

Abstract
Granular synthetic hydrogels are useful bioinks for their compatibility with a variety of chemistries, affording printable, stimuli-responsive scaffolds with programmable structure and function. Additive manufacturing of microscale hydrogels, or microgels, allows for the fabrication of large cellularized constructs with percolating interstitial space, providing a platform for tissue engineering at length scales that are inaccessible by bulk encapsulation where transport of media and other biological factors are limited by scaffold density. Herein, synthetic microgels with varying degrees of degradability are prepared with diameters on the order of hundreds of microns by submerged electrospray and UV photopolymerization. Porous microgel scaffolds are assembled by particle jamming and extrusion printing, and semi-orthogonal chemical cues are utilized to tune the void fraction in printed scaffolds in a logic-gated manner. Scaffolds with different void fractions are easily cellularized post-printing and microgels can be directly annealed into cell-laden structures. Finally, high-throughput direct encapsulation of cells within printable microgels is demonstrated, enabling large-scale 3D culture in a macroporous biomaterial. This approach provides unprecedented spatiotemporal control over the properties of printed microporous annealed particle scaffolds for 2.5D and 3D tissue culture.
Keywords: PEG microgel, 3D printing, hMSC, degradable scaffolds, thioester
Graphical Abstract
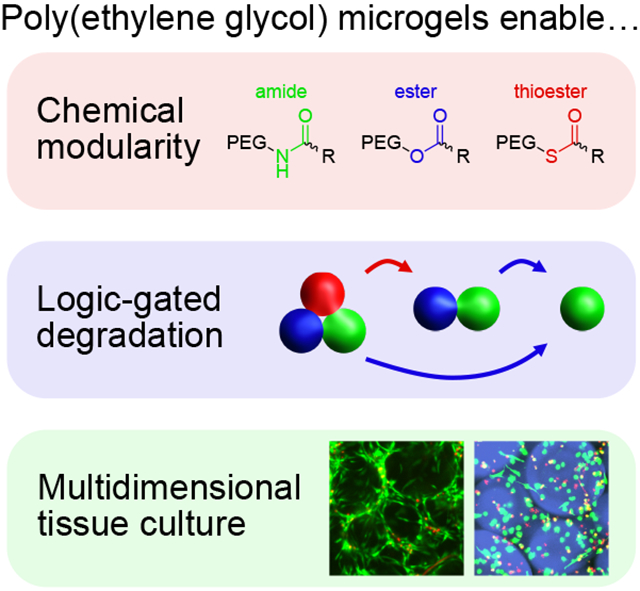
Microgel scaffolds are prepared from chemically modular synthetic polymer precursors, allowing for post-printing tuning of void fraction, logic-gated degradation, and bioprinting of cells encapsulated in 2.5D and 3D culture. Cellularized microgel constructs enable high viability on length scales that are inaccessible by bulk encapsulation, presenting opportunities for additive manufacturing of large format cell-laden biomaterials for tissue engineering and regenerative medicine.
1. Introduction
Additive manufacturing (AM) of large biomaterial-based scaffolds is an emerging method for human-scale tissue engineering. AM entails bottom-up processing, wherein individual components are sequentially or continuously deposited into or generated in a multicomponent scaffold, enabling customizable forms crucial for translational and personalized biomaterials that conform to size and shape requirements. [1] A suite of bioinks have been created for extrusion and inkjet printing, as well as photolithography, providing a range of accessible material compositions for the fabrication of 3D cell culture environments. [2] Large-scale cell-laden scaffolds have been achieved by a variety of 3D printing techniques that overcome diffusion limitations by including perfusable elements, resulting in manufactured tissues with organ-specific cellular organization and function. [3] However, these systems afford no user-directed control over scaffold properties or geometry following initial construction, limiting their ability to recapitulate processes that instruct cellular function and fate through transient biochemical, biomechanical, and biophysical interactions.
To facilitate better mimicry of the dynamic behaviors of native cellular microenvironments, next-generation bioinks composed of adaptable synthetic polymers and chemically modified biopolymers have recently been proposed and developed, resulting in multi-bioink scaffolds with diverse and spatially defined character. [4] In particular, photoactivatable chemistries permit spatiotemporal control over bioink gelation and degradation and have been employed for rapid volumetric bioprinting of high-fidelity large-scale tissue architectures. [5] Moreover, photopatterning is highly complementary to 3D printing, as this combination can yield scaffolds with intricate and biologically relevant microstructural variations in chemical design, physical and mechanical properties, and anisotropy across multiple length scales. [6] The combination of custom chemistries and emerging AM approaches can be used to study biological interactions from single-cells to macro-scale tissues, as well as assemble complex and heterogenous cellularized scaffolds for regenerative medicine. [7]
Microgels (microscale hydrogels) are a class of bioink comprising granular, water-swollen crosslinked polymer networks that can be extruded by particle jamming. [8] These materials are of special interest in biological AM for their versatile production modalities, [9] compartmentalizable reagents, and ability to form configurable annealed scaffolds with both microscale heterogeneity and well-defined superstructure. [10] Incorporation of cells in the interstitial space between connected microgels, often described as 2.5D culture (as cells are in a 3D scaffold without being fully confined by matrix), addresses important cytocompatibility limitations of bulk encapsulation due to pore sizes (microns-millimeters) orders of magnitude larger than the mesh size of polymer networks (nanometers). [11] This interstitial space acts not only as a thoroughfare for nutrient diffusion but also as an important environment for cellular colonization by spreading, growth, and migration, [12] which can collectively be influenced by chemical, mechanical, and topological cues from the microgel substrate. [13] Recently, matrices of fibrous granular hydrogels were demonstrated to undergo programmable contraction and cell-driven reorganization, marking the potential for microgel scaffolds to evolve over time in response to both intrinsic and extrinsic cues. [14]
As cells encapsulated in 2.5D culture are free to interact without intervening matrix, these platforms are excellent candidates for the study of both cell-cell and cell-matrix contacts, which contribute interdependent effects on cellular behavior. [15] Cell-cell junctions are essential for mechanosensing, nascent protein deposition, immunomodulation, and stem cell fate commitment, [16] whereas matrix characteristics and scaffold size of synthetic microgels have dramatic effects on cytoskeletal organization, secretome, and transcriptional activation. [17] Sophisticated approaches to microgel scaffold fabrication have explored macroscopic gradients of degradability and stiffness across large microgel assemblies, illustrating the possibility of spatially heterogeneous void fractions, mechanics, and biochemical functionality. [18] To add additional degrees of control over biological activity, microgels can be loaded with growth factors and nucleic acids to drive desired cellular responses, even in a combinatorial way. [19] Although existing methods to tune these features of microgel scaffolds are useful for defining initial conditions and examining subsequent cellular responses, they do not permit temporal changes associated with so-called 4D cell culture.
To exert temporal control over matrix structure and function, the integration of responsive chemistries into the network is necessary to empower on-demand and orthogonal changes in mechanics, geometry, and signaling. Degradability, stiffening, and reversible presentation of biochemical cues are key features of these systems, as native extracellular matrix (ECM) and soluble factors are constantly secreted and reorganized by local cells. [20] Incorporating various enzyme and chemical-responsive crosslinks in series or parallel allows for chemical logic-gating which meets many of these requirements, enabling programmatic stimuli-responsive delivery of specific proteins and small molecules. [21] A vast array of dynamic chemistries exist and can be readily included in synthetic hydrogels. [22] Photodegradable crosslinks have been developed for sequential release in response to at least three biocompatible, orthogonal wavelengths of light, [23] and versatile thiol-based chemistries can be used to control network integrity and viscoelasticity with pH and radical photoinitiator. [24] In recent examples, polymers and hydrogels with these functionalities have been used to release RNA and control mechanosensitive gene expression, as well as passage cells and organoids in 3D culture. [25] While simple approaches to microgel reversibility have been demonstrated, [26] granular hydrogels with orthogonal or spatiotemporal chemistries have not been developed as printable and highly tunable 4D biomaterials.
Herein, we design chemically modular microgels for logic-gated degradation to achieve user-defined void fractions in 3D-printed scaffolds at selected points in time. Using amide, ester, and thioester-linked poly(ethylene glycol) (PEG) macromers functionalized with norbornene (Figure 1A), the evolution of scaffold porosity is easily modified by mixing various ratios of these components. Ester crosslinks readily decompose in the presence of mild base, and thioester crosslinks degrade upon addition of low-concentration thiolate anion or alkaline media. Microgels are synthesized using submerged electrospray droplet generation and subsequent thiol-ene photopolymerization (Figure 1B). Thioester microgels can be eliminated from the scaffold either in series or in parallel with ester microgels by virtue of semi-orthogonal reaction kinetics between thiol-thioester exchange and thioester hydrolysis; specifically, thioester exchange is several orders of magnitude faster than hydrolysis at neutral pH. [27] Scaffold porosity is tuned by mixing various ratios of degradable and non-degradable microgels, resulting in accessible void fractions ranging from 0.2–0.7. Tuning void space in printed scaffolds allows for rapid colonization by seeded cells. Additional modularity is introduced to this system by incorporating a matrix metalloproteinase (MMP)-labile peptide dithiol crosslinker, which enables passaging of cells out of 2.5D culture using collagenase as another orthogonal degradation stimulus. Finally, the submerged electrospray technique is adapted for high-throughput encapsulation of cells, representing new opportunities for combined 2.5D and 3D co-cultures with a multitude of orthogonal and dynamic chemistries, compositions, and material properties across a variety of length scales.
