

Abstract
Installation of methyl groups can significantly improve the binding of small-molecule drugs to protein targets; however, site-selective methylation often presents a significant synthetic challenge. Metal- and S-adenosyl-methionine (SAM)-dependent methyltransferases (MTs) in natural-product biosynthetic pathways are powerful enzymatic tools for selective or chemically challenging C-methylation reactions. Each of these MTs selectively catalyzes one or two methyl transfer reactions. Crystal structures and biochemical assays of the Mn2+-dependent monomethyltransferase from the saxitoxin biosynthetic pathway (SxtA MT) revealed the structural basis for control of methylation extent. The SxtA monomethyltransferase was converted to a dimethyltransferase by modification of the metal binding site, addition of an active site base, and an amino acid substitution to provide space in the substrate pocket for two methyl substituents. A reciprocal change converted a related dimethyltransferase into a monomethyltransferase, supporting our hypothesis that steric hindrance can prevent a second methylation event. Novel understanding of MTs will accelerate the development of MT-based catalysts and MT engineering for the use in small molecule synthesis.
INTRODUCTION
Methylation is a widely used modification in drug discovery due to the so called “magic methyl effect”1, 2 that can boost drug potency up to 2000-fold. A simple methyl group can modulate many properties of a drug molecule. For instance, ibufenac is associated with high rates of idiosyncratic hepatotoxicity, while ibuprofen, which differs from ibufenac by only one methyl group, is among the most effective and widely used non-steroidal anti-inflammatory drugs3. As a result of the additional methyl substituent, ibuprofen has slower acyl glucuronide migration, reduced toxicity and increased half-life relative to ibufenac4.
Although a methyl group is a small functional group, C–H methylation is challenging to accomplish using traditional chemical methods5. For example, many compounds lack an acidic C–H at the target position so that installation of a methyl group requires the de novo synthesis route. Additional challenges exist in controlling the extent of methylation and the site- and stereoselectivity of the reaction5. In contrast to chemical methods, methyltransferases involved in natural product biosynthesis accomplish selective installation, sometimes even on a variety of substrates6 (Figure 1a). Thus, biocatalytic approaches may be valuable, particularly for methylations where de novo synthesis or deconstruction and re-assembly are impractical. C-methyltransferases (C-MT) in polyketide synthase (PKS) pathways are potential tools for this purpose. Modular type I PKSs generate bioactive natural products through successive elongation and tailoring reactions using malonyl- (Mal-) or methylmalonyl- (MeMal-) coenzyme A (CoA) building blocks7, 8. C-MTs within PKS pathways can be useful methylation tools because they selectively perform one or two methylation reactions under mild conditions and achieve high levels of site- and stereo-selectivity9–12, 6. For example, methylation by AprA MT112 from the apratoxin A pathway yielded a high level of dimethylated products. Moreover, some of these C-MTs can act on a broad range of substrates in addition to their natural substrate6, demonstrating their utility as biocatalysts.
Figure 1. C–H methylation by methyltransferases from natural product biosynthesis pathways.

(a) Selective mono- and dimethylation by representative methyltransferases in natural product biosynthesis pathways (AprA, apratoxin A; Tal, myxovirescin; GphF, gephyronic acid; BryX, bryostatin; CurJ, curacin A. (b) Reaction scheme of SxtA MT and DC domains in saxitoxin biosynthesis. The product of the SxtA MT and TaI MT is inferred to be (S)-methylmalonyl-ACP, based on the structures of AprA MT1 substrate complexes12.
Saxitoxin (STX), a paralytic shellfish toxin produced by freshwater cyanobacteria and marine dinoflagellates, is one of the most potent naturally occurring neurotoxic alkaloids known13. The positively charged guanidium groups of STX interact with negatively charged amino acids in domains I and II of voltage-gated sodium channels14, 15, causing tissue-specific paralysis16, 14, 17. SXT derivatives have potential for development as selective neuropathic pain treatments18 directed to voltage-gated channels. However, the STX biosynthetic pathway has not been fully exploited despite considerable biochemical study9, 19–24. Barriers to a synthetic biology approach include lack of an effective heterologous host and limited access to analogs25. Thus, modification of saxitoxin to generate analogs would benefit from the chemoenzymatic approach26.
STX biosynthesis is initiated by a PKS-like loading module, SxtA (Figure 1b), which includes a rare Mn2+-dependent C-MT domain for monomethylation of malonyl-acyl carrier protein (Mal-ACP) to generate methylmalonyl (MeMal)-ACP9, 27. Following the SxtA MT reaction, an SxtA decarboxylase (DC) domain converts MeMal-ACP to propionyl-ACP, the starter unit for STX biosynthesis9, 27. SxtA DC is a member of the GCN5-related N-acetyltransferase (GNAT) superfamily, which includes predominantly acyltransferases and a few decarboxylases. Like the GNAT superfamily enzymes in the biosynthesis pathways for curacin A (CurA DC) and gephyronic acid (GphF DC), SxtA DC catalyzes only decarboxylation28, 29. We previously characterized the activity and structure of a related metal-dependent C-MT, AprA MT1, an S-adenosylmethionine (SAM) and Fe3+-dependent dimethyltransferase12. AprA MT1 is followed by an MT2 domain that catalyzes a third methyl transfer coupled to decarboxylation to afford the pivaloyl-ACP starter unit for apratoxin A biosynthesis30. An AprA pseudo-decarboxylase (ΨDC) located between MT1 and MT2 lacks catalytic activity. Despite their highly similar sequences (54% identity), SxtA MT and AprA MT1 differ in their metal selectivity (Mn2+ and Fe3+, respectively) and extent of methylation (one and two methyl transfers, respectively)12. In the present study, we characterized the activity and structure of the SxtA MT. Comparison of SxtA MT and AprA MT1 uncovered features that determine the selectivity of each enzyme for transfer of one or two methyl groups. The critical discovery was an amino acid side chain in the substrate pocket that provides a steric block to a second methyl transfer. We then engineered the SxtA MT-DC didomain to transfer two methyl groups and the AprA MT1-ΨDC to transfer a single methyl group. These results provide critical insights into engineering related C-MTs for functionalization of drug scaffolds.
RESULTS AND DICUSSION
Catalytic activities of SxtA MT-DC.
The methylation and decarboxylation activities of wild type SxtA MT-DC were investigated (Figure 2a–b). We first identified the boundaries of the SxtA methyltransferase (MT, residues 1–506) and decarboxylase (DC, residues 512–710) domains by reference to a multiple sequence alignment and crystal structures of the AprA MT19, 12, CurA DC28 and GphF DC29. We produced the recombinant SxtA MT-DC didomain (1–721) for biochemical study, as the excised MT domain was unstable without the DC. The AprA ACP domain was used to deliver substrates, as the excised SxtA ACP domain was unstable. The ACP was loaded with a malonyl (Mal) or methylmalonyl (MeMal) group via a non-specific phosphopantetheinyl transferase31, 32. The MT and DC catalytic activities were assayed by monitoring the distribution of acyl-ACP species using liquid chromatography-mass spectrometry (LC-MS) with quantification of ACP-bound species using a phosphopantethine (Ppant) ejection assay33. SAM-dependent methylation of Mal-ACP was absolutely dependent on added metal. The MT exhibited a strong Mn2+ preference, with 10-fold greater methylation under our assay conditions relative to Fe3+ (Figure 2a). A metal-free enzyme that cannot catalyze methyl transfer was used to evaluate the substrate selectivity of the SxtA DC with Mal-ACP or MeMal-ACP. SxtA DC decarboxylated MeMal-ACP but had no detectable activity with Mal-ACP (Figure 2b). In accord with the chemical structure of saxitoxin, SxtA MT-DC produced no detectable dimethylation products: dimethylmalonyl- (Me2Mal-) ACP or its decarboxylated product isobutyryl-ACP.
Figure 2. SxtA MT-DC catalysis and structure.
