

Abstract
Granular hydrogels are an exciting class of microporous and injectable biomaterials that are being explored for many biomedical applications, including regenerative medicine, 3D printing, and drug delivery. Granular hydrogels often possess low mechanical moduli and lack structural integrity due to weak physical interactions between microgels. This has been addressed through covalent inter-particle crosslinking; however, covalent crosslinking often occurs through temporal enzymatic methods or photoinitiated reactions, which may limit injectability and material processing. To address this, we developed a hyaluronic acid (HA) granular hydrogel with dynamic covalent (hydrazone) inter-particle crosslinks. Extrusion fragmentation was used to fabricate microgels from photocrosslinkable norbornene-modified HA, additionally modified with either aldehyde or hydrazide groups. Aldehyde and hydrazide-containing microgels were mixed and jammed to form adhesive granular hydrogels. These granular hydrogels possessed enhanced mechanical integrity and shape stability over controls due to the covalent inter-particle bonds, while maintaining injectability due to the dynamic hydrazone bonds. We applied the adhesive granular hydrogels to 3D printing, which allowed the printing of structures that were stable without any further post-processing. Additionally, we demonstrated that adhesive granular hydrogels allowed for cell invasion in vitro. Overall, this work demonstrates the use of dynamic covalent inter-particle crosslinking to enhance injectable granular hydrogels.
Keywords: Granular hydrogel, extrusion fragmentation, injectable hydrogels, inter-particle crosslinking, dynamic covalent, extrusion printing
Graphical Abstract
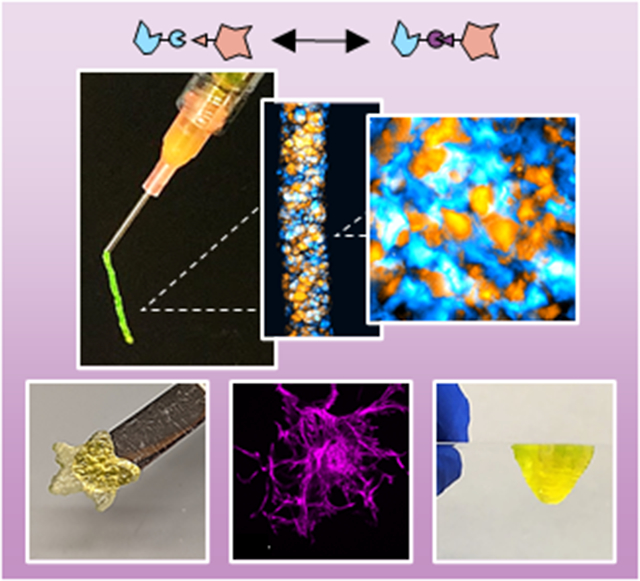
Granular hydrogels are an exciting class of microporous and injectable biomaterials. To further their properties, we introduce adhesion between particles via dynamic covalent inter-particle hydrazone crosslinking through the modification of particles with aldehyde and hydrazide chemical groups. This enhances the mechanical integrity and shape stability of granular hydrogels, including when 3D printed, while still allowing for cellular interactions.
1. Introduction
Injectable and extrudable hydrogels have many biomedical applications, from clinical use to additive manufacturing.[1,2] For these applications, many injectable hydrogels are strain-yielding to permit material flow and self-healing to retain structural stability upon removal of shear.[3] Often, shear-thinning and self-healing hydrogels are formed via reversible crosslinks (e.g., guest-host interactions, hydrogen bonding, dynamic covalent crosslinking) between polymers within a bulk, continuous hydrogel.[4-7] These bulk hydrogels have nanoscale porosity due to the inter-mesh space within the polymer network.[8] However, many biomedical processes benefit from microscale porosity, such as for cell infiltration during tissue regeneration, as well as drug and nutrient transport.[8,9]
To combine the advantages of injectability and microscale porosity into a single material, granular hydrogels have been developed that are formed through the assembly and packing of hydrogel microparticles, or “microgels”.[10,11] Granular hydrogels are strain-yielding and self-healing, as individual microgels are able to slide past one another upon exposure to shear and re-assemble upon removal of force. This allows injectability, which has been utilized to translate granular hydrogels for in vivo tissue regeneration and as extrudable inks for bioprinting.[12-15] In addition, granular hydrogels have advantageous microscale porosity due to the void space between microgels, which has been utilized to enhance cell migration and infiltration throughout scaffolds both in vitro and in vivo.[13,16-18] Further, multiple microgel populations can be combined into a single granular hydrogel system, and this “building block” approach allows for modular tuning of properties such as stiffness,[18] degradation,[12] and therapeutic delivery[19] .
To further enhance the utility of granular hydrogels, inter-particle crosslinking has been introduced to increase mechanical moduli and structural stability. Photocrosslinking has often been used to stabilize granular hydrogels through radical kinetic chain formation[20-22] and thiol-ene addition reactions.[14,23,24] For example, Pfaff et al. recently developed a PEG-based granular hydrogel system for therapeutic delivery and extrusion 3D printing, where methacrylate moieties between microgels allowed for photocrosslinking and stabilization of the construct after extrusion.[21] In another example, Xin et al. developed a PEG-based granular hydrogel bioink for 3D printing that was stabilized by thiol-norbornene inter-particle photocrosslinking after extrusion, resulting in a 1.6-fold increase in storage modulus when compared to un-crosslinked controls.[24]
In addition to photocrosslinking, other approaches have been explored to permanently stabilize microgels within a granular hydrogel, including enzymatic crosslinking[15,25,26] and click chemistry.[27-30] For example, microporous annealed particle (MAP) scaffolds consist of microgels that undergo inter-particle enzymatic annealing upon exposure to FXIII due to the presence of K and Q peptides on the microgel surface, showing promise for in vitro cell culture and in vivo tissue regeneration.[15,25,26] Others have utilized click chemistry reactions such as tetrazine-norbornene and strain-promoted azide-alkyne cycloaddition reactions to introduce inter-particle crosslinking to granular hydrogels.[27-32] For example, Jivan et al. introduced a tetrazine-functionalized PEG crosslinker in between norbornene-modified microgels, resulting in a temporal increase in storage modulus of the granular hydrogel from ~100 Pa to ~2500 Pa over 40 min.[30]
The introduction of permanent covalent crosslinks between microgels within a granular hydrogel allows for enhanced mechanical properties and structural stability; however, such permanent crosslinks often require external stimuli (e.g., light) or have a temporal component, which can complicate their processing and limit injectability. To maintain injectability of granular hydrogels while also enhancing mechanical integrity, reversible inter-particle crosslinks can be introduced. To date, these systems have relied on physical interactions such as ionic attraction, metal-ligand coordination, guest-host assembly, and hydrogen bonding.[19,33-37] For example, Hsu et al. combined positively-charged chitosan and negatively-charged gelatin microgels into an injectable granular hydrogel for neural tissue engineering applications.[34] The addition of the reversible ionic inter-particle interactions resulted in at least a 5-fold increase in storage modulus compared to neutrally charged controls, while still maintaining shear-thinning and self-healing behavior.[34] In another example, Widener et al. developed a PEG-based granular hydrogel consisting of cyclodextrin- and adamantane-modified PEG microgels that undergo guest-host inter-particle crosslinking.[36] Further, Shin et al. fabricated an injectable and conductive granular hydrogel with reversible, metal-ligand inter-particle crosslinking due to the presence of gallol moieties on the microgel surface and silver nanoparticles within the inter-particle space.[33] Lastly, Riederer et al. developed a chitosan microgel scaffold by modulating pH to induce hydrogen bonding and hydrophobic interactions between microgels, resulting in an injectable scaffold that was stable when initially submerged in saline.[37]
While physical crosslinking permits maintenance of injectability, physical interactions are significantly weaker than permanent covalent bonds.[38] Dynamic covalent bonds are a subset of crosslinking reactions where reversible covalent bonds are formed between polymers within a hydrogel, allowing shear-thinning and self-healing properties, as well as higher mechanical moduli and structural integrity than physically-crosslinked hydrogels.[3,38] Dynamic covalent bonds have been explored to form injectable bulk hydrogels with higher mechanical integrity than physically-crosslinked hydrogels.[39-42] Schiff base reactions (i.e., condensation of carbonyl and primary amine groups) represent a subset of dynamic covalent crosslinking that has been often explored for injectable hydrogel formation.[38,43] Two of the most common Schiff base reactions explored for hydrogel formation are imine and hydrazone crosslinking, the latter leading to a more stable yet still reversible crosslink formation.[43] Hydrazone crosslinks are formed by the reaction of aldehyde and hydrazide functional modifications on polymers and have been explored for multiple applications including 3D printing,[39] tissue engineering,[40,44] and therapeutic delivery.[45,46]
Herein, we developed an injectable hyaluronic acid (HA) granular hydrogel with dynamic covalent inter-particle adhesion through hydrazone bond formation. First, microgels were fabricated by extruding bulk HA hydrogels through sequentially smaller needles, resulting in fragmented microgel particles. Adhesive granular hydrogels were formed through the mixing and jamming of populations of fragmented microgels containing aldehyde and hydrazide functional groups. The porosity, mechanical properties, and flow behavior of these adhesive granular hydrogels were investigated and compared to controls of particles without these interactions. Further, we investigated the utility of adhesive granular hydrogels as 3D printable inks. Finally, we investigated cellular interactions with granular hydrogels in vitro by embedding multicellular spheroids and monitoring cell infiltration, to give insight into potential regenerative medicine strategies.
