

Abstract
Human cytomegalovirus (HCMV) replication requires a metal‐dependent endonuclease at the C‐terminus of pUL89 (pUL89‐C) for viral genome packaging and cleavage. We have previously shown that pUL89‐C can be pharmacologically inhibited with designed metal‐chelating compounds. We report herein the synthesis of a few 8‐hydroxy‐1,6‐naphthyridine subtypes, including 5‐chloro (subtype 15), 5‐aryl (subtype 16), and 5‐amino (subtype 17) variants. Analogs were studied for the inhibition of pUL89‐C in a biochemical endonuclease assay, a biophysical thermal shift assay (TSA), in silico molecular docking, and for the antiviral potential against HCMV in cell‐based assays. These studies identified eight analogs of 8‐hydroxy‐1,6‐naphthyridine‐7‐carboxamide subtypes for further characterization, most of which inhibited pUL89‐C with single‐digit μM IC50 values, and conferred antiviral activity in μM range. TSA and molecular modeling of selected analogs corroborate their binding to pUL89‐C. Collectively, our biochemical, antiviral, biophysical and in silico data suggest that 8‐hydroxy‐1,6‐naphthyridine‐7‐carboxamide subtypes can be used for designing inhibitors of HCMV pUL89‐C.
Keywords: human cytomegalovirus; pUL89-C; endonuclease; inhibitor; 8-hydroxy-1,6-naphthyridine-7-carboxamide
The replication of human cytomegalovirus (HCMV) requires a metal‐dependent endonuclease at the C‐terminus of pUL89 (pUL89‐C), which bears an RNase H / integrase‐like active site, structurally and catalytically. We report herein the synthesis and biochemical, antiviral, biophysical and in silico characterization of a few metal‐binding 8‐hydroxy‐1,6‐naphthyridine subtypes. Synthesized analogs feature structural variations at R1 and R2.
Introduction
Human cytomegalovirus (HCMV) is a β‐herpesvirus that infects the majority of the world population and causes severe diseases in newborns,[ 1 , 2 , 3 ] organ transplant recipients, [4] and human immunodeficiency virus type 1 (HIV‐1) co‐infected individuals.[ 5 , 6 ] Direct‐acting antivirals (DAAs) have been developed for treating HCMV infections (Figure 1), including polymerase inhibitors ganciclovir (GCV, 1), [7] the primary treatment option, [8] cidofovir (CDV, 2), [9] and foscarnet (FOS, 3); [10] terminase inhibitor letermovir (LTV, 4); [11] and the recently approved maribavir (MBV, 5), [12] a viral kinase pUL97 inhibitor. [13] However, polymerase inhibitors 1–3 are associated with dose‐related adverse effects, and LTV is approved only for HCMV prophylaxis in stem cell recipients. In addition, clinical resistance mutations have been observed with all these drugs, including MBV.[ 14 , 15 , 16 , 17 ] Therefore, there remains a pressing need to develop mechanistically novel HCMV drugs.
Figure 1.
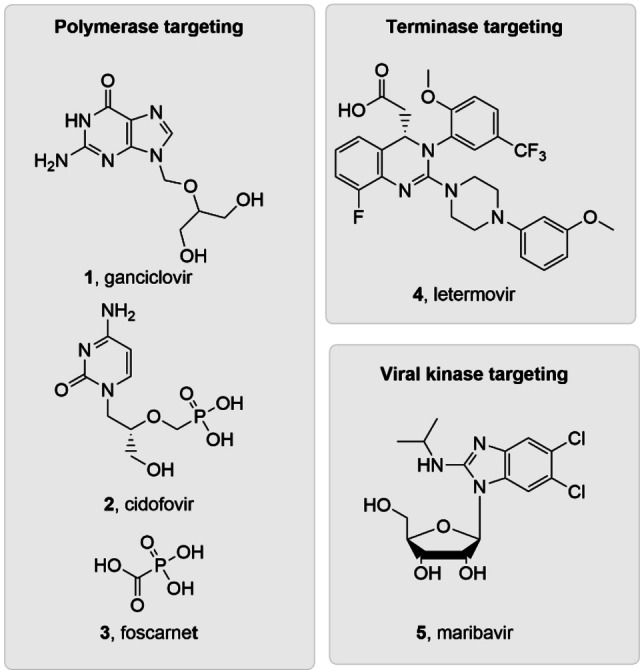
Current FDA‐approved HCMV drugs include polymerase inhibitors 1–3, terminase complex inhibitor 4 and viral kinase inhibitor 5.
