human-specific ATI antibody, HT1–56 (Suppl. Fig. 3), an antibody that detects a specific antigen of human ATI cells39. AQP5, a marker for murine ATI cells, was not detected in the cultures or in alveolar human lung tissue (Suppl. Fig. 4), despite broad detection in human airways, indicating that, in contrast to the mouse, AQP5 is not a marker of human ATI cells.
Extending the maturation in collagen I beyond day 50 allows for further maturation of the BCs present in the culture. By day 80 of the differentiation protocol (Fig. 6A) expression of KRT14 and NGFR can be detected in BCs by IF (Fig. 6B). Extending the protocol to day 80 does not compromise the differentiation and maturation of other epithelial lineages as the lineage specific markers detected at day 50 are also detected at day 80 (Fig. 7).
We also present here a strategy to rapidly assess differentiation by flow cytometry (Fig. 8, Suppl. Fig. 1) at various stages of protocol (Fig. 8A). More than 90–98% of the cells should be EPCAM+ (Fig. 8B,C,E,F, Suppl. Table 2,3) and therefore epithelial. While PDPN is expressed at low levels by DE cells and at high levels by cells undergoing lung progenitor specification at days 15 and 25 of the differentiation protocol, the number of cells expressing MUC1 increases from day 15 to 25 (Fig. 8B, Suppl. Table 3) concomitant with the increase in NKX2.1 expression occurring over the same time period (Fig. 2E,F). In adult alveoli MUC1 becomes restricted to ATII40 and PDPN to ATI41 cells while proximally PDPN is expressed in the basal cell layer42 and MUC1 coats the luminal aspect of the airways43. The differential expression of MUC1 and PDPN seen in the adult lung is also reflected in the culture at day 50. After gating on EPCAM+ cells, six major epithelial cell populations can be defined by staining for PDPN and MUC1, HT2–280, integrin beta 4 (ITGB4) and integrin alpha 6 (ITGA6) (Fig. 8C, see Suppl. Table 3 for frequency and absolute numbers of each population), which can be isolated by FACS. HT2–280 is a specific antibody for human ATII cells37, and this population can be clearly distinguished (Fig. 8C). In the MUC1/PDPN plot, 4 populations can be distinguished: MUC1+PDPN−, MUC1hiPDPN+/−, MUC1−PDPN− and MUC1lo/−PDPN+ (Fig. 8C). We assessed enrichment of lung and airway epithelial markers within each of these populations (Fig. 8D). Approximately half of the MUC1lo/−PDPN+ population expressed ITGB4 and ITGA6 (Fig. 8C). The latter two integrins always showed correlated expression, a finding consistent with the fact both form dimers. Isolation by cell sorting of ITGB4+ cells followed by IF for P63, MUC1, KRT5, SOX2, and PDPN showed co-expression of these markers13, supporting the notion that these are basal-like cells. This finding was also confirmed by strong enrichment for P63 mRNA in MUC1loPDPN+ITGB4+ population (Fig. 8D). The MUC1loPDPN+ITGB4− population also showed some enrichment for P63, most likely because, similar to all other markers, there was a continuum in ITGB4 expression. As expected MUC1+PDPN− population was enriched for the ATII markers, ABCA3 and SFTPC (Fig. 8D). SCGB3A2 and FOXJ1 were enriched in the MUC1+PDPNhi population, indicating that this population contains airway cells, including club and ciliated cells, a notion also supported by the high expression SFTPB in this population, which is expressed both in club cells and in ATII cells (Fig. 8D). ATI markers were variably distributed throughout most populations (Fig. 8D), suggesting that ATI cells at various stages of differentiation are present in the cultures. Alternatively, these findings may also suggest ATI heterogeneity that was hitherto not recognized. The MUC1−PDPN− population was not enriched for any lineage-specific markers (Fig. 8D), indicating that this population likely represents a heterogeneous transitional population.
By day 80, a population of EPCAM+NGFR+ cells can be identified by flow cytometry (Fig. 8E), while the populations addressed above, defined by the expression of PDPN, MUC1, ITGB4, ITGA6 and HT2–280, are present at roughly the same frequencies as seen at day 50 (Fig. 8C,E, Suppl. Table 3). The NGFR+ cells could be isolated and passaged in published conditions for the culture of BCs24 (Suppl. Fig. 2A) and differentiated into airway epithelial cells in air-liquid interphase cultures13 (Suppl. Fig. 2B).
