

Abstract
The etiology of neurodevelopmental disorders (NDDs) remains a challenge for researchers. However, in recent years, with the surge of clinical sequencing studies, many of these disorders have been determined to be monogenic and monoallelic in nature. Human brain development is tightly regulated, and it is sensitive to cellular alterations caused by endogenous or exogenous factors. Chromatin dysfunction was described as a molecular pathway highly dysregulated in NDDs, with many of these syndromes demonstrate phenotypic overlap with one another. Among these patients, intellectual disability appears to be a typical phenotype across disorders. Here we discuss mutations on the epigenetic machinery; writers, erasers, readers, remodelers, and even histones, that are predicted to affect gene regulation. First, we discuss disorders associated with mutations in histone acetylation enzymes. Second, we highlight syndromes involving histone methylation enzymes followed by chromatin remodeling enzymes. Lastly, we touch on recently discovered germline histone mutations and their pathogenic outcome on neurological function. Throughout this review, we discuss various animal and iPSC models of these disorders and their usefulness in determining pathomechanism and potential therapeutics. Ultimately, classifying these disorders based on their effects on the epigenome will not only aid in prognosis in patients but to elucidate the role of epigenetic machinery throughout neurodevelopment.
INTRODUCTION
Neurodevelopmental disorders (NDD) ranging from attention-deficit hyperactivity disorder (ADHD), Fragile X syndrome, autism spectrum disorder (ASD) and intellectual disability (ID) disorders, occur when the central nervous system (CNS) develops abnormally. ID, which involves defects in general cognition and adaptive behavior, has a prevalence of 3% world-wide, affecting 1 in 6 children in the US alone. NDD patients often exhibit ID as well as other comorbidities such as developmental delay (DD), epilepsy, autism spectrum disorder (ASD) and psychomotor impairments. The etiology of ID is heterogeneous as it has been linked to environmental factors during pregnancy, infections, chromosome deletions and recently de novo point mutations. Next-generation sequencing has propelled the study of these disorders through identification of 750 genes, all proposed to be causative of ID (Kochinke et al. 2016). Importantly, many of these are considered monogenic disorders, however, the pathological mechanism driving ID disorders are largely unknown. Interestingly, epigenetic determinants account for 8% of ID disorders and are starting to be recognized as major players in neurological disorders (Tjitske Kleefstra et al. 2014).
Epigenetics refers to processes that influence gene expression patterns and cellular phenotypes, but do not involve changes in DNA sequence (Dupont, Armant, and Brenner 2009). At the molecular level, epigenetic mechanisms primarily involve covalent modifications on DNA and histone proteins, however, this review will not cover DNA modifications (Dupont, Armant, and Brenner 2009). Histone modifications facilitate the condensation of chromatin, ultimately regulating DNA accessibility by transcriptional machinery. Histone post-translational modifications (PTMs), including acetylation, methylation and phosphorylation occur on the flexible N-terminus tails. These PTMs are regulated by epigenetic machinery, often protein complexes, that are responsible for the deposition (writers), removal (erasers) and recognition (readers) of the histone code (Figure 1) (Strahl and Allis 2000). Generally, histone “writers” deposits PTMs at specific genomic loci which may result in the promotion or inhibition of transcription due to chromatin accessibility. More often than not, histone acetylation defines active transcription whereas histone methylation is more complex and will be discussed in the review (Figure 2). “Eraser” proteins remove histone PTMs allowing for tight regulation of gene expression as PTM removal often results in the opposite chromatin state. “Reader” domains are often found on or in conjunction with writer proteins and chromatin remodelers, who move nucleosomes in an ATP-dependent manner. These proteins are highly regulated, resulting in a cell-type-specific steady state of histone PTMs which is malleable and allows for rapid response to cellular signals. Therefore, our understanding of the fine-tuned mechanisms that regulate the epigenetic machinery under normal conditions is critical to elucidate the mechanisms of pathogenic variants in disease.
FIGURE 1:
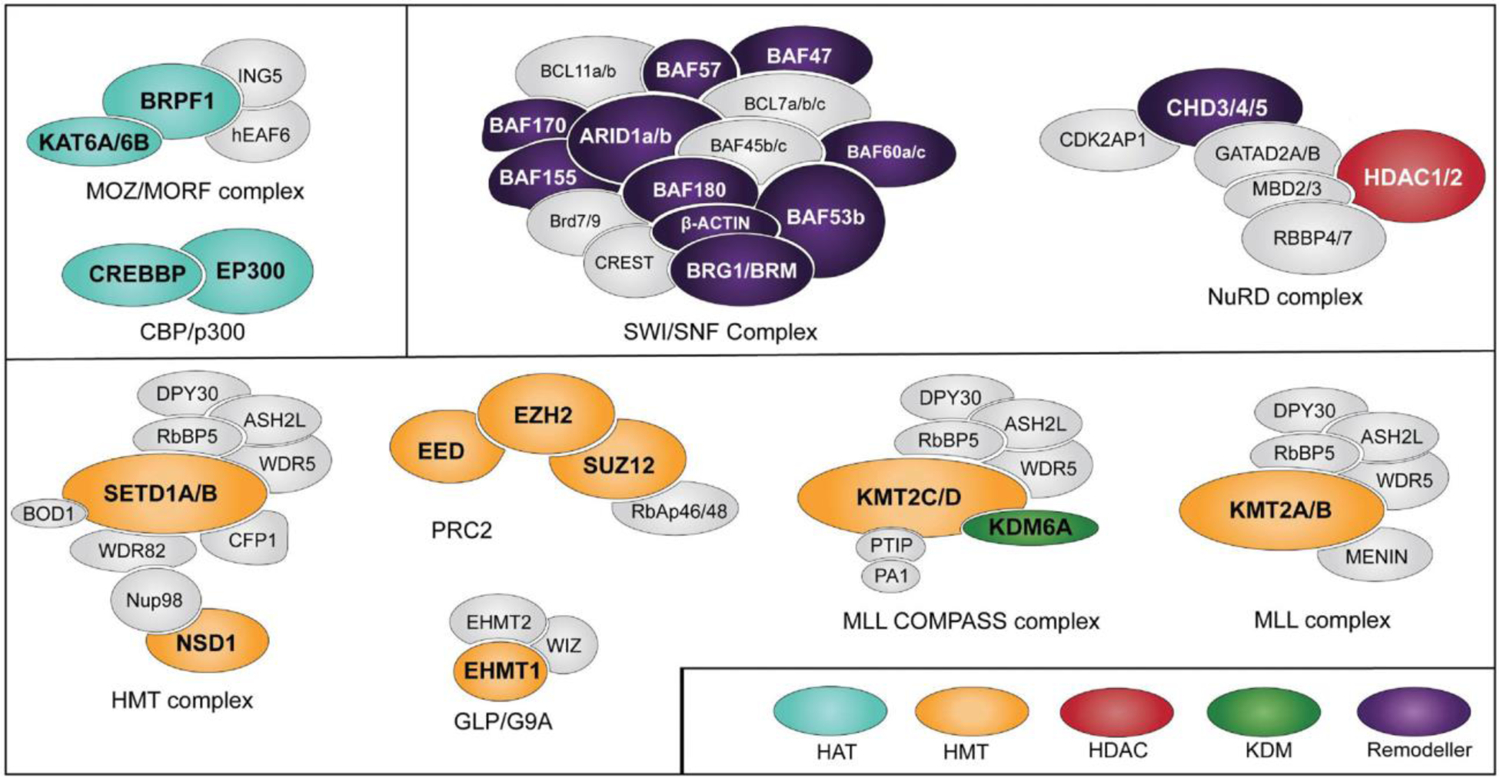
Chromatin-modifying enzymes and the complexes they belong. Proteins mutated in NDDs are shown in color.
FIGURE 2:
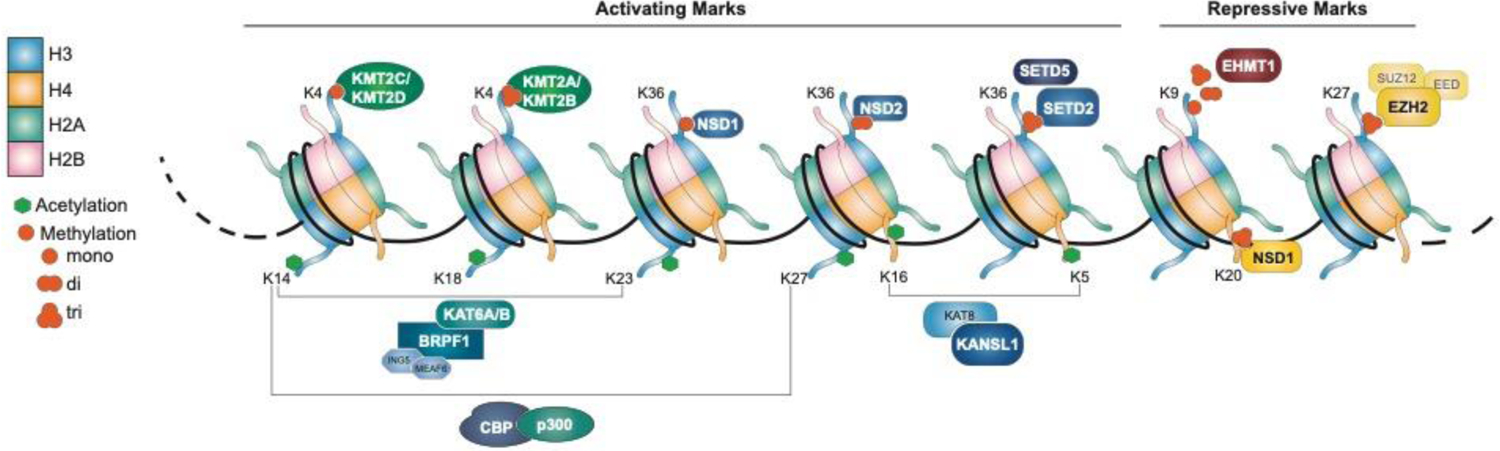
Histone post-translational modifications are classified as activating or repressing depending on the modification site and the degree of modifications. Histone acetylation is primarily activating whereas methylation can play both roles. The HATs and KMTs which are mutated in NDD, as well as their targets, are shown above.
Thanks to techniques such as tandem mass spectrometry (MS) and chromatin-immunoprecipitation sequencing (ChIP-seq), histone PTM co-occurrences, their differential abundance in a variety of in vitro and in vivo models, and their genomic localization has gained appreciation in the last decade(Vermeulen et al. 2010). Particularly, the co-occurrence of H3K4me3 (activating mark) and H3K27me3 (repressive mark) at developmental genes has been shown to “poise” these loci for rapid transcriptional activation; this specific co-ocurrance has been defined as bivalency (Voigt, Tee, and Reinberg 2013). Collectively, extensive studies have displayed the importance of establishing and maintaining histone PTMs during development and disease progression. Bivalency has been demonstrated to be necessary for proper differentiation of both neuronal and neural crest stem cells (Burney et al. 2013). Interestingly, dynamic H3K27me3 changes on transcription factor promoters mediates neocortex development, again highlighting the importance of histone PTMs in the developing brain (Albert et al. 2017). Furthermore, profiling of histone marks in neural progenitor cells reinforces this notion as PTMs were shown to influence cellular identity (Mikkelsen et al. 2007). In mature neurons, histone acetylation and methylation has been shown to drive activity-dependent modulation of neuronal networks (Bonnaud, Suberbielle, and Malnou 2016). Mice with heterozygous mutations in histone modifying enzymes exhibit altered synaptic plasticity resulting in decreased learning, short and long-term memory, and behavioral anomalies (Levenson and Sweatt 2005). Moreover, chromatin factors are ubiquitously expressed in the brain in key regions for learning, memory and movement (Figure 3).
FIGURE 3:
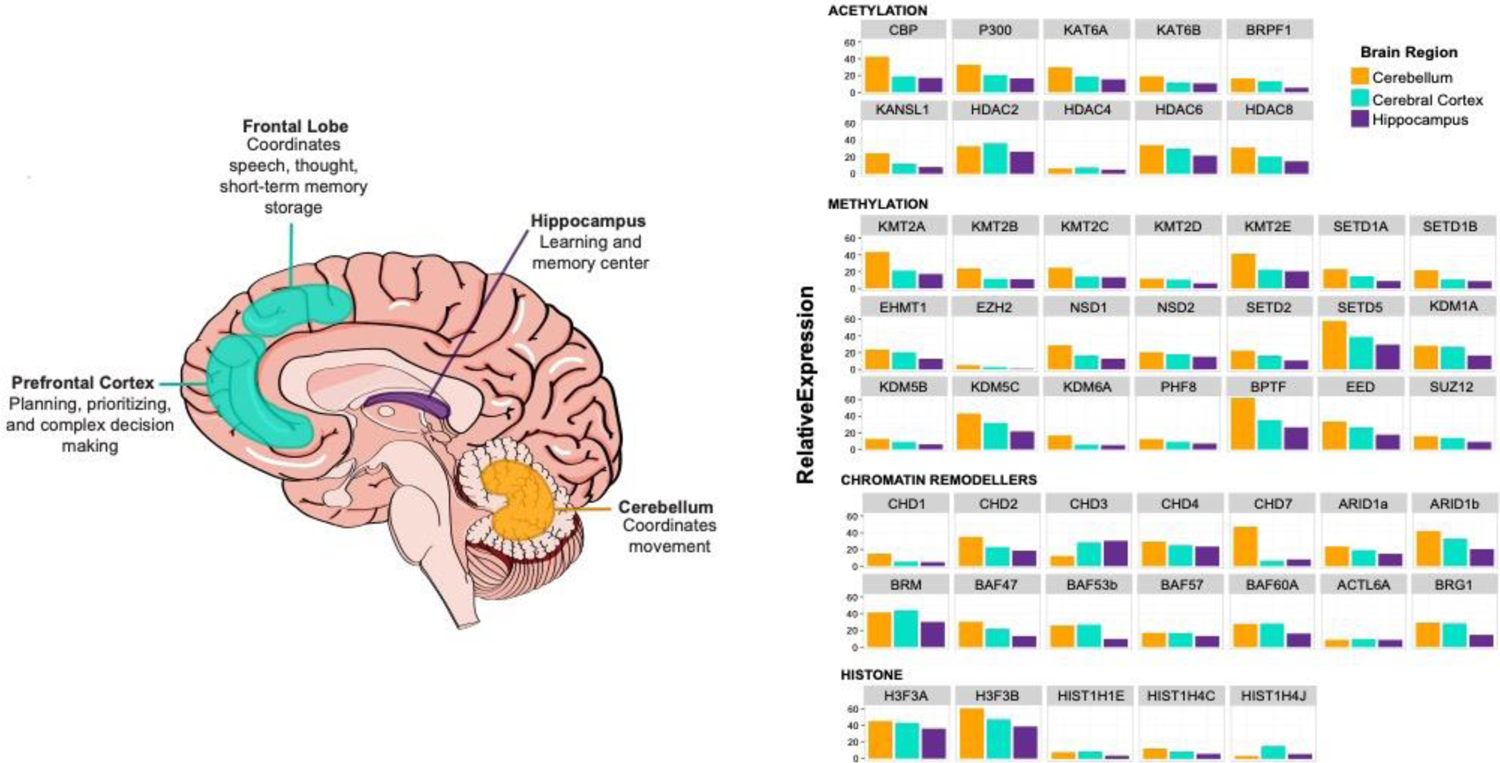
Spatial expression of epigenetic factors dysregulated in NDD. (left) Graphical representation of brain regions in which most genes are highly expressed and their respective functions. (right) Expression in these three regions according to Consensus normalized expression (NX) levels on Protein atlas. This dataset was created by combining two transcriptomic datasets (GTEx and FANTOM5).
Intriguingly, individuals carrying germline mutations in epigenetic machinery often demonstrate severe neurological dysfunction often manifesting as developmental and psychomotor delay. This suggests that the consequences of improper epigenetic regulation result in neuronal dyshomeostasis and ultimately severe impairment to the CNS. Although these germline mutations are ubiquitously expressed, it is interesting that the brain is the most affected organ in these individuals. Thus, there is sufficient evidence depicting the importance of epigenetic machinery in not only neurodevelopment but also neuronal function. However, it is imperative to illustrate how alterations to the epigenetic landscape and gene expression profiles result in observed phenotypes in these individual disorders. In this review, we focus on driver mutations of chromatin modifying enzymes in ID disorders (Table 1). Specifically, writers and erasers of histone acetylation and methylation, chromatin remodeling enzymes, and even mutations in histones themselves.
Table 1.
Driver mutations of chromatin modifying enzymes in intellectual disability disorders
| Gene | Gene OMIM | Syndrome | OMIM | Gene Function |
|---|---|---|---|---|
| CBP | 600140 | Menke-Hennekam syndrome 1/Rubinstein-Taybi syndrome 1 | 618332/180849 | Histone acetyltransferase |
| P300 | 602700 | Menke-Hennekam syndrome 2/ Rubinstein-Taybi syndrome 2 | 618333/613684 | Histone acetyltransferase |
| KAT6A | 601408 | Arboleda-Tham syndrome | 616268 | Histone acetyltransferase |
| KAT6B | 602303 | Say-Barber-Biesecker-Young-Simpson / Genitopatellar syndrome | 603736 / 606170 | Histone acetyltransferase |
| BRPF1 | 602410 | Intellectual developmental disorder with dysmorphic facies and ptosis | 617333 | Histone acetyltransferase complex subunit |
| KANSU | 612452 | Koolen-De Vries syndrome | 610443 | Histone acetyltransferase complex subunit |
| HDAC2 | 605164 | Cornelia de Lange syndrome | n/a | Histone deacetylase |
| HDAC4 | 605314 | Brachydactyly mental retardation | 600430 | Histone deacetylase |
| HDAC6 | 300272 | X-linked dominant Chondrodysplasia | 300863 | Histone deacetylase |
| HDAC8 | 300269 | Cornelia de Lange syndrome 5 | 300882 | Histone deacetylase |
| KMT2A | 159555 | Wiedemann-Steiner syndrome | 605130 | Histone lysine methytransferase |
| KMT2B | 606834 | Dystonia 28, childhood-onset | 617284 | Histone lysine methytransferase |
| KMT2C | 606833 | Kleefstra syndrome 2 | 617768 | Histone lysine methytransferase |
| KMT2D | 602113 | Kabuki syndrome 1 | 147920 | Histone lysine methytransferase |
| KMT2E | 608444 | O’Donnell-Luria-Rodan syndrome | 618512 | Histone lysine methytransferase |
| SETD1A | 611052 | Neurodevelopmental disorder with speech impairment and dysmorphic facies / Eariy-onset Epilepsy | 619056 / 618832 | Histone lysine methytransferase |
| SETD1B | 611055 | Intellectual developmental disorder with seizures and language delay | 619000 | Histone lysine methytransferase |
| EHMT1 | 607001 | Kleefstra syndrome 1 | 610253 | Histone lysine methytransferase |
| NSD1 | 606681 | Sotos syndrome 1 | 117550 | Histone lysine methytransferase |
| NSD2 | 602952 | Wolf-Hirschhom syndrome | 194190 | Histone lysine methytransferase |
| SETD2 | 612778 | Luscan-Lumish syndrome | 616831 | Histone lysine methytransferase |
| SETD5 | 615743 | Mental retardation, autosomal dominant 23 | 615761 | Histone lysine methytransferase |
| EZH2 | 601573 | Weaver syndrome | 277590 | Histone lysine methytransferase |
| SUZ12 | 606245 | Imagawa-Matsumoto syndrome | 618786 | PRC2 subunit |
| EED | 605984 | Cohen-Gibson syndrome | 617561 | PRC2 subunit |
| KDM1A | 609132 | Cleft palate, psychomotor retardation, and distinctive facial features | 616728 | Histone lysine demethylase |
| KDM5B | 605393 | Mental retardation, autosomal recessive 65 | 618109 | Histone lysine demethylase |
| KDM5C | 314690 | Mental retardation, X-linked, syndromic, Claes-Jensen type | 300534 | Histone lysine demethylase |
| KDM6A | 300128 | Kabuki syndrome 2 | 300867 | Histone lysine demethylase |
| PHF8 | 300560 | Mental retardation syndrome, X-linked, Siderius type | 300263 | Histone lysine demethylase |
| CHD1 | 602118 | Pilarowski-Bjomsson syndrome | 617682 | ATP-dependent Chromatin remodeler |
| CHD2 | 602119 | Childhood-onset Epileptic Encephalopathy | 615369 | ATP-dependent Chromatin remodeler |
| CHD3 | 602120 | Snijders Blok-Campeau syndrome | 618205 | ATP-dependent Chromatin remodeler |
| CHD4 | 603277 | Sifrim-Hitz-Weiss syndrome | 617159 | ATP-dependent Chromatin remodeler |
| CHD7 | 608892 | CHARGE syndrome | 214800 | ATP-dependent Chromatin remodeler |
| ARIDIb | 614556 | Coffin-Siris syndrome 1 | 135900 | ATP-dependent Chromatin remodeler |
| ARIDIa | 603024 | Coffin-Siris syndrome 2 | 614607 | BAF subunit |
| BAF47 | 601607 | Coffin-Siris syndrome 3 | 614608 | BAF subunit |
| BRG1 | 603254 | Coffin-Siris syndrome 4 | 614609 | ATP-dependent Chromatin remodeler |
| BAF57 | 603111 | Coffin-Siris syndrome 5 | 616938 | BAF subunit |
| ARID2 | 609539 | Coffin-Siris syndrome 6 | 617808 | BAF subunit |
| DPF2 | 601671 | Coffin-Siris syndrome 7 | 618027 | BAF subunit |
| BAF60A | 601735 | Coffin-Siris syndrome 11 | 618779 | BAF subunit |
| BRM | 600014 | Nicolaides-Baraitser syndrome | 601358 | BAF subunit |
| BAF53b | 612458 | IDD with severe speech and ambulation defects / Developmental and epileptic encephalopathy 76 | 618470 / 618468 | BAF subunit |
| ACTL6A | 604958 | n/a | n/a | BAF subunit |
| BPTF | 601819 | Neurodevelopmental disorder with dysmorphic facies and distal limb anomalies | 617755 | Chromatin remodeller |
| HIST1H1E | 142220 | Rahman syndrome | 817537 | Linker Histone |
| HIST1H4C | 602827 | unclassified | n/a | Canonical histone |
| HIST1H4J | 602826 | unclassified | n/a | Canonical histone |
| H3F3A | 601128 | Unclassified | n/a | Histone variant |
| H3F3B | 601058 | Unclassified | n/a | Histone variant |
ACETYLATION
Lysine acetylation, a ubiquitous modification found on histone proteins, is a key modulator of chromatin structure and gene expression (Allfrey, Faulkner, and Mirsky 1964). All histones, including variants and linker histones, can undergo acetylation. The negative charge of the acetyl group neutralizes the positive charge of the lysine residue, reducing the interaction with negatively charged DNA. This impedes the generation of higher order chromatin structure, ultimately resulting in an open chromatin state. Histone acetylation, commonly found in active promoters and enhancers, plays a critical role in gene activation (Kuo et al. 1998; Imhof and Wolffe 1998; Lawrence, Daujat, and Schneider 2016; Z. Wang et al. 2008). Histone acetylation is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs) (Michael Haberland, Montgomery, and Olson 2009; Gong and Miller 2013). Human HATs are classified into two groups based on their subcellular localization: type A and type B. Type A HATs reside in the nucleus and contain bromodomains that recognize acetylated lysines, and often interact with other chromatin remodeling complexes and transcription factors (Dhalluin et al. 1999; Marmorstein and Zhou 2014; Sanchez and Zhou 2009). Type A HATs are further subdivided into five homology superfamilies: (1) the Gcn5-related acetyltransferase (GNAT) family, (2) the p300/CBP family, (3) the MYST family, the basal transcription factors, and the nuclear receptor cofactors (Steunou, Rossetto, and Côté 2013). On the other hand, type B HATs, which often lack bromodomains, are located in the cytoplasm and regulate the acetylation of newly synthesized histones (Richman et al. 1988; Bannister and Kouzarides 2011). In this section, we will discuss Type A HATs, specifically within the p300/CBP and MYST family, that have been identified as causative mutations in NDD and discuss their roles in CNS development and function.
p300/CBP Family
The cyclin AMP response element-binding protein (CREB) binding protein, CBP (also known as CREBBP or KAT3A) and p300 (EP300 or KAT3B) are two members of the p300/CMB subfamily of HATs. CBP mutations are the major cause of 55–60% Rubinstein-Taybi syndrome (RSTS) 1 (MIM: 180849) cases, while p300 mutations are causative in 8% of RSTS2 (MIM: 613684) cases (Roelfsema et al. 2005; Menke et al. 2016). RSTS1 is a rare congenital disorder characterized by mild to severe intellectual disability, microencephaly, growth retardation and distinct facial features (RUBINSTEIN and TAYBI 1963). RSTS2, although similar to RSTS1, results in less severe facial dysmorphism and overall better cognitive function compared to RSTS1; however, they tend to have more severe microencephaly and malformation of facial bone structures(Bartsch et al. 2010). In the vast majority of RSTS2 cases other comorbidities exist such as psychomotor deficits, language and gastrointestinal problems (Pérez-Grijalba et al. 2019). The CBP/p300 complex primarily regulates gene expression through their combined HAT activity in acetylating H2B at residue K85, H3 on residues K9, K14, K36, K56 and H4 on residues K12, K20 and K44 (Bannister and Kouzarides 1996; Das et al. 2009; Carré et al. 2018). In addition to their collective HAT activity, they regulate gene expression through recruitment of transcription factors or basal transcription machinery. Considerably, CBP and p300 are implicated in cell differentiation, proliferation and apoptosis. CBP or p300-deficient embryos exhibit heart and neural tube defects, often resulting in gestational death (T.-P. Yao et al. 1998; Tanaka et al. 2000). Therefore, CBP/p300 are required for proper brain development, and mutations in either of these HATs can result in a neurological disorders (T.-P. Yao et al. 1998; Tsui et al. 2013; Viosca et al. 2009).
The pathogenic variants of both CBP and p300 in RSTS1/2 resulting from frameshift, nonsense, or splice-site mutations are predicted to have a loss-of-function mechanism resulting in haploinsufficiency (Alari et al. 2018). CBP haploinsufficiency leads to a decrease in the number of neurons generated and is seen to regulate neurogenesis and cortical precursor differentiation in mice (Jing Wang et al. 2010; Valor et al. 2011). Loss-of-function CBP in mice resulted in the defects in hippocampal memory which was rescued by two different HDAC inhibitions (Korzus, Rosenfeld, and Mayford 2004). Moreover, an in vitro neuronal model of RSTS1 and RSTS2 was developed using patient-derived induced pluripotent stem cells (IPSCs). In this study, they observed morphological changes and decreased neuronal excitability (Alari et al. 2018). Furthermore, these patient-derived neurons had increased number of branches; however, the length of the branches was reduced (Alari et al. 2018). Interestingly, heterozygous mutations in exons 30–31 of CBP or p300 have recently been discovered to have a substantially distinct phenotype from RSTS. Patients harboring these mutations display developmental delay, autistic behavior, short stature, microcephaly and distinct facial anomalies (Menke et al. 2016; 2018; Siddharth Banka et al. 2019). Depending on the causative gene, it is known as Menke-Hennekam syndrome (MKHK) 1 for CBP mutations and MKHK2 for p300 mutations. It is hypothesized that CBP mutations confer a gain of function mechanism and p300 mutations are specifically affecting the conformation of the zinc finger domains. It is important to note that despite similarity in causative genes and overall symptoms, RSTS and MKHK are very phenotypically distinct. Ultimately, mutations in either CBP or p300 lead to aberrant neurodevelopment through either dysregulation of its HAT activity or improper recruitment of transcription factors.
MYST Family
The MYST subfamily is comprised of five human HATs that all share a highly conserved MYST domain: Tip60, HBO1, KAT6A, KAT6B and KAT8. These HATs are involved in many cellular processes such as proliferation and differentiation, therefore it is not surprising that dysfunction of these proteins have been linked to neurological diseases. KAT6A (also known as MYST3/MOZ) or its paralog KAT6B (or MYST4/MORF) form a stable complex with ING5, EAF6 and BRPF proteins (Ullah et al. 2009). In recent years, heterozygous KAT6A mutations were identified in Arboleda-Tham syndrome (ARTHS) (MIM: 616268), formerly known as autosomal dominant mental retardation 32 (Arboleda et al. 2015; Tham et al. 2015). ARTHS patients share common features such as impaired intellectual development, dysmorphic facial features, speech delay, congenital heart disease and microecephaly (Kennedy et al. 2018). KAT6A and KAT6B both acetylate H3K9 and K18, while KAT6B also acetylates H3K23 (Lv et al. 2017; K. Yan et al. 2020). KAT6A-deficient mouse embryos lead to H3K9 hypoacetylation, which is linked to reduced transcription of Hox genes and abnormal craniofacial development (Kong et al. 2014; Voss et al. 2009). Functional studies in ARTHS patient-derived fibroblasts revealed substantial reduction in H3K9 acetylation with increased K18 acetylation as well as dysregulation in signaling pathways of p53, which is a non-histone KAT6A substrate (Arboleda et al. 2015).
Interestingly, KAT6B, the paralog of KAT6A, is known to be mutated in multiple neurodevelopmental disorders; however, it is the causative gene in Say-Barber-Biesecker-Young-Simpson variant of Ohdo syndrome (SBBYSS) (MIM:603736) and Genitopatellar syndrome (GTPTS) (MIM: 606170) (Simpson et al. 2012; Clayton-Smith et al. 2011). SBBYSS is clinically characterized by craniofacial abnormalities involving blepharophimosis (underdeveloped eyelids) and cleft palates, severe ID, hypotonia, and cardiac defects (Clayton-Smith et al. 2011). These de novo truncating KAT6B mutations mainly affect exons 16–18 of the gene and are predicted to cause haploinsufficiency (Clayton-Smith et al. 2011; Marangi et al. 2018). KAT6B is critical for mammalian CNS development as homozygous mutations of KAT6B in mice resulted in a smaller cerebral cortex and cortical plate when compared to wildtype animals (You, Yan, et al. 2015). Furthermore, these mice exhibited craniofacial abnormalities similar to the SBBYSS patients (You, Yan, et al. 2015). Although the causative gene in GTPTS is KAT6B, these patients do not exhibit distinct craniofacial abnormalities (Simpson et al. 2012; Cormier-Daire et al. 2000). However, they do exhibit severe ID, thin corpus callosum, psychomotor retardation, cardiac defects and genital anomalies. As with SBBYSS, GTPTS is delineated by truncation mutations that occur at the proximal end of exon 18; this creates KAT6B variants lacking methionine-rich domains which is known to bind to the transcription factors RUNX1 and RUNX2. The role of RUNX genes in mammalian CNS involves formation of neural circuits, specifically mediating somatosensory sensation, olfaction and motor control.
It is important to note that the border between SBBYSS and GTPTS syndromes is almost non-existent as they both have significant overlap with regards to exon 18. With this in mind, four distinct groups of KAT6B mutations have been recently suggested (Radvanszky et al. 2017; M et al. 2015). Group 1 mutations are proximal mutations, predominantly in the MYST domain and have been mainly associated with SBBYSS. Group 2 mutations, mainly seen in GTPTS, affect the acidic patch and transcription activation domain. However, the majority of these transcripts escape nonsense mediated decay. Group 3 mutations are associated with SBBYSS and GTPTS and they lack transcription activation domain with either a truncated or intact acidic patch. Lastly, group 4 mutations, exclusive to SBBYSS, contain truncations in the transcription activator domain. The majority of mutations (groups 2–4) reside after the MYST domain, suggesting that these truncated mutants retain HAT activity(M et al. 2015). However, without the remaining regulatory portions of the protein, these truncations may cause loss/gain-of-functions, altering cellular processes through dysregulation of histone acetylation or transcriptional activation.
MYST Family Co-Writer
BRPF1
Bromodomain and PHD finger containing protein 1 (BRPF1) serves as a scaffold mediating the interaction between KAT6A, KAT6B, ING5 and EAF6. BRPF1 is considered a “co-writer” as it is critical for HAT activity and transcriptional activation. It consists of multiple domains which are responsible for many of its protein-protein interactions, it also contains a PWWP domain which recognizes H3K36me3 (K. Yan et al. 2016, 1). In 2017, a cohort of 10 patients containing heterozygous BRPF1 mutations was identified, all exhibited mild to severe intellectual disability, dysmorphic facial features, ptosis, hypotonia, brain and behavioral anomalies (K. Yan et al. 2016). These mutations generated a truncated form of BRPF1, resulting in impaired H3K23 acetylation. Concurrently in 2017, another group identified 6 individuals who had what they referred to as an Intellectual Developmental Disorder with Dysmorphic Facies and Ptosis (IDDDFP) (Mattioli et al. 2017). Similar to the previous group, these patients all exhibited mild to moderate intellectual disability, growth retardation, ptosis and microencephaly (Mattioli et al. 2017). BRPF1, in this second cohort, retained the ability to bind KAT6A/B; however, these mutations prevented the formation of tetrameric complexes and had reduced HAT activity (Mattioli et al. 2017). Interestingly, aside from disrupting complex formation, a few of the pathogenic variants bearing mutations predominantly in the PWWP domain, maintain normal HAT activity. Therefore, a potential mechanism for this epigenetic dysregulation theoretically could be due to mislocalization of this HAT complex.