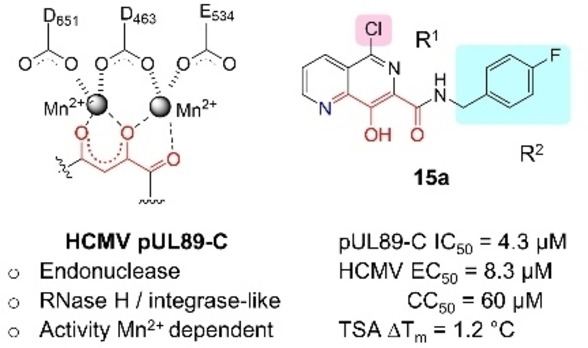
We are particularly interested in the viral terminase complex (TC), [18] a drug target [19] clinically validated by the approval of LTV. The TC minimally consists of a guide protein (pUL56) and an ATPase/endonuclease (pUL89). TC cleaves concatemeric HCMV DNA into unit‐length genomes for DNA packaging, and thus is required for productive viral replication. It is noteworthy that while key LTV resistance mutations have largely been mapped to UL56, [11] pUL89 remains underexplored for HCMV drug discovery. If developed, pUL89‐targeting antivirals will constitute a new drug class, and could be used in combination with current drugs. Interestingly, the C‐terminus of the component protein pUL89 (pUL89‐C) houses a divalent metal‐dependent endonuclease activity (Figure 2, A). [20] The RNase H‐like active site structural fold and the metal‐dependent catalytic mechanism of pUL89‐C are shared by a few other viral metallo enzymatic functions[ 21 , 22 ] we have targeted, including HIV‐1 integrase strand transfer (INST),[ 23 , 24 , 25 ] HIV‐1 reverse transcriptase‐associated ribonuclease H (RNase H),[ 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 ] hepatitis B virus (HBV) RNase H, [35] and hepatitis C virus (HCV) NS5B.[ 36 , 37 ] Along the same line, our prior research has identified and characterized a few metal‐binding pUL89‐C inhibitor types (Figure 2, B): hydroxypyridine carboxylic acid (HPCA, 6),[ 38 , 39 ] dihydroxypyrimidine (DHP) carboxylic acid (7), [40] 6‐arylamino‐3‐hydroxypyrimidine‐2,4‐dione (HPD‐NH, 8), [41] and the 6‐arylthio‐3‐hydroxypyrimidine‐2,4‐dione (HPD‐S, 9). [42] Notably, inhibitor types 6–9 all feature a chelating triad comprising three oxygen atoms for binding two Mn2+ ions (Figure 2, B, red).
Figure 2.
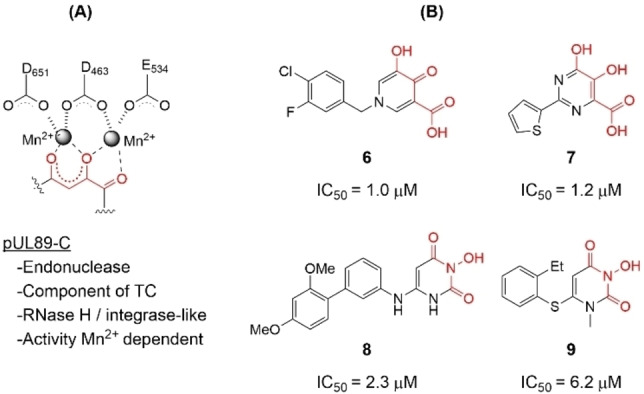
pUL89‐C as an antiviral target. (A) The metal‐dependent endonuclease activity of pUL89‐C is required by the terminase complex (TC), with the active site featuring a DDE motif chelating two Mn2+ ions; (B) Previously reported metal‐binding inhibitor types of HCMV pUL89‐C. All inhibitors entail a chelating triad comprising three O atoms (highlighted in red) and inhibit pUL89‐C in an ELISA biochemical assay with IC50 values in the low μM range.
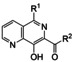
To gain better understanding on pUL89‐C inhibition, we surveyed metal‐chelating chemotypes bearing a chelating triad that may include a non‐oxygen heteroatom, such as nitrogen. Toward this end, we turned our attention to 8‐hydroxy‐1,6‐naphthyridine‐7‐carboxamide which contains one nitrogen atom in its chelating triad (Scheme 1) and has been extensively explored as an important inhibitor type of HIV‐1 INST.[ 43 , 44 , 45 , 46 , 47 , 48 , 49 ] Specifically, we synthesized and tested 5‐chloro (subtype 15), 5‐aryl (subtype 16), and 5‐amino (subtype 17) variants of the 8‐hydroxy‐1,6‐naphthyridine‐7‐carboxamide chemotype, along with an acid analog 14 (Scheme 1).
Scheme 1.

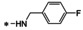
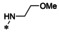
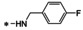
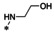
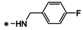
Synthesis of 8‐hydroxy‐1,6‐naphthyridine subtypes 14–17. Reagents and conditions: a) DBU, THF, 40 °C; b) HCl (Conc.), 55 °C, 18 h, 56 %, over‐2‐steps; c) POCl3, MW, 130 °C, 30 min, 85 %; d) R1NH2, AcOH, toluene, 110 °C, 18 h, 43–65 %; e) RNH2, NaOMe, DMSO, 110 °C, 43–52 %; f) ArB(OH)2, Pd(PPh3)4, K2CO3, CH3CN, MW, 150 °C, 30 min, 37–69 %; g) NH2(CH2)2OR2, 150 °C, 18 h, 39–49 %.