We have previously shown that culture in the continued presence of CHIR99021 proliferation was enhanced and differentiation inhibited13. The corresponding flow cytometric analysis showed absence of ITGB4+ITGA6+, MUC1hiPDPN+/, MUC1lo/−PDPN+ and HT2–280+ populations (Fig. 8F). These data indicate that the degree of maturation in the cultures can be assessed by flow cytometry.
As other groups have reported that their directed differentiation protocols for lung also generate a mixture of liver and gastric cells44, we reanalyzed our previously published RNA sequencing data13 to assess whether our protocol generates any non-lung cells. KeyGenes45 analysis revealed that even though there is a partial match for day 25 cultures with the transcriptional profile of 2nd trimester and adult intestine, by day 50 of the differentiation protocol, when maturation of the cultures has progressed, the cultures strongly match with lung and the transcriptional profile of intestine is lost (Suppl. Fig. 5A). We also mined single cell RNA sequencing data of day 50 cultures for a variety of identifier genes45 of other tissues and found very little to be present (Suppl. Fig. 5B). Finally, we also looked at the expression of the markers reported by McCauley et al. 2018 in our culture at day 50 and found some cells expressing TTR, a gene that encodes transthyretin, a protein mostly synthetized in the liver (Suppl. Fig. 6), a finding whose significance is unclear. We note however that TTR was identified in nasal and bronchoalveolar lavage fluids46. Taken together, although we cannot exclude that some non-lung cells might be present in the cultures at day 50, these are scant.
In Fig. 9A–D we show data demonstrating that our culture can be used as a tool to study human respiratory viruses. This also demonstrate that the culture system we describe in detail here can be used as a complementary system to that we used previously to model human parainfluenza virus (HPIV) infection 47. HPIV is a common cause of acute respiratory illness throughout life. HPIV infections are widely prevalent with 80% of children testing seropositive for HPIV1, 2, and 3 by age five. Infants, children, and the immunocompromised are the most likely to develop severe disease. HPIV1 and HPIV2 are more frequently associated with laryngotracheobronchitis while HPIV3 is more likely to spread to the smaller airways, causing bronchiolitis and pneumonia. Infections are usually self-limiting, but mortality can be high for the severely immunosuppressed. Currently there are no available effective vaccines for prevention neither are specific antiviral drugs47–49. We exposed our mature day 50 cultures to a recombinant GFP expressing-HPIV3 for 48h (Fig. 9A). After 8 days, widespread infection of the cultures was observed (Fig. 9B,C). IF analysis revealed that virus preferentially infected airway cells, as indicated by the co-localization of IF signal for the viral particle HN and markers for basal (KRT5) and ciliated (acTUB) cells, while cells expressing alveolar markers HT2–280 and CAV1 were spared (Fig. 9D). These findings are consistent with the high susceptibility of human pseudostratified mucociliary airway epithelium to HPIV50, showing that our model recapitulates features of human lung HPIV infection and thus could be utilized to study viral tropism, pathophysiology of infection and antiviral drugs.
As a further demonstration of the potential applications of our culture system, we show that our cultures can be transduced with a ZsGreen lentiviral vector at the LP specification/maturation step using standard protocols (Fig. 9E, F) and then carried through the differentiation protocol with the transduced cells being able to expand and differentiate within the collagen I gels and analyzed at the desired time points (Fig. 9G). This protocol could therefore be used in applications such as lineage tracing involving lentiviral integration site or barcode analysis or lentivirally expression of specific genes.