Functional studies of BRPF1 and its pathogenic variants elaborate on its role in brain development and function. BRPF1 is critical for forebrain development, as conditional knock-out in the forebrain results in small litter size with all mice exhibiting neurological and behavioral defects (You, Zou, et al. 2015, 1). Additionally, forebrain-specific inactivation of mouse Brpf1 lead to abnormal cerebral and hippocampal development (K. Yan et al. 2016). These anomalies were attributed to BRPF1 loss as it results in downregulation of neuronal migration, cell cycle progression and transcriptional control through the compromised status of neural stem and progenitor cells (You, Yan, et al. 2015). Moreover, despite the lack of intrinsic HAT activity in BRPF1 there is sufficient evidence demonstrating its interaction with KAT6A and KAT6B is critical for regulating histone acetylation both in vitro and in vivo (K. Yan et al. 2016). Heterozygous Brpf1 mice displayed a decrease in H3K23 acetylation, similar to the reduction seen in patient-derived fibroblasts (K. Yan et al. 2016). These findings illustrate the importance of co-writers, and accessory proteins in HAT complexes.
KANSL1
KAT8 regulatory NSL complex subunit 1 (KANSL1) is a part of the non-specific lethal (NSL) complex, which is a highly conserved protein complex that also includes KAT8/MOF, KANSL2 and WDR5. The NSL complex influences gene activation through acetylation of H4K16 and the transcription factor, p53 (Mendjan et al. 2006; X. Li et al. 2009; Dou et al. 2005). The stability and activity of the NSL complex is dependent on KANSL1(Raja et al. 2010). In 2012, two unrelated patients exhibiting characteristic symptoms of chromosome 17q21.31 deletion syndrome were discovered to have the first de novo mutations in KANSL1 (Zollino et al. 2012). Concurrently, another group performed sanger sequencing on 16 unrelated patients, identifying 16 different de novo heterozygous truncating mutations in KANSL1 in two unrelated patients. All four of the aforementioned patients were all diagnosed with Koolen-de Vries syndrome (KdVS) (MIM: 610443) which is characterized by moderate to severe intellectual disability, hypotonia, and dysmorphic facial features (Zollino et al. 2012; Koolen et al. 2012; 2016). The KANSL1 haploinsufficiency seen in KdVs is sufficient to generate a full manifestation of the chromosome 17q21.31 deletion syndrome and there were no clinically important differences between the two syndromes (Zollino et al. 2012; Koolen et al. 2016).
Whole transcriptomic sequencing on EBV-transformed patient cells bearing either a KANSL1 point mutation or deletion showed that there was a high correlation between the differential expression of similar genes between both syndromes(Koolen et al. 2012). Moreover, gene ontology (GO) analysis revealed enrichment in genes associated with neuronal and synaptic processes (Koolen et al. 2012). These results were supported using chromatin immunoprecipitation as the KANSL1 Drosophila homolog, wah, was bound to the promoter of many genes enriched for the same GO terms (Raja et al. 2010). Global knockdown of wah lead to altered synapse morphology at the neuromuscular junction and endosomal maturation, which is crucial for synaptic trafficking (Lone et al. 2010). Additionally, conditional knockdown of wah in mushroom bodies, the learning and memory center of the Drosophila brain, resulted in a 25% reduction in learning ability (Koolen et al. 2012). Functional analysis of the KANSL1 Drosophila homolog revealed the importance of this protein in complex brain functions such as learning and memory.
DEACETYLATION
HDACs are erasers of histone acetylation, in humans there are 18 identified HDACs. These enzymes are classified into two categories, “zinc-dependent” and “nicotinamide-adeneine-dinucleotide (NAD)-dependent”, based on whether they require zinc or NAD for catalysis. There are four classes of HDACs within the zinc-dependent class: class I (HDACs 1, 2, 3, 8), class IIa (HDACs 4, 5, 7, 9), class IIb (HDACs 6, 10) and class IV (HDAC 11). All of the zinc dependent HDACs are expressed in the brain; however, they have distinct cellular and subcellular localizations. Class I HDACs are primarily found in the nucleus, while class II HDACs shuttle between the cytoplasm and the nucleus. NAD-dependent deacetylases, also known as Sirtuins, are expressed throughout the brain and are highly regulated in a spatiotemporal pattern, which has been speculated to play a role in aging (Deardorff et al. 2012). To date mutations in three HDACs (HDAC4, HDAC6 and HDAC8) (4,6,8) have been determined as causative in neurodevelopmental disorders.
HDAC4
HDAC4, a class IIa HDAC, is highly expressed in the postnatal brain and regulates neuronal cell death; however, the substrates of HDAC4, and therefore the mechanism of action, remain elusive (Bolger and Yao 2005). Homozygous HDAC4 mice exhibit impaired learning, memory and long-term synaptic plasticity (M.-S. Kim et al. 2012). Haploinsufficiency due to mutations or deletions of HDAC4 were determined to cause brachydactyly mental retardation (BDMR) syndrome (MIM: 600430) characterized by developmental delay, intellectual disability, behavioral abnormalities and dysmorphic features (Williams et al., n.d.; Villavicencio-Lorini et al. 2013). Currently, there is some debate on whether or not HDAC4 is the driver for BDMR or if mutations in the TWIST2 or FLJ43879 genes are driving the phenotype (Wheeler, Huang, and Dai 2014). HDAC4 has been shown to regulate myocyte enhancer factor-2 (MEF2) proteins during development, therefore altered HDAC4 activity potentially results in BDMR through dysregulation of MEF2 (Bolger and Yao 2005). However, it remains speculative.
HDAC6
HDAC6, a class IIb HDAC, is also highly expressed in the brain and it primarily resides in the cytoplasm; however, deacetylation of its target substrates often leads to nuclear translocation (LoPresti 2020; Nagase et al. 1998; d’Ydewalle, Bogaert, and Van Den Bosch 2012). HDAC6 is a key regulation of synaptic function during acute stress in the prefrontal cortex, a region responsible for higher-order cognition(J. B. Lee et al. 2012). X-linked dominant Chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia (CPDBHM) (MIM: 300863) is caused by heterozygous mutations in HDAC6 (Chassaing et al. 2005; Simon et al. 2010). CPDBHM is characterized by macrocephaly, growth retardation, skeletal and craniofacial abnormalities and mild intellectual disabilities, these phenotypes are more pronounced in males (Chassaing et al. 2005; Simon et al. 2010). In pathological conditions, HDAC6 translocate to the nucleus, dysregulating synaptic function due to decreased expression of brain-derived neurotrophic factor (BDNF) (J. B. Lee et al. 2012). Furthermore, HDAC6 has been implicated in many neurological conditions such as Alzheimer’s’ disease, Charcot-Marie-Tooth disease and mood disorders. However, whether this regulation is mediated through deacetylation of its known substrates (β-catenin, α-tubulin) or another target of HDAC6 remains unclear.
HDAC8
HDAC8 is known to deacetylate H3 and H4 in vitro, and structural maintenance of chromosomes 3 (SMC3) in the cohesin complex (Deardorff et al. 2012; Hans Tomas Bjornsson 2015). Loss of HDAC8 activity results in increased SMC3 localization to chromatin, leading to amplified cohesin localization which results in altered transcriptional activity (Deardorff et al. 2012). Mutations in HDAC8 are responsible for Cornelia De Lange Syndrome (CdLS) 5 (MIM: 300882) (Boyle et al. 2014; Mordaunt and McLauchlan 2015; Kaiser et al. 2014). CdLS5 is defined by intellectual disability, facial dysmorphism, pre- and post-natal delay, microcephaly, heart defects, upper limb abnormalities, and genitourinary anomalies (Yuan et al. 2015). Notably, mutations in HDAC2 have been recently discovered in another form of CdLS; however, further studies are needed to compare the similarities between these class I HDAC mutations (V. F. Wagner et al. 2019). A characteristic feature of CdLS5 is abnormal skull formation. Interestingly, HDAC8 represses homeobox transcription factors, Otx2 and Lhx1, in cranial neural crest cells; thus, HDAC8 mediates patterning of the skull and deletion in mice leads to perinatal lethality due to skull instability (M Haberland et al. 2009). To this date, no in vivo studies have elucidated the role of HDAC8 in cognition or behavior despite the accumulating number of mutations in HDAC8 related to intellectual disability syndromes.
METHYLATION
Histone methylation occurs on both arginine and lysine residues and plays a critical role in transcriptional regulation (Allfrey and Mirsky 1964; Allfrey, Faulkner, and Mirsky 1964). Unlike acetylation, a binary system (acetylation or no acetylation), methylation is more complicated. Histone lysine methylation is a quaternary system as lysine residues can be mono-, di-, and trimethylated, while arginine residues can be mono- and di- methylated. Importantly, none of these modifications alter the lysine’s positive charge. Histone methylation is ambiguous as it is associated with both activation and repression of gene expression depending on the residue modified and the number of methyl groups added (Y. Zhang 2001; Black, Van Rechem, and Whetstine 2012). To this date, only methylation on histone H3 and H4 have been identified. Methylation on H3K4, H3K36, and H3K79 has been shown to mark active regions, whereas H3K9, H3K27, and H4K20 methylations are associated with repressed chromatin (Black, Van Rechem, and Whetstine 2012). There are two classes of lysine methyltransferases (KMT) in humans, and this classification depends on the presence or absence of the catalytic Su(var)3–9, E(z) (enhancer of zeste), and trithorax (SET) domain (Jenuwein 2006; Dillon et al. 2005; Nguyen and Zhang 2011). Despite having distinct catalytic domains, all KMTs utilize S-adenosyl-L-methionine (SAM) as the methyl group donor, and have high specificity to certain residues as well as the degree of methylation (Dillon et al. 2005; Nguyen and Zhang 2011). This section will discuss the role of KMTs in neurodevelopment and the implications of their dysregulation.
KMT2A
Lysine methyltransferase 2A (KMT2A), also known as mixed lineage leukemia 1 (MLL1), catalyzes the trimethylation of histone H3K4 (Dou et al. 2006; Schneider et al. 2005). KMT2A, like other SET domain-containing KMTs, is often found in a complex with four other subunits: ASH2L (absent, small, or homeotic-2 like), WDR5 (WD repeat domain 5), DPY30, and RbBP5 (retinoblastoma binding protein 5). KMT2A also interacts with Menin and HCF1, forming COMPASS-like complex (Shilatifard 2012). Importantly, H3K4 mono-, di- and trimethylation distinctly mark active genomic regions where K4me1 is enriched at enhancers, K4me2 at 5’ end of actively transcribed genes, and K4me3 at actively transcribed promoters (Heintzman et al. 2007; T. Kim and Buratowski 2009; Santos-Rosa et al. 2002). In 1989, three groups reported clinical findings from unrelated patients, all detailing hypertrichosis cubiti (abnormal amount of body hair), short stature, craniofacial abnormalities, and developmental delays(Hans-Rudolf Wiedemann et al. 1989; Flannery et al. 1989; MacDermot et al. 1989). From 1989 to 2010, seven other groups reported approximately 40 patients all exhibiting distinct facial features, developmental delays, and short stature (Edwards et al. 1994; W. D. Jones et al. 2012; Steiner and Marques 2000; Polizzi et al. 2005; Koç et al. 2007; Koenig et al. 2010). This disorder was named Weidemann-Steiner (WDSTS) (MIM 605130) and Jones et al. identified heterozygous de novo truncating mutations in the KMT2A gene in four of these WDSTS patients (W. D. Jones et al. 2012).
By employing whole-exome sequencing and confirmatory Sanger sequencing, a variety of mutations were observed: missense, nonsense, and frameshift truncating mutations. These mutations result in a loss-of-function or haploinsufficient KMT2A in patients presenting clinically similar phenotypes (Koenig et al. 2010; Y. Sun et al. 2017; Nardello et al. 2020; Stellacci et al. 2016; Jinxiu et al. 2020; N. Li et al. 2018; N. Miyake et al. 2016). Based on these findings, KMT2A was concluded to be the causative gene in WDSTS. Experimental studies revealed the critical role of KMT2A in neurogenesis in the mouse postnatal-brain, specifically with neuronal differentiation(Lim et al. 2009). In KMT2A-deficient brains, neuronal differentiation is severely impaired through reduced size and number of neurons, potentially due to the decrease in bivalent promoters in neural precursor cells (Lim et al. 2009). It also has been demonstrated that KMT2A-mediated H3K4 trimethylation is critical in prefrontal neurons as it regulates a subset of genes involved in cognition and behavior (Jakovcevski et al. 2015). Lastly, mice deficient for KMT2A had reduced dendritic spines and increased aggression (Vallianatos et al. 2020). These studies taken together reveal the role of KMT2A in neurogenesis and CNS function, and it is suspected that these functions are impaired in WDSTS patients (W. D. Jones et al. 2012). The phenotype of this syndrome continues to expand; however, the molecular mechanism remains poorly understood. To further understand this disorder, more functional assessments in model organisms must be conducted to elucidate the role of KMT2A in cognition.
KMT2B
Lysine methyltransferase 2B (KMT2B), also known as MLL4, is a paralog of KMT2A and is responsible for the trimethylation of H3K4 (FitzGerald and Diaz 1999, 2). KMT2B has been linked with gene-specific activation and functions in preimplantation development as well as embryonic genome activation(Shao et al. 2014). In 2016, an autosomal dominant neurological disorder was discovered where patients exhibited microencephaly, mild intellectual disability, and developmental delay; this disorder was classified as childhood-onset dystonia-28 (DYT28) (Zech et al. 2016). Several groups provided evidence that de novo mutations in the KMT2B gene result in haploinsufficiency, causing DYT28 (MIM 617284) (Zech et al. 2016; 2017; Kawarai et al. 2018). Mutations have been observed in over 75 patients to date. They encompass frameshift, missense, nonsense, and splice-site mutations, and all affect KMT2B function (Zech, Lam, and Winkelmann 2019). KMT2B is highly expressed in human tissue and ubiquitously expressed in the brain, with the highest expression in the cerebellum (Meyer et al. 2017). This expression is reduced in DTY28 patients; however, the levels on H3K4 methylation were not significantly altered, suggesting a potential role of KMT2B beyond K4 methylation (Meyer et al. 2017). In vitro, KMT2B knockdown results in decreased neuronal differentiation from embryonic stem cells (Lubitz et al. 2007). Moreover, KMT2B is critical in neuronal programming as ablation impairs transdifferentiation of fibroblasts to induced neuronal cells (iNs) conversion (Barbagiovanni et al. 2018). The loss of KMT2B leads to redistribution of H3K4me3 rather than reduction, explaining why previous studies found similar levels of methylation between patients and controls (Meyer et al. 2017; Barbagiovanni et al. 2018). The mechanism driving the dystonia caused by KMT2B remains elusive. However, promising studies are working to elucidate the dysregulated pathways involved in DYT28.
KMT2C
Lysine methyltransferase 2C (KMT2C), also known as MLL3, mediates H3K4 mono- and dimethylation (Ruault et al. 2002; Sedkov et al. 2003; Herz et al. 2012). KMT2C is functionally active in the COMPASS complex and is often recruited to enhancers and active gene promoters (D. Hu et al. 2013). Additionally, KMT2C is a central component of the activating signal cointegrator-2 (ASC-2) complex, which acts as a co-activator for hormone receptors (S. Lee et al. 2009; Goo et al. 2003). Knockdown of the Drosophila KMT2C, thrithorax related (trr), in the brain impaired short-term memory highlighting its role in synaptic plasticity and cognition (Koemans et al. 2017). Kleefstra et al. performed whole-exome sequencing on five individuals with the same clinical phenotype, including psychomotor delay, severe intellectual disability, dysmorphic facial features, and behavioral problems (Tjitske Kleefstra et al. 2012). They discovered a heterozygous de novo nonsense mutation in the KMT2C gene in a female patient, resulting in truncation of KMT2C(Tjitske Kleefstra et al. 2012). This truncation is predicted to reduce methyltransferase activity as it lacks the enzymatic SET domain. Another group identified five unrelated patients with de novo mutations in the KMT2C gene, all which are predicted to cause loss-of-function variants(Koemans et al. 2017). These patients, previously diagnosed with an undefined developmental disorder, were classified as all suffering from Kleefstra Syndrome-2 (KLEFS2) (MIM: 617768)(Koemans et al. 2017). It is evident that dysregulation of KMT2C’s function potentially underlies the broad spectrum of neurodevelopmental abnormalities observed. To investigate the role of KMT2C in neurons, researchers made a knockdown of its Drosophila paralog, trr, in the mushroom body of the fly brain revealing impaired short-term memory(Koemans et al. 2017). Unfortunately, specific gene targets of KMT2C remain unknown, but future research will likely reveal molecular targets of KMT2C haploinsufficiency. Furthermore, it has been recently shown that when KMT2C is promoter-bound, this represses gene transcription through recruitment of specific factors(Cheng et al. 2014). Based on these studies, KMT2C can potentially both activate and repress genes. Understanding its role in chromatin regulation will clarify its effect in transcriptional regulation during brain development, ultimately aiding in the development of therapeutics.
KMT2D
Lysine methyltransferase 2D (KMT2D), also known as MLL2, is a paralog of KMT2C and, as such, mediates mono- and dimethylation of H3K4 in mammalian cells as well(D. Hu et al. 2013; J.-E. Lee et al. 2013). Exome sequence uncovered KMT2D variants as the causative gene in Kabuki syndrome (KS1) (MIM 147920) (Ng et al. 2010). KS1 is an autosomal-dominant disorder phenotypically similar to aforementioned neurodevelopmental disorders; it is characterized by infantile hypotonia, craniofacial abnormalities, mild-moderate intellectual disability, and developmental delay (Ng et al. 2010, 2). There are dozens of clinical reports detailing over 100 unique de novo mutations of KM2D identified in individuals with KS1 (Tsukahara et al. 1997; Kobayashi and Sakuragawa 1996; Ilyina et al. 1995; Lerone et al. 1997; Kawame et al. 1999; Ewart-Toland et al. 1998; McGaughran, Donnai, and Clayton-Smith 2000; S. Banka et al. 2013; Siddharth Banka et al. 2012; Hannibal et al. 2011; Micale et al. 2011; Paulussen et al. 2011). These mutations all result in loss-of-function variants by disrupting its reader domains (PHD fingers), loss of its catalytic SET domain, or disruption of the 3D structure (S. Banka et al. 2013, 2; Dhar et al. 2012; Bögershausen and Wollnik 2013). In vitro studies determined that KMT2D mediates neural differentiation by activating neuronal differentiation genes, HOXA1–3 and NESTIN, via H3K4me3 deposition(Dhar et al. 2012). These studies suggest that the loss-of-function mutations affecting the tandem PHD domains or SET domain will likely lead to loss of H3K4me3, which is necessary for neuronal differentiation gene expression. Van Laarhoven et al. performed morpholino-mediated knockdown of KMT2D in zebrafish and observed significant craniofacial and cardiac defects (Van Laarhoven et al. 2015). Additionally, this knockdown resulted in the reduction of mature neurons and brain size despite the accumulation of neural precursor cells, suggesting the defects were due to cellular dysfunction rather than perturbed development. Further functional studies on this writer will prove beneficial for understanding its role in neuronal maturation and function.
KMT2E
Lysine methyltransferase 2E (KMT2E), also known as MLL5, diverges from the KMT2 family of methyltransferases since it lacks histone methyltransferase activity(Sebastian et al. 2009). Nonetheless, it has been reported to play critical roles in cell cycle progression, genomic stability maintenance, and differentiation (Sebastian et al. 2009, 5). Despite the lack of KMT activity, the KMT2E homolog in Drosophila, UpSET, regulates promoter-associated histone acetylation and H3K4 di- and trimethylation when in complex with Rpd/Sin3-repressor proteins (Rincon-Arano et al. 2012). O’Donnell-Luria-Rodan syndrome (ODLURO) (MIM: 618512) is a neurodevelopmental disorder characterized by developmental delay, intellectual disabilities, and subtle craniofacial abnormalities with heterozygous mutations in KMT2E being the causative gene (O’Donnell-Luria et al. 2019). Individuals with ODLURO either have protein-truncating variants or missense mutations in the KMT2E gene, with the latter resulting in increased severity (O’Donnell-Luria et al. 2019). Moreover, the individuals with missense mutation often develop drug-resistant infantile epileptic encephalopathy(O’Donnell-Luria et al. 2019). To this date, no functional analyses have been conducted; however, it has been hypothesized that haploinsufficiency is potentially the pathogenic mechanism for the truncation and missense mutations. Collectively, these mutations are hypothesized to produce loss-of-function variants.
SETD1A
SET domain-containing protein 1A (SETD1A), also known as KMT2F, mediates the trimethylation of H3K4 at promoters of active genes (J.-H. Lee and Skalnik 2008, 1). SETD1 associates with the canonical COMPASS complex, in addition to CpG-binding protein (CXXC1) and WD repeat domain 82 (WDR82) (J.-H. Lee and Skalnik 2008, 82; 2005; Deng et al. 2013). Loss-of-function variants in SETD1A have been implicated as causative in two neurodevelopmental disorders and schizophrenia(Swedish Schizophrenia Study et al. 2016). In 2019, de novo heterozygous mutations in SETD1A were discovered in individuals with early-onset epilepsy with or without developmental delay (EPEDD) (MIM: 618832) (X. Yu et al. 2019). Mutant SETD1A expression decreased the number of excitatory synapses on mouse cortical primary neurons (X. Yu et al. 2019). Significantly, these mutations did not affect general neuronal morphology which suggests a role of SETD1A in synapse formation (X. Yu et al. 2019). Kummeling et al. discovered de novo heterozygous mutations in the SETD1A gene in 15 unrelated patients with a neurodevelopmental disorder with speech impairment and dysmorphic facies (NEDSID) (MIM: 619056) (Kummeling et al. 2020). These were missense, nonsense, frameshift, and splice-site mutations, all expected to disrupt the catalytic SET domain. Patient-derived fibroblasts displayed increased activation of DNA damage response, and increased DNA degradation under stress (Kummeling et al. 2020). Additionally, when SETD1A was knocked down in Drosophila mushroom bodies to assess neural programming, results determined that SETD1A is required for proper neuronal function during the formation of memory. Given that both disorders are clinically distinct despite both deriving from pathogenic SETD1A, it will be essential to elucidate this protein’s role in neural development and neuronal function.
SETD1B
SET domain-containing protein 1B (SETD1B), also known as KMT2G, is a paralog of SETD1A and, as such, also mediates the trimethylation of H3K4(J.-H. Lee et al. 2007). Despite their homology, SETD1A and SETD1B localize to a nonoverlapping set of target genes suggesting that these methyltransferases have nonredundant contributions to the epigenetic control of gene expression (J.-H. Lee et al. 2007). Recent studies provided evidence that intellectual developmental disorder with seizures and language delay (IDDSELD) (MIM: 619000) is caused by de novo heterozygous mutations in SETD1B (Roston et al. 2020). As its name suggests, IDDSELD is characterized by global developmental delay with speech and language impairment, and the onset of myoclonic seizures early in life (Roston et al. 2020; Hiraide et al. 2019; Den et al. 2019; Hiraide et al. 2018; Palumbo et al. 2015). Additionally, individuals often exhibit behavioral abnormalities such as autism spectrum disorder and facial dysmorphism. Interestingly, the mutations discovered have been predicted to confer loss-of-function or gain-of-function effect on SETD1B. Additional functional studies on these pathogenic variants and patient cells are required to determine the pathological mechanism. Krzyzewska et al. identified a shift in the DNA methylation landscape in IDDSELD patient cells (Krzyzewska et al. 2019). Notably, SETD1B variants imposed a hypomethylated epigenetic signature which lead the authors to speculate SETD1B is enduring a loss-of-function effect (Krzyzewska et al. 2019).
EHMT1
Euchromatic histone methyltransferase (EHMT1), or G9a-like protein (GLP), catalyzes mono- and dimethylation of H3K9 and is known to recruit Polycomb Repressive Complex-2 (PRC2) (Mozzetta et al. 2014). EHMT1 predominantly deposits H3K9me2 in the genome, which promotes transcriptional repression (Peters et al. 2003; Rice et al. 2003). Heterozygous mutations on EHMT1 cause chromosome 9q34.3 deletion syndrome, better known as Kleefstra syndrome-1 (KLEFS1) (MIM: 610253) (Neas et al. 2005; Stewart et al. 2004; Harada et al. 2004; Tjitske Kleefstra et al. 2006; Iwakoshi et al. 2004, 34). Standard features in patients with KLEFS1 are severe intellectual disability, distinct facial gestalt, hypotonia, microencephaly, seizures, and cardiac defects (Tjitske Kleefstra et al. 2012; 2006). Individuals contain missense, nonsense, frameshift, and splice-site mutations (Tjitske Kleefstra et al. 2012; 2006; T Kleefstra et al. 2009). Biochemical characterization of missense mutations of EHMT1 reveals these mutations decrease its methyltransferase activity, potentially reducing H3K9me2 at specific loci, causing KLEFS1 (A. Yamada, Shimura, and Shinkai 2018). Heterozygous EHMT1 (EHMT1+/−) mice have reduced dendritic branching and a reduced number of mature spines in hippocampal neurons resulting in learning and memory deficits, specifically during stress (M. C. M. Balemans et al. 2013). Additionally, EHMT+/− mice exhibited overexcitability due to deficits in inhibitory neuron maturation and excitatory impairments (Negwer et al. 2020). To ascertain whether these effects are observed in human cells, excitatory cortical neurons were generated from IPSCs derived from KLEFS1 patients (Frega et al. 2019). Frega et al. observed similar overexcitability in patient-derived neurons. They demonstrated that these changes were due to upregulation of the NMDA receptor (NMDA-R) subunit, which correlated with reduced H3K9me2 at its promoter (Frega et al. 2019). Davis et al. suggested that EHMT1 haploinsufficiency results in disrupted NMDA-R expression promoting abnormal forebrain development, ultimately resulting in information processing deficits (Davis et al. 2020). Understanding the cognitive deficits and the pathways involved would provide a basis for targeted therapeutics, since Frega et al. were able to rescue KLEFS1 patient-derived neuronal phenotypes through pharmacological inhibition of NMDA-Rs (Frega et al. 2019).
EZH2
Enhancer of zeste 2 (EZH2) in complex with suppressor of zest 12 (SUZ12) and embryonic ectoderm development (EED) acts as the catalytic subunit of Polycomb repressive complex-2 (PRC2), which functions to repress gene expression through trimethylation of H3K27 (H3K27me3) (Cao et al. 2002; O’Carroll et al. 2001). PRC2 is essential for development as reducing a single subunit results in embryonic lethality and severe developmental defects (O’Carroll et al. 2001, 2; Pasini et al. 2004). H3K27me3 deposition and removal are critical in epigenetic inheritance of repressed transcriptional landscapes and neurogenesis (Coleman and Struhl 2017; Burgold et al. 2008; Fragola et al. 2013; Park et al. 2014). Mainly, EZH2 knockdown results in abnormal migration of cortical neurons due to overexpression of Reelin, a glycoprotein that regulates neuronal migration (Zhao et al. 2015). Furthermore, chicken embryos lacking EZH2 exhibit impaired neural tube organization due to dysregulated neural progenitor cell renewal and impaired Rho signaling (Akizu and Martínez-Balbás 2016). Weaver syndrome (WVS) (MIM: 277590), an overgrowth syndrome characterized by intellectual disability, macrocephaly, developmental delay, and characteristic facial features, is caused by heterozygous mutation EZH2 (Weaver et al. 1974; Gibson et al. 2011). Cohen et al. determined that WVS-causing EZH2 mutations reduce PRC2 activity in vitro, suggesting that WVS is caused by altered PRC2 function (A. S. A. Cohen et al. 2016). Interestingly, despite the decreased activity, PRC2 harboring WVS-causing EZH2 mutations displayed increased association with chromatin, suggesting dysregulated disengagement between the complex and chromatin (C.-H. Lee et al. 2018). Mice bearing EZH2 mutations result in perinatal lethality when homozygous, and overgrowth when heterozygotes (Lui et al. 2018). Loss of PRC2 during differentiation severely compromised neural gene induction due to marked loss of bivalent enhancers (Cruz-Molina et al. 2017). Collectively, this evidence suggests EZH2 plays a critical role in neurodevelopment; however, understanding how these mutations impart their effects on human neurogenesis remains unclear.
ASH1L
ASH1-like histone lysine methyltransferase (ASH1L), also known as KMT2H, catalyzes mono- and dimethylation of H3K36, promoting gene expression by counteracting Polycomb silencing (Miyazaki et al. 2013). Moreover, ASH1L plays a critical role in development by activating homeobox (HOX) genes and is highly expressed in both embryonic and adult brains(Okamoto et al. 2017; Miller et al. 2014). Various groups reported ASH1L as the causative gene in Autosomal dominant mental retardation-52 (MRD52) (MIM: 617796) (Okamoto et al. 2017; de Ligt et al. 2013; T. Wang et al. 2016; Stessman et al. 2017; Krumm et al. 2015). Individuals with MRD52 have mild-severe intellectual disability, autism spectrum disorder, speech delay, facial dysmorphisms, and overall developmental delay (Okamoto et al. 2017; de Ligt et al. 2013; T. Wang et al. 2016; Stessman et al. 2017). No functional analyses have been conducted on patient cells; however, knockdown of Zebrafish homolog, ash1a, resulted in a reduction in the number of neurons in the pineal gland (Cau and Wilson, Stephen W. 2003). Furthermore, mice expressing catalytically inactive ASH1L had a wide range of skeletal anomalies and impaired fertility (Miyazaki et al. 2013; Brinkmeier et al. 2015). Interestingly, ASH1L is enriched at the promoter of neurexin1α, a presynaptic adhesion molecule essential for synapse formation, upon neuronal stimulation (Zhu et al. 2016). It will be critical to determine how the ASH1L pathogenic variants affect the synaptic formation, synaptic plasticity, and ultimately learning and memory.
NSD1
Nuclear receptor SET domain-containing 1 (NSD1; also known as KMT3B) performs methylation of H3K36 as well as H4K20 (Rayasam et al. 2003). In vivo analysis demonstrated NSD1 catalyzed mono and dimethylation of H3K36 and trimethylation of H4K20 (Lucio-Eterovic et al. 2010; Qiao et al. 2011; Berdasco et al. 2009; G. G. Wang et al. 2007). Haploinsufficiency of NSD1 is the primary cause of Sotos syndrome-1 (SOTOS1) (MIM: 117550), accounting for over 75% of reported cases (Türkmen et al. 2003, 1; Kurotaki et al. 2002; Sotos et al. 1964; Hook and Reynolds 1967; Bejar et al. 1970; Douglas et al. 2003; Fryssira et al. 2010; Baujat and Cormier-Daire 2007). SOTOS1 is characterized by overgrowth, distinctive facial features, mild-severe intellectual disability, macrocephaly, and seizures (Kurotaki et al. 2002; Sotos et al. 1964). Different de novo mutations have been reported in individuals with SOTOS1, including missense, nonsense, frameshift, or deletion mutations (Türkmen et al. 2003, 1; Kurotaki et al. 2002; Douglas et al. 2003; van Haelst et al. 2005; Visser et al. 2005; Kaminsky et al. 2011). These mutations were widely distributed across the NSD1 protein, affecting the PHD domain, SET domain, and cofactor binding regions; thus, there is no mutational hot spot (Niikawa 2004; Pasillas, Shah, and Kamps 2011). Interestingly, individuals with SOTOS1 fall into two categories those due to either NSD1 haploinsufficiency or dosage effect (Niikawa 2004). Loss of NSD1 results in reduced H3K36 methylation and gene expression in vitro (Lucio-Eterovic et al. 2010). This methylation recruits DNMT3A, a DNA methyltransferase, at intergenic regions. Furthermore, blood samples from individuals with SOTOS1 exhibit hypomethylation of intergenic DNA(Weinberg et al. 2019). A mouse model for SOTOS1 with a heterozygous NSD1 mutation has been generated, which could be used in future work to elucidate the molecular mechanisms of these pathogenic variants(Oishi et al. 2020).