Results and Discussion
Chemical synthesis
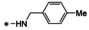
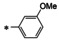
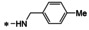
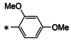
All analogs tested in the current studies were synthesized as depicted in Scheme 1 from quinolinic anhydride (10) through two key intermediates, the 5‐oxo‐naphthyridine 12 and the 5‐chloro‐naphthyridine 13 (Scheme 1). In the synthetic event, the condensation between quinolinic anhydride (10) and methyl 2‐isocyanoacetate (11) afforded the cyclized intermediate ester 12, which was converted into chloride 13 upon treatment with POCl3. Direct aminolysis of ester 13 under two distinct sets of conditions (d and e) produced 15a–d, and 15e–f, respectively. The former subtype (15a–d) was further diversified to deliver 5‐phenyl substituted analogs 16a–e via Suzuki coupling (step f). Amination of 15a–d under heating yielded 5‐amino analogs 17a–b (step g). Interestingly, when the same Suzuki coupling condition was applied to intermediate 13, a hydrolyzed product, acid 14 was obtained to serve as an important variant for structure‐activity relationship (SAR).
Biochemical and antiviral testing
Compounds were evaluated mainly in a biochemical endonuclease assay, and cell‐based antiviral and cytotoxicity assays. All analogs were first screened in these assays at 10 μM. Those with large inhibition % from both the biochemical and antiviral assays, and viability % from the cell‐based assays, were further tested in a dose‐response fashion where IC50, EC50 and CC50 values were calculated. Selected analogs were also studied in a biophysical thermal shift assay (TSA) and molecular docking to assess target binding.
The screening against pUL89‐C was conducted in an endonuclease biochemical assay measuring the cleavage of the DNA substrate. Previously reported pUL89‐C inhibitor 6 was included on each plate as the control. As shown in Table 1, non‐amide subtypes, including 5‐oxonaphthyridine ester (12, inhibition %=14), 5‐chloronaphthyridine ester (13, inhibition %=16), and 5‐phenylnaphthyridine carboxylic acid (14, inhibition %=19) all showed poor inhibition, whereas amide sub‐types, including the 5‐chloro (15a–f), 5‐aryl (16a–e) and 5‐amino analogs (17a–b) generally inhibited pUL89‐C significantly better (Table 1). However, weak inhibition was observed with a few analogs of the amide subtypes, including 5‐chloro amide 15c (inhibition %=15), 5‐aryl amides 16d (inhibition %=17) and 16e (inhibition %=8.2), and 5‐amino amide 17a (inhibition %=25). In parallel to the biochemical screening, all analogs were also screened at 10 μM in an antiviral assay in human foreskin fibroblasts (HFF) using a GFP‐expressing reporter virus, ADCREGFP. Known HCMV drug GCV (1) was used as the control. Similar to the biochemical screening results, non‐amide subtypes generally did not inhibit HCMV, whereas most 5‐chlroro amides, and all 5‐aryl and 5‐amino amides produced significant antiviral activity (inhibition %≥20) (Table 1), though a few 5‐aryl amide analogs (16c, 16d, 16e) also showed substantial cytotoxicity (viability %≤70) at 10 μM. In general, the screening assays revealed a good correlation between the biochemical and antiviral potencies.
Table 1.
Single concentration screening results of the analogs in the biochemical endonuclease assay, and cell‐based antiviral and cytotoxicity assays.
|
| |||||
|---|---|---|---|---|---|
|
Compound |
R1 |
R2 |
Nuclease inhibition % at 10 μM[a] |
Cell‐based assays [at 10 μM][b,c] |
|
|
Inhibition % |
Cell viability % |
||||
|
12 |
OH |
OMe |
14±0.5 |
11±15 |
98±2.3 |
|
13 |
Cl |
OMe |
16±2.5 |
31±2.0 |
96±1.0 |
|
14 |
|
OH |
19±0.1 |
16±018 |
97±0.9 |
|
15a |
Cl |
|
49±4.7 |
32±7.9 |
97±2.3 |
|
15b |
Cl |
|
33±5.3 |
31±14 |
95±1.0 |
|
15c |
Cl |
|
15±2.0 |
6.8±10 |
100±5.7 |
|
15d |
Cl |
|
66±13 |
51±8.2 |
96±1.5 |
|
15e |
Cl |
|
31±8.5 |
5.9±24 |
98±0.5 |
|
15f |
Cl |
|
51±14 |
7.1±7.9 |
96±2.1 |
|
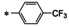
16a |
|
|
71±2.4 |
23±7.1 |
99±2.6 |
|
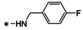
16b |
|
|
52±3.6 |
47±3.9 |
76±1.7 |
|
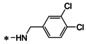
16c |
|
|
69±1.6 |
100±0.1 |
2.5±0.0 |
|
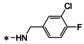
16d |
|
|
17±8.7 |
74±19 |
67±16 |
|
16e |
|
|
8.2±1.9 |
77±7.0 |
68±21 |
|
17a |
|
|
25±6.5 |
21±7.1 |
98±3.7 |
|
17b |
|
|
53±6.5 |
20±16 |
98±4.0 |
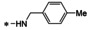
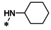
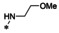
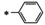
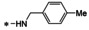
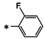
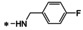
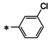
[a] Performed in duplicate and mean is shown. Control compound 6 (10 μM) was included on each plate. A minimum threshold of >80 % inhibition was established as a quality control measurement. [b] Control compound GCV (10 μM) was included on each plate. A minimum threshold of >90 % inhibition by both controls and no toxicity was established as a quality control measurement. [c] Performed in triplicate and mean is shown.
