

Abstract
We report the development of the orthogonal amber-suppressor pair Archaeoglobus fulgidus seryl-tRNA (Af-tRNASer)/Methanosarcina mazei seryl-tRNA synthetase (MmSerRS) in Escherichia coli. Furthermore, the crystal structure of MmSerRS was solved at 1.45 Å resolution, which should enable structure-guided engineering of its active site to genetically encode small, polar noncanonical amino acids (ncAAs).
Keywords: Seryl-tRNA synthetase, E. coli orthogonality, Genetic code expansion, X-ray crystallography, Non-canonical amino acids
Graphical Abstract
1. Introduction
Methods that enable the genetic incorporation of noncanonical amino acids (ncAAs) into proteins have greatly increased our ability to manipulate protein structure and function in vitro and in vivo.1 The site-selective incorporation of ncAAs into proteins in living cells is reliant on laboratory-evolved tRNA/aminoacyl-tRNA synthetase (aaRS) pairs. To ensure that the ncAA is incorporated into proteins with high fidelity and efficiency, the evolved tRNA/aaRS pair must be orthogonal to its host counterparts (i.e., not cross-react with the host tRNAs and aminoacyl-tRNA synthetases) and efficiently suppress a codon that does not encode a canonical amino acid (e.g., nonsense, four-base, etc.). Several such orthogonal tRNA/aaRS have been developed for expanding the genetic codes of prokaryotes and eukaryotes, enabling site-specific incorporation of ncAAs encompassing a wide spectrum of chemical functionalities in the context of amino acid side chains.2
In E. coli, three tRNA/aaRS pairs have been successfully engineered to incorporate ncAAs: the M. jannaschii derived tyrosyl,3 archaea-derived pyrrolysyl,4 and a yeast-derived tryptophanyl pairs.5 While these pairs have enabled the incorporation of ncAAs with a wide variety of side chains, small polar ncAAs remain challenging to genetically encode. The only exception is the successful genetic incorporation of phosphoserine using an archaea-derived pair that is naturally evolved to charge this amino acid as a biosynthetic precursor to cysteine.6,7,8,9 Additionally, recent development of heritable unnatural base pairs10 and engineered E. coli strains with compressed genetic codes11,12 promise a further expansion of available triplet codons that can be simultaneously reassigned to distinct ncAAs.
Development of an orthogonal suppressor seryl-tRNA (tRNASer)/SerRS13,14 pair would potentially enable genetic incorporation of ncAAs bearing small, polar side chains. Additionally, unlike most other pairs, SerRS does not use the anticodon of tRNASer as an identity element, making it straightforward to reassign this pair to any other codon, including the amber nonsense codon. Here, we report the development of an archaea-derived tRNASer/SerRS pair that incorporates serine in response to the amber nonsense codon in E. coli with high fidelity and efficiency. In addition, we solved the crystal structure of MmSerRS, which will facilitate the design of synthetase libraries for use in directed evolution experiments to genetically encode ncAAs with small side chains.
2. Results and discussion
2.1. Identification of orthogonal tRNA/aaRS pairs
Typically, tRNA/aaRS pairs from archaea and eukaryotes serve as starting points for developing orthogonal pairs in E. coli because of their evolutionary divergence from their bacterial counterparts.15 Archaea-derived pairs have been particularly useful to this end, since the corresponding aaRSs tend to be simpler and more robust scaffolds for directed evolution.16 Consequently, we focused on archaea-derived tRNASer/SerRS pairs for developing an orthogonal suppressor pair in E.coli.17 To this end we tested all possible combinations of six different archaeal tRNASer and five different SerRS with divergent sequences (Figure 1), since it has previously been shown that the combination of an aaRS and a tRNA from two different species can provide the optimal choice for developing an orthogonal pair.18 The SerRS genes were cloned into a pBK vector to express them using the constitutive, low-activity glnS promoter. The TAG-suppressor mutants of the tRNASer genes (anticodon mutated to CUA) were cloned into a pRepCM3b vector19 under a constitutive lpp promoter. This vector also encodes a chloramphenicol-acetyl transferase (CAT) reporter harboring a TAG mutation at a permissive site (D111TAG), which allows assessment of TAG-suppression efficiency by monitoring the degree of chloramphenicol resistance exhibited by cells hosting this plasmid.19
Figure 1.