Figure 1.

Schematized microgel preparation. A) 8-arm 20kDa PEG macromers are functionalized with norbornene with various intervening heteroatom linkers, affording photocrosslinkable hydrogels with a range of degradability. B) Granular hydrogels are prepared by submerged electrospray and subsequent thiol-ene photopolymerization. Microgels are synthesized off-stoichiometry with excess norbornene for post-printing scaffold annealing.
2. Results and Discussion
2.1. Chemically Modular Microgel Fabrication by Submerged Electrospray and UV Photopolymerization
A number of techniques have been developed to prepare microgels, including inverse suspension polymerization, microfluidics, and electrospray. [28] While granular hydrogels made with microfluidics have excellent monodispersity, these devices are low-throughput compared to electrospray, which can generate hundreds if not thousands of microgels per second. Recently, electrospray has been used to rapidly prepare photopolymerized microgels for porous annealed scaffolds and bioprinting, highlighting the utility of this technique for scalable fabrication of granular biomaterials. Moreover, electrospray affords control over particle dimension and can be used to produce anisotropic and asymmetric microgels. [29]
Norbornene-functionalized macromer precursors were prepared using HATU and DCC couplings with eight-arm 20 kDa PEG macromers terminated with amines, thiols, and alcohols to respectively yield amide, thioester, and ester crosslinks. Near-quantitative conversion of macromer end groups was confirmed by 1H NMR (Figure S1) and validated with gel rheology (Figure S2). To quickly manufacture large numbers of microgels for AM, macromers were mixed in phosphate buffered saline (PBS) with 2 mM lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) photoinitiator and four-arm 10 kDa PEG thiol at 5 weight percent total polymer and a final ratio of 0.75 thiol:ene. 800 μL of macromer and photoinitiator solution was loaded into a syringe barrel and electrosprayed at 12 mL/hour through a 23 gauge needle with a 50 mm tip to ring distance into a bath of 4 weight percent ABIL EM90 emulsifier in light paraffin oil with an applied electric field of 7 kV (Figure S3, Movie S1). Following electrospray, microgels were polymerized while dispersed in the continuous phase using 5 minutes of irradiation with 10 mW/cm2 365 nm light. Microgels were pelleted by centrifugation (4000 rcf), washed three times with hexanes, and resuspended in sterile PBS for subsequent use in scaffold construction and bioprinting.
2.2. Microgel Size Characterization and Extrusion Printing of Jammed Microgel Scaffolds
Next, we determined the size distribution of the three microgel populations (Figure 2A) by adding fluorophore to the precursor macromer solution at a final concentration of 20 μM. The average microgel size was approximately 200 μm in all three populations with low polydispersity (Figure 2B), suggesting relative insensitivity of the electrospray process to small variations in chemical structure and flexibility of this technique for microgel preparation, an essential quality for versatile bioink formulations. [30] By decreasing flow rate and increasing voltage, electrospray parameters can be altered to produce smaller microgels. [9b] To confirm printability of packed granular scaffolds, microgels were jammed by extrusion through a tapered conical tip and packed into a cylindrical rubber gasket mold with 8 mm diameter and 2 mm height. Strain yielding behavior of this scaffold was verified by rheology, with logarithmic strain sweeps from 0.01 to 300% strain resulting in crossover of storage and loss moduli near 100% (Figure 2C). Extruded filaments exhibit strong stiction forces even without secondary crosslinking between microgels, allowing for continuous extrusion of microgels without support to lengths up to 35 mm (Figure 2D). Further characterization of the printability of unannealed microgels demonstrated shear-thinning and rapid recovery following high strain (Figure S4).
Figure 2.

Characterization of granular hydrogels as an extrudable bioink. A) Fluorescein-tagged microgels were imaged and sized. Scale bar = 500 μm. B) Size distributions for amide (top), ester (middle), and thioester (bottom) microgels did not vary significantly with material chemistry. Average diameters ranged ~200-230 μm and were relatively monodisperse with PDIs <0.15. Bin widths are ~20 μm. C) Jammed microgels exhibited strain yielding behavior on oscillatory rheological testing near 100% strain, demonstrating printability. D) Microgels were printed by extrusion through an 18 gauge conical tip. Hanging filament tests indicated strong stiction forces between microgels, conferring short-term stability to printed constructs before secondary photocrosslinking. E) Printed scaffold voids were filled with fluorescent solution and imaged for analysis. Scale bar = 500 μm. F) Void fraction analysis revealed similar packing between amide (top), ester (middle), and thioester (bottom) microgels, with average post-printing void fractions near 0.2. Bin widths are ~0.02. G) Post-printing irradiation with mild UV light was used to crosslink network-tethered norbornene groups with free multi-arm PEG thiol, resulting in a ~2-fold increase in scaffold storage modulus, from ~600 Pa to >1 kPa, within seconds of light exposure. H) Annealing by secondary photocrosslinking afforded large, easy-to-handle scaffolds composed of tens of thousands of microgels.
To characterize the void space in printed and annealed scaffolds, microgels were first incubated with an equal volume of 2 mM LAP and four-arm 10 kDa PEG thiol solution (“photostiffening solution”) at a final stoichiometric equivalence of 1:1 thiol:ene for 10 minutes at 4°C. Microgels were pelleted by centrifugation (4000 rcf) and supernatant was aspirated before the pellet was transferred to a syringe barrel and extruded with either pneumatic or screw-drive dispensing at print pressures between 4–40 kPa and write speeds of 1–3 mm/second (Movie S2). After printing, scaffolds were placed under collimated 365 nm light at an intensity of 5 mW/cm2 light for 2 minutes. Scaffolds were subsequently immersed in a 1 mg/mL solution of FITC-dextran (2 MDa) and kept at room temperature for 30 minutes before imaging for pore space analysis (Figure 2E, Movie S3). As with microgel size, void fractions in the scaffolds were consistent across chemistries (Figure 2F) and averaged approximately 0.2. The effect of secondary photocrosslinking on scaffold mechanics was directly measured by in situ rheology, in which a 2 by 8 mm microgel disc pre-swollen with photostiffening solution was irradiated during an oscillatory shear measurement. Scaffold stiffening occurs rapidly upon exposure to low UV light doses, resulting in a ~2-fold increase in storage modulus (Figure 2G). Large annealed microgel scaffolds are mechanically robust and easily handled (Figure 2H), illustrating facile production of highly stable porous constructs for cell culture applications.
2.3. Logic-Gated Modification of Scaffold Void Space
To demonstrate spatiotemporal control over scaffold properties, the three microgel chemistries were labeled with orthogonal fluorophores, printed, and imaged before treatment with semi-orthogonal chemical cues for logic-gated changes in void space over time. Inspired by recent work utilizing thioester elastomers as sacrificial molds to cast complex cell-laden geometries in soft materials, [31] we sought to address existing issues with modifying the void space in printed microgel scaffolds [18a] without compromising structural integrity and exploring degradation stimuli that can be added in the presence of bioprinted cells.
Multicomponent scaffolds were prepared by mixing microgels at a ratio of 3 amide:1 ester:1 thioester before extrusion. Immediately following scaffold annealing, void space was analyzed (Figure 3A) before sequential addition of L-cysteine methyl ester (Figure 3B) and sodium hydroxide (Figure 3C) to achieve intermediate and terminal degraded fractions of 0.2 and 0.4 within the scaffold. Using a random close packing model of circles in MATLAB, [32] an approximation of the multimaterial scaffold was computationally generated using the known size distribution of our microgels (Figure 3D), and the void space in this simulation was analyzed, demonstrating good agreement with experimental results and presenting opportunities for predicting void fractions in granular scaffolds with degradable components. Thioester microgels in these annealed scaffolds degrade in the presence of either exogenous thiolate or base, enabling both series (“YES”-gated, Movie S4A) and parallel (“AND”-gated, Movie S4B) degradation of thioester and ester microgels to tune void fractions to values in excess of 0.6 (Figure 3E), which have as yet been inaccessible with microgels of diameters greater than 100 μm. [18a] Real-time monitoring of microgel erosion (Movies S4A-B) reveals the anticipated bulk degradation of thioester and ester crosslinks in thiolate-buffered and alkaline media, respectively. During degradation, microgels briefly swell but are confined by the scaffold superstructure conferred by annealing; microgels on the exterior surfaces of the scaffolds dissociate from the larger structure, and fluorescence intensity throughout the microgels homogenously dissipates.
Figure 3.

Logic gating of scaffold void fraction. A–C) Void fractions in printed filaments comprised of annealed microgels were tuned by degrading thioester (red) and ester (blue) components while retaining amide (green) microgels. Scale bars = 500 μm. D) A random close packing (RCP) model of jammed microgels simulates changes in void fraction by selective degradation of specific chemistries. E) Quantification of scaffold void fraction as the degraded fraction of microgels increased to 0.2 and 0.4 demonstrated logic-gated changes in porosity in good agreement with the RCP model.