NSD2
Nuclear receptor SET domain-containing 2 (NSD2) is also known as Wolf-Hirschhorn syndrome candidate 1 (WHSC1) or multiple myeloma SET domain (MMSET). Similar to NSD1, NSD2 mono- and di-methylates H3K36(E. J. Wagner and Carpenter 2012). Interestingly, NSD1 and NSD2 loss of function results in distinctly different phenotypes despite deposition of the same histone modification. NSD1 knockdown mice have impaired gastrulation, whereas NSD2 knockout mice showed growth retardation and developmental defects (Rayasam et al. 2003; Nimura et al. 2009). Moreover, where mutations in NSD1 result in Sotos syndrome, haploinsufficiency of NSD2 results in the majority of Wolf-Hirschhorn syndrome (WHSC) (MIM: 194190) characterized by growth abnormalities, microcephaly, intellectual disability, seizures, and severe craniofacial abnormalities (Stec et al. 1998; Paradowska-Stolarz 2014). Mutations in NSD2 attributed with WHSC result in de novo loss-of-function variants (Derar et al. 2019; Jiang et al. 2019; Barrie et al. 2019; Lozier et al. 2018). Studies reported reduced cellular proliferation and cell viability in WHSC patient-derived fibroblasts suggesting that the haploinsufficiency either reduces cellular proliferation or increases cell death (Nevado et al. 2020). Severe craniofacial abnormalities are characteristic phenotypes of WHSC. To explore this, NSD2 expression was reduced in Xenopus laevis which correlated with decreased expression of several NSD2 associated genes(Mills et al. 2019). This reduction significantly affected craniofacial patterning, specifically cartilage formation and neural crest motility (Mills et al. 2019). Additionally, NSD1 has been demonstrated to regulate gene expression through Runx2, and p300 (Y. F. Lee et al. 2014). Researchers observed that NSD1 regulates the expression of bone-related genes through modulation of H3K36 dimethylation (Y. F. Lee et al. 2014). Lastly, knockdown of zebrafish NSD2 homolog, DrWhsc1, affected endbrain enlargement, abnormal cartilage, bone reduction, and incomplete motor neuron formation (Yamada-Okabe et al. 2010). Despite all these functional studies, there are currently no therapeutics developed for WHSC, suggesting that the pathomechanism remains elusive. Currently, only symptomatic treatments are available.
SETD2
SET domain-containing protein 2 (SETD2) is the primary methyltransferase for trimethylation of H3K36 (H3K36me3) (M. Hu et al. 2010; X.-J. Sun et al. 2005). SETD2 mediates genomic imprinting and embryonic development, as SETD2−/− embryos result in embryonic lethality (Q. Xu et al. 2019). SETD2 ablation causes Luscan-Lumish syndrome (LLS) (MIM: 616831), previously known as Sotos-like syndrome. LLS, is categorized by macrocephaly, facial dysmorphism, intellectual disability, speech and language developmental delay, and behavioral problems, including autism spectrum disorder (Luscan et al. 2014; Lumish et al. 2015). Thus far, de novo SETD2 mutations are proposed to be loss-of-function either due to truncation or missense mutations (Marzin et al. 2019). SETD2 conditional knockout mice mimicked phenotypes seen in patients such as defective social interaction, reduced motor learning, and spatial memory (L. Xu et al. 2021). Xu et al. concluded that SETD2 is required for cortical arealization, the process of subdividing neocortex, and for the formation of cortical circuits (L. Xu et al. 2021). Functional studies utilizing patient samples are needed to determine the pathogenicity of the SETD2 mutations in neurodevelopment.
SETD5
SET domain-containing protein 5 (SETD5) mediates trimethylation of H3K36 (Dillon et al. 2005; Xiao, Wilson, and Gamblin 2003; Sessa et al. 2019). SETD5 mutations have been associated with neurodevelopmental disorders, including Cornelia de Lange and autism spectrum disorder (Parenti et al. 2017; Fernandes et al. 2018; Pizzo et al. 2019). Furthermore, heterozygous loss-of-function mutations in SETD5 cause autosomal dominant mental retardation-23 (MRD23) (MIM: 615761) characterized by psychomotor delay, speech impairments, dysmorphic facial features, intellectual disability, and behavioral abnormalities (Kuechler et al. 2015; Grozeva et al. 2014; Rauch et al. 2012; Green et al. 2017; Fang et al. 2019; Stur, Soares, and Louro 2017; Crippa et al. 2020). Deliu et al. demonstrated that SETD5-haploinsufficient mice have impairments in cognitive tasks due to dysregulated transcription via interaction with HDAC3 and PAF1 (Deliu et al. 2018). Haploinsufficiency of SETD5 in both mice and zebrafish resulted in global alterations to H3K36me3, impaired proliferation of neural progenitor cells, and defective synapse formation, ultimately resulting in learning and behavioral abnormalities (Sessa et al. 2019). Finally, Nakagawa et al. showed that SETD5 mediates ribosomal DNA expression through recruitment of HDAC3, and this regulation is critical for neural cell proliferation (Nakagawa et al. 2020). Sufficient evidence proves that SETD5 haploinsufficiency drives abnormal neurodevelopment. Future studies are needed to continue elucidating the pathology of MRD23 and other SETD5-related syndromes.
DEMETHYLATION
Like acetylation, methylation is removed by a group of enzymes; these are known as lysine demethylases (KDMs) (Yujiang Shi et al. 2003). For decades, lysine methylation was presumed static and irreversible due to the slow turnover of methylated histones, and the fact that no demethylases were known. In 2004, the first bonafide lysine demethylase was discovered, revealing that methylation was a dynamic mark that can be altered to mediate gene expression (Yujiang Shi et al. 2004). To this date, there are two classes of KDMs those based on the catalytic domain, either flavin adenine nucleotide (FAD)-dependent oxidase domain or Jumonji C (JmjC) domain (Yujiang Shi et al. 2004; Yang Shi and Whetstine 2007). Similar to KMTs, KDMs have a high degree of specificity for its target substrates. In this section, we will discuss clinical phenotypes that manifest when KDMs are mutated.
KDM1A
Lysine-specific demethylase 1A (KDM1A or LSD1), the first demethylase discovered, catalyzes FAD-dependent oxidative demethylation of mono and dimethylated H3K4, which results in gene repression (Shao et al. 2014). Heterozygous mutations in KDM1A are causative for a syndrome of cleft palate, psychomotor retardation, and distinctive facial features (CPRF) (MIM: 616728) (Chong et al. 2016; Tunovic et al. 2014). Individuals with pathogenic KDM1A variants present global developmental delay, intellectual disabilities, and craniofacial abnormalities (Chong et al. 2016; Tunovic et al. 2014). One such pathogenic variant contains a replacement of an evolutionarily conserved tyrosine(Y385), potentially conferring a gain-of-function mutant giving rise to decreased H3K4 methylation (Tunovic et al. 2014). Notably, KDM1A is a crucial subunit of CoREST (corepressor to the REST (RE1 silencing transcription factor/neural restrictive silencing factor) complex), which mediates repression of neuronal genes (Ballas et al. 2001; Andrés et al. 1999). Knockdown of KDM1A in mouse cortical neurons led to decreased dendritic arborizations and neurite width, suggesting it plays a critical role in neurite morphogenesis (Zibetti et al. 2010). Taken together, KDM1A regulates neuronal function and programming; however, further studies are needed to determine the factors that recruit KDM1A in specific regulatory regions in neurons and the influence of these pathogenic mutations on its function.
KDM5B
Lysine-specific demethylase 5B (KDM5B other aliases include: JARID1B, SUV420H1) is a member of the KDM5 family of JmjC domain containing demethylases and demethylates di- and trimethylated H3K4 (Xiang et al. 2007). KDM5B-mediated demethylation regulates RNA polymerase II initiation and elongation as well as alternative splicing in embryonic stem cells(He and Kidder 2017). Notably, both homozygous and heterozygous germline mutations of KDM5B have been discovered as causative in mental retardation, autosomal recessive 65 (MRT65) (MIM: 618109) (Faundes et al. 2018; Martin et al. 2018). Individuals with MRT65 display moderate-severe developmental delay, inconsistent dysmorphic facial features, and behavioral abnormalities (Faundes et al. 2018; Martin et al. 2018). KDM5B−/− embryos exhibit several neural defects due to derepression of developmental regulators in early embryogenesis(Albert et al. 2013). Moreover, a loss-of-function model mimicking MRT65 exhibited similar phenotypes as seen in patients including increased behavioral abnormalities, decreased learning and skeletal abnormalities (Martin et al. 2018, 20). Zhou et al. demonstrated that KDM5B negatively regulates neurogenesis in adult subventricular zone through regulation of Reelin expression suggesting a role of KDM5B in neuronal migration and cortical circuit organization (Zhou et al. 2016). Extensive studies have found KDM5B as a major regulator of neuronal differentiation, however the specific genes involved in this disorder and how they affect neurogenesis remains elusive.
KDM5C
KDM5C (also known as JARID1C, SMCX) catalyzes the demethylation of di- and trimethylation H3K4, recognizes H3K9me3 and negatively regulates transcription through the RE-1-silencing transcription factor (REST) complex (Lan et al. 2007; Tahiliani et al. 2007). KDM5B is highly expressed in brain and skeletal muscle, pathogenic KDM5C variants account for approximately 3% of Claes-Jensen type of X-linked syndromic mental retardation (MRXSCJ) (MIM: 300534) (Jensen et al. 2005; Peng et al. 2015). Individual present intellectual disability, microcephaly, epilepsy and abnormal facial features (Jensen et al. 2005). To date, at least 30 mutations have been identified encompassing nonsense, missense and frameshift in individuals with MRXSCJ. Functional studies of these mutations reveal decrease in KDM5C activity, suggesting a loss-of-function pathological mechanism (Tahiliani et al. 2007; Iwase et al. 2007). Patient-derived fibroblasts revealed dysregulated KDM5C genomic localization, altered DNA methylation and loci specific alterations in histone methylation (Jensen et al. 2005; Brookes et al. 2015; Grafodatskaya et al. 2013; Schenkel et al. 2018). Additionally, KDM5C is critical for neuronal development and maturation (Iwase et al. 2007; Scandaglia et al. 2017). Iwase et. al recapitulated MRXSCJ abnormalities in a condition knockout of KDM5C in mouse brains, including behavioral anomalies and memory deficits (Iwase et al. 2016). Investigation of patient mutations in Neuro2a cells revealed dysregulation of genes involved in neuronal development due to methylation changes at their promoters; the differential expression resulted in morphological changes including abnormal neurite length (Wei et al. 2016). Another promising study demonstrated that reduced functionality of KDM5C causes a lack of repression in neurodevelopmental transcripts, and a decreased KDM5C dosage resulting in neuronal defects (Poeta et al. 2019). Moreover, they demonstrated that KDM5C is a Vorinostat-sensitive gene, treatment with this HDAC inhibitor ameliorates previously compromised neuronal differentiation (Poeta et al. 2019). The role of KDM5C has been extensively studied and characterized in neuronal differentiation, its impact has been shown to be corrected in vitro and in vivo using Vorinostat, a promising therapeutic for MRXSCJ.
KDM6A
Lysine demethylase 6A (KDM6A or UTX) contains a catalytic JmjC domain and preferentially demethylates di- or trimethylated H3K27. Also, it is a member of the KMT2C/D COMPASS-like complex (Hong et al. 2007; Lan et al. 2007; C. Wang et al. 2012; Cho et al. 2007; M. G. Lee et al. 2007). As a member of the COMPASS-like complex, KDM6A regulates enhancer activation, independent of its demethylase activity (S.-P. Wang et al. 2017). Individuals presented with KS1 characteristics, described earlier, were screened for mutations in KMT2D but instead carried de novo heterozygous mutations in KDM6A (Noriko Miyake et al. 2013; Lederer et al. 2012). Like KS1, Kabuki syndrome 2 (KS2) clinically presents with developmental delay, intellectual disabilities, and craniofacial abnormalities (Lederer et al. 2012). Some features of KS1 were less common in KS2 individuals; however, all KS2 patients exhibited postnatal growth retardation and short stature (Noriko Miyake et al. 2013). Conditional knockout of KDM6A in neural stem cells led to increased anxiety-like behavior and impaired learning and memory in mice (Tang et al. 2017). Moreover, hippocampal neurons in these mice displayed impaired synaptic formation and plasticity, revealing a role of KDM6A in regulating cognition (Tang et al. 2017). Another model demonstrated that KDM6A loss increases neural stem cell proliferation and decreases terminal mitosis, resulting in aberrant neuronal differentiation (Lei and Jiao 2018). Interestingly both KS1 and KS2 stem from altered KMT2C/D COMPASS-like complex functions. Although extensive studies have been conducted in the context of cancer, specific pathways involved in neurogenesis remains ambiguous.
PHF8
PHD finger protein 8 (PHF8 also known as KDM7B) is the first monomethyl H4K20 demethylase established, interestingly it can also demethylate mono- and dimethylated H3K9 (Qi et al. 2010). PHF8 binds H3K4me3, and through this interaction, positively regulates gene expression (Feng et al. 2010; Horton et al. 2010). Reduction of PHF8 in zebrafish embryos caused cell death in the developing brain and neural tube and significant defects in craniofacial development (Qi et al. 2010). PHF8 directly controls the expression of homeodomain transcription factor MSX1/MSXB, mediating signaling and developmental pathways (Qi et al. 2010). Given its role in neurodevelopment, it’s not surprising that mutations in PHF8 results in a NDD, Siderius-type X-linked syndromic mental retardation (MRXSSD) (MIM: 300263) (Siderius et al. 1999; Laumonnier et al. 2005). MRXSSD primarily affects male individuals and they often have intellectual disability, mild dysmorphic features, and bilateral/unilateral cleft lip or palate(Koivisto et al. 2007). In vitro analysis of MRXSSD patient mutation abolished demethylase activity and transcriptional activation (Feng et al. 2010). PHF8 potentially mediates serotonin signaling and downregulation of PHF8 results in stress-induced anxiety, depression and cognitive impairment (Walsh et al. 2017). Moreover, PHF8 knockout mice displayed reduction cognition and memory due to impaired long-term potentiation. Chen et al. demonstrated PHF8 plays a role homeostasis of mTOR signaling, and suppression of mTOR recovers the cognitive deficits observed in PHF8 knockout mice. These studies provide potential targetable pathways to treat MRXSSD.
READERS
BPTF
Bromodomain PHD finger transcription factor (BPTF) is the largest subunit of the NURF complex and mediates binding to H3K4me3 (M. H. Jones, Hamana, and Shimane 2000). BPTF has been shown to promote expression of WNT8A which is crucial for neural posteriorization, through recruitment of NURF complex(Ma et al. 2015). Eight heterozygous mutations in the BPTF gene were discovered in unrelated individuals with neurodevelopmental disorder with dysmorphic facies and distal limb anomalies; NEDDFL (MIM: 617755) (Stankiewicz et al. 2017). NEDDFL is characterized by delayed psychomotor development, intellectual disability, speech impairments, microencephaly and dysmorphic features (Glinton et al. 2021). Although functional studies of patient cells have yet to be conducted, Stankiewicz et al. suggests that haploinsufficiency is the pathogenic mechanism of this disorder (Stankiewicz et al. 2017). CRISPR/Cas9-mediated knockdown of bptf gene in zebrafish resulted in increased cell death which recapitulated microencephaly and craniofacial abnormalities seen in patients (Stankiewicz et al. 2017). Due to the novelty of this disorder, there is currently no potential pathological mechanism for BPTF mutations in neurodevelopment.
EED
As mentioned above, EED is a major subunit of PRC2 complex as it recognizes previously methylated histones (Margueron et al. 2009). EED is required for maintaining neurogenesis and neurosphere formation through regulation of p21 (B. Sun et al. 2018). Heterozygous mutations in EED results in the overgrowth disorder Cohen-Gibson syndrome (COGIS) (MIM: 617561) (Cooney et al. 2017). Affected COGIS individuals have intellectual disability, dysmorphic facial features, advanced bone age and skeletal abnormalities (A. S. A. Cohen et al. 2016; Cooney et al. 2017; A. S. Cohen and Gibson 2016; Imagawa et al. 2017). EED-deficient mice exhibited postnatal lethality and diminished neuronal differentiation in the hippocampal dentate gyrus potentially explaining the pathomechanism of COGIS (Liu et al. 2019). Additionally, EED establishes a chromatin landscape that selectively represses inhibitory WNT and bone morphogenetic protein (BMP) signaling which is required for oligodendrocyte differentiation and maturation (Jiajia Wang et al. 2020). Taken together, EED plays a critical role in CNS development, through regulation of neuronal and oligodendrocytic differentiation, however, the field lacks biochemical characterization of COGIS mutations.
SUZ12
Similar to the other core PRC2 subunits, EZH2 and EED, mice with mutations in SUZ12 are embryonic lethal due to severe developmental and proliferative defects(Pasini et al. 2004). SUZ12 is essential for EZH2 activity as SUZ12 deficiency in vivo leads to loss of di- and trimethylated H3K27 (Pasini et al. 2004). Unfortunately, the SUZ12-related overgrowth syndrome, Imagawa-Matsumoto syndrome (IMMAS) (MIM: 618786) is the least characterized of the PRC2-related syndromes (Imagawa et al. 2017; 2018). Individuals with IMMASS exhibit pre- and postnatal overgrowth, dysmorphic features, intellectual disabilities, macrocephaly and developmental delay (Imagawa et al. 2017). SUZ12 is required for proper differentiation as SUZ12-deficient stem cells are de-repressed, increasing levels of differentiation specific genes (Pasini et al. 2004). Moreover, heterozygous SUZ12 mice exhibit neural tube defects and severely impaired differentiation (Miró et al. 2009).
CHROMATIN REMODELERS
Precise temporal gene expression is critical for differentiation during development. Histone PTM patterns recruit different effector proteins, such as chromatin remodelers with the ability to alter chromatin structure. Chromatin remodelers influence the spacing of nucleosomes allowing transcriptional machinery access to DNA. Remodelers are multiprotein complexes, centered around a catalytic ATPase helicase subunit, that slide, unwrap or evict nucleosomes, as well as exchange histone variants within a nucleosome. Beyond the helicase, remodelers contain subunits that enhance activity and facilitate targeting of the complex. Many remodeler members contain reader domains such as chromodomains and bromodomains, allowing for targeting to specific PTMs. The major ATPase remodeler families are BRG1 associating factors (BAF) (also known as switch/sucrose non-fermentable (SWI/SNF)), ISWI (imitation SWI), CHD (chromodomain helicase DNA-binding), and INO80 (SWI2/SNF2 related). Recently, mutations in members of ATPase remodelers have been identified in neurodevelopmental disorders giving light to the importance of these complexes in neurodevelopment.
BAF family
BAF (BRG1/BRM associating factors) complexes are the mammalian version of yeast Switch/Sucrose Non-Fermentable (SWI/SNF) remodeler (W. Wang et al. 1996; Breeden and Nasmyth 1987; Stern, Jensen, and Herskowitz 1984; Neigeborn and Carlson 1984). Canonical BAF is comprised of approximately 15 subunits, however, at least 29 isoforms of the subunits exist. This allows for multiple formations of BAF complexes in a single cell. For example, there are two mutually exclusive helicases, BRG1 and BRM. Though they appear to have some redundant roles, mouse studies have demonstrated distinct roles for BRG1 and BRM in development. Homozygotic BRG1 knockout is lethal pre-implantation where BRM knockouts are viable (Bultman et al. 2000). The variety of these isoforms allow for multiple formations of BAF complexes in a single cell. Ubiquitously expressed sub-complexes include Polybromo-1-BAF (P-BAF), which incorporated Polybromo-1, ARID2, BAF45a, and BRD7(W. Wang et al. 1996)and G-BAF that is comprised of GLTSCR1 and BRD9 (Alpsoy and Dykhuizen 2018). Cell-type-specific BAF compositions such as neuron progenitor and neuron-specific BAF (npBAF and nBAF) also exist (Lessard et al. 2007). BAF subunits exchange as neuron-progenitor cells differentiate into neurons and exit mitosis and this is critical for proper proliferation and differentiation of neural progenitor cells. Interestingly, BAF mutations have been identified in ID disorders with the majority of mutations associated with Coffin-Siris syndrome (CSS), Nicolaides-Baraiter, and Autism.
Coffin-Siris, first reported in 1970, is a rare genetic disorder with only about 140 cases reported in literature (Coffin and Siris 1970; van der Sluijs et al. 2019). Although patient phenotypes highly overlap, OMIM classifies CSS into 11 different types depending on what protein is mutated. Mutations have been identified in ARID1b (BAF250b) (MIM:135900, CSS1), ARID2 (BAF200) (MIM:609539, CSS6), BRG1 (MIM:614609,CSS4), BAF57(MIM:616938, CSS5), ARID1a (BAF250a) (MIM: 614607, CSS2), DPF2 (MIM:601671, CSS7), BAF47(MIM: 614608, CSS3), BAF60a (SMARCD1) (MIM: 618779, CSS11) (van der Sluijs et al. 2019; Kosho et al. 2014; Santen et al. 2012). Patients diagnosed with CSS exhibit intellectual disorders (IQ score 50–89), delayed sitting and walking, short stature, microcephaly, muscular hypotonia, coarse facial features, hypoplasia or aplasia of the fifth finger, bushy eyebrows, and excessive hair growth (van der Sluijs et al. 2019). AT-rich interacting domain 1b (ARID1b), the most frequently mutated gene in CSS (68%), was identified in 2012. Mutations resulted in a heterozygous deletion, causing haploinsufficiency, speculated to be the driver of CSS. The ARID subunits (ARID1a, ARID1b, and ARID2) are predicted to bridge the ATPase to the complex and their incorporation determines which BAF subtype is formed (Mashtalir et al. 2018). Interestingly, ARID1b incorporation into BAF is mutually exclusive with ARID1a (which is 60% identical). Additionally, ARID1b, but not ARID1a, represses WNT/Beta-catenin signaling, involved in proliferation, differentiation, and pluripotency (Vasileiou et al. 2015). Activation of this pathway in haploinsufficient cells may be a driving mechanism of CSS. In mouse models recapitulating the ARID1b haploinsufficiency, mice exhibited growth retardation, hypotonia, and agenesis of the corpus callosum, as well as anxiety and abnormal social behaviors (Celen et al. 2017).
Other BAF subunits are less commonly mutated in CSS and unlike ARID1b, result in expression of mutated protein and are predicted to have either loss or gain-of-function mechanisms (van der Sluijs et al. 2019). BAF47 mutations occur in exon 8 and 9 with the only reoccurring mutations being pLys364 deletion. One study utilized an inversion NesCre system to decrease BAF47 expression by 30% in mice (Filatova et al. 2019). The mutant mice were significantly smaller than the controls and their brain weight was reduced by 41%. The majority (~80%) did not survive past postnatal week 9. Intriguingly, these effects were not observed in BAF47 heterozygotic knockout mice. BRG1 mutations account for 11% of CSS cases and they cluster in the helicase/SANT-associated domains. Mutations of BRG1 as well as BAF155 lead to defects in neural tube closures and exencephaly, demonstrating the critical role of BAF complexes in neural development (Bultman et al. 2000; J. K. Kim et al. 2001). BRG1 is also involved in both neuronal and glial differentiation and BAF is recruited to cell-type-specific genes through different transcription factors. OLIG2, an oligodendrocyte-specific transcription factor, recruits BRG1 to H3K27Ac rich enhancers for myelination-specific genes(Y. Yu et al. 2013). BAF57 mutations are found in the high mobility group (HMG), a DNA binding domain, and ARID1a mutations are predicted to be truncated. ARID2 mutations have been reported in seven CSS patients (Bramswig et al. 2017; Shang et al. 2015; Van Paemel et al. 2017). DPF2 mutations that cluster in the two PHD fingers were found in eight patients (Vasileiou et al. 2018). These mutations altered DPF2 binding to H3 peptides and are predicted to have a dominant-negative mechanism. In 2019, Nixon et al., identified 5 individuals with BAF60a (SMARCD1) (MIM: 618779, CSS11) mutations(Nixon et al. 2019). Patients had some overlapping phenotypes with CSS, however, did not exhibit the same facial features such as thick eyebrows, broad noses and long eyelashes. BAF60a mutations did not disrupt incorporation into the BAF complex. Interestingly, BAF60a is important for recruiting BAF to OCT6 and KROX20 through SOX10 interaction in Schwann cells (Weider et al. 2012).
Mutations in BRM have been shown to be causative for Nicolaides-Baraitser syndrome (MIM:601358), a developmental disorder characterized by short stature, microcephaly, sparse hair, epilepsy, and intellectual disabilities (Van Houdt et al. 2012). Sousa et al. examined 59 patients with 48 missense mutations in BRM and found these mutations cluster in the highly conserved SNF2 ATPase domain and are predicted to cause gain or loss-of-function, rather than truncations (Sousa et al. 2014). Additional BAF subunits are mutated in neurodevelopmental disorders. Machol et al., found 15 patients with BAF170 (SMARCC2) mutations, also exhibiting similar phenotypes to CSS patients, including the lack of the fifth finger nail (Machol et al. 2019). Three novel mutations have also been identified in Actl6a (Marom et al. 2017).
A recent study on how BAF assembles demonstrated a sequential addition subunit modules and that the incorporation of the ARID proteins is critical in determining which BAF subcomplex will be formed (Mashtalir et al. 2018). Loss of expression or mutant BAF subunits can alter proper formation or skew the composition of the complex as well as alter the function of the complex. It is likely that the introduction of BAF mutations, disrupt BAF composition as well as altering the switch between npBAF and nBAF. Due to the importance of BAF in neurogenesis and development, this list of mutations will likely grow as we expand sequencing studies of NDD patients.
CHD Family
The CHD family of ATPase helicase remodelers is comprised of nine proteins, CHD1–9, that contain tandem chromodomains, methyl-lysine readers, and SNF2-like ATPase catalytic domain (Marfella and Imbalzano 2007). These proteins are divided into three subgroups: 1) CHD1and CHD2 contain C-terminal DNA domains that bind AT-rich regions, 2) CHD3 and CHD4 have paired N-terminal PHD zinc fingers, and 3) CHD5-CHD9 featuring Brahma and Kismet domains, CR-domains, SANT domains, and DNA binding domains25. In contrast to the other remodelers, many of the CHD proteins act without accessory proteins, however, CHD3, CHD4, and CHD5 can be incorporated in the NURD complex along with HDAC1/2, MBD2 or 3, MTA1,2 or 3, RBAP46, and RBAP48 (Hoffmann and Spengler 2019). The CHD proteins have important roles in development and differentiation. Knockdown CHD1 causes defects in self-renewal and pluripotency in ESCs. During brain development, the CHD4-containing NURD complex is critical in synapse formation by repressing genes to drive synaptogenesis (T. Yamada et al. 2014). This involves deacetylation of histone H3 at residues at K9, K14, and K27 as well as demethylation of H3K4me3. Neural crest cell migration is also mediated through CHD7 and PBAF by regulating gene expression of Sox9, Slug, and Twist (Bajpai et al. 2010; Schulz et al. 2014). Loss of CHD7 decreases the pool of neural stem cells and impairs neuron maturation (K. M. Jones et al. 2015). As sequencing of patients with intellectual disorders becomes standard, the CHD proteins have been identified as culprits in several disorders.
CHD1
CHD1, known to bind H3K4me3/H3K4me3 through its chromodomains, and is predicted to play important roles in pluripotency, transcriptional elongation, and deposition of histone variant H3.3 (Flanagan et al. 2005). CHD1, particularly the N-terminus, is critical in mouse long-term and spatial memory (Schoberleitner et al. 2019). In 2018, Pilarowski et al., identified six female patients with de novo heterozygous mutations, most of which were at highly conserved arginine residues. A larger cohort is needed to determine if there is a gender bias for Pilarowski-Bjornsson syndrome (MIM: 617682) patients (Pilarowski et al. 2018). Probands exhibited developmental delays, hypotonia, pointed chins, frontal forehead bossing, and arched eyebrows. Using patient-derived fibroblasts containing CHD1 mutation, global increases in H3K27me3 compare to wild-type cells were observed. Mice with heterozygotic loss of Chd1 are phenotypically normal(Guzman-Ayala et al. 2015), suggesting a dominant-negative mechanism for CHD1 mutants, leading to the prevention of wild-type CHD1 properly binding to the targets and regulation gene expression (Pilarowski et al. 2018). Further work is needed to determine the role of these mutations.
CHD2
CHD2 mutations have been characterized in patients with neurodevelopmental delays and epilepsy (MIM: 615369). CHD2 was identified as a driver of epilepsy when several seizure patients were identified with chromosome 15 deletions ranging from 5Mb to 511kb (Dhamija et al. 2011; Veredice et al. 2009; M. M. Li et al. 2008; Capelli et al. 2012). The smallest deletion encompassed only two genes, CHD2 and RGMA (Capelli et al. 2012). After adding CHD2 as a potential target when re-sequenced 500 individuals with epileptic encephalopathy, Carvill et al., identified six patients with CHD2 mutations (Carvill et al. 2013). As of 2021, 139 patients with neurodevelopmental disorders have been found, with ~95% of these individuals being affected by epilepsy, the onset ranging from six months to 12 years old (Carvill et al. 2013; Carvill and Mefford 1993; Suls et al. 2013; Heyne et al. 2018; Ko et al. 2018; Chen et al. 2020; Truty et al. 2019; Fitzgerald et al. 2015). Majority of CHD2 mutations lead to loss of expression or truncations, the few missense mutations do not appear to cluster in any particular region of the gene (Carvill and Mefford 1993). Mutations are mostly de novo, however, several were inherited from unaffected parents (Chen et al. 2020). CHD2 patients exhibited increased photosensitivity compared to other epileptic disorders and have varying degrees of intellectual delays. Though the mechanism of pathology is unknown, it is hypothesized that CHD2 is key in neuron differentiation by keeping bivalent genes active(Semba et al. 2017). Loss of CHD2 leads to increased H3.3 containing nucleosomes as well as elevated levels of the repressive mark, H3K27me3(Semba et al. 2017). In 2018, Kim et al., created a haploinsufficient CHD2 mouse and though this model did not recapitulate seizures, it demonstrated the importance of CHD2 in proliferation of neural progenitors, synapse transmission, hippocampus memory, and cortical synchrony, which appeared to be regulated by changes in expression of genes involved in neurogenesis, chromatin regulation and synaptic transmission(Y. J. Kim et al. 2018).
CHD3
In 2018, Blok et al., published the first report of CHD3 mutations (Snijders Blok et al. 2018). This first cohort of Snijders Blok-Campeau syndrome (MIM: 618205) consisted of 35 patients with 23 distinct mutations. These individuals suffered from ID, hypotonia, developmental delays, and severe speech and language disabilities. Many exhibited macrocephaly, with CT scans showing broad cerebrospinal fluid spaced; one patient presented with microcephaly. These probands have distinct facial features including broad foreheads, wide-spaced eyes sparse eyebrows, low set ears, and pointy chins. The majority of the CHD3 mutations clustered in the ATPase/helicase domain as well as amino acids that are evolutionarily conserved between species and other CHD proteins. Biochemical assays demonstrated several patient mutations caused loss of ATPase and remodeling activity of CHD3. The following year, Drivas et al., reported 24 additional patients with CHD3 mutations (Drivas et al. 2020). Though the majority of these mutations were found in the ATPase domain, nine were found outside of the domain. No phenotypic difference was observed between patients with mutations in or outside of the ATPase domain. Eising et al., found another CHD3 mutation in the helicase domains while genetically screening individuals with childhood apraxia of speech (Eising et al. 2019). Though, loss of catalytic activity is predicted to be the pathogenic mechanism, it is unclear the mechanism for the mutations found outside the ATPase domain.
CHD4
In 2016, two separate studies identified CHD4 mutations in patients. Sifrim et al., found five probands through screening individuals with congenital heart defects (Sifrim et al. 2016). Three patients exhibited Tetralogy of Fallot (defect of ventricular septal, pulmonary valve narrowing, misplaced aorta, and thickening of the right ventricular). In addition to cardiac defects, all five patients exhibited neurodevelopmental disorders and genital abnormalities. Weiss et al., identified an additional five patients exhibiting developmental delay, intellectual disabilities, macrocephaly, facial abnormalities, hypogonadism, hearing loss, bone fusion as well as congenital issues (Weiss et al. 2016). The five mutations were found at highly conserved residues, with four located in the C-terminal helicase domain. A follow-up study of Sifrim-Hitz-Weiss syndrome (MIM: 617159) looked at 32 individuals, and found mutations causing loss of CHD4 had less severe phenotype compared to patients with point mutations (Weiss et al. 2020). Biochemical investigation of mutations showed that some, but not all the mutations, were able to disrupt ATPase and nucleosome remodeling. Goodman et al., has seen that conditional knockout of CHD4 increases H3K27ac, as well as increasing promoter and enhancer accessibility in mouse brains(Goodman et al. 2020). Additionally, they observed increases in looping and cohesion binding, indicating reorganizing of the genome. It is yet to be determined if mutations of CHD4 will have similar effects as CHD4 knockout (Goodman et al. 2020).