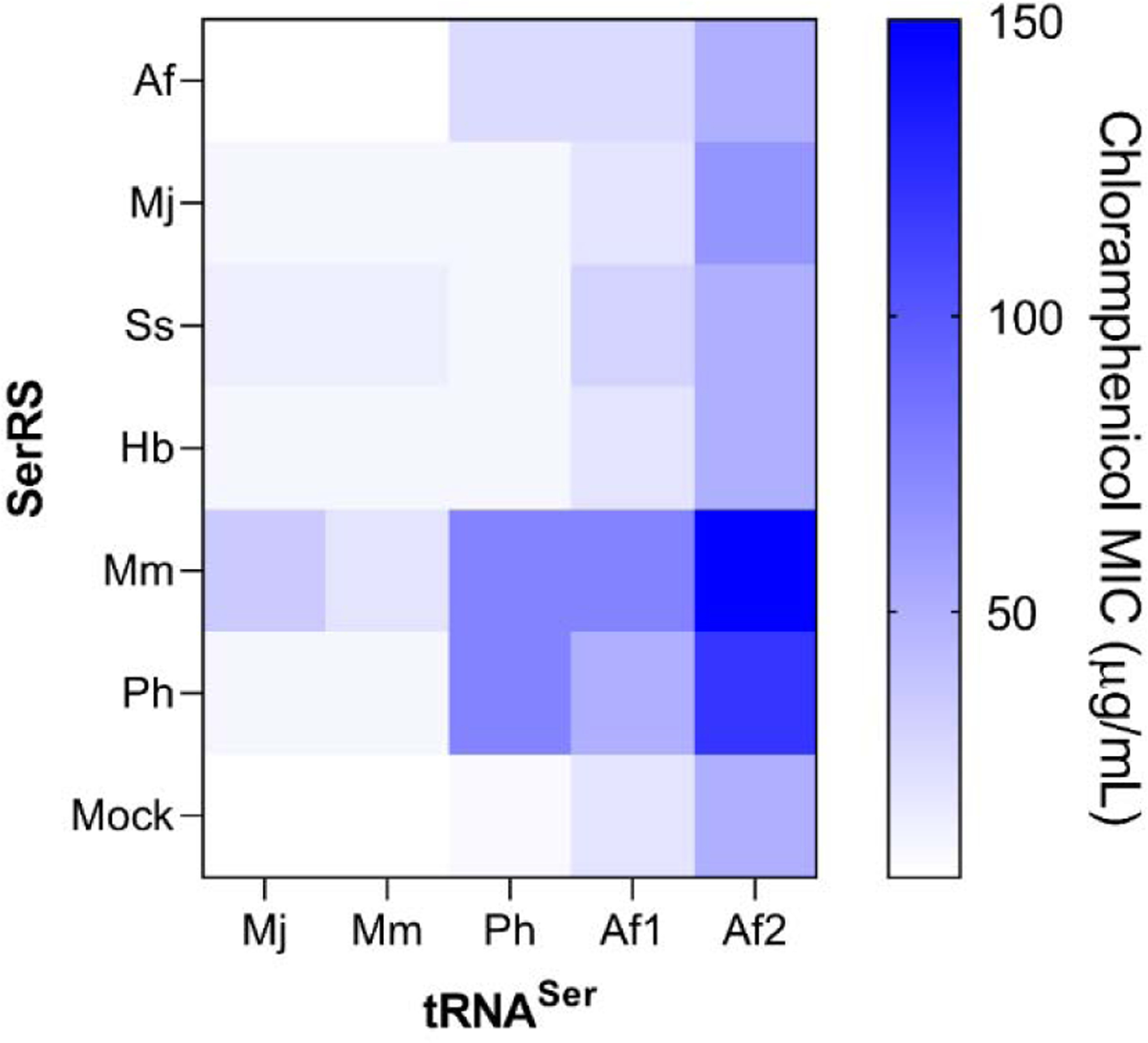
Identification of an orthogonal seryl pair. The suppression efficiency of six synthetases expressed with five orthogonal amber suppressor tRNA was measured using a chloramphenicol resistance assay. The MIC value is presented as a heatmap. Af: Archaeoglobus fulgidus, Mj: Methanocaldococcus jannaschii, Ss: Sulfolobus solfataricus, Hb: Halobacterium sp. NRC-1, Mm: Methanosarcina mazei, Ph: Pyrococcus horikoshii, Af1: Af-tRNASer(CGA), Af2: Af-tRNASer(GCU) and Mock: No synthetase expressed (empty pBK). Anticodons of tRNASers were mutated to CUA
Each of the six pRepCM3b-tRNASer plasmids were separately co-transformed into DH10b E. coli with each of the pBK-SerRS plasmids, as well as an empty pBK vector (does not express an aaRS). The resulting 35 different strains, harboring different combinations of tRNASer and SerRS variants, were individually plated on LB-agar medium supplemented with antibiotics necessary for plasmid maintenance (kanamycin and tetracycline), as well as increasing concentrations of chloramphenicol. For each distinct tRNASer, the minimal inhibitory concentration (MIC) value observed in the presence of the empty pBK plasmid is a measure of its cross-reactivity with host aaRSs, while the different MIC values exhibited in the presence of different SerRS provide an estimate of the TAG-suppression activity of the corresponding tRNASer/SerRS combination (Figure 1). We found that two different tRNASer variants from A. fulgidus and one from P. horikoshii demonstrated strong TAG suppression activity, while SerRS from M. mazei and P. horikoshii were the most active. The heterologous combination of MmSerRS and CUA mutant of Af-tRNASer(GCU) (Af2) exhibited the highest activity, enabling survival of the resulting strain at up to 150 µg/mL of chloramphenicol.