2. Results and Discussion
2.1. Synthesis and characterization of bulk hydrogels
As a first step towards the fabrication of adhesive granular hydrogels, hyaluronic acid (HA) was modified with norbornene groups (NorHA) via anhydrous esterification to conjugate norbornene carboxylic acid to HA. HA was utilized due to its extensive use in biomedical biomaterials, including materials that have translated to the clinic.[1,47] NorHA was then separately modified with either hydrazide groups (Hyd-NorHA) via amidation reaction between adipic acid dihydrazide and carboxyl groups of HA, or aldehyde groups (Ald-NorHA) via conjugation of a pendant propane diol and subsequent oxidation with sodium periodate (Figure 1a). Norbornene modification was determined to be (25 ± 2)%, (19 ± 1)%, and (24 ± 1)% for NorHA, Hyd-NorHA, and Ald-NorHA, respectively, as determined by 1H-NMR (Figure S1). There is likely some hydrolysis or consumption of the norbornene group during the hydrazide modification or purification steps, leading to the slight decrease in norbornene modification on Hyd-NorHA. These small differences were accounted for in hydrogel formulation. Hydrazide modification was determined to be ~40% by 1H-NMR (Figure S1). To determine the degree of aldehyde modification for Ald-NorHA, reductive amination was used to couple tert-butyl carbazate (TBC) to aldehydes, and peak integration of the TBC group indicated ~17% degree of modification of aldehyde groups as determined by 1H-NMR (Figure S1).
Figure 1.

Bulk hydrogel characterization. a) Overview of modified-hyaluronic acid (HA) chemical structures (left), which photocrosslink upon the addition of crosslinker (dithiothreitol, DTT) and photoinitiator (Irgacure D-2959, I2959) and exposure to UV light to form bulk hydrogels (center). Dynamic covalent hydrazone interactions form between hydrazide and aldehyde groups (right, purple box). b) Compressive moduli of bulk hydrogels. c) Schematic of the adhesion testing of bulk hydrogels (left), representative plots of tensile force v. extension (center), and quantified adhesion strengths of controls (NorHA hydrogels alone) and adhesive (Hyd-NorHA and Ald-NorHA hydrogels) hydrogels (right). Data presented as mean ± standard deviation, with a sample size of n = 3. Statistical analysis performed using a one-way ANOVA. ns = no significance, *p<0.05.
To fabricate bulk hydrogels, either NorHA, Hyd-NorHA, or Ald-NorHA was combined with dithiothreitol (DTT) crosslinker (enough to theoretically react all norbornenes with thiols) and Irgacure D-2959 (I2959) photoinitiator. Polymer concentrations were adjusted such that each hydrogel precursor solution contained a consistent norbornene concentration (12 mM), considering differences in polymer molecular weight and slight differences in degree of norbornene modification. This resulted in compressive moduli of ~30 kPa across NorHA, Hyd-NorHA, and Ald-NorHA bulk hydrogels (Figure 1b). Bulk hydrogels photo-crosslinked within 10 seconds, resulting in a storage modulus (G’) of ~10 kPa across groups (Figure S2). To investigate the adhesive properties of Hyd-NorHA and Ald-NorHA hydrogels, thin hydrogel films were formed, placed in contact, and subject to tensile forces (Figure 1c). Hyd-NorHA and Ald-NorHA hydrogel films showed an increased adhesion strength (~12 kPa) when placed in contact when compared to NorHA hydrogel controls (~5 kPa), due the presence of dynamic covalent hydrazone bond formation between hydrazide and aldehyde groups (Figure 1c). This work supports the formation of materials that present aldehydes or hydrazides and their ability to improve hydrogel adhesion strengths when placed in contact, which is important in our proposed granular hydrogel design.
Hydrogels crosslinked with both norbornene-thiol photocrosslinking and hydrazone dynamic covalent crosslinking have previously been used for both in vitro cell culture and in vivo tissue repair applications, showing high cell viability and biocompatibility.[39,46,48-50] To demonstrate the cytocompatibility of our system, human mesenchymal stromal cells (hMSCs) were encapsulated in the bulk hydrogel formulations of photocrosslinked NorHA, Hyd-NorHA, and Ald-NorHA (Figure S3). Cell viability remained above 90% after 24 h and above 85% after 48 h in culture across all groups, highlighting the cytocompatibility of the hydrogels.
2.2. Fragmented microgel fabrication and granular hydrogel formation
Microgels were formed by extrusion fragmentation methods as previously described.[51] Bulk hydrogels of NorHA, Hyd-NorHA, or Ald-NorHA were photocrosslinked within a syringe and sequentially extruded through smaller needles to form heterogeneous fragmented microgel particles with similar diameters between ~10-300 μm for all formulations (Figure 2a). The size distribution of microgels was consistent across multiple batches of microgel fabrication, demonstrating the repeatability of the extrusion fragmentation process (Figure S4a). The extrusion fragmentation microgel fabrication method was selected over emulsion techniques to allow for increased inter-particle surface contact due to rough, jagged microgel geometries.[51] Further, the fragmentation of bulk hydrogels into irregularly shaped microgels has been used to create injectable hydrogel microparticle products for clinical use, including dermal fillers and viscosupplements.[52,53]
Figure 2.

Microgel and granular hydrogel formation. a) Overview of extrusion fragmentation microgel fabrication method (left), representative fluorescent microscopy images of fragmented microgels in suspension (center), and size characterization of fragmented microgels (right). b) Overview of adhesive granular hydrogel formation (left, purple box), and formation of non-adhesive control granular hydrogels (right, grey box). Includes fluorescent microscopy images of (left) adhesive granular hydrogels (Hyd-NorHA microgels labelled with FITC-dextran [blue], Ald-NorHA microgels labelled with Rhodamine-BSA [orange]) in purple box, and (right) control granular hydrogels (NorHA microgels labelled with FITC-dextran [grey]) in grey box. c) Confocal microscopy images of fluorescently-labelled pores (white) within granular hydrogels (left) and characterization of porosity, including void space %, pore diameter, and number of pores per (500μm)2 (right). For void space (%) and number of pores per (500μm)2, data is presented as mean ± standard deviation, with a sample size of n ≥ 3. Statistical analysis performed using a one-way ANOVA. For microgel and pore diameters, data is presented as mean ± inner quartile range. ns = no significance.
We previously demonstrated that this extrusion fragmentation microgel fabrication method results in granular hydrogels with significantly increased mechanical moduli compared to assemblies of spherical microgel particles, while still maintaining microscale porosity.[51] In addition, extrusion fragmentation for microgel fabrication is rapid, scalable, and cost-effective, as only syringes and needles are required for fragmentation. Alternative fragmentation methods have included passing bulk hydrogels through steel or nylon meshes,[54,55] as well as vigorous stirring of a bulk hydrogel.[56]
To form granular hydrogels, fragmented microgel particles were jammed by vacuum-driven filtration to remove the continuous aqueous phase between particles in suspensions. Adhesive granular hydrogels were formed by mixing Hyd-NorHA and Ald-NorHA microgels in a 50/50 ratio in suspension before jamming, whereas non-adhesive (i.e., “control”) granular hydrogels were formed by jamming only NorHA fragmented microgels (Figure 2b). Granular hydrogels exhibited similar structures of packed, non-uniform particles. By labeling and imaging the space between particles, it was determined that adhesive and control granular hydrogels had similar porosity characteristics, including a total void space of ~12% in both groups, with pore diameters ranging from 100-102 μm and an average of ~120 pores per (500 μm)2 slice (Figure 2c). These similarities in granular hydrogel structures are important to assess whether the formation of inter-particle crosslinks in the adhesive granular hydrogels alters material properties when compared to control hydrogels and to decouple any changes from differences in structural features. It is important to note that the microscale porosity in the interstitial space is significantly larger than the nanoscale porosity that exists within the hydrogel mesh of the individual microgel particles, as shown by the lack of diffusion of high molecular weight fluorescent dextran into microgel particles after 24 h (Figure S4b).