CHD7
CHARGE (Coloboma of the eye, Heart defects, Atresia of the choanae, Retardation of growth and development, and Ear abnormalities and deafness) syndrome (MIM:214800) is mainly associated with heterozygotic loss of CHD7, causing haploinsufficiency. CHARGE was first described in 1979 by two separate groups (Hittner et al. 1979; Hall 1979), however, the link with CHD7 was not discovered until 2004 by Vissers et al (Vissers et al. 2004). Since then, screening for CHD7 mutations has become standard in diagnosis (van Ravenswaaij-Arts et al. 1993). Patients exhibit an extensive list of symptoms that affect multiple organs. Coloboma is the improper formation of the eye, resulting in a keyhole in the iris, causing photosensitivity (van Ravenswaaij-Arts et al. 1993). Cranial nerve abnormalities lead to loss of hearing, balance issues, swallowing difficulties, asymmetric facial palsy, lack of smell and hypogonadism (van Ravenswaaij-Arts et al. 1993). They also present with heart abnormalities, esophageal atresia, hypotonia, craniofacial abnormalities, and characteristic “CHARGE ears” (short, wide and lacking earlobes) (van Ravenswaaij-Arts et al. 1993). Analysis of expression 10 human fetuses with CHARGE, demonstrated CHD7 was localized to the spinal ganglia, cranial nerves, neural retina, pituitary (important for regulation growth and development), as well as nasal and auditory tissues (Sanlaville et al. 2006). Knockout of CHD7 in mice reduced neurogenesis in the hippocampus, dentate gyrus, which is important for memory, the sub-ventricular zone, and in the inner ear. Examining its role in differentiation, CHD7 is not essential in ESC proliferation or differentiation into neuron progenitor cells and dispensable in many cell-types (H. Yao et al. 2020). However, CHD7 is critical in development of neurons. Specifically, loss of CHD7 leads to decrease in neuroblasts and causes a shift in increases the glial cell, and neurons that are created have fewer branches with reduced length, effecting the number and the quality of neurons (Micucci et al. 2014; H. Yao et al. 2020). CHD7 is important keeping chromatin open and activating neuron differentiation genes and associates with H3K27ac rich super enhanced and cell identify specific transcription factors as well as recruiting DNA Topoisomerase IIb to long neuron genes (Feng et al. 2017; Hnisz et al. 2013). CHARGE and CHD7 has been studied more extensively than other NDDs and Trider et al. has created an extensive checklist of 49 symptoms of CHARGE patients and guidelines for treatment of the individuals as they age (Trider et al. 2017). Though this is an invaluable resource, the treatments focus on management of symptoms. Developing therapeutics to elevate the defects of neurogenesis caused by CHD7 haploinsufficiency are still needed.
HISTONES
All of the disorders previously described originate from germline mutations in enzymes that either catalyze histone post-translational modifications or chromatin remodeling. Recent studies have discovered germline mutations in histone proteins, allowing us to define their roles not only in the context of gene regulation but also development.
HIST1H1E
In 2017, Tatton-Brown et. al. conducted a study with 710 individuals all suffering from overgrowth syndrome and intellectual disability to explore the underlying genetic predispositions (Tatton-Brown et al. 2017). Utilizing exome sequencing, they reported de novo heterozygous germline mutations in the HIST1H1E gene (5/710) which encodes linker histone, histone H1.4 (H1.4). Patients bearing H1.4 mutations suffer from distinct facial features, intellectual disability, and macrocephaly (Tatton-Brown et al. 2017). This syndrome was coined Rahman syndrome (MIM: 617537), with the causative gene being HIST1H1E. Following the 2017 study, 42 patients were discovered suffering from Rahman Syndrome all exhibiting distinct facial gestalt, ID, skeletal, and congenital cardiac abnormalities (Burkardt et al. 2019; Duffney et al. 2018; n.d.). Reported frameshift mutations are centralized to the C-termini of the protein and result in a truncated H1.4 with a charge reduced from +44 to +7. Linker histones play a critical role in regulating higher-order chromatin structure through neutralization of negatively charged linker DNA via its positively charged tails. H1.4 is often sequestered in heterochromatic regions and the truncation of H1.4 ultimately impairs the linker histone’s ability to generate higher-order structures (Tatton-Brown et al. 2017; McGinty and Tan 2015; Ponte et al. 2017; Song et al. 2014).
Conducting a robust functional assessment of the HIST1H1E mutations it was determined that these mutants had proper nuclear localization and increased stability compared to wild type (Flex et al. 2019). Normally, the C-terminal tail of H1 undergoes reversible phosphorylation which mitigates chromatin compaction, chromosome segregation, and chromatin decondensation, however, these residues are all lost in HIST1H1E mutants (Flex et al. 2019; Chadee et al. 1997; Ajiro, Borun, and Cohen 1981). Interestingly, patient-derived fibroblasts showed significantly more relaxed DNA as well as a decrease in H3K4me2, H3K9me3, H3K27me3, and HP1β (Flex et al. 2019). Ultimately, the frameshift mutations disrupt chromatin structure, alter nuclear lamina organization, generate a hypomethylated DNA signature, and drive replicative senescene (Flex et al. 2019). Another study determined that the aberrant DNA methylation profile resulted in altered gene regulation, which was exacerbated in post-mitotic cells as genes involved in neural signal transduction were significantly altered (Ciolfi et al. 2020). Overall, there is sufficient evidence to start elaborating on the pathomechanism of H1.4 mutations and the role of this linker histone in neuronal development.
HIST1H4C/J
Histone H4 (H4), which is encoded by 15 distinct genes, is currently the only histone with no functionally distinct variants (Holmes et al. 2005). It is ubiquitously expressed in all cells and is a critical nucleosomal protein. In 2017, monoallelic missense heterozygous mutations of HIST1H4C were discovered via trio sequencing (Tessadori et al. 2017). These mutations result in substitutions that cause a charge switch (K->E), lose (K->Q) or retain charge (K->R) at residue 91 of H4 (Tessadori et al. 2017). These patients exhibit short stature, microencephaly, intellectual disability, craniofacial abnormalities, and foot ray anomalies. RNA sequencing utilizing patient-derived fibroblasts revealing 115 genes differentially expressed in K91 mutants. Intriguingly, 25 of these genes encoded for histone proteins. Enriched gene ontology (GO) terms for differentially regulated genes related to chromosome organization, DNA packaging, and cell cycle progress (Tessadori et al. 2017).
To assess the functional consequence of these mutations, zebrafish were injected with mRNA encoding mutations and K91R/Q injected embryos exhibited severe developmental defect (Tessadori et al. 2017). These embryos had underdeveloped brains and eyes, defective body axis growth, and dysmorphic tails which recapitulated the phenotype seen in patients (Tessadori et al. 2017). Thus, this in vivo model provided sufficient evidence that HIST1H4C mutations were causative in the observed syndrome. Zebrafish larvae expressing H4-K91R/Q exhibited increased accumulation DNA double-strand breaks (DDBs) specifically in the head and tail region and this was determined utilizing a well-known readout for DDBs, γ-H2AX signal (Tessadori et al. 2017). Additionally, these embryos had increased apoptosis in the head and tail regions (Tessadori et al. 2017). K91 is a known PTM site for H4, as acetylation has been linked to chromatin assembly regulation and ubiquitination has been linked to DNA damage response (Ye et al. 2005; Q. Yan et al. 2009; Yang et al. 2011). Co-expression of DTX3L, the E3 ligase responsible for K91 ubiquitination, with K91 mutants alleviated the increase in double-strand breaks and morphological anomalies observed (Tessadori et al. 2017). This study was able to determine that the loss of K91 ubiquitination in the mutants was sufficient to alter cell cycle progression and apoptosis.
Recent work from the Gijs van Haaften lab reported a de novo germline missense mutations in HIST1H4J, another gene encoding histone H4 (Tessadori et al. 2019). This patient had a K91E mutation and exhibited intellectual disability, microcephaly, and had dysmorphic facial features, symptoms extremely similar to the previous report HIST1H4C mutations (Tessadori et al. 2017; 2019). Here they hypothesized that the glutamic acid (E) substitution may contribute to genomic instability due to its inability to be modified as well as the addition of a negative charge. This study was critical in showing the importance of K91 residue in H4, as different substitutions at this position can result in distinct phenotypic outcomes.
H3F3A/H3F3B
The most recent discovery of histone germline missense mutations involves the two genes (H3F3A and H3F3B) encoding Histone H3 variant, Histone H3.3 (Bryant et al. 2020). Bryant et. al discovered 37 unique de novo mutations in 46 patients. These patients suffered from a common pattern of neurodevelopmental disorder encompassing microencephaly, craniofacial abnormalities, and intellectual disabilities (Bryant et al. 2020). Unlike Rahman Syndrome and the K91 mutants of H4, these H3.3 mutations are dispersed across the entire protein affecting, post-translational modification residues, chaperone interaction, protein-protein interaction, and protein-DNA contacts. Bottom-up mass spectrometry of patient lymphoblasts revealed histone PTM changes which were reproducibly altered in patients compared to controls (Bryant et al. 2020). Additionally, RNA sequencing utilizing patient-derived fibroblasts identified 323 differently expressed genes associated with mitosis and cell cycle regulation upregulated (Bryant et al. 2020). Due to the locations of these mutations along H3F3A and H3F3B it is predicted different biological processes are altered but appear to converge on similar phenotypes. This suggests that the phenotype is due to dysregulation of H3.3 in general and not limited to a single mechanism.
Here we have discussed the recent findings regarding germline variants associated with histone proteins, histone H1, histone H3.3, and histone H4. All of these patients exhibit common dysregulation of neurodevelopment clinically characterized with craniofacial abnormalities, intellectual disabilities, and developmental delay. These histone-related disorders allow a unique perspective on the role of histones specifically in the control of central nervous system development and growth.
CONCLUSION
In this review, we have highlighted the importance of epigenetic factors in neurodevelopment and how disruption of chromatin regulation at multiple levels can drive NDDs. The expansion of next-generation sequencing techniques in the diagnosis of NDD has elucidated many genes mutated in these disorders. Of particular interest are proteins involved in the regulation of DNA organization and gene expression. We have discussed mutations of histones, forming nucleosomes, the smallest unit of chromatin, to the proteins that curate PTM patterns across the genome to megadalton complexes the enforce chromatin structure. Though varying in phenotypical differences and severity, most of these disorders have devastating effects on physical and mental development. Although syndromes, such as RSTS1 and CHARGE, have been defined since the 1970s, the genetic drivers have only begun to be identified in the last couple of decades with the advances in sequencing. Sequencing has not only allowed for better classification of current syndromes; it has expanded our list of known driving mutations. Patients present with chromosomal deletions, resulting in the loss of multiple genes, or frameshift and missense mutations that can cause loss of expression, truncation, or point mutations.
For the most part, these mutations are de novo, with no familial history of the disorder as well as monogenic, indicating these mutations are causative. These disorders’ common characteristics include intellectual disabilities, hypotonia, and facial abnormalities with comorbidities such as epilepsy and cardiac abnormalities. It is essential that most of these patients need extensive medical intervention to manage the symptoms and a lifetime of care and will never achieve independence. Therefore, it is imperative that more studies are done to determine the molecular pathways disrupted in these disorders. Thus far, clinical geneticists have determined the driving mechanism of NDD involves either haploinsufficiency, loss or gain-of-function, or dominant-negative. These all point to gene dosage effects, and future studies utilizing heterozygotes will be required to elucidate these pathological mechanisms fully.
Regarding chromatin-modifying enzymes, their genes are heavily regulated as precise regulation is necessary for proper differentiation and cellular identity. Therefore, epigenetic factors are critical for accurate expression or repression and the establishment of distinct chromatin structures. As discussed previously, distinct PTMs indicate active promoters, gene bodies, enhancers, and repressed regions. Neuroepigenetic, a relatively new field, has propelled research into NDDs forward as it focuses on determining how alterations in the epigenome result in neurological disease or disorders. Furthermore, extensive work in understanding neurogenesis has demonstrated the importance of epigenetic machinery as loss of these enzymes results in alterations to precise coordination of proliferation and differentiation in neural stem cells. Deletion of chromatin proteins results in abnormal cortical neurogenesis and defective synaptic plasticity. Due to the post-mitotic nature of neurons, it raises the question of whether neurons exhibit increased sensitivity to point-mutations or deletions of the epigenetic machinery. However, this remains to be fully explored. To date, mouse models of KS1, KLEFS1, RSTS1, SOTOS1, and WVS have been generated, and defects in hippocampal memory were observed in all models (Alarcón et al. 2004; Korzus 2017; Hans T. Bjornsson et al. 2014; M. C. M. Balemans et al. 2013; Monique C. M. Balemans et al. 2014; Harrison et al. 2012; J. Zhang et al. 2014). This suggests a similar disruption to memory formation, retrieval, or overall hippocampal function in these various NDDs. Interestingly, both the RSTS1 and KS mouse model had their hippocampal defects rescued by postnatal treatment with HDAC inhibitors (Alarcón et al. 2004; Hans T. Bjornsson et al. 2014).
Interestingly one of those inhibitors, Vorinostat, is approved by the US Food and Drug Administration (FDA) as cancer therapeutic. Epigenetic therapies, including both HAT and HDAC inhibitors, are currently being studied for the treatment of neurological conditions such as spinal muscle atrophy, bipolar disorder, and Alzheimer’s disease (“Histone Acetyltransferase Inhibitors and Preclinical Studies: Expert Opinion on Therapeutic Patents: Vol 19, No 6” n.d.). As of 2021, four HDAC inhibitors are FDA-approved: Vorinostat, Panobinostat, Romidepsin, and Belinostat. These may be fruitful therapeutics in these disorders, especially those with decreased histone acetylation, which highlights the importance of classifying these disorders based on chromatin state and target gene expression. Studies of chromatin regulator proteins will offer keen insight into manipulating the epigenome in the context of disease to develop broad and potentially personalized therapeutics. Despite these promising therapeutics, one limitation remains the number of neural tissue available from patients and the overall rarity of individuals themselves. However, recent advances involving the generation of functional neurons and glia through patient-derived iPSCs or creating a patient-mimic through CRISPR/Cas9 technology allowed a new avenue to explore these etiologies NDDs (Sabitha, Shetty, and Upadhya 2021). This, coupled to the 3D self-assembled structures known as brain organoids, provides a unique opportunity not only to determine the pathways dysregulated but to assess cellular organization, transcriptional and epigenetic landscape in neurodevelopment. Collectively, utilizing both neuroscience advancements, we can begin to understand human neurodevelopment and its dysregulation using patient cells fully.
REFERENCES
- Ajiro Kozo, Borun Thaddeus W., and Cohen Leonard H.. 1981. “Phosphorylation States of Different Histone 1 Subtypes and Their Relationship to Chromatin Functions during the HeLa S-3 Cell Cycle.” Biochemistry 20 (6): 1445–54. 10.1021/bi00509a007. [DOI] [PubMed] [Google Scholar]
- Akizu Naiara, and Martínez-Balbás Marian A.. 2016. “EZH2 Orchestrates Apicobasal Polarity and Neuroepithelial Cell Renewal.” Neurogenesis 3 (1). 10.1080/23262133.2016.1250034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcón Juan M, Malleret Gaël, Touzani Khalid, Vronskaya Svetlana, Ishii Shunsuke, Kandel Eric R, and Barco Angel. 2004. “Chromatin Acetylation, Memory, and LTP Are Impaired in CBP+/− Mice: A Model for the Cognitive Deficit in Rubinstein-Taybi Syndrome and Its Amelioration.” Neuron 42 (6): 947–59. 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Alari Valentina, Russo Silvia, Terragni Benedetta, Ajmone Paola Francesca, Sironi Alessandra, Catusi Ilaria, Calzari Luciano, et al. 2018. “IPSC-Derived Neurons of CREBBP- and EP300-Mutated Rubinstein-Taybi Syndrome Patients Show Morphological Alterations and Hypoexcitability.” Stem Cell Research 30 (Stem Cells Transl. Med. 6 2017): 130–40. 10.1016/j.scr.2018.05.019. [DOI] [PubMed] [Google Scholar]
- Albert Mareike, Kalebic Nereo, Florio Marta, Lakshmanaperumal Naharajan, Haffner Christiane, Brandl Holger, Henry Ian, and Huttner Wieland B.. 2017. “Epigenome Profiling and Editing of Neocortical Progenitor Cells during Development.” The EMBO Journal 36 (17): 2642–58. 10.15252/embj.201796764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert Mareike, Schmitz Sandra U., Kooistra Susanne M., Malatesta Martina, Torres Cristina Morales, Rekling Jens C., Johansen Jens V., Abarrategui Iratxe, and Helin Kristian. 2013. “The Histone Demethylase Jarid1b Ensures Faithful Mouse Development by Protecting Developmental Genes from Aberrant H3K4me3.” PLoS Genetics 9 (4): e1003461. 10.1371/journal.pgen.1003461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allfrey VG, Faulkner R, and Mirsky AE. 1964. “ACETYLATION AND METHYLATION OF HISTONES AND THEIR POSSIBLE ROLE IN THE REGULATION OF RNA SYNTHESIS.” Proceedings of the National Academy of Sciences 51 (5): 786–94. 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allfrey VG, and Mirsky AE. 1964. “Structural Modifications of Histones and Their Possible Role in the Regulation of RNA Synthesis.” Science 144 (3618): 559–559. 10.1126/science.144.3618.559. [DOI] [PubMed] [Google Scholar]
- Alpsoy Aktan, and Dykhuizen Emily C.. 2018. “Glioma Tumor Suppressor Candidate Region Gene 1 (GLTSCR1) and Its Paralog GLTSCR1-like Form SWI/SNF Chromatin Remodeling Subcomplexes.” Journal of Biological Chemistry. 10.1074/jbc.RA117.001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrés María E., Burger Corinna, Peral-Rubio María J., Battaglioli Elena, Anderson Mary E., Grimes Julia, Dallman Julia, Ballas Nurit, and Mandel Gail. 1999. “CoREST: A Functional Corepressor Required for Regulation of Neural-Specific Gene Expression.” Proceedings of the National Academy of Sciences 96 (17): 9873–78. 10.1073/pnas.96.17.9873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arboleda Valerie A, Lee Hane, Dorrani Naghmeh, Zadeh Neda, Willis Mary, Macmurdo Colleen Forsyth, Manning Melanie A, et al. 2015. “De Novo Nonsense Mutations in KAT6A, a Lysine Acetyl-Transferase Gene, Cause a Syndrome Including Microcephaly and Global Developmental Delay.” American Journal of Human Genetics 96 (3): 498–506. 10.1016/j.ajhg.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajpai Ruchi, Chen Denise A., Rada-Iglesias Alvaro, Zhang Junmei, Xiong Yiqin, Helms Jill, Chang Ching Pin, Zhao Yingming, Swigut Tomek, and Wysocka Joanna. 2010. “CHD7 Cooperates with PBAF to Control Multipotent Neural Crest Formation.” Nature. 10.1038/nature08733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balemans MCM, Nadif Kasri N, Kopanitsa MV, Afinowi NO, Ramakers G, Peters TA, Beynon AJ, et al. 2013. “Hippocampal Dysfunction in the Euchromatin Histone Methyltransferase 1 Heterozygous Knockout Mouse Model for Kleefstra Syndrome.” Human Molecular Genetics 22 (5): 852–66. 10.1093/hmg/dds490. [DOI] [PubMed] [Google Scholar]
- Balemans Monique C. M., Ansar Muhammad, Oudakker Astrid R., van Caam Arjan P. M., Bakker Brenda, Vitters Elly L., van der Kraan Peter M., et al. 2014. “Reduced Euchromatin Histone Methyltransferase 1 Causes Developmental Delay, Hypotonia, and Cranial Abnormalities Associated with Increased Bone Gene Expression in Kleefstra Syndrome Mice.” Developmental Biology 386 (2): 395–407. 10.1016/j.ydbio.2013.12.016. [DOI] [PubMed] [Google Scholar]
- Ballas Nurit, Battaglioli Elena, Atouf Fouad, Andres Maria E., Chenoweth Josh, Anderson Mary E., Burger Corinna, et al. 2001. “Regulation of Neuronal Traits by a Novel Transcriptional Complex.” Neuron 31 (3): 353–65. 10.1016/S0896-6273(01)00371-3. [DOI] [PubMed] [Google Scholar]
- Banka S, Howard E, Bunstone S, Chandler KE, Kerr B, Lachlan K, McKee S, et al. 2013. “MLL2 Mosaic Mutations and Intragenic Deletion–Duplications in Patients with Kabuki Syndrome.” Clinical Genetics 83 (5): 467–71. 10.1111/j.1399-0004.2012.01955.x. [DOI] [PubMed] [Google Scholar]
- Banka Siddharth, Sayer Rebecca, Breen Catherine, Barton Stephanie, Pavaine Julija, Sheppard Sarah E, Bedoukian Emma, Skraban Cara, Cuddapah Vishnu A, and Clayton‐Smith Jill. 2019. “Genotype–Phenotype Specificity in Menke–Hennekam Syndrome Caused by Missense Variants in Exon 30 or 31 of CREBBP.” American Journal of Medical Genetics Part A 179 (6): 1058–62. 10.1002/ajmg.a.61131. [DOI] [PubMed] [Google Scholar]
- Banka Siddharth, Veeramachaneni Ratna, Reardon William, Howard Emma, Bunstone Sancha, Ragge Nicola, Parker Michael J., et al. 2012. “How Genetically Heterogeneous Is Kabuki Syndrome?: MLL2 Testing in 116 Patients, Review and Analyses of Mutation and Phenotypic Spectrum.” European Journal of Human Genetics 20 (4): 381–88. 10.1038/ejhg.2011.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister Andrew J, and Kouzarides Tony. 1996. “The CBP Co-Activator Is a Histone Acetyltransferase.” Nature 384 (6610): 641–43. 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- ———. 2011. “Regulation of Chromatin by Histone Modifications.” Cell Research 21 (3): 381–95. 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbagiovanni Giulia, Germain Pierre-Luc, Zech Michael, Atashpaz Sina, Riso Pietro Lo, D’Antonio-Chronowska Agnieszka, Tenderini Erika, et al. 2018. “KMT2B Is Selectively Required for Neuronal Transdifferentiation, and Its Loss Exposes Dystonia Candidate Genes.” Cell Reports 25 (4): 988–1001. 10.1016/j.celrep.2018.09.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrie Elizabeth S., Alfaro Maria P., Pfau Ruthann B., Goff Melanie J., McBride Kim L., Manickam Kandamurugu, and Zmuda Erik J.. 2019. “De Novo Loss-of-Function Variants in NSD2 (WHSC1) Associate with a Subset of Wolf-Hirschhorn Syndrome.” Cold Spring Harbor Molecular Case Studies 5 (4). 10.1101/mcs.a004044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch Oliver, Labonté Janette, Albrecht Beate, Wieczorek Dagmar, Lechno Stanislav, Zechner Ulrich, and Haaf Thomas. 2010. “Two Patients with EP300 Mutations and Facial Dysmorphism Different from the Classic Rubinstein-Taybi Syndrome.” American Journal of Medical Genetics. Part A 152A (1): 181–84. 10.1002/ajmg.a.33153. [DOI] [PubMed] [Google Scholar]
- Baujat Geneviève, and Cormier-Daire Valérie. 2007. “Sotos Syndrome.” Orphanet Journal of Rare Diseases 2 (1): 36. 10.1186/1750-1172-2-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejar RL, Smith GF, Park S, Spellacy WN, Wolfson SL, and Nyhan WL. 1970. “Cerebral Gigantism: Concentrations of Amino Acids in Plasma and Muscle.” The Journal of Pediatrics 76 (1): 105–11. 10.1016/s0022-3476(70)80138-x. [DOI] [PubMed] [Google Scholar]
- Berdasco María, Ropero Santiago, Setien Fernando, Fraga Mario F., Lapunzina Pablo, Losson Régine, Alaminos Miguel, Cheung Nai-Kong, Rahman Nazneen, and Esteller Manel. 2009. “Epigenetic Inactivation of the Sotos Overgrowth Syndrome Gene Histone Methyltransferase NSD1 in Human Neuroblastoma and Glioma.” Proceedings of the National Academy of Sciences of the United States of America 106 (51): 21830–35. 10.1073/pnas.0906831106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornsson Hans T., Benjamin Joel S., Zhang Li, Weissman Jacqueline, Gerber Elizabeth E., Chen Yi-Chun, Vaurio Rebecca G., Potter Michelle C., Hansen Kasper D., and Dietz Harry C.. 2014. “Histone Deacetylase Inhibition Rescues Structural and Functional Brain Deficits in a Mouse Model of Kabuki Syndrome.” Science Translational Medicine 6 (256): 256ra135. 10.1126/scitranslmed.3009278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornsson Hans Tomas. 2015. “The Mendelian Disorders of the Epigenetic Machinery.” Genome Research 25 (10): 1473–81. 10.1101/gr.190629.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black Joshua C., Van Rechem Capucine, and Whetstine Johnathan R.. 2012. “Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact.” Molecular Cell 48 (4): 491–507. 10.1016/j.molcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bögershausen N, and Wollnik B. 2013. “Unmasking Kabuki Syndrome: Unmasking Kabuki Syndrome.” Clinical Genetics 83 (3): 201–11. 10.1111/cge.12051. [DOI] [PubMed] [Google Scholar]
- Bolger Timothy A., and Yao Tso-Pang. 2005. “Intracellular Trafficking of Histone Deacetylase 4 Regulates Neuronal Cell Death.” Journal of Neuroscience 25 (41): 9544–53. 10.1523/JNEUROSCI.1826-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnaud Emilie M., Suberbielle Elsa, and Malnou Cécile E.. 2016. “Histone Acetylation in Neuronal (Dys)Function.” Biomolecular Concepts 7 (2): 103–16. 10.1515/bmc-2016-0002. [DOI] [PubMed] [Google Scholar]
- Boyle MI, Jespersgaard C, Brøndum-Nielsen K, Bisgaard A-M, and Tümer Z. 2014. “Cornelia de Lange Syndrome.” Clinical Genetics 88 (1): 1–12. 10.1111/cge.12499. [DOI] [PubMed] [Google Scholar]
- Bramswig Nuria C., Caluseriu O, Lüdecke HJ, Bolduc FV, Noel NCL, Wieland T, Surowy HM, et al. 2017. “Heterozygosity for ARID2 Loss-of-Function Mutations in Individuals with a Coffin–Siris Syndrome-like Phenotype.” Human Genetics. 10.1007/s00439-017-1757-z. [DOI] [PubMed] [Google Scholar]
- Breeden Linda, and Nasmyth Kim. 1987. “Similarity between Cell-Cycle Genes of Budding Yeast and Fission Yeast and the Notch Gene of Drosophila.” Nature. 10.1038/329651a0. [DOI] [PubMed] [Google Scholar]
- Brinkmeier Michelle L., Geister Krista A., Jones Morgan, Waqas Meriam, Maillard Ivan, and Camper Sally A.. 2015. “The Histone Methyltransferase Gene Absent, Small, or Homeotic Discs-1 Like Is Required for Normal Hox Gene Expression and Fertility in Mice1.” Biology of Reproduction 93 (5). 10.1095/biolreprod.115.131516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes Emily, Laurent Benoit, Õunap Katrin, Carroll Renee, Moeschler John B., Field Michael, Schwartz Charles E., Gecz Jozef, and Shi Yang. 2015. “Mutations in the Intellectual Disability Gene KDM5C Reduce Protein Stability and Demethylase Activity.” Human Molecular Genetics 24 (10): 2861–72. 10.1093/hmg/ddv046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant Laura, Li Dong, Cox Samuel G., Marchione Dylan, Joiner Evan F., Wilson Khadija, Janssen Kevin, et al. 2020. “Histone H3.3 beyond Cancer: Germline Mutations in Histone 3 Family 3A and 3B Cause a Previously Unidentified Neurodegenerative Disorder in 46 Patients.” Science Advances 6 (49). 10.1126/sciadv.abc9207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultman Scott, Gebuhr Tom, Yee Della, La Mantia Christian, Nicholson Jackie, Gilliam Anita, Randazzo Filippo, et al. 2000. “A Brg1 Null Mutation in the Mouse Reveals Functional Differences among Mammalian SWI/SNF Complexes.” Molecular Cell. 10.1016/S1097-2765(00)00127-1. [DOI] [PubMed] [Google Scholar]
- Burgold Thomas, Spreafico Fabio, De Santa Francesca, Totaro Maria Grazia, Prosperini Elena, Natoli Gioacchino, and Testa Giuseppe. 2008. “The Histone H3 Lysine 27-Specific Demethylase Jmjd3 Is Required for Neural Commitment.” PLOS ONE 3 (8): e3034. 10.1371/journal.pone.0003034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkardt Deepika D Cunha, Zachariou Anna, Loveday Chey, Allen Clare L., Amor David J., Ardissone Anna, Banka Siddharth, et al. 2019. “HIST1H1E Heterozygous Protein-Truncating Variants Cause a Recognizable Syndrome with Intellectual Disability and Distinctive Facial Gestalt: A Study to Clarify the HIST1H1E Syndrome Phenotype in 30 Individuals.” American Journal of Medical Genetics. Part A 179 (10): 2049–55. 10.1002/ajmg.a.61321. [DOI] [PubMed] [Google Scholar]
- Burney Matthew J., Johnston Caroline, Wong Kee-Yew, Teng Siaw-Wei, Beglopoulos Vassilios, Stanton Lawrence W., Williams Brenda P., Bithell Angela, and Buckley Noel J.. 2013. “An Epigenetic Signature of Developmental Potential in Neural Stem Cells and Early Neurons.” Stem Cells (Dayton, Ohio) 31 (9): 1868–80. 10.1002/stem.1431. [DOI] [PubMed] [Google Scholar]
- Cao Ru, Wang Liangjun, Wang Hengbin, Xia Li, Erdjument-Bromage Hediye, Tempst Paul, Jones Richard S., and Zhang Yi. 2002. “Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing.” Science (New York, N.Y.) 298 (5595): 1039–43. 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- Capelli Leonardo P., Krepischi Ana C.V., Gurgel-Giannetti Juliana, Mendes Mirian Fabiola S., Rodrigues Tatiane, Varela Monica C., Koiffmann Célia P., and Rosenberg Carla. 2012. “Deletion of the RMGA and CHD2 Genes in a Child with Epilepsy and Mental Deficiency.” European Journal of Medical Genetics. 10.1016/j.ejmg.2011.10.004. [DOI] [PubMed] [Google Scholar]
- Carré Gwenn-Aël, Siggers Pam, Xipolita Marilena, Brindle Paul, Lutz Beat, Wells Sara, and Greenfield Andy. 2018. “Loss of P300 and CBP Disrupts Histone Acetylation at the Mouse Sry Promoter and Causes XY Gonadal Sex Reversal.” Human Molecular Genetics 27 (1): 190–98. 10.1093/hmg/ddx398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvill Gemma L., Heavin Sinéad B., Yendle Simone C., McMahon Jacinta M., O’Roak Brian J., Cook Joseph, Khan Adiba, et al. 2013. “Targeted Resequencing in Epileptic Encephalopathies Identifies de Novo Mutations in CHD2 and SYNGAP1.” Nature Genetics. 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvill Gemma L, and Mefford Heather C. 1993. CHD2-Related Neurodevelopmental Disorders. GeneReviews®.