Even though this seryl-tRNA/aaRS pair shows high suppression activity, the relatively high MIC exhibited by Af2 in the absence of a cognate SerRS suggests that it cross-reacts significantly with a host aaRS, compromising its fidelity. The cross-reactivity of various suppressor tRNAs can be reduced – without substantially compromising their suppression efficiency in the presence of the cognate aaRS – through the use of directed evolution.5,18–20 More specifically, mutations in the acceptor and the T-stem regions have led to such improvements. To this end, we used site-saturation mutagenesis to fully randomize eleven bases in the acceptor stem of Af-tRNASer(GCU). Additionally, the adenine immediately following the anticodon (position 36) was randomized to both A and G (Figure 2A). The resulting library was cloned into a pBK vector under the lpp promoter and co-transformed into DH10b E. coli harboring a pREP plasmid that expresses the MmSerRS gene from a constitutive glnS promoter, as well as a CAT gene with a TAG codon at a permissive site (D111TAG). Growing the resulting pool of cells in the presence of 50 µg/mL chloramphenicol (on LB-agar plates) led to the enrichment of active Af-tRNASer(GCU) mutants (positive selection). The surviving tRNA mutants were isolated and co-transformed into a DH10b strain containing a pNeg plasmid that expresses an arabinose-inducible toxic barnase gene harboring two TAG nonsense codons at permissive sites (Q3TAG and D45TAG). Upon arabinose induction, any cross-reactive tRNA mutant would lead to cell death by enabling the expression of full-length barnase protein. After subjecting the library to two rounds each of alternating positive and negative selections, individual tRNA clones were tested using the chloramphenicol resistance assay described above in the presence or absence of MmSerRS. Two clones exhibited significantly attenuated MICs (<20 µg/mL) in the absence of MmSerRS, while retaining strong activity (>100 µg/mL) in its presence (Figure 2B). One of these, hit 49, had a substantially altered acceptor stem (Figure 2C), while the other (AfS.G; Figure 2D) had a wild-type acceptor stem but had an A to G mutation following the anticodon.
Figure 2.
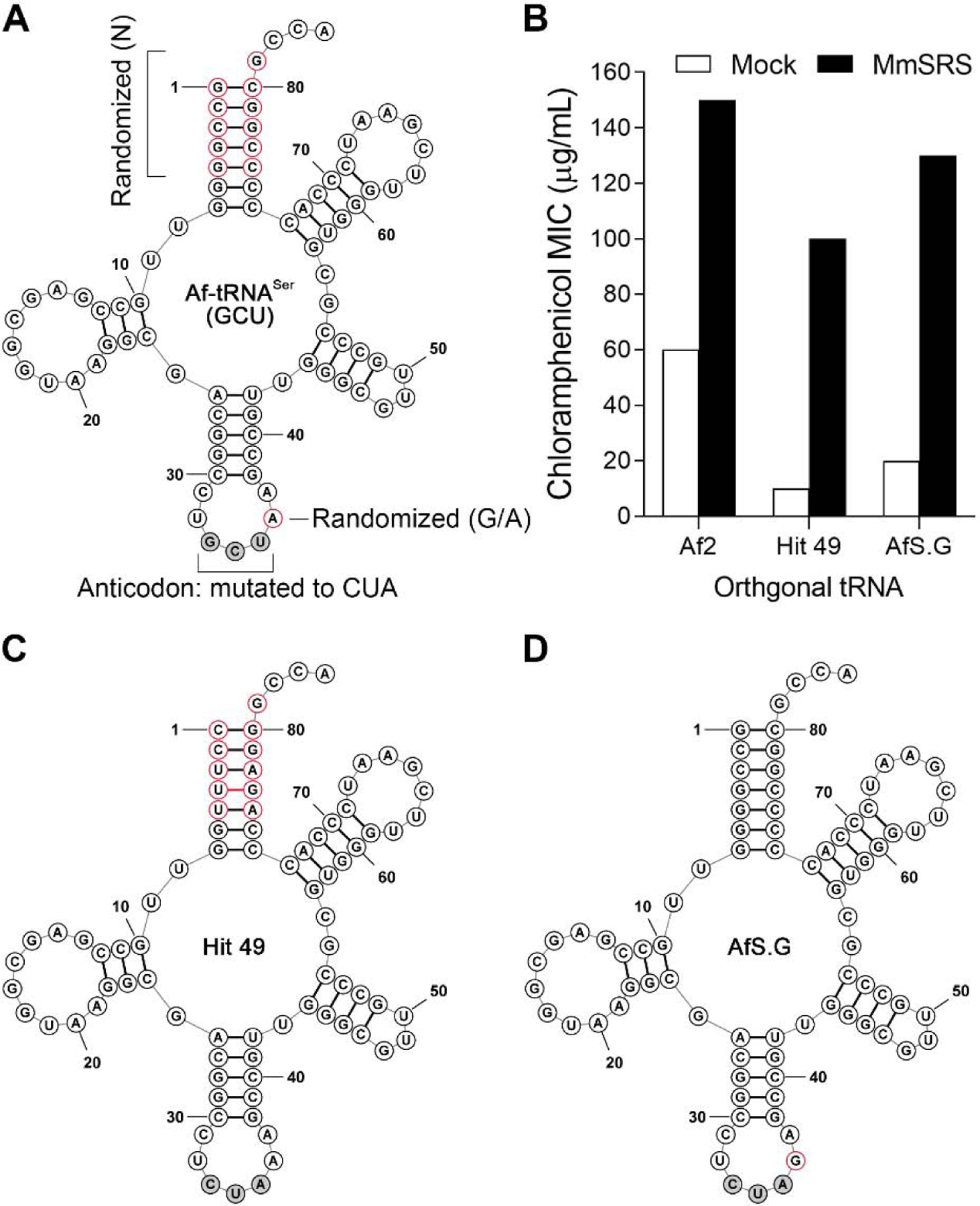
Directed evolution of an Af-tRNASer library. (A) Af-tRNASer(GCU) sequence is shown. The library sites are highlighted as red circle; N = A, G, C, and U (B) Orthogonality of synthetic tRNAs in E. coli. The chloramphenicol resistance assay was used to measure the chloramphenicol MIC values for E. coli expressing the indicated tRNA. (C) Clone 49 sequence. Modified bases are in red; the non-canonical base pair is highlighted with a red bar; the anticodon is modified to CUA. (D) AfS.G sequence. Modified base is in red; anticodon is modified to CUA.