Supplementary Material
{kind=link}
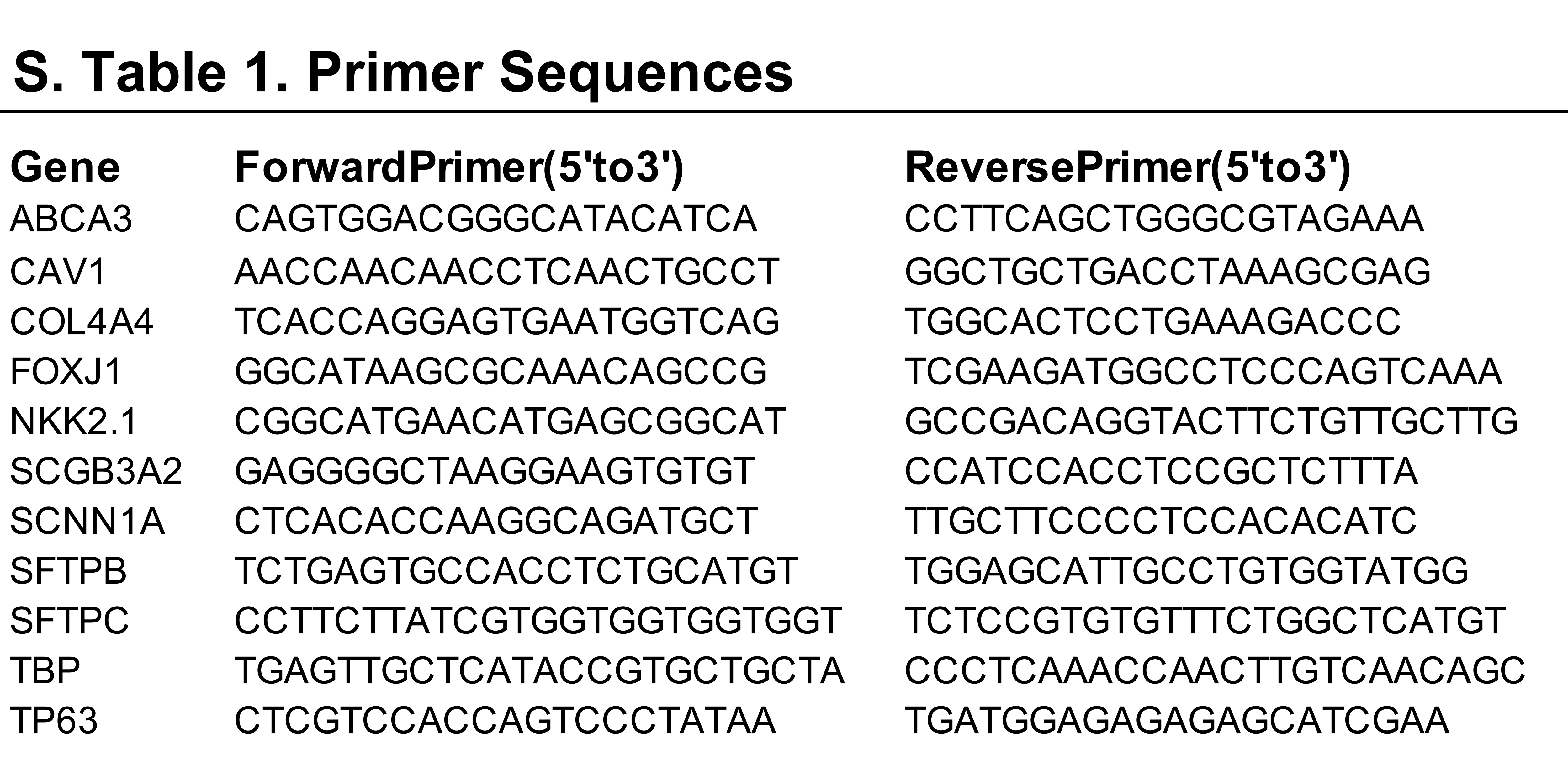{kind=link}
Key reference(s) using this protocol :
Carvalho ALRT de, Strikoudis A, Liu H-Y, et al. Glycogen synthase kinase 3 induces multilineage maturation of human pluripotent stem cell-derived lung progenitors in 3D culture. Development. 2019;146(2):dev171652. doi:10.1242/DEV.171652
Huang SXL, Green MD, de Carvalho AT, et al. The in vitro generation of lung and airway progenitor cells from human pluripotent stem cells. Nat Protoc. 2015;10(3):413–425. doi:10.1038/nprot.2015.023
Huang SXL, Islam MN, O’Neill J, et al. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat Biotechnol. 2014;32(1):84–91. doi:10.1038/nbt.2754
Acknowledgements
This work was supported by the US National Institutes of Health grants HL120046, 1U01HL134760 (to H.-W.S.) and grant AI31971 (to A.M), the Thomas R Kully IPF Research Fund (to H.-W.S.) and the Fundação para a Ciência e a Tecnologia (fellowship PD/BD/52320/2013 to A.L.R.T.d.C). Flow cytometry was performed in CCTI Flow Cytometry Core, supported in part by the Office of the Director, National Institutes of Health under awards S10RR027050 and S10OD020056.