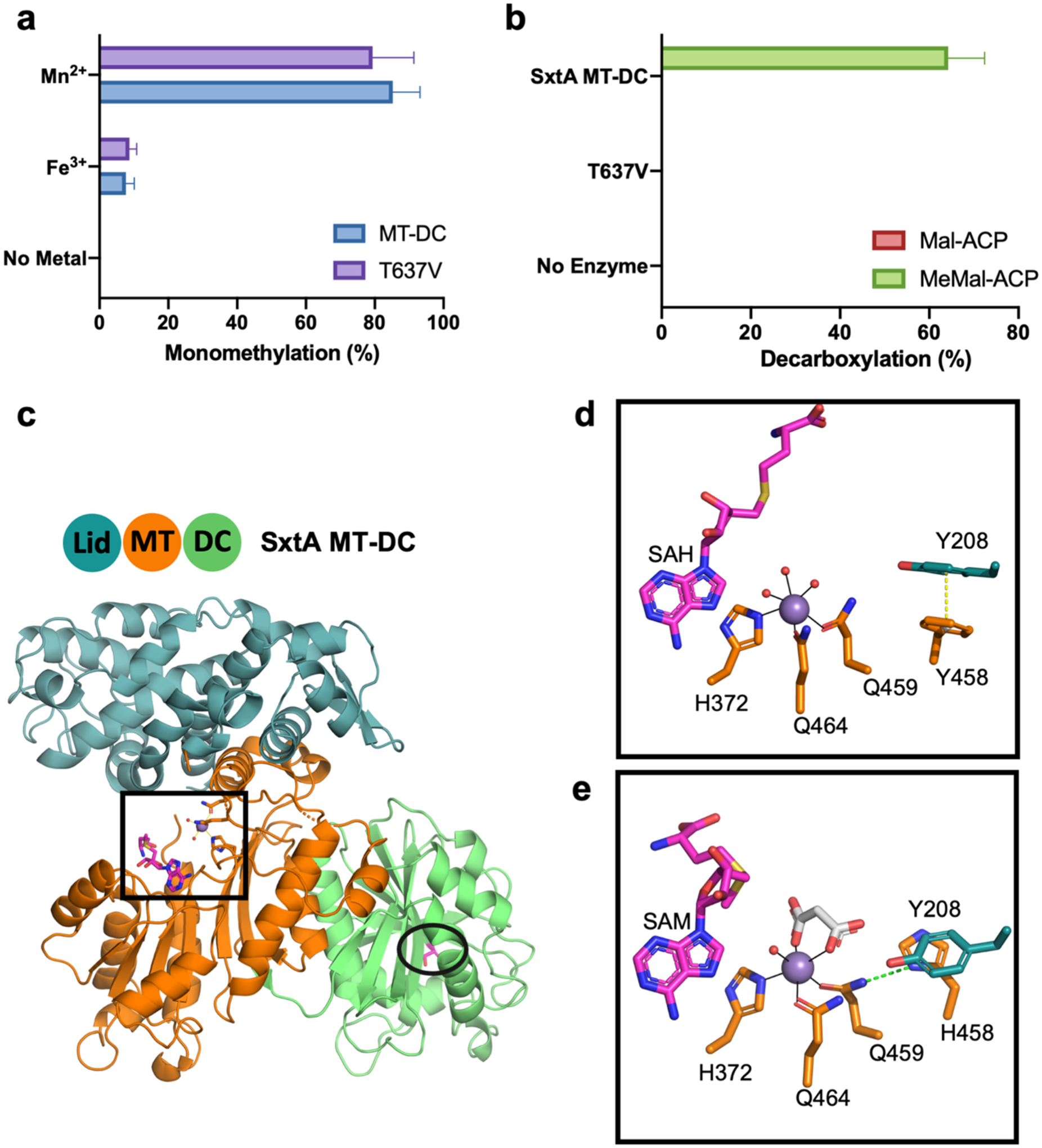
(a) SxtA MT-DC wild type and T637V methylation with added Mn2+, Fe3+ and no-metal control. Monomethylation was quantitated as percent of total Mal-ACP substrate converted to methylated products; no dimethylation products were detected. (b) SxtA MT-DC wild type and T637V decarboxylation with Mal-ACP and MeMal-ACP substrates. Decarboxylation was quantitated as percent of total Mal-ACP or MeMal-ACP substrate converted to acetyl- or propionyl-ACP. Error bars represent the standard deviation of >5 experiments. (c) Didomain ternary complex with Mn2+ (purple), SAH and Thr637 (magenta C) with domain coloring (blue MT lid, orange MT core, green DC). Active sites are boxed (MT) and circled (DC) in stick form (red O, blue N, yellow S). (d) SxtA MT active site with stacked Phe458 and Tyr208. SAH homocysteine is modeled; side chain C colored by domain of origin; waters in red. (e) MT active site of SxtA F458H. Malonate (white C) replaces two water ligands; SAM is modeled. His458 is hydrogen bonded to Gln459 ligand (dashed line).
To simplify the analysis of SxtA methylation activity, we eliminated decarboxylation activity by producing SxtA MT-DC T637V; Thr637 is analogous to the catalytic Thr in the CurA DC28. We confirmed that MT-DC T637V lacked decarboxylation activity (Figure 2b), as previously reported13, but retained methylation activity (79±12% in the standard assay) similar to the wild type MT-DC (85±8%) (Figure 2a). To facilitate quantitative comparison of methylation reactions, we determined assay conditions (time, temperature and concentration) in which the wild type enzyme methylated most of the Mal-ACP substrate (~85%). All comparative assays of wild type and mutagenized MT were performed identically under these conditions.
SxtA MT-DC wild type and F458H structures.
To identify and understand the features responsible for the single methyl transfer reaction catalyzed by SxtA MT, we solved a 2.5 Å crystal structure of the MT-DC di-domain (residues 1–710). The MT-DC was a monomer in solution and crystallized in the presence of Mn2+ and SAM with two molecules in the asymmetric unit (Table S1). The structure, solved by molecular replacement from the structures of AprA MT112 (54% sequence identity, PDB: 6B3A) and CurA DC28 (47% identity, PDB: 2REE), is a ternary complex with Mn2+ and S-adenosyl-l-homocysteine (SAH) (Figure 2c, SAM is modeled to show the predicted position of the methyl donor). We co-crystallized the MT-DC with SAM, which is notoriously unstable, so the bound species is assigned as SAH. SxtA MT has a large N-terminal lid sub-domain (1–234) and a C-terminal core (249–506) characteristic of the class I methyltransferase superfamily. The DC domain is tightly packed against the MT core domain in a manner resembling the MT1-ΨDC arrangement in AprA12. SAH and the metal bind to the MT core in a cleft between the subdomains (Figure 2c–d). The flexible sub-domain linker (235–253) wraps around the active site, but residues 235–249 were disordered in this crystal form. As a result, although electron density was strong for the metal and the SAH adenosine moiety, it was weak for the homocysteine group, which is proximal to the disordered sub-domain linker (Supplementary Figure 1a–b). The disorder also accounts for the inability to obtain crystals in this form for a complex with the substrate analog malonate. We also obtained crystals in a second form for SxtA MT-DC F458H with Mn2+ and malonate. In these crystals, which diffracted to 2.1 Å, the sub-domain linker was ordered, providing a more complete view of the MT active site (Figure 2e, Table S1, Supplementary Figures 1c–e, 2).
Analysis of the MT active site structures did not lead to an obvious mechanism for control of the extent of methylation by the monomethylating SxtA MT-DC and dimethylating AprA MT1-ΨDC. The active site structures are highly similar (RMSD = 1.4 Å), but sequence differences in and near the active site may contribute to this functional divergence. We used site-directed mutagenesis to investigate several features of the SxtA MT-DC structure that differ from AprA MT1-ΨDC. These features include the presence of an active DC domain in SxtA MT-DC, metal selectivity, and metal coordination sphere (Figure 3).
Figure 3. SxtA mutagenesis approaches and multiple sequence alignment of key MT regions.