Clone 49 exhibits very low cross-reactivity, which is ideal for developing a selection system for engineering the substrate specificity of MmSerRS. To confirm this hypothesis, the MmSerRS gene was cloned into pBK3 (Figure S1) and tRNA clone 49 was inserted into pREP (Figure S2),21 the plasmid system typically used for the directed evolution of aaRSs. These vectors were then co-transformed into DH10b E. coli and serial dilutions of an overnight culture were plated onto GMML-agar containing increasing concentrations of chloramphenicol. A pBK plasmid devoid of the synthetase (pBKempty) was used as a negative control. As expected, cells harboring tRNA clone 49/MmSerRS were able to survive high concentrations of CM, but tRNA clone 49 alone did not support cell growth in the presence of chloramphenicol (Figure S4), demonstrating the suitability of this system to enrich active MmSerRS mutants from a synthetic library.
In contrast, AfS.G shows significantly higher suppression efficiency, which makes it a better choice for developing an efficient expression system where this pair would be used for ncAA incorporation into target proteins. Previous studies suggest that the low cross-reactivity of AfS.G is unlikely to cause a loss of fidelity when the cognate MmSerRS is coexpressed.22 To demonstrate this, we cloned the tRNA-AfS.G/MmSerRS pair into the pUltra suppressor plasmid, which has been previously optimized to provide highly efficient ncAA incorporation into target proteins.23 This plasmid was co-transformed into DH10b with a pET22b plasmid expressing the sfGFP-151-TAG reporter from a strong T5-lac promoter.24 Upon induction with isopropyl 1-thio-β-D-galactopyranoside, this culture showed robust fluorescence, indicative of efficient suppression of the TAG codon in the GFP gene. The full-length reporter protein was purified by immobilized metal-ion chromatography (IMAC) with excellent yield (~250 mg/L, ~50% relative to the parent protein) using a C-terminal hexahistidine tag. The resulting protein was analyzed by SDS-PAGE and ESI-MS, which revealed a single specie with a molecular weight consistent with the incorporation of serine at position 151. These observations confirm that this orthogonal serine pair incorporates serine in response to the TAG nonsense codon with high fidelity and efficiency (Figure S6 A and B).
2.2. Protein production and crystallization of MmSerRS mutants
With an efficient and orthogonal tRNAser/SerRS pair in hand, we next determined the three-dimensional crystal structure of the MmSerRS in order to facilitate the design of active site libraries for future studies aimed at evolving synthetases with altered specificities. To express the protein, the cDNA of MmSerRS was cloned into a pET22B vector (Figure S5) under the control of an inducible T7-lac promoter. Various hexahistidine (6His)-tagged constructs were tested, including N- or C-terminal tagged proteins together with a TEV protease-cleavable N-term-6His construct (Figure S3). After protein overexpression and purification by IMAC and ion-exchange chromatography, yielding 150 mg/L of homogenous material, the different construct permutations were screened in crystallization experiments under a variety of conditions (JCSG 1–4 core suite, 24 and 4 °C). Only a handful of conditions gave rise to crystals, with the best set of conditions (LiOAc/PEG3500) yielding crystals diffracting at an unsatisfactory 6–10 Å resolution.
A systematic approach investigating the crystal packing of closely related structures of similar synthetases (PDB ID: 2DQ3, 3LSQ and 1SES) was undertaken, looking for residues or short regions that could engage in intramolecular contacts with the neighboring molecules. This search pointed to some charged residues in our target sequence that could potentially disrupt a common antiparallel β-sheet otherwise observed, therefore causing suboptimal diffraction patterns. Our analysis highlighted a noticeable sequence variety among known structures in the C-terminal portion of the enzyme, specifically, from residues 400 to 422. We therefore introduced mutations reverting two glutamic acids to valine (E402V, E416V) in order to keep the antiparallel β-sheet intact, and also shorten the peptide chain from residues 417–422 to diminish flexibility and avoid possible clashes. We then expressed and purified a set of MmSerRS C-terminal mutant constructs (Figure S7): in a preliminary crystallization screen, two mutants (Figure S7A and S7B, SerRSM1 and SerRSM2 throughout) gave rise to crystals diffracting at 3.4 Å resolution.