Semi-orthogonal cues are enabled by differences in the relative rates of degradation. For the ester and thioester chemistries, these rates are tuned by a combination of pH and thiol. At neutral pH, thiol-thioester exchange exceeds the rate of thioester hydrolysis by nearly two orders of magnitude; at pH 12, these rates are comparable and ester hydrolysis proceeds rapidly. [27] Importantly, various in vivo and in vitro microenvironments can drastically alter the degradation timescales of amide, ester, and thioester-linked hydrogels. In general, amide-linked gels do not significantly degrade in vitro or in vivo, even on longer timescales (>60 days). Ester-linked thiol-ene hydrogels degrade modestly (retaining ~85% of their initial mass) in vitro on the order of weeks, but dissolve entirely within ~24 hours in vivo. [33] Thioesters hydrolyze more quickly than esters both in vitro and in vivo. [34] New thiol-ene hydrogel chemistries have been recently introduced that further expand the range of accessible degradation times, using carbic anhydride-derived norbornene crosslinkers to form scaffolds that degel in less than 6 hours at neutral pH. [35] Additionally, hydrogels containing both hydrolytically active ester bonds and thioether crosslinks capable of relatively faster retro-Michael reactions were shown to degrade more rapidly compared to ester-only controls, further illustrating how combinations of labile bonds can be used to tune release and degradation kinetics. [36] Collectively, this toolbox of modular chemistries can be applied to thiol-ene microgels to yield annealed granular structures with spatial (i.e., microgel placement) and temporal (i.e., linker chemistry) control over micro- and macroscopic material lifetime in a variety of contexts. Scaffold degradation rates and triggers could be further altered and refined using enzyme, oxidation, or photo-responsive chemistries, as well as more elegant logic-gating combinations of these cues. [37] Additional possibilities exist for mixing chemical functionalities on different length scales, i.e., within individual microgels, between microgels within a single filament (as is demonstrated in this work), or by separating microgel populations in a multi-nozzle printer. These approaches and combinations thereof will permit straightforward construction of vascularized structures and sacrificial molds, as well as modular scaffolds with spatially defined chemical, biological, mechanical, and topological characteristics.
2.4. Cellular Colonization of 4D Printed Microporous Annealed Particle Scaffolds
Following scaffold characterization, human mesenchymal stem cells (hMSCs) were seeded on top of the granular hydrogels prepared with degraded fractions of 0, 0.2, and 0.4 to a final concentration of 200 cells per square millimeter of scaffold surface area. Porous scaffolds are effective platforms for rapid cellular colonization without direct encapsulation, and recent efforts have focused on granular materials with tunable void fractions for cell culture. [18a] In printed scaffolds without post-printing void space modification (Figure 4A), cells are primarily limited to the upper surface of the annealed microgels and do not substantially invade towards the base of the construct after three days of culture. Scaffolds with a degradable fraction of 0.2 (Figure 4B) showed more homogenous organization of cells throughout the pore network, although this distribution remained somewhat skewed towards the top of the structure after 3 days. In contrast, a degradable fraction of 0.4 (Figure 4C) resulted in a distribution of cells that became substantially more concentrated towards the bottom of the scaffold over the same time course. Cells were highly viable (>85% at all timepoints) across all conditions. Moreover, extending culture times to 7 days revealed distinct differences in the population distribution of cells between the three conditions (Figure S5). Specifically, an increased degraded fraction resulted in the population distribution moving further towards the bottom of the scaffold. However, viable fractions in these scaffolds were very similar (all ~80% viable), suggesting that the pore size in scaffolds prepared with microgels on the order of hundreds of microns in size does not significantly affect survival or proliferation. The relative distribution of viable cells in these scaffolds on day 7 remained consistent with day 3, with an increased proportion of dead cells at the bottom of the more densely packed scaffolds with degraded fractions of 0 and 0.2, potentially as a result of restricted nutrient or waste diffusion or cellular mobility. More generally, viability decreased a small amount (approximately 10%) between day 3 and 7 across all conditions, which is likely attributed to limiting media exchange to avoid perturbing the scaffolds. Fine tuning of void fraction can be achieved to greater extents by preparing multimaterial microgels with varied concentration of degradable components or introducing additional chemical functionalities (e.g., photoexchangeable crosslinks) for orthogonal stimuli-responsiveness.
Figure 4.

Seeded scaffolds with post-printing void fraction modifications support highly viable 2.5D cell culture. Void fractions in 4x1 mm disc-shaped scaffolds were tuned by degrading thioester-linked microgels mixed with amide microgels at ratios of 0:100, 20:80, and 40:60. A) Without altering porosity of jammed microgels, multi-millimeter granular constructs were not fully colonized by seeded cells within 3 days. B–C) Scaffolds retaining 80% and 60% initial microgels were rapidly colonized across the depth of the printed construct. Bin widths for all histograms are ~0.01. Labeled black lines in viability histograms represent population-wide average. x-z projection was generated from a confocal z-stack with a slice thickness of 20 μm. Live and dead cells were respectively stained with calcein AM (green) and ethidium homodimer (red). Scale bars in the top panels = 500 μm.
2.5. Annealing of Molded and Printed Cell-Laden Microgel Scaffolds
Jammed amide microgels were incubated with 2x106 cells/mL and loaded by spatula into disc-shaped molds 3 mm in width and 1 mm tall before annealing. Microgel scaffolds were loaded with a high concentration of cells as a significant fraction of cells are lost to interstitial fluid extravasation during the jamming process. [8b] Cells remained viable and relatively well-distributed throughout the scaffold immediately following photocrosslinking through the third day of culture (Figure 5A-C). Furthermore, similar microgel scaffolds were constructed by bioprinting and annealing of jammed particles with cells encapsulated in the void space, resulting in long-term stability for cell culture in high-fidelity 3D structures (Figure 5D, Figure S6). However, print parameters were not optimized for cell viability, and bioprinted scaffolds showed moderate cell death following the extrusion process from drying and shear effects (Figure S7). Taken together, these results (in combination with previous reports of microgel bioprinting) hold promise for eventual opportunities in additive manufacturing of biomaterials for cell clustering and release of specialized cell populations, [38] as well as removal of specific scaffold components to alter cell-matrix signaling and biochemical cues in 2.5D co-culture by incorporating various degradable elements in the microgel formulations. [39]
Figure 5.

Photocrosslinking molded or printed cell-laden amide microgel scaffolds enables long-term stability for 2.5D culture. A–B) Annealing molded microgels maintained scaffold architecture without decreasing cell viability. C) Maximum intensity projection of confocal z-stack demonstrating cell spreading and high viability after 3 days of culture. D) Microgels are easily printed into 3D structures with high fidelity before annealing. Inset: macroscopic view of printed microgel filament. Bin widths for all histograms are ~0.01. Scale bars = 500 μm. Live and dead cells were respectively stained with calcein AM (green) and ethidium homodimer (red).
2.6. MMP-Labile Microgels for Control of Proliferation and Cellular Release
While tuning the norbornene linkers in our thiol-ene microgels afforded semi-orthogonal degradation and tuning of void space, we introduced an additional orthogonal degradation cue by replacing the multi-arm PEG-thiol with an MMP-labile peptide dithiol. Peptide-linked microgels had similar sizing and rheological properties to the PEG-thiol-linked microgels (Figures S8, S9) but resulted in increased cellular proliferation in 2.5D culture by day 7 (Figure S10). Previous bulk hydrogel systems have used these MMP-degradable peptides for 4D cell culture, as treatment with collagenase can act as a temporal degradation cue to remove cells encapsulated in hydrogel scaffolds. [40] After 7 days in 2.5D culture between annealed MMP-labile microgels, cells remained highly viable (Figure 6A) and well-distributed throughout these constructs (Figure 6B). Following a 30-minute wash with 2 mg/mL collagenase D and gentle agitation, cells were passaged from these scaffolds by removing the collagenase wash solution, centrifuging, and resuspending pelleted cells in fresh media. Collagenase treatment did not dramatically affect viability, and cells re-plated on tissue culture dishes were nearly 100% viable after 3 days of culture (Figure 6C-D). Although the semi-orthogonality of the ester and thioester-linked microgel degradation cues afforded logic-gated remodeling of microgel scaffolds in the absence of cells, the required concentrations and exposure times of cysteine and NaOH to effectively degrade these microgels was ultimately cytotoxic. By virtue of the MMP-labile linker serving as the thiol component of these scaffolds, cytocompatible enzyme-responsiveness could be easily incorporated into any of our modular norbornene-based systems, highlighting the facile adaptability of these thiol-ene microgels. As MMP-labile microgel scaffolds had similar sizing and cellular distributions, the dimensional effect of using collagenase as an additional orthogonal logic gate in multifunctional annealed granular materials is expected to be effectively the same as was observed in the ester and thioester-linked systems.
Figure 6.

MMP-labile microgels enable on-demand release of cells in 2.5D culture. A) Maximum intensity projection of confocal z-stack demonstrating cell spreading and B) high viability after 7 days of culture in MMP-labile microgel scaffolds. C) Confocal image of cells passaged from 7 days of 2.5D culture after 3 days of 2D culture. D) Cell viability remains >80% with collagenase D treatment both 1 hour and 3 days following cell release from MMP-labile scaffolds. Scale bars = 250 μm. Live and dead cells were respectively stained with calcein AM (green) and ethidium homodimer (red).