Footnotes
Competing interests
The authors have no competing financial interests.
Data availability
The RNA sequencing and single cell RNA sequencing data are deposited in GEO under accession number GSE101558. Example flow data sets have been uploaded to the flow cytometry repository http://flowrepository.org under the following repository IDs: FR-FCM-Z2VH, FR-FCM-Z2VM, FR-FCM-Z2VN, FR-FCM-Z3Z6 and FR-FCM-Z3Z7. Raw data supporting the findings in this manuscript is available in the source data files.
References
- 1.Green MD, Huang SXL, Snoeck HW. Stem cells of the respiratory system: From identification to differentiation into functional epithelium. BioEssays. 2013;35(3):261–270. doi: 10.1002/bies.201200090 [DOI] [PubMed] [Google Scholar]
- 2.Green MD, Chen A, Nostro M-C, et al. Generation of anterior foregut endoderm from human embryonic and induced pluripotent stem cells. Nat Biotechnol. 2011;29(3):267–272. doi: 10.1038/nbt.1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mou H, Zhao R, Sherwood R, et al. Generation of Multipotent Lung and Airway Progenitors from Mouse ESCs and Patient-Specific Cystic Fibrosis iPSCs. Cell Stem Cell. 2012;10(4):385–397. doi: 10.1016/J.STEM.2012.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wong AP, Bear CE, Chin S, et al. Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nat Biotechnol. 2012;30(9):876–882. doi: 10.1038/nbt.2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang SXL, Islam MN, O’Neill J, et al. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat Biotechnol. 2014;32(1):84–91. doi: 10.1038/nbt.2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Firth AL, Dargitz CT, Qualls SJ, et al. Generation of multiciliated cells in functional airway epithelia from human induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2014;111(17):E1723–30. doi: 10.1073/pnas.1403470111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gotoh S, Ito I, Nagasaki T, et al. Generation of alveolar epithelial spheroids via isolated progenitor cells from human pluripotent stem cells. Stem cell reports. 2014;3(3):394–403. doi: 10.1016/j.stemcr.2014.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konishi S, Gotoh S, Tateishi K, et al. Directed Induction of Functional Multi-ciliated Cells in Proximal Airway Epithelial Spheroids from Human Pluripotent Stem Cells. Stem Cell Reports. 2016;6(1):18–25. doi: 10.1016/J.STEMCR.2015.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y-W, Huang SX, de Carvalho ALRT, et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat Cell Biol. 2017;19(5):542–549. doi: 10.1038/ncb3510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCauley KB, Hawkins F, Serra M, Thomas DC, Jacob A, Kotton DN. Efficient Derivation of Functional Human Airway Epithelium from Pluripotent Stem Cells via Temporal Regulation of Wnt Signaling. Cell Stem Cell. 2017;20(6):844–857.e6. doi: 10.1016/J.STEM.2017.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacob A, Morley M, Hawkins F, et al. Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell. 2017;21(4):472–488.e10. doi: 10.1016/J.STEM.2017.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamamoto Y, Gotoh S, Korogi Y, et al. Long-term expansion of alveolar stem cells derived from human iPS cells in organoids. Nat Methods. 2017;14(11):1097–1106. doi: 10.1038/nmeth.4448 [DOI] [PubMed] [Google Scholar]
- 13.Carvalho ALRT de, Strikoudis A, Liu H-Y, et al. Glycogen synthase kinase 3 induces multilineage maturation of human pluripotent stem cell-derived lung progenitors in 3D culture. Development. 2019;146(2):dev171652. doi: 10.1242/DEV.171652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dye BR, Hill DR, Ferguson MA, et al. In vitro generation of human pluripotent stem cell derived lung organoids. Elife. 2015;4. doi: 10.7554/eLife.05098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller AJ, Dye BR, Ferrer-Torres D, et al. Generation of lung organoids from human pluripotent stem cells in vitro. Nat Protoc. 2019;14(2):518–540. doi: 10.1038/s41596-018-0104-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dye BR, Miller AJ, Spence JR. How to Grow a Lung: Applying Principles of Developmental Biology to Generate Lung Lineages from Human Pluripotent Stem Cells. Curr Pathobiol Rep. 2016;4:47–57. doi: 10.1007/s40139-016-0102-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hawkins F, Kotton DN. Embryonic and Induced Pluripotent Stem Cells for Lung Regeneration. Ann Am Thorac Soc. 2015;12(Supplement 1):S50–S53. doi: 10.1513/AnnalsATS.201410-457MG [DOI] [PubMed] [Google Scholar]