- Cau Elise and Wilson Stephen W. 2003. “Ash1a and Neurogenin1 Function Downstream of Floating Head to Regulate Epiphysial Neurogenesis.” Development 130 (11): 2455–66. 10.1242/dev.00452. [DOI] [PubMed] [Google Scholar]
- Celen Cemre, Chuang Jen Chieh, Luo Xin, Nijem Nadine, Walker Angela K., Chen Fei, Zhang Shuyuan, et al. 2017. “Arid1b Haploinsufficient Mice Reveal Neuropsychiatric Phenotypes and Reversible Causes of Growth Impairment.” ELife. 10.7554/eLife.25730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadee Deborah N., Allis C. David, Wright Jim A., and Davie James R.. 1997. “Histone H1b Phosphorylation Is Dependent upon Ongoing Transcription and Replication in Normal and Ras-Transformed Mouse Fibroblasts.” Journal of Biological Chemistry 272 (13): 8113–16. 10.1074/jbc.272.13.8113. [DOI] [PubMed] [Google Scholar]
- Chassaing Nicolas, Siani Virginie, Carles Dominique, Delezoide Anne Lise, Alberti Eve Marie, Battin Jacques, Chateil Jean François, et al. 2005. “X-Linked Dominant Chondrodysplasia with Platyspondyly, Distinctive Brachydactyly, Hydrocephaly, and Microphthalmia.” American Journal of Medical Genetics Part A 136A (4): 307–12. 10.1002/ajmg.a.30570. [DOI] [PubMed] [Google Scholar]
- Chen Jiaoyang, Zhang Jing, Liu Aijie, Zhang Liping, Li Hua, Zeng Qi, Yang Zhixian, Yang Xiaoling, Wu Xiru, and Zhang Yuehua. 2020. “CHD2-Related Epilepsy: Novel Mutations and New Phenotypes.” Developmental Medicine and Child Neurology. 10.1111/dmcn.14367. [DOI] [PubMed] [Google Scholar]
- Cheng Jemmie, Blum Roy, Bowman Christopher, Hu Deqing, Shilatifard Ali, Shen Steven, and Dynlacht Brian D.. 2014. “A Role for H3K4 Monomethylation in Gene Repression and Partitioning of Chromatin Readers.” Molecular Cell 53 (6): 979–92. 10.1016/j.molcel.2014.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Young-Wook, Hong Teresa, Hong SunHwa, Guo Hong, Yu Hong, Kim Doyeob, Guszczynski Tad, et al. 2007. “PTIP Associates with MLL3- and MLL4-Containing Histone H3 Lysine 4 Methyltransferase Complex*, ♦, ♦.” Journal of Biological Chemistry 282 (28): 20395–406. 10.1074/jbc.M701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong Jessica X., Yu Joon-Ho, Lorentzen Peter, Park Karen M., Jamal Seema M., Tabor Holly K., Rauch Anita, et al. 2016. “Gene Discovery for Mendelian Conditions via Social Networking: De Novo Variants in KDM1A Cause Developmental Delay and Distinctive Facial Features.” Genetics in Medicine: Official Journal of the American College of Medical Genetics 18 (8): 788–95. 10.1038/gim.2015.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciolfi Andrea, Aref-Eshghi Erfan, Pizzi Simone, Pedace Lucia, Miele Evelina, Kerkhof Jennifer, Flex Elisabetta, et al. 2020. “Frameshift Mutations at the C-Terminus of HIST1H1E Result in a Specific DNA Hypomethylation Signature.” Clinical Epigenetics 12 (1): 7. 10.1186/s13148-019-0804-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton-Smith Jill, O’Sullivan James, Daly Sarah, Bhaskar Sanjeev, Day Ruth, Anderson Beverley, Voss Anne K, et al. 2011. “Whole-Exome-Sequencing Identifies Mutations in Histone Acetyltransferase Gene KAT6B in Individuals with the Say-Barber-Biesecker Variant of Ohdo Syndrome.” American Journal of Human Genetics 89 (5): 675–81. 10.1016/j.ajhg.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffin Grange S., and Siris Evelyn. 1970. “Mental Retardation With Absent Fifth Fingernail and Terminal Phalanx.” American Journal of Diseases of Children. 10.1001/archpedi.1970.02100050435009. [DOI] [PubMed] [Google Scholar]
- Cohen Ana S A, Yap Damian B, Suzanne M E Lewis, Chijiwa Chieko, Ramos-Arroyo Maria A, Tkachenko Natália, Milano Valentina, et al. 2016. “Weaver Syndrome-Associated EZH2 Protein Variants Show Impaired Histone Methyltransferase Function In Vitro.” Human Mutation 37 (3): 301–7. 10.1002/humu.22946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen Ana Sa, and Gibson William T.. 2016. “EED-Associated Overgrowth in a Second Male Patient.” Journal of Human Genetics 61 (9): 831–34. 10.1038/jhg.2016.51. [DOI] [PubMed] [Google Scholar]
- Coleman Rory T., and Struhl Gary. 2017. “Causal Role for Inheritance of H3K27me3 in Maintaining the OFF State of a Drosophila HOX Gene.” Science 356 (6333). 10.1126/science.aai8236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney Erin, Bi Weimin, Schlesinger Alan E., Vinson Sherry, and Potocki Lorraine. 2017. “Novel EED Mutation in Patient with Weaver Syndrome.” American Journal of Medical Genetics. Part A 173 (2): 541–45. 10.1002/ajmg.a.38055. [DOI] [PubMed] [Google Scholar]
- Cormier-Daire Valérie, Chauvet Marie-Liesse, Lyonnet Stanislas, Briard Marie-Louise, Munnich Arnold, and Le Merrer Martine. 2000. “Genitopatellar Syndrome: A New Condition Comprising Absent Patellae, Scrotal Hypoplasia, Renal Anomalies, Facial Dysmorphism, and Mental Retardation.” Journal of Medical Genetics 37 (7): 520–24. 10.1136/jmg.37.7.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crippa Milena, Bestetti Ilaria, Maitz Silvia, Weiss Karin, Spano Alice, Masciadri Maura, Smithson Sarah, et al. 2020. “SETD5 Gene Haploinsufficiency in Three Patients With Suspected KBG Syndrome.” Frontiers in Neurology 11: 631. 10.3389/fneur.2020.00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Molina Sara, Respuela Patricia, Tebartz Christina, Kolovos Petros, Nikolic Milos, Fueyo Raquel, van Ijcken Wilfred F. J., et al. 2017. “PRC2 Facilitates the Regulatory Topology Required for Poised Enhancer Function during Pluripotent Stem Cell Differentiation.” Cell Stem Cell 20 (5): 689–705.e9. 10.1016/j.stem.2017.02.004. [DOI] [PubMed] [Google Scholar]
- Das Chandrima, Lucia M Scott, Hansen Kirk C, and Tyler Jessica K. 2009. “CBP/P300-Mediated Acetylation of Histone H3 on Lysine 56.” Nature 459 (7243): 113–17. 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis Brittany A, François David, O’Regan Ciara, Adam Manal A, Harwood Adrian J, Crunelli Vincenzo, and Isles Anthony R. 2020. “Impairments in Sensory-Motor Gating and Information Processing in a Mouse Model of Ehmt1 Haploinsufficiency.” Brain and Neuroscience Advances 4 (January): 239821282092864. 10.1177/2398212820928647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deardorff Matthew A, Bando Masashige, Nakato Ryuichiro, Watrin Erwan, Itoh Takehiko, Minamino Masashi, Saitoh Katsuya, et al. 2012. “HDAC8 Mutations in Cornelia de Lange Syndrome Affect the Cohesin Acetylation Cycle.” Nature 489 (7415): 313–17. 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deliu Elena, Arecco Niccolò, Morandell Jasmin, Dotter Christoph P., Contreras Ximena, Girardot Charles, Käsper Eva-Lotta, et al. 2018. “Haploinsufficiency of the Intellectual Disability Gene SETD5 Disturbs Developmental Gene Expression and Cognition.” Nature Neuroscience 21 (12): 1717–27. 10.1038/s41593-018-0266-2. [DOI] [PubMed] [Google Scholar]
- Den Kouhei, Kato Mitsuhiro, Yamaguchi Tokito, Miyatake Satoko, Takata Atsushi, Mizuguchi Takeshi, Miyake Noriko, Mitsuhashi Satomi, and Matsumoto Naomichi. 2019. “A Novel de Novo Frameshift Variant in SETD1B Causes Epilepsy.” Journal of Human Genetics 64 (8): 821–27. 10.1038/s10038-019-0617-1. [DOI] [PubMed] [Google Scholar]
- Deng Changwang, Li Ying, Liang Shermi, Cui Kairong, Salz Tal, Yang Hui, Tang Zhanyun, et al. 2013. “USF1 and HSET1A Mediated Epigenetic Modifications Regulate Lineage Differentiation and HoxB4 Transcription.” Edited by Weissman Sherman M.. PLoS Genetics 9 (6): e1003524. 10.1371/journal.pgen.1003524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derar Nada, Al-Hassnan Zuhair N., Al-Owain Mohammed, Monies Dorota, Abouelhoda Mohamed, Meyer Brian F., Moghrabi Nabil, and Alkuraya Fowzan S.. 2019. “De Novo Truncating Variants in WHSC1 Recapitulate the Wolf-Hirschhorn (4p16.3 Microdeletion) Syndrome Phenotype.” Genetics in Medicine: Official Journal of the American College of Medical Genetics 21 (1): 185–88. 10.1038/s41436-018-0014-8. [DOI] [PubMed] [Google Scholar]
- Dhalluin Christophe, Carlson Justin E., Zeng Lei, He Cheng, Aggarwal Aneel K., Zhou Ming-Ming, and Zhou Ming-Ming. 1999. “Structure and Ligand of a Histone Acetyltransferase Bromodomain.” Nature 399 (6735): 491–96. 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- Dhamija Radhika, Breningstall Galen, Lily Wong-Kisiel Michelle Dolan, Hirsch Betsy, and Wirrell Elaine. 2011. “Microdeletion of Chromosome 15q26.1 in a Child with Intractable Generalized Epilepsy.” Pediatric Neurology. 10.1016/j.pediatrneurol.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Dhar SS, Lee S-H, Kan P-Y, Voigt P, Ma L, Shi X, Reinberg D, and Lee MG. 2012. “Trans-Tail Regulation of MLL4-Catalyzed H3K4 Methylation by H4R3 Symmetric Dimethylation Is Mediated by a Tandem PHD of MLL4.” Genes & Development 26 (24): 2749–62. 10.1101/gad.203356.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon Shane C., Zhang Xing, Trievel Raymond C., and Cheng Xiaodong. 2005. “The SET-Domain Protein Superfamily: Protein Lysine Methyltransferases.” Genome Biology 6 (8): 227. 10.1186/gb-2005-6-8-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Yali, Milne Thomas A., Ruthenburg Alexander J., Lee Seunghee, Lee Jae Woon, Verdine Gregory L., Allis C. David, and Roeder Robert G.. 2006. “Regulation of MLL1 H3K4 Methyltransferase Activity by Its Core Components.” Nature Structural & Molecular Biology 13 (8): 713–19. 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- Dou Yali, Milne Thomas A., Tackett Alan J., Smith Edwin R., Fukuda Aya, Wysocka Joanna, Allis C. David, Chait Brian T., Hess Jay L., and Roeder Robert G.. 2005. “Physical Association and Coordinate Function of the H3 K4 Methyltransferase MLL1 and the H4 K16 Acetyltransferase MOF.” Cell 121 (6): 873–85. 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Douglas Jenny, Hanks Sandra, Temple I Karen, Davies Sally, Murray Alexandra, Upadhyaya Meena, Tomkins Susan, Hughes Helen E, Cole R P Trevor, and Rahman Nazneen. 2003. “NSD1 Mutations Are the Major Cause of Sotos Syndrome and Occur in Some Cases of Weaver Syndrome but Are Rare in Other Overgrowth Phenotypes.” The American Journal of Human Genetics 72 (1): 132–43. 10.1086/345647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drivas Theodore G., Li Dong, Nair Divya, Alaimo Joseph T., Alders Mariëlle, Altmüller Janine, Barakat Tahsin Stefan, et al. 2020. “A Second Cohort of CHD3 Patients Expands the Molecular Mechanisms Known to Cause Snijders Blok-Campeau Syndrome.” European Journal of Human Genetics. 10.1038/s41431-020-0654-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffney Lara J, Valdez Purnima, Tremblay Martine W, Cao Xinyu, Montgomery Sarah, McConkie-Rosell Allyn, and Jiang Yong-Hui. 2018. “Epigenetics and Autism Spectrum Disorder: A Report of an Autism Case with Mutation in H1 Linker Histone HIST1H1E and Literature Review.” American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics : The Official Publication of the International Society of Psychiatric Genetics 177 (4): 426–33. 10.1002/ajmg.b.32631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont Cathérine, Armant D. Randall, and Brenner Carol A.. 2009. “Epigenetics: Definition, Mechanisms and Clinical Perspective.” Seminars in Reproductive Medicine 27 (5): 351–57. 10.1055/s-0029-1237423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards MJ, Crawford AE, Jammu V, and Wise G. 1994. “Hypertrichosis ‘Cubiti’ with Facial Asymmetry.” American Journal of Medical Genetics 53 (1): 56–58. 10.1002/ajmg.1320530112. [DOI] [PubMed] [Google Scholar]
- Eising Else, Carrion-Castillo Amaia, Vino Arianna, Strand Edythe A., Jakielski Kathy J., Scerri Thomas S., Hildebrand Michael S., et al. 2019. “A Set of Regulatory Genes Co-Expressed in Embryonic Human Brain Is Implicated in Disrupted Speech Development.” Molecular Psychiatry. 10.1038/s41380-018-0020-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewart-Toland A, Enns GM, Cox VA, Mohan GC, Rosenthal P, and Golabi M. 1998. “Severe Congenital Anomalies Requiring Transplantation in Children with Kabuki Syndrome.” American Journal of Medical Genetics 80 (4): 362–67. [PubMed] [Google Scholar]
- Fang Yu-Lian, Zhang Rui-Ping, Wang Yi-Zheng, Cao Li-Rong, Zhang Yu-Qin, and Cai Chun-Quan. 2019. “A Novel Mutation in a Common Pathogenic Gene (SETD5) Associated with Intellectual Disability: A Case Report.” Experimental and Therapeutic Medicine 18 (5): 3737–40. 10.3892/etm.2019.8059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faundes Víctor, Newman William G., Bernardini Laura, Canham Natalie, Clayton-Smith Jill, Dallapiccola Bruno, Davies Sally J., et al. 2018. “Histone Lysine Methylases and Demethylases in the Landscape of Human Developmental Disorders.” American Journal of Human Genetics 102 (1): 175–87. 10.1016/j.ajhg.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Weijun, Kawauchi Daisuke, Körkel-Qu Huiqin, Deng Huan, Serger Elisabeth, Sieber Laura, Lieberman Jenna Ariel, et al. 2017. “Chd7 Is Indispensable for Mammalian Brain Development through Activation of a Neuronal Differentiation Programme.” Nature Communications. 10.1038/ncomms14758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Weijun, Yonezawa Masato, Ye Jing, Jenuwein Thomas, and Grummt Ingrid. 2010. “PHF8 Activates Transcription of RRNA Genes through H3K4me3 Binding and H3K9me1/2 Demethylation.” Nature Structural & Molecular Biology 17 (4): 445–50. 10.1038/nsmb.1778. [DOI] [PubMed] [Google Scholar]
- Fernandes Isabella R, Cruz Ana C P, Ferrasa Adriano, Phan Dylan, Herai Roberto H, and Muotri Alysson R. 2018. “Genetic Variations on SETD5 Underlying Autistic Conditions.” Developmental Neurobiology 78 (5): 500–518. 10.1002/dneu.22584. [DOI] [PubMed] [Google Scholar]
- Filatova Alina, Rey Linda K., Lechler Marion B., Schaper Jörg, Hempel Maja, Posmyk Renata, Szczaluba Krzysztof, Santen Gijs W.E., Wieczorek Dagmar, and Nuber Ulrike A.. 2019. “Mutations in SMARCB1 and in Other Coffin–Siris Syndrome Genes Lead to Various Brain Midline Defects.” Nature Communications. 10.1038/s41467-019-10849-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FitzGerald KT, and Diaz MO. 1999. “MLL2: A New Mammalian Member of the Trx/MLL Family of Genes.” Genomics 59 (2): 187–92. 10.1006/geno.1999.5860. [DOI] [PubMed] [Google Scholar]
- Fitzgerald TW, Gerety SS, Jones WD, Van Kogelenberg M, King DA, McRae J, Morley KI, et al. 2015. “Large-Scale Discovery of Novel Genetic Causes of Developmental Disorders.” Nature. 10.1038/nature14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan John F., Mi Li Zhi, Chruszcz Maksymilian, Cymborowski Marcin, Clines Katrina L., Kim Youngchang, Minor Wladek, Rastinejad Fraydoon, and Khorasanizadeh Sepideh. 2005. “Double Chromodomains Cooperate to Recognize the Methylated Histone H3 Tail.” Nature. 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- Flannery David B., Fink Stacey M., Francis Gary, and Gilman Priscilla Ann. 1989. “Hypertrichosis Cubiti.” American Journal of Medical Genetics 32 (4): 482–83. 10.1002/ajmg.1320320410. [DOI] [PubMed] [Google Scholar]
- Flex Elisabetta, Martinelli Simone, Van Dijck Anke, Ciolfi Andrea, Cecchetti Serena, Coluzzi Elisa, Pannone Luca, et al. 2019. “Aberrant Function of the C-Terminal Tail of HIST1H1E Accelerates Cellular Senescence and Causes Premature Aging.” American Journal of Human Genetics 105 (3): 493–508. 10.1016/j.ajhg.2019.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragola Giulia, Germain Pierre-Luc, Laise Pasquale, Cuomo Alessandro, Blasimme Alessandro, Gross Fridolin, Signaroldi Elena, et al. 2013. “Cell Reprogramming Requires Silencing of a Core Subset of Polycomb Targets.” PLOS Genetics 9 (2): e1003292. 10.1371/journal.pgen.1003292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frega Monica, Linda Katrin, Keller Jason M, Gümüş-Akay Güvem, Mossink Britt, van Rhijn Jon-Ruben, Negwer Moritz, et al. 2019. “Neuronal Network Dysfunction in a Model for Kleefstra Syndrome Mediated by Enhanced NMDAR Signaling.” Nature Communications 10 (1): 4928. 10.1038/s41467-019-12947-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryssira H, Drossatou P, Sklavou R, Barambouti F, and Manolaki N. 2010. “Two Cases of Sotos Syndrome with Novel Mutations of the NSD1 Gene.” Genetic Counseling (Geneva, Switzerland) 21 (1): 53–59. [PubMed] [Google Scholar]
- Gibson William T, Hood Rebecca L, Zhan Shing Hei, Bulman Dennis E, Fejes Anthony P, Moore Richard, Mungall Andrew J, et al. 2011. “Mutations in EZH2 Cause Weaver Syndrome.” American Journal of Human Genetics 90 (1): 110–18. 10.1016/j.ajhg.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinton Kevin E., Hurst Anna C. E., Bowling Kevin M., Cristian Ingrid, Haynes Devon, Adstamongkonkul Dusit, Schnappauf Oskar, et al. 2021. “Phenotypic Expansion of the BPTF-Related Neurodevelopmental Disorder with Dysmorphic Facies and Distal Limb Anomalies.” American Journal of Medical Genetics. Part A, January. 10.1002/ajmg.a.62102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Fade, and Miller Kyle M. 2013. “Mammalian DNA Repair: HATs and HDACs Make Their Mark through Histone Acetylation.” Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 750 (1–2): 23–30. 10.1016/j.mrfmmm.2013.07.002. [DOI] [PubMed] [Google Scholar]
- Goo Young-Hwa, Sohn Young Chang, Kim Dae-Hwan, Kim Seung-Whan, Kang Min-Jung, Jung Dong-Ju, Kwak Eunyee, et al. 2003. “Activating Signal Cointegrator 2 Belongs to a Novel Steady-State Complex That Contains a Subset of Trithorax Group Proteins.” Molecular and Cellular Biology 23 (1): 140–49. 10.1128/MCB.23.1.140-149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman Jared V., Yamada Tomoko, Yang Yue, Kong Lingchun, Wu Dennis Y., Zhao Guoyan, Gabel Harrison W., and Bonni Azad. 2020. “The Chromatin Remodeling Enzyme Chd4 Regulates Genome Architecture in the Mouse Brain.” Nature Communications. 10.1038/s41467-020-17065-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grafodatskaya Daria, Chung Barian HY, Butcher Darci T., Turinsky Andrei L., Goodman Sarah J., Choufani Sana, Chen Yi-An, et al. 2013. “Multilocus Loss of DNA Methylation in Individuals with Mutations in the Histone H3 Lysine 4 Demethylase KDM5C.” BMC Medical Genomics 6 (1): 1. 10.1186/1755-8794-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green Claire, Willoughby Joshua, DDD Study, and Balasubramanian Meena. 2017. “De Novo SETD5 Loss-of-Function Variant as a Cause for Intellectual Disability in a 10-Year Old Boy with an Aberrant Blind Ending Bronchus.” American Journal of Medical Genetics. Part A 173 (12): 3165–71. 10.1002/ajmg.a.38461. [DOI] [PubMed] [Google Scholar]
- Grozeva Detelina, Carss Keren, Spasic-Boskovic Olivera, Parker Michael J., Archer Hayley, Firth Helen V., Park Soo-Mi, et al. 2014. “De Novo Loss-of-Function Mutations in SETD5, Encoding a Methyltransferase in a 3p25 Microdeletion Syndrome Critical Region, Cause Intellectual Disability.” American Journal of Human Genetics 94 (4): 618–24. 10.1016/j.ajhg.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman-Ayala Marcela, Sachs Michael, Koh Fong Ming, Onodera Courtney, Bulut-Karslioglu Aydan, Lin Chih Jen, Wong Priscilla, Nitta Rachel, Song Jun S., and Ramalho-Santos Miguel. 2015. “Chd1 Is Essential for the High Transcriptional Output and Rapid Growth of the Mouse Epiblast.” Development (Cambridge). 10.1242/dev.114843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Mokalled MH, Montgomery RL, and Olson EN. 2009. “Epigenetic Control of Skull Morphogenesis by Histone Deacetylase 8.” Genes & Development 23 (14): 1625–30. 10.1101/gad.1809209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland Michael, Montgomery Rusty L., and Olson Eric N.. 2009. “The Many Roles of Histone Deacetylases in Development and Physiology: Implications for Disease and Therapy.” Nature Reviews Genetics 10 (1): 32–42. 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Haelst Mieke M., Hoogeboom Jeannette J. M., Baujat Genevieve, Brüggenwirth Hennie T., Van de Laar Ingrid, Coleman Kim, Rahman Nazneen, Niermeijer Martinus F., Drop Sten L. S., and Scambler Peter J.. 2005. “Familial Gigantism Caused by an NSD1 Mutation.” American Journal of Medical Genetics. Part A 139 (1): 40–44. 10.1002/ajmg.a.30973. [DOI] [PubMed] [Google Scholar]
- Hall Bryan D. 1979. “Choanal Atresia and Associated Multiple Anomalies.” The Journal of Pediatrics. 10.1016/S0022-3476(79)80513-2. [DOI] [PubMed] [Google Scholar]
- Hannibal Mark C., Buckingham Kati J., Ng Sarah B., Ming Jeffrey E., Beck Anita E., McMillin Margaret J., Gildersleeve Heidi I., et al. 2011. “Spectrum of MLL2 (ALR) Mutations in 110 Cases of Kabuki Syndrome.” American Journal of Medical Genetics Part A 155 (7): 1511–16. 10.1002/ajmg.a.34074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann Hans-Rudolf, Kunze J, Grosse FR, and Dibbern H. 1989. Atlas of Clinical Syndromes: A Visual Aid to Diagnosis for Clinicians and Practicing Physicians. A Visual Aid to Diagnosis for Clinicians and Practicing Physicians. 2nd ed. London: Wolfe Publishing Ltd. [Google Scholar]
- Harada Naoki, Visser Remco, Dawson Angie, Fukamachi Makoto, Iwakoshi Mie, Okamoto Nobuhiko, Kishino Tatsuya, Niikawa Norio, and Matsumoto Naomichi. 2004. “A 1-Mb Critical Region in Six Patients with 9q34.3 Terminal Deletion Syndrome.” Journal of Human Genetics 49 (8): 440–44. 10.1007/s10038-004-0166-z. [DOI] [PubMed] [Google Scholar]
- Harrison SJ, Nishinakamura R, Jones KR, and Monaghan AP. 2012. “Sall1 Regulates Cortical Neurogenesis and Laminar Fate Specification in Mice: Implications for Neural Abnormalities in Townes-Brocks Syndrome.” Disease Models & Mechanisms 5 (3): 351–65. 10.1242/dmm.002873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Runsheng, and Kidder Benjamin L.. 2017. “H3K4 Demethylase KDM5B Regulates Global Dynamics of Transcription Elongation and Alternative Splicing in Embryonic Stem Cells.” Nucleic Acids Research 45 (11): 6427–41. 10.1093/nar/gkx251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman Nathaniel D., Stuart Rhona K., Hon Gary, Fu Yutao, Ching Christina W., Hawkins R. David, Barrera Leah O., et al. 2007. “Distinct and Predictive Chromatin Signatures of Transcriptional Promoters and Enhancers in the Human Genome.” Nature Genetics 39 (3): 311–18. 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Herz Hans-Martin, Mohan Man, Garruss Alexander S., Liang Kaiwei, Takahashi Yoh-hei, Mickey Kristen, Voets Olaf, Verrijzer C. Peter, and Shilatifard Ali. 2012. “Enhancer-Associated H3K4 Monomethylation by Trithorax-Related, the Drosophila Homolog of Mammalian Mll3/Mll4.” Genes & Development 26 (23): 2604–20. 10.1101/gad.201327.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyne Henrike O., Singh Tarjinder, Stamberger Hannah, Jamra Rami Abou, Caglayan Hande, Craiu Dana, De Jonghe Peter, et al. 2018. “De Novo Variants in Neurodevelopmental Disorders with Epilepsy.” Nature Genetics. 10.1038/s41588-018-0143-7. [DOI] [PubMed] [Google Scholar]
- Hiraide Takuya, Hattori Ayako, Ieda Daisuke, Hori Ikumi, Saitoh Shinji, Nakashima Mitsuko, and Saitsu Hirotomo. 2019. “De Novo Variants in SETD1B Cause Intellectual Disability, Autism Spectrum Disorder, and Epilepsy with Myoclonic Absences.” Epilepsia Open 4 (3): 476–81. 10.1002/epi4.12339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraide Takuya, Nakashima Mitsuko, Yamoto Kaori, Fukuda Tokiko, Kato Mitsuhiro, Ikeda Hiroko, Sugie Yoko, et al. 2018. “De Novo Variants in SETD1B Are Associated with Intellectual Disability, Epilepsy and Autism.” Human Genetics 137 (1): 95–104. 10.1007/s00439-017-1863-y. [DOI] [PubMed] [Google Scholar]
- “Histone Acetyltransferase Inhibitors and Preclinical Studies: Expert Opinion on Therapeutic Patents: Vol 19, No 6.” n.d. Accessed March 8, 2021. https://www.tandfonline.com/doi/full/10.1517/13543770902895727. [DOI] [PubMed]
- Hittner HM, Hirsch NJ, Kreh GM, and Rudolph AJ. 1979. “Colobomatous Microphthalmia, Heart Disease, Hearing Loss, and Mental Retardation. A Syndrome.” Journal of Pediatric Ophthalmology and Strabismus. 10.3928/0191-3913-19790301-10. [DOI] [PubMed] [Google Scholar]
- Hnisz Denes, Abraham Brian J., Lee Tong Ihn, Lau Ashley, Saint-André Violaine, Sigova Alla A., Hoke Heather A., and Young Richard A.. 2013. “XSuper-Enhancers in the Control of Cell Identity and Disease.” Cell. 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann Anke, and Spengler Dietmar. 2019. “Chromatin Remodeling Complex NuRD in Neurodevelopment and Neurodevelopmental Disorders.” Frontiers in Genetics. 10.3389/fgene.2019.00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes William F., Braastad Corey D., Mitra Partha, Hampe Cornelia, Doenecke Detlef, Albig Werner, Stein Janet L., van Wijnen Andre J., and Stein Gary S.. 2005. “Coordinate Control and Selective Expression of the Full Complement of Replication-Dependent Histone H4 Genes in Normal and Cancer Cells.” Journal of Biological Chemistry 280 (45): 37400–407. 10.1074/jbc.M506995200. [DOI] [PubMed] [Google Scholar]
- Hong SunHwa, Cho Young-Wook, Yu Li-Rong, Yu Hong, Veenstra Timothy D., and Ge Kai. 2007. “Identification of JmjC Domain-Containing UTX and JMJD3 as Histone H3 Lysine 27 Demethylases.” Proceedings of the National Academy of Sciences 104 (47): 18439–44. 10.1073/pnas.0707292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook EB, and Reynolds JW. 1967. “Cerebral Gigantism: Endocrinological and Clinical Observations of Six Patients Including a Congenital Giant, Concordant Monozygotic Twins, and a Child Who Acheived Adult Gigantic Size.” The Journal of Pediatrics 70 (6): 900–914. 10.1016/s0022-3476(67)80263-4. [DOI] [PubMed] [Google Scholar]
- Horton John R., Upadhyay Anup K., Qi Hank H., Zhang Xing, Shi Yang, and Cheng Xiaodong. 2010. “Enzymatic and Structural Insights for Substrate Specificity of a Family of Jumonji Histone Lysine Demethylases.” Nature Structural & Molecular Biology 17 (1): 38–43. 10.1038/nsmb.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Houdt Jeroen K.J., Nowakowska Beata Anna, Sousa Sérgio B., Van Schaik Barbera D.C., Seuntjens Eve, Avonce Nelson, Sifrim Alejandro, et al. 2012. “Heterozygous Missense Mutations in SMARCA2 Cause Nicolaides-Baraitser Syndrome.” Nature Genetics. 10.1038/ng.1105. [DOI] [PubMed] [Google Scholar]
- Hu Deqing, Gao Xin, Morgan Marc A., Herz Hans-Martin, Smith Edwin R., and Shilatifard Ali. 2013. “The MLL3/MLL4 Branches of the COMPASS Family Function as Major Histone H3K4 Monomethylases at Enhancers.” Molecular and Cellular Biology 33 (23): 4745–54. 10.1128/MCB.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Ming, Sun Xiao-Jian, Zhang Yuan-Liang, Kuang Ying, Hu Chao-Quan, Wu Wei-Li, Shen Shu-Hong, et al. 2010. “Histone H3 Lysine 36 Methyltransferase Hypb/Setd2 Is Required for Embryonic Vascular Remodeling.” Proceedings of the National Academy of Sciences 107 (7): 2956–61. 10.1073/pnas.0915033107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilyina H, Lurie I, Naumtchik I, Amoashy D, Stephanenko G, Fedotov V, and Kostjuk A. 1995. “Kabuki Make-up (Niikawa-Kuroki) Syndrome in the Byelorussian Register of Congenital Malformations: Ten New Observations.” American Journal of Medical Genetics 56 (2): 127–31. 10.1002/ajmg.1320560202. [DOI] [PubMed] [Google Scholar]
- Imagawa Eri, Albuquerque Edoarda V. A., Isidor Bertrand, Mitsuhashi Satomi, Mizuguchi Takeshi, Miyatake Satoko, Takata Atsushi, et al. 2018. “Novel SUZ12 Mutations in Weaver-like Syndrome.” Clinical Genetics 94 (5): 461–66. 10.1111/cge.13415. [DOI] [PubMed] [Google Scholar]
- Imagawa Eri, Higashimoto Ken, Sakai Yasunari, Numakura Chikahiko, Okamoto Nobuhiko, Matsunaga Satoko, Ryo Akihide, et al. 2017. “Mutations in Genes Encoding Polycomb Repressive Complex 2 Subunits Cause Weaver Syndrome.” Human Mutation 38 (6): 637–48. 10.1002/humu.23200. [DOI] [PubMed] [Google Scholar]
- Imhof Axel, and Wolffe Alan P. 1998. “Transcription: Gene Control by Targeted Histone Acetylation.” Current Biology 8 (12): R422–24. 10.1016/s0960-9822(98)70268-4. [DOI] [PubMed] [Google Scholar]
- Iwakoshi Mie, Okamoto Nobuhiko, Harada Naoki, Nakamura Tsuyoshi, Yamamori Shunji, Fujita Hiroko, Niikawa Norio, and Matsumoto Naomichi. 2004. “9q34.3 Deletion Syndrome in Three Unrelated Children.” American Journal of Medical Genetics 126A (3): 278–83. 10.1002/ajmg.a.20602. [DOI] [PubMed] [Google Scholar]