2.7. High-Throughput Cellular Encapsulation in Printable Microgels
Electrospray and electrically induced tip streaming are historically underutilized for the microencapsulation of cells. [28a, 41] Several alternative approaches have been well-developed to this end involving both spontaneous and photocatalyzed reactions in microfluidic systems, [42] with important translational implications of microencapsulated biophysical cues for therapeutic stem cell persistence and immunomodulation in vivo. [43]
As a concluding demonstration of the broad utility of the submerged electrospray technique for biomaterial scaffold fabrication, hMSCs were loaded into the pre-gel solution at a concentration of 106 cells/mL before electrospraying as previously described, producing microgels containing up to dozens of cells (Movie S5) of similar size to cell-free microgels, albeit with greater polydispersity (Figure S11). Microgels were pelleted (1400 rcf, 3 minutes) and directly resuspended into a 10-fold volume of sterile media, gently mixed for five minutes, pelleted again and resuspended to desired concentrations in fresh media. After 24 hours, cell-encapsulating microgels were pelleted once more, incubated with LAP and PEG-thiol as before, printed into scaffolds, and annealed. 3D cell cultures lacking porosity or vasculature are limited by nutrient diffusion at thicknesses >200 μm. [3a] To demonstrate this effect, hMSCs were suspended at a concentration of 106 cells/mL in 20 μL thioester microgel precursor solution supplemented with 1 mM CRGDS fibronectin adhesion peptide and irradiated for four minutes with 365 nm light at 2.5 mW/cm2 to yield cell-laden dome-shaped hydrogels approximately 4 millimeters in diameter and 1.5 millimeters tall. Encapsulated cells were stained with calcein AM and ethidium homodimer over the course of 3 days of culture (Figure 7A), revealing the accumulation of a necrotic core at the center of the gels beyond the ~200 μm limit and severely compromised viability by the third day (Figure 7B). In contrast, annealed printed thioester microgel scaffolds of the same dimensions but with increased permeability by virtue of their interstitial porosity (Figure 7C) support viable culture across these same timepoints (Figure 7D) and even showed signs of cellular spreading and escape from the adaptable thioester microgels. At all timepoints, both bulk and granular scaffolds showed relatively good agreement with a model of homogeneously distributed cells throughout disc-shaped constructs imaged within a square ROI (Figure S12). 3D co-cultures can also be generated using this approach for rapid manufacturing of high-throughput replicates for drug screening or disease modeling, underscoring the potential value of these granular biomaterials in a myriad of fundamental and translational science applications.
Figure 7.

Bulk vs. granular 3D encapsulation of cells. A) Bulk hydrogels supported 3D cell culture with an average viability of 80% across the thickness of the sample. B) After three days of culture, viability in bulk hydrogels decreased to ~60%, with pronounced cell death at the core of the hydrogel where nutrient diffusion is most limited. Inset: view of entire ~4 mm scaffold with visualized region outlined. C) 3D encapsulation of cells within microgels in annealed porous scaffolds afforded comparable viability; most dead cells were poorly encapsulated and retained in the voids between microgels. D) In contrast to bulk hydrogels, viability in granular scaffolds improved over three days of culture to >80% and did not depend on location within the 3D superstructure. Scale bars = 500 μm. Bin widths for all histograms are ~0.01. Labeled black lines in viability histograms represent population-wide average. Dashed blacked lines in population histograms represent modeling of a homogenous distribution of cells throughout the scaffold. Live and dead cells were respectively stained with calcein AM (green) and ethidium homodimer (red).
3. Conclusion
This platform for chemically modular microgel synthesis presents an excellent combination of versatile chemistries, facile 3D extrusion, temporal control, and the capacity to directly encapsulate cells within granular hydrogels in a high-throughput manner. Nevertheless, a number of improvements can readily be made. First, the chemistries and polymer components in this system can be expanded and revised to respond at tunable rates to a multitude of environmental stimuli and irradiation conditions, enabling reversible and repeatable addition and removal of different factors and modifications of scaffold properties. [44] Straightforward chemical modifications can further decrease the stimuli requirements for scaffold erosion, increasing the cytocompatibility of these networks in on-demand release applications. [25b, 34] Improved iterations of our materials can be used to fully elucidate the effects of 2.5D cell culture with 4D biomaterials and, by integrating dynamic chemistries, decouple the roles of stiffness and matrix viscoelasticity on cellular fate commitment, which remain unsolved in these partially confined microenvironments. [45] Moreover, combined 2.5D and 3D co-cultures may be used with bioactive matrix components to sequester growth factors secreted from 3D encapsulated cells for presentation to cells in 2.5D culture on the surface of these microgels, resulting in previously unexplored multidimensional interactions between various cell types with precise microenvironmental control. [46] Beyond sequestration, biopolymer concentration in injectable microgels has also been shown to modulate hMSC angiogenic paracrine signaling.[47] Other studies have assessed hMSC secretion as a function of scaffold-mediated cell clustering and identified optimal pore sizes on the order of 200 μm for facilitating cell-cell contacts spanning the construct to enhance secretion. [17b, 48] Porosity is a known regulator of MSC proliferation and secretion, highlighting the potential value of degradable microgel particles for tuning scaffold void fraction for secretome-engineering applications. [35b, 49] Our system is comprised of relatively large microgels with pores that exceed the optimal sizes that have been previously demonstrated for cell clustering but the size of particles fabricated by submerged electrospray can be tuned by flow rate and voltage, [9b] meaning that this approach could be easily extended to study the effects of void fraction on cell-cell contacts and MSC secretion. Excitingly, recent work used microgel scaffolds to cluster MSCs from ovariectomized vs. control rats to show a pro-resorptive secretory profile in the ovariectomized group as a mechanism for postmenopausal osteoporosis, [50] suggesting new avenues for modeling cadherin-dependent pathological processes in intrinsically porous biomaterials.
Microgels can be engineered to perform a variety of functions, including as biosensors or drug delivery vehicles. [51] In the context of more sophisticated multimaterial printing, [52] these bioinks could enable simultaneous controlled release of bioactive components and sensing of biological processes while manipulating the soluble and ECM-bound, [49, 53] mechanical, [54] topographical, [55] and geometric [56] cues necessary for the proper maturation of complex multicellular environments. Combinations of these tools with genetically engineered stem cells could approach the level of complexity required for true functional mimicry of developmental and homeostatic programming and fully realized reciprocal interactions between synthetic matrix and cellular components. [57] A final implementation of this material is for facilitated wound healing, as porous scaffolds benefit from both enhanced nutrient transport and improved cellular access. [58] Previous demonstrations of microgel scaffolds with programmable lifetimes and immunomodulatory components have shown great utility for directed tissue repair and graft acceptance, [59] which may be further refined by the inclusion of dynamic chemistries for elaborate mechanical and geometric conditioning.
Ultimately, we prepared a variety of microgel-based scaffolds using various techniques amenable to additive manufacturing of biomaterials encapsulating cells in both 2.5D and 3D culture. The degradation profiles of these materials were easily tuned using a straightforward chemical approach incorporating ester, thioester, and amide linkages in photocrosslinkable macromer precursors. Semi-orthogonal cues were demonstrated to erode annealed granular scaffolds for programmable and logic-gated post-printing tuning of scaffold void fraction. These materials enabled rapid cellularization of porous scaffolds, as well as highly viable 2.5D and 3D culture of hMSCs on length scales that are inaccessible by traditional bulk encapsulation. Our study represents a next step in the design and fabrication of 4D granular biomaterial scaffolds for advanced tissue culture and regenerative medicine.
Experimental Section
Macromer synthesis:
Macromer syntheses were performed according to previously described methods. [19b, 25a] All PEGs were purchased from JenKem Technologies and used without further purification. All other reagents were purchased from Sigma Aldrich unless otherwise noted. 2 grams (0.1 mmol) each of 8 arm PEG macromers (20 kDa, tripentaerythritol core) terminated with alcohols, amines, and thiols were added to separate flame dried, argon-filled 100 mL round bottom flasks equipped with magnetic stir bars. PEG amine and PEG alcohol were dried overnight in a vacuum oven held at 80°C. Amine and thiol-terminated macromers were dissolved in a minimal quantity of anhydrous DMF (approximately 10 mL), to which 2 equivalents per macromer arm (1.6 mmol) of 5-norbornene-2-carboxylic acid (221 mg) and HATU (608 mg) were added with an additional 2-5 mL of DMF. After 15 minutes of stirring and purging with argon, 4 equivalents per macromer arm (3.2 mmol) of N-methylmorpholine (324 mg, ~350 μL) was added. Alcohol-terminated macromer was dissolved in anhydrous dichloromethane (10 mL), to which 4 equivalents per macromer arm (3.2 mmol) of 5-norbornene-2-carboxylic acid (442 mg), N,N’-dicyclohexylcarbodiimide (660 mg), pyridine (253 mg, ~260 μL), and 0.5 equivalents per macromer arm (0.4 mmol) of 4-(dimethylamino)pyridine (49 mg) were added while stirring. All reactions were purged with argon for 30 minutes and kept overnight to stir under an argon balloon. The DCC coupling (PEG-norbornene ester) was concentrated under vacuum before being precipitated into an excess volume of ice-cold diethyl ether, while the HATU couplings (PEG-norbornene amide and thioester) were directly precipitated into an excess volume of ice-cold diethyl ether. Following centrifugation and washing of the precipitated macromers, diethyl ether was removed by air drying and macromers were dissolved in Milli-Q water (~25 mL) and dialyzed (Spectrapor tubing, 8 kDa MWCO) against excess volumes of deionized water (~4 L) with water changes approximately every hour for at least 8 cycles. Macromers were left to dialyze for no longer than 36 hours to prevent degradation of the thioester and ester macromers. Following dialysis, macromers were flash frozen in liquid nitrogen and lyophilized until complete drying was achieved (72-96 hours). Dry macromers were characterized in CDCl3 by NMR (Figure S1) to confirm >90% functionalization before being dissolved in sterile PBS to a stock concentration of 20 wt%. 1H NMR (400 MHz, CDCl3, δ): 5.8-6.3 (m, 2H, norbornene alkene), 3.4-3.9 (m, 227H; single arm of 20 kDa 8-arm PEG).
hMSC culture:
Human mesenchymal stem cells (hMSCs) were isolated from human bone marrow (Lonza), and passaged on tissue culture polystyrene (TCPS) using established protocols. Isolated hMSCs were frozen in 80% fetal bovine serum (FBS, Gibco) and 20% DMSO (Gibco). hMSCs were passaged and cultured on TCPS (Grenier) for encapsulation up to passage 5. hMSCs were cultured at 37°C and 5% CO2 in sterile-filtered growth media, which consisted of Low Glucose Dulbecco’s modified Eagle’s Medium (DMEM, Gibco) supplemented with 10% FBS, 50 U/mL penicillin (Gibco), 50μg/mL streptomycin (Gibco), and 1μg/mL amphotericin B (Gibco).