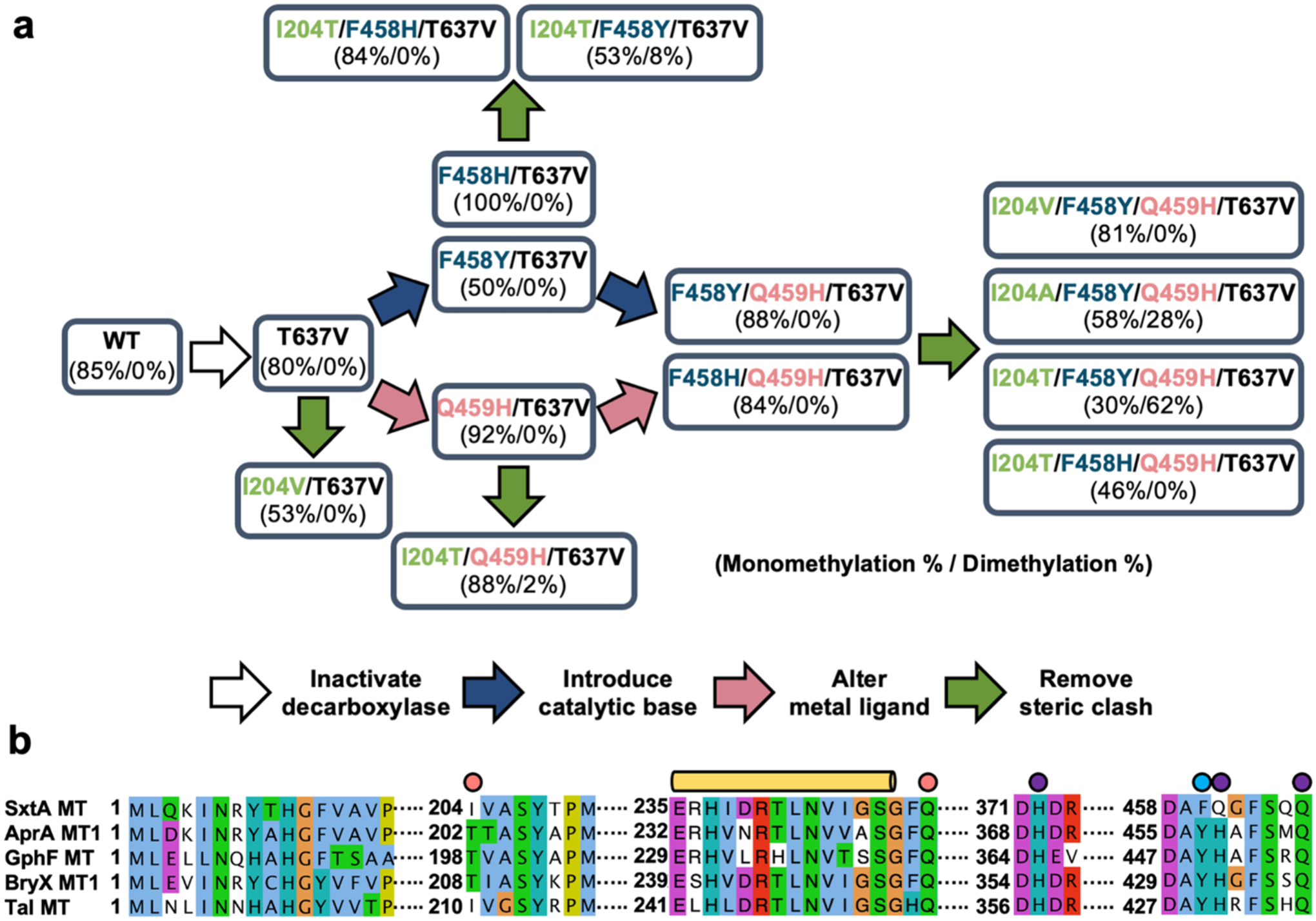
(a) Schematic of SxtA MT-DC site-directed mutagenesis. Mn2+-dependent activity of each variant is indicated (% monomethylation / % dimethylation) for reactions in the standard assay condition. (b) Sequence alignment of loading-module MTs in pathways with identified natural products. The proteins (natural product, GenBank accession) are SxtA (saxitoxin, WP_009343302.1), AprA MT1(apratoxin A, WP_075900460), GphF MT (gephyronic acid, KF479198.1), BryX MT1 (bryostatin, ABK51302.1), and TaI MT (myxovirescin A, WP_011553948.1). SxtA Ile204 and Gln250 in the active site pocket are indicated with a pink circle; Phe458, which was mutagenized to His and Tyr, by a blue circle; and metal ligands by purple circles. The disordered SxtA MT lid-core linker is highlighted with a long yellow cylinder.
The active SxtA DC may restrain the extent of methylation by SxtA MT.
The SxtA DC includes a complete DC domain, in which Thr637 and His671 are essential for catalysis28. DCs in PKS loading modules may function as gatekeepers by selectively acting on acyl-ACPs to generate starter units specific to the corresponding biosynthetic pathways: SxtA DC acts on MeMal-ACP (Figure 2b), CurA DC decarboxylates Mal-ACP28, and GphF DC on Me2Mal-ACP29. The SxtA DC domain may restrict the SxtA MT to a single methyl transfer by decarboxylating MeMal-ACP more rapidly than the MT can exchange SAH for SAM and catalyze a second methyl transfer. To investigate this possibility, we determined the extent of methylation by the DC-inactive SxtA T637V variant. This substitution eliminated decarboxylation activity, created a variant with methyl transfer activity equivalent to the wild type (Figure 2b), but led to no detectable dimethylation activity.
Effect of bound metal in the SxtA MT active site.
Whether one (SxtA MT) or two (AprA MT1 and GphF MT) methyl groups are transferred to a substrate may also be determined by the metal ion and its ligand sphere. SxtA MT is strongly selective for Mn2+ (Figure 2a) whereas AprA MT1 preferred Fe3+ and had weak activity with Mn2+ and other divalent metals12. Moreover, AprA MT1 dimethylation was Fe3+ dependent. Fe2+ supported only one methyl transfer, leading to the hypothesis that the more energetically challenging second methyl transfer may require the stronger Lewis acid of a trivalent metal ion due to the higher pKa of the MeMal-ACP α proton compared to Mal-ACP34, 12. The differing metal selectivity may be a result of different ligand spheres in SxtA MT, AprA MT1 and GphF MT (Figure 3b). In SxtA MT, the Mn2+ has two glutamine ligands (464 and 459) and one histidine (372), whereas the metal sites in AprA and GphF MTs have one glutamine ligand and two histidines, perhaps accounting for the different extent of methylation.
To test this hypothesis, we mutagenized the ligand sphere in SxtA by creating two variants (Q459H and Q459H/T637V) that mimic the AprA Fe3+ site. However, SxtA Q459H and Q459H/T637 had Mn2+-dependent methyl transfer activity similar to SxtA wild type and SxtA T637V, respectively, and we detected no dimethylated products (Figure 4a). Interestingly, the alteration of ligands from 2 Gln / 1 His to 1 Gln / 2 His substantially increased the Fe3+-dependent activity by six-fold relative to levels for the SxtA wild type and by three-fold for SxtA T637V (Figure 4b). Nevertheless, the Q459H substitution did not alter the SxtA MT preference for Mn2+.
Figure 4. Methylation activity of SxtA MT-DC variants.

(a) Mn2+-dependent activity. (b) Fe3+-dependent activity. Methylation is quantitated as the percent of input Mal-ACP converted to monomethylated products (sum of Ppant ejection fragments from MeMal-ACP and propionyl-ACP). Dimethylated products were not detected for any variant. Error bars represent standard deviation of four or more experiments.
A catalytic base for dimethylation.
Another difference in the SxtA MT and AprA MT1 active sites is Phe458 in SxtA in place of Tyr455 in AprA MT1 (Figure 3b, 2d). We previously showed that the second methyl transfer reaction of AprA MT1 was nearly abolished by substitution of Phe for Tyr45512, which is positioned toward the Mal- and MeMal-ACP and may act as a catalytic base for the second methyl transfer. Thus, we substituted Tyr or His for Phe458 in SxtA MT-DC; however, no variants with these substitutions catalyzed dimethylation. SxtA MT-DC F458Y, F458Y/T637V, F458H and F458H/T637V, under standard assay conditions, yielded 61±4%, 50±0.2%, 100%, and 100% monomethylated products, respectively, with Mn2+ (Figure 4a). With Fe3+, yields were 6±4%, 4 ±1%, 26±5%, and 28±15% mono-methylated products, respectively (Figure 4b). Interestingly, the F458H substitution led to increased product yield over the wild-type and consumed all of the Mal-ACP substrate in the standard Mn2+-dependent reaction; this substitution also increased the yield for the Fe3+-dependent reaction. We also combined the substitutions at position 458 with the more efficient Q459H substitution (F458Y/Q459H, F458H/Q459H, F458Y/Q459H/T637V, and F458H/Q459H/T637). These variants generated only monomethylated products, despite having all the features proposed to support two methylation reactions.
The F458H substitution increases monomethylation efficiency.
Although no dimethylated products were detected under our standard reaction conditions, the SxtA F458H variants consumed all of the Mal-ACP substrate, whereas SxtA wild type and T637V converted only 85±8% and 79±12%, respectively. Using SxtA MT-DC T637V to eliminate complications from decarboxylation, we measured the time course of product formation for SxtA F458H/T637V and T637V with a fixed substrate concentration and four-fold less SxtA MT-DC than for the standard assay condition (Figure 5). Under these conditions, MT-DC F458H/T637V methylated ~68% of the Mal-ACP substrate while MT-DC T637V converted only ~11%, a sixfold increase. We then quantitated the greater catalytic efficiency of MT-DC F458H/T637V compared to the parent MT-DC T637V using a 1.45 μM enzyme concentration and 38–600 μM Mal-ACP substrate (Supplementary Figure 3). The maximum attainable Mal-ACP concentration (600 μM) was not saturating for SxtA F458H/T637V, and KM could not be determined. Using initial velocities at low substrate concentrations, the catalytic efficiency (kcat/KM) was approximately fourfold greater for SxtA F458H/T637V (3.17 mM−1 min−1) than for the SxtA T637V parent (0.78 mM−1 min−1).
Figure 5. Monomethylation rate of SxtA MT-DC F458H/T637V and T637V.