The biochemical dose‐response testing was conducted with all eight compounds showing≥33 % inhibition at 10 μM in the screening assay. Known pUL89‐C inhibitor 6 was used as the control (IC50=0.5 μM). Of these eight analogs (Table 2), six (IC50=1.8‐6.1 μM) inhibited pUL89‐C in single‐digit μM range. Seven of these eight selected compounds were also tested in a dose‐response fashion in the cell‐based antiviral and cytotoxicity assays. Compound 15f was excluded due to insignificant HCMV inhibition at 10 μM. With the exception of the cytotoxic analog 16c (CC50=8.4 μM), all other six analogs conferred moderate yet consistent antiviral activity in μM range (EC50=8.3–31 μM) without considerable cytotoxicity (CC50>50 μM). Representative dose‐response curves for compounds 15a, 15d, 16a, 17b from the endonuclease assay (A), the antiviral assay (B) and cell viability assay (C) are shown in Figure 3.
Table 2.
Dose response testing of selected analogs in the biochemical endonuclease assay, and cell‐based antiviral and cytotoxicity assays.
|
Compound |
IC50 [μM][a] |
Cell‐based assays[b] |
|
|---|---|---|---|
|
EC50 [μM] |
CC50 [μM] |
||
|
15a |
4.3±1.2 |
8.3±0.27 |
60±2.4 |
|
15b |
13±3.8 |
9.7±0.14 |
>50 |
|
15d |
1.8±0.58 |
21±0.95 |
>50 |
|
15f |
5.7±0.39 |
– |
– |
|
16a |
2.8±0.56 |
31±11 |
>100 |
|
16b |
24±5.4 |
24±9.1 |
>60 |
|
16c |
6.1±2.2 |
5.5±0.42 |
8.4±0.20 |
|
17b |
2.4±0.56 |
17±0.75 |
>50 |
|
6 |
0.52±0.13 [c] |
– |
– |
|
1 (GCV) |
– |
1.6±0.17 |
>50 |
[a] Performed in duplicate. Compound 6 was used as control. [b] Performed in duplicate. GCV was used as control. [c] Previously reported value: IC50=1.0 μM [39].
Figure 3.

Biochemical, antiviral and biophysical characterization of selected analogs 15a, 15d, 16a, 17b. A) Representative dose‐response curves from the biochemical endonuclease assay. Known pUL89‐C inhibitor 6 was used as the control; B) dose‐response antiviral and (C) cell viability testing. GCV (1) was used as the control; (D) curves and (E) ΔTm from the TSA. ΔTm was determined independently at least two times with a representative curve shown in (D) and mean ΔTm plus standard deviation shown in (E). Compound 6 was used as the control.
Target protein binding
To assess target binding, pUL89‐C protein melting point was measured upon the binding of four selected compounds (15a, 15d, 16a, 17b) in the TSA. The observed thermal shift (ΔTm) indicates the change of Tm upon compound binding as compared to DMSO control. As shown in the TSA curves (Figure 3, D) all four analogs produced a right shift (ΔTm>0), denoting a stabilizing effect by the compounds on pUL89‐C. The ΔTm values are shown in Figure 3, E. The smaller shifts observed with these naphthyridine analogs when compared to control compound 6 are consistent with the lower potency in the dose‐response endonuclease assay (Table 2).
pUL89‐C features a classic DDE motif (D463, E534, and D651) at its active site which chelates two Mn2+ ions required for the endonuclease activity. [20] A reported ligand‐bound pUL89‐C structure (PDB code: 6EY7 [50] ) allowed us to predict the binding nodes of representative analogs via molecular modeling. Figure 4 depicts the preferred binding modes of 15a and 16a. Chemically, the two analogs differ mainly at the C5 position where 15a and 16a are substituted with a chloro group and a phenyl ring, respectively, resulting in substantially different binding modes (Figure 4). Strikingly, the chelating triad of compound 15a sits too far away from the two Mn2+ ions to form the featured metal binding typically expected from such chemotypes. Instead, 15a binds to the active site via two relatively weak interactions: the π‐π stacking interaction between F466 and the F‐phenyl of the amide moiety (3.3 Å, boxed and red arrow, Figure 4, A); and the π‐cation interaction between K583 and the naphthyridine core (4.6 Å, black arrow, Figure 4, A). The weak interactions amount to a poor docking score (XP GScore=−4.44 kcal/mol). By contrast, compound 16a is predicted to engage with one of the Mn2+ ions (2.4 Å and 3.1 Å, dotted black line, Figure 4, B), and form an H‐bond with T537 (3.3 Å, blue arrow, Figure 4, B). In addition, the π‐π stacking interaction with F466 and the π‐cation interaction with K583 are also predicted (box and black arrow, Figure 4, B). Collectively, these interactions confer a much better docking score (XP GScore=−7.89 kcal/mol). Interestingly, the observed docking scores reflect the same trend as the inhibitory potency from the endonuclease assay (IC50=4.3 μM for 15a vs IC50=2.8 μM for 16a, Table 2), though the molecular modeling predicts relatively weak binding at the active site, which corroborates the small shifts observed from the TSA (Figure 3, D & E).
Figure 4.