2.3. Characterizing granular hydrogels under compressive loading
The response of granular hydrogels to loading is important towards their utility in biomedical applications. Our previous work showed that granular hydrogels formed through fragmented microgels with vacuum-driven filtration exhibited significantly increased mechanical moduli when compared to granular hydrogels formed with spherical microgels.[51] This is likely due to the enhanced inter-particle contact and interdigitation due to the jagged and irregular shape of microgel particles. The high packing and contact between particles provide an opportunity to investigate the impact of inter-particle crosslinking on material properties, such as with dynamic covalent hydrazone bonds.
To determine the time required for inter-particle hydrazone bonds to form, the compressive modulus of adhesive granular hydrogels was assessed over a 3 h period. The compressive modulus of adhesive granular hydrogels was ~4 kPa immediately after jamming and then increased and plateaued to ~9 kPa after 1 h post-jamming (Figure S5). Notably, the compressive modulus of control granular hydrogels did not change over a 3 h period after jamming, maintaining a constant value of ~3 kPa (Figure S5). Thus, it is important to allow time for hydrazone bond formation after jamming prior to testing, which was maintained at 1 h for subsequent analyses. Previous studies have shown that hydrazone crosslinking in bulk hydrogels can occur on time scales of a few seconds to over an hour depending on the concentrations of components used.[39,42,57]
Adhesive granular hydrogels exhibited a ~3-fold increase in compressive modulus, ~2-fold increase in failure strain, and ~7-fold increase in failure stress when compared to non-adhesive controls (Figure 3a). Of the granular hydrogels with in situ inter-particle crosslinking reported in literature (i.e., not photocrosslinking), compressive moduli have been reported to be on the order of ~1-3 kPa.[16,27,31,33] It is interesting to note that the control granular hydrogels in this study, without any inter-particle crosslinking, have a higher compressive modulus around ~3 kPa compared to these prior reports, which is likely due to the use of fragmented hydrogels and the dense packing with vacuum-driven filtration. Further, the adhesive granular hydrogels exhibit a compressive modulus of ~9 kPa and a failure stress of ~7 kPa, which are significantly higher than values reported for other granular hydrogels.[16] In addition, when investigating the compressive properties of adhesive granular hydrogels with varied ratios of Hyd-NorHA and Ald-NorHA microgels, it was found that a 50/50 mixture resulted in maximum compressive stability (highest failure stress and strain) upon uniaxial loading (Figure S6), which motived the use of the 50/50 ratio in the adhesive granular hydrogel system. Importantly, we demonstrate that the hydrazone inter-particle bonds can be disrupted by introducing tert-butyl carbazate (TBC), an aldehyde capping molecule, into the adhesive granular hydrogel formulation. Adding TBC into adhesive granular hydrogels resulted in a reduction of compressive properties (i.e., failure stress and failure strain) to values not significantly different than control levels (Figure S7). This further provides evidence of inter-particle hydrazone bond formation in adhesive granular hydrogels.
Figure 3.

Compression testing of granular hydrogels. a) Quantified compressive moduli (5% to 10% strain, left), failure strain (center), and failure stress (right) of adhesive granular hydrogels and controls at 1 h post-jamming. b) Macroscopic images of loading and compressing granular hydrogel samples (left) and the condition of the samples after loading to 20% strain (right). c) Representative behavior of granular hydrogels upon cyclic loading to 20% compressive strain followed by return to 0 N (left), and quantified recovery of sample height after cyclic compressive loading (right). Data is presented as mean ± standard deviation, with a sample size of n = 6. Statistical analysis performed using a one-way ANOVA. ***p<0.001, ****p<0.0001.
Upon compressive loading, it was observed that adhesive granular hydrogels were able to maintain their structural integrity, whereas control granular hydrogels fell apart after loading (Figure 3b). To quantify this behavior, granular hydrogels were cyclically loaded to 20% strain, followed by removal of the compressive load to 0 N. Control granular hydrogels completely collapsed after 1 cycle of loading, whereas adhesive granular hydrogels maintained their structural integrity through multiple loading cycles, as evident by the recovery of sample height around ~80% (Figure 3c). This is likely due to the dynamic covalent inter-particle interactions that allow for adaptation upon loading, as compressive loading introduces new inter-particle contact sites within the sample, allowing for new hydrazone bond formation. The ability for hydrogel scaffolds to maintain structural integrity after multiple loading cycles is important in many biomedical applications, such as injection or implantation of a hydrogel scaffolds for regeneration of a load-bearing tissue like cartilage.[58]
To investigate the effects of pH on adhesive granular hydrogel mechanics, discs of adhesive granular hydrogels were formed and subsequently incubated in acidic (pH 3.5), neutral (pH 7.6), or basic (pH 11.1) conditions for 1 h (Figure S8). Following incubation, adhesive granular hydrogel discs were subject to compressive loading. Adhesive granular hydrogel mechanics were not significantly impacted by incubation in acidic or basic conditions (Figure S8). There were no significant differences in compressive moduli (~7 kPa) or failure stresses (~3 kPa) across pH values, and minimal changes in failure strain were observed. Further, adhesive granular hydrogels were stable under acidic and basic conditions for at least 3 weeks (Figure S8).
Our findings suggest that changes in pH do not have a significant impact on adhesive granular hydrogel mechanical properties or long-term stability. It has been previously reported that bulk hydrogels formed with hydrazone crosslinks are responsive to pH, resulting in hydrogel degradation under acidic or basic conditions. Many of these studies have utilized ring-opening oxidation of biopolymers with sodium periodate to functionalize hydrogels with two aldehyde groups (dialdehyde), which breaks the polymer backbone and increases susceptibility to degradation.[38,39,59,60] However, in this study, we conjugated aldehyde pendant functional groups to NorHA without disruption of the polymer backbone. Hozumi et al. showed that hydrazone-crosslinked HA hydrogels with such aldehyde pendant functionalization (the same chemistry as the system used herein) exhibited significantly less degradation than those crosslinked with dialdehyde back-bone-breaking functionalization.[59] The use of pendant aldehyde modification may be a reason for the pH stability observed in our materials system. In addition, the tightly-packed granular structure and the dynamic nature of hydrazone bonds may contribute to the material stability. Overall, our findings demonstrate that adhesive granular hydrogels exhibit maintenance in mechanical properties and structural stability upon exposure to both acidic and basic conditions.
2.4. Characterizing the flow behavior of granular hydrogels
The dynamic covalent nature of hydrazone bonds has been previously utilized to fabricate injectable hydrogels with shear-thinning and self-healing properties.[39,45] Thus, we sought to characterize the flow behavior of adhesive granular hydrogels where this bond is now introduced between particles. Both adhesive and control granular hydrogels exhibited strain-yielding and self-healing behavior, as assessed by oscillatory shear rheometry (Figure 4a). Interestingly, the storage modulus (G’) at low strain and low frequency (1%, 1 Hz) was ~2000 Pa for both control and adhesive granular hydrogels. Potentially, the jagged, irregular geometry of the fragmented microgels dictates the macroscale behavior under shear at low strains, thus resulting in similar storage moduli for both samples. However, adhesive granular hydrogels exhibited an increased yield strain (~40%) by 2-fold compared to the control samples (~20%), suggesting that the adhesive interactions between microgels influence flow behavior at higher strain values. Thus, additional strain is required for material yield with the introduction of inter-particle crosslinks.
Figure 4.

Granular hydrogel flow properties. a) Rheological characterization of adhesive granular hydrogels and controls showing strain sweeps from 1-500% strain at 1 Hz (left), quantified yield strain (G’ < 0.9G’initial) (center), and a rapid reduction and recovery of G′ with low (unshaded, 1% strain, 1 Hz) and high (green, 500% strain, 1 Hz) strain cycles (right). b) Macroscopic images of granular hydrogels extruded from an 18G needle and fluorescent microscopy images of extruded filaments, showing adhesive (left) and control (center) granular hydrogels, and extrusion force measurements through an 18G needle at a flow rate of 3 μL/s (right). Data is presented as mean ± standard deviation, with a sample size of n ≥ 4. Statistical analysis performed using a one-way ANOVA. *p<0.05.