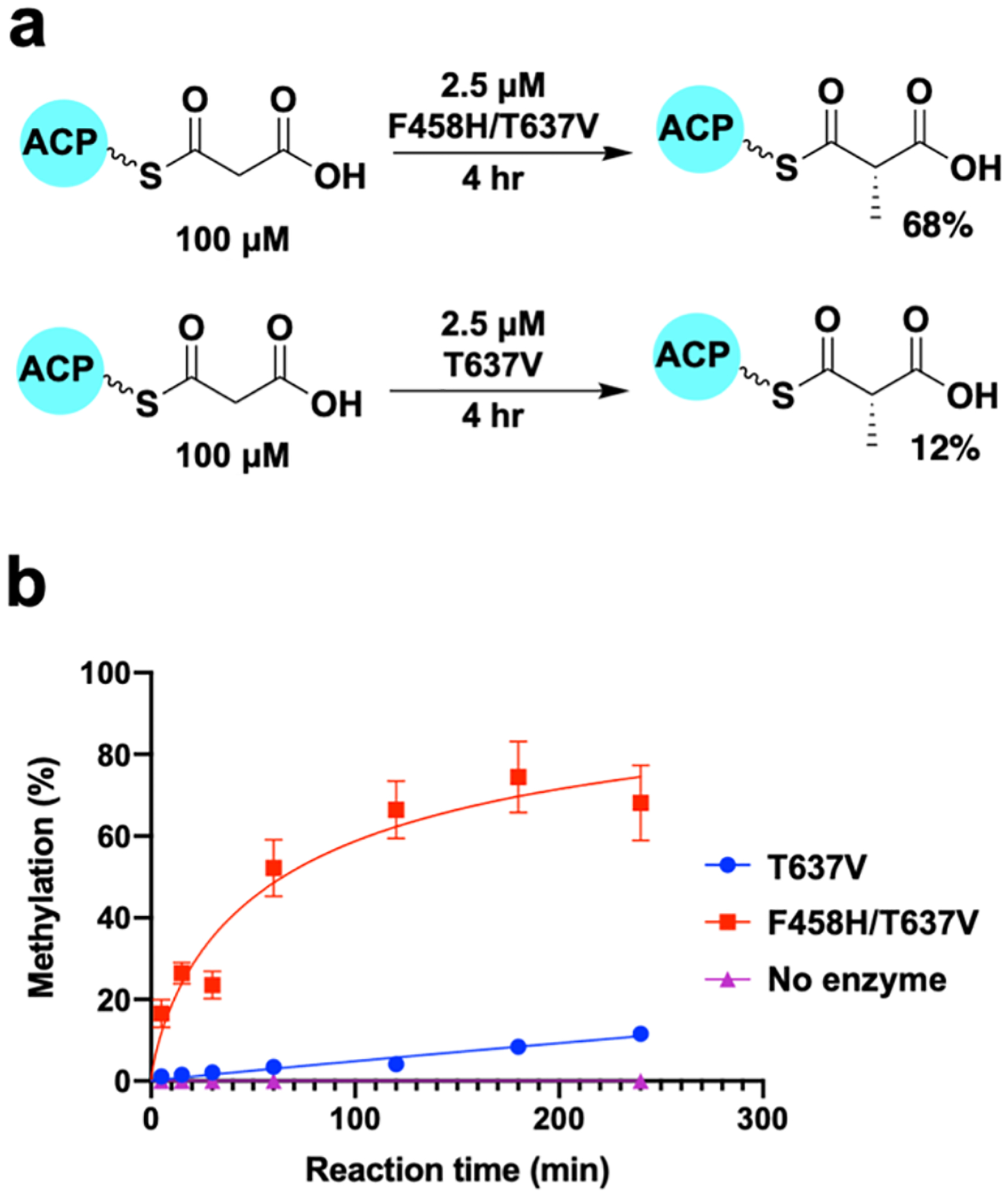
(a) Reaction of SxtA F458H/T637V and T637V on Mal-ACP. (b) Time course of methylation by SxtA F458H/T637V and T637V. SxtA concentration was fourfold lower than for the standard condition in Figure 4. Error bars represent the spread of two replicates and are too small to be seen in some cases.
Based on the crystal structure with the complete SxtA MT active site, we attribute the increased activity of SxtA MT F458H to stabilization of the metal ligand sphere (Figure 2d–e). In the SxtA MT-DC wild type structure, Phe458 forms a π-π stacking interaction with the Tyr208 side chain (centroid distance 3.7 Å). In the MT active site with His458, the side chain is not stacked with Tyr208, but instead is oriented toward the malonate substrate analog and participates in the secondary metal coordination sphere via a hydrogen bond to the side chain nitrogen of the Gln459 metal ligand (Figure 2e). His458 is not positioned to interact directly with the substrate. Second-sphere interactions can be important in supporting the active site structure and reactivity of metalloenzymes35 by stabilizing the metal site or a reaction intermediate, by orienting a ligand, or by mediating the transfer of a proton or electron36, 37. Several other amino acids in the SxtA MT (Tyr208, Asn243, Asp373, Glu434, Phe455 and Ser462) participate in the secondary coordination sphere and are conserved among annotated MTs of this family (Supplementary Figure 4), indicating the important contribution of second-sphere interactions. Here, the SxtA MT F458H structure and kinetic assay results indicate that the enhanced catalytic rate of this variant is due to the His458 hydrogen bond with the metal ligand Gln459, perhaps by stabilizing Gln459 and the metal in an optimal position for malonyl-ACP binding or for catalysis. Interestingly, the His458 substitution had no accelerating effect on a variant where the Gln459 ligand was replaced with His (F458H/Q459H/T637V, Figure 4a), consistent with the impossibility of a hydrogen bond between His458 and His459 side chains.
Other factors in the extent of methylation.
Taken together, the provision of an AprA-like metal center, addition of a putative base for the second methylation reaction, and elimination of any decarboxylation activity were insufficient for SxtA MT to catalyze two methyl transfers. Thus, we re-examined the sequence alignment and the SxtA F458H structure for other features. Among the four SxtA MT homologs with known reaction products, the TaI MT (43% identical sequence to SxtA MT, 48% identity to AprA MT1) is especially interesting. The myxovirescin loading module (TaI MT-DC-ACP) produces a propionyl starter unit38, thus its MT should be a monomethylating enzyme. However, the TaI MT active site is more similar to the dimethylating AprA MT1 than the monomethylating SxtA MT (Figure 3b). The TaI MT possesses Tyr463 (analogous to SxtA Phe458 and AprA Tyr455) and a metal coordination sphere with two histidine ligands and one glutamine. Like our results with engineered SxtA MT-DC, this suggests that other features of the enzyme control the extent of methylation.
The SxtA MT F458H structure with bound malonate and Mn2+ has an ordered sub-domain linker, providing a more complete view of the MT active site. Similar to the ternary complex of AprA MT-ΨDC with malonate and SAM (PDB: 6B3B12), the malonate in SxtA displaces two water molecules and is a bidentate Mn2+ ligand. One malonate carboxyl group hydrogen bonds with Mn2+ ligand Gln464 and represents the terminus of the Mal-ACP, and the other carboxylate is in the position of the ACP Ppant. We modeled dimethylmalonate into the malonate site of SxtA MT F458H (Figure 6). Installation of the first methyl group generates S-MeMal-ACP, with the S-methyl pointed toward the SAM/SAH pocket. A second methyl transfer would occur from the same direction as the first and would displace the first methyl group into the substrate pocket, where a steric clash with Ile204 appears inevitable. In the dimethylating AprA MT1, the analogous residue is Thr202. Moreover, like the SxtA MT, the presumed monomethyltransferase, TaI MT, also has isoleucine at this position whereas the dimethyltransferases (AprA MT1, BryX MT1 and GphF MT) have threonine (Figure 3). In crystal structures, SxtA Ile204 and AprA Thr202 are located at a kink in a long and irregular helix (SxtA 189–209; AprA 187–207). We solved a crystal structure of AprA MT-ΨDC with dimethylmalonate (Supplementary Figure 1f) and discovered that the Thr202 side chain adopts a different rotameric position than in the malonate complex where it is hydrogen bonded to the Gln248 side chain (Supplementary Figure 5a). AprA MT1 accommodates two methyl groups with a rotameric position for Thr202 in which the hydroxy group is directed into the helix kink and hydrogen bonded to the backbone carbonyl of residue 199 (Supplementary Figure 5b). This rotameric position is not accessible to SxtA Ile204 (ethyl in lieu of hydroxy) (Supplementary Figure 5c).
Figure 6. Active site of SxtA MT-DC F458H with dimethylmalonate and SAH.