Docking poses of representative analogs 15a and 16a into HCMV pUL89 active site (PDB code: 6EY7). (A) Predicted binding mode of 15a (salmon, XP GScore=−4.44 kcal/mol). (B) Predicted binding mode of 16a (cyan, XP GScore=−7.89 kcal/mol). H‐bond and metal chelation are depicted as black dotted lines. Potential π‐π and π‐cation interactions are indicated with arrows. pUL89‐C is rendered cartoon in grey. Key residues and ligands are rendered stick, with nitrogen, oxygen, fluorine and chlorine atoms are colored blue, red, cyan, and green, respectively.
Conclusion
We have synthesized and tested the 5‐chloro (subtype 15), 5‐aryl (subtype 16), and 5‐amino (subtype 17) subtypes of the 8‐hydroxy‐1,6‐naphthyridine‐7‐carboxamide chemotype. The initial endonuclease biochemical and antiviral screening assays identified eight analogs with significant pUL89‐C inhibition and antiviral activity, which were further characterized in a dose‐response fashion in these assays. Most of the selected analogs inhibited pUL89‐C with single‐digit μM IC50 values, and conferred antiviral activity in μM range. Four of these analogs were also tested in the TSA, where small yet significant right shifts were observed, suggesting that they bind and stabilize pUL89‐C. Molecular docking on two selected analogs corroborates pUL89‐C binding at the active site. Overall, these biochemical, antiviral, biophysical and in silico results support 8‐hydroxy‐1,6‐naphthyridine‐7‐carboxamide chemotype as a viable inhibitor type of HCMV pUL89‐C.
Experimental Section
Chemistry
All commercial chemicals were used as supplied unless otherwise indicated. Microwave reactions were carried out in a Biotage Initiator+ w. Robot Eight Microwave System. Compounds were purified via flash chromatography using a Teledyne Combiflash RF‐200 with RediSep columns (silica) and indicated mobile phase. All moisture sensitive reactions were performed under an inert atmosphere of ultra‐pure argon with oven‐dried glassware. 1H and 13C NMR spectra were recorded on a Varian 600 or Bruker 400 MHz spectrometers. Mass data were acquired using an Agilent 6230 TOF LC/MS spectrometer. Procedures for chemical synthesis, analytical characterization data, and original 1H and 13C NMR spectra for all tested compounds are described in Supporting Information.
In vitro pUL89‐C endonuclease assay
pUL89‐C was expressed in E. coli and purified via a C‐terminal histidine tag using nickel affinity chromatography as described.[ 20 , 39 ] The 60‐bp dsDNA substrate labeled with digoxigenin (DIG) and biotin tags (on 5’ and 3’ ends, respectively) has been described in detail.[ 39 , 42 ] pUL89‐C was pre‐incubated with compounds in 1 % DMSO and reaction buffer (3 mM MnCl2, 30 mM Tris pH 8 and 50 mM NaCl) for 10 min at room temperature. The reaction was initiated by 100 nM dsDNA substrate and incubated for 30 min at 37 °C. The addition of EDTA (final concentration 30 mM) terminated the reaction. The samples were incubated in streptavidin coated plates (Pierce Biotechnology, Rockford, IL) at room temperature with gentle shaking for 30 min and then washed three times with 150 μL wash buffer (25 mM Tris, 150 mM NaCl, 0.1 % BSA, and 0.05 % Tween‐20; pH 7.2). Anti‐DIG‐ alkaline phosphatase conjugate antibody (0.15 U/mL) (Roche Applied Sciences, Germany) was added to each well followed by 100 μL of p‐nitrophenylphosphate (1 mg/mL, Sigma‐Aldrich, Saint Louis, MO) as described.[ 39 , 40 ] Absorbance of samples were determined at 405 nm using BioTek Neo 2 plate reader. Compound percent inhibition was obtained by comparing signal at a particular dose to the signal obtained by the control sample where no cleavage was observed (EDTA added). The IC50 was determined using GraphPad Prism 9 software by comparing the percent inhibition for seven serial compound dilutions to the control samples where no cleavage occurs. The IC50 was defined as the compound concentration resulting in a 50 % reduction in substrate signal compared to undigested controls.
HCMV replication assay
HFF cells (ATCC CRL‐2088) were plated in 96‐well plates at 1.75 × 104 cells/well in DMEM supplemented with 10 % fetal bovine serum (FBS), and 1 % penicillin streptomycin (P/S). The next day HCMV ADCREGFP virus (obtained from Wade Bresnahan, University of Minnesota) was added at an MOI of 0.01 in DMEM containing 5 % FBS and 1 % P/S for 2 h. After washing with PBS, test compounds were added to each well (final DMSO concentration 0.5 %) and incubated at 37 °C and 5 % CO2 for 7 days. The cells were lysed to measure GFP fluorescence as an indication of the extent of virus replication as described.[ 39 , 40 ] GFP relative fluorescence units were determined at excitation/emission 495/515 nm in a BioTek Neo2 plate reader. Samples were per‐formed in triplicate and mean values compared to the mean value for the vehicle control (DMSO alone) wells. Mean percent inhibition for each of the nine serial compound dilutions were compared to DMSO‐treated cells and the EC50 calculated using GraphPad Prism software. EC50 represents the compound concentration resulting in a 50 % reduction in GFP fluorescence (virus replication).