Microgel fabrication by submerged electrospray and UV photopolymerization:
Poly(ethylene) glycol (PEG) microgels were synthesized via submerged electrospraying into a continuous phase. PEG-thioester-norbornene, PEG-ester-norbornene, and PEG-amide-norbornene microgels were all synthesized following the same approach. 8 arm 20 kDa PEG-norbornene (10.25 mM -ene concentration) macromer was mixed with 4 arm 10 kDa PEG-SH (JenKem) macromer to a final ratio of 0.75 thiol:ene groups in sterile phosphate-buffered saline (PBS, Gibco). Photoinitiator LAP (Sigma) was added to a final concentration of 2 mM, and cell adhesive-peptide CRGDS was added to a final concentration of 1 mM. Matrix metalloproteinase (MMP) degradable PEG microgels were synthesized using the same electrospraying approach. 8 arm 20 kDa PEG-amide-norbornene (17.95 mM -ene concentration) macromer was mixed with an MMP degradable peptide crosslinker, KCGPQG↓IWGQCK (Bachem) to a final ratio of 0.75 thiol:ene groups, and a final polymer weight percent of 4.33% in sterile PBS. LAP was added to a final concentration of 2 mM, and CRGDS was added to a final concentration of 1 mM. 800 uL of the polymer precursor solution was transferred to a 3 mL syringe (BD), loaded onto a syringe pump (New Era), and connected to a DC voltage dual-mode power supply (AmazingUnlimited) via a stainless blunt-tip 23 gauge needle (McMaster-Carr). The polymer precursor solution was then electrosprayed out of the stainless blunt-tip needle into the continuous phase, which consists of 4 weight percent ABIL EM 90 (Evonik) in Light mineral oil (Fisherbrand), at a voltage of ~7 kV and a flow rate of 12 mL/hr, at a tip-to-ring distance (in this case, a stainless-steel electrode plate) of 50 mm. A digital microscope (Dinolite) was used to qualitatively assess and record the electrospraying process (Movie S1). The dispersed polymer precursor (Figure S3) was then photopolymerized at 10mW/cm2 for 5 minutes with a UV light source (Omnicure). Photopolymerized microgels were then collected by centrifugation at 1,400 rcf for 3 minutes. Pelleted microgels were transferred to a clean conical tube, washed with hexanes (Macron) 3 times, and transitioned to sterile PBS. Microgels were stored at 4°C until ready for use.
Microgel size characterization:
For size characterization of individual populations of microgels with fluorescence microscopy, PEG-thioester-norbornene, PEG-ester-norbornene, and PEG-amide-norbornene microgels were labeled with 20 μM Alexa Fluor 546 C (Invitrogen), 20 μM Dylight 405 maleimide (Invitrogen), 20 μM and fluorescein-maleimide (Sigma), respectively. PEG-MMP microgels were labeled with 20 μM fluorescein-maleimide for size characterization. Maleimide moieties on each fluorophore react with PEG thiol via thiol-Michael addition. These fluorescent labels were mixed with the respective polymer precursor solution before electrospraying. Fluorescent PEG microgels were suspended in PBS in a multiwell glass-bottom plate and imaged on a Nikon Ti2-E inverted microscope. Images were corrected for bleaching using histogram matching and converted to binary using a local thresholding algorithm. Microgels were selected, segmented, and diameters and roundness were measured in FIJI. PDI is calculated as (standard deviation / mean)2.
Fabrication of bulk hydrogels:
Bulk PEG-thioester-norbonene, PEG-ester-norbornene, PEG-amide-norbonene, and PEG-MMP hydrogels were fabricated by mixing their respective equivalent polymer precursor solutions used for microgel electrospraying. 20 μL of polymer precursor solution was placed between Rain-X treated glass slides spaced with 0.015” (387 μm) gasket and photopolymerized with 6 mW/cm2 365 nm light for 3 minutes to produce a bulk hydrogel 8 mm in diameter.
Mechanical characterization of bulk hydrogels:
Bulk hydrogels were swollen in PBS for 30 minutes before mechanical characterization. Oscillatory shear rheology (Figure S2) was performed on bulk hydrogels using a DHR-3 rheometer (TA instruments) with an 8mm parallel plate geometry. In situ gelation kinetics were measured by oscillatory shear rheology, where the rheometer was fitted with a quartz plate adaptor to enable photoirradiation. 20 μL of polymer precursor solution was pipetted onto the quartz plate, and the solution was photopolymerized 30 seconds after the start of data acquisition with 6 mW/cm2 365 nm light. The storage and loss modulus of all bulk gels were measured using a strain of 1% and a frequency of 1 Hz.
3D printing and secondary photocrosslinking of microgel scaffolds:
PEG microgels were prepared for 3D printing by swelling microgels with photostiffening solution for 10 minutes at 4°C, pelleting by centrifugation at 4,000 rcf for 5 minutes, and aspirating the supernatant. The microgel pellet was loaded into a 1mL syringe with a gel spatula. The syringe was loaded onto either a CELLINK BIO X or Hyrel System 30M 3D printer, and extruded via pneumatic or mechanical actuation through 18 gauge syringe tips, respectively (Movie S2). 20 gauge extrusion nozzles were found to result in overjamming, while 16 gauge tips caused underjamming. [8b] PEG microgel bioinks were extruded into 35 mm glass bottom dishes or multiwell glass bottom plates. The 3D-printed microgel scaffolds were photostiffened with 2.5 mW/cm2 365 nm light for 2.5 minutes, and swollen in PBS.
Void space analysis of microgel scaffolds:
After equilibrium swelling in PBS, the printed microgel scaffolds were incubated with 1 mg/mL 2MDa fluorescein-isothiocyanate dextran in PBS for 30 minutes to visualize the void spaces with fluorescence microscopy (Movie S3). As with microgel sizing, void space images were bleach corrected, thresholded, and analyzed to calculate the relative ratio of overall print area to microgel area.
Mechanical characterization of microgel scaffolds:
PEG microgel granular scaffolds were prepared for granular rheology by swelling microgels with a 1:1 volume ratio of photostiffening solution for 10 minutes at 4°C. The photostiffening solution consists of 4 arm, 10 kDa PEG-SH (2.56 mM thiol concentration) and 2 mM LAP in sterile PBS. Microgels were then pelleted by centrifugation at 4,000 rcf for 5 minutes, and the supernatant aspirated. The microgel pellet was loaded into a 1 mL syringe with a gel spatula. Microgels were prepared using the same approach for all following experiments. Microgels were extruded through an 18 or 20 gauge tapered conical tip (Nordson) into a custom circular mold (8 mm diameter, 2.2 mm height) on the plate of the rheometer. After removing the mold, a continuous flow experiment was performed at a frequency of 1 Hz to determine that the granular viscosity decreases with increasing shear rate (Figure S4A). A cyclic strain sweep was performed by cycling between a low strain of 0.1% and a high strain of 300% at a frequency of 1 Hz, to show the yielding behavior at high strain, followed by recovery at low strain (Figure S4B). A strain sweep was performed from 0.01% strain to 300% strain, at a frequency of 1 Hz. Following the strain sweep, the storage and loss modulus were measured during photostiffening of granular scaffolds using a strain of 1% and a frequency of 1 Hz. The microgel scaffold was photostiffened 20 seconds after the start of data acquisition with 6 mW/cm2 365 nm light.
Degradation of microgel scaffolds:
PEG-thioester-norbornene, PEG-ester-norbornene, and PEG-amide-norbornene microgels were labeled with 20 μM Alexa Fluor 546 C, 20 μM Dylight 405 Maleimide, 20 μM and fluorescein-maleimide respectively to visualize the individual microgel chemistries. Microgels were mixed 3:1:1 amide:thioester:ester by volume (2:1:1 mixtures with a degraded fraction of 0.5 resulted in unstable scaffolds that failed to consistently maintain overall filament integrity), and were prepared for 3D printing by swelling microgels with photostiffening solution for 10 minutes at 4°C, pelleting by centrifugation at 4,000 rcf for 5 minutes, and aspirating the supernatant. The microgel pellet was loaded into a 1 mL syringe with a gel spatula. The mixed microgel populations were printed into a multiwell glass-bottom plate, photostiffened for 2.5 minutes with 2.5 mW/cm2 365 nm light, and swollen in PBS. Logic gated degradation of microgel scaffolds was visualized by fluorescence microscopy (Movie S4). Thioester microgels were degraded by adding 5 mM L-cysteine methyl ester (Sigma), 10% sucrose (Sigma) in PBS, pH 7.4 to the printed constructs. Ester microgels were degraded by adding 20 mM NaOH (Sigma) in PBS, pH 12 to the printed constructs. Microgels were degraded overnight in a humidified chamber at 37°C.