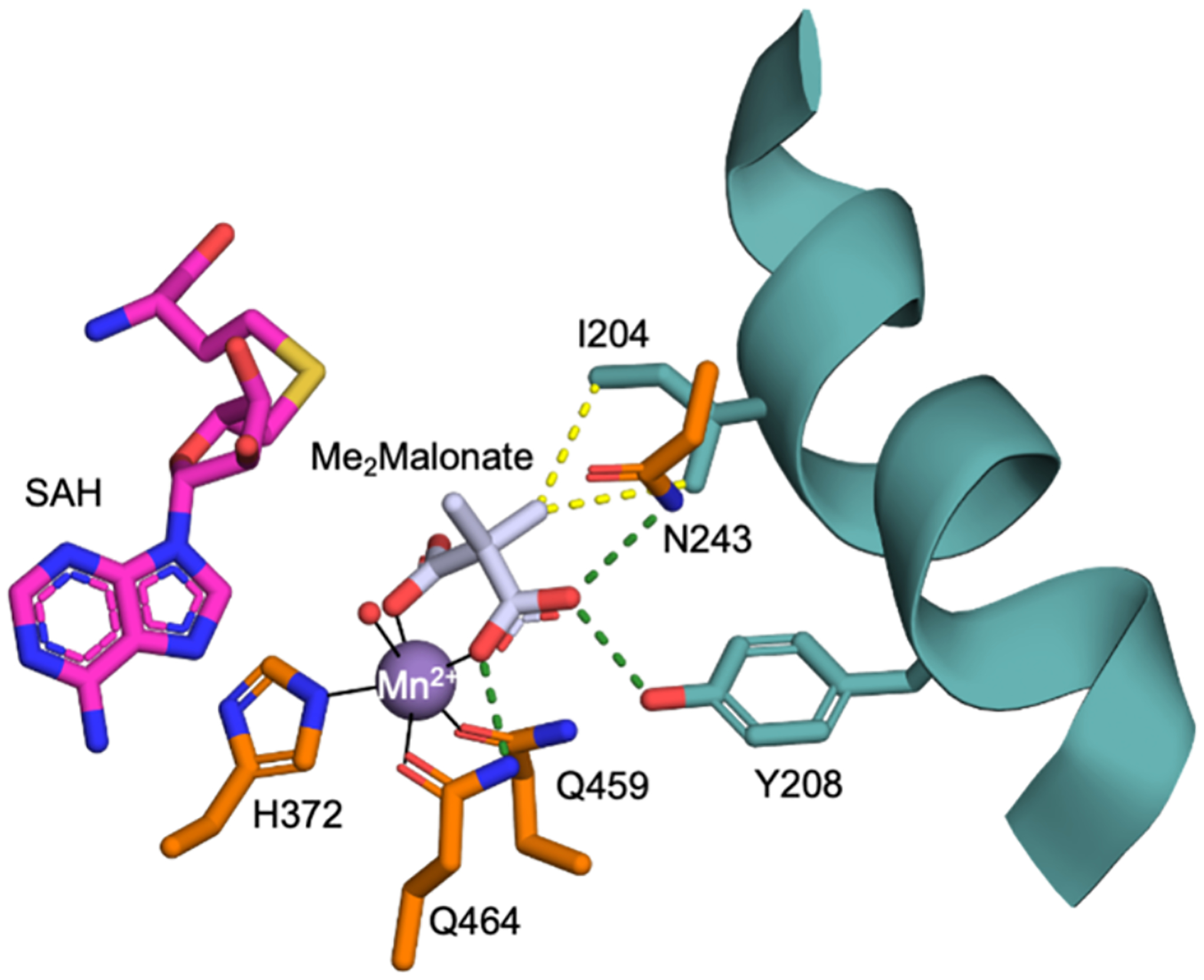
SAH and the methyl groups of dimethylmalonate are modeled, based on the structures in Figure 2d,e. The pro(R) methyl of dimethylmalonate clashes with the Ile204 side chain. Metal ligands (His372, Gln459, and Gln464), Asn243, Ile204, and Tyr208 are colored according to the domain of origin (Figure 2c), dimethylmalonate with white C and SAH with magenta C, metal coordination bonds as solid black lines, hydrogen bonds as dashed green lines and the steric clash as dotted yellow lines.
Selectivity filter for the extent of methylation.
We investigated the role of Ile204 in methyl transfer by replacing Ile204 with Thr or other small amino acids (Ala and Val) in an SxtA MT-DC background with a candidate base for a second methyl transfer (F458Y), a ligand sphere like AprA MT1 (Q459H), and no decarboxylase activity (T637V) (Figure 3a). In this background, the Thr204 and Ala204 variants had significant levels of Mn2+-dependent dimethylation activity (Figure 7). In the standard assay, the dimethylated product predominated for the Thr substitution (62±2% Me2Mal-ACP, 30±3% MeMal-ACP), whereas the Ala substitution yielded more monomethylated product (30±3% Me2Mal-ACP, 58±2% MeMal-ACP). Despite the similar size of the Val and Thr side chains, we detected no dimethylation with the I204V variant. This is consistent with the crystal structures: unlike Thr, the hydrophobic Val side chain would be unable form a hydrogen bond with the protein backbone and unable to remove its methyl substituents from the substrate pocket.
Figure 7. Impact of Ile204 on the extent of methylation.

(a-b) Mono- and dimethylation activity of SxtA MT-DC wild type and variants that include I204T/A/V in the presence of Mn2+ (a) and Fe3+ (b). (c-d) Mono- and dimethylation activity of AprA wild type and variants in the presence of Mn2+ (c) and Fe3+ (d). Monomethylation (sum of Ppant ejection fragments from MeMal-ACP and propionyl-ACP) and dimethylation (sum of Ppant ejection fragments from Me2Mal-ACP and isobutyryl-ACP) were quantitated as a percent of total Mal-ACP. Error bars represent standard errors of more than three experiments.
We confirmed that an analogous selectivity filter for the extent of methylation exists in AprA by creating AprA MT1-ΨDC T202I. In contrast to the wild type enzyme, AprA MT-ΨDC T202I had no detectable dimethylation activity, but retained monomethylation activity (60±8% MeMal-ACP), confirming the critical role of position 202 in controlling methylation extent (Figure 7c–d).
The presence of an active decarboxylase in SxtA reduced the yield of dimethylated products by about twofold (27.7% total Me2Mal- and isobutyryl-ACP for I204T/F458Y/Q459H/T637 vs. 62.2% for I204T/F458Y/Q459H/T637V, Supplementary Figure 6). Decarboxylation prior to a second methyl transfer is influenced by the availability of SAM and by conformational effects that would differ for an ACP substrate in cis, as in full-length SxtA, rather than in trans, as in our assay. This situation does not pertain to AprA MT1 because the AprA ΨDC lacks catalytic activity, and decarboxylation is coupled to a third methyl transfer reaction catalyzed by the AprA MT2 domain, which is selective for Me2Mal-ACP30.
SxtA MT metal preference.
The His substitution for Gln at SxtA position 459 created a first-shell ligand field identical to that in AprA MT1. This substitution was essential for high levels of dimethylated product in the Mn2+-dependent SxtA MT reaction, yielding eightfold greater dimethylation activity relative to the variant with Gln459 (62±6% Me2Mal-ACP for I204T/F458Y/Q459H/T637V vs. 8±2% for I204T/F458Y/Q459/T637V) (Figure 7a). However, the Q459H substitution did not switch the metal preference of SxtA from Mn2+ to match the Fe3+ preference of AprA MT1. The SxtA I204T and I204A variants in the F458Y/Q459H/T637V background formed mono- and dimethylated products in an Fe3+-dependent reaction, but levels of both products were more than twofold lower with Fe3+ than with Mn2+ (Figure 7b). The Q459H substitution led to no detectable dimethylation activity either metal unless combined with I204T. In contrast, AprA MT1 supported the second methyl transfer reaction with Fe3+, but not with divalent metal ions (Mn2+ or Fe2+)12. We attempted a reciprocal experiment by substituting Gln for His at the analogous position in AprA MT-ΨDC (H456Q); however, the protein was unstable and could not be purified. No obvious structural feature of SxtA MT or AprA MT1 explains their differential metal preferences, which we conclude are due to subtle effects outside the immediate metal environment.
CONCLUSION
Our study revealed how two metal-dependent C-MTs control the extent of methylation. The discovery of a selectivity filter for one vs. two methyl transfers is directly applicable to related C-MTs in several other biosynthetic pathways. Crystal structures of SxtA MT-DC wild type and F458H together with biochemical assays of an extensive mutagenesis panel provided insights into MT function. A single substitution in the C-MT metal ligand field (Q459H) enhanced catalytic activity. A His substitution for Phe at position 458 accelerated methyl transfer by stabilizing the ligand sphere of the catalytic metal and may be useful for engineering efficient monomethylation catalysts. However, dimethylation required both a base at position 458 (Tyr) and a dimethylmalonyl-permissive filter at position 204 (Thr). The new understanding of how the C-MTs control methylation extent will aid future methyltransferase engineering for use in drug optimization and inform the engineering of valuable methyltransferase catalysts.
MATERIALS AND METHODS
Plasmid preparation:
Previously reported plasmids were used: pMCSG7-SxtA_1_7219 (encoding the SxtA MT-DC, residues 1–721 in SxtA; GenBank WP_009343302.1) and pMAS284 AprA_ACP (encoding the AprA ACP, residues 1058–1138; GenBank WP_075900460)12. For a truncated SxtA MT-DC (residues 1–710), pMCSG7-SxtA_1_710 was sub-cloned using primers in Table S1. Site-directed mutagenesis was done via QuikChange (Agilent) using primers in Table S1.