We then turned our attention to the synthesis and co-crystallization of both SerRSM1 and SerRSM2 with a non-hydrolysable serine-AMP analogue (SerAMS, 1) to map the key active site interactions and potentially enhance crystal formation. 25,26,27,28,29
2.3. Ligand synthesis and SerM2 co-crystal
The synthesis of SerAMS 1 (5’-O-[N-(L-seryl)-sulfamoyl]adenosine) was carried out following a previously reported scheme,30 with slight variations to improve yields and avoid palladium impurities.26 Briefly (Scheme 1), 2’,3’-O-isopropylideneadenosine was heated to reflux with a molar excess of bis(tributyltin) oxide giving its 5’-O-tributyltin ether (2);31 the mixture was then treated with an excess of sulfamoyl chloride yielding the crystalline intermediate 3 after column chromatography. Compound 3 was reacted with the NHS ester of methyl (S)-(−)-3-Boc-2,2-dimethyl-4-oxazolidinecarboxylate (4) in the presence of DBU to afford compound 5, which was subjected to global acidic deprotection in aqueous TFA yielding SerAMS (1).
Scheme 1.
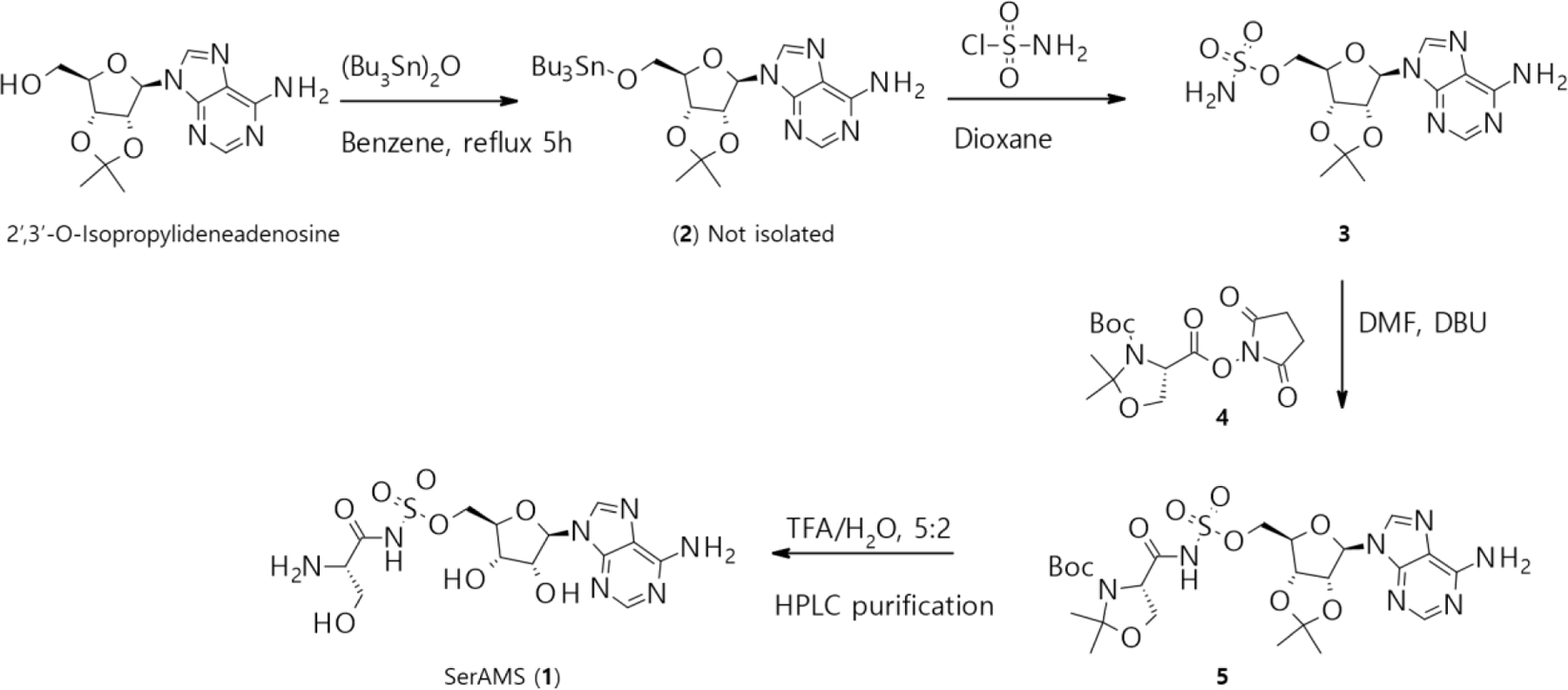
Synthesis of SerAMS (1): 2’−3’-O-isopropylideneadenosine (1.0 equiv.), bis(tributyltin) oxide (2.0 equiv.), benzene 80 °C; sulfamoyl chloride (5.0 equiv.) 0.3 M in dioxane 0 °C to RT. Intermediate 3 (1.0 equiv.) 0.4 M in DMF, intermediate 4 (2.0 equiv.), DBU (2.5 equiv.), RT, 12 hours. Intermediate 5, in TFA/H2O 5:2, lyophilize.