Cell viability assay
HFF cells were plated into 96‐well plates at 1.75×104 cells/well. Cells were treated the next day with compound in DMSO as described above in the HCMV replication assay except no virus was added. Cellular viability was determined after 7 days using the MTS‐based tetrazolium reduction assay CellTiter 96 Aqueous Non‐Radioactive cell proliferation assay (Promega) as described. [40] Samples were conducted in triplicate and mean values were compared to that for DMSO alone. Mean percent cell viability for each of nine serial compound dilutions was compared to DMSO‐treated cells and the CC50 calculated using GraphPad Prism software. CC50 was defined as the compound concentration resulting in a 50 % reduction in viability.
pUL89‐C thermal shift assay
Test compounds (20 μM) were combined with 4 μg of purified pUL89‐C in reaction buffer (5 mM MnCl2, 30 mM Tris pH 8 and 50 mM NaCl) for 25 min at room temperature. Sypro Orange Protein Gel Stain (Sigma Aldrich) was added and the assay conducted as previously described.[ 40 , 42 ] All samples were tested in triplicate at a final DMSO concentration of 1 %. Protein Thermal Shift Software Version 1.4 from Applied Biosystems was used to determine melting points and evaluate data. The mean derivative Tm for the DMSO vehicle control was compared to the mean derivative Tm of each test compound to calculate the ▵Tm.[ 50 , 51 ]
Molecular modeling
Docking was performed based on the crystal structure of an inhibitor‐bound HCMV pUL89‐C (PDB code: 6EY7 [50] ). Analogs 15a and 16a were docked using Maestro [52] (Schrödinger; LLC: New York, NY, USA) of the Schrödinger small molecule drug discovery suite 2021–3. [53] Docking protocols used the standard steps including 1) protein preparation using Protein preparation wizard [54] (Schrödinger; LLC: New York, NY, USA), in which only chain A of the tetrameric protein was prepared and minimized using the OPLS3e force field [55] to optimize the hydrogen bonding net‐work and converge the heavy atoms to an RMSD of 0.3 Å; 2) receptor grid generation, where the grid was defined in Maestro around the native α, γ‐diketoacid ligand, covering all the residues within 12 Å; 3) Ligand Preparation, where structures of both analogs were drawn in ChemDraw, saved as sdf, and subjected to LigPrep to generate conformers; and 4) Ligand Docking, where prepared ligands were docked using Glide XP [56] (Glide version 8.2). Docked poses were further processed using PyMOL [57] (Schrödinger; LLC: New York, NY, USA)
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This research was supported by the National Institute of Allergy and Infectious Diseases, the National Institutes of Health, grant number R01AI136982 (to RJG and ZW). We thank the Minnesota Supercomputing Institute (MSI) at the University of Minnesota for providing molecular modeling resources.
E. Jung, R. Majima, T. C. Edwards, R. Soto-Acosta, R. J. Geraghty, Z. Wang, ChemMedChem 2022, 17, e202200334.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Britt W. J., J. Virol. 2017, 91(15), doi: 10.1128/JVI.02392–02316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lazzarotto T., Blazquez-Gamero D., Delforge M. L., Foulon I., Luck S., Modrow S., Leruez-Ville M., Front. Pediatr. 2020, 8, doi:10.3389/fped.2020.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Revello M. G., Gerna G., J. Clin. Virol. 2004, 29(2), 71–83. [DOI] [PubMed] [Google Scholar]
- 4. Limaye A. P., Babu T. M., Boeckh M., Clin. Microbiol. Rev. 2020, 34(1), doi:10.1128/CMR.00043–00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gianella S., Letendre S., J. Infect. Dis. 2016, 214 Suppl. 2, S67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Perello R., Vergara A., Monclus E., Jimenez S., Montero M., Saubi N., Moreno A., Eto Y., Inciarte A., Mallolas J., Martinez E., Marcos M. A., BMC Infect. Dis. 2019, 19(1), doi:10.1186/s12879-12019-14643–12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Faulds D., Heel R. C., Drugs 1990, 39(4), 597–638. [DOI] [PubMed] [Google Scholar]
- 8. Martson A. G., Edwina A. E., Burgerhof J. G. M., Berger S. P., de Joode A., Damman K., Verschuuren E. A. M., Blokzijl H., Bakker M., Span L. F., van der Werf T. S., Touw D. J., Sturkenboom M. G. G., Knoester M., Alffenaar J. W. C., J. Antimicrob. Chemother. 2021, 76(9), 2356–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Clercq E., Clin. Microbiol. Rev. 2003, 16(4), 569–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zeichner S. L., Pediatr. Rev. 1998, 19(12), 399–400. [DOI] [PubMed] [Google Scholar]