2D seeding of hMSCs on microgel scaffolds:
PEG microgels were prepared for 2D seeding by swelling microgels with photostiffening solution for 10 minutes at 4°C, pelleting by centrifugation at 4,000 rcf for 5 minutes, and aspirating the supernatant. The microgel pellet was loaded into a 1 mL syringe with a gel spatula. PEG microgels were then extruded into rubber gasket molds (3 mm diameter, 1 mm height) in 35 mm glass bottom dishes. PEG microgel molds were photostiffened for 2.5 minutes with 2.5 mW/cm2 365 nm light, and swollen in sterile PBS for 1 hour. Microgel scaffolds of varying void fraction were prepared by mixing PEG-amide microgels with PEG-thioester microgels and degrading the thioester component before seeding cells. Microgels were mixed in ratios (v/v of jammed microgels) of 100:0, 80:20, and 60:40 PEG-amide microgels to PEG-thioester microgels. These mixed microgel scaffolds were then photostiffened the same as for 2D seeding. Photostiffened scaffolds were then swollen with 15 mM L-cysteine methyl ester in sterile PBS, and incubated at 37°C and 5% CO2 for 16 hours to degrade the PEG-thioester microgels, creating PEG-amide microgel scaffolds of varying void fraction. After incubation, the microgel molds were washed and swollen with sterile PBS for 1 hour before seeding with 7 μL of hMSCs (2x105 cells/mL) for a seeding density of 200 cells/mm2, and the microgel molds were incubated for 45 minutes at 37°C and 5% CO2 to facilitate cell adhesion to the microgels. After incubation, 2 mL of fresh hMSC growth media was added to each 35 mm glass bottom dish. Live/dead assay was performed by staining cells with calcein AM (Thermo) / ethidium homodimer (Thermo) diluted 1:1000 in FluoroBrite DMEM (Gibco) to a final concentration of 2 μM for 30 minutes and imaging on either a Nikon Ti2-E inverted fluorescence or a Nikon A1R laser-scanning confocal microscope. EdU assay was performed by adding EdU (Invitrogen) to PEG-amide-norbornene and PEG-MMP scaffolds at a final concentration of 10 μM on day 5. The EdU was incubated with cell scaffolds for 48 hours, and the samples were then fixed on day 7 with 4% formaldehyde (Electron Microscopy Sciences) for 30 minutes at room temperature. Samples were washed three times with PBS for 10 minutes on a shaker plate. Samples were then washed with 3% bovine serum albumin (BSA, Fisher) in PBS, and subsequently permeabilized with 0.1% Triton X-100 in PBS for 1 hour at room temperature. After permeabilization, the scaffolds were washed twice with 3% BSA in PBS, and 0.5 mL of Click-iT (Invitrogen) reaction cocktail was added to each scaffold at room temperature for 30 minutes in the dark. Lastly, each sample was washed with 1 mL of PBS, and stained with DAPI (Sigma) at a concentration of 1:500 for 1 hour at room temperature in the dark. Samples were imaged on a Nikon A1R laser scanning confocal microscope with a 10x objective. Image analysis was performed in FIJI by normalizing the height of scaffolds (which were approximately 1 mm in height and contained in rubber gasket) and counting the number of live and dead cells at each z-height. Population fraction is calculated by the number of cells in a slice divided by the total number of cells in the scaffold, and viable fraction is determined by the number of live cells in a slice divided by the total number of cells in the same slice.
2.5D encapsulation of hMSCs in microgel scaffolds:
PEG microgels were prepared for 2.5D encapsulation by swelling microgels with photostiffening solution for 10 minutes at 4°C, pelleting by centrifugation at 4,000 rcf for 5 minutes, and aspirating the supernatant. The microgel pellet was loaded into a 1mL syringe with a gel spatula. hMSCs were trypsinized and detached from TCPS, resuspended in fresh growth media and resuspended to a concentrated suspension of approximately 3x107 cells/mL. The cell suspension was mixed with the microgel pellet at a volume ratio of 1:30 with respect to microgel pellet volume, for a final cell concentration of 106 cells/mL. The microgel and cell mixture were transferred to a 1mL syringe for printing and extruded from 18-20 gauge tapered conical tips into a 35 mm glass bottom dish or multiwell glass-bottom plate. After extrusion, microgel scaffolds were photostiffened for 2.5 minutes with 2.5 mW/cm2 365 nm light. Fresh hMSC growth media was added and the cell-laden scaffolds were incubated at 37°C and 5% CO2. Scaffold population and viability distributions are calculated in the same manner as 2D-seeded scaffolds.
Collagenase D-induced passaging of hMSCs from MMP microgel scaffolds:
Collagenase-D was added to a final concentration of 2 mg/mL in FluoroBrite DMEM. This solution was added to PEG-MMP microgel scaffolds on day 7, and the plates were incubated on a shaker at 60 rpm for 30 minutes at 37°C and 5% CO2. After incubation, the supernatant containing passaged cells was transferred to a fresh conical tube supplemented with complete media to quench the collagenase-D. The passaged cell supernatant was pelleted and resuspended in fresh growth media, and plated onto a 35 mm TCPS plate for expansion. On day 3 after passaging and plating, hMSCs were stained with calcein AM / ethidium homodimer diluted 1:1000 in FluoroBrite DMEM to a final concentration of 2 μM for 30 minutes, and imaged on a Nikon Ti2-E inverted fluorescence microscope with a 4x objective.
3D encapsulation of hMSCs in bulk hydrogels:
For bulk hydrogel hMSC encapsulation, thioester, ester, and amide hydrogels were fabricated by mixing the equivalent polymer precursor solution used for microgel electrospraying. hMSCs were trypsinized and detached from TCPS, resuspended in fresh growth media and counted for an initial Trypan blue (Invitrogen) live/dead viability assay via automatic cell counter (Countess). Cells were then pelleted and resuspended in the polymer precursor solution to a final concentration of 106 cells/mL. 20 μL of hydrogel precursor and cell suspension were pipetted onto a Rain-X treated glass slide, and photopolymerized for 4 minutes with 2.5 mW/cm2 365 nm light. Bulk hydrogels were then swollen in fresh hMSC growth media and cultured in a multiwell glass bottom plate at 37°C and 5% CO2.
3D encapsulation of hMSCs in microgels by submerged electrospray and UV photopolymerization:
For hMSC encapsulation in microgels by electrospraying, PEG-thioester microgels were fabricated by mixing the equivalent polymer precursor solution used for microgel electrospraying and resuspending hMSCs in 800 uL of polymer precursor solution to a final concentration of 106 cells/mL. The polymer precursor solution includes 20 μM Dylight 405 Maleimide to visualize microgels with fluorescence microscopy. The polymer precursor and cell suspension was transferred to a 3mL syringe, loaded onto a syringe pump, and connected to 23 gauge blunt tip needle and voltage supply for electrospraying. Cell-laden PEG-thioester microgels were electrosprayed into the continuous phase at a voltage of ~7 kV and a flow rate of 12 mL/hr, at a tip-to-ring distance of 50 mm. Cell-laden microgels were photopolymerized with 10 mW/cm2 365 nm light for 3 minutes, and collected from the continuous phase by centrifugation at 1,400 rcf for 3 minutes. Pelleted microgels were then transferred to a clean conical tube and transitioned to fresh hMSC growth media. Cell-laden microgels (Movie S5) were cultured in multiwell glass bottom plates at 37°C and 5% CO2.
3D bioprinting of hMSC-encapsulating microgels:
For bioprinting hMSC-laden microgels, PEG-thioester microgels were prepared using the methods described above. Cell-laden microgels were collected and prepared for 3D bioprinting after being cultured for 24 hours by swelling microgels with a 1:1 volume ratio of photostiffening solution for 10 minutes at 37°C and 5% CO2. Microgels were then pelleted by centrifugation at 3,000 rcf for 5 minutes, and the supernatant was aspirated. The microgel pellet was loaded into a 1 mL syringe with a gel spatula and extruded from 18-20 gauge tapered conical tips into a 35 mm glass bottom dish or multiwell glass-bottom plate; paired scaffolds were prepared without extrusion by loading jammed microgels directly into a mold with a gel spatula before photocrosslinking. PEG microgel scaffolds were photostiffened for 2.5 minutes with 2.5 mW/cm2 365 nm light, swollen in fresh hMSC growth media, and incubated at 37°C and 5% CO2. Live/dead assay was performed as described above.
Statistical Analysis:
Data shown in population histograms was normalized to the total number of cells in culture to calculate the fraction of the total cell count present across the height (or from the centroid) of the scaffold. All experiments were repeated in at least triplicate with a minimum of three technical replicates per condition. Where error bars are shown, data is presented as mean ± SD. Data tabulation, analysis, and plotting were all performed in Python 3.9.