- 18.Swarr DT, Morrisey EE. Lung endoderm morphogenesis: gasping for form and function. Annu Rev Cell Dev Biol. 2015;31:553–573. doi: 10.1146/annurev-cellbio-100814-125249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang SXL, Green MD, de Carvalho AT, et al. The in vitro generation of lung and airway progenitor cells from human pluripotent stem cells. Nat Protoc. 2015;10(3):413–425. doi: 10.1038/nprot.2015.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rock JR, Onaitis MW, Rawlins EL, et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci U S A. 2009;106(31):12771–12775. doi: 10.1073/pnas.0906850106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hawkins F, Kramer P, Jacob A, et al. Prospective isolation of NKX2–1–expressing human lung progenitors derived from pluripotent stem cells. J Clin Invest. 2017;127(6):2277–2294. doi: 10.1172/JCI89950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strikoudis A, Cieślak A, Loffredo L, et al. Modeling of Fibrotic Lung Disease Using 3D Organoids Derived from Human Pluripotent Stem Cells. Cell Rep. 2019;27(12):3709–3723.e5. doi: 10.1016/j.celrep.2019.05.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dye BR, Dedhia PH, Miller AJ, et al. A bioengineered niche promotes in vivo engraftment and maturation of pluripotent stem cell derived human lung organoids. Elife. 2016;5. doi: 10.7554/eLife.19732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Butler CR, Hynds RE, Gowers KHC, et al. Rapid Expansion of Human Epithelial Stem Cells Suitable for Airway Tissue Engineering. Am J Respir Crit Care Med. 2016;194(2):156–168. doi: 10.1164/rccm.201507-1414OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramani S, Crawford SE, Blutt SE, Estes MK. Human organoid cultures: transformative new tools for human virus studies. Curr Opin Virol. 2018;29:79–86. doi: 10.1016/J.COVIRO.2018.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ciancanelli MJ, Huang SXL, Luthra P, et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science. 2015;348(6233):448–453. doi: 10.1126/science.aaa1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rafeeq MM, Murad HAS. Cystic fibrosis: current therapeutic targets and future approaches. J Transl Med. 2017;15(1):84. doi: 10.1186/s12967-017-1193-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaur A, Mathai SK, Schwartz DA. Genetics in Idiopathic Pulmonary Fibrosis Pathogenesis, Prognosis, and Treatment. Front Med. 2017;4:154. doi: 10.3389/fmed.2017.00154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whitsett JA, Wert SE, Weaver TE. Alveolar Surfactant Homeostasis and the Pathogenesis of Pulmonary Disease. Annu Rev Med. 2010;61(1):105–119. doi: 10.1146/annurev.med.60.041807.123500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim CFB, Jackson EL, Woolfenden AE, et al. Identification of Bronchioalveolar Stem Cells in Normal Lung and Lung Cancer. Cell. 2005;121(6):823–835. doi: 10.1016/J.CELL.2005.03.032 [DOI] [PubMed] [Google Scholar]
- 31.Xu X, Rock JR, Lu Y, et al. Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proc Natl Acad Sci U S A. 2012;109(13):4910–4915. doi: 10.1073/pnas.1112499109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen HJ, Poran A, Unni AM, et al. Generation of pulmonary neuroendocrine cells and SCLC-like tumors from human embryonic stem cells. J Exp Med. 2019;216(3):674–687. doi: 10.1084/JEM.20181155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Z, Ren X, Xu X, et al. Genetic Engineering of Human Embryonic Stem Cells for Precise Cell Fate Tracing during Human Lineage Development. Stem cell reports. 2018;11(5):1257–1271. doi: 10.1016/j.stemcr.2018.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.An WF, Germain AR, Bishop JA, et al. Discovery of Potent and Highly Selective Inhibitors of GSK3b. National Center for Biotechnology Information (US); 2010. [PubMed] [Google Scholar]