Bacterial expression:
All plasmids were transformed into Escherichia coli strain BL21(DE3). Cell cultures were grown at 37 °C in 0.5 L Terrific Broth (TB) with 100 μg mL−1 ampicillin until OD600 reached 1–2, then cooled to 20°C over 0.5–1 hr, induced with 200 μM IPTG, and grown overnight. In cultures producing apo-ACP, 0.5 mL of filter-sterilized 1000X trace metals mix39 was added to the 0.5 L culture to repress E. coli entD, which encodes a phosphopantetheinyl transferase that can act on many heterologous ACPs.
Purification of SxtA MT-DC and AprA ACP:
Cell pellets from 0.5 L cultures were resuspended in 35 mL Buffer A (50 mM Tris pH 7.4, 300 mM NaCl, 10% (v/v) glycerol) with 0.1 mg mL−1 lysozyme, 0.05 mg mL−1 DNase, 2 mM MgCl2, 20 mM imidazole. Resuspended cells were incubated on ice for 30 min, lysed by sonication and centrifuged at 38,650 × g, for 20 min. The soluble fraction was then filtered through a 0.45 μm MILLEX-HP membrane and loaded onto a 5 mL His trap column (cytiva) pre-equilibrated with Buffer A. The column was washed with 50 mL Buffer A and eluted with a 50 mL gradient of 20–400 mM imidazole in Buffer A. For SxtA MT-DC, the N-terminal His tag was removed by overnight dialysis at 4°C in the presence of tobacco etch virus (TEV) protease40 and 2–5 mM dithiothreitol (DTT) at a 50:1 molar ratio of MT-DC:protease. The mixture was reapplied to the His-trap column to remove uncleaved MT-DC and the protease. SxtA MT-DC and AprA ACP proteins were further purified via HiLoad Superdex S200 or S75 (Cytiva) gel filtration chromatography with Buffer B (50 mM Tris pH 7.4, 50 mM NaCl, 10% (v/v) glycerol). The SxtA MT-DC eluted as a monomer with apparent molecular weight 80 kDa. The purified SxtA MT-DC was concentrated to 12–14 mg mL−1 and stored in aliquots at −80°C.
Production of acyl-ACPs:
AprA apo-ACP (180 μM) was incubated with 20 μM Streptomyces verticillus phosphopantetheinyl transferase (Svp)32 or Bacillus subtilis phosphopantetheinyl transferase (Sfp)31, 20 mM MgCl2, and a 4-fold molar excess of Mal-CoA or MeMal-CoA for 4 hr at 30°C in Buffer B. Acyl-ACPs were purified via Superdex S75 gel filtration chromatography with Buffer B.
Protein crystallization and structure determination:
Tag-free SxtA MT-DC (residues 1–710) was crystallized at 4°C by vapor diffusion in a 1:1 mixture of protein stock (12 – 14 mg mL−1 in Buffer B with 5 mM S-adenosylmethionine (SAM), 5 mM MnCl2) and well solution (5–8% polyethylene glycol (PEG) 20K, 0.1 M MES pH 6.25–6.5). Crystals were cryo-protected by a 5-min soak in cryo-solution (20% 1,2-propanediol, 0.1 M MES pH 6.5, 5 mM MnCl2, 5 mM SAM and 10% PEG 20K), then harvested and stored in liquid nitrogen. Diffraction data were collected at the Advanced Photon Source (APS) beamline 23ID-B (GM/CA@APS) and processed using XDS41. The structure (space group P1 with two polypeptides in the asymmetric unit) was solved by molecular replacement from the structures of CurA DC (PDB 2REE28) and AprA MT1 (PDB 6B3A2) using Phaser42 in the Phenix software suite43. The SxtA MT-DC structure was built using Coot44, and the model was refined with phenix.refine43. The model is nearly complete, including a few cloning-artifact residues. A few regions were disordered and are not included in the model (228–249 and 706–710 in chain A; 149–159, 181–186, 228–248 and 706–710 in chain B). Wild type SxtA MT-DC formed mosaic crystals with limited reproducibility. Due to crystal packing, density was stronger for chain A than chain B (mean B-factor 61.7 Å2 for chain A, 71.3 Å2 for chain B). Electron density for chain B residues 268–390 was weak, so we built this region from the corresponding region of chain A and retained the side chains. TLS groups for refinement were: in chain A, N-terminus through 189, 190–267, 268–530, 531–705; in chain B, N-terminus through 252 and 253–705. The crystals were grown in the presence of SAM, but density was very weak for the methionine moiety. Typically SAM degrades to SAH during crystallization, so we included atomic coordinates for SAH in the deposited structure.
Tag-free SxtA MT-DC F458H (residues 1–721) was crystallized at 20°C by vapor diffusion from a 1:1 mixture of protein stock (12–14 mg/mL in Buffer B with 5 mM SAM, 2 mM sodium malonate, 5 mM (R)-pantetheine, 5 mM MnCl2) and well solution (18% PEG 3350, 0.16 M Li2SO4, 0.1 M Bis-Tris pH 6.5). Crystals were cryo-protected by 5-min soak in cryo-solution (32% PEG 3350, 0.2 M Li2SO4, 0.1 M Tris pH 7), then harvested and stored in liquid nitrogen. Diffraction data were collected at beamline 23ID-B (GM/CA@APS) and processed using XDS. The structure (space group P21 with two polypeptides in the asymmetric unit) was solved by molecular replacement from the SxtA MT-DC structure using Phaser. Model building was done with Coot and refinement with phenix.refine. Electron density was complete for the MT domain, including the 228–249 loop near the active site. However, in the DC domain, residues 574–588 were disordered, and the map had no density for the C-terminus (residues 627–710 in chain A and 628–710 in chain B). SDS-PAGE analysis of dissolved crystals and protein stock showed that the crystallized protein was truncated, apparently due to proteolysis during the 14 days of crystal growth (Supplementary Figure 2).
His-tagged AprA MT- ΨDC S274I/Q548P was purified as previously described12 and crystallized at 20°C by vapor diffusion from a 2:2 μL mixture of protein stock (11 mg mL−1 in Buffer B with 1 mM SAH, 5 mM MnCl2) and well solution (15% PEG 3350, 0.1 M dimethylmalonate). Microseeding was used to obtain single crystals. Harvested crystals were cryoprotected with well solution supplemented with 15% (v/v) glycerol. Crystals were isomorphous with those of Mn-bound MT- ΨDC S274I/Q548P (PDB ID 6B3A12). Electron density was complete except for the histidine tag and residues 149–159 in the MT lid.
Structure validation was done with MolProbity45, figures were prepared in PyMol, and sequence alignments were created using Clustal in Jalview46, 47
Methylation assay:
For quantitative comparison of the activities of wild type SxtA MT-DC and its mutagenized variants, a standard assay was designed in which the wild type enzyme consumed most, but not all, of the Mal-ACP substrate, and all assays were conducted identically under these conditions. Reaction mixtures contained 10 μM SxtA MT-DC, 100 μM Mal-ACP, 1 mM SAM and 0.5 mM metal (either (NH4)2Fe(SO4)2 or MnCl2) in 50 mM HEPES pH 7.8, 150 mM NaCl. The reaction was initiated by the addition of SxtA MT-DC. The mixtures were incubated at 30°C for 4 h and quenched with 10% (v/v) formic acid.
Decarboxylation assay:
Reaction mixtures and assay conditions were identical to those for the methylation assay above, excepting the omission of metal to prevent any methylation activity.
Methylation time course assay:
Reaction mixtures contained 2.5 μM SxtA MT-DC T637V or F458H/T637V (residues 1–721), 100 μM Mal-ACP, 1 mM SAM and 0.5 mM MnCl2 in 50 mM HEPES pH 7.8, 150 mM NaCl. Reactions were initiated by addition of SxtA MT-DC, and the mixtures were incubated at 30°C. Aliquots were removed at several time points (5 min,15 min, 30 min, 1 h, 2 h, 3 h and 4 h) and quenched with 10% (v/v) formic acid.
Methylation kinetic assay:
Reaction mixtures contained 1.45 μM SxtA MT-DC T637V or F458H/T637V (residues 1–721), varying concentrations of Mal-ACP (38, 60, 120, 150, 187, 250, 300, 400, or 600 μM), 0.75 μM SAM and 0.38 mM MnCl2 in 50 mM HEPES pH 7.8, 150 mM NaCl. Reactions were initiated by addition of enzyme, mixtures were incubated at 30 °C, and aliquots (10 μL) were collected at time points (5, 15 and 30 min) and quenched with 10% (v/v) formic acid. Initial velocities were obtained from the slope of product vs. reaction time (5, 15 and 30 min). The initial velocity/enzyme concentration vs. concentration curves were fit to the classic and refined Michaelis-Menten equations48 using PRISM 9 (GraphPad).