Rheological characterization provides valuable insight into the macroscopic flow behavior of hydrogels; however, many biomedical applications (e.g., injectable biomaterials for tissue regeneration, 3D printing) require hydrogel flow through a narrow outlet (i.e., syringe and needle).[1,2] Hence, contraction flows become increasingly important to investigate with granular hydrogels, as individual microgel diameters can approach the size scale of needle diameters.[10,61] Both control and adhesive granular hydrogels were extrudable through an 18G needle, resulting in filaments with structural integrity, as depicted by macroscopic assessment (Figure 4b). An 18G needle was selected for extrusion studies to ensure the inner needle diameter (838 μm) was higher than the maximum fragmented microgel size (~300 μm), yet still small enough to maintain biomedical relevance for clinical injections and extrusion printing. Using an Arduino-based force sensor setup, we determined the steady-state extrusion force required to flow granular hydrogels through a 1mL syringe and 18G needle at a flow rate of 3 μL/s (10 mL/h) (Figure 4b). Adhesive granular hydrogels required a slightly higher extrusion force (~4 N) than control granular hydrogels (~3 N), which is likely due to the additional force required to overcome the adhesive inter-particle crosslinks.
While investigating extrusion forces, we observed that adhesive granular hydrogels initially exhibited a bursting flow through the needle before flowing smoothly at steady state, whereas this bursting behavior was not observed in the control granular hydrogels without inter-particle crosslinking. This is akin to stick-slip behavior observed in dry granular materials where the fluctuations arise from bulk stresses.[62] To examine this behavior, we used the Arduino-based force sensor setup to look at the extrusion force over time for control and adhesive granular hydrogels (Figure 5a). At a flow rate of 3 μL/s (“slow”), the control hydrogels showed a smooth increase in extrusion force to steady state (~3 N), whereas the adhesive granular hydrogels exhibited cyclic dips and increases in extrusion force for the first ~30 s of flow before reaching steady state (~4 N). When the flow rate was increased to 9 μL/s (“fast”), adhesive granular hydrogels still exhibited dips and increases in extrusion force initially; however, steady-state was reached in about 10 s at a value of ~4.5 N. This behavior is further evidence of the inter-particle crosslinking that exists in the adhesive granular hydrogels.
Figure 5.

Contraction flow of granular hydrogels. a) Extrusion force measurements over time for the control and adhesive granular hydrogel samples at slow (3 μL/s) and fast (9 μL/s) flow rates. b) Overview of contraction flow device, showing device dimensions (top) and fluorescently labelled granular hydrogel flowing through device (bottom). c) Representative trajectories of tracked particle position in the contraction flow device as a function of time, including representative trajectories at multiple time intervals after starting flow in the device (0 to 10 s, 10 to 20 s, and 20 to 30 s).
To further characterize this behavior, we sought to visualize the flow of granular hydrogels over time. To do so, we developed a PDMS contraction flow device that mimicked the dimensions of a 1 mL syringe and 18G needle (Figure 5b). Granular hydrogels were fabricated where 5% of microgels contained a fluorescent dye, which was a sufficiently low density such that individual particles could be optically tracked while flowing through the device. The particle tracking data allowed us to determine the position of microgels over time in the contraction flow device, where stark jumps in microgel position indicate a bursting flow (Figure 5c, S9-S11). Particle trajectories and flow properties were examined at three time intervals (each 10 s long) after flow began. Tracked particles in control granular hydrogels at a flow rate of 3 μL/s exhibited smooth flow and particle trajectories throughout the time intervals assessed, with steady increases in position followed by smooth flow at faster velocity through the contraction channel. This smooth flowing behavior was consistent across multiple independent trials (Figure S9).
In contrast, tracked particles in adhesive granular hydrogels at a flow rate of both 3 and 9 μL/s (slow and fast, respectively) exhibited abrupt jumps in particle position, indicative of bursting flow (Figure 5c). At a flow rate of 3 μL/s, adhesive granular hydrogels exhibited bursting flow throughout the 30 s sampling time after flow initiated, consistent across three independent trials (Figure S10). At an increased flow rate of 9 μL/s, adhesive granular hydrogels were partially able to overcome the bursting behavior to exhibit smooth flow through the contraction channel by ~10-20 s after starting flow, as indicated by the disappearance of dramatic increases in position in the particle trajectories. However, bursting flow behavior in adhesive granular hydrogels was not always overcome at the higher flow rate (Figure S11).
We believe that the bursting flow behavior is due to clogging of the granular material at the bottleneck point as well as increased inter-particle adhesion due to the hydrazone interactions. The clogging and jamming behavior of wet granular materials through a contraction channel is dependent on the ratio of the contraction channel diameter (D) and the granule particle size (d). The heterogeneous fragmented microgel particles have diameters ranging from ~10-300 μm, meaning the ratio D/d ranges from ~2.8 – 84. Souzy et al. showed that, for wet, uniform, non-adhesive packed suspensions of hard spherical microparticles, intermittent bursting flow behavior is observed through a contraction channel for 2.43 < D/d < 5.26.[63] For D/d > 5.26, continuous smooth flow of particles is observed, and for D/d < 2.43, permanent clogging occurs, hindering any flow.[63] It is possible that, for our material system, the addition of inter-particle adhesive causes the fragmented granular hydrogel system flowing through an 18G needle to transition to an intermittent bursting flow regime. As adhesive interactions increase during clogging due to increased microgel-microgel contact, pressure buildup must be overcome to expel the granular hydrogel through the contraction point.
The visualization of granular hydrogel flow as well as tracking of individual microgels enables direct observation of the bursting flow behavior that was measured in the extrusion force measurements. Videos comparing control and adhesive granular hydrogels (SI Video 1) as well as adhesive granular hydrogels at varied flow rates (3, 6, or 9 μL/s; SI Video 2) are included in the Supplementary Information. Gaining insight into biomedically relevant contraction flow behavior using the PDMS device, extrusion force monitoring, and particle tracking provides valuable insight into the translation of adhesive granular hydrogels for biomedical applications. The experiments helped to identify that an increased flow rate may help to overcome adhesive inter-particle interactions more quickly to obtain smooth flow through the contraction point. These observations also highlight some considerations for designing injectable granular hydrogels reinforced with inter-particle crosslinks. While significant increases in mechanical moduli and structural stability may be imparted with inter-particle adhesion, the adhesive interactions will also alter flow behavior in crucial ways that must be considered for biomedical applications. Further, the observations of contraction flow behavior could not have been elucidated from bulk rheological measurements alone. This emphasizes the critical importance of not only characterizing granular hydrogel behavior at the bulk scale, but also investigating how granular hydrogels flow through biomedically relevant contraction channels.
2.5. Extrusion printing with granular hydrogels
One application of interest for granular hydrogels is 3D printing, leveraging their strain-yielding, self-healing behavior. As mentioned previously, printed structures typically need a secondary crosslinking step of inter-particle crosslinking for stabilization; however, the dynamic covalent inter-particle crosslinks with adhesive granular hydrogels may be sufficient to hold printed structures together without the need for post-processing. Filaments of the adhesive granular hydrogel were printed with various needle gauges (18G or 21G), flow rates (9-25 μL/s), and print speeds (8-16 mm/s) (Figure S12). A flow rate of 9 μL/s and print speed of 8 mm/s with an 18G needle yielded filaments with the least variation in filament diameter (~1.5 mm), and thus was used for extrusion printing of granular hydrogel structures. Further, as shown in Figure 5, a flow rate of 9 μL/s is high enough to mostly overcome significant bursting flow behavior for adhesive granular hydrogels flowing through a contraction channel the size of an 18G needle. To account for the initial period of bursting flow, print configurations were adjusted to begin printing ~10-20 s after initiating extrusion. There are numerous parameters that can be adjusted in the materials for use as inks, such as particle size to influence printed filament resolution.
Extrusion printing was used to fabricate star-shaped structures from both control and adhesive granular hydrogels (Figure 6a). Control granular hydrogels resulted in structures that could not be manipulated without losing shape integrity (Figure 6a, SI Video 3). In contrast, printed structures from adhesive granular hydrogels could easily be handled while remaining intact with either a spatula or tweezers (Figure 6a, SI Video 3). When placed into phosphate-buffered saline (PBS), control granular hydrogel stars immediately disassembled upon gentle swirling, whereas adhesive granular hydrogel stars remained adhered and intact (Figure 6a, SI Video 4).
Figure 6.

Extrusion printing with granular hydrogels. a) Macroscopic images depicting the stability of extrusion printed stars using control (top) or adhesive (bottom) granular hydrogels, showing printed star, manipulation, shape stability in upon agitation in phosphate-buffered saline (PBS), and long-term stability of adhesive granular hydrogel constructs (36 days). b) Macroscopic images of extrusion structures fabricated with adhesive granular hydrogels, including an outline of the LOVE statue found in Philadelphia (left), a 2 cm-tall hollow cylinder (center), a 1 cm-tall pyramid that can be inverted and maintain its shape (center), and a suspended star outline supported by the tops of five microcentrifuge tubes (right).