- 35.Gonzales LW, Guttentag SH, Wade KC, Postle AD, Ballard PL. Differentiation of human pulmonary type II cells in vitro by glucocorticoid plus cAMP. Am J Physiol Cell Mol Physiol. 2002;283(5):L940–L951. doi: 10.1152/ajplung.00127.2002 [DOI] [PubMed] [Google Scholar]
- 36.Hynds RE, Butler CR, Janes SM, Giangreco A. Expansion of human airway basal stem cells and their differentiation as 3D tracheospheres. In: Methods in Molecular Biology. Vol 1576. Humana Press Inc.; 2016:43–53. doi: 10.1007/7651_2016_5 [DOI] [PubMed] [Google Scholar]
- 37.Gonzalez RF, Allen L, Gonzales L, Ballard PL, Dobbs LG. HTII-280, a biomarker specific to the apical plasma membrane of human lung alveolar type II cells. J Histochem Cytochem. 2010;58(10):891–901. doi: 10.1369/jhc.2010.956433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barkauskas CE, Cronce MJ, Rackley CR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123(7):3025–3036. doi: 10.1172/JCI68782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dobbs LG, Gonzalez RF, Allen L, Froh DK. HTI 56, an Integral Membrane Protein Specific to Human Alveolar Type I Cells. J Histochem Cytochem. 1999;47(2):129–137. doi: 10.1177/002215549904700202 [DOI] [PubMed] [Google Scholar]
- 40.Jarrard JA, Linnoila RI, Lee H, Steinberg SM, Witschi H, Szabo E. MUC1 is a novel marker for the type II pneumocyte lineage during lung carcinogenesis. Cancer Res. 1998;58(23):5582–5589. [PubMed] [Google Scholar]
- 41.Ramirez MI, Millien G, Hinds A, Cao Y, Seldin DC, Williams MC. T1α, a lung type I cell differentiation gene, is required for normal lung cell proliferation and alveolus formation at birth. Dev Biol. 2003;256(1):62–73. doi: 10.1016/S0012-1606(02)00098-2 [DOI] [PubMed] [Google Scholar]
- 42.Rock JR, Gao X, Xue Y, Randell SH, Kong Y-Y, Hogan BLM. Notch-dependent differentiation of adult airway basal stem cells. Cell Stem Cell. 2011;8(6):639–648. doi: 10.1016/j.stem.2011.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim KC, Lillehoj EP. MUC1 mucin: a peacemaker in the lung. Am J Respir Cell Mol Biol. 2008;39(6):644–647. doi: 10.1165/rcmb.2008-0169TR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCauley KB, Alysandratos KD, Jacob A, et al. Single-Cell Transcriptomic Profiling of Pluripotent Stem Cell-Derived SCGB3A2+ Airway Epithelium. Stem Cell Reports. 2018;10(5):1579–1595. doi: 10.1016/j.stemcr.2018.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roost MS, vanIperen L, Ariyurek Y, et al. KeyGenes, a Tool to Probe Tissue Differentiation Using a Human Fetal Transcriptional Atlas. Stem Cell Reports. 2015;4(6):1112–1124. doi: 10.1016/J.STEMCR.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lindahl M, Ståhlbom B, Tagesson C. Newly identified proteins in human nasal and bronchoalveolar lavage fluids: Potential biomedical and clinical applications. Electrophoresis. 1999;20(18):3670–3676. doi: [DOI] [PubMed] [Google Scholar]
- 47.Branche A, Falsey A. Parainfluenza Virus Infection. Semin Respir Crit Care Med. 2016;37(04):538–554. doi: 10.1055/s-0036-1584798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Henrickson KJ. Parainfluenza viruses. Clin Microbiol Rev. 2003;16(2):242–264. doi: 10.1128/CMR.16.2.242-264.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schomacker H, Schaap-Nutt A, Collins PL, Schmidt AC. Pathogenesis of acute respiratory illness caused by human parainfluenza viruses. Curr Opin Virol. 2012;2(3):294–299. doi: 10.1016/j.coviro.2012.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang L, Bukreyev A, Thompson CI, et al. Infection of ciliated cells by human parainfluenza virus type 3 in an in vitro model of human airway epithelium. J Virol. 2005;79(2):1113–1124. doi: 10.1128/JVI.79.2.1113-1124.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing and single cell RNA sequencing data are deposited in GEO under accession number GSE101558. Example flow data sets have been uploaded to the flow cytometry repository http://flowrepository.org under the following repository IDs: FR-FCM-Z2VH, FR-FCM-Z2VM, FR-FCM-Z2VN, FR-FCM-Z3Z6 and FR-FCM-Z3Z7. Raw data supporting the findings in this manuscript is available in the source data files.