As expected, the kcat/KM values derived from either the classic or refined Michaelis-Menten equation are similar. For example, for MT-DC T637V, the values are 0.76 ± 0.47 mM−1 min−1 (classic) and 0.78 ± 0.45 mM−1 min−1 (refined).
Analysis of reaction products by liquid chromatography-mass spectrometry (LC-MS):
Reaction mixtures were analyzed using a simultaneous intact protein and phosphopantetheine (Ppant) ejection method49, 33 on an Agilent Q-TOF 6545 instrument (Supplemental Figure 7). Insoluble material in the quenched reaction mixture was removed by centrifugation (13,000 rpm for 15 min), and the clarified reaction mixture (0.5 μL) was loaded onto a reverse-phase high-performance liquid chromatography (HPLC) column (Phenomenex Aeris widepore C4 column 3.6 μM, 50 × 2.10 mm) at flow rate of 0.5 mL/min in 0.2% (v/v) formic acid and separated with a gradient of 5–100% acetonitrile in 0.2% (v/v) formic acid over 8 min. MS operating conditions were 300 V fragment voltage, 75 V skimmer voltage, 100 V nozzle voltage, 350 °C sheath gas temperature, 325 °C drying gas temperature. The MassHunter Qualitative Analysis Software (Agilent) was used for analysis of mass spectra, based on extracted ion counts (EIC) for acetyl- (Ac-), Mal-, propionyl-, MeMal-, isobutyryl-, and Me2Mal-ACPs and their Ppant ejection fragments. Quantitation of methylation in each reaction, where Mal-ACP was the substrate, was the summed EIC for methylated Ppant ejection species (propionyl-, MeMal-, isobutyryl- and Me2Mal-Ppant) relative to summed EIC (Mal-, propionyl-, MeMal-, isobutyryl- and Me2Mal-Ppant). Similarly, the extent of mono-methylation was the total EIC for propionyl- and MeMal-Ppant relative to the overall total, and the extent of di-methylation was the total EIC for isobutyryl- and Me2Mal-Ppant relative to the overall total.
We detected Ac-ACP in all samples, even those with an inactivated DC (MT-DC T637V). The presence of Ac-ACP was due in part to a low level of ionization-induced decarboxylation of Mal-ACP in the mass spectrometer and in part to low levels of contaminating Ac-ACP in Mal-ACP preparations. The level of contaminating Ac-ACP varied with Mal-ACP preparation and time, but the in-source decarboxylation was constant given the uniform MS protocol used for all assays. We quantitated the level of in-source decarboxylation to correct the observed Mal-Ppant EIC values. For this, we used an MT-DC variant (F458H) that methylated all Mal-ACP during the 4 h reaction time, leaving no Mal-ACP for in-source decarboxylation. (We also determined that no spontaneous decarboxylation of Mal-ACP was detectable during the 4 h reaction period.) Parallel incubations were conducted with MT-DC F458H and with a no-enzyme control and ACP species were quantitated by LC/MS (see the schematic below).
As expected, Ac-ACP levels were higher in the no-enzyme control due to in-source decarboxylation of Mal-ACP, whereas the Ac-ACP in the sample incubated with MT-DC F458H was due to contamination only. The excess Ac-ACP in the no-enzyme control was 6.2±1.4% (n=7), and all Mal-ACP values were corrected for this effect (EIC × 1.062). Similarly, low levels of in-source decarboxylation of MeMal- and Me2Mal-ACP were established by detection of propionyl-ACP in samples reacted with MT-DC T637V and isobutyryl-ACP in samples of MT-DC I204T/F458Y/Q459H/T637V where enzymatic decarboxylation was absent. In-source decay values were 10.7 ± 1.1% (n=3) for MeMal-ACP and 6.0 ± 0.7 % (n=6) for Me2Mal-ACP. The quantification of decarboxylation, EIC of propionyl- or isobutyryl-ACP over the sum of MeMal- and propionyl, and of isobutyryl-ACP and Me2Mal-ACP, was corrected according to the in-source decay values.
Supplementary Material
ACKNOWLEDGEMENT
This work was supported by National Institutes of Health (NIH) grants DK042303 and CA108874 to J.L.S and GM124880 to A.R.H.N. Y.L was supported by a Rackham Merit Fellowship from the University of Michigan Rackham Graduate School. M.A.S was supported by a predoctoral fellowships from an NIH Cellular Biotechnology Training Program (GM008353) and a University of Michigan Rackham Predoctoral Fellowship. We thank P.J. O’Brien for advice on the kinetic analysis. Y.L thanks M. Rankin and R. Benisch for critical reading of the manuscript, R. Torres for kinetics discussion, and M. Rankin for assistance with mass spectrometry. GM/CA@APS was supported by the NIH National Institute of General Medical Sciences (AGM-12006) and the National Cancer Institute (ACB-12002); the Eiger 16M X-ray detector was purchased with funds from NIH grant S10-OD012289.
ABBREVIATIONS
- Ac
acetyl
- ACP
acyl carrier protein
- APS
Advanced Photon Source
- C-MT
C-methyltransferase
- CoA
coenzyme A
- DC
decarboxylase
- EIC
extracted ion count
- GNAT
GCN5-related N-acetyltransferase
- HPLC
high-performance liquid chromatography
- LC-MS
liquid chromatography - mass spectrometry
- Me2Mal
dimethylmalonyl
- MeMal
methylmalonyl
- MT
methyltransferase
- PEG
polyethylene glycol
- PKS
polyketide synthase
- Ppant
phosphopantetheine
- RMSD
root-mean-square deviation
- SAH
S-adenosyl-l-homocysteine
- SAM
S-adenosyl-l-methionine
- SXT
saxitoxin
- ΨDC
pseudo decarboxylase
Footnotes
Supporting Information
The Supporting Information is available free of charge at http://pubs.acs.org.
Seven supplemental figures providing details of the crystal structures, electron densities, partial sequence alignment, kinetic assay data and representative mass spectra.
Two supplemental tables with crystallographic summary and primer sequences for mutagenesis.
Accession Codes
The atomic coordinates and structure factors are available in the Protein Data Bank (PDB) with ID 7UCI for the SxtA MT-DC complex with SAH, 7UCL for the SxtA MT-DC F458H complex with malonate, and 7UCH for the AprA MT1-ψDC complex with SAM and dimethylmalonate.
REFERENCES
- 1.Barreiro EJ; Kümmerle AE; Fraga CA (2011) The methylation effect in medicinal chemistry. Chem Rev 111, 5215–5246. [DOI] [PubMed] [Google Scholar]
- 2.Schönherr H; Cernak T (2013) Profound methyl effects in drug discovery and a call for new C-H methylation reactions. Angew Chem Int Ed Engl 52, 12256–12267. [DOI] [PubMed] [Google Scholar]
- 3.Castillo M; Smith PC (1995) Disposition and reactivity of ibuprofen and ibufenac acyl glucuronides in vivo in the rhesus monkey and in vitro with human serum albumin. Drug Metab Dispos 23, 566–572. [PubMed] [Google Scholar]
- 4.Johnson CH; Wilson ID; Harding JR; Stachulski AV; Iddon L; Nicholson JK; Lindon JC (2007) NMR spectroscopic studies on the in vitro acyl glucuronide migration kinetics of Ibuprofen ((+/−)-(R,S)-2-(4-isobutylphenyl) propanoic acid), its metabolites, and analogues. Anal Chem 79, 8720–8727. [DOI] [PubMed] [Google Scholar]
- 5.Aynetdinova D; Callens MC; Hicks HB; Poh CYX; Shennan BDA; Boyd AM; Lim ZH; Leitch JA; Dixon DJ (2021) Installing the “magic methyl”- C-H methylation in synthesis. Chem Soc Rev 50, 5517–5563. [DOI] [PubMed] [Google Scholar]
- 6.Stevens DC; Wagner DT; Manion HR; Alexander BK; Keatinge-Clay AT (2016) Methyltransferases excised from trans-AT polyketide synthases operate on N-acetylcysteamine-bound substrates. J Antibiot (Tokyo) 69, 567–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fischbach MA; Walsh CT (2006) Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem Rev 106, 3468–3496. [DOI] [PubMed] [Google Scholar]
- 8.Keatinge-Clay AT (2012) The structures of type I polyketide synthases. Nat Prod Rep 29, 1050–1073. [DOI] [PubMed] [Google Scholar]