- Iwase Shigeki, Brookes Emily, Agarwal Saurabh, Badeaux Aimee I., Ito Hikaru, Vallianatos Christina N., Tomassy Giulio Srubek, et al. 2016. “A Mouse Model of X-Linked Intellectual Disability Associated with Impaired Removal of Histone Methylation.” Cell Reports 14 (5): 1000–1009. 10.1016/j.celrep.2015.12.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase Shigeki, Lan Fei, Bayliss Peter, de la Torre-Ubieta Luis, Huarte Maite, Qi Hank H., Whetstine Johnathan R., Bonni Azad, Roberts Thomas M., and Shi Yang. 2007. “The X-Linked Mental Retardation Gene SMCX/JARID1C Defines a Family of Histone H3 Lysine 4 Demethylases.” Cell 128 (6): 1077–88. 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Jakovcevski Mira, Ruan Hongyu, Shen Erica Y., Dincer Aslihan, Javidfar Behnam, Ma Qi, Peter Cyril J., et al. 2015. “Neuronal Kmt2a/Mll1 Histone Methyltransferase Is Essential for Prefrontal Synaptic Plasticity and Working Memory.” Journal of Neuroscience 35 (13): 5097–5108. 10.1523/JNEUROSCI.3004-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen Lars Riff, Amende Marion, Gurok Ulf, Moser Bettina, Gimmel Verena, Tzschach Andreas, Janecke Andreas R., et al. 2005. “Mutations in the JARID1C Gene, Which Is Involved in Transcriptional Regulation and Chromatin Remodeling, Cause X-Linked Mental Retardation.” American Journal of Human Genetics 76 (2): 227–36. 10.1086/427563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein Thomas. 2006. “The Epigenetic Magic of Histone Lysine Methylation.: Delivered on 6 July 2005 at the 30th FEBS Congress in Budapest, Hungary.” FEBS Journal 273 (14): 3121–35. 10.1111/j.1742-4658.2006.05343.x. [DOI] [PubMed] [Google Scholar]
- Jiang Yanrui, Sun Huizhen, Lin Qingmin, Wang Zengge, Wang Guanghai, Wang Jian, Jiang Fan, and Yao Ruen. 2019. “De Novo Truncating Variant in NSD2gene Leading to Atypical Wolf-Hirschhorn Syndrome Phenotype.” BMC Medical Genetics 20 (1): 134. 10.1186/s12881-019-0863-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinxiu Liu, Shuimei Liang, Ming Xue, Jonathan Liu Cs, Xiangju Liu, and Wenyuan Duan. 2020. “Wiedemann-Steiner Syndrome with a de Novo Mutation in KMT2A: A Case Report.” Medicine 99 (16): e19813. 10.1097/MD.0000000000019813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones Kieran M., Saric Nemanja, Russell John P., Andoniadou Cynthia L., Scambler Peter J., and Basson M. Albert. 2015. “CHD7 Maintains Neural Stem Cell Quiescence and Prevents Premature Stem Cell Depletion in the Adult Hippocampus.” Stem Cells. 10.1002/stem.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones Michael H., Hamana Naeko, and Shimane Miyuki. 2000. “Identification and Characterization of BPTF, a Novel Bromodomain Transcription Factor.” Genomics 63 (1): 35–39. 10.1006/geno.1999.6070. [DOI] [PubMed] [Google Scholar]
- Jones Wendy D., Dafou Dimitra, McEntagart Meriel, Woollard Wesley J., Elmslie Frances V., Holder-Espinasse Muriel, Irving Melita, et al. 2012. “De Novo Mutations in MLL Cause Wiedemann-Steiner Syndrome.” American Journal of Human Genetics 91 (2): 358–64. 10.1016/j.ajhg.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser Frank J, Ansari Morad, Braunholz Diana, Concepción Gil-Rodríguez María, Decroos Christophe, Wilde Jonathan J, Fincher Christopher T, et al. 2014. “Loss-of-Function HDAC8 Mutations Cause a Phenotypic Spectrum of Cornelia de Lange Syndrome-like Features, Ocular Hypertelorism, Large Fontanelle and X-Linked Inheritance.” Human Molecular Genetics 23 (11): 2888–2900. 10.1093/hmg/ddu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminsky Erin B., Kaul Vineith, Paschall Justin, Church Deanna M., Bunke Brian, Kunig Dawn, Moreno-De-Luca Daniel, et al. 2011. “An Evidence-Based Approach to Establish the Functional and Clinical Significance of Copy Number Variants in Intellectual and Developmental Disabilities.” Genetics in Medicine 13 (9): 777–84. 10.1097/GIM.0b013e31822c79f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawame H, Hannibal MC, Hudgins L, and Pagon RA. 1999. “Phenotypic Spectrum and Management Issues in Kabuki Syndrome.” The Journal of Pediatrics 134 (4): 480–85. 10.1016/s0022-3476(99)70207-6. [DOI] [PubMed] [Google Scholar]
- Kawarai Toshitaka, Miyamoto Ryosuke, Nakagawa Eiji, Koichihara Reiko, Sakamoto Takashi, Mure Hideo, Morigaki Ryoma, et al. 2018. “Phenotype Variability and Allelic Heterogeneity in KMT2B-Associated Disease.” Parkinsonism & Related Disorders 52 (July): 55–61. 10.1016/j.parkreldis.2018.03.022. [DOI] [PubMed] [Google Scholar]
- Kennedy Joanna, Goudie David, Blair Edward, Chandler Kate, Joss Shelagh, McKay Victoria, Green Andrew, et al. 2018. “KAT6A Syndrome: Genotype-Phenotype Correlation in 76 Patients with Pathogenic KAT6A Variants.” Genetics in Medicine : Official Journal of the American College of Medical Genetics 21 (4): 850–60. 10.1038/s41436-018-0259-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Joong K., Huh Sung-Oh, Choi Heonsik, Lee Kee-Sook, Shin Dongho, Lee Changjin, Nam Ju-Suk, et al. 2001. “Srg3, a Mouse Homolog of Yeast SWI3, Is Essential for Early Embryogenesis and Involved in Brain Development.” Molecular and Cellular Biology. 10.1128/mcb.21.22.7787-7795.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Mi-Sung, Akhtar M. Waseem, Adachi Megumi, Mahgoub Melissa, Bassel-Duby Rhonda, Kavalali Ege T., Olson Eric N., and Monteggia Lisa M.. 2012. “An Essential Role for Histone Deacetylase 4 in Synaptic Plasticity and Memory Formation.” Journal of Neuroscience 32 (32): 10879–86. 10.1523/JNEUROSCI.2089-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TaeSoo, and Buratowski Stephen. 2009. “Dimethylation of H3K4 by Set1 Recruits the Set3 Histone Deacetylase Complex to 5′ Transcribed Regions.” Cell 137 (2): 259–72. 10.1016/j.cell.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Young J., Khoshkhoo Sattar, Frankowski Jan C., Zhu Bingyao, Abbasi Saad, Lee Sunyoung, Wu Ye Emily, and Hunt Robert F.. 2018. “Chd2 Is Necessary for Neural Circuit Development and Long-Term Memory.” Neuron. 10.1016/j.neuron.2018.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleefstra T, van Zelst-Stams WA, Nillesen WM, Cormier-Daire V, Houge G, Foulds N, van Dooren M, et al. 2009. “Further Clinical and Molecular Delineation of the 9q Subtelomeric Deletion Syndrome Supports a Major Contribution of EHMT1 Haploinsufficiency to the Core Phenotype.” Journal of Medical Genetics 46 (9): 598–606. 10.1136/jmg.2008.062950. [DOI] [PubMed] [Google Scholar]
- Kleefstra Tjitske, Brunner Han G., Amiel Jeanne, Oudakker Astrid R., Nillesen Willy M., Magee Alex, Geneviève David, et al. 2006. “Loss-of-Function Mutations in Euchromatin Histone Methyl Transferase 1 (EHMT1) Cause the 9q34 Subtelomeric Deletion Syndrome.” The American Journal of Human Genetics 79 (2): 370–77. 10.1086/505693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleefstra Tjitske, Kramer Jamie M., Neveling Kornelia, Willemsen Marjolein H., Koemans Tom S., Vissers Lisenka E. L. M., Wissink-Lindhout Willemijn, et al. 2012. “Disruption of an EHMT1-Associated Chromatin-Modification Module Causes Intellectual Disability.” American Journal of Human Genetics 91 (1): 73–82. 10.1016/j.ajhg.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleefstra Tjitske, Schenck Annette, Kramer Jamie M., and van Bokhoven Hans. 2014. “The Genetics of Cognitive Epigenetics.” Neuropharmacology, Neuroepigenetic Disorders, 80 (May): 83–94. 10.1016/j.neuropharm.2013.12.025. [DOI] [PubMed] [Google Scholar]
- Ko Ara, Youn Song Ee, Kim Se Hee, Lee Joon Soo, Kim Sangwoo, Choi Jong Rak, Kim Heung Dong, Lee Seung Tae, and Kang Hoon Chul. 2018. “Targeted Gene Panel and Genotype-Phenotype Correlation in Children with Developmental and Epileptic Encephalopathy.” Epilepsy Research. 10.1016/j.eplepsyres.2018.02.003. [DOI] [PubMed] [Google Scholar]
- Kobayashi O, and Sakuragawa N. 1996. “Inheritance in Kabuki Make-up (Niikawa-Kuroki) Syndrome.” American Journal of Medical Genetics 61 (1): 92–93. 10.1002/ajmg.1320610105. [DOI] [PubMed] [Google Scholar]
- Koç A, Karaer K, Ergün MA, Cinaz P, and Perçin EF. 2007. “A New Case of Hairy Elbows Syndrome (Hypertrichosis Cubiti).” Genetic Counseling (Geneva, Switzerland) 18 (3): 325–30. [PubMed] [Google Scholar]
- Kochinke Korinna, Zweier Christiane, Nijhof Bonnie, Fenckova Michaela, Cizek Pavel, Honti Frank, Keerthikumar Shivakumar, et al. 2016. “Systematic Phenomics Analysis Deconvolutes Genes Mutated in Intellectual Disability into Biologically Coherent Modules.” The American Journal of Human Genetics 98 (1): 149–64. 10.1016/j.ajhg.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koemans Tom S., Kleefstra Tjitske, Chubak Melissa C., Stone Max H., Reijnders Margot R. F., de Munnik Sonja, Willemsen Marjolein H., et al. 2017. “Functional Convergence of Histone Methyltransferases EHMT1 and KMT2C Involved in Intellectual Disability and Autism Spectrum Disorder.” PLOS Genetics 13 (10): e1006864. 10.1371/journal.pgen.1006864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig Rainer, Meinecke Peter, Kuechler Alma, Schäfer Dieter, and Müller Dietmar. 2010. “Wiedemann-Steiner Syndrome: Three Further Cases.” American Journal of Medical Genetics Part A 152A (9): 2372–75. 10.1002/ajmg.a.33587. [DOI] [PubMed] [Google Scholar]
- Koivisto AM, Ala-Mello S, Lemmelä S, Komu HA, Rautio J, and Järvelä I. 2007. “Screening of Mutations in the PHF8 Gene and Identification of a Novel Mutation in a Finnish Family with XLMR and Cleft Lip/Cleft Palate.” Clinical Genetics 72 (2): 145–49. 10.1111/j.1399-0004.2007.00836.x. [DOI] [PubMed] [Google Scholar]
- Kong Yawei, Grimaldi Michael, Curtin Eugene, Dougherty Max, Kaufman Charles, White Richard M, Zon Leonard I, and Liao Eric C. 2014. “Neural Crest Development and Craniofacial Morphogenesis Is Coordinated by Nitric Oxide and Histone Acetylation.” Chemistry & Biology 21 (4): 488–501. 10.1016/j.chembiol.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolen David A., Kramer Jamie M., Neveling Kornelia, Nillesen Willy M., Moore-Barton Heather L., Elmslie Frances V., Toutain Annick, et al. 2012. “Mutations in the Chromatin Modifier Gene KANSL1 Cause the 17q21.31 Microdeletion Syndrome.” Nature Genetics 44 (6): 639–41. 10.1038/ng.2262. [DOI] [PubMed] [Google Scholar]
- Koolen David A., Pfundt Rolph, Linda Katrin, Beunders Gea, Veenstra-Knol Hermine E., Conta Jessie H., Fortuna Ana Maria, et al. 2016. “The Koolen-de Vries Syndrome: A Phenotypic Comparison of Patients with a 17q21.31 Microdeletion versus a KANSL1 Sequence Variant.” European Journal of Human Genetics: EJHG 24 (5): 652–59. 10.1038/ejhg.2015.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzus Edward. 2017. “Advances in Experimental Medicine and Biology.” Advances in Experimental Medicine and Biology 978: 39–62. 10.1007/978-3-319-53889-1_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzus Edward, Rosenfeld Michael G., and Mayford Mark. 2004. “CBP Histone Acetyltransferase Activity Is a Critical Component of Memory Consolidation.” Neuron 42 (6): 961–72. 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosho Tomoki, Okamoto Nobuhiko, Imai Yoko, Ohashi Hirofumi, van Eerde Albertien M., Chrzanowska Krystyna, Clayton-Smith Jill, et al. 2014. “Genotype-Phenotype Correlation of Coffin-Siris Syndrome Caused by Mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A.” American Journal of Medical Genetics, Part C: Seminars in Medical Genetics. 10.1002/ajmg.c.31407. [DOI] [PubMed] [Google Scholar]
- Krumm Niklas, Turner Tychele N, Baker Carl, Vives Laura, Mohajeri Kiana, Witherspoon Kali, Raja Archana, et al. 2015. “Excess of Rare, Inherited Truncating Mutations in Autism.” Nature Genetics 47 (6): 582–88. 10.1038/ng.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzyzewska IM, Maas SM, Henneman P, Lip K. v. d., Venema A, Baranano K, Chassevent A, et al. 2019. “A Genome-Wide DNA Methylation Signature for SETD1B-Related Syndrome.” Clinical Epigenetics 11 (1): 156. 10.1186/s13148-019-0749-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuechler Alma, Zink Alexander M., Wieland Thomas, Lüdecke Hermann-Josef, Cremer Kirsten, Salviati Leonardo, Magini Pamela, et al. 2015. “Loss-of-Function Variants of SETD5 Cause Intellectual Disability and the Core Phenotype of Microdeletion 3p25.3 Syndrome.” European Journal of Human Genetics: EJHG 23 (6): 753–60. 10.1038/ejhg.2014.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummeling Joost, Stremmelaar Diante E., Raun Nicholas, Reijnders Margot R. F., Willemsen Marjolein H., Ruiterkamp-Versteeg Martina, Schepens Marga, et al. 2020. “Characterization of SETD1A Haploinsufficiency in Humans and Drosophila Defines a Novel Neurodevelopmental Syndrome.” Molecular Psychiatry, April. 10.1038/s41380-020-0725-5. [DOI] [PubMed] [Google Scholar]
- Kuo M-H, Zhou J, Jambeck P, Churchill MEA, and Allis CD. 1998. “Histone Acetyltransferase Activity of Yeast Gcn5p Is Required for the Activation of Target Genes in Vivo.” Genes & Development 12 (5): 627–39. 10.1101/gad.12.5.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurotaki Naohiro, Imaizumi Kiyoshi, Harada Naoki, Masuno Mitsuo, Kondoh Tatsuro, Nagai Toshiro, Ohashi Hirofumi, et al. 2002. “Haploinsufficiency of NSD1 Causes Sotos Syndrome.” Nature Genetics 30 (4): 365–66. 10.1038/ng863. [DOI] [PubMed] [Google Scholar]
- Lan Fei, Bayliss Peter E., Rinn John L., Whetstine Johnathan R., Wang Jordon K., Chen Shuzhen, Iwase Shigeki, et al. 2007. “A Histone H3 Lysine 27 Demethylase Regulates Animal Posterior Development.” Nature 449 (7163): 689–94. 10.1038/nature06192. [DOI] [PubMed] [Google Scholar]
- Laumonnier F, Holbert S, Ronce N, Faravelli F, Lenzner S, Schwartz CE, Lespinasse J, et al. 2005. “Mutations in PHF8 Are Associated with X Linked Mental Retardation and Cleft Lip/Cleft Palate.” Journal of Medical Genetics 42 (10): 780–86. 10.1136/jmg.2004.029439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence Moyra, Daujat Sylvain, and Schneider Robert. 2016. “Lateral Thinking: How Histone Modifications Regulate Gene Expression.” Trends in Genetics 32 (1): 42–56. 10.1016/j.tig.2015.10.007. [DOI] [PubMed] [Google Scholar]
- Lederer Damien, Grisart Bernard, Digilio Maria Cristina, Benoit Valérie, Crespin Marianne, Ghariani Sophie Claire, Maystadt Isabelle, Dallapiccola Bruno, and Verellen-Dumoulin Christine. 2012. “Deletion of KDM6A, a Histone Demethylase Interacting with MLL2, in Three Patients with Kabuki Syndrome.” American Journal of Human Genetics 90 (1): 119–24. 10.1016/j.ajhg.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Chul-Hwan, Yu Jia-Ray, Kumar Sunil, Jin Ying, LeRoy Gary, Bhanu Natarajan, Kaneko Syuzo, Garcia Benjamin A., Hamilton Andrew D., and Reinberg Danny. 2018. “Allosteric Activation Dictates PRC2 Activity Independent of Its Recruitment to Chromatin.” Molecular Cell 70 (3): 422–434.e6. 10.1016/j.molcel.2018.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Janine B., Wei Jing, Liu Wenhua, Cheng Jia, Feng Jian, and Yan Zhen. 2012. “Histone Deacetylase 6 Gates the Synaptic Action of Acute Stress in Prefrontal Cortex.” The Journal of Physiology 590 (7): 1535–46. 10.1113/jphysiol.2011.224907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Jeong-Heon, and Skalnik David G.. 2005. “CpG-Binding Protein (CXXC Finger Protein 1) Is a Component of the Mammalian Set1 Histone H3-Lys4 Methyltransferase Complex, the Analogue of the Yeast Set1/COMPASS Complex.” Journal of Biological Chemistry 280 (50): 41725–31. 10.1074/jbc.M508312200. [DOI] [PubMed] [Google Scholar]
- ———. 2008. “Wdr82 Is a C-Terminal Domain-Binding Protein That Recruits the Setd1A Histone H3-Lys4 Methyltransferase Complex to Transcription Start Sites of Transcribed Human Genes.” Molecular and Cellular Biology 28 (2): 609–18. 10.1128/MCB.01356-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Jeong-Heon, Tate Courtney M., You Jin-Sam, and Skalnik David G.. 2007. “Identification and Characterization of the Human Set1B Histone H3-Lys4 Methyltransferase Complex.” Journal of Biological Chemistry 282 (18): 13419–28. 10.1074/jbc.M609809200. [DOI] [PubMed] [Google Scholar]
- Lee Ji-Eun, Wang Chaochen, Xu Shiliyang, Cho Young-Wook, Wang Lifeng, Feng Xuesong, Baldridge Anne, et al. 2013. “H3K4 Mono- and Di-Methyltransferase MLL4 Is Required for Enhancer Activation during Cell Differentiation.” Edited by Glass Christopher. ELife 2 (December): e01503. 10.7554/eLife.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Min Gyu, Villa Raffaella, Trojer Patrick, Norman Jessica, Yan Kai-Ping, Reinberg Danny, Di Croce Luciano, and Shiekhattar Ramin. 2007. “Demethylation of H3K27 Regulates Polycomb Recruitment and H2A Ubiquitination.” Science 318 (5849): 447–50. 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- Lee Seunghee, Kim Dae-Hwan, Goo Young Hwa, Lee Young Chul, Lee Soo-Kyung, and Lee Jae W.. 2009. “Crucial Roles for Interactions between MLL3/4 and INI1 in Nuclear Receptor Transactivation.” Molecular Endocrinology 23 (5): 610–19. 10.1210/me.2008-0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Yu Fei, Nimura Keisuke, Lo Wan Ning, Saga Kotaro, and Kaneda Yasufumi. 2014. “Histone H3 Lysine 36 Methyltransferase Whsc1 Promotes the Association of Runx2 and P300 in the Activation of Bone-Related Genes.” PloS One 9 (9): e106661. 10.1371/journal.pone.0106661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Xuepei, and Jiao Jianwei. 2018. “UTX Affects Neural Stem Cell Proliferation and Differentiation through PTEN Signaling.” Stem Cell Reports 10 (4): 1193–1207. 10.1016/j.stemcr.2018.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerone M, Priolo M, Naselli A, Vignolo M, Romeo G, and Silengo MC. 1997. “Ectodermal Abnormalities in Kabuki Syndrome.” American Journal of Medical Genetics 73 (3): 263–66. . [DOI] [PubMed] [Google Scholar]
- Lessard Julie, Wu Jiang I., Ranish Jeffrey A., Wan Mimi, Winslow Monte M., Staahl Brett T., Wu Hai, Aebersold Ruedi, Graef Isabella A., and Crabtree Gerald R.. 2007. “An Essential Switch in Subunit Composition of a Chromatin Remodeling Complex during Neural Development.” Neuron. 10.1016/j.neuron.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson Jonathan M., and Sweatt J. David. 2005. “Epigenetic Mechanisms in Memory Formation.” Nature Reviews Neuroscience 6 (2): 108–18. 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- Li Marilyn M., Nimmakayalu Manjunath A., Mercer Danielle, Andersson Hans C., and Emanuel Beverly S.. 2008. “Characterization of a Cryptic 3.3 Mb Deletion in a Patient with a ‘Balanced t(15;22) Translocation’ Using High Density Oligo Array CGH and Gene Expression Arrays.” American Journal of Medical Genetics, Part A. 10.1002/ajmg.a.32116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Niu, Wang Yirou, Yang Yu, Wang Pengpeng, Huang Hui, Xiong Shiyi, Sun Luming, et al. 2018. “Description of the Molecular and Phenotypic Spectrum of Wiedemann-Steiner Syndrome in Chinese Patients.” Orphanet Journal of Rare Diseases 13 (1): 178. 10.1186/s13023-018-0909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Xiangzhi, Wu Lipeng, Corsa Sprunger, Ann Callie, Kunkel Steve, and Dou Yali. 2009. “Two Mammalian MOF Complexes Regulate Transcription Activation by Distinct Mechanisms.” Molecular Cell 36 (2): 290–301. 10.1016/j.molcel.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ligt Joep, Willemsen Marjolein H., van Bon Bregje W. M., Kleefstra Tjitske, Yntema Helger G., Kroes Thessa, Vulto-van Silfhout Anneke T., et al. 2013. “Diagnostic Exome Sequencing in Persons With Severe Intellectual Disability:” Obstetrical & Gynecological Survey 68 (3): 191–93. 10.1097/01.ogx.0000428160.59063.a6. [DOI] [PubMed] [Google Scholar]
- Lim Daniel A., Huang Yin-Cheng, Swigut Tomek, Mirick Anika L., Garcia-Verdugo Jose Manuel, Wysocka Joanna, Ernst Patricia, and Alvarez-Buylla Arturo. 2009. “Chromatin Remodelling Factor Mll1 Is Essential for Neurogenesis from Postnatal Neural Stem Cells.” Nature 458 (7237): 529–33. 10.1038/nature07726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Pei-Pei, Xu Ya-Jie, Dai Shang-Kun, Du Hong-Zhen, Wang Ying-Ying, Li Xing-Guo, Teng Zhao-Qian, and Liu Chang-Mei. 2019. “Polycomb Protein EED Regulates Neuronal Differentiation through Targeting SOX11 in Hippocampal Dentate Gyrus.” Stem Cell Reports 13 (1): 115–31. 10.1016/j.stemcr.2019.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lone M, Kungl T, Koper A, Bottenberg W, Kammerer R, Klein M, Sweeney ST, Auburn RP, O’Kane CJ, and Prokop A. 2010. “The Nuclear Protein Waharan Is Required for Endosomal-Lysosomal Trafficking in Drosophila.” Journal of Cell Science 123 (14): 2369–74. 10.1242/jcs.060582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPresti Patrizia. 2020. “HDAC6 in Diseases of Cognition and of Neurons.” Cells 10 (1). 10.3390/cells10010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozier Ekaterina R., Konovalov Fedor A., Kanivets Ilya V., Pyankov Denis V., Koshkin Philip A., Baleva Larisa S., Sipyagina Alla E., Yakusheva Elena N., Kuchina Anastasiya E., and Korostelev Sergey A.. 2018. “De Novo Nonsense Mutation in WHSC1 ( NSD2 ) in Patient with Intellectual Disability and Dysmorphic Features.” Journal of Human Genetics 63 (8): 919–22. 10.1038/s10038-018-0464-5. [DOI] [PubMed] [Google Scholar]
- Lubitz Sandra, Glaser Stefan, Schaft Julia, Stewart A. Francis, and Anastassiadis Konstantinos. 2007. “Increased Apoptosis and Skewed Differentiation in Mouse Embryonic Stem Cells Lacking the Histone Methyltransferase Mll2.” Molecular Biology of the Cell 18 (6): 2356–66. 10.1091/mbc.E06-11-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucio-Eterovic Agda Karina, Singh Melissa M., Gardner Jeffrey E., Veerappan Chendhore S., Rice Judd C., and Carpenter Phillip B.. 2010. “Role for the Nuclear Receptor-Binding SET Domain Protein 1 (NSD1) Methyltransferase in Coordinating Lysine 36 Methylation at Histone 3 with RNA Polymerase II Function.” Proceedings of the National Academy of Sciences 107 (39): 16952–57. 10.1073/pnas.1002653107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui Julian C., Barnes Kevin M., Dong Lijin, Yue Shanna, Graber Evan, Rapaport Robert, Dauber Andrew, Nilsson Ola, and Baron Jeffrey. 2018. “Ezh2 Mutations Found in the Weaver Overgrowth Syndrome Cause a Partial Loss of H3K27 Histone Methyltransferase Activity.” The Journal of Clinical Endocrinology and Metabolism 103 (4): 1470–78. 10.1210/jc.2017-01948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumish Heidi S., Wynn Julia, Devinsky Orrin, and Chung Wendy K.. 2015. “Brief Report: SETD2 Mutation in a Child with Autism, Intellectual Disabilities and Epilepsy.” Journal of Autism and Developmental Disorders 45 (11): 3764–70. 10.1007/s10803-015-2484-8. [DOI] [PubMed] [Google Scholar]
- Luscan Armelle, Laurendeau Ingrid, Malan Valérie, Francannet Christine, Odent Sylvie, Giuliano Fabienne, Lacombe Didier, et al. 2014. “Mutations in SETD2 Cause a Novel Overgrowth Condition.” Journal of Medical Genetics 51 (8): 512–17. 10.1136/jmedgenet-2014-102402. [DOI] [PubMed] [Google Scholar]
- Lv Deguan, Jia Feng, Hou Yanli, Sang Youzhou, Alvarez Angel A, Zhang Weiwei, Gao Wei-Qiang, et al. 2017. “Histone Acetyltransferase KAT6A Upregulates PI3K/AKT Signaling through TRIM24 Binding.” Cancer Research 77 (22): 6190–6201. 10.1158/0008-5472.can-17-1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlckova M, Simandlova M, Zimmermann P, Stranecky V, Hartmannova H, Hodanova K, Havlovicova M, Hancarova M, Kmoch S, and Sedlacek Z. 2015. “A Patient Showing Features of Both SBBYSS and GPS Supports the Concept of a KAT6B-Related Disease Spectrum, with Mutations in Mid-Exon 18 Possibly Leading to Combined Phenotypes.” European Journal of Medical Genetics 58 (10): 550–55. 10.1016/j.ejmg.2015.09.004. [DOI] [PubMed] [Google Scholar]
- Ma Yuanqing, Liu Xiuli, Liu Zhaoting, Wei Shi, Shang Hanqiao, Xue Yu, Cao Yu, Meng Anming, and Wang Qiang. 2015. “The Chromatin Remodeling Protein Bptf Promotes Posterior Neuroectodermal Fate by Enhancing Smad2-Activated Wnt8a Expression.” Journal of Neuroscience 35 (22): 8493–8506. 10.1523/JNEUROSCI.0377-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermot KD, Patton MA, Williams MJ, and Winter RM. 1989. “Hypertrichosis Cubiti (Hairy Elbows) and Short Stature: A Recognisable Association.” Journal of Medical Genetics 26 (6): 382–85. 10.1136/jmg.26.6.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machol Keren, Rousseau Justine, Ehresmann Sophie, Garcia Thomas, Nguyen Thi Tuyet Mai, Spillmann Rebecca C., Sullivan Jennifer A., et al. 2019. “Expanding the Spectrum of BAF-Related Disorders: De Novo Variants in SMARCC2 Cause a Syndrome with Intellectual Disability and Developmental Delay.” American Journal of Human Genetics. 10.1016/j.ajhg.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangi Giuseppe, Di Giacomo Marilena C., Lattante Serena, Orteschi Daniela, Patrizi Sara, Doronzio Paolo N., Riviello Francesco N., Vaisfeld Alessandro, Frangella Silvia, and Zollino Marcella. 2018. “A Novel Truncating Variant within Exon 7 of KAT6B Associated with Features of Both Say–Barber–Bieseker–Young–Simpson Syndrome and Genitopatellar Syndrome: Further Evidence of a Continuum in the Clinical Spectrum of KAT6B‐related Disorders.” American Journal of Medical Genetics Part A 176 (2): 455–59. 10.1002/ajmg.a.38571. [DOI] [PubMed] [Google Scholar]
- Marfella Concetta G.A., and Imbalzano Anthony N.. 2007. “The Chd Family of Chromatin Remodelers.” Mutation Research - Fundamental and Molecular Mechanisms of Mutagenesis. 10.1016/j.mrfmmm.2006.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron Raphael, Justin Neil, Ohno Katsuhito, Sharpe Miriam L, Son Jinsook, Drury William J, Voigt Philipp, et al. 2009. “Role of the Polycomb Protein Eed in the Propagation of Repressive Histone Marks.” Nature 461 (7265): 762–67. 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein Ronen, and Zhou Ming-Ming. 2014. “Writers and Readers of Histone Acetylation: Structure, Mechanism, and Inhibition.” Cold Spring Harbor Perspectives in Biology 6 (7): a018762. 10.1101/cshperspect.a018762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marom Ronit, Jain Mahim, Burrage Lindsay C., Song I. Wen, Graham Brett H., Brown Chester W., Stevens Servi J.C., et al. 2017. “Heterozygous Variants in ACTL6A, Encoding a Component of the BAF Complex, Are Associated with Intellectual Disability.” Human Mutation. 10.1002/humu.23282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin Hilary C., Jones Wendy D., McIntyre Rebecca, Sanchez-Andrade Gabriela, Sanderson Mark, Stephenson James D., Jones Carla P., et al. 2018. “Quantifying the Contribution of Recessive Coding Variation to Developmental Disorders.” Science (New York, N.Y.) 362 (6419): 1161–64. 10.1126/science.aar6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzin Pauline, Rondeau Sophie, Aldinger Kimberly A., Alessandri Jean-Luc, Isidor Bertrand, Heron Delphine, Keren Boris, Dobyns William B., and Cormier-Daire Valérie. 2019. “SETD2 Related Overgrowth Syndrome: Presentation of Four New Patients and Review of the Literature.” American Journal of Medical Genetics. Part C, Seminars in Medical Genetics 181 (4): 509–18. 10.1002/ajmg.c.31746. [DOI] [PubMed] [Google Scholar]
- Mashtalir Nazar, D’Avino Andrew R., Michel Brittany C., Luo Jie, Pan Joshua, Otto Jordan E., Zullow Hayley J., et al. 2018. “Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes.” Cell. 10.1016/j.cell.2018.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattioli Francesca, Schaefer Elise, Magee Alex, Mark Paul, Mancini Grazia M., Dieterich Klaus, Von Allmen Gretchen, et al. 2017. “Mutations in Histone Acetylase Modifier BRPF1 Cause an Autosomal-Dominant Form of Intellectual Disability with Associated Ptosis.” The American Journal of Human Genetics 100 (1): 105–16. 10.1016/j.ajhg.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaughran JM, Donnai D, and Clayton-Smith J. 2000. “Biliary Atresia in Kabuki Syndrome.” American Journal of Medical Genetics 91 (2): 157–58. [PubMed] [Google Scholar]
- McGinty Robert K., and Tan Song. 2015. “Nucleosome Structure and Function.” Chemical Reviews 115 (6): 2255–73. 10.1021/cr500373h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendjan Sascha, Taipale Mikko, Kind Jop, Holz Herbert, Gebhardt Philipp, Schelder Malgorzata, Vermeulen Michiel, et al. 2006. “Nuclear Pore Components Are Involved in the Transcriptional Regulation of Dosage Compensation in Drosophila.” Molecular Cell 21 (6): 811–23. 10.1016/j.molcel.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Menke Leonie A., van Belzen Martine J., Alders Marielle, Cristofoli Francesca, Ehmke Nadja, Fergelot Patricia, Foster Alison, et al. 2016. “CREBBP Mutations in Individuals without Rubinstein–Taybi Syndrome Phenotype.” American Journal of Medical Genetics Part A 170 (10): 2681–93. 10.1002/ajmg.a.37800. [DOI] [PubMed] [Google Scholar]
- Menke Leonie A, The DDD study, Gardeitchik Thatjana, Hammond Peter, Heimdal Ketil R, Houge Gunnar, Hufnagel Sophia B, et al. 2018. “Further Delineation of an Entity Caused by CREBBP and EP300 Mutations but Not Resembling Rubinstein-Taybi Syndrome.” American Journal of Medical Genetics Part A 176 (4): 862–76. 10.1002/ajmg.a.38626. [DOI] [PubMed] [Google Scholar]