- 11. Goldner T., Hewlett G., Ettischer N., Ruebsamen-Schaeff H., Zimmermann H., Lischka P., J. Virol. 2011, 85(20), 10884–10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mullard A., Nat. Rev. Drug Discovery 2021, 21, doi:10.1038/d41573-41021-00208–41572. [Google Scholar]
- 13. Prichard M. N., Rev. Med. Virol. 2009, 19(4), 215–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chou S., Antiviral Res. 2020, 176, 104711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chou S., Song K., Wu J., Bo T., Crumpacker C., J. Infect. Dis. 2020, 10.1093/infdis/jiaa1462. [Google Scholar]
- 16. Chou S. W., Van Wechel L. C., Marousek G. I., J. Infect. Dis. 2007, 196(1), 91–94. [DOI] [PubMed] [Google Scholar]
- 17. Douglas C. M., Barnard R., Holder D., Leavitt R., Levitan D., Maguire M., Nickle D., Teal V., Wan H., van Alewijk D., van Doorn L. J., Chou S., Strizki J., J. Infect. Dis. 2020, 221(7), 1117–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Neuber S., Wagner K., Goldner T., Lischka P., Steinbrueck L., Messerle M., Borst E. M., J. Viorl. 2017, 91(12), doi:10.1128/JVI.02384–02316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ligat G., Cazal R., Hantz S., Alain S., FEMS Microbiol. Rev. 2018, 42(2), 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nadal M., Mas P. J., Blanco A. G., Arnan C., Sola M., Hart D. J., Coll M., Proc. Natl. Acad. Sci. USA 2010, 107(37), 16078–16083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Majorek K. A., Dunin-Horkawicz S., Steczkiewicz K., Muszewska A., Nowotny M., Ginalski K., Bujnicki J. M., Nucleic Acids Res. 2014, 42(7), 4160–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang W., Q. Rev. Biophys. 2011, 44(1), 1–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tang J., Maddali K., Metifiot M., Sham Y. Y., Vince R., Pommier Y., Wang Z., J. Med. Chem. 2011, 54(7), 2282–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Z., Tang J., Salomon C. E., Dreis C. D., Vince R., Bioorg. Med. Chem. 2010, 18(12), 4202–4211. [DOI] [PubMed] [Google Scholar]
- 25. Wu B., Tang J., Wilson D. J., Huber A. D., Casey M. C., Ji J., Kankanala J., Xie J., Sarafianos S. G., Wang Z., J. Med. Chem. 2016, 59(13), 6136–6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang J., Do H. T., Huber A. D., Casey M. C., Kirby K. A., Wilson D. J., Kankanala J., Parniak M. A., Sarafianos S. G., Wang Z., Eur. J. Med. Chem. 2019, 166, 390–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tang J., Kirby K. A., Huber A. D., Casey M. C., Ji J., Wilson D. J., Sarafianos S. G., Wang Z., Eur. J. Med. Chem. 2017, 128, 168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tang J., Liu F., Nagy E., Miller L., Kirby K. A., Wilson D. J., Wu B., Sarafianos S. G., Parniak M. A., Wang Z., J. Med. Chem. 2016, 59(6), 2648–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tang J., Vernekar S. K. V., Chen Y. L., Miller L., Huber A. D., Myshakina N., Sarafianos S. G., Parniak M. A., Wang Z., Eur. J. Med. Chem. 2017, 133, 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vernekar S. K., Liu Z., Nagy E., Miller L., Kirby K. A., Wilson D. J., Kankanala J., Sarafianos S. G., Parniak M. A., Wang Z., J. Med. Chem. 2015, 58(2), 651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vernekar S. K. V., Tang J., Wu B., Huber A. D., Casey M. C., Myshakina N., Wilson D. J., Kankanala J., Kirby K. A., Parniak M. A., Sarafianos S. G., Wang Z., J. Med. Chem. 2017, 60(12), 5045–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang L., Sarafianos S. G., Wang Z., Acc. Chem. Res. 2020, 53(1), 218–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang L., Tang J., Huber A. D., Casey M. C., Kirby K. A., Wilson D. J., Kankanala J., Parniak M. A., Sarafianos S. G., Wang Z., Eur. J. Med. Chem. 2018, 156, 680–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang L., Tang J., Huber A. D., Casey M. C., Kirby K. A., Wilson D. J., Kankanala J., Xie J., Parniak M. A., Sarafianos S. G., Wang Z., Eur. J. Med. Chem. 2018, 156, 652–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huber A. D., Michailidis E., Tang J., Puray-Chavez M. N., Boftsi M., Wolf J. J., Boschert K. N., Sheridan M. A., Leslie M. D., Kirby K. A., Singh K., Mitsuya H., Parniak M. A., Wang Z., Sarafianos S. G., Antimicrob. Agents Chemother. 2017, 61(6), doi: 10.1128/AAC.00245–00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen Y. L., Tang J., Kesler M. J., Sham Y. Y., Vince R., Geraghty R. J., Wang Z., Bioorg. Med. Chem. 2012, 20(1), 467–479. [DOI] [PubMed] [Google Scholar]