- 9.Chun SW; Hinze ME; Skiba MA; Narayan ARH (2018) Chemistry of a Unique Polyketide-like Synthase. J. Am. Chem. Soc 140, 2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meinke JL; Mehaffey MR; Wagner DT; Sun N; Zhang Z; Brodbelt JS; Keatinge-Clay AT (2018) Structural and Functional Studies of a gem-Dimethylating Methyltransferase from a trans-Acyltransferase Assembly Line. ACS Chem Biol 13, 3306–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skiba MA; Sikkema AP; Fiers WD; Gerwick WH; Sherman DH; Aldrich CC; Smith JL (2016) Domain Organization and Active Site Architecture of a Polyketide Synthase C-methyltransferase. ACS Chem Biol 11, 3319–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skiba MA; Sikkema AP; Moss NA; Tran CL; Sturgis RM; Gerwick L; Gerwick WH; Sherman DH; Smith JL (2017) A Mononuclear Iron-Dependent Methyltransferase Catalyzes Initial Steps in Assembly of the Apratoxin A Polyketide Starter Unit. ACS Chem Biol 12, 3039–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wiese M; D’Agostino PM; Mihali TK; Moffitt MC; Neilan BA (2010) Neurotoxic alkaloids: saxitoxin and its analogs. Mar Drugs 8, 2185–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llewellyn LE (2006) Saxitoxin, a toxic marine natural product that targets a multitude of receptors. Nat Prod Rep 23, 200–222. [DOI] [PubMed] [Google Scholar]
- 15.Shen H; Liu D; Wu K; Lei J; Yan N (2019) Structures of human Na(v)1.7 channel in complex with auxiliary subunits and animal toxins. Science 363, 1303–1308. [DOI] [PubMed] [Google Scholar]
- 16.Dusek RJ; Smith MM; Van Hemert C; Shearn-Bochsler VI; Hall S; Ridge CD; Hardison DR; Kaler RSA; Bodenstein BL; Hofmeister EK; et al. (2021) Acute oral toxicity and tissue residues of saxitoxin in the mallard (Anas platyrhynchos). Harmful Algae 109, 102109. [DOI] [PubMed] [Google Scholar]
- 17.Thottumkara AP; Parsons WH; Du Bois J (2014) Saxitoxin. Angew Chem Int Ed Engl 53, 5760–5784. [DOI] [PubMed] [Google Scholar]
- 18.Lukowski AL; Denomme N; Hinze ME; Hall S; Isom LL; Narayan ARH (2019) Biocatalytic Detoxification of Paralytic Shellfish Toxins. ACS Chem Biol 14, 941–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cullen A; D’Agostino PM; Mazmouz R; Pickford R; Wood S; Neilan BA (2018) Insertions within the Saxitoxin Biosynthetic Gene Cluster Result in Differential Toxin Profiles. ACS Chem Biol 13, 3107–3114. [DOI] [PubMed] [Google Scholar]
- 20.Kellmann R; Mihali TK; Jeon YJ; Pickford R; Pomati F; Neilan BA (2008) Biosynthetic intermediate analysis and functional homology reveal a saxitoxin gene cluster in cyanobacteria. Appl Environ Microbiol 74, 4044–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kellmann R; Mihali TK; Neilan BA (2008) Identification of a saxitoxin biosynthesis gene with a history of frequent horizontal gene transfers. J Mol Evol 67, 526–538. [DOI] [PubMed] [Google Scholar]
- 22.Mihali TK; Carmichael WW; Neilan BA (2011) A putative gene cluster from a Lyngbya wollei bloom that encodes paralytic shellfish toxin biosynthesis. PLoS One 6, e14657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsuchiya S; Cho Y; Yoshioka R; Konoki K; Nagasawa K; Oshima Y; Yotsu-Yamashita M (2017) Synthesis and Identification of Key Biosynthetic Intermediates for the Formation of the Tricyclic Skeleton of Saxitoxin. Angew Chem Int Ed Engl 56, 5327–5331. [DOI] [PubMed] [Google Scholar]
- 24.Wang DZ; Zhang SF; Zhang Y; Lin L (2016) Paralytic shellfish toxin biosynthesis in cyanobacteria and dinoflagellates: A molecular overview. J Proteomics 135, 132–140. [DOI] [PubMed] [Google Scholar]
- 25.Luo Y; Li BZ; Liu D; Zhang L; Chen Y; Jia B; Zeng BX; Zhao H; Yuan YJ (2015) Engineered biosynthesis of natural products in heterologous hosts. Chem Soc Rev 44, 5265–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chakrabarty S; Romero EO; Pyser JB; Yazarians JA; Narayan ARH (2021) Chemoenzymatic Total Synthesis of Natural Products. Acc. Chem. Res 54, 1374–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chun SW; Narayan ARH (2019) Biocatalytic synthesis of α-amino ketones. Synlett 30, 1269–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu L; Geders TW; Wang B; Gerwick WH; Håkansson K; Smith JL; Sherman DH (2007) GNAT-like strategy for polyketide chain initiation. Science 318, 970–974. [DOI] [PubMed] [Google Scholar]
- 29.Skiba MA; Tran CL; Dan Q; Sikkema AP; Klaver Z; Gerwick WH; Sherman DH; Smith JL (2020) Repurposing the GNAT Fold in the Initiation of Polyketide Biosynthesis. Structure 28, 63–74.e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skiba MA; Sikkema AP; Moss NA; Lowell AN; Su M; Sturgis RM; Gerwick L; Gerwick WH; Sherman DH; Smith JL (2018) Biosynthesis of t-Butyl in Apratoxin A: Functional Analysis and Architecture of a PKS Loading Module. ACS Chem Biol 13, 1640–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quadri LE; Weinreb PH; Lei M; Nakano MM; Zuber P; Walsh CT (1998) Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry 37, 1585–1595. [DOI] [PubMed] [Google Scholar]
- 32.Sánchez C; Du L; Edwards DJ; Toney MD; Shen B (2001) Cloning and characterization of a phosphopantetheinyl transferase from Streptomyces verticillus ATCC15003, the producer of the hybrid peptide-polyketide antitumor drug bleomycin. Chem Biol 8, 725–738. [DOI] [PubMed] [Google Scholar]
- 33.Meluzzi D; Zheng WH; Hensler M; Nizet V; Dorrestein PC (2008) Top-down mass spectrometry on low-resolution instruments: characterization of phosphopantetheinylated carrier domains in polyketide and non-ribosomal biosynthetic pathways. Bioorg Med Chem Lett 18, 3107–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arnett EM; Maroldo SG; Schilling SL; Harrelson JA (1984) Ion pairing and reactivity of enolate anions. 5. Thermodynamics of ionization of β-di- and tricarbonyl compounds in dimethyl sulfoxide solution and ion pairing of their alkali salts. J Am Chem Soc 106, 6759–6767. [Google Scholar]
- 35.Span EA; Suess DLM; Deller MC; Britt RD; Marletta MA (2017) The Role of the Secondary Coordination Sphere in a Fungal Polysaccharide Monooxygenase. ACS Chem Biol 12, 1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dudev T; Lin; Dudev M; Lim C (2003) First−Second Shell Interactions in Metal Binding Sites in Proteins: A PDB Survey and DFT/CDM Calculations. J Am Chem Soc 125, 3168–3180. [DOI] [PubMed] [Google Scholar]
- 37.Mazmanian K; Dudev T; Lim C (2018) How First Shell-Second Shell Interactions and Metal Substitution Modulate Protein Function. Inorg Chem 57, 14052–14061. [DOI] [PubMed] [Google Scholar]
- 38.Simunovic V; Zapp J; Rachid S; Krug D; Meiser P; Müller R (2006) Myxovirescin A biosynthesis is directed by hybrid polyketide synthases/nonribosomal peptide synthetase, 3-hydroxy-3-methylglutaryl-CoA synthases, and trans-acting acyltransferases. Chembiochem 7, 1206–1220. [DOI] [PubMed] [Google Scholar]
- 39.Skiba MA; Maloney FP; Dan Q; Fraley AE; Aldrich CC; Smith JL; Brown WC (2018) PKS-NRPS Enzymology and Structural Biology: Considerations in Protein Production. Methods Enzymol 604, 45–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kapust RB; Tözsér J; Copeland TD; Waugh DS (2002) The P1’ specificity of tobacco etch virus protease. Biochem Biophys Res Commun 294, 949–955. [DOI] [PubMed] [Google Scholar]
- 41.Kabsch W (2010) Xds. Acta Crystallogr D Biol Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liebschner D; Afonine PV; Baker ML; Bunkóczi G; Chen VB; Croll TI; Hintze B; Hung LW; Jain S; McCoy AJ; et al. (2019) Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emsley P; Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 45.Chen VB; Arendall WB 3rd; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Larkin MA; Blackshields G; Brown NP; Chenna R; McGettigan PA; McWilliam H; Valentin F; Wallace IM; Wilm A; Lopez R; et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. [DOI] [PubMed] [Google Scholar]
- 47.Waterhouse AM; Procter JB; Martin DM; Clamp M; Barton GJ (2009) Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson KA (2019) New standards for collecting and fitting steady state kinetic data. Beilstein J Org Chem 15, 16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dorrestein PC; Bumpus SB; Calderone CT; Garneau-Tsodikova S; Aron ZD; Straight PD; Kolter R; Walsh CT; Kelleher NL (2006) Facile detection of acyl and peptidyl intermediates on thiotemplate carrier domains via phosphopantetheinyl elimination reactions during tandem mass spectrometry. Biochemistry 45, 12756–12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.