Adhesive granular hydrogel stars maintained their structure and shape stability for at least 36 days of incubation in PBS (Figure 6a). The stability of printed structures incubated in PBS was quantified by showing minimal mass loss and maintenance of shape fidelity over a period of 36 days (Figure S13). In addition, there was minimal release (<20%, cumulative) of encapsulated high molecular weight fluorescently-labelled dextran (2 MDa) from the printed adhesive granular hydrogel structures over 36 days. This work further highlights the enhanced stability of adhesive granular hydrogels.
To further demonstrate the printability of adhesive granular hydrogels, additional structures were printed. These included a continuous filament in the shape of the famous LOVE statue in Philadelphia, a hollow cylinder (height of 2 cm), and a square pyramid with a height of 1 cm were extrusion printed with adhesive granular hydrogel (Figure 6b). The structures were stable without additional supports and could reversibly be inverted without losing shape stability. Although this is just a proof-of-concept, adhesive granular hydrogels can be extrusion printed without the need for a support bath or any additional post-processing steps (i.e., photocrosslinking, incubation in additional crosslinkers). To further highlight the stability and integrity of the adhesive granular hydrogel, the outline of a star was continuously extruded onto the tops of five microcentrifuge tubes spaced ~3-5 cm apart, where suspended overhangs maintained structural stability in mid-air under the force of gravity without any bottom support (Figure 6b). These features further highlight the utility of the adhesive granular hydrogel.
2.6. Cellular invasion into granular hydrogels
Injectable granular hydrogels can be designed and used as biomaterials for endogenous tissue repair, a promising regenerative medicine approach where host cells invade into the microporous granular hydrogel space to promote tissue healing.[15,34,64,65] Spheroids can be embedded within granular hydrogels and cultured in vitro to gain insight into how granular hydrogel material properties influence cell invasion.[66] We embedded multicellular spheroids consisting of a 2-to-1 ratio of human umbilical vein endothelial cells (HUVECs) and human mesenchymal stromal cells (hMSCs) within control and adhesive granular hydrogel scaffolds (Figure 7a). In addition, the effect of covalently attaching RGD peptide to microgels prior to introducing spheroids was explored.
Figure 7.

In vitro cell invasion in granular hydrogels. a) Schematic overview of embedding multicellular spheroids (HUVECs + hMSCs) in a fragmented granular hydrogel, with or without the addition RGD. b) Representative confocal microscopy images of F-Actin staining, showing spheroid outgrowth into granular hydrogels (top: control, bottom: adhesive), without RGD (left) and with RGD (right) after 3 days. c) Quantification of spheroid coverage area by percent (top) and absolute area in μm2 (bottom). Data is presented as mean ± standard deviation, with a sample size of n ≥ 3. Statistical analysis performed using a one-way ANOVA. ns = no significance, **p<0.01.
After 3 days in culture, spheroids embedded in both granular hydrogel groups containing RGD had significant outgrowth into the surrounding granular hydrogel, as indicated by an increase in both percent spheroid coverage area and total spheroid area (Figure 7b,c). There was no significant difference between spheroid outgrowth in control and adhesive granular hydrogel groups. This demonstrates that the hydrazone inter-particle crosslinking does not hinder cell invasion, further highlighting the cytocompatibility and potential use of the material.
Hydrogels crosslinked with hydrazone bonds have been extensively explored for in vitro cell encapsulation, where the dynamic nature and stress relaxation of the hydrazone bond can be more conducive to cell invasion and migration than a purely covalently crosslinked hydrogel.[67] The dynamic behavior of the inter-particle hydrazone bond in the adhesive granular hydrogels likely contributes to an environment that allows for the cell invasion between microgels. In addition, the tight packing of the vacuum-packed fragmented granular hydrogels used in this study still allows for cell invasion despite lower porosity and pore sizes than loosely packed and spherical granular assembles.[51,66] Overall, these results suggest that both granular hydrogel groups explored herein are favorable for cell invasion upon the addition of RGD into the scaffolds. Combined with the enhanced mechanical properties and shape stability, this further highlights the utility of adhesive granular hydrogels for regenerative medicine applications.
3. Conclusions
In summary, we have developed an injectable granular hydrogel with dynamic covalent inter-particle crosslinks from the formation of hydrazone bonds through aldehyde- and hydrazide-modified microgels. The adhesive interactions between microgels resulted in granular hydrogels with significantly increased mechanical moduli and structural integrity upon compressive loading. Further, the dynamic nature of the hydrazone bond allows for flowability of the adhesive granular hydrogel, though tuning of flow rates is required to ensure enough force is provided to overcome bursting flow through a needle. In addition, we showed that the adhesive granular hydrogel could be extrusion printed into stable structures, without the need for support baths or post-crosslinking steps to support the structures. Lastly, we showed that, upon the addition of RGD, both control and adhesive granular hydrogels allowed cell invasion from embedded cellular spheroids in vitro. Overall, this work highlights the utility of dynamic covalent inter-particle crosslinking to improve granular hydrogel properties for their biomedical use, including in regenerative medicine.
4. Experimental Section
Materials:
Sodium hyaluronic acid (HA, MW = 60 kDa) was purchased from Lifecore Biomedical. Unless otherwise indicated, all other reagents were obtained from Sigma-Aldrich.
Chemical modification of hyaluronic acid (HA):
Norbornene-modified HA (NorHA) was prepared as previously described.[68] Briefly, HA modified with tetrabutylammonium salt (HA-TBA) was dissolved in anhydrous dimethylsulfoxide (DMSO). Dimethyl aminopyridine (DMAP), norbornene carboxylic acid, and ditertbutyl dicarbonate (Boc2O) were added to the DMSO and allowed to react overnight. NorHA product was then dialyzed for 14 days in DI water, frozen at −80 °C, and subsequently lyophilized for 6 days. The polymer was stored at −20 °C until further use.
Hydrazide-NorHA (Hyd-NorHA) synthesis was adapted from previously described protocols.[39] Briefly, 1 g of NorHA and 13 g adipic acid dihydrazide (ADH) were combined in dH2O. A mixture of 1.55 g of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and 1.53 g of hydroxybenzotriazole (HoBT) were separately dissolved in a 1:1 mixture of acetonitrile: DI water (10 mL each) and added dropwise to the reaction. The pH of the mixture was maintained at 6.8 for 4 h, followed by overnight reaction for 18 h. Hyd-NorHA product was then dialyzed for 5 days in DI water, frozen at −80 °C, and subsequently lyophilized for 6 days. The polymer was stored at −20 °C until further use.
Aldehyde-modified NorHA (Ald-NorHA) was synthesized using pendant aldehyde modification adapted from previous protocols.[69,70] First, 1 g of NorHA was dissolved in 233 mL of MES buffer at a pH of 4.75. 3-Amino-1,2-propanediol (910 μL), HoBT (383 mg dissolved in 10 mL of 1:1 acetonitrile: DI water) and EDC (125 μL) were added to the mixture, which was allowed to react for 18 h. The product was dialyzed against DI water for 3 days. Following dialysis, 150 mL of the reaction mixture was added to a 500 mL round bottom flask (RBF) covered with tin foil. A volume of 2.5 mL of 100 mg/mL sodium periodate in DI water was added to the mixture and allowed to react for 10 min. The reaction was quenched by adding 375 μL of ethylene glycol to the RBF and stirring for 2 h. The product was dialyzed against DI water for 2 days, subsequently frozen at −80 °C, and then lyophilized for 6 days. The polymer was stored at −20 °C until further use.
Lyophilized polymers were dissolved in deuterium oxide (D2O) at a concentration of 10 mg/mL and analyzed using 1H-NMR (Bruker NEO400) to determine degree of modification. Norbornene degree of modification was determined to be (25 ± 2)%, (24 ± 1)%, and (19 ± 1)% for NorHA, Ald-NorHA, and Hyd-NorHA, respectively. Degree of hydrazide modification was determined to be (40 ± 5)% for Hyd-NorHA.