Binary complex formation was carried out by mixing protein and ligand at a 1:10 molar ratio to a final concentration of 0.45 mM for the protein and 5 mM for the ligand. Single crystals appeared after 3 days of hanging drop crystallization and grew to their maximum size within a week. Homogenous crystals grew in 18% PEG3350 and 200 mM thiocyanate at 22 °C. Single crystals were flash-frozen in liquid nitrogen before they were mounted for data collection. The structure of the binary complex MmSerRS:SerAMS was then determined and refined at 1.45 Å resolution (Figure 3A).
Figure 3.
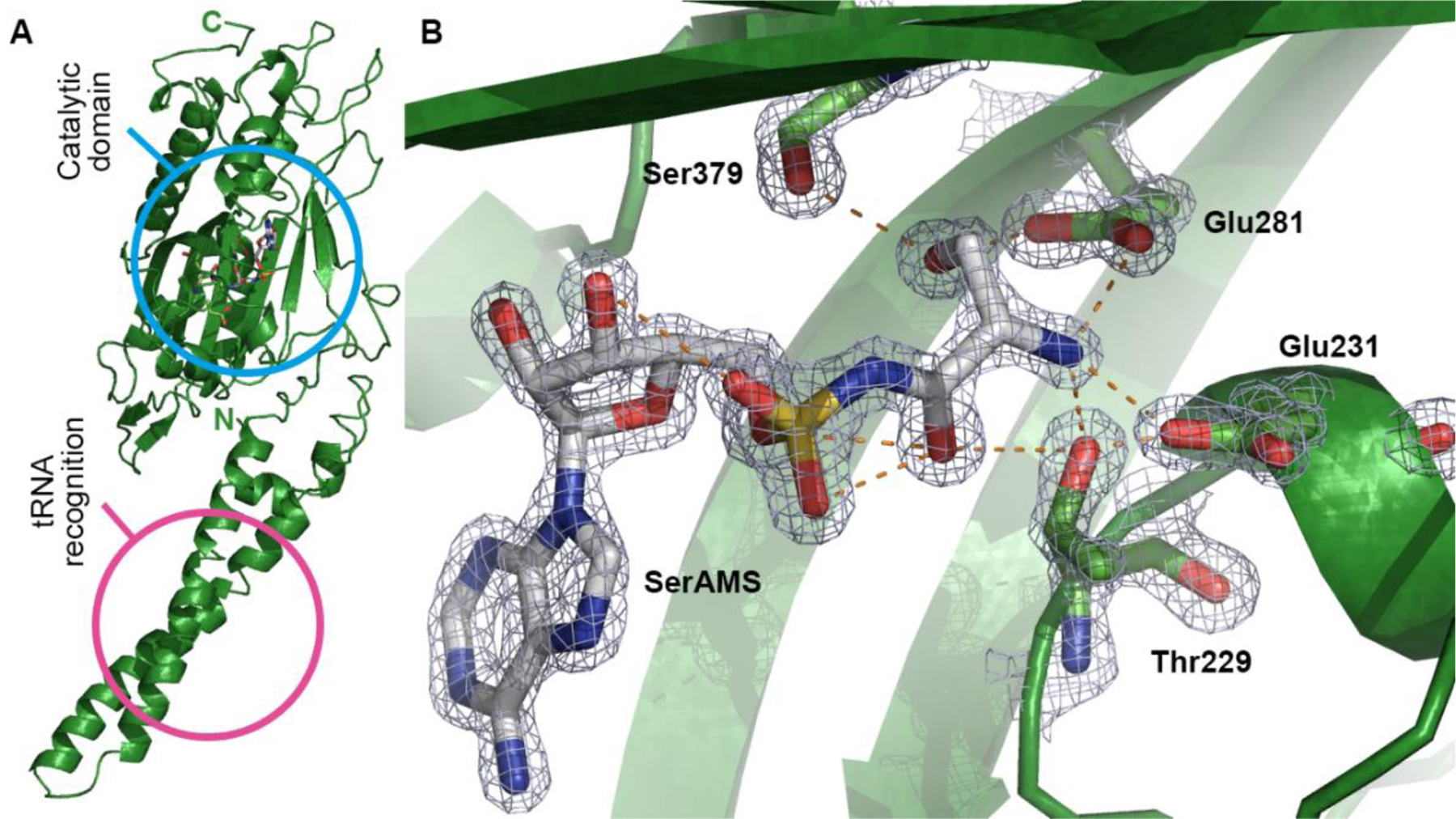
Co-crystal structure of MmSerRS:SerAMS (1) (PDB ID: NNNN). (A) Overall structure. Catalytic site (sky blue) and tRNA recognition site (pink) are highlighted with circles. (B) Amino acid binding site. SerAMS (1) forms a strong hydrogen bonding (orange dashed lines) network with surrounding residues (Thr229, Glu231, Glu281, and Ser379) at the amino acid binding site. Electron density as a 2F0Fc contour map (3s) is shown in meshed surface around SerAMS and amino acid binding residues. Other residues are omitted for clarity.