Supplementary Material
Acknowledgements
The authors gratefully acknowledge Dr. Shangjing Xin, Dr. Matthew Davidson, Dr. Joselle McCracken, Grant Bauman, and Dr. Joseph Dragavon for helpful discussions. Funding was provided by NIH R01 DE16523. N.P.S. and B.E.K. contributed equally to this work. Some imaging work was performed at the BioFrontiers Institute Advanced Light Microscopy Core (RRID: SCR_018302) on a Nikon A1R microscope supported by NIST-CU Cooperative Agreement award number 70NANB15H226.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Connor E. Miksch, Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO, 80303, USA; The BioFrontiers Institute, University of Colorado Boulder, Boulder, CO, 80303, USA
Nathaniel P. Skillin, Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO, 80303, USA; The BioFrontiers Institute, University of Colorado Boulder, Boulder, CO, 80303, USA; Medical Scientist Training Program, School of Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO, 80045, USA
Bruce E. Kirkpatrick, Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO, 80303, USA; The BioFrontiers Institute, University of Colorado Boulder, Boulder, CO, 80303, USA; Medical Scientist Training Program, School of Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO, 80045, USA
Grace K. Hach, Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO, 80303, USA
Varsha V. Rao, Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO, 80303, USA; The BioFrontiers Institute, University of Colorado Boulder, Boulder, CO, 80303, USA
Timothy J. White, Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO, 80303, USA
Kristi S. Anseth, Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO, 80303, USA; The BioFrontiers Institute, University of Colorado Boulder, Boulder, CO, 80303, USA
References
- [1].a) Guzzi EA; Tibbitt MW, Adv. Mater 2020, 32, 1901994; [DOI] [PubMed] [Google Scholar]; b) Zhang YS; Haghiashtiani G; Hübscher T; Kelly DJ; Lee JM; Lutolf M; McAlpine MC; Yeong WY; Zenobi-Wong M; Malda J, Nature Reviews Methods Primers 2021, 1, 75. [Google Scholar]
- [2].a) Bejoy AM; Makkithaya KN; Hunakunti BB; Hegde A; Krishnamurthy K; Sarkar A; Lobo CF; Keshav DVS; G D; S DD; Mascarenhas S; Chakrabarti S; Kalepu SRRD; Paul B; Mazumder N; Bioprinting 2021, 24, 00176; [Google Scholar]; b) Ouyang L; Armstrong JPK; Lin Y; Wojciechowski JP; Lee-Reeves C; Hachim D; Zhou K; Burdick JA; Stevens MM, Sci Adv 2020, 6, abc5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Kang HW; Lee SJ; Ko IK; Kengla C; Yoo JJ; Atala A, Nat. Biotechnol 2016, 34, 312; [DOI] [PubMed] [Google Scholar]; b) Kolesky DB; Homan KA; Skylar-Scott MA; Lewis JA, PNAS 2016, 113, 3179; [DOI] [PMC free article] [PubMed] [Google Scholar]; c)Skylar-Scott MA; Uzel SGM; Nam LL; Ahrens JH; Truby RL; Damaraju S; Lewis JA, Sci Adv 2019, 5, aaw2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Hull SM; Brunel LG; Heilshorn SC, Adv. Mater 2021, 2103691; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hull SM; Lindsay CD; Brunel LG; Shiwarski DJ; Tashman JW; Roth JG; Myung D; Feinberg AW; Heilshorn SC, Adv. Funct. Mater 2021, 31, 2007983; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Morgan FLC; Moroni L; Baker MB, Adv. Healthc. Mater 2020, 9, 1901798. [DOI] [PubMed] [Google Scholar]
- [5].a) Bernal PN; Delrot P; Loterie D; Li Y; Malda J; Moser C; Levato R, Adv. Mater 2019, 31, 1904209; [DOI] [PubMed] [Google Scholar]; b) Lee M; Rizzo R; Surman F; Zenobi-Wong M, Chem. Rev 2020, 120, 10950. [DOI] [PubMed] [Google Scholar]
- [6].a) Bretherton RC; DeForest CA, ACS Biomater. Sci. Eng 2021, 7, 01549; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Primo GA; Mata A, Adv. Funct. Mater 2021, 31, 2009574; [Google Scholar]; c) Yang C; DelRio FW; Ma H; Killaars AR; Basta LP; Kyburz KA; Anseth KS, PNAS 2016, 113, E4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Bertassoni LE, Adv. Mater 2021, 2101321; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Daly AC; Davidson MD; Burdick JA, Nat. Commun 2021, 12, 753; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang P; Abate AR, Adv. Mater 2020, 32, 2005346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Highley CB; Song KH; Daly AC; Burdick JA, Adv. Sci 2019, 6, 1801076; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xin S; Deo KA; Dai J; Pandian NKR; Chimene D; Moebius RM; Jain A; Han A; Gaharwar AK; Alge DL, Sci Adv 2021, 7, abk3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Muir VG; Qazi TH; Shan J; Groll J; Burdick JA, ACS Biomater. Sci. Eng 2021, 7, 4269; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xin S; Chimene D; Garza JE; Gaharwar AK; Alge DL, Biomater. Sci 2019, 7, 1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Amstad E; Harrington MJ, Philos. Trans. A Math. Phys. Eng. Sci 2021, 379, 0338; [DOI] [PubMed] [Google Scholar]; b) Caldwell AS; Aguado BA; Anseth KS, Adv. Funct. Mater 2020, 30, 1907670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vernerey FJ; Lalitha Sridhar S; Muralidharan A; Bryant SJ, Chem. Rev 2021, 121, 11085. [DOI] [PubMed] [Google Scholar]
- [12].a) Chaudhuri O; Cooper-White J; Janmey PA; Mooney DJ; Shenoy VB, Nature 2020, 584, 535; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Parisi C; Qin K; Fernandes FM, Philos. Trans. A Math. Phys. Eng. Sci 2021, 379, 0344. [DOI] [PubMed] [Google Scholar]
- [13].Iturriaga L; Van Gordon KD; Larrañaga-Jaurrieta G; Camarero-Espinosa S, Adv. NanoBio. Res 2021, 2100068. [Google Scholar]
- [14].Davidson MD; Prendergast ME; Ban E; Xu KL; Mickel G; Mensah P; Dhand A; Janmey PA; Shenoy VB; Burdick JA, Sci Adv 2021, 7, 8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Barcelona-Estaje E; Dalby MJ; Cantini M; Salmeron-Sanchez M, Adv. Healthc. Mater 2021, 10, 2002048. [DOI] [PubMed] [Google Scholar]
- [16].a) Cosgrove BD; Mui KL; Driscoll TP; Caliari SR; Mehta KD; Assoian RK; Burdick JA; Mauck RL, Nat. Mater 2016, 15, 4725; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Loebel C; Mauck RL; Burdick JA, Nat. Mater 2019, 18, 883; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xie AW; Zacharias NA; Binder BYK; Murphy WL, Stem. Cells. Transl. Med 2021, 10, 1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Caldwell AS; Campbell GT; Shekiro KMT; Anseth KS, Adv. Healthc. Mater 2017, 6, 1700254; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Caldwell AS; Rao VV; Golden AC; Anseth KS, Biomaterials 2020, 232, 119725; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xin S; Wyman OM; Alge DL, Adv. Healthc. Mater 2018, 7, 1800160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Seymour AJ; Shin S; Heilshorn SC, Adv. Healthc. Mater 2021, 10, 2100644; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xin S; Dai J; Gregory CA; Han A; Alge DL, Adv. Funct. Mater 2019, 30, 1907102; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xin S; Gregory CA; Alge DL, Acta Biomater. 2020, 101, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Jivan F; Alge DL, Adv. Ther 2019, 3, 1900148; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jivan F; Yegappan R; Pearce H; Carrow JK; McShane M; Gaharwar AK; Alge DL, Biomacromolecules 2016, 17, 3516; [DOI] [PubMed] [Google Scholar]; c) Kurt E; Segura T, Adv. Healthc. Mater 2021, 2101867; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Isaac A; Jivan F; Xin S; Hardin J; Luan X; Pandya M; Diekwisch TGH; Alge DL, ACS Biomater. Sci. Eng 2019, 5, 6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Huang G; Li F; Zhao X; Ma Y; Li Y; Lin M; Jin G; Lu TJ; Genin GM; Xu F, Chem. Rev 2017, 117, 12764; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang K; Feng Q; Fang Z; Gu L; Bian L, Chem. Rev 2021, 121, 11149. [DOI] [PubMed] [Google Scholar]
- [21].a) Badeau BA; Comerford MP; Arakawa CK; Shadish JA; DeForest CA, Nat. Chem 2018, 10, 251; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gawade PM; Shadish JA; Badeau BA; DeForest CA, Adv. Mater 2019, 31, 1902462; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ruskowitz ER; Comerford MP; Badeau BA; DeForest CA, Biomater. Sci 2019, 7, 542. [DOI] [PubMed] [Google Scholar]