To determine the degree of modification of aldehydes for Ald-NorHA, a reductive amination reaction was used as previously described.[71] Briefly, Ald-NorHA (30 mg) was dissolved in 3 mL of acetate buffer (pH 5.2). Separately, 80 mg of tert-butyl carbazate (TBC) was dissolved in 3 mL of the same buffer. The TBC solution was added dropwise to the Ald-NorHA solution and allowed to react for 24 h. The next day, a solution of 30 mg of sodium cyanoborohydride (NaBH3CN) was dissolved in 3 mL of acetate buffer and added dropwise to the mixture. The reaction proceeded overnight. The product was obtained by precipitating in acetone three times, followed by dialysis against DI water for 3 days. The product was frozen at −80 °C, lyophilized for 6 days, and then stored at −20 °C until further use. The authors would like to note that great care should be taken when handling NaBH3CN and a full safety procedure should be developed before performing this reaction. The degree of aldehyde modification for Ald-NorHA was determined by 1H-NMR as stated above by quantifying the carbazate t-Boc peak. The degree of aldehyde modification was determined to be 17% for Ald-NorHA.
Hydrogel precursor solutions and bulk hydrogel fabrication:
The norbornene functional group concentration in hydrogel precursor solutions was standardized at 12 mM, resulting in 1.97 wt.%, 2.89 wt.%, and 2.05 wt.% solutions for NorHA, Hyd-NorHA, and Ald-NorHA solutions, respectively. Each hydrogel precursor solution also contained 6 mM of dithiothreitol (DTT), to match the concentrations of norbornenes with thiols, and 0.05 wt.% Irgacure D-2959 (I2959). Hydrogels were photocrosslinked in bulk upon exposure to UV light (Omnicure S2000 lamp) for 5 min at an intensity of 10 mW/cm2.
Compression testing of bulk hydrogels:
A volume of 50 μL of hydrogel precursor was placed into a cylindrical mold (4.7 mm diameter) and crosslinked with light. Mechanical testing was performed (TA Instruments, DMA Q800) to determine the compressive moduli of the bulk hydrogel samples. Samples were subject to 0.01 N pre-load force and subsequently compressed until 30% strain at a rate of 0.5 N/min. The compressive moduli were calculated as the slope of the stress-strain curve from 5-10% strain.
Adhesion testing of bulk hydrogels:
Thiolated glass microscope slides were prepared by adapting previously described protocols.[72] Glass microscope slides (Fisher Scientific) were submerged in 1 M NaOH for 30 min, subsequently rinsed twice with DI water, and allowed to air dry. Glass slides were incubated in a solution of 0.5 v/v% solution of (3-Mercaptopropyl) trimethoxy silane in toluene for 18 h, then subsequently rinsed with fresh toluene twice and allowed to air dry. The glass slides were incubated at 100°C for 1 h and stored in sealed containers purged with N2 at −20°C until further use.
A volume of 30 μL of hydrogel precursor solution was placed onto a thiolated glass slide, covered with a piece of cover glass and a 1 cm2 photomask, and exposed to UV light for 5 min to undergo gelation and covalent attachment to the glass slide. To assess adhesion, bulk hydrogel films that were covalently attached to thiolated glass slides were placed in contact for 1 h and secured using binder clips. Uniaxial tensile testing was performed (Instron 5848, 5N load cell), where stresses were exerted on hydrogels via the extension of glass slide substrates at a rate of 3 mm/min until failure of the inter-hydrogel interface. The load (N) and the extension (mm) were recorded (Instron Bluehill software), and the adhesive strength was calculated as the maximum load divided by the sample cross-sectional area.
Rheological characterization of bulk hydrogels:
Rheological properties of bulk hydrogels were characterized using an oscillatory shear rheometer (AR2000, TA Instruments) fitted with a 20 mm diameter cone and plate geometry and 27 μm gap. Time sweeps (1% strain, 1 Hz) were performed at 25 °C to characterize bulk gelation upon exposure to light at an intensity of 10 mW/cm2.
Cell viability in bulk hydrogels:
Cells (hMSCs, passage 3) were trypsinized, washed with PBS, and gently mixed with the polymer precursor solution (NorHA, DTT, LAP, and 0.5 mM thiolated RGD [GCGYGRGDSPG, Genscript]) at a final concentration of 10x106 cells/mL. This mixture was then transferred to a cut-off syringe barrel and crosslinked under UV (10 mW/cm2) for 2 minutes. The crosslinked samples were washed with culture media to remove unreacted components and transferred to a well plate with fresh media and cultured at 37°C. To evaluate viability, samples were incubated with Calcein AM and Ethidium homodimer for 30 minutes following the manufacturer’s instructions (Live/Dead kit; ThermoFisher), followed by washing and fixation with 4% PFA. Images (z-stacks) of live and dead cells were acquired on a Leica SP5 upright confocal microscope at randomly chosen regions. Viability was quantified by calculating the ratio of live cells per total cells in maximum projection images of each region using ImageJ.
Microgel fabrication:
To fabricate microgels, an extrusion fragmentation method was used, as previously described.[51] A volume of 1 mL of hydrogel precursor solution was added to a 3 mL syringe (BD). The syringe was exposed to UV light for 5 min. The bulk hydrogel was then extruded by hand through 18G, 23G, 27G, and 30G needles (McMaster-Carr), sequentially. Excess PBS (1 mL) was added after extruding through the 18G needle to reduce the extrusion forces needed. Microgels were then suspended in pure PBS and centrifuged at 10,000 rpm for 3 min, and the supernatant was removed. This washing step was repeated 3 times to remove any unreacted polymer and isolate microgels.
Microgel characterization:
Prior to fabrication, a trace amount of high molecular weight FITC-dextran (2 MDa) was added to hydrogel precursor solutions. Fluorescence microscopy (Olympus BX51) was used to image the microgels after fabrication, and Image J was used to quantify microgel size. Microgel diameters were calculated by treating the area of the microgel as a circle and determining the equivalent circular diameter. To demonstrate the nanoscale porosity within the microgels, un-labelled microgels were incubated in a 1 wt.% FITC-dextran PBS solution overnight at room temperature. Fluorescence microscopy was used to image the microgels after incubation.
Granular hydrogel formation:
Microgels suspended in PBS were jammed by vacuum-driven filtration (Steriflip, 0.22μm-pores, Millipore) to form granular hydrogels. To form non-adhesive (control) granular hydrogels, NorHA microgels were used. To form adhesive granular hydrogels, Hyd-NorHA and Ald-NorHA microgels were combined in suspension in a 50/50 ratio and mixed by vortexing prior to jamming, unless otherwise specified. The adhesive granular hydrogel was visualized using fluorescent microscopy by labelling Hyd-NorHA microgels with FITC-dextran and Ald-NorHA microgels with rhodamine-bovine serum albumin (Rhodamine-BSA).
Confocal imaging and porosity characterization:
Microgels were suspended in a solution of 5 mg/mL FITC-dextran (2 MDa) and jammed by vacuum-driven filtration. Granular hydrogel samples were loaded into a cylindrical PDMS mold and covered with a glass cover slip. An upright confocal microscope (Leica TCS SP5) fitted with a water immersion 25x objective lens was used to visualize the FITC-dextran distributed within the interstitial pores of the granular hydrogels. Volumetric stacks measuring 500 μm x 500 μm x 100 μm were taken at randomly selected regions with a z-spacing of 5 μm between successive stack slices. Pores were analyzed using ImageJ software. Void space (%), pore diameter, and number of pores per area were analyzed as an average of the values in each z-stack.
Compression testing:
A volume of 50 μL of granular hydrogel was secured in a cylindrical mold. Mechanical testing was performed to determine the compressive moduli, failure stress, and failure strain of samples. Granular hydrogels were secured with a 0.001 N pre-load and compressed until failure at a rate of 0.1 N/min. The compressive moduli were calculated as the slope of the stress-strain curve from 5-10% strain. To assess recovery of sample height after loading, samples cyclically underwent compression to 20% strain at a rate of 0.1 N/min followed by return to 0 N force at a rate of −0.1 N/min. Recovery of sample height was determined after each cycle of loading by assessing the sample strain at 0 N. All samples were assessed 1 h post-jamming unless otherwise noted.
pH stability testing:
A volume of 50 μL of granular hydrogel was secured in a cylindrical mold for 1 h. Then, granular hydrogel discs were transferred to PBS baths adjusted to acidic (pH 3.5), neutral (7.6), or basic (pH 11.1) conditions, and allowed to incubate for 1 h. Granular hydrogels were then tested under compression as described above. For long-term stability studies, granular hydrogels were photographed after 1 and 22 days incubation in various pH conditions.
Rheological characterization of granular hydrogels:
Rheological properties of granular hydrogels were assessed using an oscillatory shear rheometer (AR2000, TA Instruments) with a 20 mm parallel steel plate geometry set at a 1 mm gap. Strain sweeps (1 – 500 % strain, 1 Hz) were used to assess shear yielding properties. For shear recovery experiments, low (1%) and high (500%) strains were periodically applied at 1 Hz.