- 37. Chen Y. L., Zacharias J., Vince R., Geraghty R. J., Wang Z., Bioorg. Med. Chem. 2012, 20(15), 4790–4800. [DOI] [PubMed] [Google Scholar]
- 38. Kankanala J., Wang Y., Geraghty R. J., Wang Z., ChemMedChem 2018, 13(16), 1658–1663. [DOI] [PubMed] [Google Scholar]
- 39. Wang Y., Mao L., Kankanala J., Wang Z., Geraghty R. J., J. Virol. 2017, 91(3), doi:10.1128/JVI.02152–02116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. He T., Edwards T. C., Xie J., Aihara H., Geraghty R. J., Wang Z., J. Med. Chem. 2022, 65(7), 5830–5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Y., Tang J., Wang Z., Geraghty R. J., Antiviral Res. 2018, 152, 10–17. [DOI] [PubMed] [Google Scholar]
- 42. Wang L., Edwards T. C., Sahani R. L., Xie J. S., Aihara H., Geraghty R. J., Wang Z. Q., Eur. J. Med. Chem. 2021, 222, 10.1016/j.ejmech.2021.113640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Embrey M. W., Wai J. S., Funk T. W., Homnick C. F., Perlow D. S., Young S. D., Vacca J. P., Hazuda D. J., Felock P. J., Stillmock K. A., Witmer M. V., Moyer G., Schleif W. A., Gabryelski L. J., Jin L. X., Chen I. W., Ellis J. D., Wong B. K., Lin J. H., Leonard Y. M., Tsou N. N., Zhuang L. H., Bioorg. Med. Chem. Lett. 2005, 15(20), 4550–4554. [DOI] [PubMed] [Google Scholar]
- 44. Guare J. P., Wai J. S., Gomez R. P., Anthony N. J., Jolly S. M., Cortes A. R., Vacca J. P., Felock P. J., Stillmock K. A., Schleif W. A., Moyer G., Gabryelski L. J., Jin L. X., Chen I. W., Hazuda D. J., Young S. D., Bioorg. Med. Chem. Lett. 2006, 16(11), 2900–2904. [DOI] [PubMed] [Google Scholar]
- 45. Hazuda D. J., Anthony N. J., Gomez R. P., Jolly S. M., Wai J. S., Zhuang L. H., Fisher T. E., Embrey M., Guare J. P., Egbertson M. S., Vacca J. P., Huff J. R., Felock P. J., Witmer M. V., Stillmock K. A., Danovich R., Grobler J., Miller M. D., Espeseth A. S., Jin L. X., Chen I. W., Lin J. H., Kassahun K., Ellis J. D., Wong B. K., Xu W., Pearson P. G., Schleif W. A., Cortese R., Emini E., Summa V., Holloway M. K., Young S. D., Proc. Natl. Acad. Sci. USA 2004, 101(31), 11233–11238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Melamed J. Y., Egbertson M. S., Varga S., Vacca J. P., Moyer G., Gabryelski L., Felock P. J., Stillmock K. A., Witmer M. V., Schleif W., Hazuda D. J., Leonard Y., Jin L. X., Ellis J. D., Young S. D., Bioorg. Med. Chem. Lett. 2008, 18(19), 5307–5310. [DOI] [PubMed] [Google Scholar]
- 47. Thomas M. G., De Rycker M., Wall R. J., Spinks D., Epemolu O., Manthri S., Norval S., Osuna-Cabello M., Patterson S., Riley J., Simeons F. R. C., Stojanovski L., Thomas J., Thompson S., Naylor C., Fiandor J. M., Wyatt P. G., Marco M., Wyllie S., Read K. D., Miles T. J., Gilbert I. H., J. Med. Chem. 2020, 63(17), 9523–9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wall R. J., Moniz S., Thomas M. G., Norval S., Ko E. J., Marco M., Miles T. J., Gilbert I. H., Horn D., Fairlamb A. H., Wyllie S., Antimicrob. Agents Chemother. 2018, 62(8), e00235–00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhuang L. C., Wai J. S., Embrey M. W., Fisher T. E., Egbertson M. S., Payne L. S., Guare J. P., Vacca J. P., Hazuda D. J., Felock P. J., Wolfe A. L., Stillmock K. A., Witmer M. V., Moyer G., Schleif W. A., Gabryelski L. J., Leonard Y. M., Lynch J. J., Michelson S. R., Young S. D., J. Med. Chem. 2003, 46(4), 453–456. [DOI] [PubMed] [Google Scholar]
- 50. Bongarzone S., Nadal M., Kaczmarska Z., Machon C., Alvarez M., Albericio F., Coll M., ACS Omega 2018, 3(8), 8497–8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huber A. D., Pineda D. L., Liu D., Boschert K. N., Gres A. T., Wolf J. J., Coonrod E. M., Tang J., Laughlin T. G., Yang Q., Puray-Chavez M. N., Ji J., Singh K., Kirby K. A., Wang Z., Sarafianos S. G., ACS Infect. Dis. 2019, 5(5), 750–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schrödinger Schrödinger Release 2021–3: Maestro; Schrödinger, LLC: New York, NY, USA, 2021.
- 53.Schrödinger Schrödinger Small-Molecule Drug Discovery Suite 2021–3: Schrödinger, LLC: New York, NY, USA, 2021.
- 54. Sastry G. M., Adzhigirey M., Day T., Annabhimoju R., Sherman W., J. Comput.-Aided Mol. Des. 2013, 27(3), 221–234. [DOI] [PubMed] [Google Scholar]
- 55. Jorgensen W. L., Maxwell D. S., Tirado-Rives J., J. Am. Chem. Soc. 1996, 118(45), 11225–11236. [Google Scholar]
- 56. Friesner R. A., Murphy R. B., Repasky M. P., Frye L. L., Greenwood J. R., Halgren T. A., Sanschagrin P. C., Mainz D. T., J. Med. Chem. 2006, 49(21), 6177–6196. [DOI] [PubMed] [Google Scholar]
- 57.Schrödinger The PyMOL Molecular Graphics System; Version 2.0: Schrödinger, LLC: New York, NY, USA, 2021.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.