- [22].Podgorski M; Fairbanks BD; Kirkpatrick BE; McBride M; Martinez A; Dobson A; Bongiardina NJ; Bowman CN, Adv. Mater 2020, 32, 1906876. [DOI] [PubMed] [Google Scholar]
- [23].Pelloth JL; Tran PA; Walther A; Goldmann AS; Frisch H; Truong VX; Barner-Kowollik C, Adv. Mater 2021, 33, 2102184. [DOI] [PubMed] [Google Scholar]
- [24].a) Brown TE; Carberry BJ; Worrell BT; Dudaryeva OY; McBride MK; Bowman CN; Anseth KS, Biomaterials 2018, 178, 496; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Brown TE; Marozas IA; Anseth KS, Adv. Mater 2017, 29, 1605001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Carberry BJ; Rao VV; Anseth KS, Ann. Biomed. Eng 2020, 48, 2053; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ishikawa S; Kamata H; Chung U.-i.; Sakai T; RSC Adv. 2021, 11, 23637; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mavila S; Culver HR; Anderson AJ; Prieto TR; Bowman CN, Angew. Chem 2021, 10741; [DOI] [PubMed] [Google Scholar]; d) Yavitt FM; Brown TE; Hushka EA; Brown ME; Gjorevski N; Dempsey PJ; Lutolf MP; Anseth KS, Adv. Mater 2020, 32, 1905366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Kesselman LR; Shinwary S; Selvaganapathy PR; Hoare T, Small 2012, 8, 1092; [DOI] [PubMed] [Google Scholar]; b) Mealy JE; Chung JJ; Jeong HH; Issadore D; Lee D; Atluri P; Burdick JA, Adv. Mater 2018, 30, 1705912. [DOI] [PubMed] [Google Scholar]
- [27].Bracher PJ; Snyder PW; Bohall BR; Whitesides GM, Orig Life Evol Biosph 2011, 41, 399. [DOI] [PubMed] [Google Scholar]
- [28].a) Young CJ; Poole-Warren LA; Martens PJ, Biotechnol Bioeng 2012, 109, 1561; [DOI] [PubMed] [Google Scholar]; b) Fraser AK; Ki CS; Lin C-C, Macromol. Chem. Phys 2014, 215, 507. [Google Scholar]
- [29].He T; Jokerst JV, Biomater. Sci 2020, 8, 5555. [DOI] [PubMed] [Google Scholar]
- [30].Lee SC; Gillispie G; Prim P; Lee SJ, Chem. Rev 2020, 120, 10834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Carberry BJ; Hergert JE; Yavitt FM; Hernandez JJ; Speckl KF; Bowman CN; McLeod RR; Anseth KS, Biofabrication 2021, 13, 044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xu T, GitHub 2021. [Google Scholar]
- [33].Hunckler MD; Medina JD; Coronel MM; Weaver JD; Stabler CL; Garcia AJ, Adv. Healthc. Mater 2019, 8, 1900371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Konieczynska MD; Grinstaff MW, Acc Chem Res 2017, 50, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].a) Lin F-Y; Lin C-C, ACS Macro Letters 2021, 10, 3; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jiang Z; Lin F-Y; Jiang K; Nguyen H; Chang C-Y; Lin C-C, Materials Science and Engineering: C 2022. 112712. [Google Scholar]
- [36].Kharkar PM; Kloxin AM; Kiick KL, J Mater Chem B 2014, 2 (34), 5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shahi S; Roghani-Mamaqani H; Talebi S; Mardani H, Polym. Chem 2022, 13, 161. [Google Scholar]
- [38].Wong SW; Lenzini S; Giovanni R; Knowles K; Shin JW, Acta Biomater. 2021, 133, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lewis KJ; Tibbitt MW; Zhao Y; Branchfield K; Sun X; Balasubramaniam V; Anseth KS, Biomater. Sci 2015, 3, 821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].a) Gonzalez Rodriguez A; Schroeder ME; Grim JC; Walker CJ; Speckl KF; Weiss RM; Anseth KS, FASEB J 2021, 35, e21382; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gonzalez Rodriguez A; Schroeder ME; Walker CJ; Anseth KS, APL Bioeng 2018, 2, 046104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pan Z; Bui L; Yadav V; Fan F; Chang HC; Hanjaya-Putra D, Biomater. Sci 2021, 9, 3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].a) Kamperman T; Henke S; Visser CW; Karperien M; Leijten J, Small 2017, 13, 1603711; [DOI] [PubMed] [Google Scholar]; b) Truong VX; Li F; Forsythe JS, ACS Macro Letters 2017, 6, 657. [DOI] [PubMed] [Google Scholar]
- [43].a) Choe G; Park J; Park H; Lee JY, Polymers 2018, 10, 997; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Guerzoni LPB; Rose JC; Gehlen DB; Jans A; Haraszti T; Wessling M; Kuehne AJC; De Laporte L, Small 2019, 15, 1900692; [DOI] [PubMed] [Google Scholar]; c) Mao AS; Ozkale B; Shah NJ; Vining KH; Descombes T; Zhang L; Tringides CM; Wong SW; Shin JW; Scadden DT; Weitz DA; Mooney DJ, PNAS 2019, 116, 15392; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wong SW; Tamatam CR; Cho IS; Toth PT; Bargi R; Belvitch P; Lee JC; Rehman J; Reddy SP; Shin JW, Nat. Biomed. Eng 2022, 6, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].a) Shieh P; Hill MR; Zhang W; Kristufek SL; Johnson JA, Chem. Rev 2021, 121, 7059; [DOI] [PubMed] [Google Scholar]; b) Jiang Y; Chen J; Deng C; Suuronen EJ; Zhong Z, Biomaterials 2014, 35, 4969; [DOI] [PubMed] [Google Scholar]; c) Fairbanks BD; Macdougall LJ; Mavila S; Sinha J; Kirkpatrick BE; Anseth KS; Bowman CN, Chem. Rev 2021, 121, 6915; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Bongiardina NJ; Long KF; Podgórski M; Bowman CN, Macromolecules 2021, 54, 8341. [Google Scholar]
- [45].a) Zonderland J; Moroni L, Biomaterials 2021, 268, 120572; [DOI] [PubMed] [Google Scholar]; b) Wang Z; Zhu X; Cong X, Biofabrication 2021, 13, 035051. [DOI] [PubMed] [Google Scholar]
- [46].Gionet-Gonzales M; Casella A; Diloretto D; Ginnell C; Griffin KH; Bigot A; Leach JK, Adv Healthc Mater 2021, 10, 2101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Thomas D; Fontana G; Chen X; Sanz-Nogues C; Zeugolis DI; Dockery P; O'Brien T; Pandit A, Biomaterials 2014, 35, 8757. [DOI] [PubMed] [Google Scholar]
- [48].Qazi TH; Tytgat L; Dubruel P; Duda GN; Van Vlierberghe S; Geissler S, ACS Biomater Sci Eng 2019, 5, 5348. [DOI] [PubMed] [Google Scholar]
- [49].Wechsler ME; Rao VV; Borelli AN; Anseth KS, Adv Healthc Mater 2021, 10, 2001948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Rao VV; Wechsler ME; Cravens E; Wojda SJ; Caldwell AS; Kirkpatrick BE; Donahue SW; Anseth KS, Acta Biomater 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].a) Shin DS; Tokuda EY; Leight JL; Miksch CE; Brown TE; Anseth KS, ACS Biomater. Sci. Eng 2018, 4, 378; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen J; Huang K; Chen Q; Deng C; Zhang J; Zhong Z, ACS Appl. Mater. Interfaces 2018, 10, 3929. [DOI] [PubMed] [Google Scholar]
- [52].Skylar-Scott MA; Mueller J; Visser CW; Lewis JA, Nature 2019, 575, 330. [DOI] [PubMed] [Google Scholar]
- [53].Caldwell AS; Rao VV; Golden AC; Bell DJ; Grim JC; Anseth KS, Bioeng Transl Med 2021, 6, e10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wong SW; Lenzini S; Cooper MH; Mooney DJ; Shin JW, Sci Adv 2020, 6, aaw0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Riegert J; Topel A; Schieren J; Coryn R; Dibenedetto S; Braunmiller D; Zajt K; Schalla C; Rutten S; Zenke M; Pich A; Sechi A, PLoS One 2021, 16, e0257495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].a) Kilian KA; Bugarija B; Lahn BT; Mrksich M, PNAS 2010, 107, 4872; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dudaryeva OY; Bucciarelli A; Bovone G; Huwyler F; Jaydev S; Broguiere N; al-Bayati M; Lütolf M; Tibbitt MW, Adv. Funct. Mater 2021, 2104098. [Google Scholar]
- [57].a) Shariati K; Ling AS; Fuchs S; Dillenburger B; Liu W; Ma M, Adv. Funct. Mater 2021, 2108057; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Skylar-Scott MA; Huang JY; Lu A; Ng AHM; Duenki T; Liu S; Nam LL; Damaraju S; Church GM; Lewis JA, Nat Biomed Eng 2022, 6, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Correa S; Grosskopf AK; Lopez Hernandez H; Chan D; Yu AC; Stapleton LM; Appel EA, Chem. Rev 2021, 121, 11385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].a) Headen DM; Woodward KB; Coronel MM; Shrestha P; Weaver JD; Zhao H; Tan M; Hunckler MD; Bowen WS; Johnson CT; Shea L; Yolcu ES; Garcia AJ; Shirwan H, Nat. Mater 2018, 17, 732; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Griffin DR; Weaver WM; Scumpia PO; Di Carlo D; Segura T, Nat. Mater 2015, 14, 737; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Griffin DR; Archang MM; Kuan CH; Weaver WM; Weinstein JS; Feng AC; Ruccia A; Sideris E; Ragkousis V; Koh J; Plikus MV; Di Carlo D; Segura T; Scumpia PO, Nat. Mater 2021, 20, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.