Extrusion force measurements:
Granular hydrogels were loaded into a 3 mL syringe (BD) with an 18G needle (McMaster-Carr). The syringe was loaded onto a syringe pump, and a round force-sensitive resistor (Interlink 402) was placed in between the syringe plunger and the pump. Data was acquired using a setup built with an Arduino Uno Rev3. The syringe pump was extruded at the specified rate for at least 1 min to reach a steady plateau in extrusion force. Voltage output was recorded using the Arduino IDE serial monitor and converted to Newtons using a force calibration curve.
Visualization of contraction flow and particle tracking:
To visualize contraction flow of granular hydrogels through a needle, a polydimethylsiloxane (PDMS, Sylgard 184, Dow Corning) device was constructed. A mold for PDMS devices was designed in SolidWorks and exported as STL files. The mold dimensions were based on the inner dimensions of a 1-mL Luer-Lock syringe (BD) connected to an 18G needle (McMaster-Carr) and printed with Accura ClearVue using stereolithography. The PDMS for the devices was mixed using a 1:9 agent to base ratio, poured over the molds, degassed under vacuum for one and a half hours, and then placed in a 37°C oven for 72 h to cure. Once cured, the device was cut out of the PDMS, cleaned with isopropyl alcohol, and biopsy punches (Integra Miltex) were used to create the inlet channels. The devices were plasma bonded to a glass slide to seal the channels. Tefzel (ETFE) Tubing (Idex) with an inner diameter of 2.36 mm (0.093”) was used to connect a 3 mL syringe (BD) loaded with granular hydrogel to the PDMS device through the inlet channel. A Luer adapter, nut short, and ferrule with a stainless steel ring (Idex) were used to secure the inlet tubing to the syringe.
Granular hydrogels were fabricated such that 5% of microgels were fluorescent due to incorporation of 2 MDa FITC-dextran before fragmentation. The 3 mL syringe was placed in a syringe pump for steady extrusion of the granular hydrogel through the device, and the Arduino-based force sensor setup was used to monitor the extrusion force over time. Particle flow was captured using slow-motion video on an iPhone 8+ at a frame rate of 240 fps. Particle tracking was conducted using Blender 2.91 by recording the pixel position of a tracked particle at each frame and subsequently converting to distance (mm) over time (s).
Extrusion printing:
Structures were printed using a custom-modified 3D FDM printer (Velleman K8200). Standard software was used to generate G-code (Slic3r) based on 3D CAD models (Blender 2.91) and to control hardware (Repetier). Granular hydrogels were loaded into 3 mL syringes with specified needle gauges (18 or 21G) and print speeds (8-16 mm/s) and flow rates (9-25 μL/s) were controlled using Repetier. Star-shaped structures made from adhesive granular hydrogels with encapsulated 2 MDa FITC-dextran were incubated in PBS for 36 days. The mass and shape (photographed areas quantified with ImageJ) of printed structures were recorded throughout the 36-day time to assess mass loss and printed shape fidelity, respectively. To assess FITC-dextran release, the PBS was collected at designated times throughout the study and replaced with fresh PBS. At day 36, the printed structures were degraded with hyaluronidase and a plate reader was used to determine FITC-dextran release.
Cell Invasion In Vitro Assay:
PDMS slabs measuring 20 mm x 15 mm x 1.5 mm (10:1, Sylgard 184, Dow Corning) were fabricated by curing for 2 hours at 60°C. A 10 mm biopsy punch was used to create a through hole, and the PDMS was plasma-bonded onto a glass slide. A piece of fabric mesh (BioDesign Cell MicroSieves, 50 μm pore size) was glued over the PDMS covering the hole to prevent sample loss. This mold setup was immersed in PBS overnight and sterilized under a UV germicidal lamp for 1 hour prior to use. Multicellular spheroids were formed by seeding a cell suspension consisting of HUVECs and hMSCs in the ratio 2:1 onto AggreWell-400 templated agarose microwell array and culturing for 24 hours, as previously described.[17] Cell seeding density was adjusted to target ~ 1000 cells per spheroid. Fragmented microgels (NorHA +/− RGD, Ald-NorHA +/− RGD, Hyd-NorHA +/− RGD) were separately prepared as described above. To create spheroid embedded granular hydrogels, spheroids were harvested from the microwell array and mixed with the appropriate fragmented microgel suspension (~50 spheroids per 0.5 mL of granular hydrogel), jammed using vacuum filtration as described above, transferred to a 1 mL syringe, and injected into the PDMS-based mold through a 19G blunt needle. The setup was transferred to 4-well plates, covered with endothelial cell growth media (EGM-2 bullet kit), and cultured on an orbital rocker at 37°C with daily media changes. After 3 days of culture, samples were washed with PBS and fixed in 4% PFA for 2 hours. Cells were permeabilized with 0.1 % Triton X-100, blocked with 3% horse serum, and incubated with Alexa Fluor 555 Phalloidin for 3 hours at room temperature. Volumetric z-stacks of cell outgrowth from the spheroids were acquired on a Leica S5 upright confocal microscope and maximum projection images were created using ImageJ. Spheroid area was quantified by adjusting threshold and using the particle analyzer function.
Statistical analysis:
Data is presented as mean ± standard deviation, unless otherwise indicated. Statistical analysis was conducted in GraphPad Prism 8 using a one-sided ANOVA and a Tukey’s post hoc comparison, unless otherwise indicated. For all samples, n ≥ 3 for statistical analysis, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns = not significant. There was no pre-processing of data used herein.
Supplementary Material
Acknowledgements
This work was supported by the National Science Foundation (graduate research fellowship to V.G.M., UPenn MRSEC: DMR-1720530), the German Research Foundation (QA 58/1-1 to T.H.Q.) and the National Institutes of Health (R01AR077362). The NSF Major Research Instrumentation Program (award NSF CHE-1827457) and Vagelos Institute for Energy Science and Technology supported the purchase of the NMRs used in this study. The authors would like to thank A. Dhand for assistance with tensile testing, M. Prendergast for assistance with 3D printing and video capture, and R. Pifer for assistance with CAD modeling and particle tracking.
Footnotes
Supporting information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Mandal A, Clegg JR, Anselmo AC, Mitragotri S, Bioeng. Transl. Med 2020, 5, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lee SC, Gillispie G, Prim P, Lee SJ, Chem. Rev 2020, 120, 10834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Uman S, Dhand A, Burdick JA, J. Appl. Polym. Sci 2019, 48668. [Google Scholar]
- [4].Qu J, Zhao X, Ma PX, Guo B, Acta Biomater. 2017, 58, 168. [DOI] [PubMed] [Google Scholar]
- [5].Sun Y, Huang Y, J. Mater. Chem. B 2016, 4, 2768. [DOI] [PubMed] [Google Scholar]
- [6].Hou S, Wang X, Park S, Jin X, Ma PX, Adv. Healthc. Mater 2015, 4, 1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rowland MJ, Parkins CC, McAbee JH, Kolb AK, Hein R, Loh XJ, Watts C, Scherman OA, Biomaterials 2018, 179, 199. [DOI] [PubMed] [Google Scholar]
- [8].Li J, Mooney DJ, Nat. Rev. Mater 2016, 1, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Annabi N, Nichol JW, Zhong X, Ji C, Koshy S, Khademhosseini A, Dehghani F, Tissue Eng. - Part B Rev 2010, 16, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Daly AC, Riley L, Segura T, Burdick JA, Nat. Rev. Mater 2020, 5, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Riley L, Schirmer L, Segura T, Curr. Opin. Biotechnol 2019, 60, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Seymour AJ, Shin S, Heilshorn SC, Adv. Healthc. Mater 2021, 10, 2100644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Feng Q, Li Q, Wen H, Chen J, Liang M, Huang H, Lan D, Dong H, Cao X, Adv. Funct. Mater 2019, 29, 1906690. [Google Scholar]