2.4. Structural analysis
Crystals belong to space group C 2 2 21 and contain one molecule per asymmetric unit. However, data from size exclusion chromatography (Figure S9) confirmed the protein is dimeric in solution; the extensive intermolecular contacts across a crystallographic two fold axis also indicates its dimeric nature, similar to other available structures from human, bacteria and archaea.32,33,34 As depicted in figure 3A, MmSerRS shows the distinctive features of a typical class II synthetase: the nucleotide binding module is composed of antiparallel β sheets and within this module lie three conserved motifs (motif 1,2 and 3) that are characteristic of class II synthetases. 14,35,36,37,38
The overall structural fold of this protein is highly similar to other known structures of SerRS family members. A protein sequence comparison identified bacterial Aquifex aeolicus VF5 (PDB ID: 2DQ3) as the protein with the highest sequence similarity (57% sequence identity and 74% sequence similarity). The superimposition of Cα atoms of the full-length structure shows a low root mean square deviation (rmsd) of 0.52, underscoring the high degree of similarity between the two macromolecules. As shown in the crystal structure, each monomeric unit is composed of a solvent exposed N-terminal antiparallel coiled-coil arm; this 57 Å long helical arm has tRNA-binding function, specifically for the variable arm recognition.38 The catalytic site is located within the C-terminal portion of the monomer and is structured around seven-stranded antiparallel β sheets (Figure 3A).
There are 13 residues contributing 16 hydrogen bonds to the ligand in both MmSerRS and its homologue from Aquifex aeolicus, where only one residue is not identical, suggestive of a highly conserved Ser-AMP binding pocket. The serine recognition site comprises Thr229, Glu231, Glu281, and Ser379 which form an extensive hydrogen bonding network with serine. With respect to Ser hydroxyl side chain recognition, hydrogen bonding with Glu281 and Ser379 appears to be essential, and the gamma-carboxyl side chains of Glu231 and Glu281 form salt bridges with the amino group of the serine. In addition to the charged residues, Thr229 plays a key role in accommodating both the amino group as well as the carbonyl group of SerAMS (1) (Figure 3B). In fact, two residues that appear to be the main determinants in accommodating Ser side chain, Glu281 and Ser379, were sensitive to Ala mutation as assessed by the chloramphenicol resistance assay (Figure S11). These results suggest a possible rationale for library design to create a pocket to accommodate ncAAs by mutating residues surrounding the Ser side chain including Thr229, Cys256, Lys279, Glu281, Ser348, Ser350, Asn377, and Ser379.
3. Conclusion
We have generated an orthogonal amber suppressor pair Af-tRNASer/MmSerRS of archaeal origin that can be used for ncAA mutagenesis in E. coli. Directed evolution experiments showed that the acceptor stem and/or the anticodon loop of suppressor tRNA can be modified to enhance orthogonality of the tRNA. In addition, we have solved the X-ray co-crystal structure of MmSerRS:SerAMS at high resolution. This structural information will be useful for generating active site mutants of SerRS that accommodate small and/or polar ncAAs into proteins in E. coli. These latter experiments are currently underway.
Supplementary Material
Acknowledgments
Kristen Williams for assistance in manuscript preparation, Dr. Sean A. Reed, Dr. Angad P. Mehta and Dr. Michael J. Bollong for helpful discussions and manuscript revision. We would like to acknowledge Dr. Marc-André Elsliger from the Wilson lab at Scripps Research for initial crystallization screening efforts, together with Dr. Bernhard Kuhle from the Schimmel lab for invaluable assistance throughout the project. This work is supported by NIH R01 GM062159 (Schultz).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare no competing financial interest
References
- 1.Young DD; Schultz PG ACS Chem Biol 2018, 13, 854–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dumas A; Lercher L; Spicer CD; Davis BG Chem Sci 2015, 6, 50–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang L; Brock A; Herberich B; Schultz PG Science 2001, 292, 498–500. [DOI] [PubMed] [Google Scholar]
- 4.Wan W; Tharp JM; Liu WR Biochim Biophys Acta 2014, 1844, 1059–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatterjee A; Xiao H; Yang PY; Soundararajan G; Schultz PG Angew Chem Int Ed Engl 2013, 52, 5106–9. [DOI] [PubMed] [Google Scholar]
- 6.Fukunaga R; Yokoyama S Nat Struct Mol Biol 2007, 14, 272–9. [DOI] [PubMed] [Google Scholar]
- 7.Lee S; Oh S; Yang A; Kim J; Soll D; Lee D; Park HS Angew Chem Int Ed Engl 2013, 52, 5771–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park HS; Hohn MJ; Umehara T; Guo LT; Osborne EM; Benner J; Noren CJ; Rinehart J; Soll D Science 2011, 333, 1151–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rogerson DT; Sachdeva A; Wang K; Haq T; Kazlauskaite A; Hancock SM; Huguenin-Dezot N; Muqit MM; Fry AM; Bayliss R; Chin JW Nat Chem Biol 2015, 11, 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malyshev DA; Dhami K; Lavergne T; Chen T; Dai N; Foster JM; Correa R Jr.; Romesberg FE Nature 2014, 509, 385–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lajoie MJ; Rovner AJ; Goodman DB; Aerni HR; Haimovich AD; Kuznetsov G; Mercer JA; Wang HH; Carr PA; Mosberg JA; Rohland N; Schultz PG; Jacobson JM; Rinehart J; Church GM; Isaacs FJ Science 2013, 342, 357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang K; Fredens J; Brunner SF; Kim SH; Chia T; Chin JW Nature 2016, 539, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartlein M; Madern D; Leberman R Nucleic Acids Res 1987, 15, 1005–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cusack S; Berthet-Colominas C; Hartlein M; Nassar N; Leberman R Nature 1990, 347, 249–55. [DOI] [PubMed] [Google Scholar]
- 15.Woese CR; Olsen GJ; Ibba M; Soll D Microbiol Mol Biol Rev 2000, 64, 202–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grasso KT; Yeo MJR; Hillenbrand CM; Ficaretta ED; Italia JS; Huang RL; Chatterjee A bioRxiv 2019, 829028.