- Meyer Esther, Carss Keren J., Rankin Julia, Nichols John M. E., Grozeva Detelina, Joseph Agnel P., Mencacci Niccolo E., et al. 2017. “Mutations in the Histone Methyltransferase Gene KMT2B Cause Complex Early-Onset Dystonia.” Nature Genetics 49 (2): 223–37. 10.1038/ng.3740. [DOI] [PubMed] [Google Scholar]
- Micale Lucia, Augello Bartolomeo, Fusco Carmela, Selicorni Angelo, Loviglio Maria N., Silengo Margherita Cirillo, Reymond Alexandre, et al. 2011. “Mutation Spectrum of MLL2 in a Cohort of Kabuki Syndrome Patients.” Orphanet Journal of Rare Diseases 6 (1): 1–8. 10.1186/1750-1172-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micucci Joseph A., Layman Wanda S., Hurd Elizabeth A., Sperry Ethan D., Frank Sophia F., Durham Mark A., Swiderski Donald L., et al. 2014. “CHD7 and Retinoic Acid Signaling Cooperate to Regulate Neural Stem Cell and Inner Ear Development in Mouse Models of CHARGE Syndrome.” Human Molecular Genetics. 10.1093/hmg/ddt435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen Tarjei S., Ku Manching, Jaffe David B., Issac Biju, Lieberman Erez, Giannoukos Georgia, Alvarez Pablo, et al. 2007. “Genome-Wide Maps of Chromatin State in Pluripotent and Lineage-Committed Cells.” Nature 448 (7153): 553–60. 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller Jeremy A., Ding Song-Lin, Sunkin Susan M., Smith Kimberly A., Ng Lydia, Szafer Aaron, Ebbert Amanda, et al. 2014. “Transcriptional Landscape of the Prenatal Human Brain.” Nature 508 (7495): 199–206. 10.1038/nature13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills Alexandra, Bearce Elizabeth, Cella Rachael, Kim Seung Woo, Selig Megan, Lee Sangmook, and Lowery Laura Anne. 2019. “Wolf-Hirschhorn Syndrome-Associated Genes Are Enriched in Motile Neural Crest Cells and Affect Craniofacial Development in Xenopus Laevis.” Frontiers in Physiology 10: 431. 10.3389/fphys.2019.00431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miró Xavier, Zhou Xunlei, Boretius Susann, Michaelis Thomas, Kubisch Christian, Alvarez-Bolado Gonzalo, and Gruss Peter. 2009. “Haploinsufficiency of the Murine Polycomb Gene Suz12 Results in Diverse Malformations of the Brain and Neural Tube.” Disease Models & Mechanisms 2 (7–8): 412–18. 10.1242/dmm.001602. [DOI] [PubMed] [Google Scholar]
- Miyake N, Tsurusaki Y, Koshimizu E, Okamoto N, Kosho T, Brown NJ, Tan TY, et al. 2016. “Delineation of Clinical Features in Wiedemann–Steiner Syndrome Caused by KMT2A Mutations.” Clinical Genetics 89 (1): 115–19. 10.1111/cge.12586. [DOI] [PubMed] [Google Scholar]
- Miyake Noriko, Mizuno Seiji, Okamoto Nobuhiko, Ohashi Hirofumi, Shiina Masaaki, Ogata Kazuhiro, Tsurusaki Yoshinori, et al. 2013. “KDM6A Point Mutations Cause Kabuki Syndrome.” Human Mutation 34 (1): 108–10. 10.1002/humu.22229. [DOI] [PubMed] [Google Scholar]
- Miyazaki Hitomi, Higashimoto Ken, Yada Yukari, Endo Takaho A., Sharif Jafar, Komori Toshiharu, Matsuda Masashi, et al. 2013. “Ash1l Methylates Lys36 of Histone H3 Independently of Transcriptional Elongation to Counteract Polycomb Silencing.” Edited by Cavalli Giacomo. PLoS Genetics 9 (11): e1003897. 10.1371/journal.pgen.1003897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordaunt DA, and McLauchlan A. 2015. “HDAC8-Deficiency Causes an X-Linked Dominant Disorder with a Wide Range of Severity.” Clinical Genetics 88 (1): 98–98. 10.1111/cge.12588. [DOI] [PubMed] [Google Scholar]
- Mozzetta Chiara, Pontis Julien, Fritsch Lauriane, Robin Philippe, Portoso Manuela, Proux Caroline, Margueron Raphaël, and Ait-Si-Ali Slimane. 2014. “The Histone H3 Lysine 9 Methyltransferases G9a and GLP Regulate Polycomb Repressive Complex 2-Mediated Gene Silencing.” Molecular Cell 53 (2): 277–89. 10.1016/j.molcel.2013.12.005. [DOI] [PubMed] [Google Scholar]
- Nagase T, Ishikawa K, Suyama M, Kikuno R, Hirosawa M, Miyajima N, Tanaka A, Kotani H, Nomura N, and Ohara O. 1998. “Prediction of the Coding Sequences of Unidentified Human Genes. XII. The Complete Sequences of 100 New CDNA Clones from Brain Which Code for Large Proteins in Vitro.” DNA Research: An International Journal for Rapid Publication of Reports on Genes and Genomes 5 (6): 355–64. 10.1093/dnares/5.6.355. [DOI] [PubMed] [Google Scholar]
- Nakagawa Tadashi, Hattori Satoko, Nobuta Risa, Kimura Ryuichi, Nakagawa Makiko, Matsumoto Masaki, Nagasawa Yuko, et al. 2020. “The Autism-Related Protein SETD5 Controls Neural Cell Proliferation through Epigenetic Regulation of RDNA Expression.” IScience 23 (4): 101030. 10.1016/j.isci.2020.101030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardello Rosaria, Mangano Giuseppe Donato, Fontana Antonina, Gagliardo Cesare, Midiri Federico, Borgia Paola, Brighina Filippo, Raieli Vincenzo, Mangano Salvatore, and Salpietro Vincenzo. 2020. “Broad Neurodevelopmental Features and Cortical Anomalies Associated with a Novel de Novo KMT2A Variant in Wiedemann-Steiner Syndrome.” European Journal of Medical Genetics 64 (2): 104133. 10.1016/j.ejmg.2020.104133. [DOI] [PubMed] [Google Scholar]
- Neas Katherine R., Smith Janine M., Chia Nicole, Huseyin Suna, Heaps Luke St, Peters Greg, Sholler Gary, Tzioumi Dimitra, Sillence David O., and Mowat David. 2005. “Three Patients with Terminal Deletions within the Subtelomeric Region of Chromosome 9q.” American Journal of Medical Genetics Part A 132A (4): 425–30. 10.1002/ajmg.a.30496. [DOI] [PubMed] [Google Scholar]
- Negwer Moritz, Piera Karol, Hesen Rick, Lütje Lukas, Aarts Lynn, Schubert Dirk, and Kasri Nael Nadif. 2020. “EHMT1 Regulates Parvalbumin-Positive Interneuron Development and GABAergic Input in Sensory Cortical Areas.” Brain Structure and Function 225 (9): 2701–16. 10.1007/s00429-020-02149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neigeborn L, and Carlson M. 1984. “Genes Affecting the Regulation of SUC2 Gene Expression by Glucose Repression in Saccharomyces Cerevisiae.” Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevado Julián, Ho Karen S., Zollino Marcella, Blanco Raquel, Cobaleda César, Golzio Christelle, Beaudry‐Bellefeuille Isabelle, et al. 2020. “International Meeting on Wolf-Hirschhorn Syndrome: Update on the Nosology and New Insights on the Pathogenic Mechanisms for Seizures and Growth Delay.” American Journal of Medical Genetics Part A 182 (1): 257–67. 10.1002/ajmg.a.61406. [DOI] [PubMed] [Google Scholar]
- Ng Sarah B., Bigham Abigail W., Buckingham Kati J., Hannibal Mark C., McMillin Margaret J., Gildersleeve Heidi I., Beck Anita E., et al. 2010. “Exome Sequencing Identifies MLL2 Mutations as a Cause of Kabuki Syndrome.” Nature Genetics 42 (9): 790–93. 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Anh Tram, and Zhang Yi. 2011. “The Diverse Functions of Dot1 and H3K79 Methylation.” Genes & Development 25 (13): 1345–58. 10.1101/gad.2057811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niikawa Norio. 2004. “Molecular Basis of Sotos Syndrome.” Hormone Research 62 Suppl 3: 60–65. 10.1159/000080501. [DOI] [PubMed] [Google Scholar]
- Nimura Keisuke, Ura Kiyoe, Shiratori Hidetaka, Ikawa Masato, Okabe Masaru, Schwartz Robert J., and Kaneda Yasufumi. 2009. “A Histone H3 Lysine 36 Trimethyltransferase Links Nkx2-5 to Wolf-Hirschhorn Syndrome.” Nature 460 (7252): 287–91. 10.1038/nature08086. [DOI] [PubMed] [Google Scholar]
- Nixon Kevin C.J., Rousseau Justine, Stone Max H., Sarikahya Mohammed, Ehresmann Sophie, Mizuno Seiji, Matsumoto Naomichi, et al. 2019. “A Syndromic Neurodevelopmental Disorder Caused by Mutations in SMARCD1, a Core SWI/SNF Subunit Needed for Context-Dependent Neuronal Gene Regulation in Flies.” American Journal of Human Genetics. 10.1016/j.ajhg.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Carroll Dónal, Erhardt Sylvia, Pagani Michaela, Barton Sheila C., Surani M. Azim, and Jenuwein Thomas. 2001. “The Polycomb-Group Gene Ezh2 Is Required for Early Mouse Development.” Molecular and Cellular Biology 21 (13): 4330–36. 10.1128/MCB.21.13.4330-4336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell-Luria Anne H., Pais Lynn S., Faundes Víctor, Wood Jordan C., Sveden Abigail, Luria Victor, Jamra Rami Abou, et al. 2019. “Heterozygous Variants in KMT2E Cause a Spectrum of Neurodevelopmental Disorders and Epilepsy.” The American Journal of Human Genetics 104 (6): 1210–22. 10.1016/j.ajhg.2019.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi Sabrina, Zalucki Oressia, Vega Michelle S, Harkins Danyon, Harvey Tracey J, Kasherman Maria, Davila Raul A, et al. 2020. “Investigating Cortical Features of Sotos Syndrome Using Mice Heterozygous for Nsd1.” Genes, Brain and Behavior 19 (4): e12637. 10.1111/gbb.12637. [DOI] [PubMed] [Google Scholar]
- Okamoto Nobuhiko, Miya Fuyuki, Tsunoda Tatsuhiko, Kato Mitsuhiro, Saitoh Shinji, Yamasaki Mami, Kanemura Yonehiro, and Kosaki Kenjiro. 2017. “Novel MCA/ID Syndrome with ASH1L Mutation.” American Journal of Medical Genetics Part A 173 (6): 1644–48. 10.1002/ajmg.a.38193. [DOI] [PubMed] [Google Scholar]
- Van Paemel Ruben, De Bruyne Pauline, van der Straaten Saskia, D’hondt Marleen, Fränkel Urlien, Dheedene Annelies, Menten Björn, and Callewaert Bert. 2017. “Confirmation of an ARID2 Defect in SWI/SNF-Related Intellectual Disability.” American Journal of Medical Genetics, Part A. 10.1002/ajmg.a.38407. [DOI] [PubMed] [Google Scholar]
- Palumbo Orazio, Palumbo Pietro, Delvecchio Maurizio, Palladino Teresa, Stallone Raffaella, Crisetti Matteo, Zelante Leopoldo, and Carella Massimo. 2015. “Microdeletion of 12q24.31: Report of a Girl with Intellectual Disability, Stereotypies, Seizures and Facial Dysmorphisms.” American Journal of Medical Genetics Part A 167 (2): 438–44. 10.1002/ajmg.a.36872. [DOI] [PubMed] [Google Scholar]
- Paradowska-Stolarz Anna M. 2014. “Wolf-Hirschhorn Syndrome (WHS) - Literature Review on the Features of the Syndrome.” Advances in Clinical and Experimental Medicine: Official Organ Wroclaw Medical University 23 (3): 485–89. 10.17219/acem/24111. [DOI] [PubMed] [Google Scholar]
- Parenti Ilaria, Teresa-Rodrigo María E., Pozojevic Jelena, Gil Sara Ruiz, Bader Ingrid, Braunholz Diana, Bramswig Nuria C., et al. 2017. “Mutations in Chromatin Regulators Functionally Link Cornelia de Lange Syndrome and Clinically Overlapping Phenotypes.” Human Genetics 136 (3): 307–20. 10.1007/s00439-017-1758-y. [DOI] [PubMed] [Google Scholar]
- Park Dae Hwi, Hong Sung Jun, Salinas Ryan D., Liu Siyuan John, Sun Shawn W., Sgualdino Jacopo, Testa Giuseppe, Matzuk Martin M., Iwamori Naoki, and Lim Daniel A.. 2014. “Activation of Neuronal Gene Expression by the JMJD3 Demethylase Is Required for Postnatal and Adult Brain Neurogenesis.” Cell Reports 8 (5): 1290–99. 10.1016/j.celrep.2014.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasillas Martina P., Shah Meera, and Kamps Mark P.. 2011. “NSD1 PHD Domains Bind Methylated H3K4 and H3K9 Using Interactions Disrupted by Point Mutations in Human Sotos Syndrome.” Human Mutation 32 (3): 292–98. 10.1002/humu.21424. [DOI] [PubMed] [Google Scholar]
- Pasini Diego, Bracken Adrian P, Jensen Michael R, Denchi Eros Lazzerini, and Helin Kristian. 2004. “Suz12 Is Essential for Mouse Development and for EZH2 Histone Methyltransferase Activity.” The EMBO Journal 23 (20): 4061–71. 10.1038/sj.emboj.7600402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulussen Aimée D. C., Stegmann Alexander P. A., Blok Marinus J., Tserpelis Demis, Posma‐Velter Crool, Detisch Yvonne, Smeets Eric E. J. G. L., et al. 2011. “MLL2 Mutation Spectrum in 45 Patients with Kabuki Syndrome.” Human Mutation 32 (2): E2018–25. 10.1002/humu.21416. [DOI] [PubMed] [Google Scholar]
- Peng Yunhui, Suryadi Jimmy, Yang Ye, Kucukkal Tugba G., Cao Weiguo, and Alexov Emil. 2015. “Mutations in the KDM5C ARID Domain and Their Plausible Association with Syndromic Claes-Jensen-Type Disease.” International Journal of Molecular Sciences 16 (11): 27270–87. 10.3390/ijms161126022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez‐Grijalba Virginia, García‐Oguiza Alberto, López María, Armstrong Judith, García‐Miñaur Sixto, Mesa‐Latorre Jose María, O’Callaghan Mar, et al. 2019. “New Insights into Genetic Variant Spectrum and Genotype–Phenotype Correlations of Rubinstein‐Taybi Syndrome in 39 CREBBP‐positive Patients.” Molecular Genetics & Genomic Medicine 7 (11): e972. 10.1002/mgg3.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters Antoine H.F.M., Kubicek Stefan, Mechtler Karl, O’Sullivan Roderick J., Derijck Alwin A.H.A., Perez-Burgos Laura, Kohlmaier Alexander, et al. 2003. “Partitioning and Plasticity of Repressive Histone Methylation States in Mammalian Chromatin.” Molecular Cell 12 (6): 1577–89. 10.1016/S1097-2765(03)00477-5. [DOI] [PubMed] [Google Scholar]
- Pilarowski Genay O., Vernon Hilary J., Applegate Carolyn D., Boukas Leandros, Cho Megan T., Gurnett Christina A., Benke Paul J., et al. 2018. “Missense Variants in the Chromatin Remodeler CHD1 Are Associated with Neurodevelopmental Disability.” Journal of Medical Genetics. 10.1136/jmedgenet-2017-104759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzo Lucilla, Jensen Matthew, Polyak Andrew, Rosenfeld Jill A., Mannik Katrin, Krishnan Arjun, McCready Elizabeth, et al. 2019. “Rare Variants in the Genetic Background Modulate Cognitive and Developmental Phenotypes in Individuals Carrying Disease-Associated Variants.” Genetics in Medicine: Official Journal of the American College of Medical Genetics 21 (4): 816–25. 10.1038/s41436-018-0266-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poeta Loredana, Padula Agnese, Attianese Benedetta, Valentino Mariaelena, Verrillo Lucia, Filosa Stefania, Shoubridge Cheryl, et al. 2019. “Histone Demethylase KDM5C Is a SAHA-Sensitive Central Hub at the Crossroads of Transcriptional Axes Involved in Multiple Neurodevelopmental Disorders.” Human Molecular Genetics 28 (24): 4089–4102. 10.1093/hmg/ddz254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polizzi A, Pavone P, Ciancio E, La Rosa C, Sorge G, and Ruggieri M. 2005. “Hypertrichosis Cubiti (Hairy Elbow Syndrome): A Clue to a Malformation Syndrome.” Journal of Pediatric Endocrinology and Metabolism 18 (10). 10.1515/JPEM.2005.18.10.1019. [DOI] [PubMed] [Google Scholar]
- Ponte Inma, Romero Devani, Yero Daniel, Suau Pedro, and Roque Alicia. 2017. “Complex Evolutionary History of the Mammalian Histone H1.1–H1.5 Gene Family.” Molecular Biology and Evolution, January, msw241. 10.1093/molbev/msw241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Hank H., Sarkissian Madathia, Hu Gang-Qing, Wang Zhibin, Bhattacharjee Arindam, Gordon D. Benjamin, Gonzales Michelle, et al. 2010. “Histone H4K20/H3K9 Demethylase PHF8 Regulates Zebrafish Brain and Craniofacial Development.” Nature 466 (7305): 503–7. 10.1038/nature09261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Qi, Li Yan, Chen Zhi, Wang Mingzhu, Reinberg Danny, and Xu Rui-Ming. 2011. “The Structure of NSD1 Reveals an Autoregulatory Mechanism Underlying Histone H3K36 Methylation.” The Journal of Biological Chemistry 286 (10): 8361–68. 10.1074/jbc.M110.204115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radvanszky J, Hyblova M, Durovcikova D, Hikkelova M, Fiedler E, Kadasi L, Turna J, Minarik G, and Szemes T. 2017. “Complex Phenotypes Blur Conventional Borders between Say–Barber–Biesecker–Young–Simpson Syndrome and Genitopatellar Syndrome.” Clinical Genetics 91 (2): 339–43. 10.1111/cge.12840. [DOI] [PubMed] [Google Scholar]
- Raja Sunil Jayaramaiah, Charapitsa Iryna, Conrad Thomas, Vaquerizas Juan M., Gebhardt Philipp, Holz Herbert, Kadlec Jan, Fraterman Sven, Luscombe Nicholas M., and Akhtar Asifa. 2010. “The Nonspecific Lethal Complex Is a Transcriptional Regulator in Drosophila.” Molecular Cell 38 (6): 827–41. 10.1016/j.molcel.2010.05.021. [DOI] [PubMed] [Google Scholar]
- Rauch Anita, Wieczorek Dagmar, Graf Elisabeth, Wieland Thomas, Endele Sabine, Schwarzmayr Thomas, Albrecht Beate, et al. 2012. “Range of Genetic Mutations Associated with Severe Non-Syndromic Sporadic Intellectual Disability: An Exome Sequencing Study.” Lancet (London, England) 380 (9854): 1674–82. 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- Ravenswaaij-Arts, van Conny M, Hefner Meg, Blake Kim, and Martin Donna M. 1993. CHD7 Disorder. GeneReviews®.
- Rayasam Geetha Vani, Wendling Olivia, Angrand Pierre-Olivier, Mark Manuel, Niederreither Karen, Song Luyan, Lerouge Thierry, Hager Gordon L., Chambon Pierre, and Losson Régine. 2003. “NSD1 Is Essential for Early Post-Implantation Development and Has a Catalytically Active SET Domain.” The EMBO Journal 22 (12): 3153–63. 10.1093/emboj/cdg288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice Judd C., Briggs Scott D., Ueberheide Beatrix, Barber Cynthia M., Shabanowitz Jeffrey, Hunt Donald F., Shinkai Yoichi, and Allis C. David. 2003. “Histone Methyltransferases Direct Different Degrees of Methylation to Define Distinct Chromatin Domains.” Molecular Cell 12 (6): 1591–98. 10.1016/S1097-2765(03)00479-9. [DOI] [PubMed] [Google Scholar]
- Richman R, Chicoine LG, Collini MP, Cook RG, and Allis CD. 1988. “Micronuclei and the Cytoplasm of Growing Tetrahymena Contain a Histone Acetylase Activity Which Is Highly Specific for Free Histone H4.” The Journal of Cell Biology 106 (4): 1017–26. 10.1083/jcb.106.4.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon-Arano Hector, Halow Jessica, Delrow Jeffrey J., Parkhurst Susan M., and Groudine Mark. 2012. “UpSET Recruits HDAC Complexes and Restricts Chromatin Accessibility and Acetylation at Promoter Regions.” Cell 151 (6): 1214–28. 10.1016/j.cell.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelfsema Jeroen H, White Stefan J, Ariyürek Yavuz, Bartholdi Deborah, Niedrist Dunja, Papadia Francesco, Bacino Carlos A, et al. 2005. “Genetic Heterogeneity in Rubinstein-Taybi Syndrome: Mutations in Both the CBP and EP300 Genes Cause Disease.” The American Journal of Human Genetics 76 (4): 572–80. 10.1086/429130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roston Alexandra, Evans Dan, Gill Harinder, McKinnon Margaret, Isidor Bertrand, Cogné Benjamin, Mwenifumbo Jill, et al. 2020. “SETD1B -Associated Neurodevelopmental Disorder.” Journal of Medical Genetics, June, jmedgenet-2019–106756. 10.1136/jmedgenet-2019-106756. [DOI] [PubMed] [Google Scholar]
- Ruault Myriam, Brun Marie Elisabeth, Ventura Mario, Roizès Gérard, and De Sario Albertina. 2002. “MLL3, a New Human Member of the TRX/MLL Gene Family, Maps to 7q36, a Chromosome Region Frequently Deleted in Myeloid Leukaemia.” Gene 284 (1–2): 73–81. 10.1016/s0378-1119(02)00392-x. [DOI] [PubMed] [Google Scholar]
- RUBINSTEIN JACKH, and TAYBI HOOSHANG. 1963. “Broad Thumbs and Toes and Facial Abnormalities: A Possible Mental Retardation Syndrome.” American Journal of Diseases of Children 105 (6): 588. 10.1001/archpedi.1963.02080040590010. [DOI] [PubMed] [Google Scholar]
- Sabitha KR, Shetty Ashok K., and Upadhya Dinesh. 2021. “Patient-Derived IPSC Modeling of Rare Neurodevelopmental Disorders: Molecular Pathophysiology and Prospective Therapies.” Neuroscience and Biobehavioral Reviews 121 (February): 201–19. 10.1016/j.neubiorev.2020.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Roberto, and Zhou Ming-Ming. 2009. “The Role of Human Bromodomains in Chromatin Biology and Gene Transcription.” Current Opinion in Drug Discovery & Development 12 (5): 659–65. [PMC free article] [PubMed] [Google Scholar]
- Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, Clément-Ziza M, Delezoide AL, Aubry MC, et al. 2006. “Phenotypic Spectrum of CHARGE Syndrome in Fetuses with CHD7 Truncating Mutations Correlates with Expression during Human Development.” Journal of Medical Genetics. 10.1136/jmg.2005.036160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santen Gijs W.E., Aten Emmelien, Sun Yu, Almomani Rowida, Gilissen Christian, Nielsen Maartje, Kant Sarina G., et al. 2012. “Mutations in SWI/SNF Chromatin Remodeling Complex Gene ARID1B Cause Coffin-Siris Syndrome.” Nature Genetics. 10.1038/ng.2217. [DOI] [PubMed] [Google Scholar]
- Santos-Rosa Helena, Schneider Robert, Bannister Andrew J., Sherriff Julia, Bernstein Bradley E., Emre Tolga N. C., Schreiber Stuart L., Mellor Jane, and Kouzarides Tony. 2002. “Active Genes Are TriMethylated at K4 of Histone H3.” Nature 419 (6905): 407–11. 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- Scandaglia Marilyn, Lopez-Atalaya Jose P., Medrano-Fernandez Alejandro, Lopez-Cascales Maria T., Blanco Beatriz Del, Lipinski Michal, Benito Eva, et al. 2017. “Loss of Kdm5c Causes Spurious Transcription and Prevents the Fine-Tuning of Activity-Regulated Enhancers in Neurons.” Cell Reports 21 (1): 47–59. 10.1016/j.celrep.2017.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel Laila C., Aref-Eshghi Erfan, Skinner Cindy, Ainsworth Peter, Lin Hanxin, Paré Guillaume, Rodenhiser David I., Schwartz Charles, and Sadikovic Bekim. 2018. “Peripheral Blood Epi-Signature of Claes-Jensen Syndrome Enables Sensitive and Specific Identification of Patients and Healthy Carriers with Pathogenic Mutations in KDM5C.” Clinical Epigenetics 10 (1): 21. 10.1186/s13148-018-0453-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider Jessica, Wood Adam, Lee Jung-Shin, Schuster Rebecca, Dueker Jeff, Maguire Courtney, Swanson Selene K., Florens Laurence, Washburn Michael P., and Shilatifard Ali. 2005. “Molecular Regulation of Histone H3 Trimethylation by COMPASS and the Regulation of Gene Expression.” Molecular Cell 19 (6): 849–56. 10.1016/j.molcel.2005.07.024. [DOI] [PubMed] [Google Scholar]
- Schoberleitner Ines, Mutti Anna, Sah Anupam, Wille Alexandra, Gimeno-Valiente Francisco, Piatti Paolo, Kharitonova Maria, et al. 2019. “Role for Chromatin Remodeling Factor Chd1 in Learning and Memory.” Frontiers in Molecular Neuroscience. 10.3389/fnmol.2019.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz Yvonne, Wehner Peter, Opitz Lennart, Salinas-Riester Gabriela, Bongers Ernie M.H.F., Van Ravenswaaij-Arts Conny M.A., Wincent Josephine, et al. 2014. “CHD7, the Gene Mutated in CHARGE Syndrome, Regulates Genes Involved in Neural Crest Cell Guidance.” Human Genetics. 10.1007/s00439-014-1444-2. [DOI] [PubMed] [Google Scholar]
- Sebastian S, Sreenivas P, Sambasivan R, Cheedipudi S, Kandalla P, Pavlath GK, and Dhawan J. 2009. “MLL5, a Trithorax Homolog, Indirectly Regulates H3K4 Methylation, Represses Cyclin A2 Expression, and Promotes Myogenic Differentiation.” Proceedings of the National Academy of Sciences 106 (12): 4719–24. 10.1073/pnas.0807136106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedkov Yurii, Cho Elizabeth, Petruk Svetlana, Cherbas Lucy, Smith Sheryl T., Jones Richard S., Cherbas Peter, Canaani Eli, Jaynes James B., and Mazo Alexander. 2003. “Methylation at Lysine 4 of Histone H3 in Ecdysone-Dependent Development of Drosophila.” Nature 426 (6962): 78–83. 10.1038/nature02080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba Yuichiro, Harada Akihito, Maehara Kazumitsu, Oki Shinya, Meno Chikara, Ueda Jun, Yamagata Kazuo, et al. 2017. “Chd2 Regulates Chromatin for Proper Gene Expression toward Differentiation in Mouse Embryonic Stem Cells.” Nucleic Acids Research. 10.1093/nar/gkx475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa Alessandro, Fagnocchi Luca, Mastrototaro Giuseppina, Massimino Luca, Zaghi Mattia, Indrigo Marzia, Cattaneo Stefano, et al. 2019. “SETD5 Regulates Chromatin Methylation State and Preserves Global Transcriptional Fidelity during Brain Development and Neuronal Wiring.” Neuron 104 (2): 271–289.e13. 10.1016/j.neuron.2019.07.013. [DOI] [PubMed] [Google Scholar]
- Shang Linshan, Cho Megan T., Retterer Kyle, Folk Leandra, Humberson Jennifer, Rohena Luis, Sidhu Alpa, et al. 2015. “Mutations in ARID2 Are Associated with Intellectual Disabilities.” Neurogenetics. 10.1007/s10048-015-0454-0. [DOI] [PubMed] [Google Scholar]
- Shao Gen-Bao, Chen Jun-Chao, Zhang Liu-Ping, Huang Pan, Lu Hong-Yan, Jin Jie, Gong Ai-Hua, and Sang Jian-Rong. 2014. “Dynamic Patterns of Histone H3 Lysine 4 Methyltransferases and Demethylases during Mouse Preimplantation Development.” In Vitro Cellular & Developmental Biology. Animal 50 (7): 603–13. 10.1007/s11626-014-9741-6. [DOI] [PubMed] [Google Scholar]
- Shi Yang, and Whetstine Johnathan R.. 2007. “Dynamic Regulation of Histone Lysine Methylation by Demethylases.” Molecular Cell 25 (1): 1–14. 10.1016/j.molcel.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Shi Yujiang, Lan Fei, Matson Caitlin, Mulligan Peter, Whetstine Johnathan R., Cole Philip A., Casero Robert A., and Shi Yang. 2004. “Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1.” Cell 119 (7): 941–53. 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Shi Yujiang, Sawada Jun-ichi, Sui Guangchao, Affar El Bachir, Whetstine Johnathan R., Lan Fei, Ogawa Hidesato, Po-Shan Luke Margaret, Nakatani Yoshihiro, and Shi Yang. 2003. “Coordinated Histone Modifications Mediated by a CtBP Co-Repressor Complex.” Nature 422 (6933): 735–38. 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- Shilatifard Ali. 2012. “The COMPASS Family of Histone H3K4 Methylases: Mechanisms of Regulation in Development and Disease Pathogenesis.” Annual Review of Biochemistry 81 (1): 65–95. 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siderius LE, Hamel BC, van Bokhoven H, de Jager F, van den Helm B, Kremer H, Heineman-de Boer JA, Ropers HH, and Mariman EC. 1999. “X-Linked Mental Retardation Associated with Cleft Lip/Palate Maps to Xp11.3-Q21.3.” American Journal of Medical Genetics 85 (3): 216–20. [PubMed] [Google Scholar]
- Sifrim Alejandro, Hitz Marc Phillip, Wilsdon Anna, Breckpot Jeroen, Al Turki Saeed H., Thienpont Bernard, McRae Jeremy, et al. 2016. “Distinct Genetic Architectures for Syndromic and Nonsyndromic Congenital Heart Defects Identified by Exome Sequencing.” Nature Genetics. 10.1038/ng.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon Delphine, Laloo Benoit, Barillot Malika, Barnetche Thomas, Blanchard Camille, Rooryck Caroline, Marche Michèle, et al. 2010. “A Mutation in the 3’-UTR of the HDAC6 Gene Abolishing the Post-Transcriptional Regulation Mediated by Hsa-MiR-433 Is Linked to a New Form of Dominant X-Linked Chondrodysplasia.” Human Molecular Genetics 19 (10): 2015–27. 10.1093/hmg/ddq083. [DOI] [PubMed] [Google Scholar]
- Simpson Michael A, Deshpande Charu, Dafou Dimitra, Vissers Lisenka E L M, Woollard Wesley J, Holder Susan E, Gillessen-Kaesbach Gabriele, et al. 2012. “De Novo Mutations of the Gene Encoding the Histone Acetyltransferase KAT6B Cause Genitopatellar Syndrome.” American Journal of Human Genetics 90 (2): 290–94. 10.1016/j.ajhg.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Sluijs Pleuntje J., Jansen Sandra, Vergano Samantha A., Adachi-Fukuda Miho, Alanay Yasemin, AlKindy Adila, Baban Anwar, et al. 2019. “The ARID1B Spectrum in 143 Patients: From Nonsyndromic Intellectual Disability to Coffin–Siris Syndrome.” Genetics in Medicine. 10.1038/s41436-018-0330-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blok Snijders, Lot Justine Rousseau, Twist Joanna, Ehresmann Sophie, Takaku Motoki, Venselaar Hanka, Rodan Lance H., et al. 2018. “CHD3 Helicase Domain Mutations Cause a Neurodevelopmental Syndrome with Macrocephaly and Impaired Speech and Language.” Nature Communications. 10.1038/s41467-018-06014-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F, Chen P, Sun D, Wang M, Dong L, Liang D, Xu R-M, Zhu P, and Li G. 2014. “Cryo-EM Study of the Chromatin Fiber Reveals a Double Helix Twisted by Tetranucleosomal Units.” Science 344 (6182): 376–80. 10.1126/science.1251413. [DOI] [PubMed] [Google Scholar]
- Sotos JF, Dodge PR, Muirhead D, Crawford JD, and Talbot NB. 1964. “CEREBRAL GIGANTISM IN CHILDHOOD. A SYNDROME OF EXCESSIVELY RAPID GROWTH AND ACROMEGALIC FEATURES AND A NONPROGRESSIVE NEUROLOGIC DISORDER.” The New England Journal of Medicine 271 (July): 109–16. 10.1056/NEJM196407162710301. [DOI] [PubMed] [Google Scholar]
- Sousa Sérgio B., Hennekam Raoul C., Abdul-Rahman Omar, Alders Marielle, Azzarello-Burri Silvia, Bottani Armand, Bowdin Sarah, et al. 2014. “Phenotype and Genotype in Nicolaides-Baraitser Syndrome.” American Journal of Medical Genetics, Part C: Seminars in Medical Genetics. 10.1002/ajmg.c.31409. [DOI] [PubMed] [Google Scholar]