- [14].Highley CB, Song KH, Daly AC, Burdick JA, Adv. Sci 2019, 6, 1801076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Griffin DR, Weaver WM, Scumpia PO, Di Carlo D, Segura T, Nat. Mater 2015, 14, 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Darling NJ, Xi W, Sideris E, Anderson AR, Pong C, Carmichael ST, Segura T, Adv. Healthc. Mater 2020, 9, 1901391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Qazi T, Wu J, Muir VG, Weintraub S, Gullbrand SE, Lee D, Issadore D, Burdick JA, Adv. Mater 2021, DOI 10.1002/adma.202109194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].de Rutte JM, Koh J, Di Carlo D, Adv. Funct. Mater 2019, 29, 1900071. [Google Scholar]
- [19].Mealy JE, Chung JJ, Jeong HH, Issadore D, Lee D, Atluri P, Burdick JA, Adv. Mater 2018, 30, 1800248. [DOI] [PubMed] [Google Scholar]
- [20].Pruett L, Ellis R, McDermott M, Roosa C, Griffin D, J. Mater. Chem. B 2021, 9, 7132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pfaff BN, Pruett LJ, Cornell NJ, De Rutte J, Di Carlo D, Highley CB, Griffin DR, ACS Biomater. Sci. Eng 2021, 7, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sheikhi A, de Rutte J, Haghniaz R, Akouissi O, Sohrabi A, Di Carlo D, Khademhosseini A, Biomaterials 2019, 192, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Xin S, Wyman OM, Alge DL, Adv. Healthc. Mater 2018, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xin S, Chimene D, Garza JE, Gaharwar AK, Alge DL, Biomater. Sci 2019, 7, 1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Koh J, Griffin DR, Archang MM, Feng AC, Horn T, Margolis M, Zalazar D, Segura T, Scumpia PO, Di Carlo D, Small 2019, 15, 1903147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nih LR, Sideris E, Carmichael ST, Segura T, Adv. Mater 2017, 29, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Caldwell AS, Campbell GT, Shekiro KMT, Anseth KS, Adv. Healthc. Mater 2017, 6, 1700254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Caldwell AS, Rao VV, Golden AC, Bell DJ, Grim JC, Anseth KS, Bioeng. Transl. Med 2021, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Isaac A, Jivan F, Xin S, Hardin J, Luan X, Pandya M, Diekwisch TGH, Alge DL, ACS Biomater. Sci. Eng 2019, 5, 6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jivan F, Alge DL, Adv. Ther 2020, 3, 1900148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Caldwell AS, Rao VV, Golden AC, Anseth KS, Biomaterials 2020, 232, 119725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Truong NF, Lesher-Pérez SC, Kurt E, Segura T, Bioconjug. Chem 2019, 30, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Shin M, Song KH, Burrell JC, Cullen DK, Burdick JA, Adv. Sci 2019, 6, 1901229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hsu RS, Chen PY, Fang JH, Chen YY, Chang CW, Lu YJ, Hu SH, Adv. Sci 2019, 6, 1900520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yu Z, Liu J, Tan CSY, Scherman OA, Abell C, Angew. Chemie - Int. Ed 2018, 57, 3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Widener AE, Bhatta M, Angelini TE, Phelps EA, Biomater. Sci 2021, 9, 2480. [DOI] [PubMed] [Google Scholar]
- [37].Riederer MS, Requist BD, Payne KA, Way JD, Krebs MD, Carbohydr. Polym 2016, 152, 792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Muir VG, Burdick JA, Chem. Rev 2021, 121, 10908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wang LL, Highley CB, Yeh YC, Galarraga JH, Uman S, Burdick JA, J. Biomed. Mater. Res. - Part A 2018, 106, 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Domingues RMA, Silva M, Gershovich P, Betta S, Babo P, Caridade SG, Mano JF, Motta A, Reis RL, Gomes ME, Bioconjug. Chem 2015, 26, 1571. [DOI] [PubMed] [Google Scholar]
- [41].Luo P, Liu L, Xu W, Fan L, Nie M, Carbohydr. Polym 2018, 199, 170. [DOI] [PubMed] [Google Scholar]
- [42].Uman S, Wang LL, Thorn SL, Liu Z, Duncan JS, Sinusas AJ, Burdick JA, Adv. Healthc. Mater 2020, 9, 2000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang Z, He C, Chen X, Mater. Chem. Front 2018, 2, 1765. [Google Scholar]
- [44].Dahlmann J, Krause A, Möller L, Kensah G, Möwes M, Diekmann A, Martin U, Kirschning A, Gruh I, Dräger G, Biomaterials 2013, 34, 940. [DOI] [PubMed] [Google Scholar]
- [45].Wang LL, Liu Y, Chung JJ, Wang T, Gaffey AC, Lu M, Cavanaugh CA, Zhou S, Kanade R, Atluri P, Morrisey EE, Burdick JA, Nat. Biomed. Eng 2018, 1, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sharma PK, Taneja S, Singh Y, ACS Appl. Mater. Interfaces 2018, 10, 30936. [DOI] [PubMed] [Google Scholar]
- [47].Highley CB, Prestwich GD, Burdick JA, Curr. Opin. Biotechnol 2016, 40, 35. [DOI] [PubMed] [Google Scholar]
- [48].Galarraga JH, Kwon MY, Burdick JA, Sci. Rep 2019, 9, 19987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang LL, Chung JJ, Li EC, Uman S, Atluri P, Burdick JA, J. Control. Release 2018, 285, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Truong NF, Kurt E, Tahmizyan N, Lesher-Pérez SC, Chen M, Darling NJ, Xi W, Segura T, Acta Biomater. 2019, 94, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Muir VG, Qazi T, Shen J, Groll J, Burdick J, ACS Biomater. Sci. Eng 2021, 7, 4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chan KMC, Li RH, Chapman JW, Trac EM, Kobler JB, Zeitels SM, Langer R, Karajanagi SS, Acta Biomater. 2014, 10, 2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kablik J, Monheit GD, Yu LP, Chang G, Gershkovich J, Dermatologic Surg. 2009, 35, 302. [DOI] [PubMed] [Google Scholar]
- [54].Sinclair A, O’Kelly MB, Bai T, Hung HC, Jain P, Jiang S, Adv. Mater 2018, 30, 1803087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gehlen DB, Jürgens N, Omidinia-Anarkoli A, Haraszti T, George J, Walther A, Ye H, De Laporte L, Macromol. Rapid Commun 2020, 41, 2000191. [DOI] [PubMed] [Google Scholar]
- [56].Feig VR, Santhanam S, Mcconnell KW, Liu K, Azadian M, Brunel LG, Huang Z, Tran H, George PM, Bao Z, Adv. Mater. Technol 2021, 2100162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Davidson MD, Ban E, Schoonen ACM, Lee MH, D’Este M, Shenoy VB, Burdick JA, Adv. Mater 2020, 32, 1905719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Balakrishnan B, Banerjee R, Chem. Rev 2011, 111, 4453. [DOI] [PubMed] [Google Scholar]
- [59].Hozumi T, Kageyama T, Ohta S, Fukuda J, Ito T, Biomacromolecules 2018, 19, 288. [DOI] [PubMed] [Google Scholar]
- [60].Zhu D, Wang H, Trinh P, Heilshorn SC, Yang F, Biomaterials 2017, 127, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Xin S, Deo KA, Dai J, Krishna N, Pandian R, Chimene D, Moebius RM, Jain A, Han A, Gaharwar AK, Alge DL, Sci. Adv 2021, 7, eabk3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Albert I, Tegzes P, Kahng B, Albert R, Sample JG, Pfeifer M, Barabási AL, Vicsek T, Schiffer P, Phys. Rev. Lett 2000, 84, 5122. [DOI] [PubMed] [Google Scholar]
- [63].Souzy M, Zuriguel I, Marin A, Phys. Rev. E 2020, 101, 060901. [DOI] [PubMed] [Google Scholar]
- [64].Qazi TH, Burdick JA, Biomater. Biosyst 2021, 1, 100008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Fang J, Koh J, Fang Q, Qiu H, Archang MM, Hasani-Sadrabadi MM, Miwa H, Zhong X, Sievers R, Gao DW, Lee R, Di Carlo D, Li S, Adv. Funct. Mater 2020, 2004307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Qazi TH, Muir VG, Burdick JA, ACS Biomater. Sci. Eng 2022, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Rizwan M, Baker AEG, Shoichet MS, Adv. Healthc. Mater 2021, 10, 2100234. [DOI] [PubMed] [Google Scholar]
- [68].Gramlich WM, Kim IL, Burdick JA, Biomaterials 2013, 34, 9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bermejo-Velasco D, Kadekar S, Tavares Da Costa MV, Oommen OP, Gamstedt K, Hilborn J, Varghese OP, ACS Appl. Mater. Interfaces 2019, 11, 38232. [DOI] [PubMed] [Google Scholar]
- [70].Martínez-Sanz E, Ossipov DA, Hilborn J, Larsson S, Jonsson KB, Varghese OP, J. Control. Release 2011, 152, 232. [DOI] [PubMed] [Google Scholar]
- [71].Jia X, Burdick JA, Kobler J, Clifton RJ, Rosowski JJ, Zeitels SM, Langer R, Macromolecules 2004, 37, 3239. [Google Scholar]
- [72].Seo JH, Shin D, Mukundan P, Revzin A, Colloids Surfaces B Biointerfaces 2012, 98, 1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.