- 17.Lesjak S; Weygand-Durasevic I FEMS Microbiol Lett 2009, 294, 111–8. [DOI] [PubMed] [Google Scholar]
- 18.Anderson JC; Schultz PG Biochemistry 2003, 42, 9598–608. [DOI] [PubMed] [Google Scholar]
- 19.Chatterjee A; Xiao H; Schultz PG Proc Natl Acad Sci U S A 2012, 109, 14841–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo J; Melancon CE 3rd; Lee HS; Groff D; Schultz PG Angew Chem Int Ed Engl 2009, 48, 9148–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melancon CE 3rd; Schultz PG Bioorg Med Chem Lett 2009, 19, 3845–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Javahishvili T; Manibusan A; Srinagesh S; Lee D; Ensari S; Shimazu M; Schultz PG ACS Chem Biol 2014, 9, 874–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chatterjee A; Sun SB; Furman JL; Xiao H; Schultz PG Biochemistry 2013, 52, 1828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koh M; Cho HY; Yu C; Choi S; Lee KB; Schultz PG Bioconjug Chem 2019, 30, 2102–2105. [DOI] [PubMed] [Google Scholar]
- 25.Landeka I; Filipic-Rocak S; Zinic B; Weygand-Durasevic I Biochim Biophys Acta 2000, 1480, 160–70. [DOI] [PubMed] [Google Scholar]
- 26.Belrhali H; Yaremchuk A; Tukalo M; Larsen K; Berthet-Colominas C; Leberman R; Beijer B; Sproat B; Als-Nielsen J; Grubel G; et al. Science 1994, 263, 1432–6. [DOI] [PubMed] [Google Scholar]
- 27.Dock-Bregeon A; Sankaranarayanan R; Romby P; Caillet J; Springer M; Rees B; Francklyn CS; Ehresmann C; Moras D Cell 2000, 103, 877–84. [DOI] [PubMed] [Google Scholar]
- 28.Bilokapic S; Maier T; Ahel D; Gruic-Sovulj I; Soll D; Weygand-Durasevic I; Ban N EMBO J 2006, 25, 2498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rocha R; Barbosa Pereira PJ; Santos MA; Macedo-Ribeiro S Acta Crystallogr Sect F Struct Biol Cryst Commun 2011, 67, 153–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ueda H; Shoku Y; Hayashi N; Mitsunaga J; In Y; Doi M; Inoue M; Ishida T Biochim Biophys Acta 1991, 1080, 126–34. [DOI] [PubMed] [Google Scholar]
- 31.Jenkins ID; Verheyden JP; Moffatt JG J Am Chem Soc 1976, 98, 3346–57. [DOI] [PubMed] [Google Scholar]
- 32.Vincent C; Borel F; Willison JC; Leberman R; Hartlein M Nucleic Acids Res 1995, 23, 1113–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Itoh Y; Sekine S; Kuroishi C; Terada T; Shirouzu M; Kuramitsu S; Yokoyama S RNA Biol 2008, 5, 169–77. [DOI] [PubMed] [Google Scholar]
- 34.Xu X; Shi Y; Yang XL Structure 2013, 21, 2078–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cusack S; Hartlein M; Leberman R Nucleic Acids Res 1991, 19, 3489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruff M; Krishnaswamy S; Boeglin M; Poterszman A; Mitschler A; Podjarny A; Rees B; Thierry JC; Moras D Science 1991, 252, 1682–9. [DOI] [PubMed] [Google Scholar]
- 37.Mosyak L; Safro M Biochimie 1993, 75, 1091–8. [DOI] [PubMed] [Google Scholar]
- 38.Biou V; Yaremchuk A; Tukalo M; Cusack S Science 1994, 263, 1404–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.