- Stankiewicz Paweł, Khan Tahir N., Szafranski Przemyslaw, Slattery Leah, Streff Haley, Vetrini Francesco, Bernstein Jonathan A., et al. 2017. “Haploinsufficiency of the Chromatin Remodeler BPTF Causes Syndromic Developmental and Speech Delay, Postnatal Microcephaly, and Dysmorphic Features.” American Journal of Human Genetics 101 (4): 503–15. 10.1016/j.ajhg.2017.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stec I, Wright TJ, van Ommen GJ, de Boer PA, van Haeringen A, Moorman AF, Altherr MR, and den Dunnen JT. 1998. “WHSC1, a 90 Kb SET Domain-Containing Gene, Expressed in Early Development and Homologous to a Drosophila Dysmorphy Gene Maps in the Wolf-Hirschhorn Syndrome Critical Region and Is Fused to IgH in t(4;14) Multiple Myeloma.” Human Molecular Genetics 7 (7): 1071–82. 10.1093/hmg/7.7.1071. [DOI] [PubMed] [Google Scholar]
- Steiner CE, and Marques AP. 2000. “Growth Deficiency, Mental Retardation and Unusual Facies:” Clinical Dysmorphology 9 (2): 155–56. 10.1097/00019605-200009020-00021. [DOI] [PubMed] [Google Scholar]
- Stellacci Emilia, Onesimo Roberta, Bruselles Alessandro, Pizzi Simone, Battaglia Domenica, Leoni Chiara, Zampino Giuseppe, and Tartaglia Marco. 2016. “Congenital Immunodeficiency in an Individual with Wiedemann–Steiner Syndrome Due to a Novel Missense Mutation in KMT2A.” American Journal of Medical Genetics Part A 170 (9): 2389–93. 10.1002/ajmg.a.37681. [DOI] [PubMed] [Google Scholar]
- Stern Michael, Jensen Robert, and Herskowitz Ira. 1984. “Five SWI Genes Are Required for Expression of the HO Gene in Yeast.” Journal of Molecular Biology. 10.1016/0022-2836(84)90315-2. [DOI] [PubMed] [Google Scholar]
- Stessman Holly A F,, Xiong Bo, Coe Bradley P, Wang Tianyun, Hoekzema Kendra, Fenckova Michaela, Kvarnung Malin, et al. 2017. “Targeted Sequencing Identifies 91 Neurodevelopmental-Disorder Risk Genes with Autism and Developmental-Disability Biases.” Nature Genetics 49 (4): 515–26. 10.1038/ng.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steunou Anne-Lise, Rossetto Dorine, and Côté Jacques. 2013. “Fundamentals of Chromatin,” 147–212. 10.1007/978-1-4614-8624-4_4. [DOI] [Google Scholar]
- Stewart Douglas R., Huang Alina, Faravelli Francesca, Anderlid Britt-Marie, Medne Livija, Ciprero Karen, Kaur Maninder, et al. 2004. “Subtelomeric Deletions of Chromosome 9q: A Novel Microdeletion Syndrome: Subtelomeric 9q Deletion Syndrome.” American Journal of Medical Genetics Part A 128A (4): 340–51. 10.1002/ajmg.a.30136. [DOI] [PubMed] [Google Scholar]
- Strahl Brian D., and Allis C. David. 2000. “The Language of Covalent Histone Modifications.” Nature 403 (6765): 41–45. 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Stur E, Soares LA, and Louro ID. 2017. “SETD5 Gene Variant Associated with Mild Intellectual Disability - a Case Report.” Genetics and Molecular Research: GMR 16 (2). 10.4238/gmr16029615. [DOI] [PubMed] [Google Scholar]
- Suls Arvid, Jaehn Johanna A., Kecskés Angela, Weber Yvonne, Weckhuysen Sarah, Craiu Dana C., Siekierska Aleksandra, et al. 2013. “De Novo Loss-of-Function Mutations in CHD2 Cause a Fever-Sensitive Myoclonic Epileptic Encephalopathy Sharing Features with Dravet Syndrome.” American Journal of Human Genetics. 10.1016/j.ajhg.2013.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Bin, Chang Eunhyuk, Gerhartl Anna, and Szele Francis G. 2018. “Polycomb Protein Eed Is Required for Neurogenesis and Cortical Injury Activation in the Subventricular Zone.” Cerebral Cortex (New York, NY) 28 (4): 1369–82. 10.1093/cercor/bhx289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Xiao-Jian, Wei Ju, Wu Xin-Yan, Hu Ming, Wang Lan, Wang Hai-Hong, Zhang Qing-Hua, Chen Sai-Juan, Huang Qiu-Hua, and Chen Zhu. 2005. “Identification and Characterization of a Novel Human Histone H3 Lysine 36-Specific Methyltransferase.” The Journal of Biological Chemistry 280 (42): 35261–71. 10.1074/jbc.M504012200. [DOI] [PubMed] [Google Scholar]
- Sun Yu, Hu Guorui, Liu Huili, Zhang Xia, Huang Zhuo, Yan Hui, Wang Lili, Fan Yanjie, Gu Xuefan, and Yu Yongguo. 2017. “Further Delineation of the Phenotype of Truncating KMT2A Mutations: The Extended Wiedemann-Steiner Syndrome.” American Journal of Medical Genetics. Part A 173 (2): 510–14. 10.1002/ajmg.a.38025. [DOI] [PubMed] [Google Scholar]
- Swedish Schizophrenia Study, INTERVAL Study, DDD Study, UK10 K Consortium, Singh Tarjinder, Kurki Mitja I, Curtis David, et al. 2016. “Rare Loss-of-Function Variants in SETD1A Are Associated with Schizophrenia and Developmental Disorders.” Nature Neuroscience 19 (4): 571–77. 10.1038/nn.4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani Mamta, Mei Pinchao, Fang Rui, Leonor Thiago, Rutenberg Michael, Shimizu Fumiko, Li Jing, Rao Anjana, and Shi Yujiang. 2007. “The Histone H3K4 Demethylase SMCX Links REST Target Genes to X-Linked Mental Retardation.” Nature 447 (7144): 601–5. 10.1038/nature05823. [DOI] [PubMed] [Google Scholar]
- Tanaka Yasunori, Naruse Ichiro, Hongo Takuya, Xu Ming-Jiang, Nakahata Tatsutoshi, Maekawa Toshio, and Ishii Shunsuke. 2000. “Extensive Brain Hemorrhage and Embryonic Lethality in a Mouse Null Mutant of CREB-Binding Protein.” Mechanisms of Development 95 (1–2): 133–45. 10.1016/s0925-4773(00)00360-9. [DOI] [PubMed] [Google Scholar]
- Tang Gang-Bin, Zeng Yu-Qiang, Liu Pei-Pei, Mi Ting-Wei, Zhang Shuang-Feng, Dai Shang-Kun, Tang Qing-Yuan, et al. 2017. “The Histone H3K27 Demethylase UTX Regulates Synaptic Plasticity and Cognitive Behaviors in Mice.” Frontiers in Molecular Neuroscience 10. 10.3389/fnmol.2017.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton-Brown Katrina, Loveday Chey, Yost Shawn, Clarke Matthew, Ramsay Emma, Zachariou Anna, Elliott Anna, et al. 2017. “Mutations in Epigenetic Regulation Genes Are a Major Cause of Overgrowth with Intellectual Disability.” American Journal of Human Genetics 100 (5): 725–36. 10.1016/j.ajhg.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessadori Federico, Giltay Jacques C., Hurst Jane A., Massink Maarten P., Duran Karen, Vos Harmjan R., van Es Robert M, et al. 2017. “Germline Mutations Affecting the Histone H4 Core Cause a Developmental Syndrome by Altering DNA Damage Response and Cell Cycle Control.” Nature Genetics 49 (11): 1642–46. 10.1038/ng.3956. [DOI] [PubMed] [Google Scholar]
- Tessadori Federico, Rehman Atteeq U, Giltay Jacques C, Xia Fan, Streff Haley, Duran Karen, Bakkers Jeroen, Lalani Seema R, and van Haaften Gijs. 2019. “A de Novo Variant in the Human HIST1H4J Gene Causes a Syndrome Analogous to the HIST1H4C-Associated Neurodevelopmental Disorder.” European Journal of Human Genetics 28 (5): 674–78. 10.1038/s41431-019-0552-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tham Emma, Lindstrand Anna, Santani Avni, Malmgren Helena, Nesbitt Addie, Dubbs Holly A, Zackai Elaine H, et al. 2015. “Dominant Mutations in KAT6A Cause Intellectual Disability with Recognizable Syndromic Features.” American Journal of Human Genetics 96 (3): 507–13. 10.1016/j.ajhg.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trider Carrie Lee, Arra-Robar Angela, van Ravenswaaij-Arts Conny, and Blake Kim. 2017. “Developing a CHARGE Syndrome Checklist: Health Supervision across the Lifespan (from Head to Toe).” American Journal of Medical Genetics, Part A. 10.1002/ajmg.a.38085. [DOI] [PubMed] [Google Scholar]
- Truty Rebecca, Patil Nila, Sankar Raman, Sullivan Joseph, Millichap John, Carvill Gemma, Entezam Ali, et al. 2019. “Possible Precision Medicine Implications from Genetic Testing Using Combined Detection of Sequence and Intragenic Copy Number Variants in a Large Cohort with Childhood Epilepsy.” Epilepsia Open. 10.1002/epi4.12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui David, Voronova Anastassia, Gallagher Denis, Kaplan David R, Miller Freda D, and Wang Jing. 2013. “CBP Regulates the Differentiation of Interneurons from Ventral Forebrain Neural Precursors during Murine Development.” Developmental Biology 385 (2): 230–41. 10.1016/j.ydbio.2013.11.005. [DOI] [PubMed] [Google Scholar]
- Tsukahara M, Kuroki Y, Imaizumi K, Miyazawa Y, and Matsuo K. 1997. “Dominant Inheritance of Kabuki Make-up Syndrome.” American Journal of Medical Genetics 73 (1): 19–23. [PubMed] [Google Scholar]
- Tunovic Sanjin, Barkovich James, Sherr Elliott H., and Slavotinek Anne M.. 2014. “De Novo ANKRD11 and KDM1A Gene Mutations in a Male with Features of KBG Syndrome and Kabuki Syndrome.” American Journal of Medical Genetics. Part A 164A (7): 1744–49. 10.1002/ajmg.a.36450. [DOI] [PubMed] [Google Scholar]
- Türkmen Seval, Gillessen-Kaesbach Gabriele, Meinecke Peter, Albrecht Beate, Neumann Luitgard M., Hesse Volker, Palanduz Sükrü, et al. 2003. “Mutations in NSD1 Are Responsible for Sotos Syndrome, but Are Not a Frequent Finding in Other Overgrowth Phenotypes.” European Journal of Human Genetics: EJHG 11 (11): 858–65. 10.1038/sj.ejhg.5201050. [DOI] [PubMed] [Google Scholar]
- Ullah Mukta, Pelletier Nadine, Xiao Lin, Zhao Song Ping, Wang Kainan, Degerny Cindy, Tahmasebi Soroush, et al. 2009. “Molecular Architecture of Quartet MOZ/MORF Histone Acetyltransferase Complexes.” Molecular and Cellular Biology 29 (3): 942–942. 10.1128/mcb.01901-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallianatos Christina N., Raines Brynne, Porter Robert S., Bonefas Katherine M., Wu Michael C., Garay Patricia M., Collette Katie M., et al. 2020. “Mutually Suppressive Roles of KMT2A and KDM5C in Behaviour, Neuronal Structure, and Histone H3K4 Methylation.” Communications Biology 3 (1): 1–14. 10.1038/s42003-020-1001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valor LM, Pulopulos MM, Jimenez-Minchan M, Olivares R, Lutz B, and Barco A. 2011. “Ablation of CBP in Forebrain Principal Neurons Causes Modest Memory and Transcriptional Defects and a Dramatic Reduction of Histone Acetylation But Does Not Affect Cell Viability.” Journal of Neuroscience 31 (5): 1652–63. 10.1523/jneurosci.4737-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laarhoven Peter M., Neitzel Leif R., Quintana Anita M, Geiger Elizabeth A., Zackai Elaine H., Clouthier David E., Artinger Kristin B., Ming Jeffrey E., and Shaikh Tamim H.. 2015. “Kabuki Syndrome Genes KMT2D and KDM6A : Functional Analyses Demonstrate Critical Roles in Craniofacial, Heart and Brain Development.” Human Molecular Genetics 24 (15): 4443–53. 10.1093/hmg/ddv180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasileiou Georgia, Ekici Arif B., Uebe Steffen, Zweier Christiane, Hoyer Juliane, Engels Hartmut, Behrens Jürgen, Reis André, and Hadjihannas Michel V.. 2015. “Chromatin-Remodeling-Factor ARID1B Represses Wnt/β-Catenin Signaling.” American Journal of Human Genetics. 10.1016/j.ajhg.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasileiou Georgia, Vergarajauregui Silvia, Endele Sabine, Popp Bernt, Büttner Christian, Ekici Arif B., Gerard Marion, et al. 2018. “Mutations in the BAF-Complex Subunit DPF2 Are Associated with Coffin-Siris Syndrome.” American Journal of Human Genetics. 10.1016/j.ajhg.2018.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veredice Chiara, Bianco Flaviana, Contaldo Ilaria, Orteschi Daniela, Stefanini Maria Chiara, Battaglia Domenica, Lettori Donatella, Guzzetta Francesco, and Zollino Marcella. 2009. “Early Onset Myoclonic Epilepsy and 15q26 Microdeletion: Observation of the First Case.” Epilepsia. 10.1111/j.1528-1167.2009.02078.x. [DOI] [PubMed] [Google Scholar]
- Vermeulen Michiel, Eberl H. Christian, Matarese Filomena, Marks Hendrik, Denissov Sergei, Butter Falk, Lee Kenneth K., et al. 2010. “Quantitative Interaction Proteomics and Genome-Wide Profiling of Epigenetic Histone Marks and Their Readers.” Cell 142 (6): 967–80. 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- Villavicencio-Lorini Pablo, Klopocki Eva, Trimborn Marc, Koll Randi, Mundlos Stefan, and Horn Denise. 2013. “Phenotypic Variant of Brachydactyly-Mental Retardation Syndrome in a Family with an Inherited Interstitial 2q37.3 Microdeletion Including HDAC4.” European Journal of Human Genetics: EJHG 21 (7): 743–48. 10.1038/ejhg.2012.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viosca Jose, Lopez-Atalaya Jose P, Olivares Roman, Eckner Richard, and Barco Angel. 2009. “Syndromic Features and Mild Cognitive Impairment in Mice with Genetic Reduction on P300 Activity: Differential Contribution of P300 and CBP to Rubinstein-Taybi Syndrome Etiology.” Neurobiology of Disease 37 (1): 186–94. 10.1016/j.nbd.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Visser Remco, Shimokawa Osamu, Harada Naoki, Kinoshita Akira, Ohta Tohru, Niikawa Norio, and Matsumoto Naomichi. 2005. “Identification of a 3.0-Kb Major Recombination Hotspot in Patients with Sotos Syndrome Who Carry a Common 1.9-Mb Microdeletion.” The American Journal of Human Genetics 76 (1): 52–67. 10.1086/426950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers Lisenka E.L.M., Van Ravenswaaij Conny M.A., Admiraal Ronald, Hurst Jane A., De Vries Bert B.A., Janssen Irene M., Van Der Vliet Walter A., et al. 2004. “Mutations in a New Member of the Chromodomain Gene Family Cause CHARGE Syndrome.” Nature Genetics. 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- Voigt Philipp, Tee Wee-Wei, and Reinberg Danny. 2013. “A Double Take on Bivalent Promoters.” Genes & Development 27 (12): 1318–38. 10.1101/gad.219626.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss Anne K, Collin Caitlin, Dixon Mathew P, and Thomas Tim. 2009. “Moz and Retinoic Acid Coordinately Regulate H3K9 Acetylation, Hox Gene Expression, and Segment Identity.” Developmental Cell 17 (5): 674–86. 10.1016/j.devcel.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Wagner Eric J., and Carpenter Phillip B.. 2012. “Understanding the Language of Lys36 Methylation at Histone H3.” Nature Reviews. Molecular Cell Biology 13 (2): 115–26. 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner Victoria F., Hillman Paul R., Britt Allison D., Ray Joseph W., and Farach Laura S.. 2019. “A De Novo HDAC2 Variant in a Patient with Features Consistent with Cornelia de Lange Syndrome Phenotype.” American Journal of Medical Genetics. Part A 179 (5): 852–56. 10.1002/ajmg.a.61101. [DOI] [PubMed] [Google Scholar]
- Walsh Ryan M., Shen Erica Y., Bagot Rosemary C., Anselmo Anthony, Jiang Yan, Javidfar Behnam, Wojtkiewicz Gregory J., et al. 2017. “Phf8 Loss Confers Resistance to Depression-like and Anxiety-like Behaviors in Mice.” Nature Communications 8 (May): 15142. 10.1038/ncomms15142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Chaochen, Lee Ji-Eun, Cho Young-Wook, Xiao Ying, Jin Qihuang, Liu Chengyu, and Ge Kai. 2012. “UTX Regulates Mesoderm Differentiation of Embryonic Stem Cells Independent of H3K27 Demethylase Activity.” Proceedings of the National Academy of Sciences 109 (38): 15324–29. 10.1073/pnas.1204166109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Gang G., Cai Ling, Pasillas Martina P., and Kamps Mark P.. 2007. “NUP98-NSD1 Links H3K36 Methylation to Hox-A Gene Activation and Leukaemogenesis.” Nature Cell Biology 9 (7): 804–12. 10.1038/ncb1608. [DOI] [PubMed] [Google Scholar]
- Wang Jiajia, Yang Lijun, Dong Chen, Wang Jincheng, Xu Lingli, Qiu Yueping, Weng Qinjie, Zhao Chuntao, Xin Mei, and Lu Q. Richard. 2020. “EED-Mediated Histone Methylation Is Critical for CNS Myelination and Remyelination by Inhibiting WNT, BMP, and Senescence Pathways.” Science Advances 6 (33): eaaz6477. 10.1126/sciadv.aaz6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Jing, Weaver Ian C G, Gauthier-Fisher Andrée, Wang Haoran, He Ling, Yeomans John, Wondisford Frederic, Kaplan David R, and Miller Freda D. 2010. “CBP Histone Acetyltransferase Activity Regulates Embryonic Neural Differentiation in the Normal and Rubinstein-Taybi Syndrome Brain.” Developmental Cell 18 (1): 114–25. 10.1016/j.devcel.2009.10.023. [DOI] [PubMed] [Google Scholar]
- Wang Shu-Ping, Tang Zhanyun, Chen Chun-Wei, Shimada Miho, Koche Richard P., Wang Lan-Hsin, Nakadai Tomoyoshi, et al. 2017. “A UTX-MLL4-P300 Transcriptional Regulatory Network Coordinately Shapes Active Enhancer Landscapes for Eliciting Transcription.” Molecular Cell 67 (2): 308–321.e6. 10.1016/j.molcel.2017.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Tianyun, Guo Hui, Xiong Bo, Stessman Holly A.F., Wu Huidan, Coe Bradley P., Turner Tychele N., et al. 2016. “De Novo Genic Mutations among a Chinese Autism Spectrum Disorder Cohort.” Nature Communications 7 (1): 13316. 10.1038/ncomms13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Côté J, Xue Y, Zhou S, Khavari PA, Biggar SR, Muchardt C, et al. 1996. “Purification and Biochemical Heterogeneity of the Mammalian SWI-SNF Complex.” The EMBO Journal. 10.1002/anie.200805483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Zhibin, Zang Chongzhi, Rosenfeld Jeffrey A, Schones Dustin E, Barski Artem, Cuddapah Suresh, Cui Kairong, et al. 2008. “Combinatorial Patterns of Histone Acetylations and Methylations in the Human Genome.” Nature Genetics 40 (7): 897–903. 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver DD, Graham CB, Thomas IT, and Smith DW. 1974. “A New Overgrowth Syndrome with Accelerated Skeletal Maturation, Unusual Facies, and Camptodactyly.” The Journal of Pediatrics 84 (4): 547–52. 10.1016/s0022-3476(74)80675-x. [DOI] [PubMed] [Google Scholar]
- Wei Gengze, Deng Xinxian, Agarwal Saurabh, Iwase Shigeki, Disteche Christine, and Xu Jun. 2016. “Patient Mutations of the Intellectual Disability Gene KDM5C Downregulate Netrin G2 and Suppress Neurite Growth in Neuro2a Cells.” Journal of Molecular Neuroscience: MN 60 (1): 33–45. 10.1007/s12031-016-0770-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weider Matthias, Küspert Melanie, Bischof Melanie, Vogl Michael R., Hornig Julia, Loy Kristina, Kosian Thomas, et al. 2012. “Chromatin-Remodeling Factor Brg1 Is Required for Schwann Cell Differentiation and Myelination.” Developmental Cell. 10.1016/j.devcel.2012.05.017. [DOI] [PubMed] [Google Scholar]
- Weinberg Daniel N., Papillon-Cavanagh Simon, Chen Haifen, Yue Yuan, Chen Xiao, Rajagopalan Kartik N., Horth Cynthia, et al. 2019. “The Histone Mark H3K36me2 Recruits DNMT3A and Shapes the Intergenic DNA Methylation Landscape.” Nature 573 (7773): 281–86. 10.1038/s41586-019-1534-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss Karin, Lazar Hayley P., Kurolap Alina, Martinez Ariel F., Paperna Tamar, Cohen Lior, Smeland Marie F., et al. 2020. “The CHD4-Related Syndrome: A Comprehensive Investigation of the Clinical Spectrum, Genotype–Phenotype Correlations, and Molecular Basis.” Genetics in Medicine. 10.1038/s41436-019-0612-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss Karin, Terhal Paulien A., Cohen Lior, Bruccoleri Michael, Irving Melita, Martinez Ariel F., Rosenfeld Jill A., et al. 2016. “De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms.” American Journal of Human Genetics. 10.1016/j.ajhg.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler Patricia G., Huang Dongli, and Dai Zunyan. 2014. “Haploinsufficiency of HDAC4 Does Not Cause Intellectual Disability in All Affected Individuals.” American Journal of Medical Genetics. Part A 164A (7): 1826–29. 10.1002/ajmg.a.36542. [DOI] [PubMed] [Google Scholar]
- Williams Stephen R, Aldred Micheala A, Der Kaloustian Vazken M, Halal Fahed, Gowans Gordon, McLeod D Ross, Zondag Sara, Toriello Helga V, Magenis R Ellen, and Elsea Sarah H. n.d. “Haploinsufficiency of HDAC4 Causes Brachydactyly Mental Retardation Syndrome, with Brachydactyly Type E, Developmental Delays, and Behavioral Problems.” American Journal of Human Genetics 87 (2): 219–28. 10.1016/j.ajhg.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Yang, Zhu Ziqi, Han Gang, Ye Xiaolei, Xu Bo, Peng Zhouchun, Ma Yuanjun, et al. 2007. “JARID1B Is a Histone H3 Lysine 4 Demethylase Up-Regulated in Prostate Cancer.” Proceedings of the National Academy of Sciences of the United States of America 104 (49): 19226–31. 10.1073/pnas.0700735104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Bing, Wilson Jonathan R, and Gamblin Steven J. 2003. “SET Domains and Histone Methylation.” Current Opinion in Structural Biology 13 (6): 699–705. 10.1016/j.sbi.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Xu Lichao, Zheng Yue, Li Xuejing, Wang Andi, Huo Dawei, Li Qinglan, Wang Shikang, et al. 2021. “Abnormal Neocortex Arealization and Sotos-like Syndrome-Associated Behavior in Setd2 Mutant Mice.” Science Advances 7 (1). 10.1126/sciadv.aba1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Qianhua, Xiang Yunlong, Wang Qiujun, Wang Leyun, Brind’Amour Julie, Bogutz Aaron Blair, Zhang Yu, et al. 2019. “SETD2 Regulates the Maternal Epigenome, Genomic Imprinting and Embryonic Development.” Nature Genetics 51 (5): 844–56. 10.1038/s41588-019-0398-7. [DOI] [PubMed] [Google Scholar]
- Yamada Ayumi, Shimura Chikako, and Shinkai Yoichi. 2018. “Biochemical Validation of EHMT1 Missense Mutations in Kleefstra Syndrome.” Journal of Human Genetics 63 (5): 555–62. 10.1038/s10038-018-0413-3. [DOI] [PubMed] [Google Scholar]
- Yamada Tomoko, Yang Yue, Hemberg Martin, Yoshida Toshimi, Cho Ha Young, Murphy J. Patrick, Fioravante Diasynou, et al. 2014. “Promoter Decommissioning by the NuRD Chromatin Remodeling Complex Triggers Synaptic Connectivity in the Mammalian Brain.” Neuron. 10.1016/j.neuron.2014.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada-Okabe Toshiko, Imamura Kentaro, Kawaguchi Nanami, Sakai Haruya, Yamashita Michiaki, and Matsumoto Naomichi. 2010. “Functional Characterization of the Zebrafish WHSC1-Related Gene, a Homolog of Human NSD2.” Biochemical and Biophysical Research Communications 402 (2): 335–39. 10.1016/j.bbrc.2010.10.027. [DOI] [PubMed] [Google Scholar]
- Yan Kezhi, Rousseau Justine, Littlejohn Rebecca Okashah, Kiss Courtney, Lehman Anna, Rosenfeld Jill A, Stumpel Constance T R, et al. 2016. “Mutations in the Chromatin Regulator Gene BRPF1 Cause Syndromic Intellectual Disability and Deficient Histone Acetylation.” American Journal of Human Genetics 100 (1): 91–104. 10.1016/j.ajhg.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Kezhi, Rousseau Justine, Machol Keren, Cross Laura A, Agre Katherine E, Gibson Cynthia Forster, Goverde Anne, et al. 2020. “Deficient Histone H3 Propionylation by BRPF1-KAT6 Complexes in Neurodevelopmental Disorders and Cancer.” Science Advances 6 (4): eaax0021. 10.1126/sciadv.aax0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Qingsheng, Dutt Shilpee, Xu Rong, Graves Katherine, Juszczynski Przemyslaw, Manis John P., and Shipp Margaret A.. 2009. “BBAP Monoubiquitylates Histone H4 at Lysine 91 and Selectively Modulates the DNA Damage Response.” Molecular Cell 36 (1): 110–20. 10.1016/j.molcel.2009.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Xiaohan, Yu Wenhua, Shi Lei, Sun Luyang, Liang Jing, Yi Xia, Li Qian, et al. 2011. “HAT4, a Golgi Apparatus-Anchored B-Type Histone Acetyltransferase, Acetylates Free Histone H4 and Facilitates Chromatin Assembly.” Molecular Cell 44 (1): 39–50. 10.1016/j.molcel.2011.07.032. [DOI] [PubMed] [Google Scholar]
- Yao Hui, Hannum Douglas F., Zhai Yiwen, Hill Sophie F., Albanus Ricardo D.’Oliveira, Lou Wenjia, Skidmore Jennifer M., et al. 2020. “CHD7 Promotes Neural Progenitor Differentiation in Embryonic Stem Cells via Altered Chromatin Accessibility and Nascent Gene Expression.” Scientific Reports. 10.1038/s41598-020-74537-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Tso-Pang, Oh Suk P, Fuchs Miriam, Zhou Nai-Dong, Ch’ng Lian-Ee, Newsome David, Bronson Roderick T, Li En, Livingston David M, and Eckner Richard. 1998. “Gene Dosage–Dependent Embryonic Development and Proliferation Defects in Mice Lacking the Transcriptional Integrator P300.” Cell 93 (3): 361–72. 10.1016/s0092-8674(00)81165-4. [DOI] [PubMed] [Google Scholar]
- Constantin d’ Ydewalle, Bogaert Elke, and Van Den Bosch Ludo. 2012. “HDAC6 at the Intersection of Neuroprotection and Neurodegeneration.” Traffic (Copenhagen, Denmark) 13 (6): 771–79. 10.1111/j.1600-0854.2012.01347.x. [DOI] [PubMed] [Google Scholar]
- Ye Jianxin, Ai Xi, Eugeni Ericka E., Zhang Liwen, Carpenter Laura Rocco, Jelinek Mary A., Freitas Michael A., and Parthun Mark R.. 2005. “Histone H4 Lysine 91 Acetylation: A Core Domain Modification Associated with Chromatin Assembly.” Molecular Cell 18 (1): 123–30. 10.1016/j.molcel.2005.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Linya, Yan Kezhi, Zhou Jinfeng, Zhao Hong, Bertos Nicholas R., Park Morag, Wang Edwin, and Yang Xiang-Jiao. 2015. “The Lysine Acetyltransferase Activator Brpf1 Governs Dentate Gyrus Development through Neural Stem Cells and Progenitors.” PLOS Genetics 11 (3): e1005034. 10.1371/journal.pgen.1005034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Linya, Zou Jinfeng, Zhao Hong, Bertos Nicholas R., Park Morag, Wang Edwin, and Yang Xiang-Jiao. 2015. “Deficiency of the Chromatin Regulator Brpf1 Causes Abnormal Brain Development.” Journal of Biological Chemistry 290 (11): 7114–29. 10.1074/jbc.m114.635250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Xiuya, Yang Lin, Li Jin, Li Wanxing, Li Dongzhi, Wang Ran, Wu Kai, et al. 2019. “De Novo and Inherited SETD1A Variants in Early-Onset Epilepsy.” Neuroscience Bulletin 35 (6): 1045–57. 10.1007/s12264-019-00400-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Yang, Chen Ying, Kim Bongwoo, Wang Haibo, Zhao Chuntao, He Xuelian, Liu Lei, et al. 2013. “Olig2 Targets Chromatin Remodelers to Enhancers to Initiate Oligodendrocyte Differentiation.” Cell. 10.1016/j.cell.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Bo, Pehlivan Davut, Karaca Ender, Patel Nisha, Charng Wu-Lin, Gambin Tomasz, Gonzaga-Jauregui Claudia, et al. 2015. “Global Transcriptional Disturbances Underlie Cornelia de Lange Syndrome and Related Phenotypes.” The Journal of Clinical Investigation 125 (2): 636–51. 10.1172/JCI77435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zech Michael, Boesch Sylvia, Maier Esther M., Borggraefe Ingo, Vill Katharina, Laccone Franco, Pilshofer Veronika, et al. 2016. “Haploinsufficiency of KMT2B, Encoding the Lysine-Specific Histone Methyltransferase 2B, Results in Early-Onset Generalized Dystonia.” American Journal of Human Genetics 99 (6): 1377–87. 10.1016/j.ajhg.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zech Michael, Jech Robert, Havránková Petra, Fečíková Anna, Berutti Riccardo, Urgošík Dušan, Kemlink David, et al. 2017. “KMT2B Rare Missense Variants in Generalized Dystonia.” Movement Disorders: Official Journal of the Movement Disorder Society 32 (7): 1087–91. 10.1002/mds.27026. [DOI] [PubMed] [Google Scholar]
- Zech Michael, Lam Daniel D., and Winkelmann Juliane. 2019. “Update on KMT2B-Related Dystonia.” Current Neurology and Neuroscience Reports 19 (11): 92. 10.1007/s11910-019-1007-y. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ji F, Liu Y, Lei X, Li H, Ji G, Yuan Z, and Jiao J. 2014. “Ezh2 Regulates Adult Hippocampal Neurogenesis and Memory.” Journal of Neuroscience 34 (15): 5184–99. 10.1523/JNEUROSCI.4129-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y 2001. “Transcription Regulation by Histone Methylation: Interplay between Different Covalent Modifications of the Core Histone Tails.” Genes & Development 15 (18): 2343–60. 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- Zhao Linnan, Li Jun, Ma Yuanlin, Wang Jiutao, Pan Wen, Gao Kai, Zhang Zhengrong, et al. 2015. “Ezh2 Is Involved in Radial Neuronal Migration through Regulating Reelin Expression in Cerebral Cortex.” Scientific Reports 5 (1): 15484. 10.1038/srep15484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Qiong, Obana Edwin A., Radomski Kryslaine L., Sukumar Gauthaman, Wynder Christopher, Dalgard Clifton L., and Doughty Martin L.. 2016. “Inhibition of the Histone Demethylase Kdm5b Promotes Neurogenesis and Derepresses Reln (Reelin) in Neural Stem Cells from the Adult Subventricular Zone of Mice.” Molecular Biology of the Cell 27 (4): 627–39. 10.1091/mbc.E15-07-0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Tao, Liang Chen, Li Dongdong, Tian Miaomiao, Liu Sanxiong, Gao Guanjun, and Guan Ji-Song. 2016. “Histone Methyltransferase Ash1L Mediates Activity-Dependent Repression of Neurexin-1α.” Scientific Reports 6 (1): 26597. 10.1038/srep26597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zibetti Cristina, Adamo Antonio, Binda Claudia, Forneris Federico, Toffolo Emanuela, Verpelli Chiara, Ginelli Enrico, Mattevi Andrea, Sala Carlo, and Battaglioli Elena. 2010. “Alternative Splicing of the Histone Demethylase LSD1/KDM1 Contributes to the Modulation of Neurite Morphogenesis in the Mammalian Nervous System.” Journal of Neuroscience 30 (7): 2521–32. 10.1523/JNEUROSCI.5500-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zollino Marcella, Orteschi Daniela, Murdolo Marina, Lattante Serena, Battaglia Domenica, Stefanini Chiara, Mercuri Eugenio, Chiurazzi Pietro, Neri Giovanni, and Marangi Giuseppe. 2012. “Mutations in KANSL1 Cause the 17q21.31 Microdeletion Syndrome Phenotype.” Nature Genetics 44 (6): 636–38. 10.1038/ng.2257. [DOI] [PubMed] [Google Scholar]
- N.d. 10.1002/ajmg.a.38616. [DOI]